

Abstract
A model linear oppositely charged polyelectrolyte complex exhibits phase separation upon heating consistent with lower critical solution temperature (LCST) behavior. The LCST coexistence curves narrow with increasing monovalent salt concentration (Cs) that reduces the polymer concentration (Cp) in the polymer-rich phase. The polymer-rich phase exhibits less hydration with increasing temperature, while an increase in Cs increases the hydration extent. The apparent critical temperature, taken as the minimum in the phase diagram, occurs only for a narrow range of Cs. Mean field theory suggests an increasing Bjerrum length with temperature can lead to an electrostatic-driven LCST; however, the temperature dependence of the Flory−Huggins interaction parameter and solvation effects must also be considered.
Graphical Abstract
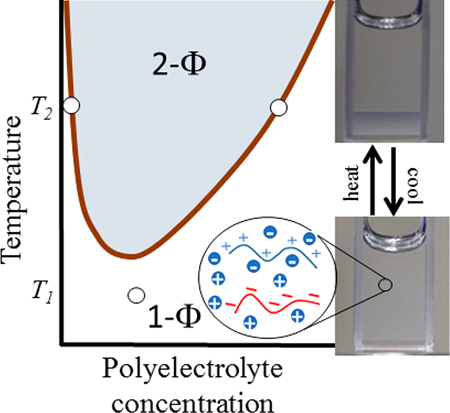
Associative phase separation of oppositely charged macromolecules (coacervation) leads to a dense polymer-rich liquid phase in coexistence with a dilute polymer phase. The miscibility envelope is typically shown as salt concentration (Cs) versus total polymer concentration (Cp). The Voorn−Overbeek (VO) extension of the Flory−Huggins (FH) theory qualitatively predicts several aspects of the coacervation phase behavior.1 Theoretical2–7 and simulation8–10 models that go beyond the VO theory are able to describe the molecular mass dependence11,12 of the Cs−Cp phase boundary, effects of mixing the ratio of oppositely charged polyelectrolytes,13 and an unequal salt partitioning between coexisting phases12 on the Cs−Cp plane.
Coacervation reflects a complex interplay between charge and van der Waals interactions, and solvation (hydration) in addition to the microscopic details of the association equilibria between oppositely charged polymers that couples to the entropic release of counterions. The complexation between oppositely charged polymer segments may lead to polar segments.14 The topological distribution of such ion pairs, chemical identity, stoichiometry, and host−guest length are additional important details.15–17 It is known that aqueous soluble polar polymers, such as polyethylene glycol, display temperature-dependent hydration that naturally leads to lower critical solution temperature behavior. Mean field models for polymer solvation have found agreement with data for polyethylene glycol.18,19 Such specific interactions and polymer solvation go beyond solvent continuum descriptions.20,21 For instance, the standard FH theory does not predict a lower critical solution temperature through an inverse temperature dependence22 of the FH parameter (χ) that characterizes the van der Waals interaction energy between the polymer and the solvent.23,24 Generally, χ > χc determines the state of miscibility, where χc is the critical condition that defines the critical temperature in the original FH theory or a critical salt concentration in the VO theory.
Predictions for the critical conditions in polyelectrolyte complex solutions were summarized in a recent Macromolecules Perspective by Muthukumar.14 Due to the presence of charges, additional parameters beyond van der Waals govern the interactions: the Coulomb strength parameter defined by the ratio of the Bjerrum length (lB) to charge separation distance (l), along with the Debye screening length (κ−1). Therefore, the role of temperature (T) appears in χ, lB, and κ−1, complicating the analysis of the phase behavior in polyelectrolyte complexes.
Considering this theoretical work, does associative phase separation of oppositely charged polyelectrolytes exhibit lower critical, upper critical, or mixed solution temperatures? This Letter experimentally demonstrates phase separation upon heating in mixtures of common strong polyelectrolytes25 (Figure 1a): potassium-poly(styrenesulfonate) (KPSS) and poly(diallyl dimethylammonium bromide) (PDADMAB) with added KBr salt. Charge stoichiometric (1:1) mixtures of KPSS:PDADMAB with an initial polymer concentration (Cpi) of 0.3 mol/L exhibit stable one-phase (1-Φ) at CKBr ≥ 2.15 mol/L at 20 °C (ref 26), with CKBr defined by the concentration of polyelectrolyte counterions and added KBr. For CKBr ≤ 2.10 mol/L, the mixture exhibits two phases (2-Φ). The separate polyelectrolytes are stable and soluble in aqueous KBr solutions, so phase separation occurs due to charge complexation.
Figure 1.

Temperature dependence of the transmittance for (a) KPSS/PDADMAB prepared at CKBr = 2.15 mol/L and Cpi = 0.3 mol/L. The cloud point temperature Tcp is shown by the vertical dotted line and photos at (b) 15 °C and (c) 32 °C. The laser scanning confocal micrograph in (d) shows coacervate droplets in the turbid phase. (e) Shows the coexisting phases at 32 °C.
Figure 1a shows the laser light transmittance % (power transmitted/incident power) as a function of T for CKBr = 2.15 mol/L and Cpi = 0.3 mol/L. The transmittance plateaus between 5 and 10 °C, indicating a stable single phase, Figure 1b. However, the transmittance smoothly decreases before the sharp drop at the cloud point temperature (Tcp). At T > Tcp, the cloudy sample, Figure 1c, exhibits micron-sized spherical droplets, as shown by confocal microscopy, Figure 1d. The meniscus between the coexisting phases forms over time with rate depending upon the sample composition and temperature. The kinetics of this process can be accelerated by centrifugation to form clear phases. This clear dense phase exhibits a viscoelastic liquid response (G′′ > G′) with terminal relaxation exhibiting storage and loss moduli scaling as G′ ∼ ω1.7 and G′′ ∼ ω1, respectively, from frequency (ω) sweep small-amplitude rheology.26 These results show that macroscopic phase separation occurs upon heating. Further heating within the two-phase region again causes the transparent coacervate to become turbid, rather than an instantaneous movement of the meniscus. This is due to the formation of micron-sized polymer-poor droplets, as measured by ultrasmall angle neutron scattering and confocal microscopy (Supporting Information).
Figure 2a summarizes the effect of CKBr on Tcp at fixed Cpi = 0.3 mol/L. Mixtures prepared at CKBr = 1.75 mol/L and Cpi = 0.3 mol/L remain phase separated down to 0 °C. Tcp initially increases slowly, then sharply increases at higher CKBr values. The cloud point shifts by 50 °C, with a 0.25 mol/L change in salt concentration. Figure 2b shows a 20 °C increase in Tcp for varied Cpi from 0.01 to 0.3 mol/L at fixed CKBr = 2.15 mol/L. All samples exhibit two phases above the Tcp.
Figure 2.

Cloud-point temperature vs (a) KBr concentration with fixed Cpi = 0.3 mol/L and (b) initial polymer concentration with fixed CKBr = 2.15 mol/L. Dotted lines are guide to the eye. Error bars represent one standard deviation estimated from measured uncertainty in the sampling micropipette volumes and molar extinction coefficient. While error bars are shown, they may be smaller than the symbols used.
Figure 2a can be rationalized with respect to the Cs−Cp diagrams at a fixed temperature. The 1-Φ is reached with an increase in salt concentration. Similarly, Figure 2b at a fixed temperature, an increase in polymer concentration at fixed Cs leads to 1-Φ. However, the trends of temperature are the key finding. Figure 2b has the appearance of a lower critical solution temperature. Tcp corresponds to the point where the kinetics of phase separation becomes observable, rather than an exact thermodynamic phase boundary. A kinetics study demonstrates that these observations are not strongly dependent upon the heating rate from 0.05 to 0.4 °C/min. These cloud point data were further examined as coexistence curves by analysis of the liquid−liquid phase separated solutions.
The coexistence curves in Figure 3 on the T−CPSS plane for CPSS = 0.15 mol/L (Cpi = 0.3 mol/L) at three salt concentrations are consistent with lower critical solution behavior. As expected, above the apparent critical temperature, taken as the minimum, the solution phase separates into polymer-poor and polymer-rich phases. At CKBr = 1.75 mol/L (Figure 3a), the apparent critical temperature was not experimentally accessible. The ratio of the CPSS between the coacervate and supernatant is ≈60 at 5 °C for Figure 3a. The concentration of PSS generally increases in the dense phase as temperature increases. This implies the extent of solvation (hydration) decreases as temperature is increased. The shape of the phase boundaries depends on the salt concentration. The PSS concentration in the supernatant increases with increasing CKBr, while it decreases in the dense phase. This increased hydration of the coacervate is consistent with the observations of Schlenoff et al.27 The coexistence curves appear more symmetric at the highest CKBr in Figure 3c.
Figure 3.

Coexistence curves for KPSS/PDADMAB mixture at CKBr = 1.75, (b) 2.0, and (c) 2.05 mol/L showing the two-phase (2Φ) and one-phase (1Φ) region. The initial PSS concentration was 0.15 mol/L (circles) and 0.3 mol/L (squares) that phase separates into the supernatant (open symbols) and concentrated phases (filled symbols) at the given temperature. Measured Tcp at CPSS = 0.15 mol/L (▲) and 0.3 mol/L (▼) are indicated in (b) and (c). Error bars represent one standard deviation estimated from the uncertainty in the sampling micropipette volumes. While error bars are shown, they may be smaller than the symbols used.
The initial polymer concentration does influence the equilibrium phase concentrations. Figure 3b includes initial CPSS = 0.3 mol/L (Cpi = 0.6 mol/L) that phase separates into a slightly higher coacervate concentration. Since the coacervate concentration is dependent on the initial polymer concentration, one cannot treat the system as pseudobinary and draw a tie line. This is not surprising, since the polycation concentration and salt concentrations are also required for the multicomponent analysis. We note that a nonequal salt partitioning on the Cs−Cp plane12 may be responsible for these observations. As shown in the Supporting Information, at fixed temperature and salt concentration an initial polymer concentration increased by a factor of 12 changes the coacervate CPSS by 37%.
What molecular parameters are responsible for phase separation upon heating? By considering one concentration within the 2-Φ region of the Cs−Cp diagram, the phase envelope must shift down to recover 1-Φ upon cooling. The cloud point temperatures (Figure 2a) and critical temperature (Figure 3) increase with increasing added salt. The key temperature-dependent parameters are χ, the Bjerrum length, lB = e2/(4πεεokBT), and the Debye screening length (κ−1) for monovalent salt, κ−1 = (4π lBCs)−1/2, where e is the elementary charge, κB is Boltzmann’s constant, and εo and ε are the permittivity of free space and dielectric constant of the medium, respectively. Therefore, changing temperature has implications in the binary interaction parameter and treatment of ionic interactions, when considering mean field theory.
Thermodynamic models for polyelectrolyte complexation were recently reviewed.28 Salehi and Larson4 considers a mean field treatment of ion condensation upon polyelectrolytes, ion pairing of charged segments, and charge regulation as reversible equilibria that lead to laws of mass action. They find that a decrease in the ion pairing equilibrium constant (Kip) lowers the Cs−Cp phase envelope (Figure 4 in ref 4). Therefore, the temperature-dependence of the ion pairing equilibrium constant can lead to upper critical or lower critical solution temperature behavior depending upon the Gibbs free energy of formation. However, as they point out, counterion condensation and ion pairing are inter-related, since they have common ions. Independent measurements of these equilibrium constants are challenging since enthalpic contributions may arise from intermediate steps, such as solvation.
Fu and Schlenoff25 estimated the enthalpy of complexation by isothermal titration calorimetry as exothermic for KPSS and PDADMAB at fixed salt concentration. If the standard free enthalpy of ion pair formation were negative (exothermic), then Kip would decrease with increasing temperature via the Van’t Hoff equation,
| (1) |
When combined with the predictions by Salehi and Larson, lowering temperature raises the ion pairing equilibrium constant (eq 1), causing the phase envelope to shift upward, maintaining the 2-Φ region. This predicts that the LCST may not be due to the temperature dependence of the coupled ion pairing and counterion condensation.
Dipolar attractions by ion pairs was considered by Adhikari et al.7 with strength proportional to lB2 that enhance the effective hydrophobicity when combined with χ. They predict that lowering temperature raises the Cs−Cp envelope (Figure 3 in ref 7) by considering an inverse temperature dependence on χ and dipolar attractions. While this does not recover the behavior in Figure 3, the inverse-temperature dependence of χ counteracts the added effective hydrophobicity from dipolar attractions. However, the dielectric constant was fixed in that calculation. The temperature dependence of the dielectric constant of water increases the Bjerrum length from 0.696 to 0.738 nm between 0 and 50 °C. This suggests increased immiscibility with increasing temperature via dipolar attractions. A related study by Kudlay et al. predicts an increasing χ increases the coacervate concentration.2 It would be desirable to independently measure the temperature dependence of χ to properly evaluate these predictions.
The liquid state theory by Zhang et al.6 predicts that an increase in the Bjerrum length raises the Cs−Cp phase envelope (Figure 3 in ref 6), consistent with the current experimental trends. Similarly, Riggleman et al.29 predict an increasing Bjerrum length or reduced excluded volume parameter increases the driving force for phase separation. These theoretical studies show that phase separation of electrostatic origin is intimately tied to the Bjerrum length.
Phase separation upon heating must also include the correct temperature dependence of the nonelectrostatic aspects of the models. One may reconsider the temperature dependence of χ, such as χ = A/T + B + C ln T,30 or separation into enthalpic and entropic terms.31 This may be an oversimplification due to the multicomponent polyelectrolyte solutions, since the system may not be described by one interaction parameter. The observations of the coacervate exhibiting less hydration with increasing temperature is suggestive of less polymer and ion solvation. An increase in salt concentration increases the extent of hydration. Further characterization of the solvation characteristics23 should provide independent evidence for the molecular origins of the phase separation. These results will vary with the specific chemical composition of the polymers. In general, one needs to know the temperature dependence of χ to compare to predictions for both lower and upper critical solution behavior.
EXPERIMENTAL SECTION
Polyelectrolytes with mass-average relative molecular mass (Mw) of KPSS (Mw ≈ 200 kg/mol) and PDADMAB (Mw ≈ 150 kg/mol) were prepared at predetermined polymer and KBr concentrations. The component solutions are then mixed at room temperature, followed by vortex mixing and stored at 5 °C in sealed vials for 24 h. The mixtures are cooled to ≈3 °C and vortex mixed repeatedly to observe uniform solutions. All samples were stored at 5 °C before performing further measurements.
Ultraviolet−visible (UV−vis) absorption spectroscopy was used to quantify the PSS polymer concentrations as functions of temperature and salt concentrations. A phase separated mixture is equilibrated and centrifuged in a temperature-controlled environment. This accelerates the coalescence of dilute droplets and leads to the coexisting supernatant and transparent coacervate at T > Tcp. Next, known volumes of supernatant and coacervate are sampled and diluted using a 2.5 mol/L KBr solution at room temperature to quantify the PSS concentration based on the absorbance calibration at the absorbance peak at 261 nm. The measured values of CPSS are used to construct the coexistence curves.
Sample transmission was measured by a solid-state 532 nm laser or 632 nm He−Ne laser light with transmitted intensity measured by a Si photodiode (PM100 Thorlab). Samples in spectroscopic glass cuvettes were measured in a Quantum Northwest QPod unit with the temperature controlled to 0.01 °C precision. Different heating rates were applied from 0.05 to 0.4 °C/min with the Peltier heating element having temperature resolution of 0.02 °C. The ramping rate was slow enough to minimize the effect of kinetics during phase separation. The turbidity is reversible with little hysteresis, as observed during cooling scans. Samples were stirred continuously by a magnetic stir bar to minimize temperature gradients and prevent sedimentation of the droplets of the dense liquid phase.
Supplementary Material
ACKNOWLEDGMENTS
Access to USANS BT5 was provided by the Center for High Resolution Neutron Scattering, a partnership between NIST and the National Science Foundation under Agreement No. DMR-1508249. We thank Dr. J. Warren, director of the NIST Materials Genome Initiative, for funding. Certain commercial equipment and materials are identified in this paper in order to specify adequately the experimental procedure. In no case does such identification imply recommendations by the National Institute of Standards and Technology (NIST) nor does it imply that the material or equipment identified is necessarily the best available for this purpose.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmacrolett.8b00952.
Material source, sample preparation, and ion-exchange process, ultrasmall angle neutron scattering, rheology measurement method, confocal microscopy method, transmission profiles, heating and cooling transmittance scans, UV−vis absorption method, and concentration calculation tables (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).Overbeek J; Voorn M Phase Separation in Polyelectrolyte Solutions; Theory of Complex Coacervation. J. Cell. Comp. Physiol 1957, 49 (Suppl 1), 7–22 discussion, 22–26. [PubMed] [Google Scholar]
- (2).Kudlay A; Ermoshkin AV; de la Cruz MO Complexation of Oppositely Charged Polyelectrolytes: Effect of Ion Pair Formation. Macromolecules 2004, 37 (24), 9231–9241. [Google Scholar]
- (3).Qin J; Priftis D; Farina R; Perry SL; Leon L; Whitmer J; Hoffmann K; Tirrell M; de Pablo JJ Interfacial Tension of Polyelectrolyte Complex Coacervate Phases. ACS Macro Lett 2014, 3 (6), 565–568. [DOI] [PubMed] [Google Scholar]
- (4).Salehi A; Larson RG A Molecular Thermodynamic Model of Complexation in Mixtures of Oppositely Charged Polyelectrolytes with Explicit Account of Charge Association/Dissociation. Macromolecules 2016, 49 (24), 9706–9719. [Google Scholar]
- (5).Radhakrishna M; Basu K; Liu Y; Shamsi R; Perry SL; Sing CE Molecular Connectivity and Correlation Effects on Polymer Coacervation. Macromolecules 2017, 50 (7), 3030–3037. [Google Scholar]
- (6).Zhang P; Alsaifi NM; Wu J; Wang Z-G Polyelectrolyte Complex Coacervation: Effects of Concentration Asymmetry. J. Chem. Phys 2018, 149 (16), 163303. [DOI] [PubMed] [Google Scholar]
- (7).Adhikari S; Leaf MA; Muthukumar M Polyelectrolyte Complex Coacervation by Electrostatic Dipolar Interactions. J. Chem. Phys 2018, 149 (16), 163308. [DOI] [PubMed] [Google Scholar]
- (8).Ou ZY; Muthukumar M Entropy and Enthalpy of Polyelectrolyte Complexation: Langevin Dynamics Simulations. J. Chem. Phys 2006, 124 (15), 154902. [DOI] [PubMed] [Google Scholar]
- (9).Lytle TK; Radhakrishna M; Sing CE High Charge Density Coacervate Assembly via Hybrid Monte Carlo Single Chain in Mean Field Theory. Macromolecules 2016, 49 (24), 9693–9705. [Google Scholar]
- (10).Andreev M; Prabhu VM; Douglas JF; Tirrell M; de Pablo JJ Complex Coacervation in Polyelectrolytes from a Coarse-Grained Model. Macromolecules 2018, 51, 6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Spruijt E; Westphal AH; Borst JW; Cohen Stuart MA; van der Gucht J Binodal Compositions of Polyelectrolyte Complexes. Macromolecules 2010, 43 (15), 6476–6484. [Google Scholar]
- (12).Li L; Srivastava S; Andreev M; Marciel AB; de Pablo JJ; Tirrell MV Phase Behavior and Salt Partitioning in Polyelectrolyte Complex Coacervates. Macromolecules 2018, 51 (8), 2988–2995. [Google Scholar]
- (13).Priftis D; Xia X; Margossian KO; Perry SL; Leon L; Qin J; de Pablo JJ; Tirrell M Ternary, Tunable Polyelectrolyte Complex Fluids Driven by Complex Coacervation. Macromolecules 2014, 47 (9), 3076–3085. [Google Scholar]
- (14).Muthukumar M 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions. Macromolecules 2017, 50 (24), 9528–9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kabanov VA; Zezin AB A New Class of Complex Water-Soluble Polyelectrolytes. Makromol. Chem 1984, 6 (6), 259–276. [Google Scholar]
- (16).Izumrudov V; Bronich T; Zezin A; Kabanov V The Kinetics and Mechanism of Intermacromolecular Reactions in Poly-Electrolyte Solutions. J. Polym. Sci., Polym. Lett. Ed 1985, 23 (8), 439–444. [Google Scholar]
- (17).Philipp B; Dautzenberg H; Linow K-J; Kötz J; Dawydoff W Polyelectrolyte Complexes Recent Developments and Open Problems. Prog. Polym. Sci 1989, 14 (1), 91–172. [Google Scholar]
- (18).Matsuyama A; Tanaka F Theory of Solvation-Induced Reentrant Phase Separation in Polymer Solutions. Phys. Rev. Lett 1990, 65 (3), 341–344. [DOI] [PubMed] [Google Scholar]
- (19).Dormidontova EE Role of Competitive PEO−Water and Water−Water Hydrogen Bonding in Aqueous Solution PEO Behavior. Macromolecules 2002, 35 (3), 987–1001. [Google Scholar]
- (20).Chremos A; Douglas JF Communication: Counter-Ion Solvation and Anomalous Low-Angle Scattering in Salt-Free Polyelectrolyte Solutions. J. Chem. Phys 2017, 147 (24), 241103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Andreev M; de Pablo JJ; Chremos A; Douglas JF Influence of Ion Solvation on the Properties of Electrolyte Solutions. J. Phys. Chem. B 2018, 122 (14), 4029–4034. [DOI] [PubMed] [Google Scholar]
- (22).Strobl G The Physics of Polymers; Springer-Verlag: Berlin, 1996. [Google Scholar]
- (23).Dudowicz J; Freed KF; Douglas JF Solvation of Polymers as Mutual Association. I. General Theory. J. Chem. Phys 2013, 138 (16), 164901. [DOI] [PubMed] [Google Scholar]
- (24).Dudowicz J; Freed KF; Douglas JF Solvation of Polymers as Mutual Association. II. Basic Thermodynamic Properties. J. Chem. Phys 2013, 138 (16), 164902. [DOI] [PubMed] [Google Scholar]
- (25).Fu J; Schlenoff JB Driving Forces for Oppositely Charged Polyion Association in Aqueous Solutions: Enthalpic, Entropic, but Not Electrostatic. J. Am. Chem. Soc 2016, 138 (3), 980–990. [DOI] [PubMed] [Google Scholar]
- (26).Ali S; Prabhu VM Relaxation Behavior by Time-Salt and Time-Temperature Superpositions of Polyelectrolyte Complexes from Coacervate to Precipitate. Gels 2018, 4 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Schlenoff JB; Rmaile AH; Bucur CB Hydration Contributions to Association in Polyelectrolyte Multilayers and Complexes: Visualizing Hydrophobicity. J. Am. Chem. Soc 2008, 130 (41), 13589–13597. [DOI] [PubMed] [Google Scholar]
- (28).Sing CE Development of the Modern Theory of Polymeric Complex Coacervation. Adv. Colloid Interface Sci 2017, 239, 2–16. [DOI] [PubMed] [Google Scholar]
- (29).Riggleman RA; Kumar R; Fredrickson GH Investigation of the Interfacial Tension of Complex Coacervates Using Field-Theoretic Simulations. J. Chem. Phys 2012, 136 (2), 024903. [DOI] [PubMed] [Google Scholar]
- (30).Qian C; Mumby SJ; Eichinger BE Phase Diagrams of Binary Polymer Solutions and Blends. Macromolecules 1991, 24 (7), 1655–1661. [Google Scholar]
- (31).Schwahn D; Pipich V Aqueous Solutions of Poly(Ethylene Oxide): Crossover from Ordinary to Tricritical Behavior. Macromolecules 2016, 49 (21), 8228–8240. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.