

Abstract
The incorporation of exogenous molecules into live cells is essential for both biological research and therapeutic applications. In particular, for the emerging field of super-resolution microscopy of live mammalian cells, it remains a challenge to deliver tailored, often cell-impermeable, fluorescent probes into live cells for target labeling. Here, utilizing the outstanding mechanical, electrical, and optical properties of graphene, we report a facile approach that enables both high-throughput delivery of fluorescent probes into adherent mammalian cells and in situ super-resolution microscopy on the same device. ~90% delivery efficiencies are achieved for free dyes and dye-tagged affinity probes, short peptides, and whole antibodies, thus enabling high-quality super-resolution microscopy. Moreover, we demonstrate good spatiotemporal controls, which, in combination with the ready patternability of graphene, allow for the spatially selective delivery of two different probes for cells at different locations on the same substrate.
Keywords: intracellular delivery, graphene, electroporation, live-cell labeling, super-resolution microscopy
The delivery of exogenous genes or probes into live cells is critically important for both biological research and therapeutic applications.1–3 For example, in bioimaging, it is often necessary to deliver exogenous genes or probes into cells to visualize the targets of interest. In particular, for the emerging field of super-resolution microscopy (SRM) of live mammalian cells,4–8 although the intracellular gene expression of fluorescent proteins (FPs) provides a relatively straightforward approach,4 FPs are large in size and offer limited brightness and photostability when compared to organic dyes.9,10 On the other hand, high-performance dye-based probes for SRM often do not readily cross the strong barrier created by the cell plasma membrane,6,8,10 and so rely on membrane-disruption techniques for intracellular delivery.
Although chemical permeabilization, including the use of mild detergents and toxins,2,11–13 provides relatively easy means of intracellular delivery, recovery of membrane integrity is often challenging. Microinjection provides a controlled means for intracellular delivery,2 but is limited in throughput and highly dependent on the operator’s skill. Electroporation creates small, resealable pores on the cell membrane via applied electric fields, and is nowadays routinely used for intracellular delivery owing to its high efficiency and low cell toxicity.2,3,14 However, electroporation is typically performed for detached and (re-)suspended cells. For the delivery of external fluorescent probes, the long (~10 h) subsequent re-plating time,6,8 which is essential for the cells to re-adhere to the coverslip for high-resolution imaging, is inconvenient and potentially gives rise to undesired side effects due to the prolonged introduction of probes. Recent advances in nanotechnology and microfluidics have led to the exciting development of numerous new intracellular-delivery methods,1,2,15–21 each overcoming certain limits of traditional approaches. However, so far these technologies have not been designed to enable high-resolution microscopy on the same device after high-throughput delivery.
Here we introduce a graphene-based, facile approach for the direct, high-throughput delivery of fluorescent probes into adherent cells to enable in situ live-cell SRM on the same device within minutes. Recent years have witnessed rising research interest in interfacing graphene with cell biology.22–29 We previously demonstrated the use of graphene to encapsulate wet mammalian cells to enable facile electron microscopy.30 In this work, we employ graphene’s outstanding properties as both a good substrate for adherent cell growth25–29 and a good conductor to enable electroporation-based probe delivery into adhered mammalian cell lines and primary cells, and then utilize the high optical transmission and ultimate thinness of graphene to achieve in situ SRM imaging on the same device using an oil-immersion objective lens. Moreover, through the ready patternability of graphene, we demonstrate the spatially selective delivery of different probes into cells at different parts of the same substrate.
RESULTS AND DISCUSSION
Graphene-enabled dye delivery into adherent cells.
Monolayer graphene, as produced by chemical vapor deposition on copper foils,31 was deposited onto regular glass coverslips as ~10×5 mm2 pieces, sealed with a small plastic tube, and contacted at both edges (Fig. 1a and Fig. S1). Two-point measurements showed resistances of a few kΩ across the as-prepared devices. Raman spectroscopy, atomic force microscopy, and interference reflection microscopy (IRM)32 confirmed that graphene in the final devices to be continuous monolayers with minimal defects (Fig. S2). Adherent mammalian cells, including both cell lines (A549 and PtK2 cells) and primary cells (rat hippocampal neurons and neural stem cells), were cultured on the graphene surface under standard tissue culture conditions. Previous work has shown graphene-based substrates to be suitable for the growth, development, and proper physiology of many cell types.25–29 We likewise found the four cell types investigated in this work grew and developed well on our graphene electrodes, for which cases the two cell lines were cultured on uncoated graphene, whereas for the primary neurons and neural stem cells, the graphene surface was coated with poly-D-lysine or poly-L-ornithine/laminin following established protocols (Methods).33,34
Figure 1.
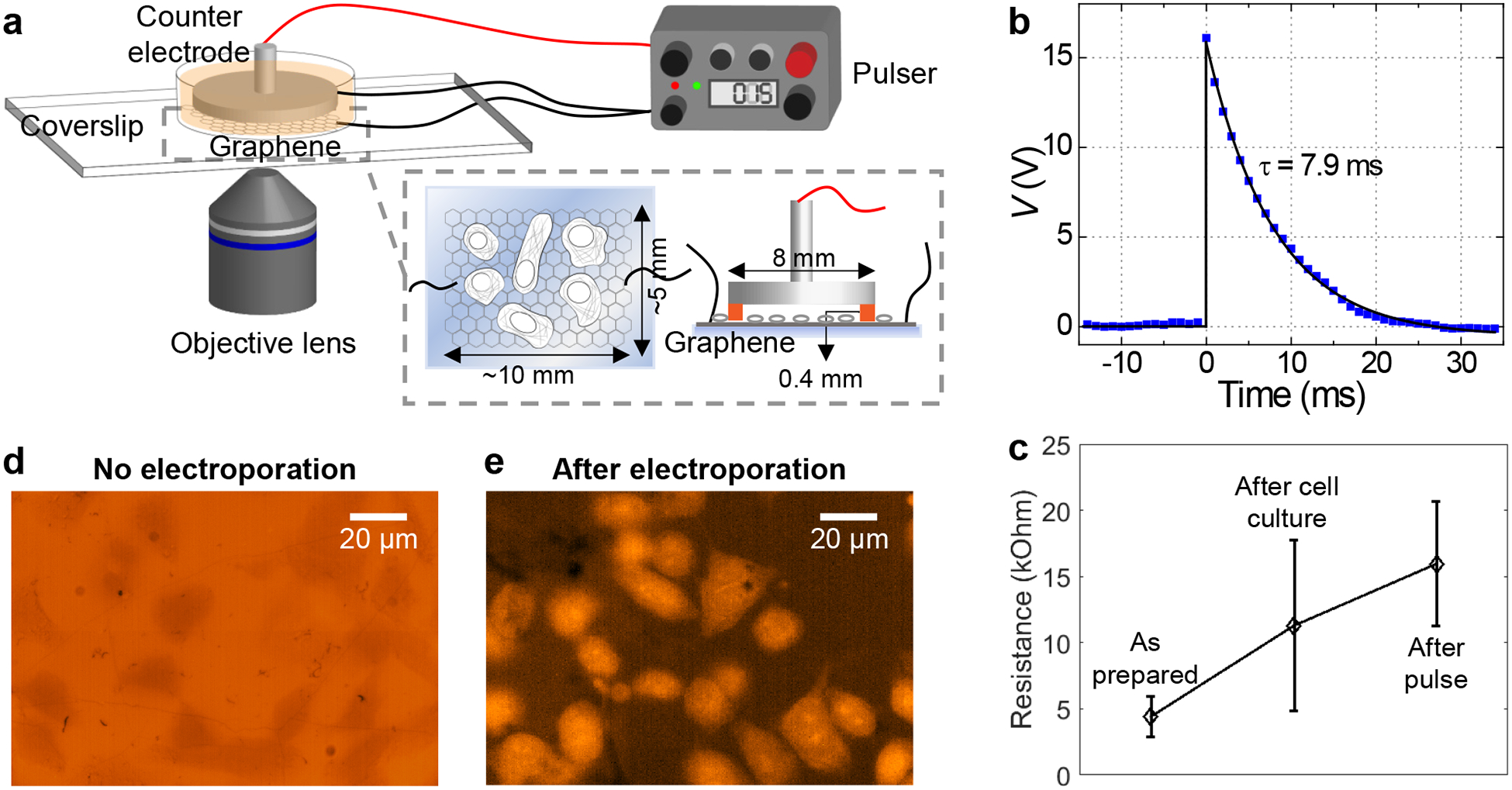
Electroporation of adherent cells on a graphene-covered glass coverslip. (a) Schematic of the experimental setup. (b) A representative pulse shape, as measured from an oscilloscope. (c) Measured two-point resistance of graphene for the as-prepared device, after culturing of adherent cells, and after electroporation (N = 4 measured devices). Error bars: standard deviation between samples. (d) Fluorescence micrograph of PtK2 cells incubated in an SR101-containing medium for 10 minutes; cells appear as darker regions due to the physical exclusion of the dye. (e) Fluorescence micrograph of PtK2 cells on graphene after the application of the voltage pulse, 10 min incubation, and wash-out of free dyes.
At the time of probe delivery, the culture medium was replaced with a commercial electroporation buffer solution, to which we added the desired fluorescent probes. Counter electrode was made out of a metal pin-stub mount commonly used in scanning electron microscopy (SEM), which was mounted up-side-down with the application of ~400 μm-thick Teflon tape on the rim as an insulating spacer (Fig. 1a and Fig. S1). This counter electrode was gently placed into the device, and the assembled device was mounted onto an inverted fluorescence microscope. Scanning the microscope focus indicated that the distance between the graphene and counter-electrode surfaces was ~500 μm.
For electroporation, we applied a voltage pulse across the graphene and counter electrodes through capacitor discharging using a commercial electroporator. Efficient probe delivery was readily achieved at low voltages (~15 V) using a small (10 μF) capacitor, with typical pulse halftimes of ~5–10 ms (Fig. 1b). Although the voltage we applied was substantially lower than what is optimized for suspended mammalian cells in commercial systems (e.g., ~1000 V for a 3 cm electrode separation35), the electrical field experienced by the cells was comparable (~300 V/cm based on our ~500 μm electrode separation). Similar electroporation results are thus expected despite the very different absolute voltages. Only a mild increase in graphene resistance was noted after the electroporation process (Fig. 1c), suggesting that the graphene electrode remained mostly intact.
We started with the delivery of an organic dye, sulforhodamine 101 (SR101, molecular weight: 606.7). As SR101 is not normally taken up by PtK2 cells, microscopy of cells in a medium containing this probe showed lower fluorescence for cell-occupied areas owing to physical exclusion (Fig. 1d). After electroporation in the graphene device, 10 min of incubation, and then washout of free dyes, we found the cytoplasm of most cells became fluorescent due to the incorporation of SR101 (Fig. 1e). See also Figs. 2 and 4 below for a direct comparison of the labeling of various probes for cells in the same devices with and without graphene electroporation.
Figure 2.
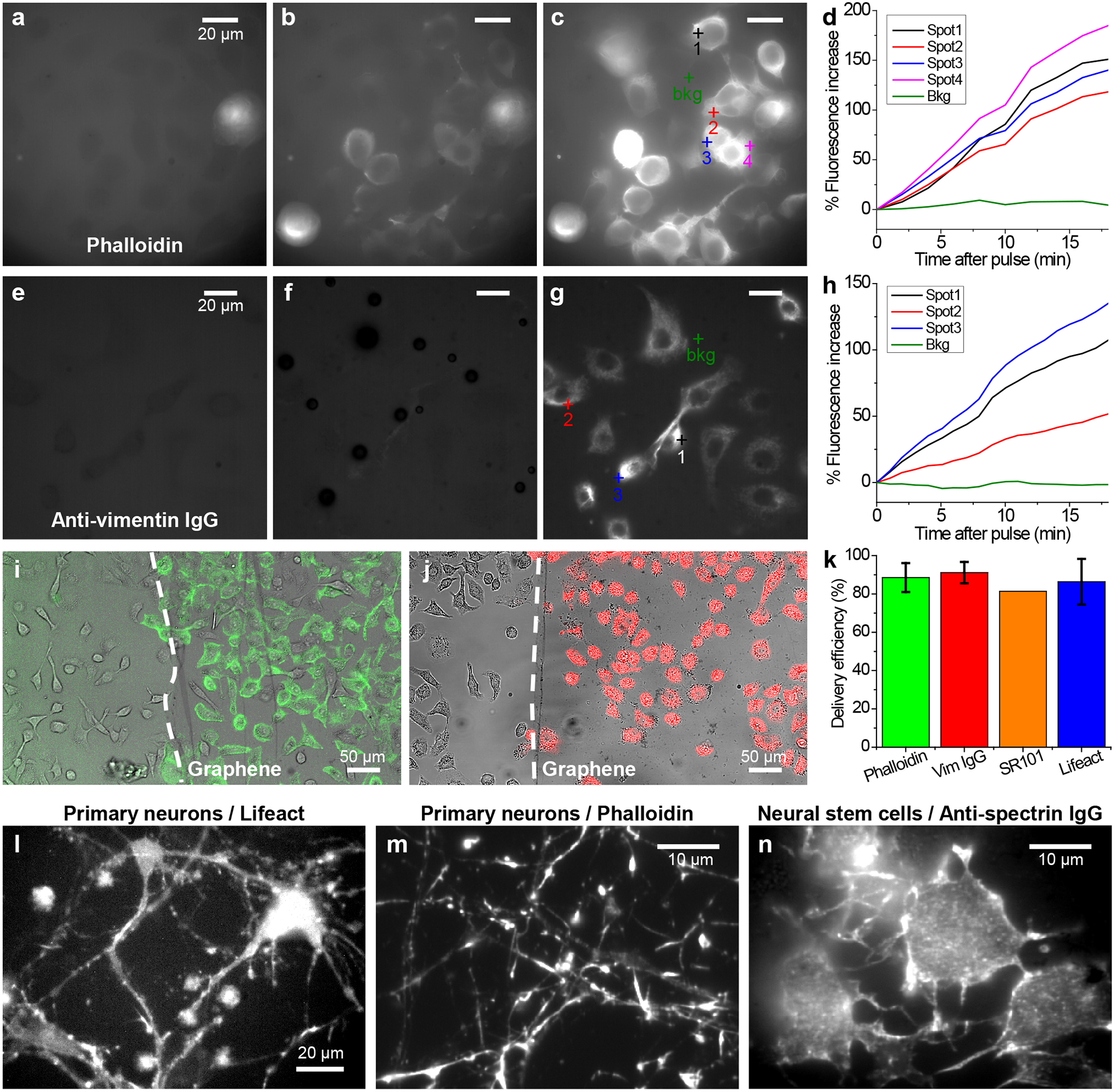
Delivery of dye-tagged probes that bind to specific intracellular targets. (a-c) Delivery of phalloidin-AF488 into A549 cells through graphene electroporation. (a) Fluorescence micrograph of the sample before the application of the voltage pulse. (b,c) Fluorescence micrographs for the same area 1 min (b) and 20 min (c) after the voltage pulse. (d) Change in local fluorescence intensity over time after the voltage pulse, for the different spots marked in (c). (e-g) Delivery of AF647-conjugated anti-vimentin full IgG antibody into A549 cells. (e) Fluorescence micrograph before the voltage pulse. (f,g) Fluorescence micrographs for the same area 1 min (f) and 20 min (g) after the voltage pulse. (h) Change in local fluorescence intensity over time after the voltage pulse, for the different spots marked in (g). (i) Merged transmission (grayscale) and fluorescence (green) micrographs for the spatially controlled delivery of phalloidin in A549 cells adhered to graphene (to the right of the dashed line) vs. no delivery to cells on the bare glass surface without graphene (left of the dashed line). (j) Similar results for the spatially controlled delivery of anti-vimentin IgG (red). (k) Percentage of cells being labeled for phalloidin (N = 7 devices), anti-vimentin IgG (N = 4 devices), SR101 (N = 1 device), and Lifeact (N = 4 devices), as determined from correlated transmission and fluorescence micrographs. Error bars: standard deviation between samples. (l,m) Fluorescence micrographs for the graphene-electroporation delivery of Lifeact-Cy5 (l) and phalloidin-AF647 (m) into primary hippocampal neurons that were cultured in graphene devices for 14 days. (n) Delivery of CF647-conjugated anti-spectrin IgG into primary neural stem cells that have been differentiating in a graphene device for 3 days.
Figure 4.
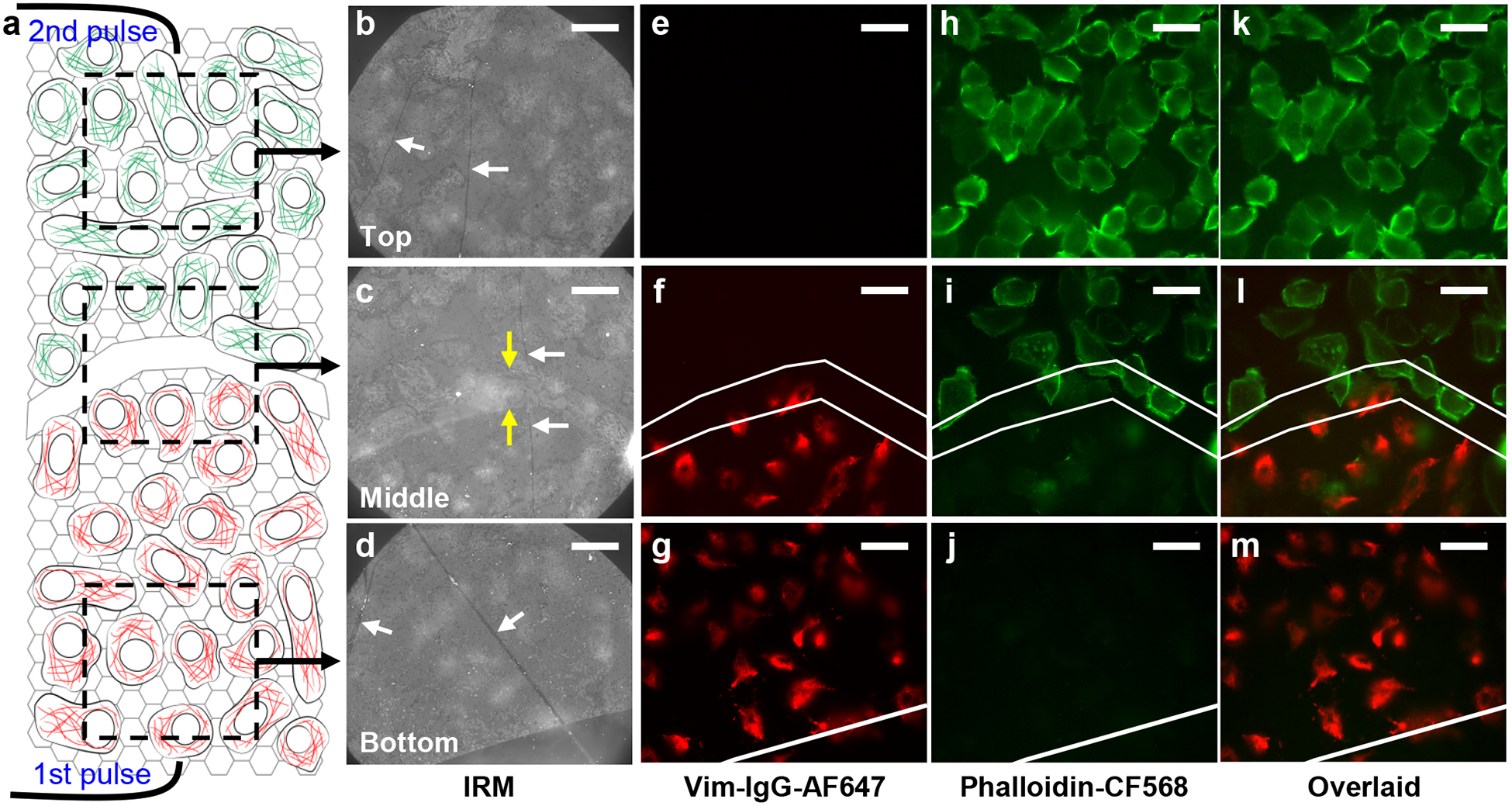
Patterned delivery of two different probes for cells adhered to different regions of the same substrate. (a) Schematic of the sample, with a scratch through the graphene electrode that divided it into two halves. A voltage pulse was first applied to the bottom half in the presence of the first (red) fluorescent probe, cells were recovered for ~20 min, and then a second voltage pulse was applied to the top half in the presence of the second (green) fluorescent probe. (b-d) IRM images of A549 cells on graphene for the top (b), middle (c), and bottom (d) parts of the sample, as schematized in (a). White arrows point to wrinkles in graphene, which give high IRM contrast. Yellow arrows point to edges of the scratch. (e-g) Fluorescence micrographs for the same areas as (b-d), for the channel of the first fluorescent probe (AF647-tagged anti-vimentin IgG antibody). (h-j) Fluorescence micrographs for the same areas for the second fluorescent probe (phalloidin-CF568). (k-m) Overlay of the two color channels. White lines in (f,i,l) mark the edges of the top and bottom halves of graphene. Scale bars: 20 μm.
Delivery of small-to-large targeting probes into cell lines and primary cells.
We next examined dye-tagged probes that bind to specific intracellular targets, starting with phalloidin-Alexa Fluor 488 (phalloidin-AF488), a small (~1.3 kDa), highly specific fluorescent marker for the actin cytoskeleton. As expected, the probe itself was non-permeable for A549 cells, and so cells initially appeared as darker regions in fluorescence micrographs due to physical exclusion (Fig. 2a). After the application of an electroporation voltage pulse, the cells quickly lit up in ~1 min (Fig. 2bd), and continued to rise in fluorescence signal (Fig. 2cd). This rise slowed down after ~15 min (Fig. S3), at which point the labeling was often sufficient for SRM (below). Low-magnification images showed, over large areas, most cells on the graphene surface to be efficiently labeled, whereas in the same view, cells on the bare glass substrate remained unlabeled (Fig. 2i). We further found that the cells well maintained their viability after electroporation (Fig. S4). In comparison, chemical permeabilization through Triton X-100 caused notable cell death before efficient probe delivery could be achieved (Fig. S4).
We next turn to the more challenging task of dye-tagged whole immunoglobulin G (IgG) antibodies. Whereas antibody-based fluorescence labeling (immunofluorescence) is routine for SRM of fixed and permeabilized cells and is favorable for its ease and versatility,8,9 its use in live-cell microscopy and SRM has been rare13 due to difficulties in delivering the sizeable (~155 kDa) IgG molecules into the cell. We found graphene-based electroporation enabled efficient delivery of dye-tagged IgGs, e.g., Alexa Fluor 647 (AF647)-tagged IgG against vimentin (Fig. 2e–g), although the increase in intracellular fluorescence was slower (Fig. 2e–h) when compared to that of phalloidin, possibly attributable to slower diffusion of the heavy IgGs. Low-magnification images showed that similar to that with phalloidin, highly efficient and selective labeling was achieved for cells on the graphene surface (Fig. 2j). Correlating transmission and fluorescence micrographs showed that, consistently, ~80–90% of cells on the graphene surface to be successfully labeled (Fig. 2k) for the cases of phalloidin, anti-vimentin IgG, SR101, as well as Cy5-tagged Lifeact, a 17-amino-acid peptide that reversely binds to actin filaments.36
We further validated our method for primary neurons and neural stem cells: these cells require extended adherent culture before maturation, at which stage intracellular delivery is difficult with conventional methods. Even though the positively charged poly-D-lysine and poly-L-ornithine coating likely shifted the surface potential, this effect is likely small when compared to the 15 V voltage applied to our devices. Consequently, with the same graphene-based electroporation procedure as above, we achieved good delivery results for the mature primary rat hippocampal neurons after 14-day adherent culture (Fig. 2l and 2m for Lifeact-Cy5 and phalloidin-AF647, respectively), as well as for the differentiating primary neural stem cells after 3-day adherent culture (Fig. 2n for CF647-conjugated anti-spectrin IgG). Together, we have shown that our graphene-based electroporation allowed for the intracellular delivery of small to large non-permeable probes into both adherent cell lines and primary cells with high efficiency.
In situ live-cell STORM super-resolution microscopy.
Based on the above good delivery results, we next achieved in situ live-cell STORM (stochastic optical reconstruction microscopy37,38) SRM in the same device immediately after probe delivery. Here we utilized the outstanding properties of monolayer graphene that, although it is highly conductive (to enable electroporation), it is also highly transparent to light (~98% trasmission39) and is only a single layer of atoms, such that high-resolution microscopy could be performed directly through graphene using an oil-immersion objective lens with no potential image deteriorations due to mismatched index of refractivity. Thus, after electroporation delivery of STORM-compatible probes, we replaced the cell medium with a live-cell STORM imaging buffer,6 and mounted the device on a 3D-STORM system equipped with a 100x, oil-immersion objective.
Conventional epi-fluorescence images taken at low illumination powers, e.g., for vimentin filaments labeled in live A549 cells through the graphene-electroporation delivery of AF647-tagged IgG, showed no signs of distortion as we imaged through the graphene electrode (Fig. 3a). By next increasing the illumination power, we photoswitched most of the labeled probe molecules into a non-emitting dark state. The reversible photoswitching of these molecules between the dark and emitting states led to well-resolved, bright single-molecule fluorescence (Fig. 3b) that “blinked” stochastically in space and time, which we recorded continuously at 110 frames per second using an EM-CCD. An average of ~3,500 photons were collected for each emitting single molecule, in agreement with that typically obtained with AF647 in live-cell STORM.6 Accumulating the 3D localizations38 obtained from 47,128 frames of single-molecule images led to 3D-STORM SRM images of high resolution (Fig. 3c). For the single vimentin intermediate filament pointed to by the red arrow in Fig. 3c, a cross-sectional profile gave an FWHM (full width at half maximum) of 30 nm (Fig. 3d), consistent with a convolution of the ~20 nm spatial resolution of STORM6,37,38 with a ~20 nm diameter of IgG-labeled vimentin filament. Figure 3e further shows a case (magenta arrow in Fig. 3c) in which two filaments were clearly resolved at a center-to-center distance of 144 nm, well below the diffraction limit. Subdividing the collected frames of single-molecule images to construct a sequence of STORM images further enabled the scrutiny of nanoscale structural changes over time (Fig. 3fg). Good live-cell STORM SRM was also achieved for the actin cytoskeleton labeled by Lifeact-Cy5 (Fig. 3hi) and phalloidin-AF647 (Fig. S5).
Figure 3.
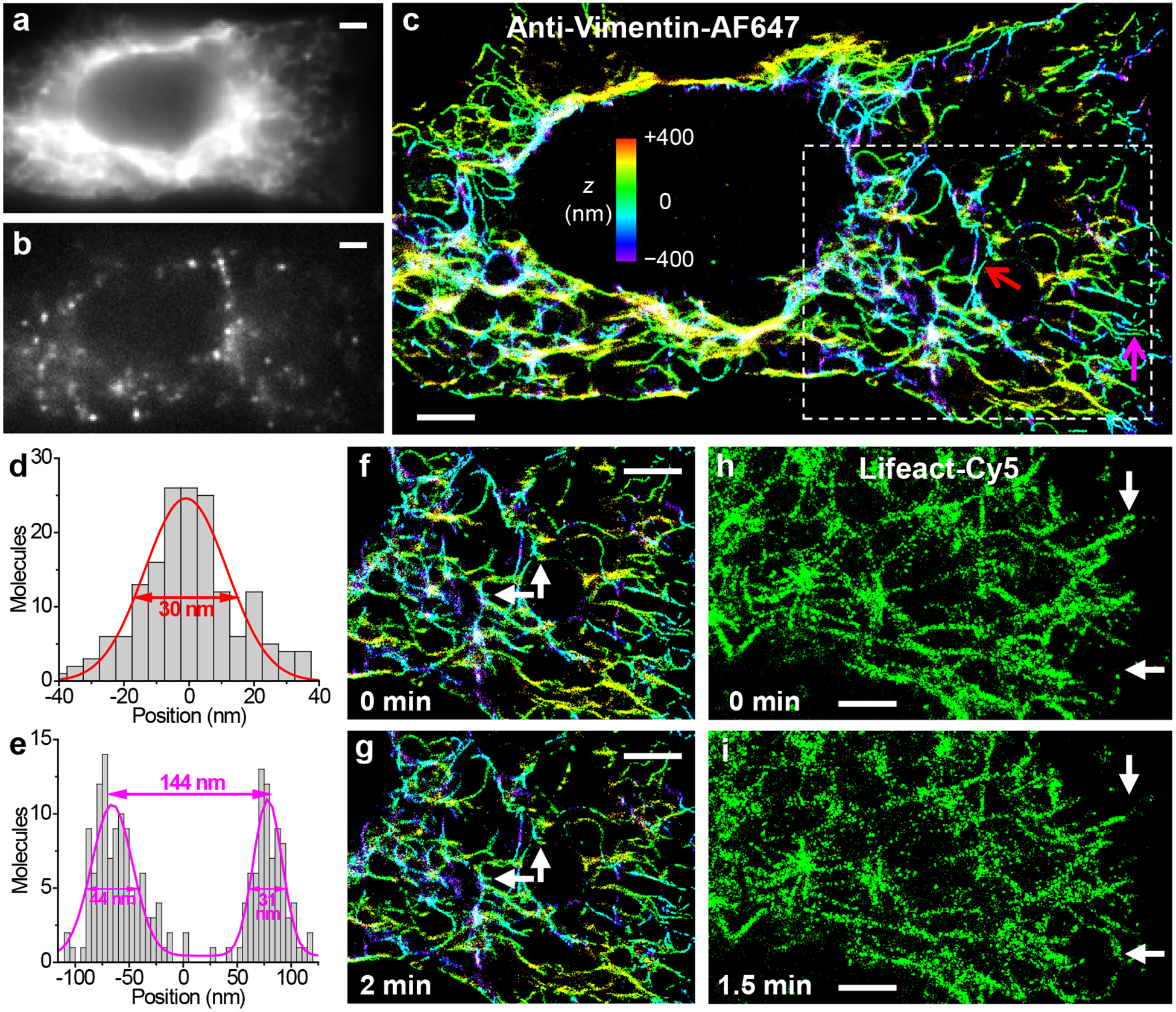
In situ live-cell STORM SRM through fluorescent probes delivered by graphene electroporation. (a) Diffraction-limited image of vimentin in a live A549 cell labeled through the graphene-electroporation delivery of an AF647-tagged IgG antibody. (b) A typical frame of single-molecule images during STORM acquisition. (c) Resultant 3D-STORM image. Color presents the depth (z) information (color scale bar). (d) Cross-sectional profile of the single vimentin filament pointed to by the red arrow in (c). Fit to a normal distribution gave FWHM of 30 nm. (e) Cross-sectional profile for two adjacent filaments pointed to by the magenta arrow in (c). (f,g) A sequence of two STORM images at 0 min (f) and 2 min (g). Arrows point to structural changes at the nanoscale. (h,i) A sequence of two STORM images of actin filaments in a live A549 cell labeled through the graphene-electroporation delivery of Lifeact-Cy5, at 0 min (h) and 1.5 min (i). Arrows point to notable structural changes. Scale bars: 2 μm.
Patterned delivery of two different probes.
We next further exploit the spatial and temporal control of our graphene-based approach to enable patterned delivery of two different probes. As a first demonstration, we made a scratch at the center of graphene to divide it into two (top and bottom) halves, which were each separately contacted by a metal wire (Fig. 4a). IRM32 (Fig. 4b–d) showed that the scratch was ~20 μm-wide, for which region graphene was fully removed, and conductance measurements indicated that the two halves were electrically isolated. After plating cells, we first replaced the culture medium with an electroporation buffer that contained AF647-conjugated anti-vimentin IgG, and applied a 15 V pulse only to the bottom half of the graphene electrode against the counter electrode. After ~15 min incubation, the cells were allowed to recover in a medium containing 2 mM ATP and 2% glucose at 37 °C for ~20 min.13 The medium was replaced by another electroporation buffer that contained phalloidin-CF568, and then another 15 V pulse was applied across the top half of the graphene electrode and the counter electrode.
As expected, the above sequential electroporation procedure enabled patterned delivery, so that fluorescent micrographs taken in the AF647 and CF568 channels showed that the former was selectively delivered into cells on the bottom half of the graphene electrode (Fig. 4e–g;k–m), whereas the latter was selectively delivered into cells in the top half of the device (Fig. 4h–m). Interestingly, we further found that the spatial specificity for the second probe relied on the proper recovery (sealing) of the plasma membrane after the first electroporation step. Skipping this recovery step led to nonspecific delivery into the unsealed cells due to the first electroporation pulse (Fig. S6).
CONCLUSION
In summary, we have developed an integrated system that enables the facile electroporation delivery of fluorescent probes into adherent mammalian cell lines and primary cells for immediate single-molecule detection and SRM on the same platform. High (~90%) delivery efficiency was achieved with low pulse voltages for probes ranging in size from free dye molecules up to full IgG antibodies, and the outstanding optical properties of graphene enabled SRM using an oil-immersion objective. Moreover, we demonstrated good spatial and temporal controls, achieving patterned delivery of two probes for different regions of the same substrate with high selectivity. By removing the need to detach and then re-adhere the cells to coverslips for high-resolution microscopy, as required by typical electroporation methods, our in situ approach greatly expedites labeling and reduces the potential adverse effects due to prolonged retention of external probes inside live cells, and enabled electroporation of the highly demanding primary neurons and neural stem cells after extended periods of adherent culture. Whereas in this work we have focused on probes based on organic dyes for their ease of visualization, our approach may also enable the delivery of other probes or chemicals, including drugs, into live cells. By being able to deliver different chemicals to different, spatially predefined subsets of cells on the same substrate under the same conditions, a well-controlled, multiplexed platform may thus be constructed for the quantitative examination of drug effects through high-resolution microscopy.
MATERIALS AND METHODS
Graphene deposition and device preparation.
Monolayer graphene grown on copper foils (Graphene Supermarket) was transferred onto cleaned, thickness #1.5 glass coverslips (24×60 mm2, VWR) as ~10×5 mm2 rectangular pieces following the standard wet-transfer method with polymethyl methacrylate protection.31 The graphene piece was electrically connected at both ends with silver paint (16031, Ted Pella) (Fig. S1), and two-point resistance across the graphene electrode was measured using a multimeter. The graphene electrode was mounted with a clear plastic tube (cut from a 1.6 mL microcentrifuge tube, inner diameter ~9 mm) using Cytoseal 60 (8310–16, Thermo Scientific) or epoxy, which functioned as the chamber for cell growth (below). The device was air-dried and sterilized under a UV lamp for >30 min. For experiments on hippocampal neurons, the graphene surface was coated with a solution of 200 μg/mL poly-D-lysine hydrobromide (Sigma-Aldrich) in Dulbecco’s phosphate-buffered saline (DPBS; Gibco) at 37 °C overnight. For experiments on neural stem cells, the graphene surface was coated with poly-L-ornithine hydrobromide (Sigma-Aldrich) at 10 μg/mL and natural mouse laminin (Invitrogen) at 5 μg/mL.
Cell culture in the graphene device.
A549 and PtK2 adherent mammalian cells were cultured following standard protocols in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with GlutaMAX-I (10566–016, Gibco) and 10% fetal bovine serum. Cells were kept in a T25 flask at 37 °C with 5% CO2 in a humidified incubator, and passaged 2~3 times each week. 16~24 h before electroporation, cells were detached with TrypLE (12605–010, Gibco) and harvested. ~75,000 cells were plated into each device, which directly adhered to the uncoated graphene surface and yielded ~60–95% confluency after 16~24 h. Primary rat hippocampal neurons33 were from BrainBits (Springfield, IL). ~10,000 cells were plated into the above poly-D-lysine-coated device in a neuron medium [10 mL B27 and 5 mL GlutaMAX to 500 mL Neurobasal medium (Invitrogen)]. Half of the medium was replaced every 3–4 days. Rat adult hippocampal neural stem cells were a kind gift from the David V. Schaffer Lab, and were isolated, cultured, and differentiated as previously described.34 ~20,000 cells were plated into the above poly-L-ornithine and laminin-coated device. The culturing medium was DMEM/F-12 supplemented with 1% (v/v) N-2 Supplement (Invitrogen), 1 μM retinoic acid, and 1% (v/v) fetal bovine serum.
Assembly of device for electroporation.
For the silver paint contacts on each side, a fine copper wire was laid on top and gently affixed to the coverslip surface with tape. A new drop of silver paint was applied on top to complete connection to the graphene electrode (Fig. S1). Meanwhile, a counter electrode was made out of a metal pin-stub mount commonly used in scanning electron microscopy, which was mounted upside-down in the device (Fig. S1). This counter electrode is reusable, and was sterilized with ethanol and water before use. A ~400 μm-thick Teflon tape was used to cover the rim of the metal stub, which acted as an insulating spacer to define the distance between the metal surface and the graphene electrode (Fig. 1a). Cell culture medium was gently replaced with ~150 μL of electroporation buffer that contained the fluorescent probes for delivery (below). The device was mounted onto an inverted microscope (below), and the above-described metal counter electrode was gently placed into the device. Scanning the focus of the microscope indicated that the distance between the graphene and counter-electrode surfaces was ~500 μm. A commercial capacitor-discharge pulser (Cell-Porator 1600, Gibco BRL Life Technologies) was employed to apply a voltage pulse across the graphene electrode and the counter electrode, and the applied voltage was monitored with an oscilloscope (TDS210, Tektronix). The pulser capacitor was set to 10 μF, charged to the set voltage (~15 V typical), and triggered.
Fluorescent probes.
All dye dilutions were freshly prepared before electroporation. Alexa Fluor 488 (AF488)-labeled phalloidin (A12379, Invitrogen) and phalloidin-CF568 (00044-F, Biotium) were diluted in the electroporation buffer (1652676, Bio-Rad) to a final concentration of ~50 nM. Alexa Fluor 647 (AF647)-labeled phalloidin (A22287, Invitrogen) was diluted to ~100 nM. Lifeact-Cy5 was custom-ordered from Sigma-Aldrich, and diluted in the electroporation buffer to ~42 nM. AF647-conjugated rabbit anti-vimentin monoclonal IgG antibody (9856S, Cell Signaling Technology) was diluted at 1/50~1/100 to a final concentration of ~2–4 μg/mL. CF647-conjugated anti-spectrin IgG antibody was prepared by reacting a monoclonal mouse anti-βII-spectrin antibody (612563, BD Biosciences) with the Mix-n-Stain CF647 Antibody Labeling Kit (#92238, Biotium), and was diluted in the electroporation buffer to a final concentration of ~2 μg/mL.
Conventional, diffraction-limited microscopy.
Conventional, diffraction-limited microscopy was performed on an Olympus IX73 inverted wide-field epifluorescence microscope equipped with a standard lamp (U-HGLGPS). Three different filter cubes were used for the deep-red (AF647 and Cy5), orange (CF568), and green (AF488) fluorescence channels, respectively, all assembled from optical filters from Chroma (Bellows Falls, VT). For the deep-red channel, excitation filter was ET620/60x, dichroic mirror was zt647rdc-UF1, and emission filter was ET700/75m. For the orange channel, excitation filter was ET545/25x, dichroic mirror was zt561rdc-UF1, and emission filter was ET605/70m. For the green channel, excitation filter was ET470/40x, dichroic mirror was T495LPXR, and emission filter was ET525/50m. Interference reflection microscopy (IRM) was performed on the same setup with a filter cube that had a 50/50 beam splitter (Chroma 21000) at the dichroic-mirror position, a narrow bandpass filter (Chroma D532/10x) as the excitation filter, and no emission filter, as previously described.32 Wide-field fluorescence and IRM images were obtained with either an UplanSapo 60x water-immersion objective lens (NA 1.2) or an UplanSapo 20x objective lens. Images were recorded with an Andor Zyla 4.2 sCMOS camera at ~100 ms integration time (~10 frames per second).
Live-cell STORM SRM.
Live-cell STORM SRM was performed on a homebuilt setup based on a Nikon Eclipse Ti-E inverted optical microscope using a CFI Plan Apochromat λ 100x oil-immersion objective lens (NA 1.45), as described previously.30 After electroporation and ~15 min incubation, the electroporation buffer was replaced with a live-cell STORM buffer (L15 medium + 2% glucose + 20 mM HEPES+ 5 mM MEA + 0.8 mg/mL glucose oxidase + 40 μg/mL catalase).6 The sample was continuously illuminated with a 647 nm laser, which was introduced through the back focal plane of the objective lens to illuminate a few micrometers into the cells at a typical power density of ~1 kW cm−2. Resultant blinking single-molecule fluorescence was recorded in the wide-field using an EM-CCD camera (iXon Ultra 897, Andor) at ~9 ms integration time (~110 frames per second) for ~50,000 frames. For 3D-STORM, a cylindrical lens was added to the optical path to encode the depth (z) position into the ellipticity of the single-molecule images.38 The recorded single-molecule images were then processed into STORM data and images using previously described methods.37,38
Patterned electroporation.
Monolayer graphene was transferred to a pre-cleaned glass coverslip as described above, and contacted with silver paint at both ends. A micropipette tip was used to make a scratch through the center of graphene. Conductance measurements indicated that the resultant two halves were electrically isolated. A device was assembled as described above for cell culturing. For the sequential delivery of different fluorescent probes to the two halves of the device, the culture medium was replaced with an electroporation buffer that contained the first probe, and a voltage pulse was applied across the bottom half of the graphene electrode and the counter electrode to enable labeling for ~15 min. The cells were recovered in a culture medium that was supplemented with 2% glucose (w/v) and 2 mM ATP (BSA04, Cytoskeleton Inc.) in the incubator for ~20–30 min.13 The medium was replaced by an electroporation buffer that contained the second probe, and a second voltage pulse was applied to the top half of the graphene electrode. After ~15 min, the buffer was replaced with a regular L15 imaging buffer for microscopy.
Supplementary Material
ACKNOWLEDGMENT
We thank Limin Xiang for assistance and discussion. We acknowledge support from the National Institute Of General Medical Sciences of the National Institutes of Health (DP2GM132681), the Bakar Fellows Award, and the Packard Fellowships for Science and Engineering. K.X. is a Chan Zuckerberg Biohub investigator. S.M. acknowledges support from Samsung Scholarship.
Footnotes
A pre-print version of this work was deposited to bioRxiv.40
The Supporting Information is available free of charge on the ACS Publications website.
Photographs of the device, characterization of the graphene sheets, labeling results over longer time scales, cell viability tests for Triton treatment vs. graphene-based electroporation, 3D-STORM SRM of the phalloidin-AF647-labeled actin cytoskeleton in live A549 cells, unspecific delivery of the second probe with incomplete recovery of the plasma membrane (PDF)
REFERENCES
- (1).Stewart MP; Sharei A; Ding X; Sahay G; Langer R; Jensen KF In Vitro and Ex Vivo Strategies for Intracellular Delivery. Nature 2016, 538, 183–192. [DOI] [PubMed] [Google Scholar]
- (2).Stewart MP; Langer R; Jensen KF Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev 2018, 118, 7409–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yarmush ML; Golberg A; Sersa G; Kotnik T; Miklavcic D Electroporation-Based Technologies for Medicine: Principles, Applications, and Challenges. Annu. Rev. Biomed. Eng 2014, 16, 295–320. [DOI] [PubMed] [Google Scholar]
- (4).Shroff H; Galbraith CG; Galbraith JA; Betzig E Live-Cell Photoactivated Localization Microscopy of Nanoscale Adhesion Dynamics. Nat. Methods 2008, 5, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wombacher R; Heidbreder M; van de Linde S; Sheetz MP; Heilemann M; Cornish VW; Sauer M Live-Cell Super-Resolution Imaging with Trimethoprim Conjugates. Nat. Methods 2010, 7, 717–719. [DOI] [PubMed] [Google Scholar]
- (6).Jones SA; Shim SH; He J; Zhuang XW Fast, Three-Dimensional Super-Resolution Imaging of Live Cells. Nat. Methods 2011, 8, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).van de Linde S; Heilemann M; Sauer M Live-Cell Super-Resolution Imaging with Synthetic Fluorophores. Annu. Rev. Phys. Chem 2012, 63, 519–540. [DOI] [PubMed] [Google Scholar]
- (8).Xu K; Shim S-H; Zhuang X Super-Resolution Imaging through Stochastic Switching and Localization of Single Molecules: An Overview In Far-Field Optical Nanoscopy; Tinnefeld P, Eggeling C, Hell S, Eds.; Springer: Berlin, 2015; pp 27–64. [Google Scholar]
- (9).Li HL; Vaughan JC Switchable Fluorophores for Single-Molecule Localization Microscopy. Chem. Rev 2018, 118, 9412–9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang L; Frei MS; Salim A; Johnsson K Small-Molecule Fluorescent Probes for Live-Cell Super-Resolution Microscopy. J. Am. Chem. Soc 2019, 141, 2770–2781. [DOI] [PubMed] [Google Scholar]
- (11).Ahnert-Hilger G; Bhakdi S; Gratzl M Minimal Requirements for Exocytosis. A Study Using Pc 12 Cells Permeabilized with Staphylococcal Alpha-Toxin. J. Biol. Chem 1985, 260, 12730–12734. [PubMed] [Google Scholar]
- (12).Walev I; Bhakdi SC; Hofmann F; Djonder N; Valeva A; Aktories K; Bhakdi S Delivery of Proteins into Living Cells by Reversible Membrane Permeabilization with Streptolysin-O. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Teng KW; Ishitsuka Y; Ren P; Youn Y; Deng X; Ge P; Lee SH; Belmont AS; Selvin PR Labeling Proteins inside Living Cells Using External Fluorophores for Microscopy. eLife 2016, 5, e20378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shi JF; Ma YF; Zhu J; Chen YX; Sun YT; Yao YC; Yang ZG; Xie J A Review on Electroporation-Based Intracellular Delivery. Molecules 2018, 23, 3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kim W; Ng JK; Kunitake ME; Conklin BR; Yang PD Interfacing Silicon Nanowires with Mammalian Cells. J. Am. Chem. Soc 2007, 129, 7228–7229. [DOI] [PubMed] [Google Scholar]
- (16).Shalek AK; Robinson JT; Karp ES; Lee JS; Ahn DR; Yoon MH; Sutton A; Jorgolli M; Gertner RS; Gujral TS; MacBeath G; Yang EG; Park H Vertical Silicon Nanowires as a Universal Platform for Delivering Biomolecules into Living Cells. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 1870–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Boukany PE; Morss A; Liao WC; Henslee B; Jung HC; Zhang XL; Yu B; Wang XM; Wu Y; Li L; Gao KL; Hu X; Zhao X; Hemminger O; Lu W; Lafyatis GP; Lee LJ Nanochannel Electroporation Delivers Precise Amounts of Biomolecules into Living Cells. Nat. Nanotechnol 2011, 6, 747–754. [DOI] [PubMed] [Google Scholar]
- (18).Xie X; Xu AM; Leal-Ortiz S; Cao YH; Garner CC; Melosh NA Nanostraw-Electroporation System for Highly Efficient Intracellular Delivery and Transfection. ACS Nano 2013, 7, 4351–4358. [DOI] [PubMed] [Google Scholar]
- (19).Han X; Liu ZB; Jo MC; Zhang K; Li Y; Zeng ZH; Li N; Zu YL; Qin LD Crispr-Cas9 Delivery to Hard-to-Transfect Cells via Membrane Deformation. Sci. Adv 2015, 1, e1500454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cao YH; Hjort M; Chen HD; Birey F; Leal-Ortiz SA; Han CM; Santiago JG; Pasca SP; Wu JC; Melosh NA Nondestructive Nanostraw Intracellular Sampling for Longitudinal Cell Monitoring. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E1866–E1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cao Y; Ma E; Cestellos-Blanco S; Zhang B; Qiu R; Su Y; Doudna JA; Yang P Nontoxic Nanopore Electroporation for Effective Intracellular Delivery of Biological Macromolecules. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 7899–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nguyen P; Berry V Graphene Interfaced with Biological Cells: Opportunities and Challenges. J. Phys. Chem. Lett 2012, 3, 1024–1029. [DOI] [PubMed] [Google Scholar]
- (23).Kostarelos K; Novoselov KS Exploring the Interface of Graphene and Biology. Science 2014, 344, 261–263. [DOI] [PubMed] [Google Scholar]
- (24).Cheng C; Li S; Thomas A; Kotov NA; Haag R Functional Graphene Nanomaterials Based Architectures: Biointeractions, Fabrications, and Emerging Biological Applications. Chem. Rev 2017, 117, 1826–1914. [DOI] [PubMed] [Google Scholar]
- (25).Kalbacova M; Broz A; Kong J; Kalbac M Graphene Substrates Promote Adherence of Human Osteoblasts and Mesenchymal Stromal Cells. Carbon 2010, 48, 4323–4329. [Google Scholar]
- (26).Li N; Zhang XM; Song Q; Su RG; Zhang Q; Kong T; Liu LW; Jin G; Tang ML; Cheng GS The Promotion of Neurite Sprouting and Outgrowth of Mouse Hippocampal Cells in Culture by Graphene Substrates. Biomaterials 2011, 32, 9374–9382. [DOI] [PubMed] [Google Scholar]
- (27).Fabbro A; Scaini D; Leon V; Vazquez E; Cellot G; Privitera G; Lombardi L; Torrisi F; Tomarchio F; Bonaccorso F; Bosi S; Ferrari AC; Ballerini L; Prato M Graphene-Based Interfaces Do Not Alter Target Nerve Cells. ACS Nano 2016, 10, 615–623. [DOI] [PubMed] [Google Scholar]
- (28).Pampaloni NP; Lottner M; Giugliano M; Matruglio A; D’Amico F; Prato M; Garrido JA; Ballerini L; Scaini D Single-Layer Graphene Modulates Neuronal Communication and Augments Membrane Ion Currents. Nature Nanotechnol. 2018, 13, 755–764. [DOI] [PubMed] [Google Scholar]
- (29).Wang R; Shi MJ; Brewer B; Yang LJ; Zhang YC; Webb DJ; Li DY; Xu YQ Ultrasensitive Graphene Optoelectronic Probes for Recording Electrical Activities of Individual Synapses. Nano Lett. 2018, 18, 5702–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wojcik M; Hauser M; Li W; Moon S; Xu K Graphene-Enabled Electron Microscopy and Correlated Super-Resolution Microscopy of Wet Cells. Nature Commun. 2015, 6, 7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li XS; Cai WW; An JH; Kim S; Nah J; Yang DX; Piner R; Velamakanni A; Jung I; Tutuc E; Banerjee SK; Colombo L; Ruoff RS Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper Foils. Science 2009, 324, 1312–1314. [DOI] [PubMed] [Google Scholar]
- (32).Li W; Moon S; Wojcik M; Xu K Direct Optical Visualization of Graphene and Its Nanoscale Defects on Transparent Substrates. Nano Lett. 2016, 16, 5027–5031. [DOI] [PubMed] [Google Scholar]
- (33).Kaech S; Banker G Culturing Hippocampal Neurons. Nat. Protoc 2006, 1, 2406–2415. [DOI] [PubMed] [Google Scholar]
- (34).Peltier J; Agrawal S; Robertson MJ; Schaffer DV In Vitro Culture and Analysis of Adult Hippocampal Neural Progenitors In Protocols for Adult Stem Cells; Conboy IM, Schaffer DV, Barcellos-Hoff MH, Li S, Eds.; Humana Press: Totowa, NJ, 2010; pp 65–87. [DOI] [PubMed] [Google Scholar]
- (35).Kim JA; Cho KC; Shin MS; Lee WG; Jung NC; Chung CI; Chang JK A Novel Electroporation Method Using a Capillary and Wire-Type Electrode. Biosens. Bioelectron 2008, 23, 1353–1360. [DOI] [PubMed] [Google Scholar]
- (36).Riedl J; Crevenna AH; Kessenbrock K; Yu JH; Neukirchen D; Bista M; Bradke F; Jenne D; Holak TA; Werb Z; Sixt M; Wedlich-Soldner R Lifeact: A Versatile Marker to Visualize F-Actin. Nat. Methods 2008, 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rust MJ; Bates M; Zhuang X Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (Storm). Nat. Methods 2006, 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Huang B; Wang W; Bates M; Zhuang X Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science 2008, 319, 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Nair RR; Blake P; Grigorenko AN; Novoselov KS; Booth TJ; Stauber T; Peres NMR; Geim AK Fine Structure Constant Defines Visual Transparency of Graphene. Science 2008, 320, 1308. [DOI] [PubMed] [Google Scholar]
- (40).Moon S; Li W; Xu K Graphene-Enabled, Spatially Controlled Electroporation of Adherent Cells for Live-Cell Super-Resolution Microscopy. 2019, 642868 bioRxiv. 10.1101/642868 (accessed Mar 6, 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.