

Abstract
Pituitary adenomas (PA) and pheochromocytomas/paragangliomas (PHEO/PGL) are rare tumors. Although they may co-exist by coincidence, there is mounting evidence that genes predisposing in PHEO/PGL development, may play a role in pituitary tumorigenesis. In 2012, we described a GH-secreting PA caused by an SDHD mutation in a patient with familial PGLs and found loss of heterozygosity at the SDHD locus in the pituitary tumor, along with increased hypoxia-inducible factor 1α (HIF-1α) levels. Additional patients with PAs and SDHx defects have since been reported. Overall, prevalence of SDHx mutations in PA is very rare (0.3–1.8 % in unselected cases) but we and others have identified several cases of PAs with PHEOs/PGLs, like our original report, a condition which we termed the 3 P association (3PAs). Interestingly, when 3PAs is found in the sporadic setting, no SDHx defects were identified, whereas in familial PGLs, SDHx mutations were identified in 62.5–75 % of the reported cases. Hence, pituitary surveillance is recommended among patients with SDHx defects. It is possible that the SDHx germline mutation-negative 3PAs cases may be due to another gene, epigenetic changes, mutations in modifier genes, mosaicism, somatic mutations, pituitary hyperplasia due to ectopic hypothalamic hormone secretion or a coincidence. PA in 3PAs are mainly macroadenomas, more aggressive, more resistant to somatostatin analogues, and often require surgery. Using the Sdhb + / − mouse model, we showed that hyperplasia may be the first abnormality in tumorigenesis as initial response to pseudohypoxia. We also propose surveillance and follow-up approach of patients presenting with this association.
Keywords: pituitary adenoma, pheochromocytoma, paraganglioma, succinate dehydrogenase
Introduction
Pheochromocytomas and paragangliomas (PHEOs/PGLs) are rare neuroendocrine tumors that produce catecholamines and arise from three structures derived from the neural crest: the adrenal medulla (PHEOs) and the sympathetic and parasympathetic paraganglia (PGLs) [1]. Most PHEOs are sporadic, unicentric and unilateral, but up to half (or even more, depending on the age of presentation) may be familial, multicentric and bilateral [2]. In recent years, substantial progress has been accomplished in the field of genetics and pathogenesis of PHEO/PGL and we know now that there are more than 20 susceptibility genes [3] including the genes encoding the four succinate dehydrogenase complex (SDH) subunits (SDHA, -B, -C, -D or SDHx collectively) [4–7]. Mutations/functional variants of SDHx have also been implicated in Carney–Stratakis dyad or syndrome and, rarely, in Carney triad [8], renal cancer [9–12], pancreatic neuroendocrine tumors [13] and Cowden-like syndrome [14].
Frequency of SDHx mutations in pituitary adenomas and the new syndromic association (3PAs)
In 2012, we described a family with multiple PGLs and PHEOs caused by a germline SDHD mutation; the index case also had an aggressive GH-secreting pituitary adenoma (PA) [15]. We found loss of heterozygosity (LOH) at the SDHD locus in the pituitary tumor, along with increased hypoxia-inducible factor 1α (HIF-1α) levels. These findings indicated that the SDH defect was most likely causatively linked to the development of the pituitary tumor and that pseudohypoxia pathways were activated, as shown in PHEOs/PGLs that bear SDHx mutations [15, 16].
However, what was also significant, was that the case of this patient was similar to several other cases previously reported since 1952; in most of these reports, the co-existence of PHEOs/PGLs and PAs was thought to represent a mere coincidence [17]. Following our 2012 publication, additional cases of PAs among patients with SDHx defects were described; today, it is widely accepted that the association of PAs with PHEOs and/or PGLs, or the 3 P association (3PAS) represents a new inherited form of a predisposition to multiple endocrine tumors caused by SDHx defects [18–25].
Indeed, to investigate the frequency of germline SDHx mutations in PAs we sequenced 168 patients with sporadic and familial PAs. Overall, SDHx mutations were rare: 1.8 % of the studied cases [26], in accordance with other reports [18, 25, 27]. However, among the patients included in the cohort, there were 7 that had presented with medical or family history of PHEOs/PGLs [26]; these patients had SDHx sequence pathogenic defects. Whenever family samples were available, we were able to show that SDHx defects were associated with either PHEOs/PGLs or PAs or both. On the other hand, in the few patients where PHEOs/PGLs and PAs were found in the sporadic setting (there was no family history or prior medical history of other SDHx-related defects), no SDHx mutations were identified. The difference was highly significant with more than 75 % of patients with 3PAs and positive family history bearing SDHx mutations [26]. The overall prevalence of 3PAs with SDHx mutations irrespective of family history was 42.8 % (3 out of 7 detected cases). Similar results were obtained in a different cohort of patients with PAs. Denes et al. studied 39 patients with sporadic or familial PHEO/PGL and/or PAs. Eight patients with SDHx mutations or variants within an international cohort of 19 patients with both tumors, were identified, which accounted for 42.1 % of the affected subjects. Five out of the 8 cases (62.5 %) were found within families with familial PHEOs/PGLs [22]. There was also a single patient with an SDHAF2 variant located in the 5′-UTR.
Interestingly, in the cohort studied by Denes et al., there were also 4 cases who presented with PHEO/PGLs and PAs but were found to harbor VHL or MEN1 gene mutations [22]. These 2 cases of PA and PHEO/PGL co-existence in patients with VHL mutations are the only ones reported in the literature, so far. Considering the frequency with which patients with VHL undergo regular surveillance imaging of the brain, the low frequency may indicate that the association of VHL and PA probably does not represent a true (genetically linked) predisposition. On the other hand in the 2 cases with MEN1 mutations, LOH was identified in the available PHEO tissue, indicating that PHEOs and/or PGLs can be part of the MEN 1 syndrome (as the Men1 knock-out animal model also suggests); thus, genetic testing for menin mutations should be considered in patients with PHEO/PGL if there are other suggestive signs of MEN 1. MEN 4 (the CDKN1B gene, coding for p27) may also be screened for in patients with an MEN 1-like phenotype, PAs, and PHEOs/PGLs that do not have menin or SDHx defects [22, 28–32].
We reviewed all the reported cases so far of the combination of PAs and PHEOs/PGLs since 1952 (eighty-two in total), including a recent report published while our manuscript was under review [33]. Literature search was done using PubMed, Scopus, and Google Scholar as search engines and “pituitary adenoma”, “3PAs”, “pheochromocytoma”, “paraganglioma” and combinations as search terms. Thirty-one (37.80 %) of these cases harbored mutations in predisposing PHEOs/PGLs or PA genes. Twenty-two patients (26.82 %) of all 3PAs cases had a personal or family history suggestive of a hereditary endocrine syndrome, whereas thirty-seven of all cases (45.1 %) were isolated; for the rest 28 % no information regarding family history was available (Table 1, 2). Among the 82 3PAs cases described so far, 17 % had both identified genetic mutations and family history. This co-existence was much higher (48.38 %) if we consider only the cases with a genetic defect. Regarding the frequency of the identified genetic defects it is obvious that the majority of cases carry SDHx defects (19 out of 31 cases, 61.3 %), with MEN1 and MAX being the second and third most frequently related genes. Of course, due to the retrospective nature of this review and the lack of genetic screening in the majority of 3PAs cases, generalizations regarding the overall frequency of the implicated genes should be avoided.
Table 1.
Patients with pituitary adenoma and pheochromocytoma/paraganglioma syndrome (3PAs) with identified germline genetic defects.
| Pituitary | Pheo/PGL | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Nr | Sex | Type | Size | Treatment | Age | Type | Treatment | Age | Family history | Mutation | LOH/ICH in PA | Reference |
| 1 | F | PRL | NK | NK | 27 | Pheo | NK | NK | No | SDHA c.91C > T p.Arg31Ter, VHL c.589G > A p.Asp197Asn | Not performed | Dénes et al. (2015) [22] |
| 2 | F | PRL | Macro | NK | 49 | PGLs | NK | 49 | NK | SDHA p.Arg31 * c.91C > T | SDHA/SDHB negative staining | Niemeijer et al. (2015) [13] |
| 3 | M | GH | Macro | SSA | 84 | PGL | No | 84 | No | SDHAF2 c.−52T > C | Not performed | Dénes et al. (2015) [22] |
| 4 | M | PRL | Macro | DA, surgery | 33 | PGL | Surgery | 33 | Mother: PRL, brother: PGL | SDHB c.298 T > C p.Ser100Pro | LOH at SDBH locus, intracytoplasmic vacuoles | Dénes et al. (2015) [22] |
| 5 | F | NFPA | Macro | Surgery x3, RT | 53 | PGL | RT | 28 | Sister: glioma | SDHB c.587 G > A p.Cys196Tyr | LOH at SDBH locus/SDHB staining: diffuse/intracytoplasmic vacuoles | Dénes et al. (2015) [22] |
| 6 | F | PRL | Macro | DA | 38 | PGLs carotid and mediastinal | Carotid: surgery, mediastinal: inoperable | 35 | Brother PGLs | SDHB mutation, (actual genetic defect not available) | Not performed | Gorospe et al. (2017) [76] |
| 7 | F | PRL | Macro | DA, RT | 60 | PGL | RT | 60 | NK | SDHB c.423 + 1 G > A | Not performed | Dénes et al. (2015) [22] |
| 8 | F | NFPA | Micro | No | 50 | Pheo | Surgery | 50 | NK | SDHB c.770dupT p.Asn-258GlufsTer17 | Not performed | Dénes et al. (2015) [22] |
| 9 | M | GH | NK | SSA | 72 | PGL | No | 70 | Brother & niece: PA, sister: bilateral HNPGL | SDHB c.689 G > A p.Arg230His | Not performed | Xekouki et al. (2015) [26] |
| 10 | F | PRL | Micro | NK | 50 | PGL | NK | 47 | Brother: HNPGL, grandmother: GIST | SDHB c.642 + 1 G > A, splice site alteration | Not performed | Xekouki et al. (2015) [26] |
| 11 | F | PRL | Micro | DA | 33 | PGL | Surgery | 43 | Brain tumor | SDHB c.18 C > A p.Ala6Alaa 3 PTEN polymorphisms | Not performed | Efstathiadou et al. (2014) [77] |
| 12 | F | PRL | Macro | DA | 38 | PGL | SSA | NK | Brother index case: PGL. Mother and sister positive for region of ex.1 of SDHB | deletion affecting ex. 1 of SDHB | Not performed | Guerrero Pérez et al. (2016) [21] |
| 13 | M | PRL | Macro | DA | 53 | PGL | Surgery | 38 | Cousin: PA, brother: PGL | SDHC c.380 A > G p.His127Arg | Not performed | Dénes et al. (2015) [22] |
| 14 | F | PRL | Macro | NK | 60 | PGL | NK | 60 | No | SDHC c256–257insTTTp-Phe85dup | Not performed | López-Jiménez et al. (2008) [78] |
| 15 | F | PRL | Macro | Surgery, DA | 23 | PGL | Surgery | 32 | Sister, aunt and grandmother: PA; sister: bilateral HNPGL | SDHD c.242 C > T, p.Pro81Leu | Not performed | Xekouki et al. (2015) [26] |
| 16 | M | PRL | Macro | DA, surgery | 60 | PGL, Pheo | Surgery (Pheo) | 62 | NK | SDHD c.274 G > T pAsp92Tyr | LOH at SDHD locus/SDHB positive ICH, SDHA IHC positive | Papathomas et al. (2014) [25] |
| 17 | F | GH | Macro | Surgery, SSA | 56 | PGL | NK | 56 | Father and 2 sisters: HNPGL; sister: GIST | SDHD c.274 G > T p.Asp92Tyr | LOH at SDHD locus/SDHB positive ICH, SDHA IHC positive | Papathomas et al. (2014) [25] |
| 18 | F | PRL | Macro | DA, surgery | 33 | PGL | Surgery x2 | 39 | Aunt, uncle, brother: HNPGL | SDHD c.242 C > T p.Pro81Leu | Not performed | Varsavsky et al. (2013) [79] |
| 19 | M | GH | Macro | SSA, surgery | 37 | PGL, Pheo | Surgery | 37 | Sister and paternal uncle neck PGLs HNPGL | SDHD c.298_301 del, premature stop at codon 133 AIP & CDKN1B polymorphism | PA: LOH at SDHD locus, reduced SDHD protein/patchy SDHB staining | Xekouki et al. (2012) [15,17] |
| 20 | M | GH/PRL | Macro | Surgery, RT, DA | 27 | Pheo | Surgery | 31 | No | MEN1 c.1452delGp.Thr557Ter | Menin staining of the Pheo: no menin positive cells | Dénes et al. (2015) [22] |
| 21 | F | NFPA | Macro | Surveillance | 45 | PGL | NK | 45 | No | MEN1 c.196_200dupAG-CCC frameshift (pathogenic), polymorphism C423T no amino acid change | Not performed in Pheo | Jeong et al. (2014) [80] |
| 22 | M | PRL | NK | NK | 41 | Pheo | Surgery | 48 | NK | MEN1 K119X, RET WT | Not performed in Pheo | Langer et al. (2002) [81] |
| 23 | NK | NK | NK | NK | NK | Pheo | NK | NK | Hyperparathy-roidism and pancreatic islet cell tumor | MEN1 c.320dell2 | Not performed in Pheo | Dackiw et al. (1999) [32] |
| 24 | NK | NK | NK | NK | NK | Pheo | NK | NK | Pancreatic islet cell tumor and rectal leiomyoma | MEN1 1325insA | Not performed in Pheo | Dackiw et al. (1999) [32] |
| 25 | M | PRL | NK | NK | 29 | Pheo | NK | 32 | MEN1 | Not performed, other family members MEN1 mutation | HPTH at age 21 years | Carty et al. (1998) [82] |
| 26 | M | GH | Macro | Surgery | 62 | Pheo | Surgery | 62 | No | RET p.Cys618Ser | Not performed in PA | Heinlen et al. (2011) [83] |
| 27 | M | ACTH | Micro | Surgery x2 | 48 | Pheo | Surgery | 66 | Son: HPTH | RET c.1900T > C, p.Cys634Arg. Negative for MEN1 mutations | Not performed in Pheo | Naziat et al. (2013) [84] |
| 28 | F | PRL | Macro | DA | 49 | Pheo | Bilateral adrenalectomy | 49 | No | MAX c.296 G > T, Neg for MEN1, VHL, SDHB, SDHC, SDHD, SDHAF2, or TMEM127 genes | Not performed | Roszko et al. (2017) [34] |
| 29 | M | PRL | Micro | DA | 49 | Pheo | Surgery | 32 | No | Germinal heterozygous deletion of exon 3 of MAX (detected by MLPA). Neg for RET, VHL, SDHx, CDKN1B, AIP, MEN1 | Not performed | Daly et al., (2018) [33] |
| 30 | F | GH | Macro | SSA, DA, Pegvisomant, RT | 26 | Bilateral Pheos | Bilateral adrenalectomy | 35 | No | Germinal heterozygous deletion of exons 1–3 and intron 3 of MAX. Neg for RET, VHL, SDHx, CDKN1B, AIP, MEN1 | Not performed | Daly et al. (2018) [33] |
| 31 | M | GH | Macro | Surgery, RT | 16 | Bilateral Pheos | Bilateral adrenalectomy | 22 | No | Germinal heterozygous deletion of exon 3 of MAX (detected by MLPA). Neg for RET, VHL, SDHx, CDKN1B, AIP, MEN1 | Not performed | Daly et al. (2018) [33] |
M: Male; F: Female; NFPA: Non-functional pituitary adenoma; PRL: Prolactinoma; GH: Acromegaly; Macro: Macroadenoma; Micro: Microadenoma; DA: Dopamine agonist; RT: Radiotherapy; SSA: Somatostatin analogue; Pheo: Pheochromocytoma; PGL: Paraganglioma; HNPGL: Head and neck paraganglioma; PTC: Papillary thyroid cancer; GIST: Gastrointestinal stromal tumor; pNET: Pancreatic neuroendocrine tumor; MTC: Medullary thyroid carcinoma; HPTH: Hyperparathyoidism; NK: Not known; MEN1: Multiple endocrine neoplasia type 1; NF1: Neurofibromatosis type 1; A: Single nucleotide polymorphism with a minor allele frequency of 0.2% and a genotype frequency of 0.5% (1000 Genomes Project Consortium, 2012) [85].
Table 2.
Patients with pituitary adenoma and pheochromocytoma/paraganglioma without identified germline genetic defects.
| Pituitary | Pheo/PGL | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Nr | Sex | Type | Size | Treatment | Age | Type | Treatment | Age | Family history | Mutation | Other Info (when reported) | Reference |
| 1 | M | PRL | Micro | Surgery | 54 | Pheo | Bilateral adrenalectomy | NK | No | Neg for RET, VHL, SDHB, SDHD mutations | Guerrero Pérez et al. (2016) [21] | |
| 2 | F | GH | Micro | Surgery | 56 | Pheo | Bilateral adrenalectomy | NK | No | Neg for RET, VHL, MEN-1 mutations | Bilateral nodules on parathyroid glands, asymptomatic | Guerrero Pérez et al. (2016) [21] |
| 3 | M | PRL | Macro | Cabergoline | 38 | Pheo/PGL | α-Adrenergic blockade | NK | NK | Neg for MEN1 | Catecholamine-mediated cardiac toxicity | Koshy et al. (2016) [86] |
| 4 | F | ACTH | Micro | Surgery | 61 | PGL | Surveillance | 61 | No | Negative for SDHA-D, MEN1, RET, AIP | Bilateral HNPGL | Xekouki et al. (2015) [26] |
| 5 | F | PRL | Macro | DA, surgery | 35 | Pheo | Surgery | 55 | No | Negative for SDHA-D, MEN1, RET, AIP | Bilateral Pheo | Xekouki et al. (2015) [26] |
| 6 | F | GH | Macro | Surgery | 35 | PGL | Surgery | 58 | No | Negative for SDHA-D, MEN1, RET, AIP | Bladder PGL | Xekouki et al. (2015) [26] |
| 7 | F | NFPA | Macro | Surgery | 39 | Pheo | Surgery | 34 | No | Negative for SDHA-D, MEN1, RET, AIP | Xekouki et al. (2015) [26] | |
| 8 | F | GH | Macro | Surgery, RT, DA, SSA | 56 | Pheo | Surgery | 66 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B | GIST, thyroid follicular adenoma | Boguszewski et al. (2012) [87], Dénes et al. (2015) [22] |
| 9 | M | NFPA | Macro | Surgery | 53 | PGL | Surgery | 50 | Father: PA | SDHA c.969 C > T p.Cly323GlyaSDHB-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B all normal | Abdominal PGL Wilms tumor, liposarcoma, renal oncocytoma; PA: no LOH at SDHA locus, intracytoplasmic vacuoles, SDHA and B staining preserved | Dénes et al. (2015) [22] |
| 10 | F | GH | Macro | Surgery, RT, SSA | 39 | Pheo | Surgery | 20 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B | Dénes et al. (2015) [22] | |
| 11 | F | NFPA | Macro | Surgery, RT | 73 | PGL | RT | 73 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEMU7, MAX, FH, CDKN1B | HNPGL | Dénes et al. (2015) [22] |
| 12 | M | GH | Macro | Infarcted | 16 | Pheo | NK | 16 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B | Dénes et al. (2015) [22] | |
| 13 | M | PRL | Macro | Surgery | 40 s | PGL | NK | 52 | No | Negative for SDHA-D, AF2, MEN1, RET, AIR VHL, TMEM127, MAX, FH, CDKN1B | HNPGL | Dénes et al. (2015) [22] |
| 14 | F | PRL | NK | NK | 27 | Pheo | NK | 41 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B | Dénes et al. (2015) [22] | |
| 15 | M | NK | NK | NK | NK | Pheo/PGL | NK | NK | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B | Denes etal. (2015) [22] | |
| 16 | F | PRL | Micro | DA | 40 | Pheo | Surgery | 38 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKNW | Dénes et al. (2015) [22] | |
| 17 | M | PRL | Micro | DA | 56 | Pheo | Surgery | 56 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B | Dénes et al. (2015) [22] | |
| 18 | F | PRL | Macro | DA | 61 | Pheo | Surgery | 61 | No | Negative for SDHA-D, AF2, MEN1, RET, AIP, VHL, TMEM127, MAX, FH, CDKN1B | Dénes et al. (2015) [22] | |
| 19 | F | PRL | Macro | NK | 60 | PGL | RT | 60 | No | Negative for SDHB | Parghane et al. (2014) [88] | |
| 20 | F | NFPA | Macro | No | 52 | Pheo | Surgery | 52 | No | Negative for SDHA-D, AF2, RET, MAX, TMEMT27, VHL | GHRH secreting Pheo | Mumby et al. (2014) [89] |
| 21 | M | GH | Macro | Surgery | 29 | Pheo | Surgery | 29 | No | Not performed | Bilateral Pheo lipoma, metastatic PTC | Sisson et al. (2012) [90] |
| 22 | NK | GH | NK | NK | NK | Pheo | NK | NK | MEN1 | Not performed | Bilateral Pheo HPTH, pNET Clinical features NF1 | Gatta-Cherifi et al. (2012) [91] |
| 23 | M | GH | Macro | Surgery | 45 | PGL, Pheo | Surgery × 3 | 54 | Father HNPGL, Sister: adrenal abnormality | Not performed | Abdominal, HN, cardiac PGLs | Zhang et al. (2011) [92] |
| 24 | M | NFPA | Micro | No | 64 | Pheo | Surgery | 64 | No | Not performed | High cortisol (cured post-adrenalectomy) | Yaylali et al. (2008) [93] |
| 25 | M | NFPA | Macro | Surgery | 59 | Pheo | Surgery | 59 | No | Not performed | Breckenridge et al. (2003) [94] | |
| 26 | M | NFPA | Macro | Surgery | 56 | Pheo | No | 56 | No | Not performed | Dünser et al. (2002) [95] | |
| 27 | F | GH | Macro | Surgery | 57 | Pheo | Surgery | 57 | No | Not performed | Sleilati et al. (2002) [96] | |
| 28 | M | NK | Micro | No | 43 | Pheo | Surgery | 43 | No | Negative for RET | Lipoma, pectus excavatum, pleomor-phic parotid adenoma. GH levels responded to OGTT post adrenalectomy | Baughan et al. (2001) [97] |
| 29 | F | NK | Micro | No | 44 | Pheo | Surgery | 44 | No | Not performed | Cushing’s (cured post-adrenalectomy) | Khalil et al. (1999) [98] |
| 30 | M | GH | Macro | Surgery | 41 | Pheo, PGLs | Surgery | 20 | NK | Negative for RET | Teh et al. (1996) [99] | |
| 31 | M | PRL | Macro | Surgery | 20 | PGL | Surgery | 20 | NK | Not performed | HNPGL | Azzarelli et al. (1988) [100] |
| 32 | M | PRL | Macro | DA | 26 | Pheo | Surgery | 26 | Father: metastatic MTC and probably Pheo | Not performed | Bertrand et al. (1987) [101] | |
| 33 | F | NFPA | NK | NK | 70 | PGL | NK | 70 | Daughter and granddaughter: PA, bilateral HNPGL | Not performed | HPTH, PTC, gastric leiomyoma, amyloidosis | Larraza-Hernandez et al. (1982) [102] |
| 34 | F | PRL | Micro | NK | 35 | Pheo | NK | 35 | NK | Not performed | Meyers (1982) [103] | |
| 35 | M | NFPA | Macro | Surgery | 66 | PGL | Surgery | 63 | NK | Not performed | Blumenkopf & Boekelheide (1982) [104] | |
| 36 | F | GH | NK | Surgery, RT | 53 | Pheo | Surgery | 58 | Brother: hypertension | Not performed | Anderson et al. (1981) [105] | |
| 37 | F | GH | NK | RT | 33 | Pheo | None | 45 | No | Not performed | Multinodular goiter | Anderson et al. (1981) [105] |
| 38 | F | GH | Macro | NK | 53 | Pheo | NK | 53 | NK | Not performed | HPTH | Myers & Eversman (1981) [106] |
| 39 | F | PRL | NK | NK | 23 | Pheo | NK | 23 | NK | Not performed | HPTH, gastrinoma, adrenal adenoma | Alberts et al. (1980) [107] |
| 40 | F | NK | Macro | NK | 22 | Pheo | NK | 22 | Granddaughter: unilateral Pheo | Not performed | Islet cell tumor/ renal adenoma | Janson et al. (1978) [108] |
| 41 | F | GH | Macro | NK | 15 | Pheo | NK | 15 | NK | Not performed | HPTH | Manger & Clifford (1977) [109] |
| 42 | F | NFPA | NK | NK | 49 | Pheo | None | 49 | NK | Not performed | Papillary carcinoma of thyroid | Melicow (1977) [110] |
| 43 | M | GH | NK | NK | 21 | Pheo | NK | 44 | NK | Not performed | Kadowaki et al. (1976) [111] | |
| 44 | F | GH | NK | NK | 19 | PGL | NK | 19 | NK | Not performed | PGL (HN, pelvis), HPTH | Farhi etal. (1976) [112] |
| 45 | F | GH | NK | NK | 36 | PGL | NK | 36 | NK | Not performed | HNPGL, HPTH, hyperplasia of antral and duodenal gastrin cells | Berg et al. (1976) [113] |
| 46 | F | NK | Micro | NK | 43 | Pheo | NK | 43 | NK | Not performed | MTC | Wolf et al. (1972) [114] |
| 47 | F | GH | NK | RT | 36 | Pheo | Surgery | 36 | NK | Not performed | toxic nodular, goiter, endometriosis, and diabetes, DM | Miller &Wynn (1971) [115] |
| 48 | M | ACTH | NK | NK | NK | Pheo | Surgery | 41 | VI generations with MEN | Not performed | MTC | Steiner et al. (1968) [116] |
| 49 | M | GH | Macro | RT | 23 | Pheo | None | 41 | NK | Not performed | Kahn&Mullon (1964) [117] | |
| 50 | M | GH | NK | NK | NK | Pheo | NK | NK | NK | Not performed | German & Flanigan (1964) [118] | |
| 51 | M | GH | NK | NK | 44 | Pheo | NK | NK | NK | Not performed | Iversen (1952) [119] | |
M: Male; F: Female; NFPA: Non-functional pituitary adenoma; PRL: Prolactinoma; GH: Acromegaly; Macro: Macroadenoma; Micro: Microadenoma; DA: Dopamine agonist; RT: Radiotherapy; SSA: Somatostatin analogue; Pheo: Pheochromocytoma; PGL: Paraganglioma; HNPGL: Head and neck paraganglioma; PTC: Papillary thyroid cancer; GIST: Gastrointestinal stromal tumor; pNET: Pancreatic neuroendocrine tumor; MTC: Medullary thyroid carcinoma; HPTH: Hyperparathyoidism; NK: Not known; MEN1: Multiple endocrine neoplasia type 1; NF1: Neurofibromatosis type 1; GOTT: Glucose oral tolerance test; a Single nucleotide polymorphism with a frequency of 3.5% (Bayley et al. 2005) [120].
In a most recent publication Daly et al., reported 3 patients who presented with 3PAs and were found to have MAX exon/intragenic deletions using multiplex ligation-dependent probe amplification (MLPA), confirming a previous report of Roszko et al. [34]. The authors recommend that MAX MLPA should be considered in 3PAs and in PHEO cases in individuals screened negative with Sanger sequencing for the reported genetic causes including MAX mutations.
Despite the new identified genetic mutations/variants associated with 3PAs, for the majority of cases we do not know what other PHEOs/PGLs-causing genetic defects may be associated with a predisposition to PAs. At this point, what one screens for (beyond SDHx, menin, and possibly p27) remains unknown but it should be guided by detailed medical and family history, the latter extending to previous generations and even distant relatives, as these mutations often have weak penetrance. One should also avoid the effort to “fit” unusual cases into the known conditions: it is evident that the known classification of MEN syndromes does not always cover rare individual cases that present with significant overlap in their phenotype or, even, seemingly unrelated clinical signs that may represent new associations. Regarding the 51 cases of 3PAs with no known genetic defect (Table 2) most of them are older and genetic testing was not available. Therefore, we cannot exclude the possibility that they harbor a genetic defect in any of the implicated genes; for some of them and based on the clinical presentation and family history the genetic defect is obvious. However, in the rest of the cases and particularly where genetic screening was negative, the co-existence of a PA and a PHEO or PGL may indeed represent an extremely rare coincidence due to mutations in co-segregating genetic defects or epigenetic changes; few cases may be explained by ectopic hormone secretion by a PHEO/PGL mimicking a functioning PA. Regarding the reported variations in sporadic PAs in Table 3, most of them are variants of unknown significance and some of them are predicted to be damaging using the available prediction tools. However, one should perform functional studies to prove their deleterious effect.
Table 3.
SDHx variants/mutations in pituitary adenomas.
| Patient Nr | Gene | Sex | Age | Type | Size/Treatment | ICH/LOH | Other tumors/conditions | Family history | Genetics tested/Prediction | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | SDHB | F | 49 | PRL | Micro/NK | SDHB positive | PHPT | Aunt: ACTH PA | c.5 C > T; p.A2V [Deleterious (SIFT)/benign (PolyPhen)] | de Sousa et al. (2017) [27] |
| 2 | SDHC | F | 34 | PRL | Micro/NK | SDHB positive | None | Brother: PRL PA | c.403 G > C; p.E110Q (Deleterious (SIFT)/possibly damaging (PolyPhen)) | de Sousa et al. (2017) [27] |
| 3 | SDHC | F | 63 | GH | NK | SDHB positive | Pituitary gangliocytoma, PHPT | No | p.E110Q [Deleterious (SIFT)/possibly damaging (PolyPhen)] | de Sousa et al. (2017) [27] |
| 4 | SDHA | M | 62 | NFPA | Macro/TSS | Loss of staining for both SDHA and SDHB | None | No | c.725_736del/c.989_990insTA (double hit) | Gill et al. (2014) [18] |
| 5 | SDHB | F | 35 | PRL | Macro/TSS | intracytoplasmic vacuoles | None | Mother: Prolactinoma, brother: PGL | c.298 T > C (damaging mutation) | Dénes et al. (2015) [22] |
| 6 | SDHA | M | 30 | NFPA | Macro/TSS | ICH negative for SDHA and SDHB | None | Mother carotid body paraganglioma | c.1873C > T p.His625Tyr | Dwight et al. (2013) [24] |
| 7 | SDHB | F | 15 | Nl< | NK | NK | NK | NK | ex. 7 c.761insC p.254fsX255 | Benn et al. (2006) [121] |
| 8 | SDHD | M | 12 | Cushing | Micro/TSS | Not studied | Thyroid | No | ex. 2 c.53 C > T, p.Ala18Val (very rare SNP (allele frequency <0.001) Benign (PSIC 0.004) (by PolyPhen) Deleterious (Score 0.01) (by SHIFT) | Xekouki et al. (2015) [26] |
| 9 | SDHB | F | 31 | PRL | Macro/TSS | LOH at SDHB locus in the PA/SDHB staining: loss of expression of SDHB | None | Grandmother’s first cousin PGL | del ex. 6 to 8 | Dénes et al. (2015) [22] |
| 10 | SDHD | F | 16 | Cushing | Micro/TSS | Not studied | None | No | ex. 2c.149 A > G/p.His50Arg (Probably damaging (PSIC 0.993) (Polyphen) Benign (Score 0.48) (SHIFT) | Xekouki et al. (2015) [26] |
| 11 | SDHB | F | 14 | Cushing | Micro/TSS | Not studied | None | No | ex. 5c.487 T > C, p.Ser163Pro (reported as potentially pathogenic by Ni et al. 2012) [122] | Xekouki et al. (2015) [26] |
| 12 | SDHB | M | 10 | Cushing | Micro/TSS | Not studied | Brachydactyly/dysmorphic features | No | ex. 5c.487 T > C, p.Ser163Pro (reported as potentially pathogenic by Ni et al. 2012) [122] | Xekouki et al. (2015) [26] |
M: Male; F: Female; NK: Not known; PRL: Prolactinoma; GH: Acromegaly; NFPA: Non-functional pituitary adenoma; Macro: Macroadenoma; Micro: Microadenoma; PA: Pituitary adenoma; PGL: Paraganglioma; PHPT: Primary hyperparathyroidism, TSS: Transphenoidal surgery.
Phenotypic and pathological characteristics of PAs with SDHx mutations
We attempted to see whether PAs with SDHx mutations appear to have a different progression as previously described [21, 22, 25, 26]. Unfortunately, not all clinical data were available to make a safe distinction between the PAs as part of 3PAs with and without germline mutations. However, based on the available data from the reported cases we looked for any difference in phenotypic characteristics between PAs in the context of 3PAs with (Table 1) and without genetic mutations (Table 2) and the isolated PAs with SDHx mutations/variants (Table 3). The SDHx-related PAs in 3PAs were more common among familial cases, more frequently macroadenomas, they often led to multiple phenotypes within the same family (somatotropinomas, prolactinomas, and nonfunctioning adenomas) and required more than one modes of treatment (Table 1 & Supl. Table 1S). Patients with isolated PAs and SDHx mutations/variants (Table 3) were significantly younger compared to the ones in cases with 3PAs regardless of the presence of a genetic mutation (Table 1, 2), and they all required surgery (regardless of the size of their tumors) but did not require multiple treatments (Supl. Table 4). A sub-analysis between the isolated PAs with SDHx mutations/variants and their counterparts in 3PAs, revealed that the latter were more frequently macroadenomas and required more than one treatment modality, which may suggest that the presence of PHEOs/PGLs may have contributed to the increased size and treatment resistance (Supl. Table 2S).
At this point, there does not appear to exist an apparent phenotype-genotype correlation, although very few families with 3PAS have other tumors associated with SDHx mutations such as gastrointestinal stromal tumors (GISTs) or renal cancer. The limited number of cases and the lack of prospective studies also do not allow for an accurate estimate of the expected age of presentation: based on the available cases (Table 1, 3) age at diagnosis ranged from as early as 15 years to as late as 72 years old. PHEOs or PGLs in these patients were bilateral and/or multiple, with a tendency to recur. The latter observation is one of the characteristics of SDHx-related PHEOs/PGLs [35].
Interestingly, in our first report, the growth hormone (GH) receptor (GHR) gene was found to be expressed in PHEO samples from our patient with the SDHD mutation, as well as in tumor samples from other patients harboring SDHB or SDHD mutations [15]. Based on this finding together with the clinical observation that there was a noticeable decrease (almost three-fold) of plasma and urinary metanephrines after pituitary transsphenoidal surgery (that was greater than the one noted following bilateral adrenalectomy for the patient’s PHEOs), we assumed that normalization of GH levels after TSS contributed significantly to such biochemical changes. This was the first and only study so far reporting the expression of GHR in PHEOs whereas there are reports of the differential expression of ghrelin and GH-releasing hormone (GHRH) receptors in various adrenal tumors, including PHEOs [36–38]. The role of GHR in SDHx-mutant tumors needs to be investigated further.
One interesting histological phenotype reported by Dénes et al. [22], in pituitary gland tissues from patients with SDHx mutations was the presence of intracytoplasmic vacuoles in their PAs. Although electron microscopy was not used to identify the exact nature of the vacuoles [39], there is a possibility that they represented autophagic bodies. The relationship between hypoxia-related pathways and autophagy activation is well established [40, 41] and autophagy has been shown to contribute to chemo- and radio-therapy resistance [42, 43]. We have described a similar morphological finding in the PA tissue of the original case with the SDHD mutation (Fig. 1a) [15].
Fig. 1.
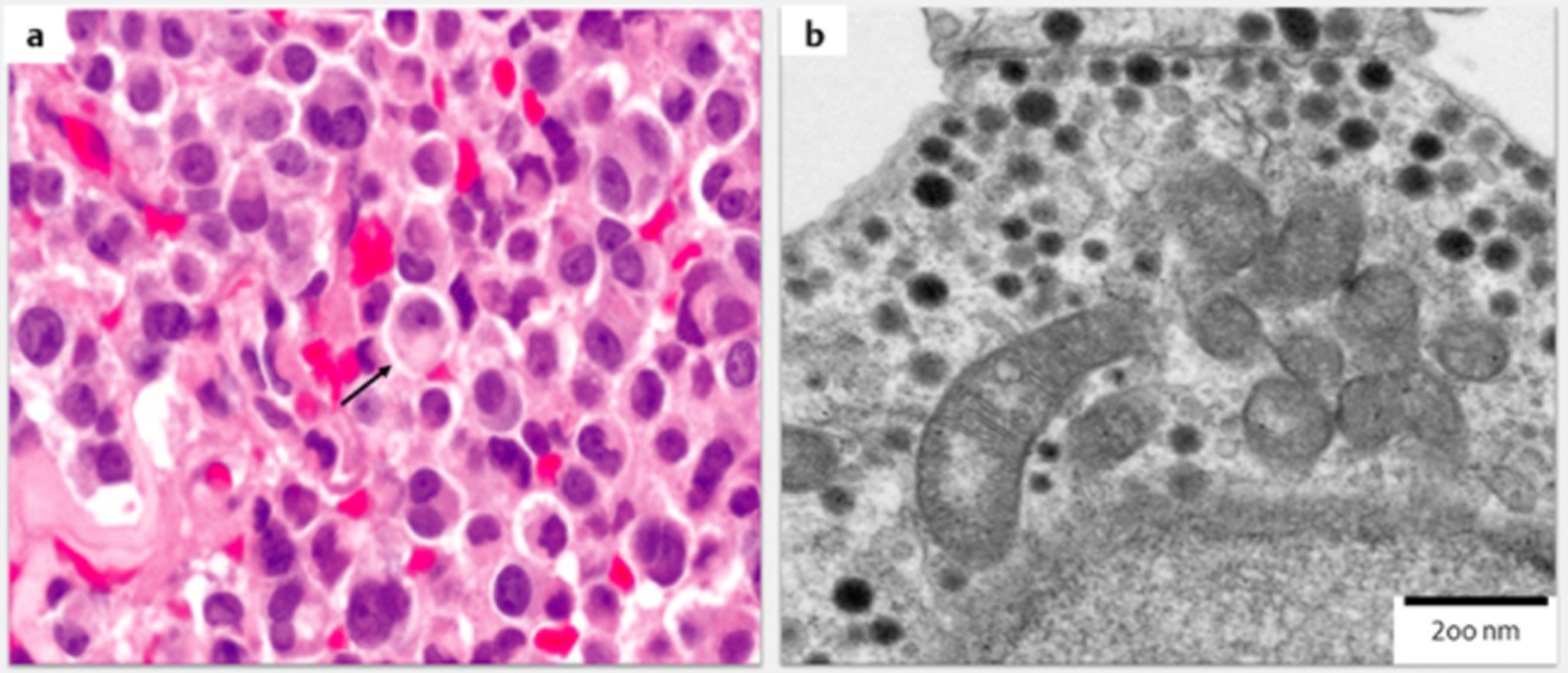
a: H & E staining of the pituitary adenoma of the SDHD patient reported by Xekouki et al., 2012 [15, 17] showing cytoplasmic vacuoles (arrow) (× 40) b: Enlarged mitochondria with marked swelling and abnormal and/or missing internal cristae in pituitary cells of Sdhb + / − mice (electron microscopy 200 nm).
Loss of SDHB immunochemistry has been shown to be an excellent indicator of germline or somatic mutations in the SDHx genes [1, 44, 45]. In most of the SDHx-related PAs reported so far, completely absent or weak diffuse staining of SDHB was found (in those cases that this was performed) (Table 1, 3). Therefore, as in PHEOs/PGLs, SDHB staining may be used as an additional diagnostic tool to screen for SDHx mutations. Furthermore, as shown by Richter et al. [46], profiling of Krebs cycle metabolites, such as ratios of succinate:fumarate with the use of mass spectrometry, is another useful method to identify patients for testing of SDHx mutations and to assess functionality associated with SDHx variants of uncertain significance in PGLs. Although this method has not been used in any of the 3PAs cases it would be interesting to see whether it could predict the presence of SDHx mutations or distinguish damaging mutations from nonfunctional polymorphisms in pituitary adenomas in the context of 3PAs as in PGLs.
Traditionally LOH has been used in oncology to confirm the causative association between a tumor and the loss of a tumor suppressor gene [47]. As shown in Table 1, in some cases no LOH studies were performed; in few cases no LOH of the SDHx mutated locus was identified. Does this mean that the absence of consistent LOH in PAs indicates lack of association with the identified SDHx mutations? Although this is difficult to answer with certainty at this time, bilateral adrenal medullary hyperplasia associated with a germline SDHB mutation showed retention of heterozygosity [48], and PHEOs without loss of the normal SDHD allele have been shown in patients with pathogenic SDHD mutations [49]. Additionally, cases of “paradoxical” loss of the mutant SDHx allele have been shown [24, 50], pointing to the suggestion that the SDHx defects may not always lead to tumorigenesis in the classical tumor suppressor gene way. Finally, epigenetic alterations such as somatic SDHC promoter methylation and postzygotic somatic mosaicism could provide another explanation for those cases negative for germline mutations [51, 52].
Molecular mechanisms of SDHx mutations in the development of a PA
To further understand the causal relationship between PA and SDHx mutations, we analyzed the pituitaries of adult Sdhb+ / − mice. Although no gross pituitary tumors were detected, the pituitary of 12-month old Sdhb + /− mice was hypercellular, mainly due to the increased number of prolactin (PRL)-secreting cells and to a lesser extend GH-secreting cells. There were also blood-filled lakes, abnormal mitochondria (Fig. 1b), nuclear inclusions of unknown nature, and abnormal and poorly defined heterochromatin forming a ring-shaped pattern rather than being centrally located as in pituitaries of the normal control animals [26]. Pituitary hyperplasia likely preceding the formation of adenomas, was found in 3- and 12-month-old Aip-deficient male mice [53] as well as in those lacking p19 (arf) or p27(kip–1), and those overexpressing the beta-catenin or the pituitary tumor-transforming gene (CTNNB1 and PTTG, respectively) [54–57]. In humans, pituitary hyperplasia has been reported in patients with germline mutations in the PRKAR1A or AIP tumor suppressor genes, and in those with gigantism and chromo-some Xq26 duplications [58–60].
Like in other SDHx-deficient PHEOs/PGLs [61], there is evidence that hypoxic signalling is activated in SDHx-mutated pituitary tumors [15]. This finding was supported by the HIF-1α strong cytoplasmic (and occasionally nuclear) expression in the pituitaries of Sdhb + /− mice [26]. Other evidence of hypoxia-activated pathway in these mice was the enlarged mitochondria with destructed cristae found in the Sdhb + /− animals [26, 62]. Similar mitochondrial findings have been reported in tumor samples from patients with Carney triad, as well as in paragangliomas from patients with SDHC and SDHD mutations [56]. Fragmented and defective mitochondria have been found in many different cancers and alterations in mitochondrial dynamics are associated with tumor progression or resistance to therapy [63]. The altered chromatin pattern that we also observed in Sdhb + /− mice, like the one observed in mice overexpressing PTTG and in hypoxic cells [55, 64] is another indication that chronic activation of pseudohypoxia in SDHx-deficient pituitary cells can drive genetic instability and eventually lead to tumor formation.
Although electron microscopy failed to identify the actual nature of the nuclear inclusions seen in Sdhb + /− pituitary cells with light microscopy, it has revealed that these appeared to be fused to the nucleus rather than being intranuclear. These inclusions resembled the vacuoles described by Denes et al., in SDHx-deficient pituitary tumors and may represent late autophagic vacuoles previously filled with digested abnormal mitochondria. Thus, we may speculate that PA formation from SDHx mutations may follow a long process, whereby hyperplasia is the initial response to a prolonged stimulation due to activation of pseudo-hypoxia signals (Fig. 2).
Fig. 2.
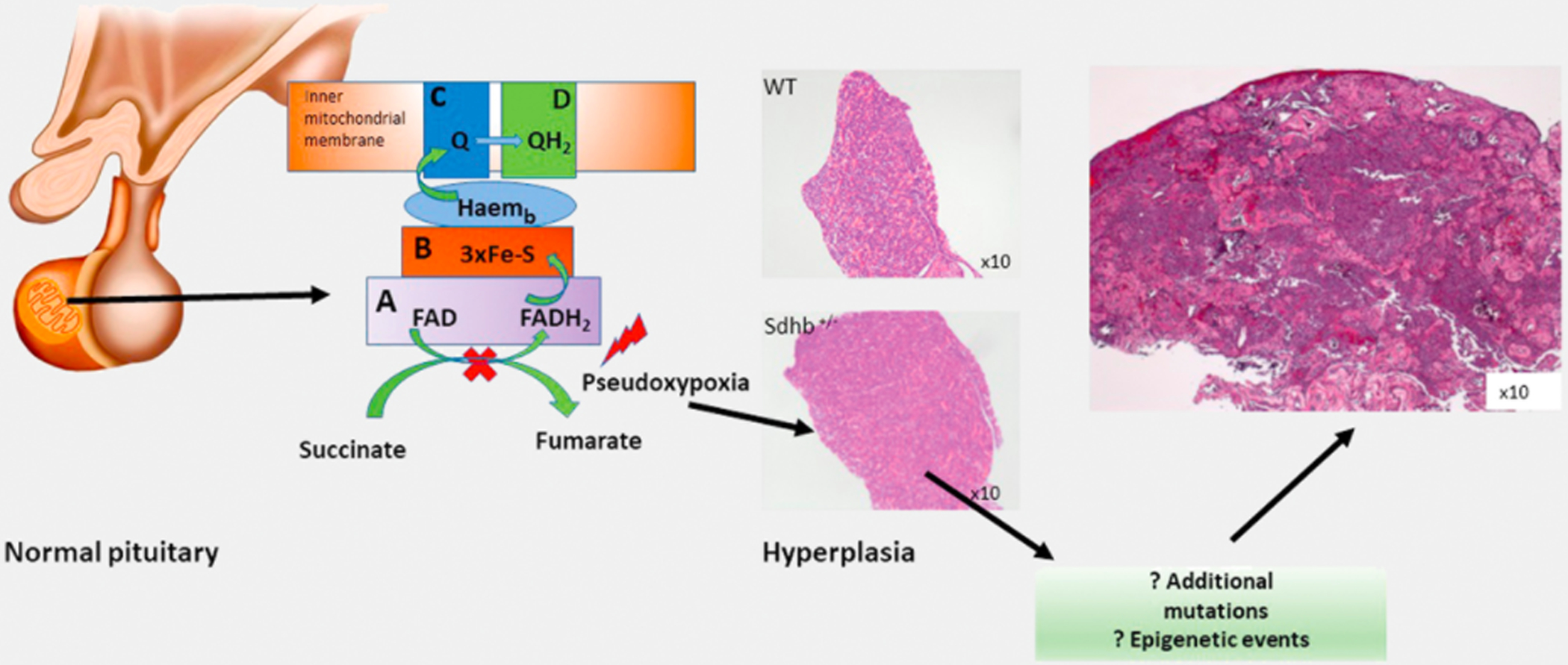
Proposed mechanism of tumor development in SDHx-deficient pituitary adenomas; SDHx mutations may initially lead to hyperplasia, as it was shown in Sdhb + / − mice, which eventually either at the presence of another mutation or epigenetic events it will lead to pituitary tumor development (H & E staining of the original case reported in 2012).
Recommendations for genetic screening and follow-up of patients with 3PAs
Although cases of 3PAs have been reported since 1952, it was only in 2012 when we showed that SDHx mutations may be involved in pituitary tumorigenesis [15]. So far there are 28 cases of confirmed SDHx mutation-related pituitary adenomas (with the variants of unknown significance excluded). Currently, we recommend that a detailed baseline medical and family history is taken from all these patients along with careful physical examination to detect signs of other tumors associated with SDHx defects (for examplle, GISTs) (Fig. 3). Hormonal testing should be obtained, once familial PHEOs and/or PGLs are recognized in the index case and their family members. Attention should be paid to any symptoms that would indicate GH or PRL hypersecretion and visual disturbance, as most of the pituitary adenomas in the context of 3PAs are PRL- or GH-secreting macro-adenomas or NFPAs. If there are no suspicious symptoms or findings and biochemistry is normal then annual biochemical surveillance should include testing for PHEOs/PGLs, as per the most recent recommendations [65, 66]. If at any time, clinical findings or biochemistry indicate hormonal hypersecretion, a pituitary MRI should be performed.
Fig. 3.

Recommended clinical investigation and follow-up in patients with 3PAs.
Treatment of PAs due to SDHx mutations should not differ from the recommended treatment for sporadic tumors [67–71]. However, it is possible that more than one treatment modalities will be required, as these tumors are often more aggressive and tend to recur more frequently than their sporadic counterparts (although this impression may change as more cases are now identified by screening and are followed prospectively).
Special attention should be given to patients that may have MEN1, MEN2, or MEN4, as now we know that all three syndromes may be associated with both PAs and PHEOs/PGLs. Thus, plasma or urine metanephrine measurements may be needed prior to surgeries in patients with suggestive signs and symptoms of a PHEO or a PGL. Finally, in case of a family history of a PHEO/PGL due to MAX mutation, careful medical history and physical examination should be performed in all mutation carriers for the presence of other tumors related to MAX mutations such as PA and renal cancer as recently described [33, 72].
Genetic testing for SDHx mutations should be performed in any patient who presents with 3PAs particularly if there is family history of PHEOs/PGLs in other family members (the latter may not only include first-degree relatives). In the absence of a family history of PHEOs/or PGLs, screening for SDHx mutations in patients with apparently sporadic 3PAS may also be performed, as these genetic defects are known to have low penetrance in affected families. Alternatively, and/or if genetic screening is not readily available, SDHB staining and succinate:fumarate ratio maybe another way to look for SDHx defects. The presence of intracytoplasmic vacuoles is another pathology finding strongly suggestive of SDHx gene defect. If screening for SDHx mutations is negative or pathology and biochemistry are not suggestive of SDHx defects, then screening for menin or RET mutations should be based on the presence of clinical features and/or tumors that would indicate the presence of the MEN1 or MEN2 syndromes. Screening for MEN4 is not considered routine at this point, as is the case for other genes known to predispose to PA development [73]. Finally, screening for MAX mutations should be considered if SDHx and menin are screened negative.
Final remarks
In the era of next generation, sequencing and new molecular techniques hundreds of new genes responsible for tumors have been discovered [74]. The opposite is also true: many new associations are found for mutations of the same gene(s). Indeed, today is the era of physician-researchers working like crime scene investigation (CSI) agents, gathering data carefully, connecting seemingly unrelated facts, and discovering new phenotypes for known gene defects [75]. The SDHx genes are very well suited for the investigation of new associations, as they are expressed in all cells, have an essential role in mitochondrial function, and their defects have low, overall, penetrance.
Supplementary Material
Footnotes
Supplementary material for this article is available online at http://www.thieme-connect.de/products.
Conflict of Interest
The authors declare no relevant conflicts of interest. This work was supported by the NICHD intramural research program.
References
- [1].WHO Classification of Tumours of Endocrine Organs WHO/IARC Classification of Tumours. Lloyd RV, Osamura RY, Klöppel GR. (eds) Volume 10: 4th Edition WHO; 2017 [Google Scholar]
- [2].Kantorovich V, Koch CA, Pacak K. Pheochromocytoma and Paraganglioma Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, Ed Source Endotext. Available from https://www.ncbi.nlm.nih.gov/pubmed/25905361
- [3].Vicha A, Musil Z, Pacak K. Genetics of pheochromocytoma and paraganglioma syndromes: New advances and future treatment options. Curr Opin Endocrinol Diabetes Obes 2013; 20: 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, Van der Mey A, Taschner PEM, Rubinstein WS, Myers EN, Richard CW III, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000; 287: 848–851 [DOI] [PubMed] [Google Scholar]
- [5].Niemann S, Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 2000; 26: 268–270 [DOI] [PubMed] [Google Scholar]
- [6].Astuti D, Latif F, Dallol A, Dahia PLM, Douglas F, George E, Sköldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 2001; 69: 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Burnichon N, Brière J-J, Libé R, Vescovo L, Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-Roqueplo A-P. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010; 19: 3011–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med 2007; 357: 1054–1056 [DOI] [PubMed] [Google Scholar]
- [9].Gill AJ, Hes O, Papathomas T, Šedivcová M, Tan PH, Agaimy A, Andresen PA, Kedziora A, Clarkson A, Toon CW, Sioson L, Watson N, Chou A, Paik J, Clifton-Blight RJ, Robinson BG, Benn DE, Hills K, Maclean F, Niemeijer ND, Vlatkovic L, Hartmann A, Corssmit EPM, van Leenders GJLH, Przbycin C, McKenney J, Magi-Galluzzi CM, Yilmaz A, Yu D, Nicoli KD, Yong JL, Sibony M, Yakirevich E, Fleming S, Chow CW, Miettinen M, Michal M, Trpkov K. Succinate dehydrogenase (SDH)-deficient renal carcinoma: A morphologically distinct entity: A clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol 2014; 38: 1588–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ozluk Y, Taheri D, Matoso A, Sanli O, Berker NK, Yakirevich E, Yakirevich E, Balasubramanian S, Ross JS, Ali SM, Netto GJ. Renal carcinoma associated with a novel succinate dehydrogenase A mutation: A case report and review of literature of a rare subtype of renal carcinoma. Hum Pathol 2015; 46: 1951–1955 [DOI] [PubMed] [Google Scholar]
- [11].Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peçzkowska M, Morrison CD, Lehtonen R, Januszewicz A, Järvinen H, Juhola M, Mecklin J-P, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HPH, Eng C. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 2004; 74: 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 2008; 100: 1260–1262 [DOI] [PubMed] [Google Scholar]
- [13].Niemeijer ND, Papathomas TG, Korpershoek E, De Krijger RR, Oudijk L, Morreau H, Bayley J-P, Hes FJ, Jansen JC, Dinjens WNM, Corssmit EPM. Succinate dehydrogenase (SDH)-deficient pancreatic neuroendocrine tumor expands the SDH-related tumor spectrum. J Clin Endocrinol Metab 2015; 100: E1386–E1393 [DOI] [PubMed] [Google Scholar]
- [14].Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, Platzer P, Orloff MS, Waite KA, Eng C. Germline mutations and mariants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet 2008; 83: 261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xekouki P, Pacak K, Almeida M, Wassif CA, Rustin P, Nesterova M, Sierra MDLL, Matro J, Ball E, Azevedo M, Horvath A, Lyssikatos C, Quezado M, Patronas N, Ferrando B, Pasini B, Lytras A, Tolis G, Stratakis CA. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: A new association for SDH? J Clin Endocrinol Metab 2012; 97: E357–E366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jochmanová I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J Natl Cancer Inst 2013; 105: 1270–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xekouki P, Stratakis CA. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: Could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer 2012; 19: C33–C40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gill AJ, Toon CW, Clarkson A, Sioson L, Chou A, Winship I, Robinson BG, Benn DE, Clifton-Bligh RJ, Dwight T. Succinate dehydrogenase deficiency is rare in pituitary adenomas. Am J Surg Pathol 2014; 38: 560–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Evenepoel L, Papathomas TG, Krol N, Korpershoek E, de Krijger RR, Persu A, Dinjens WNM. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet Med 2015; 17: 610–620 [DOI] [PubMed] [Google Scholar]
- [20].Niemeijer ND, Rijken JA, Eijkelenkamp K, van der Horst-Schrivers ANA, Kerstens MN, Tops CM, van Berkel A, Timmers HJ, Kunst HPM, Leemans CR, Bisschop PH, Dreijerink KM, van Dooren M, Bayley J-P, Pereira AM, Jansen J, Hes FJ, Hense E, Corssmit EP. The phenotype of SDHB germline mutation carriers: A nationwide study. Eur J Endocrinol 2017; 177: 115–125 [DOI] [PubMed] [Google Scholar]
- [21].Guerrero Pérez F, Lisbona Gil A, Robledo M, Iglesias P, Vilabona Artero C. Pituitary adenoma associated with pheochromocytoma/paraganglioma: A new form of multiple endocrine neoplasia. Endocrinol Nutr 2016; 63: 506–508 [DOI] [PubMed] [Google Scholar]
- [22].Dénes J, Swords F, Rattenberry E, Stals K, Owens M, Cranston T, Xekouki P, Moran L, Kumar A, Wassif C, Fersht N, Baldeweg SE, Morris D, Lightman S, Agha A, Rees A, Grieve J, Powell M, Boguszewski CL, Dutta P, Thakker RV, Srirangalingam U, Thompson CJ, Druce M, Higham C, Davis J, Eeles R, Stevenson M, O’Sullivan B, Taniere P, Skordilis K, Gabrovska P, Barlier A, Webb SM, Aulinas A, Drake WM, Bevan JS, Preda C, Dalantaeva N, Ribeiro-Oliveira A, Garcia IT, Yordanova G, Iotova V, Evanson J, Grossman AB, Trouillas J, Ellard S, Stratakis CA, Maher ER, Roncaroli F, Korbonits M. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J Clin Endocrinol Metab 2015; 100: E531–E541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dénes JSF, Xekouki P, Kumar AV, Maher ER, Ferscht N, Grieve J, Baldeweg SE, Stratakis CA, Korbonits M. Familial pituitary adenoma and paraganglioma syndrome—a novel type of multiple endocrine neoplasia. In: 94th Annual Meeting of The Endocrine Society: 2012; 203 (OR41–2) [Google Scholar]
- [24].Dwight T, Mann K, Benn DE, Robinson BG, McKelvie P, Gill AJ, Winship I, Clifton-Bligh RJ. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J Clin Endocrinol Metab 2013; 98: E1103–E1108 [DOI] [PubMed] [Google Scholar]
- [25].Papathomas TG, Gaal J, Corssmit EPM, Oudijk L, Korpershoek E, Heimdal K, Bayley JP, Morreau H, van Dooren M, Papaspyrou K, Schreiner T. Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC-PGL syndromes: A clinicopathological and molecular analysis. Eur J Endocrinol 2014; 170: 1–1224096523 [Google Scholar]
- [26].Xekouki P, Szarek E, Bullova P, Giubellino A, Quezado M, Mastroyannis SA, Mastorakos P, Wassif CA, Raygada M, Rentia N, Dye L. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in human and mice. J Clin Endocrinol Metab 2015; 100: E710–E719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].De Sousa SM, McCabe MJ, Wu K, Roscioli T, Gayevskiy V, Brook K, Rawlings L, Scott HS, Thompson TJ, Earls P, Gill AJ. Germline variants in familial pituitary tumour syndrome genes are common in young patients and families with additional endocrine tumours. Eur J Endocrinol 2017; 176: 635–644 [DOI] [PubMed] [Google Scholar]
- [28].Marx S, Spiegel AM, Skarulis MC, Doppman JL, Collins FS, Liotta LA. Multiple endocrine neoplasia type 1: Clinical and genetic topics. Annals of internal medicine 1998; 129: 484–494 [DOI] [PubMed] [Google Scholar]
- [29].Okada R, Shimura T, Tsukida S, Ando J, Kofunato Y, Momma T, Yashima R, Koyama Y, Suzuki S, Takenoshita S. Concomitant existence of pheochromocytoma in a patient with multiple endocrine neoplasia type 1. Surg Case Rep 2016; 2: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, Edwards CR, Heath DA, Jackson CE, Jansen S, Lips K. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM 1996; 89: 653–670 [DOI] [PubMed] [Google Scholar]
- [31].Jamilloux Y, Favier J, Pertuit M, Delage-Corre M, Lopez S, Teissier M-P, Mathonnet M, Galinat S, Barlier A, Archambeaud F. A MEN1 syndrome with a paraganglioma. Eur J Hum Genet 2014; 22: 283–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dackiw APB, Cote GJ, Fleming JB, Schultz PN, Stanford P, Vassilopoulou-Sellin R, Evans DB, Gagel RF, Lee JE. Screening for MEN1 mutations in patients with atypical endocrine neoplasia. Surgery 1999; 126: 1097–1104 [DOI] [PubMed] [Google Scholar]
- [33].Daly AF, Castermans E, Oudijk L, Guitelman MA, Beckers P, Potorac I, Neggers SJ, Sacre N, van der Lely AJ, Bours V, de Herder WW. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr Relat Cancer 2018; 25: L37–L42 [DOI] [PubMed] [Google Scholar]
- [34].Roszko KL, Blouch E, Blake M, Powers JF, Tischler AS, Hodin R, Sadow P, Lawson EA. Case Report of a prolactinoma in a patient with a novel MAX mutation and bilateral pheochromocytomas. J Endoc Soc 2017; 1: 1401–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kantorovich V, King KS, Pacak K. SDH-related pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab 2010; 24: 415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ziegler CG, Brown JW, Schally AV, Erler A, Gebauer L, Treszl A, Young L, Fishman LM, Engel JB, Willenberg HS, Petersenn S. Expression of neuropeptide hormone receptors in human adrenal tumors and cell lines: antiproliferative effects of peptide analogues. Proc Natl Acad Sci U S A 2009; 106: 15879–15884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Freddi S, Arnaldi G, Fazioli F, Scarpelli M, Appolloni G, Mancini T, Kola B, Bertagna X, Mantero F, Collu R, Boscaro M. Expression of growth hormone-releasing hormone receptor splicing variants in human primary adrenocortical tumours. Clin Endocrinol 2005; 62: 533–538 [DOI] [PubMed] [Google Scholar]
- [38].Ueberberg B, Unger N, Sheu SY, Walz MK, Schmid KW, Saeger W, Mann K, Petersenn S. Differential expression of ghrelin and its receptor (GHS-R1a) in various adrenal tumors and normal adrenal gland. Horm Metab Res 2008; 40: 181–188 [DOI] [PubMed] [Google Scholar]
- [39].O’Toole SM, Dénes J, Robledo M, Stratakis CA, Korbonits M. The association of pituitary adenomas and phaeochromocytomas or paragangliomas. Endocr Relat Cancer 2015; 22: T105–T122 [DOI] [PubMed] [Google Scholar]
- [40].Zhang H, Bosch-Marce M, Shimoda LA, Yee ST, Jin HB, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283: 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [41].Zakeri Z, Lockshin RA. Cell death: History and future. Adv Exp Med Biol 2008; 615: 1–11 [DOI] [PubMed] [Google Scholar]
- [42].Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ, Kim YS, Chung KY. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir Res 2015; 16: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chaachouay H, Fehrenbacher B, Toulany M, Schaller M, Multhoff G, Rodemann HP. AMPK-independent autophagy promotes radioresistance of human tumor cells under clinical relevant hypoxia in vitro. Radiother Oncol 2015; 116: 409–416 [DOI] [PubMed] [Google Scholar]
- [44].van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, Sleddens HF, Derkx P, Rivière J, Dannenberg H, Petri BJ. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol 2009; 10: 764–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, Richardson AL, Sidhu SB, Robinson BG, Clifton-Bligh RJ. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol 2010; 41: 805–814 [DOI] [PubMed] [Google Scholar]
- [46].Richter S, Peitzsch M, Rapizzi E, Lenders JW, Qin N, De Cubas AA, Schiavi F, Rao JU, Beuschlein F, Quinkler M, Timmers HJ. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J Clin Endocrinol Metab 2014; 99: 3903–3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ryland GL, Doyle MA, Goode D, Boyle SE, Choong DYH, Rowley SM, Li J, Bowtell DD, Tothill RW, Campbell IG, Gorringe KL. Loss of heterozygosity: what is it good for? BMC Med Genom 2015; 8: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Grogan RH, Pacak K, Pasche L, Huynh TT, Greco RS. Bilateral Adrenal Medullary Hyperplasia Associated With an SDHB Mutation. J Clin Oncol 2011; 29: E200–E202 [DOI] [PubMed] [Google Scholar]
- [49].Weber A, Hoffmann MM, Neumann HPH, Erlic Z. Somatic mutation analysis of the SDHB, SDHC, SDHD, and RET genes in the clinical assessment of sporadic and hereditary pheochromocytoma. Horm Cancer 2012; 3: 187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, Baudin E. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008; 16: 79–88 [DOI] [PubMed] [Google Scholar]
- [51].Haller F, Moskalev EA, Faucz FR, Barthelmeß S, Wiemann S, Bieg M, Assie G, Bertherat J, Schaefer IM, Otto C, Rattenberry E. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer 2014; 21: 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Buffet A, Smati S, Mansuy L, Ménara M, Lebras M, Heymann MF, Simian C, Favier J, Murat A, Cariou B, Gimenez-Roqueplo AP. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab 2014; 99: E369–E373 [DOI] [PubMed] [Google Scholar]
- [53].Lecoq AL, Zizzari P, Hage M, Decourtye L, Adam C, Viengchareun S, Veldhuis JD, Geoffroy V, Lombès M, Tolle V, Guillou A. Mild pituitary phenotype in 3- and 12-month-old Aip-deficient male mice. J Endocrinol 2016; 231: 59–69 [DOI] [PubMed] [Google Scholar]
- [54].Lloyd RV, Ruebel KH, Zhang S, Jin L. Pituitary hyperplasia in glycoprotein hormone alpha subunit-, p18(INK4C)-, and p27(kip-1)-null mice: Analysis of proteins influencing p27(kip-1) ubiquitin degradation. Am J Pathol 2002; 160: 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Donangelo I, Gutman S, Horvath E, Kovacs K, Wawrowsky K, Mount M, Melmed S. Pituitary tumor transforming gene overexpression facilitates pituitary tumor development. Endocrinology 2006; 147: 4781–4791 [DOI] [PubMed] [Google Scholar]
- [56].Bai F, Chan HL, Smith MD, Kiyokawa H, Pei X-H. p19Ink4d is a tumor suppressor and controls pituitary anterior lobe cell proliferation. Mol Cell Biol 2014; 34: 2121–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci U S A 2011; 108: 11482–11487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, Schernthaner-Reiter MH, Szarek E, Leal LF, Caberg JH, Castermans E. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med 2014; 371: 2363–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Stergiopoulos SG, Abu-Asab MS, Tsokos M, Stratakis CA. Pituitary pathology in Carney complex patients. Pituitary 2004; 7: 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Villa C, Lagonigro MS, Magri F, Koziak M, Jaffrain-Rea ML, Brauner R, Bouligand J, Junier MP, Di Rocco F, Sainte-Rose C, Beckers A. Hyperplasia-adenoma sequence in pituitary tumorigenesis related to aryl hydrocarbon receptor interacting protein gene mutation. Endocr Relat Cancer 2011; 18: 347–356 [DOI] [PubMed] [Google Scholar]
- [61].Jochmanová I, Zhuang Z, Pacak K. Pheochromocytoma: Gasping for air. Horm Cancer 2015; 6: 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Szarek E, Ball ER, Imperiale A, Tsokos M, Faucz FR, Giubellino A, Moussallieh FM, Namer IJ, Abu-Asab MS, Pacak K, Taïeb D. Carney triad, SDH-deficient tumors, and Sdhb + /− mice share abnormal mitochondria. Endocr Relat Cancer 2015; 22: 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Grandemange S, Herzig S, Martinou JC. Mitochondrial dynamics and cancer. Seminars Cancer Biol 2009; 19: 50–56 [DOI] [PubMed] [Google Scholar]
- [64].Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 2008; 8: 180–192 [DOI] [PubMed] [Google Scholar]
- [65].Lenders JWM, Duh Q-Y, Eisenhofer G, Gimenez-Roqueplo A-P, Grebe SKG, Murad MH, Naruse M, Pacak K, Young WF Jr.. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99: 1915–1942 [DOI] [PubMed] [Google Scholar]
- [66].Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JWM, Lussey-Lepoutre C, Steichen O. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016; 174: G1–G10 [DOI] [PubMed] [Google Scholar]
- [67].Aghi MK, Chen CC, Fleseriu M, Newman SA, Lucas JW, Kuo JS, Barkhoudarian G, Farrell CJ, Sheehan J, Ziu M, Dunn IF. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on the Management of Patients with Nonfunctioning Pituitary Adenomas: Executive Summary. Neurosurgery 2016; 79: 521–523 [DOI] [PubMed] [Google Scholar]
- [68].Katznelson L, Laws ER, Melmed S, Molitch ME, Murad MH, Utz A, Wass JA. Acromegaly: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99: 3933–3951 [DOI] [PubMed] [Google Scholar]
- [69].Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, Wass JA. Diagnosis and treatment of hyperprolactinemia: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011; 96: 273–288 [DOI] [PubMed] [Google Scholar]
- [70].Nieman LK, Biller BMK, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A. Treatment of cushing’s syndrome: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2015; 100: 2807–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].The Endocrine Society. Pituitary incidentaloma: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2011; 96: 894–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Casey RT, Warren AY, Martin JE, Challis BG, Rattenberry E, Whitworth J, Andrews KA, Roberts T, Clark GR, West H, Smith PS. Clinical and Molecular Features of Renal and Pheochromocytoma/Paraganglioma Tumor Association Syndrome (RAPTAS): Case Series and Literature Review. J Clin Endocrinol Metab 2017; 102: 4013–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rostomyan L, Beckers A. Screening for genetic causes of growth hormone hypersecretion. Growth Horm IGF Res 2016; 30: 52–57 [DOI] [PubMed] [Google Scholar]
- [74].Goodwin S, McPherson JD, McCombie WR. Coming of age: Ten years of next-generation sequencing technologies. Nat Rev Genet 2016; 17: 333–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Beckers A. Means, motive, and opportunity: SDH mutations are suspects in pituitary tumors. J Clin Endocrinol Metab 2013; 98: 2274–2276 [DOI] [PubMed] [Google Scholar]
- [76].Gorospe L, Cabañero-Sánchez A, Muñoz-Molina GM, Pacios-Blanco RE, Ureña Vacas A, García-Santana E. An unusual case of mediastinal paraganglioma and pituitary adenoma. Surgery 2017; 162: 1338–1339 [DOI] [PubMed] [Google Scholar]
- [77].Efstathiadou ZA, Sapranidis M, Anagnostis P, Kita MD, Rosenthal EL. Unusual case of Cowden-like syndrome, neck paraganglioma, and pituitary adenoma. Head Neck 2014; 36: E12–E16 [DOI] [PubMed] [Google Scholar]
- [78].López-Jiménez E, De Campos JM, Kusak EM, Landa I, Leskelä S, Montero-Conde C, Leandro-Garcia LJ, Vallejo LA, Madrigal B, Rodríguez-Antona C, Robledo M, Cascón A. SDHC mutation in an elderly patient without familial antecedents. Clin Endocrinol 2008; 69: 906–910 [DOI] [PubMed] [Google Scholar]
- [79].Varsavsky M, Sebastián-Ochoa A, Torres Vela E. Coexistence of a pituitary macroadenoma and multicentric paraganglioma: A strange coincidence. Endocrinología y Nutrición 2013; 60: 154–156 [DOI] [PubMed] [Google Scholar]
- [80].Jeong YJ, Oh HK, Bong JG. Multiple endocrine neoplasia type 1 associated with breast cancer: A case report and review of the literature. Oncol Lett 2014; 8: 230–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Langer P, Cupisti K, Bartsch DK, Nies C, Goretzki PE, Rothmund M, Röher HD. Adrenal Involvement in Multiple Endocrine Neoplasia Type 1. World J Surg 2002; 26: 891. [DOI] [PubMed] [Google Scholar]
- [82].Carty SE, Helm AK, Amico JA, Clarke MR, Foley TP, Watson CG, Mulvihill JJ, Marx S, Skogseid B. The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery 1998; 124: 1106–1114 [DOI] [PubMed] [Google Scholar]
- [83].Heinlen JE, Buethe DD, Culkin DJ, Slobodov G. Multiple endocrine neoplasia 2a presenting with pheochromocytoma and pituitary macroadenoma. ISRN Oncology 2011; 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Naziat A, Karavitaki N, Thakker R, Ansorge O, Sadler G, Gleeson F, Cranston T, McCormack A, Grossman AB, Shine B. Confusing genes: A patient with MEN2A and Cushing’s disease. Clin Endocrinol 2013; 78: 966–968 [DOI] [PubMed] [Google Scholar]
- [85].1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Koshy AN, Cheng VE, Sajeev JK, Venkataraman P, Profitis K. Bilateral pheochromocytoma and paraganglioma: A rare cause of cardiomyopathy. Eur Heart J Cardiovasc Imag 2016; 17: 705. [DOI] [PubMed] [Google Scholar]
- [87].Boguszewski CL, Fighera TM, Bornschein A, Marques FM, Dénes J, Rattenbery E, Maher ER, Stals K, Ellard S, Korbonits M. Genetic studies in a coexistence of acromegaly, pheochromocytoma, gastrointestinal stromal tumor (GIST) and thyroid follicular adenoma. Arq Bras Endocrinol Metabol 2012; 56: 507–512 [DOI] [PubMed] [Google Scholar]
- [88].Parghane RV, Agrawal K, Mittal BR, Shukla J, Bhattacharya A, Mukherjee KK. 68Ga DOTATATE PET/CT in a rare coexistence of pituitary macroadenoma and multiple paragangliomas. Clin Nuc Med 2014; 39: 91–93 [DOI] [PubMed] [Google Scholar]
- [89].Mumby C, Davis JRE, Trouillas J, Higham CE. Phaeochromocytoma and acromegaly: A unifying diagnosis. Endocrinol Diabetes Metab Case Rep 2014; 140036: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Sisson JC, Giordano TJ, Avram AM. Three endocrine neoplasms: an unusual combination of pheochromocytoma, pituitary adenoma, and papillary thyroid carcinoma. Thyroid 2012; 22: 430–436 [DOI] [PubMed] [Google Scholar]
- [91].Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, Young J, Delemer B, Du Boullay H, Verger MF, Kuhn JM, Sadoul JL, Ruszniewski P, Beckers A, Monsaingeon M, Baudin E, Goudet P, Tabarin A. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d’étude des Tumeurs Endocrines database. Eur J Endocrinol 2012; 166: 269–279 [DOI] [PubMed] [Google Scholar]
- [92].Zhang C, Ma G, Liu X, Zhang H, Deng H, Nowell J, Miao Q. Primary cardiac pheochromocytoma with multiple endocrine neoplasia. J Cancer Res Clin Oncol 2011; 137: 1289–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yaylali GF, Akin F, Bastemir M, Yaylali YT, Ozden A. Phaeochromocytoma combined with subclinical Cushing’s syndrome and pituitary microadenoma. Clin Invest Med 2008; 31: 176–181 [DOI] [PubMed] [Google Scholar]
- [94].Breckenridge SM, Hamrahian AH, Faiman C, Suh J, Prayson R, Mayberg M. Coexistence of a pituitary macroadenoma and pheochromocytoma – a case report and review of the literature. Pituitary 2003; 6: 221–225 [DOI] [PubMed] [Google Scholar]
- [95].Dünser MW, Mayr AJ, Gasser R, Rieger M, Friesenecker B, Hasibeder WR. Cardiac failure and multiple organ dysfunction syndrome in a patient with endocrine adenomatosis. Acta Anaesthesiol Scand 2002; 46: 1161–1164 [DOI] [PubMed] [Google Scholar]
- [96].Sleilati GG, Kovacs KT, Honasoge M. Acromegaly and pheochromocytoma: Report of a rare coexistence. Endocr Pract 2002; 8: 54–60 [DOI] [PubMed] [Google Scholar]
- [97].Baughan J, De Gara C, Morrish D. A rare association between acromegaly and pheochromocytoma. Am J Surg 2001; 182: 185–187 [DOI] [PubMed] [Google Scholar]
- [98].Khalil WKA, Vadasz J, Rigo E, Kardos L, Tiszlavicz L, Gaspar L. Pheochromocytoma combined with unusual form of Cushing’s syndrome and pituitary microadenoma. Eur J Endocrinol 1999; 141: 653–654 [DOI] [PubMed] [Google Scholar]
- [99].Teh BT, Hansen J, Svensson PJ, Hartley L. Bilateral recurrent phaeochromocytoma associated with a growth hormone-secreting pituitary tumour. Br J Surg 1996; 83: 1132. [DOI] [PubMed] [Google Scholar]
- [100].Azzarelli B, Felten S, Muller J, Miyamoto R, Purvin V. Dopamine in paragangliomas of the glomus jugulare. Laryngoscope 1988; 98: 573–578 [DOI] [PubMed] [Google Scholar]
- [101].Bertrand JH, Ritz P, Reznik Y, Grollier G, Potier JC, Evrad C, Mahoudeau JA. Sipple’s syndrome associated with a large prolactinoma. Clin Endocrinol 1987; 27: 607–614 [DOI] [PubMed] [Google Scholar]
- [102].Larraza-Hernandez O, Albores-Saavedra J, Benavides G, Krause LG, Perez-Merizaldi JC, Ginzo A. Multiple endocrine neoplasia. Pituitary adenoma, multicentric papillary thyroid carcinoma, bilateral carotid body paraganglioma, parathyroid hyperplasia, gastric leiomyoma, and systemic amyloidosis. Am J Clin Pathol 1982; 78: 527–532 [DOI] [PubMed] [Google Scholar]
- [103].Meyers DH. Association of phaeochromocytoma and prolactinoma. MJA 1982; 1: 13–14 [DOI] [PubMed] [Google Scholar]
- [104].Blumenkopf B, Boekelheide K. Neck paraganglioma with a pituitary adenoma. Case report. J Neurosurg 1982; 57: 426–429 [DOI] [PubMed] [Google Scholar]
- [105].Anderson RJ, Lufkin EG, Sizemore GW, Carney JA, Sheps SG, Silliman YE. Acromegaly and pituitary adenoma with phaeochromocytoma: A variant of multiple endocrine neoplasia. Clin Endocrinol 1981; 14: 605–612 [DOI] [PubMed] [Google Scholar]
- [106].Myers JH, Eversman JJ. Acromegaly, hyperparathyroidism, and pheochromocytoma in the same patient. A multiple endocrine disorder. Arch Intern Med 1981; 141: 1521–1522 [PubMed] [Google Scholar]
- [107].Alberts WM, McMeekin JO, George JM. Mixed multiple endocrine neoplasia syndromes. JAMA 1980; 244: 1236–1237 [PubMed] [Google Scholar]
- [108].Janson KL, Roberts JA, Varela M. Multiple endocrine adenomatosis: in support of the common origin theories. J Urol 1978; 119: 161–165 [DOI] [PubMed] [Google Scholar]
- [109].Manger WM, Gifford RW. Studies on 38 patients with pheochromocytoma in whom catecholamines were determined In: Pheochromocytoma. New York, NY: Springer; 1977: 276–295 [Google Scholar]
- [110].Melicow MM. One hundred cases of pheochromocytoma (107 tumors) at the Columbia-Presbyterian Medical Center, 1926–1976: A clinicopathological analysis. Cancer 1977; 40: 1987–2004 [DOI] [PubMed] [Google Scholar]
- [111].Kadowaki S, Baba Y, Kakita T, Yamamoto H, Fukase M, Goto Y, Seino Y, Kato Y, Matsukara S, Imura H. A case of acromegaly associated with pheochromocytoma [in Japanese]. Saishin Igaku 1976; 31: 1402–1409 [Google Scholar]
- [112].Farhi F, Dikman SH, Lawson W, Cobin RH, Zak FG. Paragangliomatosis associated with multiple endocrine adenomas. Arch Pathol Lab Med 1976; 100: 495–498 [PubMed] [Google Scholar]
- [113].Berg B, Biörklund A, Grimelius L, Ingemansson S, Larsson LI, Stenram U, Akerman M. A new pattern of multiple endocrine adenomatosis: Chemodectoma, bronchial carcinoid, GH-producing pituitary adenoma, and hyperplasia of the parathyroid glands, and antral and duodenal gastrin cells. Acta Med Scand 1976; 200: 321–326 [PubMed] [Google Scholar]
- [114].Wolf LM, Duduisson M, Schrub JC, Metayer J, Laumonier R. Sipple’s syndrome associated with pituitary and parathyroid adenomas. Ann Endocrinol 1972; 33: 455–463 [PubMed] [Google Scholar]
- [115].Miller GL, Wynn J. Acromegaly, pheochromocytoma, toxic goiter, diabetes mellitus, and endometriosis. Arch Intern Med 1971; 127: 299–303 [PubMed] [Google Scholar]
- [116].Steiner A, Goodman A, Powers S. Study of a kindred with pheochromocytoma, medullary thyroid carcinoma, hyperparathyroidism and Cushing’s disease: multiple endocrine neoplasia, type 2. Medicine 1968; 47: 371–409 [DOI] [PubMed] [Google Scholar]
- [117].Kahn MT, Mullon DA. Phoechromocytoma without hypertension. Report of a patient with acromegaly JAMA 1964; 188: 74–75 [PubMed] [Google Scholar]
- [118].German WJ, Flanigan S. Pituitary adenomas: A follow-up study of the Cushing series. Clin Neurosurg 1964; 10: 72–81 [DOI] [PubMed] [Google Scholar]
- [119].Iversen K Acromegaly associated with phaeochromocytoma. Acta Med Scand 1952; 142: 1–5 [DOI] [PubMed] [Google Scholar]
- [120].Bayley JP, Devilee P, Taschner PEM. The SDH mutation database: An online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med Genet 2005; 6: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Benn DE, Gimenez-Roqueplo A-P, Reilly JR, Bertherat J, Burgess J, Byth K, Croxson M, Dahia PLM, Elston M, Henley OGD, Herman P, Murday V, Niccoli-Sire P, Pasieka JL, Rohmer V, Tucker K, Jeunemaitre X, Marsh DJ, Plouin PF, Robinson BG. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006; 91: 827–836 [DOI] [PubMed] [Google Scholar]
- [122].Ni Y, He X, Chen J, Moline J, Mester J, Orloff MS, Ringel MD, Eng C. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum Mol Genet 2012; 21: 300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.