

Abstract
Epoxyeicosatrienoic acids (EETs) are epoxy fatty acids that have biological actions that are essential for maintaining water and electrolyte homeostasis. An inability to increase EETs in response to a high salt diet results in salt-sensitive hypertension. Vasodilation, inhibition of epithelial sodium channel, and inhibition of inflammation are the major EET actions that are beneficial to the heart, resistance arteries, and kidneys. Genetic and pharmacological means to elevate EETs demonstrated anti-hypertensive, anti-inflammatory, and organ protective actions. Therapeutic approaches to increase EETs were then developed for cardiovascular diseases. Soluble epoxide hydrolase (sEH) inhibitors were developed and progressed to clinical trials for hypertension, diabetes, and other diseases. EET analogs were another therapeutic approach taken and these drugs are entering the early phases of clinical development. Even with the promise for these therapeutic approaches, there are still several challenges, unexplored areas, and opportunities for epoxy fatty acids.
Keywords: epoxyeicosatrienoic acid, natriuresis, endothelium, inflammation, sodium channels
INTRODUCTION
Kidney and cardiovascular diseases are inextricably linked: the kidney is essential for renin secretion, as well as water and electrolyte homeostasis. Human and animal studies conducted by Dr. Lewis K. Dahl demonstrated that excessive salt intake contributed to hypertension.1,2 Over a half century since these key findings by Dr. Dahl and other investigators connecting salt, kidney, and hypertension there has been strides in the treatment of cardiovascular and kidney diseases. Salt restriction and diuretics represented successful approaches to treating hypertension and preventing cardiovascular events.3,4 Nevertheless, the cardiovascular disease hypertension still afflicts one in every four people and remains the leading cause of death worldwide.5,6 Chronic hypertension is a leading contributor to cardiovascular, renal, and cerebral disease morbidity, and mortality, and prevalence and multiple medical and socio-economic consequences make it a major health challenge. While it is widely accepted that early clinical intervention minimizes the devastating consequences of hypertension, a lack of obvious symptoms complicates its timely diagnosis and the prevalence of uncontrolled hypertension is approximately 50%.7,8 A significant portion of patients with hypertension develop chronic kidney disease (CKD) and progress to end-stage renal disease.9,10 Kidney diseases such as CKD afflict one out of every ten Americans and continue to lack proper treatments.9 Accordingly, there continues to be a significant need for finding new approaches to treat cardiovascular and kidney diseases.
Common dysfunctional mechanisms that contribute to cardiovascular and kidney diseases include water and electrolyte regulation, inflammation, and endothelial function. Humans over recent centuries have adapted to a dramatic increase in sodium intake and a reduction in potassium intake.11,12 The kidney’s relative inability to adapt to this change in dietary salt has led to a substantial increase in salt-sensitive hypertension.12 Evidence from genetic polymorphisms and monogenic hypertension has led to the postulate that hypertension is the consequence of multiple genetic polymorphisms that in aggregate result in defective kidney sodium excretion.13,14,15 Direct evidence that impaired kidney function contributes to hypertension has been demonstrated in kidney transplantation studies both in rats and humans.16,17 Inflammation – in particular kidney inflammation – has been demonstrated to contribute to hypertension and kidney diseases.18,19,20 Likewise, endothelial dysfunction contributes to salt-sensitive hypertension and is a strong indicator of poor kidney and cardiovascular health.21,22 These dysfunctional mechanisms are likely interrelated and act in concert to cause and contribute to the progression of cardiovascular and kidney diseases.
Epoxy fatty acids provide a pathway that can improve water and electrolyte regulation, decrease inflammation, and improve endothelial function in salt-sensitive hypertension and CKD.23,24 Likewise, genetic manipulation decreasing epoxy fatty acids and decreasing kidney epoxy fatty acids have been associated with salt-sensitive hypertension.24,25 Experimental evidence in animal models and humans revealed that genetic or pharmacological manipulation increasing bioavailability of epoxy fatty acids, epoxyeicosatrienoic acids (EETs), improves cardiovascular and renal function.25,26,27 The focus of this review is to present how evaluating EET actions in salt regulation evolved into the development of therapeutics that demonstrate great promise for treating kidney and cardiovascular diseases.
EPOXY FATTY ACIDS – BIOLOGICAL ACTIONS
Cytochrome P450 (CYP) epoxygenase enzymes form epoxides by acting on carbon double bonds of fatty acids. Arachidonic acid is the most abundant fatty acid in mammals and is acted upon by CYP epoxygenase enzymes to form four regioisomeric EETs (Table 1).28 The major CYP epoxygenase enzymes are the CYP2C and CYP2J; CYP2C is the main epoxygenase enzyme responsible for kidney and endothelial EET generation.29,30 Of the four EET regioisomers, 11,12-EET and 14,15-EET are the major products of the human CYP2C8 and CYP2C9, the mouse Cyp2C44 and the rat CYP2C23.29,30,31 Following generation, EETs can then be acted upon by soluble epoxide hydrolase (sEH), which adds water across the epoxide bond forming a diol (DHET). The rank order affinity for EETs for sEH is: 14,15-EET > 11,12-EET > 8,9-EET > 5,6-EET.23 In general, EETs are biologically active, whereas DHETs are less active or inactive in terms of vascular, renal, and inflammatory actions.22,23
Table 1.
Epoxy fatty acid and therapeutic structures
| Classes of Eicosanoid Molecules | Molecules | Structure Activity Relationship | References | |
|---|---|---|---|---|
| Regioisomeric Epoxyeicosatrienoic Acids (EETs) Dihydroxyeicosatrienoic Acids (DHETs) |
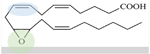
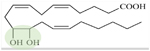
5,6-EET 8,9-EET 11,12-EET 14,15-EET 5,6-DHET 8,9-DHET 11,12-DHET 14,15-DHET |
34–39, 46, 49–61, 117, 158 | ||
| DHETs are sEH products of epoxide bond converted to diol (11,12-DHET, green). | ||||
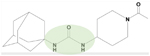
| Soluble Epoxide Hydrolase (sEH) Inhibitors | AUDA AR9281 GSK2256294 EC5026 |
AUDA is a urea-based sEH inhibitor (green). sEH inhibitors include amide, piperidyl, and aminoheteroaryl inhibitors. | 82–91, 94–96, 98–101, 103–109, 111–116, 142, 146 | |
| EET Analogs | NUDSA EET-A EET-B |
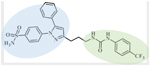
EET-A is 14,15-EET analog with a urea (green) that mimics the epoxide and resist she activity and a double bond at the 8,9 position (blue). | 119–133, 135, 166 | |
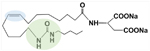
| Dual Modulator Soluble Epoxide Hydrolase (sEH) Inhibitors | PTUPB RB394 DM509 |
PTUPB is a dual sEH inhibitor (green) and COX-2 inhibitor (blue) with a linker. | 143, 144, 165, 167, 168 | |
The finding that EETs are endothelium-derived hyperpolarizing factors (EDHFs) resulted in extensive evaluation in several organ vasculatures.32,33 Early studies demonstrated that EETs – in particular 11,12-EET and 14,15-EET – vasodilate renal, coronary, cerebral, and mesenteric arterioles (Figure 1).23 Renal afferent arterioles dilated to 11,12-EET and 14,15-EET but failed to respond to their corresponding DHETs.34 Finding that DHETs were inactive provided initial evidence that inhibiting sEH could increase the EET-mediated vasodilation. Although 8,9-EET and 5,6-EET have been demonstrated to be vasodilatory in some vasculatures, it was found that in renal afferent arterioles 8,9-EET was inactive and 5,6-EET caused cyclooxygenase-dependent (COX) constriction of renal afferent arterioles.34,35,36,37 As for the cellular site of action, dilation in renal arterioles and other arterioles by EETs was determined to be due to a direct action on vascular smooth muscle cells.23,32,33,34 Cell signaling mechanisms responsible for EET EDHF dilation include vascular smooth muscle cell protein kinase A (PKA) activating large-conductance K+ (BKCa) channels to cause hyperpolarization.38,39 Another important aspect of EET vascular action is interactions with hormonal and paracrine vasodilators and vasoconstrictors. Vasodilation by bradykinin depends on endothelial EET release.33,40 Importantly, an EET contribution to bradykinin vasodilation has been verified in humans.41,42 EETs also contribute to the bradykinin-dependent vascular actions under conditions of angiotensin converting enzyme (ACE) inhibition.43,44 EETs also oppose the vasoconstrictor actions of endothelin and angiotensin II.45,46 The findings that EETs act as EDHFs and oppose vasoconstrictors involved in renal and cardiovascular diseases provided impetus for testing if increasing EETs could provide beneficial actions in these disease states.
Figure 1.
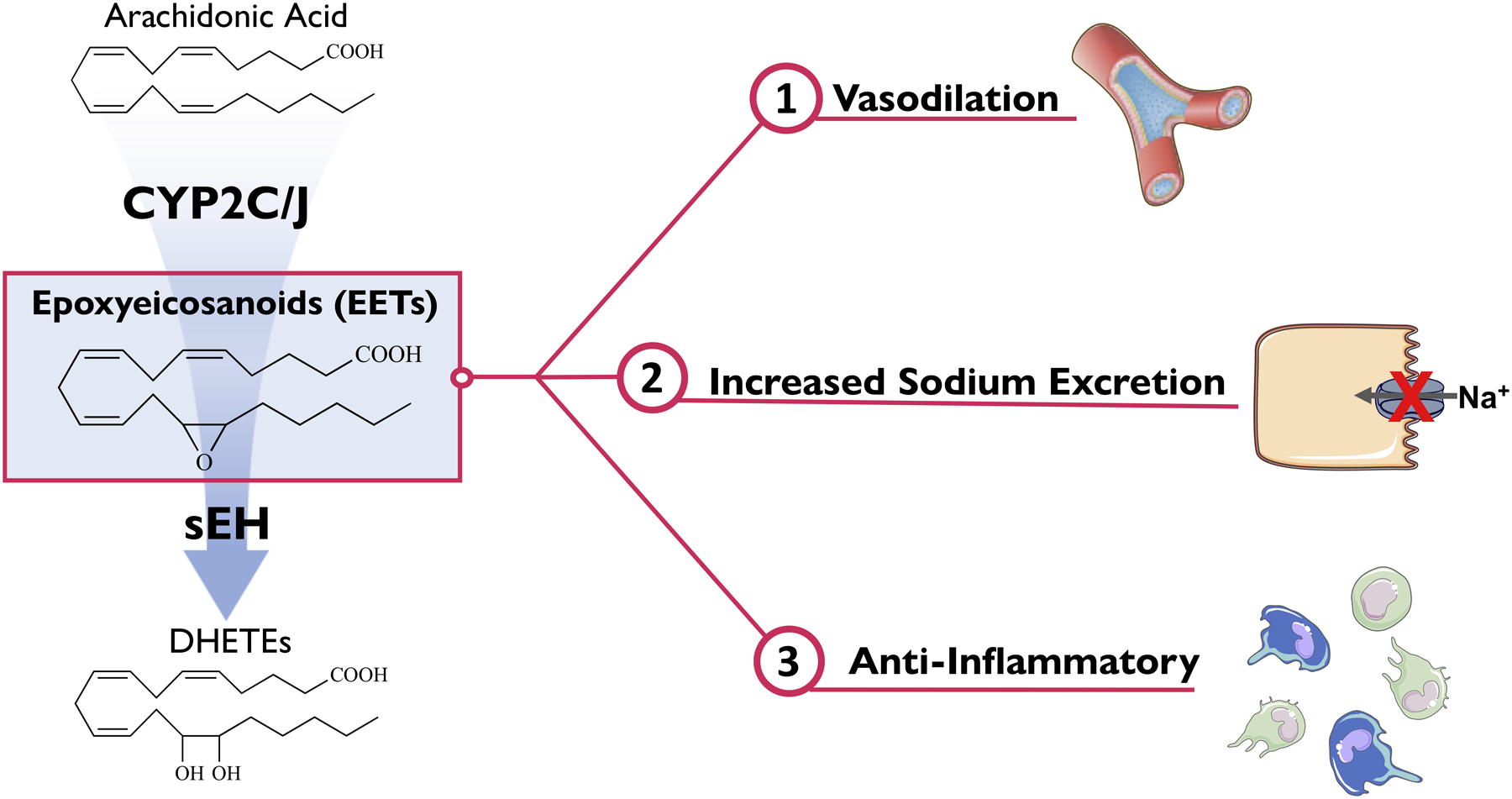
Epoxyeicosanoids (EETs) cause vasodilation, increased sodium excretion, and are anti-inflammatory.
Renal epithelial EET actions promote natriuresis to maintain water and electrolyte homeostasis (Figure 1). EETs have been demonstrated to have epithelial actions on proximal and distal tubules.24,47 EETs have been demonstrated to have epithelial actions on proximal and distal tubules.48,49,50,51 Initial studies found that EETs inhibit the proximal tubule Na+-K+ ATPase.49,52 EETs were also demonstrated to mediate the angiotensin II decrease Na+/H+ exchange in proximal tubule cells.49 Early studies on distal nephron segments demonstrated that 5,6-EET inhibited apical sodium transport in collecting duct cells.53 These 5,6-EET epithelial actions were COX-dependent similarly to 5,6-EET’s vascular actions.53 More recently, experimental studies have focused on the significant collecting duct epithelial actions of 11,12-EET.50,51,54 11,12-EET induces natriuresis through extracellular signal regulated kinase (ERK1/2)-dependent effects on the collecting duct epithelial sodium channel (ENaC).54 Although 11,12-EET has been consistently demonstrated to inhibit ENaC using electrophysiological approaches, 14,15-EET was inactive when evaluated in isolated rat collecting ducts but 14,15-EET inhibited ENaC when evaluated in immortalized mpk-CCDc14 collecting duct cells.48,51 Epithelial basolateral inward rectifying K+ channels located along the convoluted tubule and collecting duct are inhibited by EETs.47,55 11,12-EET inhibits the basolateral K+ channels resulting in cell membrane depolarization and reduction in the driving force for apical Na+ reabsorption.55 11,12-EET could contribute to renal K+ secretion by stimulating epithelial collecting duct principal cell BKCa channels.56 Consequently, EETs renal epithelial actions are important in the regulation of plasma Na+ and K+ levels to maintain fluid homeostasis and blood pressure.
Anti-inflammatory EET actions are critical in combating hypertension and progressive kidney diseases (Figure 1). Initial studies described the ability for 11,12-EET to decrease adherent mononuclear cells in mice carotid arteries after TNFα administration57. Comparison between 11,12-EET to 14,15-EET and VCAM-1 blocking antibody demonstrated that 11,12-EET was superior to 14,15-EET and was as effective as VCAM-1 blocking antibody in decreasing adherent inflammatory cells.57 11,12-EET was also found to decrease endothelial cell VCAM-1 expression and NFκB promoter activity.57 In rat pulmonary artery endothelial cell 11,12-EET and 14,15-EET suppressed oxidized-LDL leukotriene B4 production and activity through inhibiting p38 MAPK phosphorylation NFκB activation and inhibiting 5-lipoxygenase (5-LO).58 11,12-EET also opposes lipopolysaccharide induced M1 macrophage polarization and pro-inflammatory cytokines at the transcriptional and post-transcriptional level.59 Lung inflammation during ischemia reperfusion injury or induced by cigarette smoke is decreased by 11,12-EET or 14,15-EET.60,61 EETs act by decreasing cytokines via inhibiting NFκB activation and increasing anti-inflammatory proteins Nrf2 and heme oxygenase-1 (HO-1).61 These anti-inflammatory actions support the notion that increasing EET levels could combat those cardiovascular and renal diseases which have a significant inflammatory contribution.
The vascular, renal epithelial, and inflammatory biological actions supported the overarching idea that decreased EETs could contribute significantly to hypertension, vascular inflammatory diseases, and acute and progressive kidney diseases (Figure 2). There have been several studies that link vascular dysfunction, inflammation, and impaired natriuresis to salt-sensitive hypertension.15,24,47 For example, the vasoconstrictor agents can induce renal microvascular and interstitial inflammation to impair epithelial transport and sodium excretion.15,24 Angiotensin II hypertension is salt-sensitive. Remarkably, anti-inflammatory agents improve renal microvascular function, prevent the renal interstitial inflammatory response, and eliminate salt-sensitive hypertension.18,19 Interestingly, EET generation and regulation by sEH at the endothelial cell, epithelial cell, and inflammatory cell level could influence renal and cardiovascular function and blood pressure regulation.23,25 Interactions between inflammation, vascular function, and epithelial sodium transport can be regulated by EETs.23,25 In IgA nephropathy the level of renal epithelial cell sEH expression positively correlates with proteinuria and macrophage infiltration.62 Likewise, decreased endothelial cell EET production causes dysfunction and contributes to infiltration of inflammatory cells into the vascular smooth muscle cells.25,63 Macrophages also have the ability to generate EETs and reduced macrophage EET generation results in a profibrotic macrophage transcriptome.64 Several studies in animal disease models and humans have consistently found that decreased EETs or increased sEH activity contributes to hypertension and the progression of cardiovascular and renal diseases.23,24 Therefore, decreases in EET generation or increases in sEH activity can impact vascular and kidney function as well as the inflammatory state in a manner linked to water and electrolyte homeostasis and blood pressure control.
Figure 2. Left Panel:
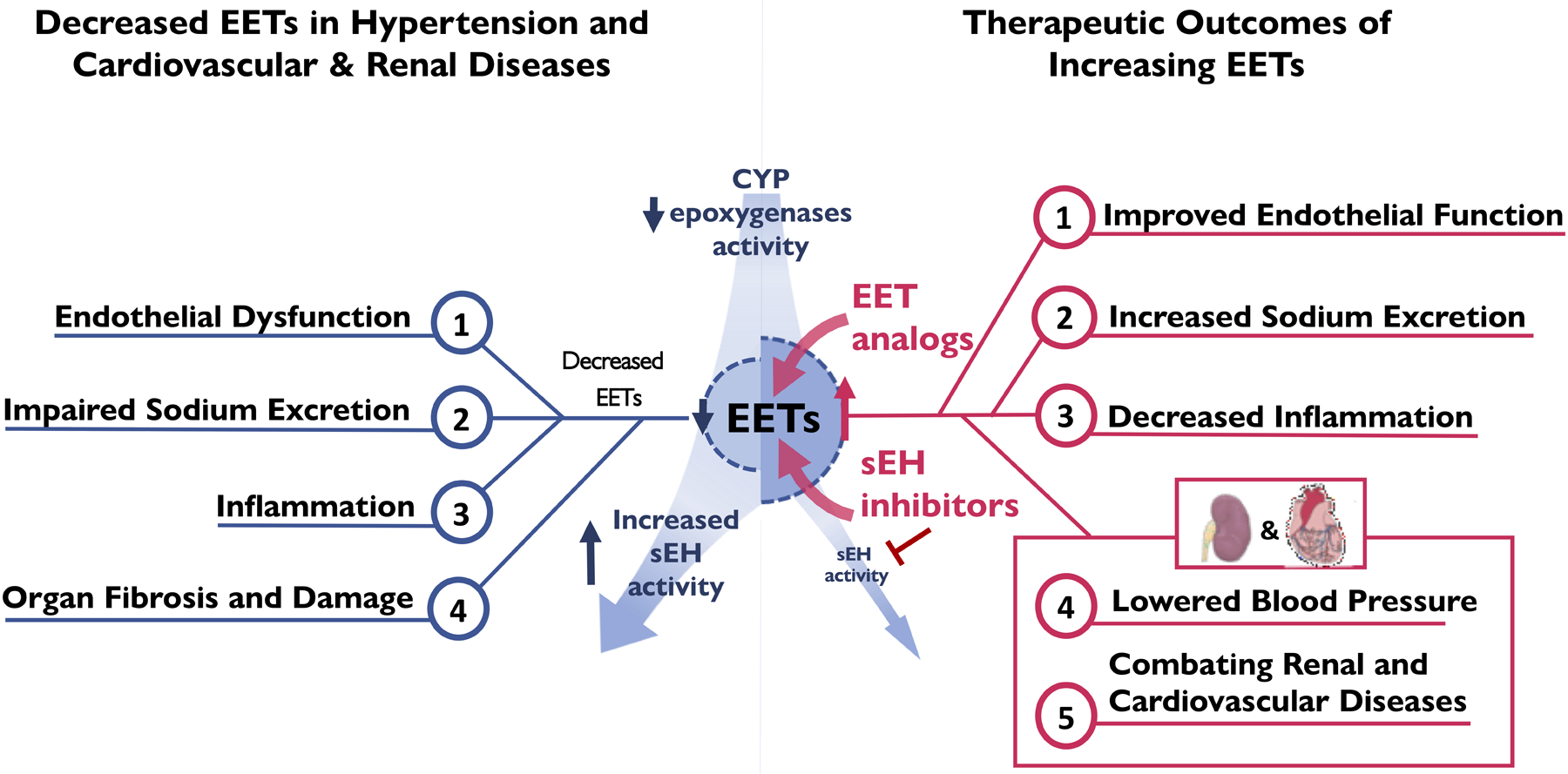
Decreased CYP epoxygenase activity or increased sEH activity results in decreased epoxyeicosatrienoic acids (EETs) resulting in vascular dysfunction, inflammation, impaired pressure natriuresis, organ fibrosis, and hypertension mediated renal and cardiovascular (CV) complications.
Right Panel: Epoxyeicosatrienoic acid (EET) analogs and soluble epoxide hydrolase (sEH) improve vascular function, decrease inflammation, increase natriuresis, combat cardiovascular (CV) diseases, lower blood pressure, combat kidney diseases.
SOLUBLE EPOXIDE HYDROLASE INHIBITION
The concept that increased sEH activity converting EETs to DHETs could contribute to hypertension and salt-sensitivity accelerated the therapeutic development of sEH inhibitors (Figure 2). Human studies revealed that genetic variants in EPHX2 the gene that encodes for sEH can influence endothelial function.65,66,67,68 The EPHX2 Arg55 genetic variant results in increased sEH activity and reduces human forearm blood flow responses to bradykinin.68 In addition, this EPHX2 Arg55 polymorphism is associated with greater risks for coronary artery disease.67 Acute kidney disease following cardiac surgery is associated with the EPHX2 Arg 55 polymorphism and a decreased plasma epoxide to diol ratio.69 Endothelial dysfunction in humans has been found in instances where capacity for inducing EET release is impaired.66 These findings in humans showed that increased sEH activity (and decreased EETs) contribute to endothelial dysfunction which is a precursor of salt-sensitive hypertension.
Evidence for epoxygenase CYP2C and CYP2J genetic variants that decrease EET and hypertension are inconclusive.66,70,71,72 Several CYP2C and CYP2J2 gene variants demonstrate reduced epoxygenase activity.66,70,71,72 As an example, CYPJ2 gene G-50 single nucleotide polymorphism is associated with increased coronary artery disease risk and decreased plasma EET levels.70 Analyses of ethnic cohorts have demonstrated no association with hypertension or an increased risk for essential hypertension.71,72,73,74 On the other hand, animal studies on salt regulation and blood pressure control have consistently demonstrated that an impaired ability to increase CYP2C generation of EETs in response to dietary NaCl contributes to salt-sensitive hypertension.24,47,54 Decreased renal epoxygenase activity and EETs have been associated with hypertension in angiotensin hypertension and Lyon hypertensive rats.75,76,77 Cyp2c44 −/− and Cyp4a10 −/− mice exhibit decreased renal epoxygenase activity in response to a high salt diet and develop salt-sensitive hypertension (Figure 2).48,54,78 These findings provided compelling reasons for developing sEH inhibitors as a means to increase renal EET levels and to treat salt-sensitive hypertension and cardiovascular diseases.
Initial studies determining kidney CYP2C epoxygenase and sEH regulation were conducted in angiotensin II hypertension.75,76,79,80 These studies discovered that angiotensin II hypertension was associated with an increased kidney sEH expression that resulted in a decrease in the EET to DHET ratio.75,79 In contrast, rat angiotensin II salt-sensitive hypertension did not have a change in sEH expression but did have an inability to increase kidney and renal microvascular CYP2C expression in response to a high salt diet.76 The decrease in CYP2C expression was also found to be partly responsible for the persistence of salt-sensitive hypertension following cessation of angiotensin II infusion.76 Thus, these experimental studies support the notion that the increase in kidney CYP2C expression in response to a high salt diet in normotensive rats contributes to water and electrolyte homeostasis and blood pressure control. In addition, inhibiting sEH to increase EETs in angiotensin II and salt-sensitive hypertension improved natriuresis, reduced afferent arteriolar responses to angiotensin II, improved endothelial-dependent dilation, decreased renal inflammation, and slowed progressive kidney damage.76,79,80 Thus, the evaluation of sEH inhibitors supported the notion that increasing kidney EET levels had therapeutic potential in hypertension.81
The development of sEH inhibitors progressed rapidly due to extensive knowledge concerning the enzymatic binding and activity, the ability to synthesize carbamate urea sEH inhibitors in large quantities, and development of the first orally active sEH inhibitor, AUDA (Table 1).81 Current orally active sEH inhibitor structures include urea-based sEH inhibitors, piperidyl urea sEH inhibitors, an amide series of sEH inhibitors and animoheteroarlyl sEH inhibitors.81,82,83 Experimental studies with sEH inhibitors determined anti-hypertensive actions in in a number of models: spontaneously hypertensive rats (SHR), angiotensin II hypertension, DOCA-salt hypertension, Lyon hypertensive rats, and Ren-2 transgenic hypertension.75,84,85,86 These studies included sEH inhibitors administered in preventive and clinically relevant interventional manner to rat and mice hypertension models. A consistent finding was that sEH inhibitors lowered blood pressure in these hypertension animals (Figure 2). EPHX2 gene deletion resulted in decreased blood pressure in DOCA-salt hypertension and sEH inhibitors did not have additional actions in EPHX2 gene deleted mice.85 The decrease in blood pressure in response to sEH inhibitors or genetic EPHX2 deletion was associated with an increase in the EET to DHET ratio, natriuresis, improved renal microvascular function, and decreased renal inflammation.85 Interestingly, in several instances the decrease in kidney damage following chronic sEH inhibition was greater than what would be expected based on the degree of blood pressure lowering.79,85,87 Subsequent studies found that sEH inhibitors could decrease progressive kidney disease independent of blood pressure lowering actions.88,89,90 Findings in these hypertension animal models resulted in extensive investigation of sEH inhibitors to treat renal and cardiovascular diseases.
Kidney-protective actions of sEH inhibitors have been demonstrated to be independent of the anti-hypertensive actions. Diabetic and hypertensive kidney injury has been clearly demonstrated to be decreased by the sEH inhibitor AUDA in the Goto-Kakizaki diabetic rat.88 The decrease in renal inflammation, glomerular injury, and tubular damage in the Goto-Kakizaki rats treated with an sEH inhibitor was not associated with a lowering of blood pressure, blood glucose, cholesterol, or triglyceride levels.88 Kidney-protective actions of sEH inhibitors or EPHX2 genetic deletion have also been demonstrated in unilateral ureter obstruction (UUO) induced kidney fibrosis.89,90 Administration of sEH inhibitors or EPHX2 genetic deletion decreased kidney inflammation by decreasing neutrophil infiltration and decreasing cytokine TNFα and ICAM-1 levels.89,90 The anti-inflammatory sEH inhibitor cell signalling mechanisms of actions in UUO were determined to be via NFκB downregulation and decreased transforming growth factor-β1 (TGF-β1)/Smad 3 mediated inflammation.89,90 Additional evaluation revealed that sEH inhibition anti-fibrotic actions were due to suppressing the epithelial to mesenchymal transition.89,90 Renal epithelial cell culture studies determined that sEH inhibition increases E-cadherin resulting in decreased α-smooth muscle actin to prevent transition from an epithelial cell to myofibroblast cell phenotype.89,90 Similar results were obtained in drug-induced nephrotoxicity studies.91,92 Administration of sEH inhibitors decreased NFκB activation and TNFα inflammation to reduce cisplatin induced acute kidney injury.91 Taken together, these studies demonstrated that sEH inhibitors decrease progressive renal disease through anti-inflammatory and anti-fibrotic mechanisms (Figure 2).
Cardiovascular diseases represent another area for which sEH inhibitors hold an exciting promise. Left ventricular cardiac hypertrophy and cardiac ischemia reperfusion injury are two animal disease models where sEH inhibitors have been extensively tested.25 Decreased EET generation and increased sEH activity is evident in several rodent cardiac hypertrophy models.93,94,95 Administration of sEH inhibitors prevents and reverses pressure overload left ventricular hypertrophy.93,96,97 Angiotensin II hypertension mediated cardiac hypertrophy is attenuated by sEH inhibition and the decrease in hypertrophy is partially independent of blood pressure lowering.93,98 Decreased cardiac hypertrophy in response to sEH inhibition is mediated by anti-inflammatory and anti-fibrotic actions.95,99 Like left ventricular hypertrophy, myocardial infarction is also improved by sEH inhibitors.100,101,102 Several dog, rat, and mice studies have demonstrated positive short-term and long-term outcomes for sEH inhibitors to treat cardiac ischemia reperfusion injury.99 The short-term positive effects on cardiac ischemia reperfusion injury are mediated through KATP channel activation, PI3 kinase signalling, and delays mitochondrial permeability transition pore (mPTP) opening to oppose cardiac cell apoptosis and necrosis.103,104,105 The long-term cardiac fibrotic and hypertrophy following myocardial infarction is also reduced by sEH inhibitors.99,105 Atrial fibrillation and cardiac arrhythmias in ischemia reperfusion and cardiac hypertrophy animal models are attenuated sEH inhibitors.99,106,107 These long-term cardiac protective actions for sEH inhibitors have significant anti-inflammatory actions.99 Treatment with sEH inhibitors decreases circulating inflammatory cytokines and inhibits cardiac NFκB activation.96,108 Likewise, chronic sEH inhibitor administration decreases cardiac fibrosis and hypertrophy by decreasing MAPK and endoplasmic reticulum stress activation.99,109 The sEH inhibition cerebral protective effects from stroke also involve reduction in MAPK and PI3 kinase signaling to prevent neuronal cell apoptosis.110,111,112 Intriguingly, sEH inhibitor anti-inflammatory actions are a major mechanism for protective actions in vascular diseases.25 The increase in cytokines, chemokines, and adhesion molecules that occurs in atherosclerotic and vascular remodeling animal models is greatly reduced by sEH inhibition.113,114,115 More recently, sEH inhibition demonstrated anti-inflammatory coronary artery actions in a Kawasaki disease mouse model that is an acquired heart disease in pediatric patients.116 These finding provide evidence for broad cardiovascular protective actions for sEH inhibitors that utilize EET anti-inflammatory, anti-apoptotic, and anti-fibrotic actions (Figure 2).
The overwhelming body of evidence from preclinical animal models for sEH inhibition having therapeutic value in hypertension, renal diseases, and cardiovascular diseases has allowed for rapid advancement of sEH inhibitors to clinical trials. Two sEH inhibitors, AR9281 and GSK2256294, have advanced to human clinical trials.25,27,79 Another sEH inhibitor, EC5026, is advancing to a Phase 1 clinical trial for chronic pain management. Phase I clinical trials for AR9281 and GSK2256294 were positive for safety and pharmacokinetics.117,118 AR9281 entered into a Phase IIa clinical trial for hypertension and type 2 diabetes but did not provide sufficient efficacy.81 Although GSK2256294 has not advanced to Phase II clinical trials, there were positive cardiovascular findings. Endothelial function assessed by forearm blood flow responses to bradykinin was improved in male obese smokers.27 This human study confirms the sEH inhibitor cardiovascular beneficial actions observed in copious animal studies. There are two human clinical trials that are in the recruiting and beginning stages for evaluating the sEH inhibitor GSK2256294 for treatment of insulin resistance and subarachnoid hemorrhage. There appears to be a bright and exciting future for sEH inhibitors to one day be approved for treating human disease.
EET ANALOGS
Given the successful development of the sEH inhibitors to clinical trials, one might ask what the benefit would be for developing EET analogs to target the epoxygenase pathway. There are three primary reasons to develop EET analogs. The first reason is that sEH inhibitors could be less effective in situations where decreased EET levels are due to decreased CYP2C epoxygenase EET generation. As previously mentioned, decreased CYP2C generation has been found in animals with salt-sensitive hypertension.76 Administration of sEH inhibitors to these salt-sensitive hypertension animals are not always as effective in lowering blood pressure.75,88 A second reason for developing EET analogs is that sEH inhibition will increase epoxy fatty acids in general rather than specific EETs.23,81 The contribution of epoxy fatty acids such as linoleic epoxy fatty acids to the beneficial effects for sEH inhibitors has not been extensively evaluated. EET analogs represent a direct method to mimic arachidonic acid epoxy fatty acids (EETs).25,26 The third reason is the possibility of developing EET analogs that resist metabolism by sEH and other pathways.25,26 Several biosteres that mimic epoxide bonds, replace the carboxylic acid and mimic double bonds present in 11,12-EET and 14,15-EET result in EET analogs that retain biological activity and resist metabolism (Table 1).25,26 The last reason for developing EET analogs is that regiospecific 11,12-EET and 14,15-EET mimics can be developed. There are four regioisomeric EETs and in several circumstances these EETs have variable biological actions.23,29,47 In addition, the sEH preferred regioisomeric EET substrate is 14,15-EET with lesser preference for 11,12-EET and 8,9-EET and 5,6-EET as a poor substrate for sEH.23,81 Kidney glomerular permeability studies provides evidence where the EET regioisomeric analog action is important. Focal segmental glomerular sclerosis circulating factor or angiotensin II induced increases in glomerular permeability are blocked by 8,9-EET and 8,9-EET analogs.119,120 In contrast, by 5,6-EET, 11,12-EET, 14,15-EET, 11,12-EET analogs and 14,15-EET analogs are ineffective in blocking the increase in glomerular permeability.119 Consequently, EET regioisomeric analogs have been developed and evaluated in preclinical animal models for renal and cardiovascular diseases.
Like sEH inhibitors, EET analogs were developed and initially tested in hypertension and salt-sensitive hypertension.121,122 Genetic manipulation to increase EET levels was evaluated prior to testing EET analogs in salt-sensitive hypertension. Mice with endothelial-specific overexpression of the human CYP2J2 (Tie2-CYP2J2) and CYP2C8 (Tie2-CYP2C8) epoxygenases showed attenuated afferent arteriole constrictor responses to endothelin-1 and enhanced dilator responses to acetylcholine.45 Angiotensin II salt-sensitive hypertension and renal injury was significantly attenuated in Tie2-CYP2J2 and Tie2-CYP2C8 mice.45 Importantly, we found that primary renal endothelial cells isolated from Tie2-CYP2J2 and Tie2-CYP2C8 mice produced significantly higher concentrations of 11,12-EET and 14,15-EET compared to renal endothelial cells isolated from wild-type controls.45 The subsequent development of orally bioavailable EET analogs allowed for testing in rodent hypertension. EET analogs for 11,12-EET and 14,15-EET lowered blood pressure in SHR, angiotensin hypertension, and salt-sensitive hypertension.121,122 The 11,12-EET analog, NUDSA, was also demonstrated to decrease blood pressure and improve endothelial function in mice with metabolic syndrome.123 Likewise, 14,15-EET analogs, EET-A and EET-B, decreased blood pressure in angiotensin hypertension.121,122 Additional studies in angiotensin hypertension revealed that EET-A improved mesenteric resistance artery endothelial function and inhibited ENaC to promote sodium excretion.122 Malignant angiotensin hypertension in Cyp1a1-Ren transgenic rats was also prevented by chronic EET-A administration.124 EET analogs also decreased renal inflammation and macrophage infiltration in hypertension.122 EET analogs are anti-hypertensive and improve renal function through HO-1 signalling and inhibition of the sodium-chloride transporter (NCC).125 Hypertension resulting from cyclosporine administration to rats for one month was prevented by EET analog treatment.126 EET-B treatment decreased renal inflammation, apoptosis, and fibrosis associated with cyclosporine.126 Cyp2c44 −/− mice have been extensively studied because Cyp2c44 is the major kidney epoxygenase and Cyp2c44 expression is increased in response to a high K+ or high Na+ salt diet to generate EETs in the cortical collecting duct.47 More importantly, mice with Cyp2c44 epoxygenase genetic deficiency and decreased EET levels develop salt sensitive hypertension.54,122 Hyperactive ENaC and a reduction in ERK1/2 ENaC subunit phosphorylation contribute to the salt-sensitive hypertension in Cyp2c44 −/− mice.54 EET-A prevented salt-sensitive hypertension and decreased renal injury in Cyp2c44 −/− mice.122 The anti-hypertensive actions of EET-A in Cyp2c44 −/− mice were likely a result of the ability for EET-A to inhibit ENaC in cortical collecting duct cells.122 These findings support the notion that EET analogs lower blood pressure in hypertension through the known EET vascular, epithelial transport, and anti-inflammatory actions (Figure 2).
Renal protective actions have been demonstrated for EET analogs that are independent of systemic actions.25 Acute and chronic renal disease animal models provide evidence that EET analogs anti-inflammatory, anti-fibrotic, and anti-apoptotic actions decrease renal disease progression.25 EET analogs administered in a preventive or interventional mode can decrease renal vascular, glomerular, and tubular injury.25 Initial studies evaluated EET analogs in drug-induced nephrotoxicity.127 Treatment with EET analogs decreased the tubular injury in response to the chemotherapeutic agent cisplatin.127 Cisplatin induced nephrotoxicity renal inflammation and apoptosis was reduced by EET analog treatment.127 Renal ischemia reperfusion injury is decreased by EET analog treatment.128 EET analog treatment improved renal oxygenation, decreased inflammatory cell infiltration, and acted via PI3 kinase Akt, GSK-3β signalling to reduce epithelial cell apoptosis.128 EET analogs also protect from long-term progressive renal injury.25 Kidney fibrosis in UUO mice is prevented by EET-A through actions on epithelial to mesenchymal transition.129 EET-A treatment to UUO mice resulted in a decrease in epithelial to mesenchymal transducers and myofibroblast markers resulting in decreased renal α-smooth muscle actin and collagen levels.129 Progressive renal injury due to radiation exposure is also prevented by EET analogs.130 EET-A given two days following radiation exposure to rats mitigated the progressive renal microvascular and tubular damage.130 The decrease in radiation induced renal damage by EET-A was associated with reductions in the p53/Fas/FasL apoptotic pathway.130 Likewise, renal injury associated with lupus nephritis is decreased by EET analog treatment.131 Systemic lupus nephritis mice treated with EET-A had markedly diminished CXC chemokines and receptors, reduced renal TNF-α, IL-6, and IL-1β levels, and reduced renal immune cell infiltration resulting in decreased renal damage.131 As a whole, EET analogs have been consistently demonstrated to decrease acute and chronic kidney disease progression through anti-apoptotic, anti-inflammatory, and anti-fibrotic mechanisms (Figure 2).
EET analogs cardiovascular protective actions have been evaluated in several cardiac disease animal models.25 Initial studies were conducted with EET analogs in cardiac ischemia reperfusion injury.132,133,134 These studies revealed that NUDSA administered after myocardial infarction resulted in improved cardiac contractile function.134 Likewise, left ventricular function one month following left anterior descending coronary artery ligation in mice was improved by EET analog treatment started five days after ligation.134 EET-B treatment for two months improved cardiac function in SHR subjected to 30 minutes of left coronary artery occlusion to induce myocardial infarction.133 The improved cardiac function in SHR was linked to increased HO-1 cardiomyocyte levels and decreased inflammation and fibrosis.133 Obesity-induced diabetic cardiomyopathy in mice was reduced by EET analog treatment.135 EET analog administration resulted in peroxisome proliferator-activated receptor coactivator (PGC-1α) induction to control mitochondrial function, induce HO-1, and reduce inflammation in the heart of obese diabetic mice.131 A contribution of EET activation in heart vascular endothelial cells to cardiac protection has been demonstrated in endothelial specific CYP2J2 overexpressing mice.136 Myocardial perfusion following myocardial infarction in mice was increased by increased endothelial cell EET levels.136 Endothelial cell EETs increased angiogenesis via Jagged1/Notch 1 signalling in mice following cardiac ischemia.136 Cardiac hypertrophy was also decreased in hypertensive rats treated with EET-A for two weeks.137,138 Ischemia reperfusion induced ventricular fibrillation in hypertension was also greatly diminished by EET analogs.138 The primary signalling mechanisms that appear to mediate EET analog cardiac protective actions include opposing apoptosis and improving mitochondrial function.25,132,139 In line with EET biological actions, EET analogs activate cardiac myocyte mitochondrial KATP channels resulting in an increase in mitochondrial cristae density.99,140 Anti-apoptotic EET analog actions are mediated through MAP kinase signalling in the heart to prevent ischemia reperfusion injury.25,99 These findings support the notion that EET analogs are a potential therapeutic strategy for cardiac diseases (Figure 2).
FUTURE FOR EPOXY FATTY ACID THERAPEUTICS
We have made significant strides on the journey to developing EET-based therapeutics for treating renal and cardiovascular diseases. We have gone from the initial description of EETs as regulators of renal hemodynamics and epithelial transport to the development of sEH inhibitors and EET analogs. In less than a decade sEH inhibitors were in clinal trials for hypertension and diabetes.81 On the other hand, EET analogs developed at a slower pace due to the lack of a protein target and ability to obtain orally bioavailable EET analogs.26 EET analogs are now being advanced towards clinical trials for kidney diseases. The journey continues as the potential for an EET pathway drug being approved for treating renal or cardiovascular diseases appears likely.
Challenges and headwinds still exist for epoxy fatty acids to reach a point to benefit human health. As with every hormonal and paracrine factor and cell signalling pathway, there is an upside and downside to therapeutically targeting EETs and sEH. Although the positives appear to outweigh the negatives for sEH inhibitors and EET analogs, there has been concerns with their potential due to effects on angiogenesis and tumor growth and metastasis in certain cancers.141,142,143 In contrast, however, sEH inhibitors given alone or in combination with COX inhibitors can have anti-cancer actions.144,145,146 EET angiogenesis could be a detrimental action for tumors but be very useful when increased vascularization of ischemic tissue is required.147,148 In addition, the EET actions on the pulmonary vasculature is vasoconstriction.149,150 This opposite pulmonary vascular EET actions is like that found for prostaglandins.149 Even with these potential challenges, sEH inhibitors have demonstrated beneficial cardiovascular effects in human clinical trials to combat chronic diseases.27 Opportunities for therapeutically targeting epoxy fatty acids remain abundant.
Dietary factors like diets enriched with ω−3 fatty acids can influence ω−3 epoxy fatty acid levels and consumption of specific plants can influence sEH activity. There has been considerable investigation into the actions for ω−3 eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) fatty acids. Increases in dietary ω−3 EPA and DHA have been found to have cardiovascular beneficial actions.151,152 Intriguingly, epoxygenase enzymes generated EPA epoxyeicosatetraenoic acids (EEQs) and DHA epoxydocosapentaenoic acids (EDPs) metabolites that have also been found to have kidney and cardiovascular actions.153,154,155,156 Interestingly, 17,18-EEQ analogs have been developed, tested, and reached Phase 1 clinical trials for treating atrial fibrillation and cardiac arrhythmias.152,154 Other dietary means can elevate epoxy fatty acids. Plants from the order Brassicales and Cimicifuga dahurica roots have been found to have decrease sEH activity.157,158 Eating flaxseed has been demonstrated to inhibit sEH and lower blood pressure in humans with hypertension.159 There is great potential and much to be explored for the kidney and cardiovascular dietary ω−3 epoxy fatty acids and plants with sEH inhibitory activity.
Another major area open for opportunities are to better define the contribution for EET regioisomers. 11,12-EET and 14,15-EET have been the most extensively studied EET regioisomers; however, 5,6-EET and 8,9-EET have been demonstrated to have vascular and kidney actions.23,25 Vascular endothelial cell actions have been described for 5,6-EET to activate transient receptor potential vanilloid receptor 4 (TRPV4) channels.160 An EET-binding pocket mediates 5,6-EET TRPV4 activation and can control hypotonic cell swelling.160 Likewise, 8,9-EET and 8,9-EET analogs have selective actions on glomerular permeability induced by a circulating permeability factor.119 Regioisomeric actions on flow induced arteriolar dilation have been determined for with 11,12-EET and 14,15-EET selective antagonists.161 These studies revealed that 14,15-EET but not 11,12-EET contributed to mesenteric resistance artery flow induced dilation.161 Interestingly, decreased 14,15-EET levels are significantly associated with abdominal aortic calcification in patients with primary aldosteronism.162 Another interesting aspect for EETs is that there is significant evidence for the existence of EET receptors.163,164 Given the difference in EET regioisomeric renal and cardiovascular actions and the potential for EET receptors, there are still significant gaps in knowledge pertaining to EETs that require experimental investigation.
Bifunctional molecules that alter epoxy fatty acids is another emerging area for further development and exploration.165,166 The development of bifunctional molecules has revolved around having sEH inhibition as a focal activity for the molecules (Table 1).165,167 EET analogs capable of inhibiting sEH are an example of fused pharmacophore dual modulators.25,168 Although these have been described based on in vitro effects on vascular function, there has been no significant evaluation of these bifunctional molecules in animal disease models. One of the first dual modulators was a linked pharmacophore with combined sEH and COX-2 inhibition.145,167 The dual sEH and COX-2 inhibitor, PTUPB combats diabetic kidney injury in Zucker Diabetic Fatty rats.167 In addition, PTUPB has been demonstrated to have anti-cancer and anti-metastatic actions in mice.145,146 Finally, merged pharmacophore sEH inhibitors with nuclear receptor agonism have demonstrated great potential. RB394 is a dual sEH inhibitor and PPARγ agonist that has been demonstrated to combat diabetes and associated liver and kidney injury.169 Another merged pharmacophore combines sEH inhibition with FXR agonism.170 The dual acting sEH inhibitor and FXR agonist, DM509 has been demonstrated to combat non-alcoholic liver disease to a greater extent than an FXR agonist given alone.170 These findings demonstrate that bifunctional molecules with sEH inhibitor activity could be used to treat heart, kidney, liver, and metabolic diseases.
CONCLUSION
Epoxy fatty acids contribution to renal, vascular, and heart function led to the notion that increasing EETs could combat disease. EETs were initially found to be vasodilatory and natriuretic; their importance in salt-sensitive hypertension was clearly established. The combination of pharmacological and genetic tools determined the importance for the EETs and sEH in hypertension, diabetes, ischemic kidney and heart diseases, stroke, and others. Human studies provided additional support to the importance of the epoxygenase pathway in these diseases. Development of sEH inhibitors and EET analogs has progressed to a point where their exciting therapeutic potential to treat kidney and cardiovascular diseases is being actively investigated.
ACKNOWLEDGEMENTS
Servier Medical Art was used to generate Figures and is licensed by Servier under a Creative Commons Attribution 3.0 Unported License. ChemDraw was utilized for drawing the chemical structures.
SOURCE OF FUNDING
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases DK103616 and Dr. Ralph and Marian Falk Medical Research Trust Bank of America, N.A., Trustee.
Footnotes
CONFLICT OF INTEREST STATEMENT
Dr. Imig has patents that cover the composition of matter for EET analogs, sEH inhibitors, bifunctional sEH inhibitors. There are no other conflicts of interest, financial or otherwise, are declared by the author.
REFERENCES
- 1.Knudsen KD, Dahl LK, Thompson K, Iwai J, Heine M, Leitl G. Effects of chronic excess salt ingestion. Inheritance of hypertension in the rat. J Exp Med. 1970;132:976–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dahl LK. Salt and hypertension. Am J Clin Nutr. 1972;25:231–244. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda M, Kimura G Pathophysiology of Antihypertensive Therapy with Diuretics. Hypertens Res. 2006;29:645–653. [DOI] [PubMed] [Google Scholar]
- 4.Bibbins-Domingo K, Chertow GM, Coxson PG, et al. Projected effect of dietary salt reductions on future cardiovascular disease. N Engl J Med. 2010;362:590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet. 2014;383:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth GA, Johnson C, Abajobir A, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedrinelli R, Dell’omo G, Cameli M, et al. Resistant hypertension: an overview. Minerva Cardioangiol. 2018;66:337–348. [DOI] [PubMed] [Google Scholar]
- 8.Carey RM, Calhoun DA, Bakris GL, et al. Resistant Hypertension: Detection, Evaluation, and Management: A Scientific Statement From the American Heart Association. Hypertension. 2018;72:e53–e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barri YM. Hypertension and kidney diseases: a deadly connection. Curr Hypertens Rep. 2008;10: 39–45. [DOI] [PubMed] [Google Scholar]
- 10.Chen TK, Knicely DH, Grams ME. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA. 2019;322:1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frassetto L, Morris RC Jr, Sellmeyer DE, Todd K, Sebastian A. Diet, evolution and aging--the pathophysiologic effects of the post-agricultural inversion of the potassium-to-sodium and base-to-chloride ratios in the human diet. Eur J Nutr. 2001;40:200–213. [DOI] [PubMed] [Google Scholar]
- 12.Singh RB, Singh NK, Mody R, Cameron EA. Does sodium play an adverse role in hypertension? Acta Cardiol. 1987;42:187–206. [PubMed] [Google Scholar]
- 13.Arnett DK, Claas SA. Omics of Blood Pressure and Hypertension. Circ Res. 2018;122:1409–1419. [DOI] [PubMed] [Google Scholar]
- 14.Padmanabhan S, Aman A, Dominiczak AF. Genomics of hypertension. Pharmacol Res. 2017;121:219–229. [DOI] [PubMed] [Google Scholar]
- 15.Johnson RJ, Lanaspa MA, Gabriela Sánchez-Lozada L, Rodriguez-Iturbe B. The discovery of hypertension: evolving views on the role of the kidneys, and current hot topics. Am J Physiol Renal Physiol. 2015;308:F167–F178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rettig R, Schmitt B, Pelzl B, Speck T. The kidney and primary hypertension: contributions from renal transplantation studies in animals and humans. J Hypertens. 1993;11:883–891. [DOI] [PubMed] [Google Scholar]
- 17.Rettig R, Folberth C, Stauss H, Kopf D, Waldherr R, Unger T. Role of the kidney in primary hypertension: a renal transplantation study in rats. Am J Physiol. 1990;258:F606–F611. [DOI] [PubMed] [Google Scholar]
- 18.Mattson DL. Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat Rev Nephrol. 2019;15:290–300. [DOI] [PubMed] [Google Scholar]
- 19.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116:1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens. 2006;15:152–158. [DOI] [PubMed] [Google Scholar]
- 21.Choi HY, Park HC, Ha SK. Salt Sensitivity and Hypertension: A Paradigm Shift from Kidney Malfunction to Vascular Endothelial Dysfunction. Electrolyte Blood Press. 2015;13:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aaron KJ, Sanders PW. Role of dietary salt and potassium intake in cardiovascular health and disease: a review of the evidence. Mayo Clin Proc. 2013;88:987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imig JD. Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension. 2015;65:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imig JD. Prospective for Cytochrome P450 Epoxygenase Cardiovascular and Renal Therapeutics. Pharmacology & Therapeutics. 2018;192:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell WB, Imig JD, Schmitz JM, Falck JR. Orally active epoxyeicosatrienoic acid analogs. J Cardiovasc Pharmacol. 2017;70: 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Cheriyan J, Gutterman DD, Mayer RJ, Ament Z, Griffin JL, Lazaar AL, Newby DE, Tal-Singer R, Wilkinson IB. Mechanisms of Vascular Dysfunction in COPD and Effects of a Novel Soluble Epoxide Hydrolase Inhibitor in Smokers. Chest. 2017;151:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capdevila JH, Falck JR, Estabrook RW. Cytochrome P450 and the arachidonate cascade. FASEB J. 1992;6:731–736. [DOI] [PubMed] [Google Scholar]
- 29.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. [DOI] [PubMed] [Google Scholar]
- 30.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41:163–181. [PubMed] [Google Scholar]
- 31.DeLozier TC, Tsao CC, Coulter SJ, et al. CYP2C44, a new murine CYP2C that metabolizes arachidonic acid to unique stereospecific products. J Pharmacol Exp Ther. 2004;310:845–854. [DOI] [PubMed] [Google Scholar]
- 32.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. [DOI] [PubMed] [Google Scholar]
- 33.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. [DOI] [PubMed] [Google Scholar]
- 34.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol. 1996;7:2364–2370. [DOI] [PubMed] [Google Scholar]
- 35.Cazade M, Bidaud I, Hansen PB, Lory P, Chemin J. 5,6-EET potently inhibits T-type calcium channels: implication in the regulation of the vascular tone. Pflugers Arch. 2014;466:1759–1768. [DOI] [PubMed] [Google Scholar]
- 36.Carroll MA, Garcia MP, Falck JR, McGiff JC. 5,6-epoxyeicosatrienoic acid, a novel arachidonate metabolite. Mechanism of vasoactivity in the rat. Circ Res. 1990;67:1082–1088. [DOI] [PubMed] [Google Scholar]
- 37.Sacerdoti D, Bolognesi M, Di Pascoli M, et al. Rat mesenteric arterial dilator response to 11,12-epoxyeicosatrienoic acid is mediated by activating heme oxygenase. Am J Physiol Heart Circ Physiol. 2006;291:H1999–H2002. [DOI] [PubMed] [Google Scholar]
- 38.Imig JD, Dimitropoulou C, Reddy DS, White RE, Falck JR. Afferent arteriolar dilation to 11,12-EET analogs involves PP2A activity and Ca2+-activated K+ Channels. Microcirculation. 2008;15: 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imig JD, Inscho EW, Deichmann PC, Reddy KM, Falck JR. Afferent arteriolar vasodilation to the sulfonimide analog of 11, 12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension. 1999;33:408–413. [DOI] [PubMed] [Google Scholar]
- 40.Imig JD, Falck JR, Wei S, Capdevila JH. Epoxygenase metabolites contribute to nitric oxide-independent afferent arteriolar vasodilation in response to bradykinin. J Vasc Res. 2001;38:247–255. [DOI] [PubMed] [Google Scholar]
- 41.Taddei S, Versari D, Cipriano A, et al. Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpolarizing factor in essential hypertensive patients. J Am Coll Cardiol. 2006;48:508–515. [DOI] [PubMed] [Google Scholar]
- 42.Batenburg WW, Garrelds IM, van Kats JP, Saxena PR, Danser AH. Mediators of bradykinin-induced vasorelaxation in human coronary microarteries. Hypertension. 2004;43:488–492. [DOI] [PubMed] [Google Scholar]
- 43.Gauthier KM, Cepura CJ, Campbell WB. ACE inhibition enhances bradykinin relaxations through nitric oxide and B1 receptor activation in bovine coronary arteries. Biol Chem. 2013;394:1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ito O, Omata K, Ito S, Hoagland KM, Roman RJ. Effects of converting enzyme inhibitors on renal P-450 metabolism of arachidonic acid. Am J Physiol Regul Integr Comp Physiol. 2001;280:R822–R830. [DOI] [PubMed] [Google Scholar]
- 45.Lee CR, Imig JD, Edin ML, et al. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24:3770–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imig JD, Zhao X, Falck JR, Wei S, Capdevila JH. Enhanced renal microvascular reactivity to angiotensin II in hypertension is ameliorated by the sulfonimide analog of 11,12-epoxyeicosatrienoic acid. J Hypertens. 2001;19:983–992. [DOI] [PubMed] [Google Scholar]
- 47.Capdevila J, Wang W. Role of cytochrome P450 exoxygenase in regulating renal membrane transport and hypertension. Curr Opin Nephrol Hypertens. 2013;22:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang WH, Zhang C, Lin DH, et al. Cyp2c44 epoxygenase in the collecting duct is essential for the high K+ intake-induced antihypertensive effect. Am J Physiol Renal Physiol. 2014;307:F453–F460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romero MF, Madhun ZT, Hopfer U, Douglas JG. An epoxygenase metabolite of arachidonic acid 5,6 epoxy-eicosatrienoic acid mediates angiotensin-induced natriuresis in proximal tubular epithelium. Adv Prostaglandin Thromboxane Leukot Res. 1991;21A:205–208. [PubMed] [Google Scholar]
- 50.Sun P, Liu W, Lin DH, et al. Epoxyeicosatrienoic acid activates BK channels in the cortical collecting duct. J Am Soc Nephrol. 2009;20:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pavlov TS, Ilatovskaya DV, Levchenko V, Mattson DL, Roman RJ, Staruschenko A. Effects of cytochrome P-450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC). Am J Physiol Renal Physiol. 2011;301:F672–F681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madhun ZT, Goldthwait DA, McKay D, Hopfer U, Douglas JG. An epoxygenase metabolite of arachidonic acid mediates angiotensin II-induced rises in cytosolic calcium in rabbit proximal tubule epithelial cells. J Clin Invest. 1991;88:456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakairi Y, Jacobson HR, Noland TD, Capdevila JH, Falck JR, Breyer MD. 5,6-EET inhibits ion transport in collecting duct by stimulating endogenous prostaglandin synthesis. Am J Physiol. 1995;268:F931–F939. [DOI] [PubMed] [Google Scholar]
- 54.Capdevila JH, Pidkovka N, Mei S, Gong Y, Falck JR, Imig JD, Harris RC, Wang WH. The Cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. J Biol Chem. 2014;289:4377–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z, Wei Y, Falck JR, Atcha KR, Wang WH. Arachidonic acid inhibits basolateral K channels in the cortical collecting duct via cytochrome P-450 epoxygenase-dependent metabolic pathways. Am J Physiol Renal Physiol. 2008;294:F1441–F1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun P, Liu W, Lin DH, et al. Epoxyeicosatrienoic acid activates BK channels in the cortical collecting duct. J Am Soc Nephrol. 2009;20:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang JX, Zhang SJ, Xiong YK, et al. EETs Attenuate Ox-LDL-Induced LTB4 Production and Activity by Inhibiting p38 MAPK Phosphorylation and 5-LO/BLT1 Receptor Expression in Rat Pulmonary Arterial Endothelial Cells. PLoS One. 2015;10:e0128278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai M, Wu L, He Z, et al. Epoxyeicosatrienoic acids regulate macrophage polarization and prevent LPS-induced cardiac dysfunction. J Cell Physiol. 2015;230:2108–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen W, Yang S, Ping W, Fu X, Xu Q, Wang J. CYP2J2 and EETs protect against lung ischemia/reperfusion injury via anti-inflammatory effects in vivo and in vitro. Cell Physiol Biochem. 2015;35:2043–2054. [DOI] [PubMed] [Google Scholar]
- 61.Li Y, Yu G, Yuan S, et al. 14,15-Epoxyeicosatrienoic acid suppresses cigarette smoke condensate-induced inflammation in lung epithelial cells by inhibiting autophagy. Am J Physiol Lung Cell Mol Physiol. 2016;311:L970–L980. [DOI] [PubMed] [Google Scholar]
- 62.Wang Q, Liang Y, Qiao Y, et al. Expression of soluble epoxide hydrolase in renal tubular epithelial cells regulates macrophage infiltration and polarization in IgA nephropathy. Am J Physiol Renal Physiol. 2018;315:F915–F926. [DOI] [PubMed] [Google Scholar]
- 63.Fleming I Vascular cytochrome p450 enzymes: physiology and pathophysiology. Trends Cardiovasc Med. 2008;18:20–25. [DOI] [PubMed] [Google Scholar]
- 64.Behmoaras J, Diaz AG, Venda L, et al. Macrophage epoxygenase determines a profibrotic transcriptome signature. J Immunol. 2015;194:4705–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bellien J, Iacob M, Remy-Jouet I, Lucas D, Monteil C, Gutierrez L, Vendeville C, Dreano Y, Mercier A, Thuillez C, Joannides R. Epoxyeicosatrienoic acids contribute with altered nitric oxide and endothelin-1 pathways to conduit artery endothelial dysfunction in essential hypertension. Circulation. 2012;125:1266–1275. [DOI] [PubMed] [Google Scholar]
- 66.Bellien J, Joannides R. Epoxyeicosatrienoic acid pathway in human health and diseases. J Cardiovasc Pharmacol. 2013;61:188–196. [DOI] [PubMed] [Google Scholar]
- 67.Lee CR, North KE, Bray MS, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet. 2006;15:1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee CR, Pretorius M, Schuck RN, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with forearm vasodilator responses in humans. Hypertension. 2011;57:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shuey MM, Billings FT 4th, Wei S, et al. Association of gain-of-function EPHX2 polymorphism Lys55Arg with acute kidney injury following cardiac surgery. PLoS One. 2017;12:e0175292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spiecker M, Darius H, Hankeln T, et al. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation. 2004;110(15):2132–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dreisbach AW, Japa S, Sigel A, Parenti MB, Hess AE, Srinouanprachanh SL, Rettie AE, Kim H, Farin FM, Hamm LL, Lertora JJ. The Prevalence of CYP2C8, 2C9, 2J2, and soluble epoxide hydrolase polymorphisms in African Americans with hypertension. Am J Hypertens. 2005;18: 1276–1281. [DOI] [PubMed] [Google Scholar]
- 72.King LM, Gainer JV, David GL, Dai D, Goldstein JA, Brown NJ, Zeldin DC. Single nucleotide polymorphisms in the CYP2J2 and CYP2C8 genes and the risk of hypertension. Pharmacogenet Genomics. 2005;15:7–13. [DOI] [PubMed] [Google Scholar]
- 73.Yu BN, Luo CH, Wang D, et al. CYP2C9 allele variants in Chinese hypertension patients and healthy controls. Clin Chim Acta. 2004;348:57–61. [DOI] [PubMed] [Google Scholar]
- 74.Polonikov AV, Ivanov VP, Solodilova MA, et al. A common polymorphism G-50T in cytochrome P450 2J2 gene is associated with increased risk of essential hypertension in a Russian population. Dis Markers. 2008;24:119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. [DOI] [PubMed] [Google Scholar]
- 76.Zhao X, Pollock DM, Inscho EW, Zeldin DC, Imig JD. Decreased renal cytochrome P450 2C enzymes and impaired vasodilation are associated with salt-sensitive hypertension. Hypertension. 2003;41:709–714. [DOI] [PubMed] [Google Scholar]
- 77.Messer-Létienne I, Bernard N, Roman RJ, Sassard J, Benzoni D. Cytochrome P-450 arachidonate metabolite inhibition improves renal function in Lyon hypertensive rats. Am J Hypertens. 1999;12:398–404. [DOI] [PubMed] [Google Scholar]
- 78.Nakagawa K, Holla VR, Wei Y, et al. Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J Clin Invest. 2006;116(6):1696–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 80.Jung O, Brandes RP, Kim IH, et al. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. [DOI] [PubMed] [Google Scholar]
- 81.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marino JP Jr., Soluble epoxide hydrolase, a target with multiple opportunities for cardiovascular drug discovery. Curr Top Med Chem. 2009;9:452–463. [DOI] [PubMed] [Google Scholar]
- 83.Duflot T, Roche C, Lamoureux F, Guerrot D, Bellien J. Design and discovery of soluble epoxide hydrolase inhibitors for the treatment of cardiovascular diseases. Expert Opin Drug Discov. 2014;9:229–243. [DOI] [PubMed] [Google Scholar]
- 84.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. [DOI] [PubMed] [Google Scholar]
- 85.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297:F740–F448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Honetschlagerova Z, Huskova Z, Vanourkova Z, Sporkova A, Kramer HJ, Hwang SH, Tsai HJ, Hammock BD, Imig JD, Cervenka L, Kopkan L. Renal mechanisms contributing to the antihypertensive action of soluble epoxide hydrolase inhibition in REN-2 transgenic rats with inducible. J Physiol. 2011;589:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kujal P, Certikova Chabova V, Skaroupkova P, Huskova Z, Vernerova Z, Kramer HJ, Walkowska A, Kompanowska-Jezierska E, Sadowski J, Kitada K, Nishiyama A, Hwang SH, Hammock BD, Imig JD, Cervenka L. Inhibition of soluble epoxide hydrolase is renoprotective in 5/6 nephrectomized Ren-2 transgenic hypertensive rats. Clin Exp Pharmacol Physiol. 2014;41:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Olearczyk JJ, Quigley JE, Mitchell B, Yamamoto T, Kim IH, Newman JW, Lauria A, Hammock BD, Imig JD. Inhibition of the soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clinical Science. 2009;116:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol Renal Physiol. 2014;307:F971–F980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim J, Yoon SP, Toews ML, et al. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol. 2015;308:F131–F139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Y, Webb HK, Fukushima H, et al. Attenuation of cisplatin-induced renal injury by inhibition of soluble epoxide hydrolase involves nuclear factor κB signaling. J Pharmacol Exp Ther. 2012;341:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, Hammock BD. Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol Toxicol. 2009;25:217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ai D, Pang W, Li N, et al. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:564–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Althurwi HN, Maayah ZH, Elshenawy OH, El-Kadi AO. Early Changes in Cytochrome P450s and Their Associated Arachidonic Acid Metabolites Play a Crucial Role in the Initiation of Cardiac Hypertrophy Induced by Isoproterenol. Drug Metab Dispos. 2015;43:1254–1266. [DOI] [PubMed] [Google Scholar]
- 95.Alsaad AM, Zordoky BN, Tse MM, El-Kadi AO. Role of cytochrome P450-mediated arachidonic acid metabolites in the pathogenesis of cardiac hypertrophy. Drug Metab Rev. 2013;45:173–195. [DOI] [PubMed] [Google Scholar]
- 96.Sirish P, Li N, Liu JY, et al. Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc Natl Acad Sci U S A. 2013;110:5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roche C, Besnier M, Cassel R, et al. Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am J Physiol Heart Circ Physiol. 2015;308:H1020–H1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang H, Zhang K, Liang J, et al. Soluble epoxide hydrolase inhibitor, TUPS, attenuates isoproterenol/angiotensin II-induced cardiac hypertrophy through mammalian target of rapamycin-mediated autophagy inhibition. J Pharm Pharmacol. 2019;71:1291–1300. [DOI] [PubMed] [Google Scholar]
- 99.Oni-Orisan A, Alsaleh N, Lee CR, Seubert JM. Epoxyeicosatrienoic acids and cardioprotection: the road to translation. J Mol Cell Cardiol. 2014;74:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Islam O, Patil P, Goswami SK, et al. Inhibitors of soluble epoxide hydrolase minimize ischemia-reperfusion-induced cardiac damage in normal, hypertensive, and diabetic rats. Cardiovasc Ther. 2017;35: 10.1111/1755-5922.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kompa AR, Wang BH, Xu G, et al. Soluble epoxide hydrolase inhibition exerts beneficial anti-remodeling actions post-myocardial infarction. Int J Cardiol. 2013;167:210–219. [DOI] [PubMed] [Google Scholar]
- 102.Guo Y, Luo F, Zhang X, et al. TPPU enhanced exercise-induced epoxyeicosatrienoic acid concentrations to exert cardioprotection in mice after myocardial infarction. J Cell Mol Med. 2018;22:1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Batchu SN, Lee SB, Samokhvalov V, et al. Novel soluble epoxide hydrolase inhibitor protects mitochondrial function following stress. Can J Physiol Pharmacol. 2012;90:811–823. [DOI] [PubMed] [Google Scholar]
- 104.Gross GJ, Gauthier KM, Moore J, et al. Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am J Physiol Heart Circ Physiol. 2008;294:H2838–H2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Seubert JM, Sinal CJ, Graves J, et al. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gui Y, Li D, Chen J, et al. Soluble epoxide hydrolase inhibitors, t-AUCB, downregulated miR-133 in a mouse model of myocardial infarction. Lipids Health Dis. 2018;17:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu D, Li N, He Y, et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:18733–18738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li N, Liu JY, Timofeyev V, et al. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J Mol Cell Cardiol. 2009;47:835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guglielmino K, Jackson K, Harris TR, et al. Pharmacological inhibition of soluble epoxide hydrolase provides cardioprotection in hyperglycemic rats. Am J Physiol Heart Circ Physiol. 2012;303:H853–H862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simpkins AN, Rudic RD, Schreihofer DA, et al. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol. 2009;174:2086–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hung TH, Shyue SK, Wu CH, et al. Deletion or inhibition of soluble epoxide hydrolase protects against brain damage and reduces microglia-mediated neuroinflammation in traumatic brain injury. Oncotarget. 2017;8:103236–103260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang W, Iliff JJ, Campbell CJ, Wang RK, Hurn PD, Alkayed NJ. Role of soluble epoxide hydrolase in the sex-specific vascular response to cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhou C, Huang J, Li Q, Nie J, Xu X, Wang DW. Soluble Epoxide Hydrolase Inhibition Protected against Angiotensin II-induced Adventitial Remodeling. Sci Rep. 2017;7:6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Q, Huo L, He J, et al. Soluble epoxide hydrolase is involved in the development of atherosclerosis and arterial neointima formation by regulating smooth muscle cell migration. Am J Physiol Heart Circ Physiol. 2015;309:H1894–H1903. [DOI] [PubMed] [Google Scholar]
- 115.Simpkins AN, Rudic RD, Roy S, Tsai HJ, Hammock BD, Imig JD. Soluble epoxide hydrolase inhibition modulates vascular remodeling. Am J Physiol Heart Circ Physiol. 2010;298:H795–H806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dai N, Zhao C, Kong Q, Li D, Cai Z, Wang M. Vascular repair and anti-inflammatory effects of soluble epoxide hydrolase inhibitor. Exp Ther Med. 2019;17:3580–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen D, Whitcomb R, MacIntyre E, et al. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol. 2012;52:319–328. [DOI] [PubMed] [Google Scholar]
- 118.Lazaar AL, Yang L, Boardley RL, et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016;81:971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sharma M, McCarthy ET, Reddy DS, et al. 8,9-Epoxyeicosatrienoic acid protects the glomerular filtration barrier. Prostaglandins Other Lipid Mediat. 2009;89:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McCarthy ET, Sharma M. Indomethacin protects permeability barrier from focal segmental glomerulosclerosis serum. Kidney Int. 2002;61:534–541. [DOI] [PubMed] [Google Scholar]
- 121.Imig JD, Elmarakby A, Nithipatikom K, Wei S, Capdevila JH, Tuniki VR, Sangra B, Anjaiah S, Manthati VL, Reddy DS, Falck JR. Development of epoxyeicosatrienoic acid analogs with in vivo anti-hypertensive actions. Frontiers in Vascular Physiology. 2010;1:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Khan MA, Pavlov TS, Christian SV, Neckar J, Staruschenko A, Gauthier KM, Capdevila JH, Falck JR, Campbell WB, Imig JD. Epoxyeicosatrienoic acid (EET) analog lowers blood pressure through vasodilation and sodium channel inhibition. Clinical Science. 2014;127:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sodhi K, Puri N, Inoue K, Falck JR, Schwartzman ML, Abraham NG. EET agonist prevents adiposity and vascular dysfunction in rats fed a high fat diet via a decrease in Bach 1 and an increase in HO-1 levels. Prostaglandins Other Lipid Mediat. 2012;98:133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jichova S, Kopkan L, Huskova Z, Dolezelova S, Neckar J, Kujal P, Vernerova Z, Kramer HJ, Sadowski J, Kompanoowska-Jezierska E, Reddy NR, Falck JR, Imig JD, Cervenka L. Epoxyeicosatrienoic analog attenuates development of malignant hypertension, but does not reverse it once established: a study in Cyp1a1-Ren-2 transgenic rats. Journal of Hypertension. 2016;34: 2008–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schragenheim J, Bellner L, Cao J, et al. EET enhances renal function in obese mice resulting in restoration of HO-1-Mfn1/2 signaling, and decrease in hypertension through inhibition of sodium chloride co-transporter. Prostaglandins Other Lipid Mediat. 2018;137:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yeboah MM, Hye Khan MA, Chesnik MA, et al. The epoxyeicosatrienoic acid analog PVPA ameliorates cyclosporine-induced hypertension and renal injury in rats. Am J Physiol Renal Physiol. 2016;311:F576–F585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013;27:2946–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hoff U, Bubalo G, Fechner M, et al. A synthetic epoxyeicosatrienoic acid analogue prevents the initiation of ischemic acute kidney injury. Acta Physiol (Oxf). 2019;227:e13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Skibba M, Hye Khan MA, Kolb LL, et al. Epoxyeicosatrienoic Acid Analog Decreases Renal Fibrosis by Reducing Epithelial-to-Mesenchymal Transition. Front Pharmacol. 2017;8:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hye Khan MA, Fish B, Wahl G, et al. Epoxyeicosatrienoic acid analogue mitigates kidney injury in a rat model of radiation nephropathy. Clin Sci (Lond). 2016;130:587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hye Khan MA, Stavniichuk A, Sattar MA, Falck JR, Imig JD. Epoxyeicosatrienoic Acid Analog EET-A Blunts Development of Lupus Nephritis in Mice. Front Pharmacol. 2019;10:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Batchu SN, Lee SB, Qadhi RS, et al. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br J Pharmacol. 2011;162:897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Neckář J, Hsu A, Hye Khan MA, et al. Infarct size-limiting effect of epoxyeicosatrienoic acid analog EET-B is mediated by hypoxia-inducible factor-1α via downregulation of prolyl hydroxylase 3. Am J Physiol Heart Circ Physiol. 2018;315:H1148–H1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cao J, Tsenovoy PL, Thompson EA, et al. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat. 2015;116–117:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Singh SP, McClung JA, Bellner L, et al. CYP-450 Epoxygenase Derived Epoxyeicosatrienoic Acid Contribute To Reversal of Heart Failure in Obesity-Induced Diabetic Cardiomyopathy via PGC-1 α Activation. Cardiovasc Pharm Open Access. 2018;7:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhao Q, Huang J, Wang D, Chen L, Sun D, Zhao C. Endothelium-specific CYP2J2 overexpression improves cardiac dysfunction by promoting angiogenesis via Jagged1/Notch1 signaling. J Mol Cell Cardiol. 2018;123:118–127. [DOI] [PubMed] [Google Scholar]
- 137.Khan MAH, Manthati V, Errabelli R, Pavlov TS, Staruschenko A, Falck JR, Imig JD. An orally active epoxyeicosatrienoic acid analog attenuates kidney injury in hypertensive Dahl salt sensitive rat. Hypertension. 2013;62:905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Červenka L, Husková Z, Kopkan L, Kikerlová S, Sedláková L, Vaňourková Z, Alánová P, Kolář F, Hammock BD, Hwang SH, Imig JD, Falck JR, Sadowski J, Kompanowska-Jezierska E, Neckář J. Two pharmacological epoxyeicosatrienoic acids-enhancing therapies effectively anti-hypertensive and reduce the severity of ischemic arrhythmias in rats with angiotensin II-dependent hypertension. Journal of Hypertension. 2018;36:1326–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Romashko M, Schragenheim J, Abraham NG, McClung JA. Epoxyeicosatrienoic Acid as Therapy for Diabetic and Ischemic Cardiomyopathy. Trends Pharmacol Sci. 2016;37:945–962. [DOI] [PubMed] [Google Scholar]
- 140.El-Sikhry HE, Alsaleh N, Dakarapu R, Falck JR, Seubert JM. Novel Roles of Epoxyeicosanoids in Regulating Cardiac Mitochondria. PLoS One. 2016;11:e0160380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fleming I The cytochrome P450 pathway in angiogenesis and endothelial cell biology. Cancer Metastasis Rev. 2011;30:541–555. [DOI] [PubMed] [Google Scholar]
- 142.Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC. EET signaling in cancer. Cancer Metastasis Rev. 2011;30:525–540. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 143.Panigrahy D, Edin ML, Lee CR, et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012;122:178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bettaieb A, Chahed S, Bachaalany S, Griffey S, Hammock BD, Haj FG. Soluble Epoxide Hydrolase Pharmacological Inhibition Ameliorates Experimental Acute Pancreatitis in Mice. Mol Pharmacol. 2015;88:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang G, Panigrahy D, Hwang SH, et al. Dual inhibition of cyclooxygenase-2 and soluble epoxide hydrolase synergistically suppresses primary tumor growth and metastasis. Proc Natl Acad Sci U S A. 2014;111:11127–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li J, Zhou Y, Wang H, et al. COX-2/sEH dual inhibitor PTUPB suppresses glioblastoma growth by targeting epidermal growth factor receptor and hyaluronan mediated motility receptor. Oncotarget. 2017;8:87353–87363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sander AL, Sommer K, Neumayer T, et al. Soluble epoxide hydrolase disruption as therapeutic target for wound healing. J Surg Res. 2013;182:362–367. [DOI] [PubMed] [Google Scholar]
- 148.Dai N, Zhao C, Kong Q, Li D, Cai Z, Wang M. Vascular repair and anti-inflammatory effects of soluble epoxide hydrolase inhibitor. Exp Ther Med. 2019;17:3580–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kylhammar D, Rådegran G. The principal pathways involved in the in vivo modulation of hypoxic pulmonary vasoconstriction, pulmonary arterial remodelling and pulmonary hypertension. Acta Physiol (Oxf). 2017;219:728–756. [DOI] [PubMed] [Google Scholar]
- 150.Kandhi S, Zhang B, Froogh G, et al. EETs promote hypoxic pulmonary vasoconstriction via constrictor prostanoids. Am J Physiol Lung Cell Mol Physiol. 2017;313:L350–L359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Swanson D, Block R, Mousa SA. Omega-3 fatty acids EPA and DHA: health benefits throughout life. Adv Nutr. 2012;3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schunck WH, Konkel A, Fischer R, Weylandt KH. Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases. Pharmacol Ther. 2018;183:177–204. [DOI] [PubMed] [Google Scholar]
- 153.Schunck WH. EPA and/or DHA? A test question on the principles and opportunities in utilizing the therapeutic potential of omega-3 fatty acids. J Lipid Res. 2016;57:1608–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Adebesin AM, Wesser T, Vijaykumar J, et al. Development of Robust 17(R),18(S)-Epoxyeicosatetraenoic Acid (17,18-EEQ) Analogues as Potential Clinical Antiarrhythmic Agents. J Med Chem. 2019;62:10124–10143. [DOI] [PubMed] [Google Scholar]
- 155.Sharma A, Hye Khan MA, Levick SP, Lee KS, Hammock BD, Imig JD. Novel Omega-3 Fatty Acid Epoxygenase Metabolite Reduces Kidney Fibrosis. Int J Mol Sci. 2016;17:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Arnold C, Markovic M, Blossey K, et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}−3 fatty acids. J Biol Chem. 2010;285:32720–32733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kitamura S, Morisseau C, Harris TR, Inceoglu B, Hammock BD. Occurrence of urea-based soluble epoxide hydrolase inhibitors from the plants in the order Brassicales. PLoS One. 2017;12:e0176571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thao NP, Luyen BTT, Lee JS, Kim JH, Dat NT, Kim YH. Inhibition Potential of Cycloartane-Type Glycosides from the Roots of Cimicifuga dahurica against Soluble Epoxide Hydrolase. J Nat Prod. 2017;80:1867–1875. [DOI] [PubMed] [Google Scholar]
- 159.Caligiuri SP, Aukema HM, Ravandi A, Guzman R, Dibrov E, Pierce GN. Flaxseed consumption reduces blood pressure in patients with hypertension by altering circulating oxylipins via an α-linolenic acid-induced inhibition of soluble epoxide hydrolase. Hypertension. 2014;64:53–59. [DOI] [PubMed] [Google Scholar]
- 160.Berna-Erro A, Izquierdo-Serra M, Sepúlveda RV, et al. Structural determinants of 5’,6’-epoxyeicosatrienoic acid binding to and activation of TRPV4 channel. Sci Rep. 2017;7:10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bukhari IA, Shah AJ, Gauthier KM, et al. 11,12,20-Trihydroxy-eicosa-8(Z)-enoic acid: a selective inhibitor of 11,12-EET-induced relaxations of bovine coronary and rat mesenteric arteries. Am J Physiol Heart Circ Physiol. 2012;302:H1574–H1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu P, Zhang S, Gao J, et al. Downregulated Serum 14, 15-Epoxyeicosatrienoic Acid Is Associated With Abdominal Aortic Calcification in Patients With Primary Aldosteronism. Hypertension. 2018;71:592–598. [DOI] [PubMed] [Google Scholar]
- 163.Park SK, Herrnreiter A, Pfister SL, et al. GPR40 is a low-affinity epoxyeicosatrienoic acid receptor in vascular cells. J Biol Chem. 2018;293:10675–10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liu X, Qian ZY, Xie F, et al. Functional screening for G protein-coupled receptor targets of 14,15-epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2017;132:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hiesinger K, Wagner KM, Hammock BD, Proschak E, Hwang SH. Development of multitarget agents possessing soluble epoxide hydrolase inhibitory activity. Prostaglandins Other Lipid Mediat. 2019;140:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Achenbach J, Klingler FM, Blöcher R, et al. Exploring the chemical space of multitarget ligands using aligned self-organizing maps. ACS Med Chem Lett. 2013;4:1169–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hye Khan MA, Hwang SH, Sharma A, Corbett JA, Hammock BD, Imig JD. A dual COX-2/sEH inhibitor improves the metabolic profile and reduces kidney injury in Zucker diabetic fatty rat. Prostaglandins Other Lipid Mediat. 2016;125:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Falck JR, Koduru SR, Mohapatra S, Manne R, Atcha R, Manthati VL, Capdevila JH, Christian S, Imig JD, Campbell WB. 14,15-Epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates: carboxylate modifications. J Med Chem. 2014;57:6965–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hye Khan MA, Kolb L, Skibba M, et al. A novel dual PPAR-γ agonist/sEH inhibitor treats diabetic complications in a rat model of type 2 diabetes. Diabetologia. 2018;61:2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hye Khan MA, Schmidt J, Stavniichuk A, Imig JD, Merk D. A dual farnesoid X receptor/soluble epoxide hydrolase modulator treats non-alcoholic steatohepatitis in mice. Biochem Pharmacol. 2019;166:212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]