

Abstract
The development of a concise total synthesis of (±)-phyllantidine (1), a member of the securinega family of alkaloids containing an unusual oxazabicyclo[3.3.1]nonane core, is described. The synthesis employs a unique synthetic strategy featuring the ring expansion of a substituted cyclopentanone to a cyclic hydroxamic acid as a key step that allows facile installation of the embedded nitrogen-oxygen (N—O) bond. The optimization of this sequence to effect the desired regiochemical outcome and its mechanistic underpinnings were assessed both computationally and experimentally. This synthetic approach also features an early-stage diastereoselective aldol reaction to assemble the substituted cyclopentanone, a mild reduction of an amide intermediate without N—O bond cleavage, and the rapid assembly of the butenolide found in (1) via use of the Bestmann ylide.
Keywords: computational chemistry, nitrogen heterocycles, phyllantidine, ring expansion, total synthesis
Introduction
The securinega alkaloids, isolated from the Euphorbiaceae family of plants, are a class comprised of over 70 known members featuring unique bridged tetracyclic or pentacyclic cores (e.g., 1–11; Figure 1).[1,2] The diverse molecular architectures found within this class of natural products, combined with their wide-ranging biological activities, have inspired dozens of synthetic efforts aimed at their preparation, most of which have focused on the parent alkaloids securinine (2), allosecurinine (3), and their enantiomers (4 and 5).[1–3] As illustrated in Figure 1, a subset of the securinega alkaloids contain an embedded nitrogen-oxygen (N—O) bond (e.g., 1 and 6–11), which offers an additional synthetic challenge not found in 2–5.
Figure 1.

Members of the Securinega family of alkaloids.
Phyllantidine (a.k.a. “Phyllanthidine”) (1, Figure 1), the parent N—O bond-containing securinega alkaloid, was first isolated in 1965 from the root bark of Phyllanthus discoides.[4] Its structure was incorrectly assigned upon isolation, but later corrected in 1972 to that of (−)-phyllantidine (1).[5] Its enantiomer, (+)-phyllantidine (1), was isolated in 1992 from the leaves of Breynia coronata.[6] Phyllantidine possesses a unique oxazabicyclo[3.3.1]nonane structure and exhibits leishmanicidal activity and anti-inflammatory properties.[7,8] Interestingly, Flueggeacosine B (11), a heterodimer of phyllantidine (1) and a novel securinega alkaloid fragment containing a azabicyclo[3.2.1]octane system, was isolated recently in 2018 from the roots of Flueggea suffruticosa and displays potent neuronal differentiation activity.[9]
Securinega alkaloids containing a N—O bond are among the most structurally complex members of the family, and have thus been the subject of far fewer synthetic efforts.[10,11] Biosynthetically, the isoxazolidine cores of the Virosaines (8 and 9) and Flueggine A (10) are postulated to arise via 1,3-dipolar nitrone-alkene cycloadditions,[12] a transformation that served to inspire the reported Virosaine A (8),[10a,b] Virosaine B (9),[10c] and Flueggine A (10) syntheses.[10c,d] Since the isolation of phyllantidine (1) over five decades ago, only one total synthesis has been reported.[11] In contrast to 8–10, the tetrahydro-1,2-oxazine residing at the core of 1 is not accessible via a classical 1,3-dipolar cycloaddition. Thus, Carson and Kerr turned to an analogous homo [3+2] cycloaddition between a nitrone and an electron-deficient, chiral cyclopropane. Given the limited accessibility of this ring system[1a,13] and our general interest in securinega alkaloid synthesis,[3] we sought to develop an innovative, modular approach that allows for the installation of N—O bonds embedded in densely functionalized scaffolds, such as those found in 1.
Efforts toward 1 began by examining ways to furnish the tetrahydro-1,2-oxazine core. We recognized that cyclic hydroxamic acids are versatile synthetic intermediates as they can serve as precursors to cyclic hydroxamic esters, hydroxylamines, and nitrones.[14] However, the ambident nucleophilicity, acidity, and inherent lability of the hydroxamic acid N—O bond can complicate installation within highly functionalized systems.[15] In searching for preparative methods that would avoid these inherent limitations, we were drawn to a report by King and co-workers describing a mild ring-expansion sequence to afford cyclic hydroxamic acids from cycloalkanones.[16a] This unique reactivity was discovered serendipitously during their work developing small-molecule HNO surrogates from compounds bearing an acyloxy nitroso moiety (Scheme 1).[16b] Interestingly, King noted that upon treatment with base, acyloxy nitroso moieties on ring systems with six or greater carbons (i.e., 12) are effective HNO donors, whereas strain relief in ring systems containing four or five carbons (i.e., 14) drives an efficient ring expansion producing five- and six-membered cyclic hydroxamic acids (14 → 15; Scheme 1).[16a,17] Importantly, when unsymmetrical systems were employed (e.g. 14), migration of the more electron-rich alkyl group predominates yielding a single regiomer (e.g., 15), reactivity akin to that observed in Baeyer–Villiger oxidations.[18]
Scheme 1.

Reported ring expansion to cyclic hydroxamic acids by King and co-workers.[16a]
Given the ease of accessing cyclic ketone precursors and the efficiency of this expansion when performed on substituted systems, we viewed this as a promising approach to the synthesis of complex N—O-containing molecules. If generalizable, this chemistry would allow for the installation of N—O bonds into functionally dense architectures, such as 1, in a manner that is both orthogonal and complementary to more traditional nucleophilic substitution-based strategies which are better suited for generating unsubstituted cyclic hydroxamic acids.[15] In the context of preparing 1 (Scheme 2), we envisioned employing this ring expansion in the stereo- and regioselective conversion of ketone 18 to key intermediate 17. Setting the stage for this event would require the stereoselective aldol addition of cyclopentanone to the ketone derived from oxidation of known TBS-protected diol 19.[19] Harnessing the nucleophilicity of the hydroxamic acid moiety in the derived ring expansion product (17) would then allow construction of the B-ring via intramolecular haloetherification to afford 16. Selective amide reduction and butenolide formation would then complete the synthesis.
Scheme 2.

Initial retrosynthetic analysis of 1. TBS= tert-butyldimethylsilyl.
Results and Discussion
Efforts toward 1 began as shown in Scheme 3 with preparation of known mono TBS-protected diol 19[19] from 1,4-cyclohexadiene (20). Upjohn dihydroxylation of 20[20] and subsequent protection with tert-butyldimethylsilyl chloride (TBSCl) proved to be the most effective sequence to deliver multi-gram quantities of 19. Oxidation of alcohol 19 with Dess–Martin periodinane (DMP) gave the desired a-silyloxy ketone 22 in 87% yield, without any observable isomerization of the olefin. Subsequent aldol addition of the lithium enolate, derived from cyclopentanone, proceeded as expected to the least hindered diastereotopic face of ketone 22 and, to our delight, with complete selectivity for the illustrated relative stereochemistry between the two newly formed stereogenic centers, thus producing 18 as the only isolable product. Although, when considering the two possible chair-like Zimmerman–Traxler transition states (Scheme 4)[21] it appeared that at least one diastereomer (18; desired) would be favored over the other (24; undesired), due to the steric clash of the TBS group and solvated lithium cation, the extent of the observed selectivity was not fully anticipated. Furthermore, based on this analysis, we suspected that 18 possessed the desired stereochemistry.
Scheme 3.
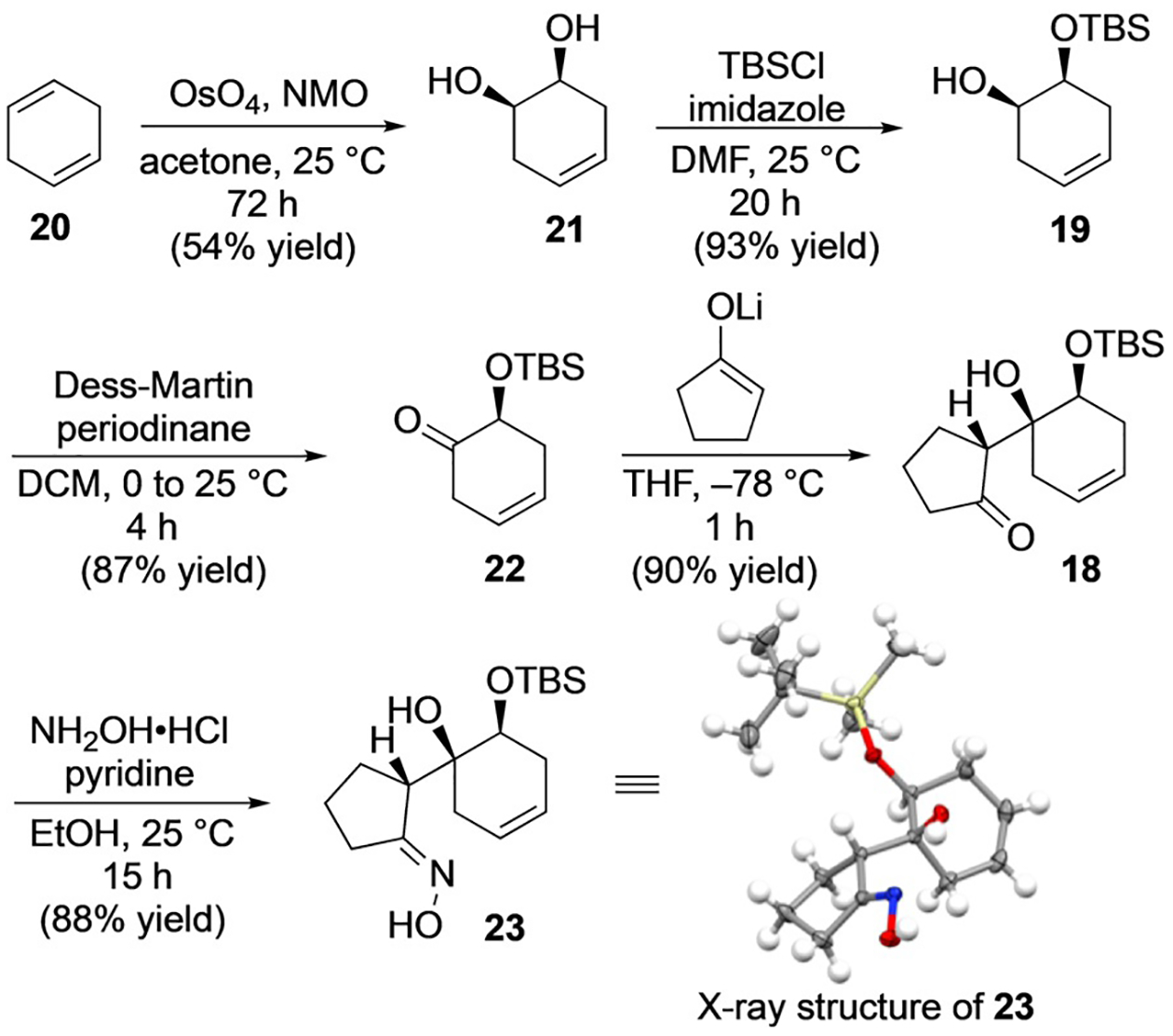
Preparation of ring expansion precursor 23. NMO=N-methylmorpholine N-oxide, DMF= dimethylformamide, DCM=dichloromethane.
Scheme 4.
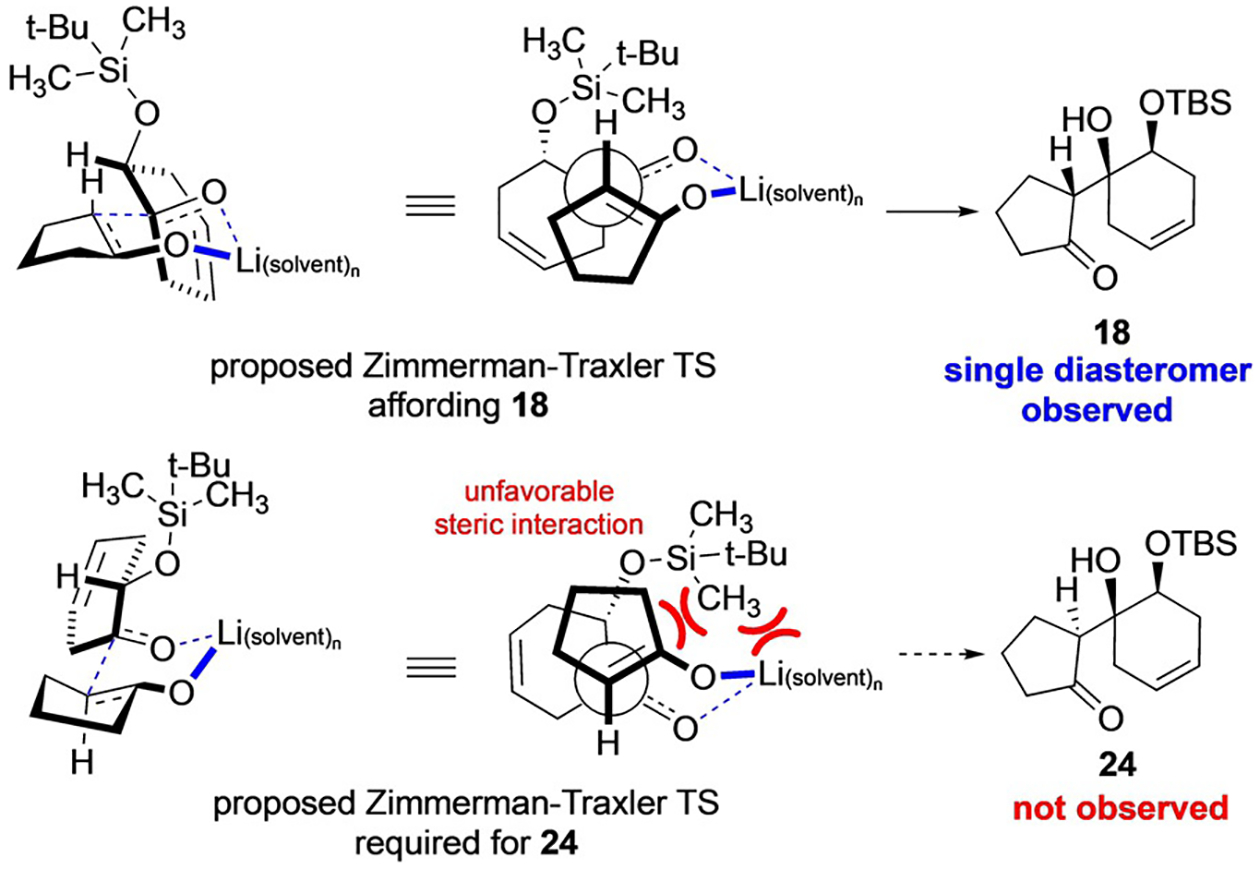
Proposed pathway leading to formation of 18.
Optimistically, 18 was carried forward to access ring expansion precursor 23 which, fortuitously, was obtained as a crystalline solid in 88% yield. Single crystal X-ray analysis of 23 confirmed that we had indeed obtained the relative stereochemistry necessary to access 1 (Scheme 3). Given ready access to multi-gram quantities of oxime 23, we turned to the key ring-expansion step.[16a] To this end, 23 was found to undergo oxidation to acyloxy nitroso intermediate 25 upon treatment with 1.5 equivalents of phenyliodonium diacetate (PIDA) in AcOH/DCM solvent while warming from 0°C to 25°C (Scheme 5).[22] Although 25 was not particularly stable at room temperature, it could be separated from excess oxidant by quickly passing the reaction mixture through a plug of silica. Subsequent cleavage of the acetate by exposure to anhydrous K2CO3 in MeOH at 0°C induced ring expansion to the desired hydroxamic acid 17. Unfortunately, this desired material was produced as the minor component of a 1:2 regiochemical mixture with undesired hydroxamic acid 26 (Scheme 5). Based on initial observations reported by King and co-workers,[16a] the predominance of undesired regiomer (26) was rather surprising but not entirely unexpected given the known regiochemical outcomes in Baeyer–Villiger oxidations; the latter also proceed by carbon bond migration to an electronically deficient heteroatom and have been demonstrated to be influenced by inductively withdrawing functionality adjacent to the migrating center. Migration of the less substituted alkyl group is uncommon,[18,23] but has been observed in the rearrangement of β-alkoxy ketones.[24]
Scheme 5.

Evaluation of the stereochemical and regiochemical outcome of the ring expansion sequence.
Although desired hydroxamic acid 17 was the minor component of the regiochemical mixture, the reasonable overall yield and facile separation of 17 from 26 via column chromatography allowed us to attempt completing the A-B-C ring system via intramolecular halocyclization of 17. To this end, we turned to conditions reported by Kim and co-workers (Scheme 6);[25] in contrast to the excellent yields observed on their substrates (e.g., 27 → 28 or 29), halocyclization of 17 produced complex mixtures of products with no trace of desired iodide 30. Commonly employed iodolactonization conditions, which utilize excess elemental iodine and sodium bicarbonate, exclusively gave oxabicyclic iodoether 31 (63% yield), thus clearly indicating a change in substrate would be required.
Scheme 6.

Competitive iodoether 31 formation during attempted iodocyclization of hydroxamic acid 17. NBS =N-bromosuccinimide.
Experimental Investigations into the Origins of the Regioselectivity Observed in the Ring Expansion
The unfavorable product mixture obtained in the ring expansion of 25 (Scheme 5), coupled with undesired reactivity of the tertiary alcohol in 17 (Scheme 6), led us to take a step back and further explore, both experimentally and computationally, the impact of the tertiary hydroxyl group on the regiochemical outcome of the ring-expansion. To confirm that this moiety was indeed playing a significant role, we prepared substrates 32 and 36 wherein only the former contains a tertiary alcohol.[26]
Exposure of 36 to either NaOH or K2CO3 in MeOH resulted in conversion to a single regiomeric product corresponding to hydroxamic acid 37, the product arising from migration of the more heavily-substituted carbon (Scheme 7). Having established the inductively withdrawing hydroxyl as the likely culprit, we reintroduced this single group by preparing acyloxy nitroso 32 via a sequence similar to that described above for 22 → 25, and subjected it to ring expansion with a small subset of bases.[26] As with 36, hydroxide bases in methanol solvent proved most effective. The counterion, however, had little effect and all conditions produced unfavorable mixtures of hydroxamic acids 33 and 34. Interestingly, in contrast to 25, the reaction of 32 was found to also produce a spirocyclic product (35). As illustrated in Scheme 8, we suspect 35 arises from 32 via initial attack of the now less sterically encumbered tertiary alcohol onto the nearby nitroso group which, in turn, induces an acyl shift that sets the stage for carbon bond migration to bicycle 32C. The latter undergoes facile methanolysis to furnish 35 due to the twisted nature of its bridgehead hydroxamic ester.[27,28] In an attempt to avoid the formation of 35 by increasing the rate of acetolysis we prepared a trifluoroacetate derivative of 32.[26] As expected this variant was found to react much more rapidly and produced only 33 and 34 (1:1.3). Unfortunately, the latter were isolated in low yield and regeneration of the starting ketone through loss of HNO was observed as the predominant event.[16]
Scheme 7.

Ring expansion of model acyloxy nitroso substrates 32 and 36.
Scheme 8.

Ring expansion of acyloxy nitroso 32, proposed mechanism pathway I, and a proposed mechanism for the formation of spirocycle 35.
Further consideration of the mechanism by which 35 was likely arising led us to wonder if the ring expansions to 33 and 34 were also proceeding via initial attack of hydroxide on the nitroso moiety followed by acyl transfer, rather than simple collapse of an initially formed tertiary alkoxide and bond migration (Schemes 8, 9, pathway I vs. II). To evaluate the mechanistic possibilities, we conducted two isotopic labeling experiments. As illustrated (Scheme 9), incorporation of doubly 18O-labeled acetate into 36 followed by ring expansion upon exposure to 5m aqueous NaOH in methanol confirmed that the acetate derived oxygen resides at the hydroxamic acid carbonyl, as evidenced by HRMS and 13C NMR analyses.[29] Additionally, we performed the ring expansion of unlabeled substrate 36 using Na18OH/H218O solution in methanol solvent[30] and found it led to facile expansion 36 → 37 with no 18O incorporation into the product 37, which eliminates pathway II as a valid mechanistic pathway (Scheme 9).[31] Given the results of the above studies, the ring expansion appears to proceed through initial acetate cleavage followed by alkyl migration onto the nitroso moiety (pathway I; Scheme 8).
Scheme 9.

18O-labeling experiments for the expansion of 36 → 37 and possible mechanism pathway II.
Computational Investigations into the Origins of the Regiochemical Discrepancies of the Ring Expansion
To gain further insight into the undesirable regiochemical outcome of ring expansions containing the requisite b-oxygen substituent, we investigated the two model systems (32 → 33 and 34; and 36 → 37 and 38) computationally at the RB3LYP/6–311 + G* level. The calculated free energies are for the gas phase, but are expected to reasonably represent the trend in free energies in the solution phase, which are often difficult to compute due to the need to apply thermal corrections to account for diffusion, versus translational effects in the gas phase.[26,32] As illustrated in Figure 2, the calculations reveal that, in the expansion of 32, which contains a b-tertiary alcohol, the preferred reaction pathway should proceed from anion 32A through 33TS (+ 10.1 kcalmol−1) to afford product 33A (−27.0 kcalmol−1), which yields 33 upon protonation. The pathway leading to 34A (−22.6 kcalmol−1) via 34TS (+ 10.6 kcalmol−1) is only 0.5 kcalmol−1 higher in energy. Given the computed energetics, both regiomers 33 and 34 are expected to be observed, a result consistent with the experiment, albeit with the reverse regiochemical outcome, as 34 predominates over 33 experimentally (Scheme 7).
Figure 2.

Computed profile (RB3LYP/6–311+G*) for the ring expansion process of (32A → 33A and 34A; and 36A → 37A and 38A) in the gas phase. Depicted free energy differences (ΔG°) for the transition states and products are reported relative to the starting anion in kcal mol−1.
For the expansion of 36, which lacks a b-tertiary alcohol, the preferred reaction pathway proceeding from anion 36A was found to pass through 37TS (+ 9.6 kcalmol−1) to afford product 37A (−22.0 kcalmol−1). The pathway leading to 38A (−21.6 kcalmol−1) via 38TS (+ 11.5 kcalmol−1) is 1.9 kcal mol−1 higher in energy. Given the computed energetics, only product 37 would be expected, again consistent with experimental observations.
The ring expansion is computed to be fairly exothermic in both cases, as the products (37A, 38A, 34A, and 33A) are found to be > 21 kcalmol−1 more stable than starting anions 36A and 32A; the result of ring strain reduction. However, a major difference between the two model systems is the clear participation of the neighboring H-bond in both 34TS and 33TS. In the former this interaction engages the neighboring nitroso p-system in a nearly anti-periplanar fashion relative to the C–C bond undergoing migration to 34A (Figure 2); an effect that serves to stabilize 34TS relative 33TS thereby minimizing the TS energy difference. The replacement of the OH in 34TS and 33TS with an OCH3 group leads to a large transition state difference of (3.2 kcalmol−1).[26] Given the lack of a neighboring hydroxyl in 36A, the corresponding transition state energies for 37TS and 38TS do not experience a similar compression.
We also considered the computed dipole moments of the transition state pairs and found them to be substantially different for both model substrates (μ = 8.0 D, 37TS; μ = 10.4 D, 38TS and μ = 6.8 D, 33TS; μ = 8.8 D, 34TS). This ≈ 2 Debye difference between the TS pairs suggests that solvent effects could influence the regiochemical outcome, as polar solvents may offer greater stabilization to the higher dipole moment TS˛s 34TS and 38TS thereby bringing the energies of the two transition states closer. Such an effect would have a greater repercussion on expansions of β-tertiary alcohol substrates, as the TS energies are already computed to be close in energy in the gas phase (0.5 kcalmol−1).
The starting materials, products, and transition states of the model systems (32A → 33A and 34A; 36A also → 37A and 38A) were modeled using the default integral equation formalism polarizable continuum model (IEFPCM) in Gaussian16 to probe possible solvent effects. It is important to note the energies calculated here are free energies for a system confined to a predetermined, modeled solvent cavity “a continuum” based on the properties of the chosen model solvent. They are not solvation energies and do not fully account for diffusion, hydrogen-bonding and aggregative properties of the substrate etc. However, such calculations offer a reasonable platform to model the general trend of potential solvent effects in systems.[32] The calculations revealed that in a THF or methanol continuum, the transition state energies reverse with 34TS being preferred by 2.4 kcal mol−1 and 38TS by 1.1 kcalmol−1 for THF and 2.6 kcalmol−1 and 1.5 kcalmol−1, respectively for methanol. These results suggest that a solvent effect may be observed experimentally, and solvents with higher dielectric constants (i.e., methanol) may help to stabilize the transition state with the higher dipole moment (34TS and 38TS) to a greater extent. However, such calculations do not take into account the donor-acceptor or hydrogen-bonding properties of the solvent, as well as charge transfer from the solute to the solvent, which can dominate the overall solvent effect.[26,32]
An Adjusted Approach to Phyllantidine (1)
From the brief computational study, it became clear that the regiochemical course of this ring expansion was not only impacted by inductive effects but also hydrogen bonding and solvent. Thus, we continued our efforts toward 1 by exploring the protection of the β-tertiary alcohol. To this end, initial attempts to protect the remaining alcohol in aldol adduct 18, as a TBS-ether, triethylsilyl ether, or an acetate were all unsuccessful.[33] Given that these latter failures were likely due to steric hindrance by the resident TBS ether, we decided to first deprotect and then reprotect the resulting 1,2-syn diol as an acetonide. To this end, we screened conditions to cleave the TBS ether in 18 and, after considerable experimentation, found that rapid exposure to a large excess of HF-pyridine gave 39 in an excellent 95% yield (Scheme 10). In contrast, exposure of 18 to catalytic HCl in MeOH gave exceedingly efficient silyl ether cleavage, but a completely different product, tricyclic acetal 40 (Scheme 10).
Scheme 10.

Deprotection of TBS ether in aldol adduct 18.
With 1,2-syn diol 39 in hand, we explored protection as the corresponding acetonide and found that exposure to an excess of 2,2-dimethoxypropane (2,2-DMP) and pyridinium p-toluene sulfonate (PPTS) smoothly afforded acetonide 41, which was then carried forward to acyloxy nitroso 43 (Scheme 11). To our chagrin, we found that exposure of 43 to our standard ring expansion conditions afforded a 1:1.5 regiochemical mixture of hydroxamic acids (44:45) (Scheme 11). Therefore, removing the hydrogen-bond donor ability of the tertiary alcohol appeared to have only a modest positive affect on regiochemistry compared to that observed for the expansion of 25 (Scheme 5). Puzzled by the minimal change of regioselectivity, we chose to install a carbonate protecting group to explore the effect of altering the electronics and Lewis basicity of the protecting group[34] on the regioselectivity of the ring expansion.
Scheme 11.
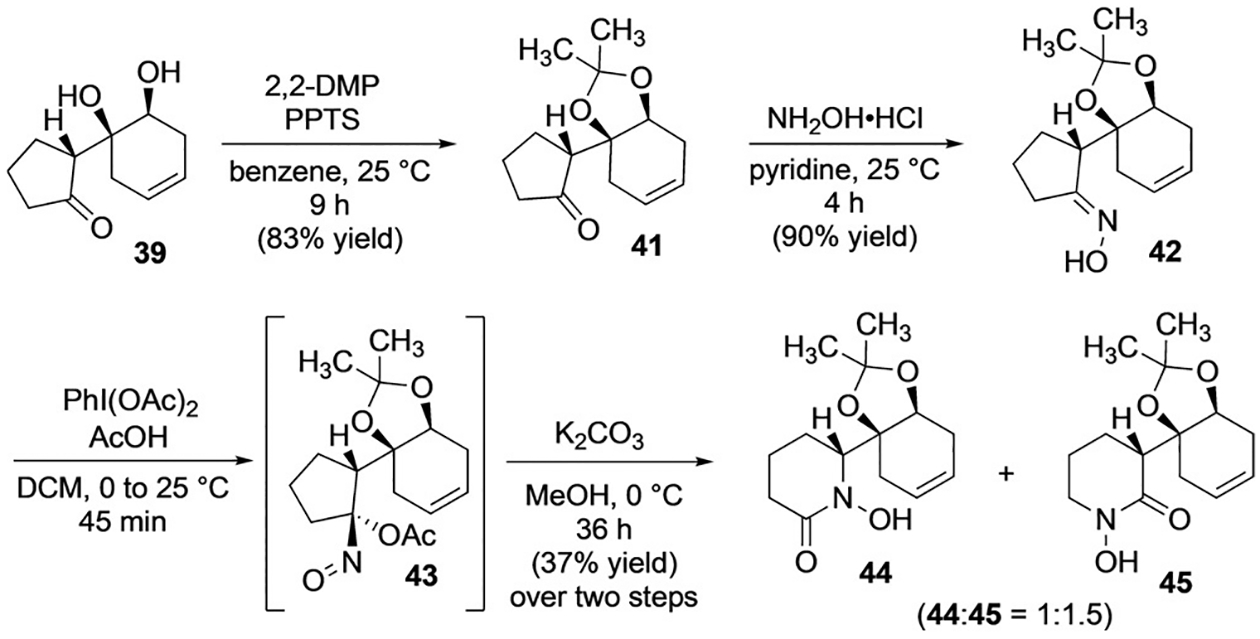
Ring expansion of acetonide-protected acyloxy nitroso 43.
To this latter end, treatment of diol 39 with phosgene and pyridine furnished cyclic carbonate 46, and oxime formation gave 47, both proceeding in excellent yields (Scheme 12). Oxidation of 47 with phenyliodonium diacetate, using the previously developed protocol (i.e., 23 → 25; Scheme 5), gave only 12% yield of 48. The oxidation was found to be promoted by AcOH, but 48 was only transiently stable to the reaction conditions. Conducting the ring expansion in AcOH solvent, and immediate separation of 48 from the reaction mixture via flash column chromatography provided acyloxy nitroso 48 in 64% yield as a single diastereomer. The illustrated relative stereochemistry of 48, a blue crystalline solid, was determined by X-ray crystallography.
Scheme 12.

Preparation of carbonate-protected acyloxy nitroso adduct 48.
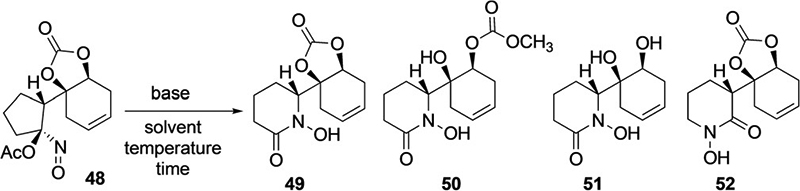
Upon treatment of 48 with K2CO3 in MeOH, we were surprised to find that the desired hydroxamic acid (49) was obtained as the major product, along with methyl carbonate 50, diol 51, and undesired hydroxamic acid 52 in a 2.4:1.6:1.3:1 ratio; (49:50:51:52) (Table 1, Entry 2). Since 50 and 51 originate from 49, the regiochemical outcome of the ring expansion is significantly altered, now favoring migration of the more heavily substituted alkyl group (ca. 5:1 ratio). This regioselectivity appears to be general among the carbonate and hydroxide bases screened in MeOH solvent (Table 1, Entries 1–7) and lithium methoxide (LiOMe), which was found to produce less of 50 and 51 (Table 1, Entries 8,9) under short reaction times.
Table 1:
The effect of various bases/solvents/temperatures on the regiochemical outcome of the ring expansion of carbonate acyloxy nitroso 48.[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Base (Equiv.) | Solvent | t [min] | T [°C] | Conv.[b] [%] | Ratio[b] 49:50:51:52 |
| 1[c] | Na2CO3 (1.6) | CH3OH | 120 | 0 | 37 | 1.3:0:1:1 |
| 2 | K2CO3 (1.2) | CH3OH | 120 | 0 | 100 | 2.4:1.6:1.3:1 |
| 3 | K2CO3 (1.6) | CH3OH | 120 | −15 | 100 | 5.3:6.0:1.8:1 |
| 4 | Cs2CO3 (1.6) | CH3OH | 30 | 0 | 63 | 3.3:2:0.8:1 |
| 5 | KOH (1.6) | CH3OH | 5 | 0 | 66 | 2.5:1.5:0.7:1 |
| 6 | NaOH (1.6) | CH3OH | 5 | 0 | 53 | 4:0:0:1 |
| 7 | LiOH (1.6) | CH3OH | 5 | 0 | 63 | 3:0:0:1 |
| 8 | 2.5 m LiOMe (1.1) | CH3OH | 5 | 0 | 100 | 3:0:0:1 |
| 9 | 1.0 m LiOMe (1.6) | CH3OH | 60 | −15 | 100 | 2.2:1:0:1 |
| 10 | 1.0 m LiOMe (1.6) | THF | 5 | 0 | 100 | 4:2:0:1 |
| 11 | 1.0 m LiOMe (1.0) | THF | 20 | −10 | 100 | 5.5:1.3:0:1 |
| 12 | 1.0 m LiOMe (2.0) | THF | 45 | −15 | 100 | > 20:1:0:1 |
| 13 | 1.0 m LiOMe (5.0) | THF | 3000 | −80 | 100 | 1:0:0:0 |
| 14[c] | 1.0 m LiOMe (1.6) | Et2O | 120 | −15 | 44 | 5:1:0:1 |
| 15[c] | 1.0 m LiOMe (1.6) | CH3CN | 120 | −15 | 17 | 1.6:0:0:1 |
| 16 | 1.0 m LiOMe (1.6) | CH2Cl2 | 15 | −15 | 90 | 1:1:0:1 |
| 17 | 1.0 m LiOMe (1.6) | CH3-C6H5 | 60 | −15 | 84 | 1:0.8:0:1 |
| 18 | 1.0 m LiOMe (1.6) | C6H5 | 5 | 10 | 100 | 1:1:0:1 |
| 19 | 1.0 m KOMe (1.6) | THF | 5 | −15 | 100 | 13:4:0:1 |
| 20 | 1.0 m NaOMe (1.6) | THF | 25 | −15 | 100 | 6:2:20:1 |
| 21 | 1.0 m LiOMe (1.6) | THF | 45 | −15 | 100 | 15:3.5:0:1 |
Reactions were conducted on 10 mg scale in 2 mL of the given solvent at the given temperature and were monitored by TLC analysis for disappearance of 48; Entries 2 and 13 were run on larger scale.[26]
Ratios and % conversions were determined by integration of the proton signals in the 1H NMR spectra of the crude mixtures.
The reaction was not complete after 120 min and the ratio was taken at this cut-off.
Given the computational observations, we decided to conduct a screen to identify any possible solvent effects. After screening various solvents using LiOMe as the base (Table 1, Entries 9, 12, and 14–18), it became clear that non-Lewis basic and non-polar solvents were ineffective in altering the regiochemical preference toward 49. Medium polarity, aprotic, ethereal solvents such as THF and Et2O were found to engender the ring expansion process, affording 49 preferentially over 52 with small amounts of 50 arising from partial carbonate cleavage (Table 1, Entries 10–14). By lowering the temperature to −80°C in THF solvent, albeit with longer reaction times (50 h), (Table 1 Entry 13) 49 could be obtained exclusively!
The last factor evaluated was whether the metal counterion was important in obtaining this high degree of regioselectivity (Table 1, Entries 19–21). The rate of reaction was found to follow the expected trend for metal alkoxides (KOMe > NaOMe > LiOMe),[35] and, since the basicity of LiOMe is moderated in ethereal solvents due to aggregation effects,[35,36] it results in less carbonate cleavage products (50 and 51). Overall, the degree of regioselectivity was found to be > 17:1 across all the metal alkoxides in THF solvent at low temperature (−15°C).
We again turned to computational studies, to gain insight into the observed switch in regioselectivity of the ring expansion process when employing a carbonate protecting group versus an acetonide, and to evaluate the positive role of the ethereal solvent. The likely intermediates formed when employing acetonide 43 and cyclic carbonate 48 as substrates were explored. In contrast to our previous calculations, we incorporated the Li counterion into the model and explored the energetics in both the presence and absence of THF. To this end, the effect of a single THF molecule on the transition state geometries was modeled in the gas phase at the RB3LYP/6–311 + G* level, which was chosen due to its ability to provide reasonable execution times for large systems combined with its general applicability for modeling organic reactions in the gas-phase.[37] Although, at the B3LYP level of theory, without dispersion, one cannot expect to precisely model experimental outcomes, the trends one observes can be helpful in developing reasonable hypotheses.[26] As described below these calculations did illustrate differences in the extent of lithium ion coordination in 43 and 48 and the potential impact of the solvent.
From an experimental perspective (Figure 3 (top)), the ring-expansion regiochemistry of acyloxy nitroso compounds mirrors that observed in Bayer–Villiger oxidations. Specifically, in the absence of any heteroatoms (i.e., 36 → 37, Figure 3) ring-expansion proceeds regioselectively via migration of the more substituted carbon; similar to that observed for the Baeyer–Villiger oxidation (e.g., 53 → 54).[38,39] Placing a β-hydroxy group adjacent to the migrating center negates this regioselective preference, as observed experimentally for both the Baeyer–Villiger oxidation (e.g., 55 → 56 and 57) and our initial model substrate (i.e., 32 → 33 and 34, Figure 3); a consequence of the inductively electron-withdrawing hydroxyl group retarding migration and, in the latter, the added effect of hydrogen-bonding to the nitroso moiety (see above).[40] Our efforts to overcome the hydrogen bonding effects by hydroxyl protection (Figure 3, bottom) unexpectedly revealed that a carbonate protecting group (e.g., 48) is more effective than an acetonide (e.g., 43) in shifting the ring expansion toward migration of the more heavily substituted (electron-rich) carbon. Our initial expectation was that the electronegative carbonyl carbon of the carbonate group would diminish the desired regiochemical outcome relative to the acetonide by further enhancing the inductively withdrawing effect of its β-oxygen.
Figure 3.

Summary of the regiochemical outcomes of the various acyloxy nitroso ring expansion substrates evaluated and comparison to the outcomes of related substrates in the Baeyer–Villiger oxidation. TFPAA=trifluoroperacetic acid.
Exploring the impact of solvent (cf. 44:45 and 49:52 in Figure 3) revealed that although both MeOH and THF are Lewis bases, MeOH, a high polarity hydrogen-bonding solvent, appears to be less effective whereas the medium polarity/aprotic THF is optimal. This observation guided our choice to model the effect of an explicit THF molecule.[26] The latter suggests that THF is able to disrupt metal-substrate coordination in the carbonate system (eg., 48Li, Figure 3), but not in the acetonide (eg., 43Li, Figure 3). These computational results are consistent with the known differences in Lewis basicities of acetonide and carbonate oxygens and disruptive effects of THF on metal coordination.[34,36] In general if one considers the impact of the metal coordination and tighter binding to the more Lewis basic acetonide versus the carbonate (cf. 43Li and 48Li, Figure 3), an enhancement of the deleterious effects of the inductively withdrawing oxygen would be expected in 43Li, resulting in the observed decrease in selectivity. This difference is predicted by the computational studies incorporating THF, which also suggests that the solvent is capable of completely disrupting coordination of the lithium ion to the carbonate in 48Li.[26] Thus, effectively increasing electron density and restoring the propensity for migration of the more substituted, electron-rich carbon to furnish the desired hydroxamic acid 49.[34–36]
Having finally identified a substrate and conditions suited for a regioselective ring expansion affording 49, we turned toward completing the synthesis of 1 (Scheme 13). Noting that halocyclization was successful at functionalizing the internal olefin in substrate 17, during previous attempts to construct the A-B-C ring system of 1 (Scheme 6), we applied those same conditions to hydroxamic acid 49. Gratifyingly, we were able to construct iodide 58 in 90% yield, establishing the A-B-C ring system of 1. Elimination of the secondary iodide in 58 was accomplished by treatment with DBU in refluxing benzene to afford 59 (90% yield) and set the stage for reduction of the hydroxamic ester (Scheme 13). The conversion of 59 to oxazine 60 was envisioned to be difficult given the potential for facile reductive cleavage of the N—O bond.[14g,41] After screening various known methods for the reduction of hydroxamic esters to O-alkyl hydroxylamines,[14b–e] conditions adapted from Reinhoudt and co-workers,[42] employing monochloroalane as the reductant, proved most effective and simultaneously removed the cyclic carbonate thereby producing the oxazabicyclo[3.3.1]nonane core of 1 in 63% yield. Subsequent oxidation of 60 with DMP gave relatively unstable enone 61, which was taken directly to the next step to minimize decomposition. Exposure of 61 to 5 equivalents of Bestmann ylide[43] in benzene with microwave heating under high dilution conditions provided (±)-phyllantidine (1) in 62% yield over the two step sequence. [44]
Scheme 13.
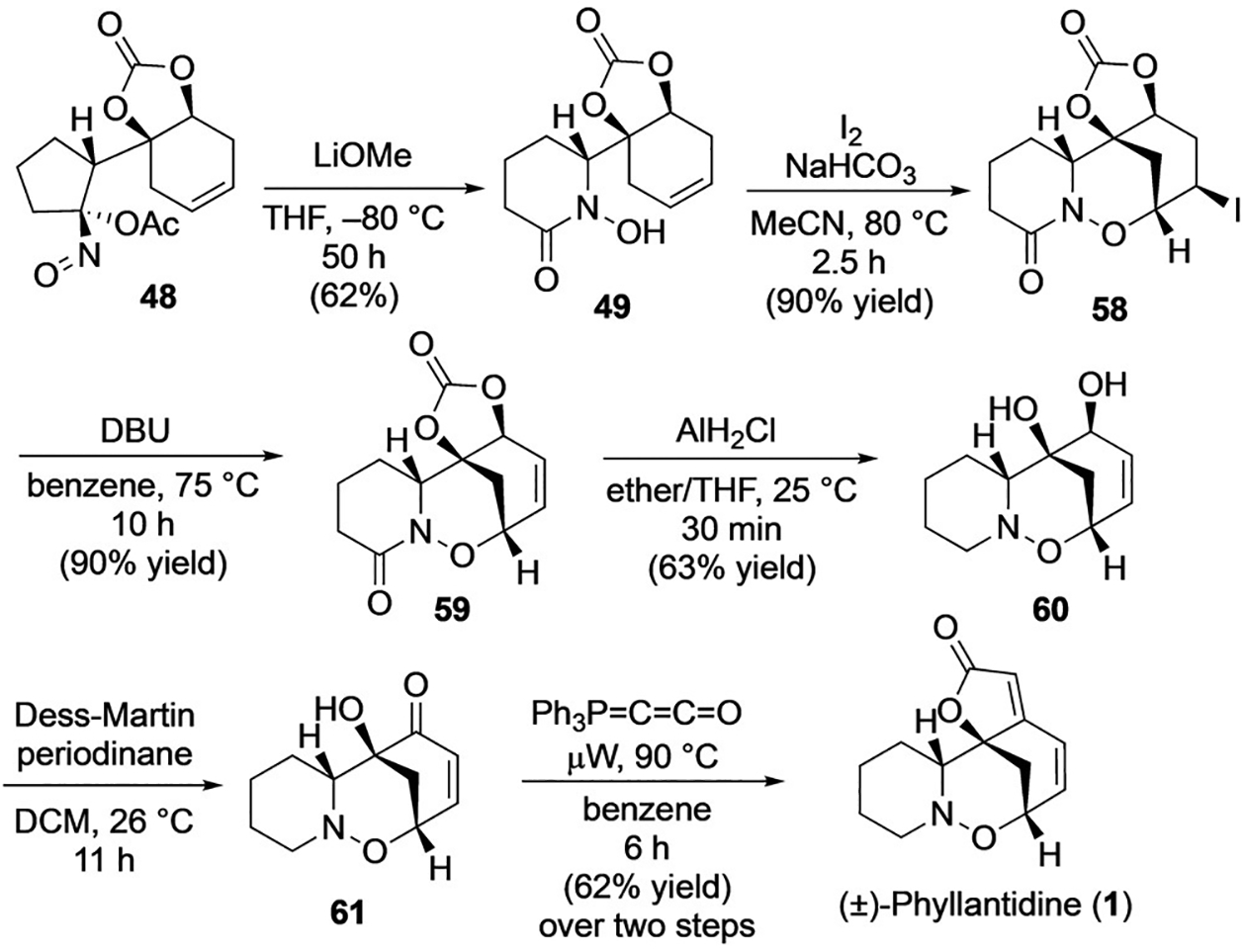
Completion of the total synthesis of (±)-phyllantidine (1). DBU= 1,8-diazabicyclo[5.4.0]undec-7-ene.
Conclusion
Phyllantidine (1) has proven to be an inspriational target for the development of a ring expansion approach for the installation of embedded N—O bonds into functionally dense architectures. The sequence involves the stereoretentive conversion of a substituted cyclopentanone to a cyclic hydroxamic acid, via an acyloxy nitroso intermediate. Efforts to apply this ring expansion protocol revealed that, akin to the Baeyer–Villiger oxidation, inductively withdrawing oxygen substituents adjacent to the migrating center can override the intrinsically favored migration of the most heavily substituted (electron-rich) carbon. Evaluation of these effects through a combined experimental and computational approach revealed that the deleterious inductive effects could be mitigated by careful selection of solvent and electronic modification of the substrate. Specifically, the introduction of functionality capable of diminishing oxygen˛s Lewis basicity coupled with the use of ethereal solvents to disrupt counterproductive metal–substrate coordination were found to completely reverse the observed migratory aptitudes. In addition to the ring expansion chemistry, the synthesis features: an early-stage diastereoselective aldol reaction to assemble the substituted cyclopentanone, a mild reduction of an amide intermediate without N—O bond cleavage, and rapid butenolide assembly via use of the Bestmann ylide. Overall, the synthesis requires 12-steps to prepare 1 from readily available, known materials (19) or 14-steps from commercially available 1,4-cyclohexadiene and is accomplished in 9% yield from 19.
Supplementary Material
Acknowledgements
We gratefully acknowledge financial support from Baylor University, the Welch Foundation (Chair, AA-006), the Cancer Prevention and Research Institute of Texas (CPRIT, R1309), the National Science Foundation (NSF, CHE-1764240), and the National Institute of General Medical Sciences of the National Institutes of Health (NIGMS-NIH, R01GM136759). K.M.L. is grateful for an NIH NRSA postdoctoral fellowship (NIGMS-NIH, F32GM129969) to explore this ring expansion chemistry.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Kyle M. Lambert, Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, TX 76798 (USA).
Joshua B. Cox, Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, TX 76798 (USA).
Lin Liu, Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, TX 76798 (USA).
Amy C. Jackson, Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, TX 76798 (USA)
Sam Yruegas, Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, TX 76798 (USA).
Kenneth B. Wiberg, Department of Chemistry, Yale University, New Haven, CT 06520 (USA)
John L. Wood, Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, TX 76798 (USA).
References
- [1]. For comprehensive reviews on the isolation, identification, and syntheses of the securinega alkaloids see:Wehlauch R, Gademann K, Asian J Org. Chem 2017, 6, 1146–1159;Chirkin E, Atkatlian W, Poreé F-H in The Alkaloids. Chemistry and Biology (Ed.: Knölker H-J), Academic Press, London, 2015, pp. 1–120;Weinreb SM, Nat. Prod. Rep 2009, 26, 758–775.
- [2]. For examples of dimeric, trimeric, tetrameric, and pentameric securinega alkaloids see:Park J, Jeon S, Kang G, Lee J, Baik M-H, Han S, J. Org. Chem 2019, 84, 1398–1406;Jeon S, Han S, J. Am. Chem. Soc 2017, 139, 6302–6305, and references therein.
- [3].a) Chen J-H, Levine SR, Buergler JF, McMahon TC, Medeiros MR, Wood JL, Org. Lett 2012, 14, 4531–4533; [DOI] [PubMed] [Google Scholar]; b) Medeiros MR, Wood JL, Tetrahedron 2010, 66, 4701–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Parello J, Munavalli S, Hebd CR. Seances Acad. Sci 1965, 260, 337–340. [PubMed] [Google Scholar]
- [5].Horii Z, Imanishi T, Yamauchi M, Hanaoka M, Parello J, Munavalli S, Tetrahedron Lett. 1972, 13, 1877–1880. [Google Scholar]
- [6].Lajis NH, Guan OB, Sargent MV, Skelton BW, White AH, Aust. J. Chem 1992, 45, 1893–1897. [Google Scholar]
- [7].Moraes LS, Donza MR, Rodrigues AP, Silva BJ, Brasil DS, Zoghbi M, Andrade EH, Guilhon GM, Silva EO, Molecules 2015, 20, 22157–22169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Park KJ, Kim CS, Khan Z, Oh J, Kim SY, Choi SU, Lee KR, J. Nat. Prod 2019, 82, 1345–1353. [DOI] [PubMed] [Google Scholar]
- [9].Wu Z-L, Huang X-J, Xu M-T, Ma X, Li L, Shi L, Wang W-J, Jiang R-W, Ye W-C, Wang Y, Org. Lett 2018, 20, 7703–7707. [DOI] [PubMed] [Google Scholar]
- [10]. Virosaine A:Miyatake-Ondozabal H, Bannwart LM, Gademann K, Chem. Commun 2013, 49, 1921–1923;Hughes JME, Gleason JL, Angew. Chem. Int. Ed 2017, 56, 10830–10834; Angew. Chem.2017, 129, 10970 – 10974; Virosaine B:Wei H, Qiao C, Liu G, Yang Z, Angew. Chem. Int. Ed 2013, 52, 620–624; Angew. Chem.2013, 125, 648 – 652; Flueggine A:Ma N, Yao Y, Zhao B-X, Wang Y, Ye W-C, Chem. Commun 2014, 50, 9284–9287, and reference [10c].
- [11].Carson CA, Kerr MA, Angew. Chem. Int. Ed 2006, 45, 6560–6563; Angew. Chem. 2006, 118, 6710 – 6713. [DOI] [PubMed] [Google Scholar]
- [12].Berthet M, Cheviet T, Dujardin G, Parrot I, Martinez J, Chem. Rev 2016, 116, 15235–15283, and references therein;Hong AY, Vanderwal CD, J. Am. Chem. Soc 2015, 137, 7306–7309.
- [13]. For methods to construct tetrahydro-1,2-oxazines see:Young IS, Kerr MA, Angew. Chem. Int. Ed 2003, 42, 3023–3026; Angew. Chem. 2003, 115, 3131 – 3134;Young IS, Kerr MA, Org. Lett 2004, 6, 139–141;Ganton MD, Kerr MA, J. Org. Chem 2004, 69, 8554–8557;Sapeta K, Kerr MA, J. Org. Chem 2007, 72, 8597–8599.
- [14]. For examples of: reduction of hydroxamic acids to nitrones, see:Katahara S, Kobayashi S, Fujita K, Matsumoto T, Sato T, Chida N, J. Am. Chem. Soc 2016, 138, 5246–5249; selective reduction to hydroxylamines, see:Bailey CL, Joh AY, Hurley ZQ, Anderson CL, Singaram B, J. Org. Chem 2016, 81, 3619–3628;Coleridge BM, Angert TP, Marks LR, Hamilton PN, Sutton CP, Matos K, Burkhardt ER, Tetrahedron Lett. 2010, 51, 5973–5976;Godjoian G, Singaram B, Tetrahedron Lett. 1997, 38, 1717–1720;Van Elburg PA, Reinhoudt DN, Heterocycles 1987, 26, 437–445; other reactions, see:Pichette S, Aubert-Nicol S, Lessard J, Spino C, J. Org. Chem 2012, 77, 11216–11226;Keck GE, Wager TT, McHardy SF, Tetrahedron 1999, 55, 11755–11772.
- [15].a) Trapencieris P, Strazdina J, Bertrand P, Chem. Heterocycl. Compd 2012, 48, 833–835; [Google Scholar]; b) Bapat JB, Black DC, Brown RFC, Adv. Heterocycl. Chem 1969, 10, 199–240; [Google Scholar]; c) The chemistry of hydroxylamines, oximes and hydroxamic acids (Eds.: Rappoport Z, Liebman JF), Wiley, Chichester, 2009. [Google Scholar]
- [16].a) Hadimani MB, Mukherjee R, Banerjee R, Shoman ME, Aly OM, King SB, Tetrahedron Lett. 2015, 56, 5870–5873; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sha X, Isabell TS, Patel RP, Day CS, King SB, J. Am. Chem. Soc 2006, 128, 9687–9692. [DOI] [PubMed] [Google Scholar]
- [17].Wiberg KB, Angew. Chem. Int. Ed. Engl 1986, 25, 312–322; Angew. Chem. 1986, 98, 312 – 322. [Google Scholar]
- [18].Renz M, Meunier B, Eur. J. Org. Chem 1999, 737–750. [Google Scholar]
- [19].Sun X, Worthy AD, Tan KL, Angew. Chem. Int. Ed 2011, 50, 8167–8171; Angew. Chem. 2011, 123, 8317 – 8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nicolaou KC, Cai Q, Qin B, Petersen MT, Mikkelsen R, Heretsch P, Angew. Chem. Int. Ed 2015, 54, 3074–3078; Angew. Chem. 2015, 127, 3117 – 3121. [DOI] [PubMed] [Google Scholar]
- [21].a) Zimmerman HE, Traxler MD, J. Am. Chem. Soc 1957, 79, 1920–1923; [Google Scholar]; b) Evans DA, Aldrichimica Acta 1982, 15, 23–32. [Google Scholar]
- [22]. A screen of oxidants revealed that PIDA was the most effective oxidant; the originally reported conditions of 1 equivalent of lead tetraacetate at 0°C in DCM solvent (Ref. [16a]) only lead to trace product. Full optimization conditions can be found in the Supporting Information.
- [23].Moulay S, Chem. Educ. Res. Pract. Eur 2002, 3, 33–64. [Google Scholar]
- [24].Coffen DL, Katonak DA, Helv. Chim. Acta 1981, 64, 1645. [Google Scholar]
- [25].Kim HR, Shin SI, Park HJ, Jeon DJ, Ryu EK, Synlett 1998, 789–791. [Google Scholar]
- [26]. See the Supporting Information for further details.
- [27].a) Tani K, Stoltz BM, Nature 2006, 441, 731–734; [DOI] [PubMed] [Google Scholar]; b) Liniger M, VanderVelde DG, Takase MK, Shahgholi M, Stoltz BM, J. Am. Chem. Soc 2016, 138, 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. The rapid hydrolysis of the trifluoroacetate variant of 32 precludes formation of 35 through such a pathway, as the trifluoroacetate moiety is lost prior to engagement of the tertiary alcohol.
- [29].a) Risley JM, Van Etten RL, J. Am. Chem. Soc 1979, 101, 252–253; [Google Scholar]; b) Everett JR, Org. Magn. Reson 1982, 19, 86–88. [Google Scholar]
- [30]. Heating of the neat reaction mixture (H218O solvent) was attempted, but required heating to 50°C before minor amounts of 37 were detected by TLC analysis (60% EtOAc/hexanes, Rf =0.44, FeCl3 stain). These conditions ultimately led to decomposition of 36 prior to expansion. THF and CH3CN were also used as co-solvents, however, heating was also required leading to substantial decomposition of 36 without any significant expansion to 37. Thus, methanol was an essential component for the use of hydroxide bases.
- [31]. In order to consider the possibility of a radical-mediated mechanism, the expansion of 36 to 37 was conducted in the presence of TEMPO.[26] No additional intermediates were observed and these conditions afforded 37 as the sole product.
- [32].a) Ho J, Klamt A, Coote ML, J. Phys. Chem. A 2010, 114, 13442–13444; [DOI] [PubMed] [Google Scholar]; b) Tomasi J, Mennucci B, Cammi R, Chem. Rev 2005, 105, 2999–3093; [DOI] [PubMed] [Google Scholar]; c) Marenich AV, Olson RM, Chamberlin AC, Cramer CJ, Truhlar DG, Chem J. Theory Comput. 2007, 3, 2055–2067. [DOI] [PubMed] [Google Scholar]
- [33]. These attempts primarily resulted in β-elimination of the tertiary alcohol accompanied by complex mixtures of decomposition products.
- [34].Laurence C, Gal J-F in Lewis Basicity and Affinity Scales: Data and Measurement, Wiley, Chichester, 2010, pp. 1–447. [Google Scholar]
- [35].a) Msayib KJ, Watt CIF, Chem. Soc. Rev 1992, 21, 237–243; [Google Scholar]; b) Bagno A, Scorrano G, J. Chem. Soc. Perkin Trans 2 1990, 1017–1027; [Google Scholar]; c) Exner JH, Steiner EC, J. Am. Chem. Soc 1974, 96, 1782–1787. [Google Scholar]
- [36].Gruver JM, Liou LR, McNeil AJ, Ramirez A, Collum DB, J. Org. Chem 2008, 73, 7743–7747;Brown CA, J. Chem. Soc. Chem. Commun 1974, 680–681, and references therein;Reich HJ, J. Org. Chem 2012, 77, 5471–5491, and references therein;Reich HJ, Sikorski WH, J. Org. Chem 1999, 64, 14–15.
- [37].a) Wiberg KB, J. Comput. Chem 2004, 25, 1342–1346; [DOI] [PubMed] [Google Scholar]; b) Tirado-Rives J, Jorgensen WL, Chem J. Theory Comput. 2008, 4, 297–306; [DOI] [PubMed] [Google Scholar]; c) Rodrigues-Oliveira AF, Ribeiro FWM, Cervi G, Correra TC, ACS Omega 2018, 3, 9075–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Krow GR, Org. React 1993, 43, 251–798. [Google Scholar]
- [39].Meinwald J, Frauenglass E, J. Am. Chem. Soc 1960, 82, 5235–5239. [Google Scholar]
- [40].a) Buchbauer G, Gabmeier J, Haslinger E, Robien W, Steindl H, Helv. Chim. Acta 1985, 68, 231–235; [Google Scholar]; b) Duddeck H, Kaiser M, Z. Naturforsch. B 1982, 37, 1672–1674. [Google Scholar]
- [41].a) Wang S, Zhao X, Zhang-Negrerie D, Du Y, Org. Chem. Front 2019, 6, 347–351; [Google Scholar]; b) Tabolin A, Loffe SL, Chem. Rev 2014, 114, 5426–5476. [DOI] [PubMed] [Google Scholar]
- [42].Van Elburg PA, Reinhoudt DN, Heterocycles 1987, 26, 437–445. [Google Scholar]
- [43].a) Bestmann HC, Sandmeier D, Angew. Chem. Int. Ed. Engl 1975, 14, 634; Angew. Chem. 1975, 87, 630; [Google Scholar]; b) Kitson RRA, Taylor RJK, Wood JL, Org. Lett 2009, 11, 5338–5341. [DOI] [PubMed] [Google Scholar]
- [44]. Phyllantidine (1), as well as compounds 35, 40, 59, 60, and 61, were selected for screening against the U.S. National Cancer Institutes˛ 60 cancer cell line panel. However, all compounds tested were found to be essentially inactive against these cell lines after the single dose screen.[26]
- [45]. CCDC 1950718 (23), 1950719 (35), 1950720 (40), 1950721 (47), and 1950722 (48) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.