

Abstract
Species Human mastadenovirus E (HAdV-E) comprises several simian types and a single human type: HAdV-E4, a respiratory and ocular pathogen. RFLP analysis for the characterization of intratypic genetic variability has previously distinguished two HAdV-E4 clusters: prototype (p)-like and a-like. Our analysis of whole genome sequences confirmed two distinct lineages, which we refer to as phylogroups (PGs). PGs I and II comprise the pand a-like genomes, respectively, and differ significantly in their G + C content (57.7% ± 0.013 vs 56.3% ± 0.015). Sequence differences distinguishing the two clades map to several regions of the genome including E3 and ITR. Bayesian analyses showed that the two phylogroups diverged approximately 602 years before the present. A relatively faster evolutionary rate was identified for PG II. Our data provide a rationale for the incorporation of phylogroup identity to HAdV-E4 strain designation to reflect the identified unique genetic characteristics that distinguish PGs I and II.
Keywords: Adenovirus type 4, Human mastadenovirus E, Genetic diversity, Evolution
1. Introduction
The more than 89 currently recognized human adenovirus (HAdV) genotypes (Dhingra et al., 2019; Kajan et al., 2018) are categorized into seven species designated Human mastadenovirus A to G (HAdV-A to HAdV-G) based on their genetic characteristics (Benko et al., 2005). The number of constituent types varies among the seven species from a single one in species HAdV-E and -G, to more than 50 in species HAdVD (Kajan et al., 2018; Walsh et al., 2011; Hashimoto et al., 2018; Yoshitomi et al., 2017).
Human adenovirus type 4 (HAdV-E4) is the only type in species Human mastadenovirus E (HAdV-E) thus far isolated from humans. Species HAdV-E also comprises several simian adenovirus types isolated from non-human primates (NHP), SAdV-21 through −26, SAdV-30, SAdV-36 through −39, and CHAdV Y25, suggesting that the emergence of HAdV-E4 as a human pathogen was the result of a zoonotic event or of an interspecies recombination process involving adenoviruses of two or more taxonomic species (Dehghan et al., 2013; Jacobs et al., 2004). Among all SAdVs in species HAdV-E, SAdV-26 is the most closely related to HAdV-E4 (Dehghan et al., 2013). In humans, HAdV-E4 infection is associated with acute respiratory disease of variable severity affecting both military recruits in basic training and civilians in various settings (Mölsa et al., 2016; Kandel et al., 2010; Narra et al., 2016; Kalimuddin et al., 2017; Kajon et al., 2007; Rogers et al., 2019) and with conjunctivitis of variable clinical manifestations, including epidemic keratoconjunctivitis, pharyngoconjunctival fever and hemorrhagic conjunctivitis (Aoki et al., 1982; Muzzi et al., 1975; Schepetiuk et al., 1993; Tullo and Higgins, 1980; Tsuzuki-Wang et al., 1997).
Extensive intratypic genetic variability manifested by the occur-rence of multiple genomic variants discriminable by restriction fragment length polymorphism (RFLP) analysis of the viral genome has been reported for HAdV-E4 since the late 1980s (Kajon et al., 2007; Li and Wadell, 1988; Adrian, 1992). By determining the percentage of comigrating restriction fragments, two major clusters of genetic homology were recognized among described genomic variants of HAdV-E4: a genomic cluster comprised of prototype (p)-like strains closely related to the prototype strain RI-67, and a second genomic cluster comprised of a-like strains (Li and Wadell, 1988).
The advent of next-generation sequencing (NGS) technologies has greatly facilitated the detailed characterization of complete HAdV genomes and their comparison. In 2005, the genome of the prototype strain of HAdV-E4, RI-67, was sequenced and annotated (Purkayastha et al., 2005). Later, the first genomic comparisons between HAdV-E4 strains were reported together with the initial observations of possible interspecies recombination events underlying the evolution of this unique HAdV type (Dehghan et al., 2013).
In the present study, we obtained whole genome sequences (WGS) for a collection of 15 new HAdV-E4 strains isolated from cases of respiratory and ocular disease in the United States and Japan, to assemble a large sample representing the spectrum of genetic diversity identified for this HAdV type. Using a global dataset of WGS from strains isolated between 1953 and 2015, we have conducted a comprehensive computational analysis of their evolutionary relationships and rates of divergence over time.
2. Material and methods
2.1. Viral strains and next generation whole genome sequencing
Whole genome sequences were determined by NGS for 15 new HAdV-E4 strains isolated from cases of acute respiratory or ocular disease. The new sequences were generated at Hokkaido University using an Ion Torrent platform similar to that described in other studies (Kajan et al., 2018), and at the Wadsworth Center, New York State Department of Health using an Illumina MiSeq platform, as previously described (Kajon et al., 2018).
Additional WGS were gathered from the NCBI GenBank (Table 1). The genome type (p- or a-like) of each strain was determined (or verified) in silico with CLC Genomics Workbench software (v10, QIAGEN, Aarhus, Denmark) for enzymes BamHI, SmaI, SspI and XhoI. In addition, to re-examine the putative recombinant origins of HAdV-E4, WGS of simian adenoviruses (SAdVs) classified within species HAdV-E, SAdV-23 (AY530877), −24 (AY530878), −25 (AF394196 and FJ025918), −26 (FJ025923), −30 (FJ025920), −36 (FJ025917), −37 (FJ025921 and FJ025919), −38 (FJ025922), −39 (FJ025924) and chimpanzee adenovirus Y25 in HAdV-E (CHAdV-E25) (JN254802), as well as WGS for HAdV-B3 (DQ086466), -B7 (KF268134), -B11 (AF532578), -B14 (FJ822614), -B16 (JN860680), -B34 (AY737797), -B35 (AY271307), -B55 (FJ643676), -B66 (JN860676), -B68 (JN860678) and -B79 (LC177352) were included in the analysis. WGS and individual coding sequences (CDS) of 36 open reading frames (ORF) were multiple-sequence aligned with MAFFT (Katoh and Standley, 2013). The nucleotide content was assessed as the percentage of guanine and cytosine (%G + C) calculated with CLC Genomics Workbench software. The %G + C for the first, second and third codon positions was estimated for each HAdV-4 sequence. The mean %G + C in species HAdV-B, –C and -D was estimated using at least one sequence for each genotype for each of these three species. The corresponding GenBank accession numbers are those reported in cited references Walsh et al., 2011; Yoshitomi et al., 2017; Hashimoto et al., 2018.
Table 1.
Origin and genomic characteristics of HAdV-E4 strains included in the study.
| Isolation | Genomic data | Restriction Fragment Length Polymorphisms (RFLP)a(# of sites) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Strain ID | Phylogroup | Genome type | Year | Place | Specimen | Accession numberb | WGS sourceb | Genome Length (bp) | G + C (%) | BamHI | SmaI | SspI | XhoI |
| 1 | RI-67 | 1953 | MO,USA | respiratory | AY594253 | GenBank | 35990 | 57.7 | 7 | 20 | 4 | 9 | ||
| prototype strain | KX384949 | GenBank | 35990 | 57.7 | 7 | 20 | 4 | 9 | ||||||
| 2 | CL 68578 | 1963 | NC,USA | respiratory | AY487947 | GenBank | 35994 | 57.7 | 7 | 20 | 4 | 9 | ||
| vaccine strain | AY594254 | GenBank | 35994 | 57.7 | 7 | 19 | 4 | 9 | ||||||
| 3 | RU2533 | 1966 | USA | respiratory | MF002043 | NYSDOH | 35975 | 57.7 | 7 | 20 | 4 | 9 | ||
| 4 | RDU2954 | 1966 | NJ,USA | respiratory | KX384948 | GenBank | 35991 | 57.7 | 7 | 20 | 4 | 9 | ||
| 5 | RU4445 | 1968 | EGP | respiratory | KX384947 | GenBank | 35991 | 57.7 | 7 | 20 | 4 | 9 | ||
| 6 | RU7872 | I | p-like | 1971 | MN,USA | respiratory | KX384950 | GenBank | 35983 | 57.7 | 7 | 20 | 4 | 9 |
| 7 | V1003 | 1981 | NY,USA | respiratory | KX384957 | GenBank | 35929 | 57.7 | 7 | 20 | 4 | 9 | ||
| 8 | V2029E | 1986 | GA,USA | respiratory | KX384946 | GenBank | 35904 | 57.7 | 7 | 20 | 4 | 9 | ||
| 9 | NHRC90255 | 2000 | NJ,USA | respiratory | AP014852 | Hokkaido | 35914 | 57.7 | 7 | 20 | 4 | 9 | ||
| 10 | NHRC90870 | 2004 | NJ,USA | respiratory | AP014853 | Hokkaido | 35914 | 57.7 | 7 | 20 | 4 | 9 | ||
| 11 | NHRC90339 | 2011 | NJ,USA | respiratory | EF371058 | GenBank | 35914 | 57.7 | 7 | 20 | 4 | 9 | ||
| 12 | NYS 15–4054 | 2015 | NY,USA | respiratory | KY996447 | GenBank | 35968 | 57.7 | 7 | 20 | 4 | 9 | ||
| 13 | V0014 | 1978 | FRA | respiratory | KX384956 | GenBank | 35960 | 56.4 | 8 | 15 | 5 | 10 | ||
| 14 | J1007 | 1981 | JPN | respiratory | KY996452 | NYSDOH | 35962 | 56.3 | 8 | 15 | 5 | 10 | ||
| 15 | NA | 1984 | JPN | ocular | AB679754 | Hokkaido | 35960 | 56.3 | 8 | 15 | 5 | 10 | ||
| 16 | V1933 | 1985 | NM,USA | respiratory | KX384955 | GenBank | 35961 | 56.3 | 8 | 15 | 5 | |||
| 17 | NA | 1991 | JPN | ocular | AB679755 | Hokkaido | 35961 | 56.3 | 8 | 15 | 5 | 10 | ||
| 18 | ZG 95–873 | 1995 | CA,USA | respiratory | KX384951 | GenBank | 35967 | 56.3 | 8 | 14 | 5 | 10 | ||
| 19 | 078Jax | 1997 | SC,USA | respiratory | KX384953 | GenBank | 35963 | 56.3 | 8 | 15 | 5 | 10 | ||
| 20 | 186Jax | 1998 | SC,USA | respiratory | KX384952 | GenBank | 35963 | 56.3 | 8 | 15 | 5 | 10 | ||
| 21 | 10Jax | 2001 | SC,USA | respiratory | KX384954 | GenBank | 35962 | 56.3 | 8 | 15 | 5 | 10 | ||
| 22 | NA | 2001 | JPN | ocular | AB679756 | Hokkaido | 35963 | 56.3 | 7 | 14 | 5 | 10 | ||
| 23 | NHRC11023 | 2001 | IL,USA | respiratory | AP014849 | Hokkaido | 35973 | 56.3 | 8 | 14 | 5 | |||
| 24 | NHRC50654 | 2001 | TX,USA | respiratory | AP014850 | Hokkaido | 35964 | 56.3 | 8 | 14 | 5 | |||
| 25 | T158 | 2002 | SC,USA | respiratory | KX384945 | GenBank | 35965 | 56.3 | 8 | 15 | 5 | 10 | ||
| 26 | NHRC3 | 2002 | TX,USA | respiratory | AY599837 | GenBank | 35965 | 56.3 | 8 | 14 | 5 | 10 | ||
| 27 | NHRC42606 | II | a-like | 2003 | SC,USA | respiratory | AY599835 | GenBank | 35965 | 56.3 | 8 | 15 | 5 | 10 |
| 28 | NHRC70935 | 2004 | SC,USA | respiratory | AP014844 | Hokkaido | 35967 | 56.3 | 8 | 14 | 5 | 10 | ||
| 29 | NHRC22650 | 2006 | CA,USA | respiratory | AP014841 | Hokkaido | 36155 | 56.3 | 8 | 15 | 5 | 10 | ||
| 30 | GZ01 | 2008 | CHN | respiratory | KF006344 | GenBank | 35960 | 56.3 | 8 | 14 | 5 | 10 | ||
| 31 | NHRC23703 | 2008 | CA,USA | respiratory | AP014842 | Hokkaido | 35959 | 56.3 | 8 | 15 | 5 | 10 | ||
| 32 | NHRC92165 | 2009 | NJ,USA | respiratory | AP014845 | Hokkaido | 35964 | 56.3 | 8 | 15 | 5 | 10 | ||
| 33 | WPAFB7 | 2009 | CA,USA | respiratory | AP014847 | Hokkaido | 35961 | 56.3 | 8 | 13 | 6 | 10 | ||
| 34 | TB071911 | 2011 | CT,USA | respiratory | KY996453 | GenBank | 35952 | 56.3 | 8 | 15 | 5 | 10 | ||
| 35 | NHRC36401 | 2011 | MO,USA | respiratory | AP014851 | Hokkaido | 35960 | 56.3 | 8 | 13 | 6 | 10 | ||
| 36 | NYS 12–12752 | 2012 | NY,USA | respiratory | KY996450 | GenBank | 35955 | 56.3 | 8 | 15 | 5 | 10 | ||
| 37 | NYS 12–27440 | 2012 | NY,USA | respiratory | KY996451 | GenBank | 35948 | 56.3 | 8 | 14 | 6 | 10 | ||
| 38 | NYS 13–5497 | 2013 | NY,USA | respiratory | KY996449 | GenBank | 35960 | 56.3 | 8 | 13 | 6 | 10 | ||
| 39 | NYS 14–4876 | 2014 | NY,USA | respiratory | KY996448 | GenBank | 35934 | 56.3 | 8 | 15 | 5 | 10 | ||
| 40 | NYS 14–38662 | 2014 | NY,USA | respiratory | KY996443 | GenBank | 35960 | 56.3 | 8 | 14 | 6 | 10 | ||
| 41 | NYS 14–38813 | 2014 | NY,USA | respiratory | KY996444 | GenBank | 35948 | 56.3 | 8 | 14 | 6 | 10 | ||
| 42 | NYS 14–33430 | 2014 | NY,USA | respiratory | KY996445 | GenBank | 35948 | 56.3 | 8 | 14 | 6 | 10 | ||
| 43 | NYS 14–9111 | 2014 | NY,USA | respiratory | KY996442 | GenBank | 35948 | 56.3 | 8 | 14 | 6 | 10 | ||
| 44 | NYS 15–3477 | 2015 | NY,USA | respiratory | KY996446 | GenBank | 35949 | 56.3 | 8 | 14 | 5 | 9 | ||
| 45 | NYS 15–1428 | 2015 | NY,USA | respiratory | MF002042 | GenBank | 35960 | 56.3 | 8 | 14 | 6 | 10 | ||
Restriction sites predicted by CLC Genomics Workbench v10.1.1.
Sequences obtained in this study are in bold font.
2.2. Sequence alignment and analysis
Phylogenetic trees were inferred with MrBayes v3.2.7 (Ronquist et al., 2012) using the general time-reversible substitution model with heterogeneity among sites, modeled under a gamma distribution and allowing for a proportion of invariable sites (GTR+Γ+I) as substitution model, chosen as the model with the highest corrected Akaike information criterion (AICc) calculated with jModelTest 2 v0.1.10 (Darriba et al., 2012) for the multiple sequence alignments. Trees were inferred with chain lengths of 106 states to assure convergence. Multiple sequence alignments comparisons were performed with Clustal X (Larkin et al., 2007).
2.3. Similarity analysis, topological testing and G + C content
Similarity analyses between groups of sequences were performed by a sliding window approach with window size 500 bp and step size 250 bp. In each window the average evolutionary distance with Kimura model between groups was calculated as the mean of the distances between sequences in both groups. In addition, the averaged %G + C difference among HAdV-4 sequences was estimated for each window.
Topological testing of the cluster of sequences was performed by comparing the Bayes factor for the likelihood of one model considering the clustering and the model with the null hypothesis without that clustering (Sasaki et al., 2016). The topological model with the highest Bayes factor and a factor > 5 difference to other models was considered as the model with the highest support. For each window, the evolutionary models were tested with jModelTest 2 to assure the GTR + I + G model was among the best fitting models.
The percent similarities among nucleotide and protein sequences were estimated by using the Sequence Demarcation Tool software v1.2 (Muhire et al., 2014).
Mean percent sequence similarities between two genotypes in the same species in HAdV-B 1, -B 2, –C and -D, were estimated by pairwise comparisons of sequences for all recognized genotypes in each of these three species using the Sequence Demarcation Tool software v1.2. The GenBank accession numbers for the analyzed sequences are those reported in cited references Walsh et al., 2011; Yoshitomi et al., 2017; Hashimoto et al., 2018.
Simplot, a sequence similarity plotting tool (Lole et al., 1999), was used to identify conserved and divergent regions along the genomes of the examined HAdV-E4 strains. WGS were aligned with MAFFT v7.388 and default parameters using the Geneious v11.1.4 software platform (Biomatters, New Zealand). A similarity plot was generated in Simplot v3.5.1 using a 200-nucleotide sliding window, a 20-nucleotide step size, GapStrip: On, Kimura distance model, and Ts/Tv = 2.0.
2.4. Bayesian estimation of the time to the most recent common ancestors
To test whether the HAdV-E4 dataset provided enough data to analyze the temporal signal, the clock-likeness was checked by performing a linear regression between the parameters ‘root-to-tip divergence’ and ‘sampling date’ with TempEst (Rambaut et al., 2016). Time to the most recent common ancestor (tMRCA) was estimated by independent Bayesian Markov Chain Monte Carlo (MCMC) coalescent analyses by BEAST v2.4.6 (Bouckaert et al., 2014) with chain lengths of 5 × 107 to ensure effective sample size (ESS) > 300 in all parameters of the models. Analyses were performed separately for WGS in the two groups of strains identified as p- and a-like genomes. Additionally, the tMRCA for both groups was estimated by analyzing a combination of CDS alignments excluding those suspected to contain effects of recombination events. Strict and relaxed exponential clock models were considered for the datasets in combination with coalescent constant, exponential and Bayesian skyline models for the populations (Drummond et al., 2005, 2006). The marginal likelihood of the combination of models and data was estimated in BEAST and with the Path-Sampler application in the BEAST package. Additionally, the distribution of the mutation rate for clades was calculated by extracting the parameter of each tree sampled every 5 × 104 states in the BEAST chain using TreeStat v1.2 (http://tree.bio.ed.ac.uk/software/treestat/). The values extracted from trees sampled along the BEAST chain were used to model the distribution of the parameter.
2.5. Statistical analyses
Statistical assessments were performed in R v3.5 (Team, 2013). The statistical significance of %G + C differences among groups of sequences was assessed with phylogenetic independent contrasts (PIC) to correct for the shared ancestry among sequences before analyzing the correlation with the assigned phylogroup. In addition, parameters such as %G + C and percent sequence identity are reported as mean values, respectively, followed by the standard deviation.
3. Results and discussion
3.1. HAdV-E4 genomic variants cluster into two separable phylogroups
WGS of HAdV-E4 strains isolated in the United States and Japan (n = 15) were combined with prior publicly available sequences (n = 32) to compile and align a total of 47 genomic sequences of 45 HAdV-E4 strains representing a diversity of genomic variants (Fig. 1), geographical locations and year of specimen collection (Table 1). The original genome typing data (p- or a-like) were confirmed by in silico RFLP analysis using recognition sequences for the restriction endonucleases BamHI, SmaI, SspI and XhoI, with 7, 19–20, 4, and 9 cleavage sites for p-like and 7–8, 13–15, 5–6 and 8–10 cleavage sites for a-like strains (Table 1 and Supplementary Fig. 1). The phylogenetic tree of WGS, including those of SAdVs in HAdV-E and HAdV genotypes classified within species HAdV-B (Fig. 1), showed two major clades of HAdV-E4 strains consistent with the original genogrouping described by Li and Wadell based on the analysis of percentage of comigrating restriction fragments (Kajon et al., 2007; Li and Wadell, 1988; Adrian, 1992). HAdV-E was rooted by a cluster containing all considered SAdV genomes. This phylogenetic position supported the previously formulated hypothesis of a zoonotic origin for HAdV-E4 (Dehghan et al., 2013; Jacobs et al., 2004). Based on the highly supported phylogenetic distinction, these clades are hereafter referred to as phylogroup I (PG I) and phylogroup II (PG II) for HAdV-E4 p- and a-like strains, respectively.
Fig. 1. HAdV-E4 comprises two distinguishable phylogroups.
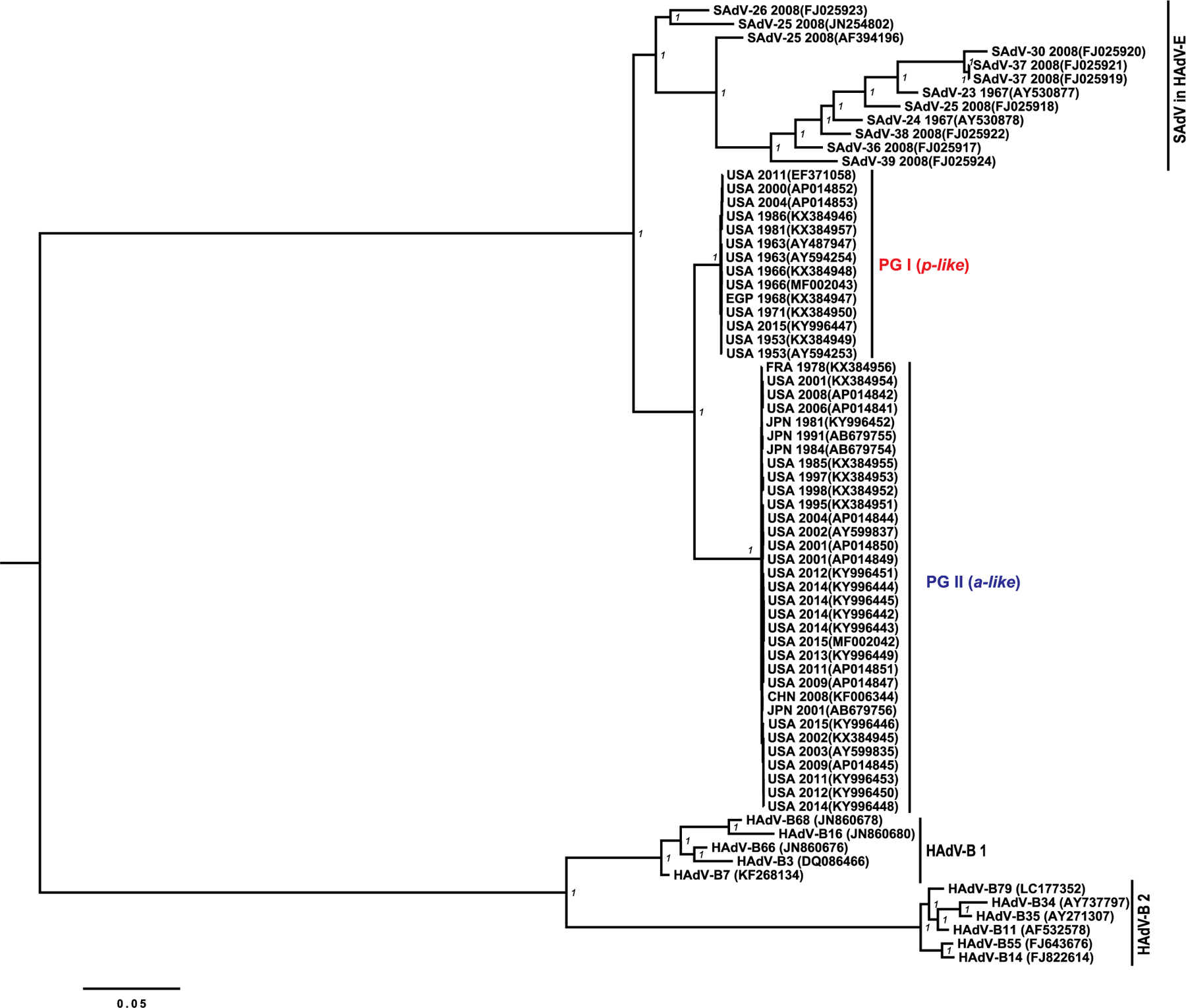
Phylogenetic tree of whole genome sequences of HAdVs of species B, E, and non-human primate adenovirus of species HAdV-E (SAdVs). Bayesian posterior probability support is shown next to the branches.
The results of our phylogenetic analysis prompted us to examine further the genetic divergence. In addition to distinct digestion profiles with various restriction endonucleases (Supplementary Fig. 1), the two phylogroups of genomic variants also differed in their mean %G + C. Genomes in PG I showed a mean %G + C of 57.7% ± 0.013 while genomes in PG II showed a significantly lower mean %G + C of 56.3% ± 0.015 (P < 2.2 × 10−16, after correction applying PIC) (Table 1). The difference in %G + C between both phylogroups is also reflected in the nucleotide content for 1st, 2nd and 3rd codon positions, where the average %G + C content for each position in PG I is 58.8% ± 0.03, 44.5% ± 0.01 and 73.1% ± 0.02, respectively. The corresponding values in PG II are 58.3% ± 0.02, 44.1% ± 0.01 and 70.6% ± 0.05, respectively. Although more pronounced in the 3rd codon position, the average %G + C for the three codon positions was significantly higher in PG I than in PG II (P < 2.2 × 10−16, after correction applying PIC). Also, the distribution along the genome of such a difference in %G + C content was assessed with a sliding window approach (Fig. 2B). Such assessment reflected an uneven but widespread difference in the G + C content. The evolutionary significance of the differences in mean %G + C between the phylogroups is expected to be tested as WGS for more HAdV-E4 strains become available. Nevertheless, the striking difference highlights the absence of intermediary PG strains, which may be attributable to a founder effect, a fitness cost for recombinants and/or insufficient sampling. It is noteworthy that the %G + C among types in other species is: 51.20% ± 0.09 in HAdV-B 1, 49.20% ± 0.83 in HAdV-B 2, 55.25% ± 0.06 in HAdV-C and 56.89% ± 0.54 in HAdV-D.
Fig. 2. Genomic differences along the two phylogroups.

The horizontal axes represent the genomic positions in HAdV-E relative to the prototype strain RI-67 USA, 1953 (KX384949). (A) Genomic annotation of HAdV-E. (B) Sliding window analysis of the average %G + C difference between PG I, and PG II across the genome. The vertical and horizontal axes show the average percentage difference between both phylogroups and the genome position, respectively. (C and D) Sliding window analyses of evolutionary distance between members of PG I and PG II to sequences of other clusters, respectively, vertical axes show the average evolutionary distance in the respective window to sequences in subspecies HAdV-B 1 (except HAdV-B16), subspecies HAdV-B 2, HAdV-B16 (JN860680) and SAdVs in species HAdV-E (see Fig. 1). (E) Sliding window analysis comparing the support for PG I and PG II cluster versus clusters of PG I and PG II with other types and species. The vertical axis shows the Bayes factor between sequences in clusters color-coded as per the key below with higher values showing higher support for the topological clustering of the groups as shown in the bottom of the panel. Regions with low topological support for the clustering of PG I and PG II are highlighted by black lines on the top of the panel.
3.2. Evolutionary divergence between both phylogroups
The branching of two distinct lineages is attributable to the accumulation of mutations along the genome and/or to recombination events occurring over time. The average inter-phylogroup evolutionary distance was 0.0413 ± 0.0002 mutations/site. To assess the distribution of this divergence, we compared the average evolutionary distance among sequences within both phylogroups to other clusters of sequences in a sliding window approach with 500 bp window and 250 bp step size (Fig. 2C and D). These clusters included subspecies HAdV-B 1 and HAdV-B 2 sequences, specifically type HAdV-B16 previously suggested to be related to HAdV-E (Dehghan et al., 2013), and sequences of SAdVs in species HAdV-E. Furthermore, the topological hypotheses of PG I clustering with PG II or with SAdVs in species HAdV-E, and PG II with SAdVs in species HAdV-E or with subspecies HAdV-B 2, were tested by a Bayesian approach (Fig. 2E). The topological testing for 60% of the genomic windows (93/154) showed high support for the clustering of PG I with PG II (Supplementary Table 1), as suggested by the complete genome phylogeny (Fig. 1). On the other hand, 40% of the genomic windows (61/154) supported the clustering of one of the phylogroups with SAdVs in HAdV-E more strongly than with the other phylogroup, suggesting possible recombination events with NHP adenoviruses, as has been proposed (Dehghan et al., 2013).
The 60% of windows showing support for the topological clustering of both phylogroups, prompted us to estimate the time to the most recent common ancestor (tMRCA) for these windows following a Bayesian approach with BEAST and calibrating the tree with the isolation year of each strain due to the lack of other temporal data. Similar approaches have been followed for other viruses as fossil data are not available (Firth et al., 2010; Zhang et al., 2017). The temporal structure was tested to assure the divergence time could be estimated in datasets including a) SAdV + HAdV-E, b) HAdV-E, c) PG I and d) PG II (Supplementary Fig. 2); the results supported strong time structure in HAdV-E (R2 = 0.68, P < 10−12), PG I (R2 = 0.86, P < 10−6) and PG II (R2 = 0.59, P < 10−7). The dataset including SAdV sequences was less conformant with a linear regression (R2 = 0.11, P < 10−2), possibly as a consequence of uncharacterized recombination events in SAdV, and hampered the inclusion of this group for the divergence analysis. Nevertheless, these estimates are expected to be further refined as new genomic sequences become available. The Bayesian analysis under different combinations of molecular clocks and population models showed that the tMRCA of PG I is longer than that for PG II (Table 2). The models involving relaxed molecular clocks, which allow for different molecular clock rates along the branches modeled under exponential or log-normal distributions, were well supported. These models showed that both phylogroups diverged approximately 600 years before the present (ybp), established as 2015, the most recent calibration point (strains #12, 44 and 45, Table 1), or around year 1400 in the absolute time scale (Fig. 3). The relaxed molecular clocks also showed a slightly higher median clock rate for PG II. The comparison of the mutation rate distributions per phylogroup extracted from the sampled trees in the Bayesian MCMC the mutation rate of PG II (median rate: 3.69 × 10−5 mutations/site/year) was 13% significantly higher than the mutation rate of PG I (median rate: 3.24 × 10−5 mutations/site/year) (P < 2 × 10−115, Student’s t-test). Furthermore, a similar trend was shown in the slopes of the linear regression for PG I and II (Supplementary Figs. 2C–D). These mutation rates were comparable to previous estimates of mutation rates of 7.20 × 10−5 and 3.46 × 10−5 for HAdV-B and HAdV-C, respectively (Firth et al., 2010). Notably, these figures were two orders of magnitude greater than those expected for other double-stranded DNA viral DNA polymerases (Duffy et al., 2008), and approximately four orders of magnitude greater than the mutation rate of primate hosts, thus providing a strong argument against the hypothesis of host-parasite co-speciation as the divergence between Homo sapiens and Pan troglodytes, which is estimated as 6.4 MYA (CI: 5.1–11.8 MYA) (derived from 79 studies in http://timetree.org/), would require an average adenoviral genome mutation rate of approximately 10−8 mutations/year/site.
Table 2.
Evaluation of clock and population models for Bayesian estimation of the divergence times.
| Both phylogroups | Phylogroup I | Phylogroup II | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clock | Population | Path Sampler Log likelihood | Bayes Factora | Median Clock rate+ | Median tMRCA (ybp) | 95% HPD | Median tMRCA (ybp) | 95% HPD | Median Clock Rate+ | Median tMRCA (ybp) | 95% HPD | Median Clock Rate+ |
| Strict | Constant | −45592 | – | 1.17E-05 | 1752 | [1333, 2268] | 86 | [72, 103] | – | 71 | [54, 92] | – |
| Exponential | −45600 | −8 | 1.20E-05 | 1735 | [1336, 2231] | 85 | [72, 101] | – | 67 | [52, 87] | – | |
| Bayesian Skyline | −45586 | 6 | 1.16E-05 | 1787 | [1359, 2312] | 85 | [72, 103] | – | 69 | [53, 90] | – | |
| Exponential | Constant | −45577 | 15 | 1.85E-05 | 447 | [241, 730] | 89 | [68, 132] | 4.84E-5 | 54 | [39, 82] | 5.79E-5 |
| Exponential | −45573 | 19 | 1.75E-05 | 602 | [270, 1217] | 91 | [67, 144] | 3.24E-5 | 54 | [40, 84] | 3.69E-5 | |
| Bayesian Skyline | −45577 | 15 | 1.75E-05 | 583 | [255, 1286] | 91 | [66, 156] | 3.17E-5 | 54 | [40, 85] | 3.82E-5 | |
| Lognormal | Constant | −45582 | 10 | 2.22E-05 | 284 | [84, 599] | 90 | [67, 135] | 4.19E-5 | 54 | [39, 81] | 1.04E-4 |
| Exponential | −45578 | 14 | 2.81E-05 | 623 | [80, 1497] | 88 | [67, 129] | 2.6E-5 | 55 | [40, 78] | 3.15E-5 | |
| Bayesian Skyline | −45582 | 10 | 2.98E-05 | 564 | [66, 1561] | 88 | [67, 135] | 2.62E-5 | 56 | [41, 81] | 3.5E-5 | |
The Bayes factor is the ratio of the likelihood of a particular model to the likelihood of the simplest model with strict clock and constant population
The clock rate is provided in mutations/site/year.
Fig. 3. Bayesian estimation of the time to the most recent common ancestor for HAdV-E4 strains in PG I and PG II.

The phylogenetic tree is annotated in the branches with years before the present. The 95% highest posterior density (HPD) ranges for tMRCAs of all sequences are shown for both phylogroups between brackets. The relative time and absolute time scales are shown in the bottom. The median relative divergence time for other branches is shown next to the branches.
The results of our analyses (Fig. 3) suggested that the currently circulating strains in PG I are descendants of an ancestral strain circulating ~91 ybp (~1924 in the absolute time scale) in the 95% highest posterior density range (95% HPD) [67, 144 ybp] while strains in PG II are descendants of an ancestral strain circulating ~54 ybp (~1961 in the absolute time scale) in the 95% HPD [40, 84 ybp].
Two independent descriptions of the 1965 Chinese strain BC129 as an “a-like” genomic variant (Li and Wadell, 1988; Adrian, 1992) date the detection of PG II to 13 years before 1978, the year of detection of the oldest strain in the examined collection, V0014 (Table 1), lending to the Bayesian estimations and suggesting that both PG I and PG II have been circulating for similar periods of time. As the genomic sequence for strain BC129 is not available we could not include 1965 as a calibration point in the analysis.
The significantly higher mutation rate in PG II under the different analysis models (Table 2) suggested: (i) a higher number of mutations accumulated during replication in PG II than in PG I, or (ii) a higher number of infections by PG II that increased the overall frequency of mutations despite a relatively similar mutation rate in each replicative cycle from both PGs. The number of samples in PG II and its frequent isolation across the world supported the second hypothesis and we hypothesize that this may be attributable to a higher viral fitness for PG II.
3.3. Detailed analysis of sequence diversity between phylogroups I and II
On average, PG I genomes were found to be ~94.5% identical to genomes in PG II. Interestingly, this level of genetic relatedness is comparable to that between any two of the currently recognized HAdV genotypes within a given species: 94% ± 5 for HAdV-B, 96% ± 1 for HAdV-C, and 94% ± 1 for HAdV-D. Many of these are also distinguishable as unique serotypes in neutralization assays.
We conducted an additional sequence identity analysis using Simplot to identify conserved and divergent regions along the genomes of PG I and PG II strains. A representative simplified plot including only 3 genomic variants from each phylogroup is shown in Fig. 4.
Fig. 4. Analysis of genomic sequences of representative strains of phylogroups I and II for regions of divergence and similarity.

A similarity plot was generated in Simplot using the whole genome sequence (WGS) for strain 1 (RI-67) as the query and the WGS for strains 2, 11, 12, 27, 31, and 37 as the references. The plot represents the percent similarity in a 200 nucleotides sliding window and 20-nucleotide step size with gapped sites removed.
The most striking differences between the genomes in PG I and PG II map to the inverted terminal repeats (ITRs) and the early region 3 (E3), with mutations with the potential to result in phenotypic differences relevant to pathogenesis are found in multiple genomic loci, including E1A, E1B, VARNA, L3 and L4, as described in detail below.
3.3.1. Inverted terminal repeat
Our analysis of 12 WGS in PG I and 33 WGS in PG II confirmed previous reports of differences in the ITR sequences between the two lineages of HAdV-E4 genomic variants including previously reported differences in length (Dehghan et al., 2013; Houng et al., 2006). PG I genomes had an average ITR length of 113.8 bp ± 3.4 and an intragroup mean percent identity of 98.6% ± 1.7, whereas the average length for PG II genomes was 206.5 ± 2.7 and their mean intragroup percent identity was 99.6% ± 0.3. As shown in Fig. 2D and in Supplementary Fig. 3, in this region of the genome PG I and the SAdVs in species HAdV-E cluster together while PG II clusters more closely with members of subspecies HAdV-B 2.
Downstream from the origin of DNA replication, the ITRs of most HAdV genomes encode binding motifs for the host cellular transcription factors NFI and NFIII, which are required for efficient genome replication (Mul et al., 1990; Rosenfeld et al., 1987; Hatfield and Hearing, 1993; Pruijn et al., 1986; Hay, 1985). As reported previously, the canonical NFI binding motif encoded in the genomes of most HAdVs is notably absent in the genomes of HAdV-E4 PG I strains (Dehghan et al., 2013; Purkayastha et al., 2005; Zhang et al., 2019). The HAdV-E4 PG I ITR only encodes the NFIII binding motif while PG II ITRs carry the same NFIII motif and an NFI recognition sequence similar to that found in the genomes of members of species HAdV-B (Dehghan et al., 2013; Zhang et al., 2019).
In our analysis, we found the NFI and NFIII binding motifs for all examined PG II strains to be identical. A variant NFIII binding motif 5′-TATGTAAATAA-3′ was identified in the genomes of PG I strains #9–11.
The terminal 8 bp section of the ITR among the examined HAdV-E4 strains was generally conserved (5′-CATCATCA-3′). PG I strain RI-67 (ATCC VR-4) had a divergent sequence (5′-CTATCTAT-3′) as reported previously (Dehghan et al., 2013; Jacobs et al., 2004; Purkayastha et al., 2005). A different sequence, 5′-CATCATCA-3′, was reported by Hang and colleagues (Hang et al., 2017) for RI-67 ATCC VR-1572 (GenBank accession KX384949) suggesting variation in RI-67 stocks among different repositories. Interestingly, we identified novel variant terminal sequences in PG I and PG II strains: 5′-ATAATATA-3′ in strain #34; 5′-AATAATAT-3′ in strains #3, 8, and 44; 5′-CAATAATA-3′ in strains #12, 36, 37, 39, and 41–43; and 5′-GCATCATC-3′ for strain #14. In addition, a 194 bp insertion by duplication of the neighboring genomic loci was found adjacent to the right hand ITR in strain NHRC22650 (#29 in Table 1).
Using vectors constructed from the HAdV-B35 background, Wunderlich and colleagues showed that the terminal ITR sequence can affect viral replication (Wunderlich et al., 2014). The elucidation of the functional significance of the variation identified in this and other studies for the ITR region among PG I and PG II strains will require experimental evaluation using engineered mutant viruses.
3.3.2. Early region 1
3.3.2.1. Early region 1A (E1A).
The HAdV genome is predicted to encode two predominant E1A polypeptides resulting from alternative splicing. E1A is an important multifunctional protein that induces transition of the host cell into the S phase of the cell cycle (Berk, 2005), and is a potent transactivator of HAdV early gene expression (Winberg and Shenk, 1984; Montell et al., 1984). Four conserved regions (CR1 to CR4) are found in the large E1A protein while the small E1A protein only includes CR1, CR2, and CR4 (Avvakumov et al., 2004). The genomes of PG I encode a 28 kDa (257 aa) polypeptide and a 24.6 kDa (226 aa) polypeptide. The genomes of PG II strains encode slightly shorter polypeptides of 27 kDa (246 aa) and 23.5 kDa (215 aa), respectively. The predicted polypeptide sequences encoded by all examined genomes in PG I are identical, while PG II sequences had an average intragroup percent identity of 99.8% ± 0.3. Collectively, all examined strains had an inter-group percent identity of 96.1% ± 6.3.
The most striking difference between the E1A polypeptides encoded by the two phylogroups is an 11 amino acid deletion (aa 82–94) between the conserved regions CR1 and CR2 found in all PG II genomes examined in this study. The deletion includes a leucine at position 91 and a threonine at position 93 which Avvakumov and colleagues described as highly conserved residues in the E1A proteins encoded by members of species HAdV-B, -D, and -E (Avvakumov et al., 2004). While no functional role has been assigned to these amino acids, this region has been shown to be a flexible linker between CR1 and CR2. This linker is important in the formation of stable ternary complexes between E1A, RB, and CBP/p300 (Ferreon et al., 2009). The minimum length of the linker required for functionality is not known. However, removal of the linker in HAdV-C5 was shown to result in failure to induce colony formation in infected BRK cells (Wang et al., 1995). Importantly, compared to other published E1A sequences (Avvakumov et al., 2004), the linker in the E1A polypeptide encoded by PG II strains ranks among the shortest linker. Interestingly, the 11 amino acid deletion described above was not present in any of the examined genomes of simian members of species HAdV-E.
3.3.2.2. Early region 1B (E1B).
The E1B transcriptional unit encodes two polypeptides, E1B 19K and E1B 55K, which are translated from two distinct initiation codons in different reading frames (Bos et al., 1981). Both proteins serve important functions in blocking p53-dependent induction of apoptosis through different mechanisms. In addition, E1B 55K in conjunction with E4 ORF6 has been shown to aid in the transport of viral mRNAs late in infection (Bridge and Ketner, 1990).
The E1B 19K protein plays a critical role in suppressing apoptosis induced by E1A and is regarded as the Bcl-2 homolog encoded by adenoviruses (reviewed in Cuconati and White, 2002). The predicted polypeptide sequence for E1B 19K is conserved among members of PG I as well as among members of PG II with a 97.6% ± 2.6 inter-phylogroup sequence identity. There are three non-synonymous mutations at positions 43, 100, and 125, using the sequence of RI-67 as a reference. Additionally, there is a 30 nucleotide (10 amino acid) in-frame insertion among PG II members located in the shared coding region for E1B 19K and E1B 55K. This insertion is also present in the coding sequence for E1B 19K in the genomes of several simian members of species HAdV-E (SAdV-23 to −26 and ChAdVY25), although only 5 of the 10 amino acids are conserved within PG II sequences.
The E1B 55K protein performs several functions critical for viral replication (Blackford and Grand, 2009). The predicted sequences for E1B 55K encoded by all examined PG I strains were identical. The predicted sequences for E1B 55K encoded by PG II strains had an average sequence identity of 98.2% ± 1.9. All of them are characterized by a 10 amino acid insertion at their N-terminus resulting from the 30-nucleotide insertion described above.
3.3.3. Virus-associated RNAs
The genomes of all examined HAdV-E4 strains encode two virus-associated (VA) RNAs, designated VA RNAI and VA RNAII. The VA RNAs are non-coding RNAs, transcribed by RNA polymerase III that fold into highly structured RNAs resembling microRNA precursors. VA RNAs function as suppressors of RNAi by interfering with the activity of the endoribonuclease Dicer (Andersson et al., 2005). While the function of VA RNAI has been well characterized as a competitive substrate that binds the interferon-inducible double-stranded RNA-dependent protein kinase (PKR) (Vachon and Conn, 2016), the role of VA RNAII in the virus life cycle is still poorly understood. Consistent with the original observations reported by Kidd and colleagues (Kidd et al., 1995), all PG II genomes examined in this study exhibit a 65 bp deletion in VA RNAII starting at position 10593 (relative to the prototype strain RI-67) that partially ablates the promoter element A and results in the complete loss of promoter element B with a predicted lack of expression of VA RNAII. The genomes of a subset of PG I strains (#8, 9, 10, and 11) have an additional 20 bp deletion starting at nucleotide position 10640, immediately downstream of promoter element B.
3.3.4. Late region 3
Our analysis focused exclusively on the hypervariable regions 1–7 of the hexon gene (HVR 1–7) which encode the serotype-specific residues displayed in loops 1 and 2 of the hexon capsid protein that projects from the surface of the virion (Crawford-Miksza and Schnurr, 1996). HVR-7 in loop 2 was recently shown to contain a conformational neutralization epitope (Tian et al., 2018).
We identified non-synonymous point mutations in HVR 1 and HVRs 3–7 that distinguish the genomes of PG I and II. The results of our sequence data analysis for this region are summarized in Fig. 5.
Fig. 5. Amino acid differences identified in the hypervariable region (HVR) 1 and HVRs 3–7 of the hexon polypeptide among examined strains of phylogroups I and II.

No differences were identified for HVR 2 so the corresponding section of the sequences is not shown.
Using a colorimetric neutralization assay and rabbit anti-HAdV-E4 strain RI-67 hyper-immune sera Crawford-Miksza and colleagues showed that strain Z-G, with sequence characteristics of a-like genomes in PG II (#18 in our sample), exhibited a four-fold reduction in neutralization titer compared to that of the prototype strain RI-67 (Crawford-Miksza et al., 1999). A reduced cross-reactivity of HAdV-16 antisera with the Z-G strain compared to the prototype RI-67 was also observed, identifying this a-like strain as an antigenic variant. Taken together, these prior findings and our data showing conservation of HVR 1–7 sequence features among strains of PG II suggest that this clade may have also drifted antigenically.
3.3.5. Late region 4 100K
Late region 4 (L4) 100K is an abundantly expressed polypeptide necessary for efficient translation of late viral mRNAs (Hayes et al., 1990; Xi et al., 2004), trimerization and nuclear localization of the hexon polypeptide (Hong et al., 2005), and also to protect infected cells from granzyme B-dependent cell death by cytotoxic lymphocytes (Andrade et al., 2001). The predicted L4–100K amino acid sequences encoded by PG I and PG II strains had an average percent identity of 98.4% ± 5.4. PG I strain #12 (Kajon et al., 2018) exhibited a single glutamine insertion (nucleotides CAG) at amino acid position 20. Interestingly, several differences were identified between PG I and PG II sequences in the glycine-arginine rich (GAR) C-terminal region of L4–100K. Arginine-glycine-glycine (RGG) motifs in GAR regions are sites of arginine methylation (reviewed by Thandapani et al., 2013; Blanc and Richard, 2017). The L4–100K GAR region contains three RGG tripeptide motifs that are conserved among various types in species HAdV-B, –C and –F as well as in PG I strain RI-67 (Iacovides et al., 2007). Mutations of these arginines in HAdV-C5 L4–100K interfere with protein interactions with late viral transcripts, possibly disrupt the role of L4–100K in hexon trimerization, and prevent shuttling of L4–100K to the nucleus, ultimately resulting in decreased viral replication (Iacovides et al., 2007; Koyuncu and Dobner, 2009). Our analysis identified a 5 amino acid deletion (glycine-glycine-glycine-arginine-serine) in all PG II strains between amino acid positions 755 and 761 (relative to RI-67) that disrupts the third RGG motif in the RGG domain. In addition, a single glycine insertion at position 743 (relative to RI-67) is encoded in the genomes of all examined strains of PG II creating an additional RGG tripeptide. The genomes of strains #18, 23, 24, 26, 28, 33, 35, 37, 38, and 40–43 also exhibit a glycine to glutamic acid replacement at position 717 that disrupts the consensus for an RGG tripeptide.
Further work is needed to elucidate whether the sequence differences detected at the C-terminus of L4–100K between PG I and PG II strains affect viral replication.
3.3.6. Early region 3
The E3 transcriptional unit comprises gene repertoires that vary considerably among HAdV species HAdV-A through -G. The conserved E3 genes encode non-structural modulators of host responses to infection (Bhat et al., 1986; Gooding and Wold, 1990; Wold et al., 1994, 1995; Burgert and Blusch, 2000; Burgert et al., 2002; Lichtenstein et al., 2004a, 2004b; Windheim et al., 2004). The variable repertoires of species-specific E3 genes are located between the highly conserved E3-gp19K and RIDα, and encode non-structural type I membrane glyco-proteins expressed at early and late times post infection (Hawkins and Wold, 1995; Frietze et al., 2010; Robinson et al., 2011). In the same location as HAdV-C E3–11.6K encoding the adenovirus death protein (ADP) (Tollefson et al., 1996), the E3 region of members of species HAdV-E encodes 2 or 3 CR1 genes (Davison et al., 2003a, 2003b) of unknown function. As shown schematically in Fig. 6A, while the SAdVs in HAdV-E genomes encode 3 CR1 genes designated CR1β, CR1γ, and CR1δ (Dehghan et al., 2013; Jacobs et al., 2004; Purkayastha et al., 2005), CR1γ is absent in the genomes of both HAdV-E4 phylogroups. Interestingly, as originally reported (Jacobs et al., 2004), a vestigial E3 CR1γ sequence lacking an initiating ATG and splice acceptor site is present in the genomes of strains of PG I due to a 326 bp deletion relative to the SAdV-26 genome. We identified in PG II genomes a unique 318 bp deletion in the 5’ region of the vestigial E3 CR1γ sequence. Additionally, while the splice acceptor sequence present in SAdV-26 is retained in these genomes, a mutation (ATG to ATA) ablates the start codon. Although unlikely to be expressed, a short 165 bp ORF evolutionarily unrelated to CR1γ annotated as E3–6.3K (Burgert et al., 2002; Li and Wold, 2000) is present in this region in the genomes of PG I strains. As a result of a deletion introducing an early stop codon, the E3–6.3K ORF is significantly truncated in the genomes of PG II strains (data not shown).
Fig. 6. Genetic content and diversity in the E3 region among members of species HAdV-E.

A. Schematic comparing the genetic content of the E3 region for SAdV-26 and strains of HAdV-E4 in phylogroups I and II. B. Alignment of amino acid sequences of E3 CR1β and E3-CR1δ. Amino acid differences between phylogroups are highlighted in blue. The transmembrane domain of the predicted type I membrane proteins is highlighted in light purple.
Marked amino acid sequence differences resulting from point mutations as well as from insertions or deletions (indels) in the N-terminal ectodomains of E3 CR1β and E3 CR1δ (Fig. 6B) distinguish the genomes of PG I and PG II and highlight the divergence of these two ORFs from those encoded by SAdV-26. These genes have very low sequence similarity to any other genes in the NCBI database, thus making sequence-based prediction of biological activity and function challenging. Using an extracellular protein array, Martinez-Martin and colleagues recently showed the interaction of the ectodomain of E3–24.8K/CR1β encoded by PG I strain RI-67 with the inhibitory receptor LILRB1, suggesting a possible immunomodulatory function for this protein (Martinez-Martin et al., 2016). No interactions were detected for E3–29.7K/CR1δ.
4. Conclusions and insights towards a more meaningful strain designation for HAdV-E4 based on genomics data
Our data from the computational analyses of 45 WGS strains of HAdV-E4 representing the spectrum of intratypic genetic variability described to date, indicate that the two phylogroups of HAdV-E4 have been circulating and evolving independently from a common ancestor, presumably a simian adenovirus as was suggested by the close genetic relationship to SAdVs in HAdV-E (Roy et al., 2009). The genomic differences between PG I and PG II identified in this study are strongly indicative of a genetic basis for probable differences in pathogenesis and fitness between the two separable evolutionary lineages. Data from molecular epidemiology studies of both respiratory and ocular disease associated with HAdV-E4 infection (Kajon et al., 2007, 2018; Rogers et al., 2019; Cooper et al., 1993; Ren et al., 1985) show that both lineages have been in circulation over the last three decades with a noticeable predominance of PG II strains among examined clinical isolates. This supports the hypothesis of a potential selective advantage and/or of an increased virulence for this clade.
The shift in the last decade towards molecular diagnosis of viral infections and the growing capabilities for molecular typing of virus strains from original clinical specimens create an opportunity for the development of assays that could discriminate PGs I and II, thus overcoming the challenges posed by the costly and labor-intensive genome typing by in silico or gel-based RFLP. The International Committee on Taxonomy of Viruses (ICTV) provides no guidelines for the classification or designation of viruses beyond the species level. The use of a designation that could reflect both the unique genetic and associated phenotypic characteristics of any given HAdV-E4 strain would be extremely informative for epidemiological and functional studies of HAdV-E4 infection and associated disease.
We propose the use of the term phylogroups I and II in the designation of HAdV-E4 strains when molecular typing data are available. The basic designation HAdV-E4 PG I or HAdV-E4 PG II will better reflect the distinct genomic characteristics identified in the present work and those reported in other studies.
The implications of the identified genetic differences between phylogroups for viral pathogenesis and fitness, and the value of phylogrouping when typing clinical isolates of HAdV-E4 merits further investigation. Moving forward, the following in vitro and in vivo phenotypes should be considered for comparison between phylogroups: serological reactivities, replication and viral progeny release kinetics, plaque size, proinflammatory responses induced by infection in cell culture, and pulmonary inflammation in rodent models of HAdV respiratory infection.
Supplementary Material
Acknowledgements
The authors wish to thank Professors Koki Aoki and Nobuyoshi Kitaichi for their support and encouragement of this collaborative effort. C.R.B. is supported by the University of New Mexico Infectious Diseases and Inflammation NIH Training Grant T32-AI007538.
Abbreviations:
- HAdV
human adenovirus
- HAdV-E
Human mastadenovirus E
- HAdV-E4
human adenovirus type 4
- SAdV
simian adenovirus
- NHP
non-human primates
- RFLP
restriction fragment length polymorphism
- NGS
next-generation sequencing
- WGS
whole genome sequences
- CDS
coding sequences
- ORF
open reading frames
- tMRCA
time to the most recent common ancestor
- PG I
phylogroup I
- PG II
phylogroup II
- PIC
phylogenetic independent contrasts
- %G + C
percentage of genomic guanine-cytosine
- 95% HPD
95% highest posterior density range
- ITR
inverted terminal repeats
- E3
early region 3
- E1A
early region 1A
- E1B
early region 1B
- VA RNA
virus-associated RNA
- L4
late region 4
Footnotes
Declaration of interests
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.virol.2019.08.028.
References
- Adrian T, 1992. Genome type Analysis of adenovirus type-4. Intervirology 34, 180–183. 10.1159/000150280. [DOI] [PubMed] [Google Scholar]
- Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, Akusjarvi G, 2005. Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol 79, 9556–9565. 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade F, Bull HG, Thornberry NA, Ketner GW, Casciola-Rosen LA, Rosen A, 2001. Adenovirus L4–100K assembly protein is a granzyme B substrate that potently inhibits granzyme B-mediated cell death. Immunity 14, 751–761. 10.1016/S1074-7613(01)00149-2. [DOI] [PubMed] [Google Scholar]
- Aoki K, Kato M, Ohtsuka H, Ishii K, Nakazono N, Sawada H, 1982. Clinical and aetiological study of adenoviral conjunctivitis, with special reference to adenovirus types 4 and 19 infections. Br. J. Ophthalmol 66, 776–780. 10.1136/bjo.66.12.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov N, Kajon AE, Hoeben RC, Mymryk JS, 2004. Comprehensive sequence analysis of the E1A proteins of human and simian adenoviruses. Virology 329, 477–492. 10.1016/j.virol.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Benko M, Harrach B, Both G, Russell W, Adair B, Adam E, De Jong J, Hess M, Johnson M, Kajon A, 2005. In: Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (Eds.), Adenoviridae Virus Taxonomy Eighth Report of the International Committee on the Taxonomy of Viruses. Elsevier Academic press, San Diego, CA, pp. 213–228. [Google Scholar]
- Berk AJ, 2005. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 24, 7673–7685. 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- Bhat BM, Brady HA, Pursley MH, Wold WS, 1986. Deletion mutants that alter differential RNA processing in the E3 complex transcription unit of adenovirus. J. Mol. Biol 190, 543–557. 10.1016/0022-2836(86)90240-8. [DOI] [PubMed] [Google Scholar]
- Blackford AN, Grand RJA, 2009. Adenovirus E1B 55-Kilodalton protein: multiple roles in viral infection and cell transformation. J. Virol 83, 4000–4012. 10.1128/JVI.02417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc RS, Richard S, 2017. Arginine methylation: the coming of age. Mol. Cell 65, 8–24. 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Bos JL, Polder LJ, Bernards R, Schrier PI, van den Elsen PJ, van der Eb AJ, van Ormondt H, 1981. The 2.2 kb E1b mRNA of human Ad12 and Ad5 codes for two tumor antigens starting at different AUG triplets. Cell 27, 121–131. 10.1016/0092-8674(81)90366-4. [DOI] [PubMed] [Google Scholar]
- Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ, 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol 10, e1003537 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge E, Ketner G, 1990. Interaction of adenoviral E4 and E1b products in late gene expression. Virology 174, 345–353. 10.1016/0042-6822(90)90088-9. [DOI] [PubMed] [Google Scholar]
- Burgert HG, Blusch JH, 2000. Immunomodulatory functions encoded by the E3 transcription unit of adenoviruses. Virus Genes 21, 13–25. [PubMed] [Google Scholar]
- Burgert HG, Ruzsics Z, Obermeier S, Hilgendorf A, Windheim M, Elsing A, 2002. Subversion of host defense mechanisms by adenoviruses. Curr. Top. Microbiol. Immunol 269, 273–318. 10.1007/978-3-642-59421-2_16. [DOI] [PubMed] [Google Scholar]
- Cooper RJ, Bailey AS, Killough R, Richmond SJ, 1993. Genome analysis of adenovirus 4 isolated over a six year period. J. Med. Virol 39, 62–66. 10.1002/jmv.1890390112. [DOI] [PubMed] [Google Scholar]
- Crawford-Miksza LK, Schnurr DP, 1996. Adenovirus serotype evolution is driven by illegitimate recombination in the hypervariable regions of the hexon protein. Virology 224, 357–367. 10.1006/viro.1996.0543. [DOI] [PubMed] [Google Scholar]
- Crawford-Miksza LK, Nang RN, Schnurr DP, 1999. Strain variation in adenovirus serotypes 4 and 7a causing acute respiratory disease. J. Clin. Microbiol 37, 1107–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuconati A, White E, 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev 16, 2465–2478. 10.1101/gad.1012702. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D, 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9 10.1038/nmeth.2109. 772–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison AJ, Akter P, Cunningham C, Dolan A, Addison C, Dargan DJ, Hassan-Walker AF, Emery VC, Griffiths PD, Wilkinson GW, 2003a. Homology between the human cytomegalovirus RL11 gene family and human adenovirus E3 genes. J. Gen. Virol 84, 657–663. 10.1099/vir.0.18856-0. [DOI] [PubMed] [Google Scholar]
- Davison AJ, Benko M, Harrach B, 2003b. Genetic content and evolution of adenoviruses. J. Gen. Virol 84, 2895–2908. 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- Dehghan S, Seto J, Liu EB, Walsh MP, Dyer DW, Chodosh J, Seto D, 2013. Computational analysis of four human adenovirus type 4 genomes reveals molecular evolution through two interspecies recombination events. Virology 443, 197–207. 10.1016/j.virol.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra A, Hage E, Ganzenmueller T, Bottcher S, Hofmann J, Hamprecht K, Obermeier P, Rath B, Hausmann F, Dobner T, Heim A, 2019. Molecular evolution of human adenovirus (HAdV) species C. Sci. Rep 9, 1039 10.1038/s41598-018-37249-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A, Shapiro B, Pybus OG, 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol 22, 1185–1192. 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Ho SY, Phillips MJ, Rambaut A, 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol 4, e88 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S, Shackelton LA, Holmes EC, 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet 9, 267–276. 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE, 2009. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc. Natl. Acad. Sci. U. S. A 106, 13260–13265. 10.1073/pnas.0906770106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth C, Kitchen A, Shapiro B, Suchard MA, Holmes EC, Rambaut A, 2010. Using time-structured data to estimate evolutionary rates of double-stranded DNA viruses. Mol. Biol. Evol 27, 2038–2051. 10.1093/molbev/msq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frietze KM, Campos SK, Kajon AE, 2010. Open reading frame E3–10.9K of subspecies B1 human adenoviruses encodes a family of late orthologous proteins that vary in their predicted structural features and subcellular localization. J. Virol 84, 11310–11322. 10.1128/JVI.00512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding LR, Wold WS, 1990. Molecular mechanisms by which adenoviruses counteract antiviral immune defenses. Crit. Rev. Immunol 10, 53–71 [PubMed] [Google Scholar]
- Hang J, Vento TJ, Norby EA, Jarman RG, Keiser PB, Kuschner RA, Binn LN, 2017. Adenovirus type 4 respiratory infections with a concurrent outbreak of coxsackievirus A21 among United States Army Basic Trainees, a retrospective viral etiology study using next-generation sequencing. J. Med. Virol 89, 1387–1394. 10.1002/jmv.24792. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Gonzalez G, Harada S, Oosako H, Hanaoka N, Hinokuma R, Fujimoto T, 2018. Recombinant type human mastadenovirus D85 associated with epidemic keratoconjunctivitis since 2015 in Japan. J. Med. Virol 90, 881–889. 10.1002/jmv.25041. [DOI] [PubMed] [Google Scholar]
- Hatfield L, Hearing P, 1993. The Nfiii/Oct-1 binding-site stimulates adenovirus DNA-replication in vivo and is functionally redundant with adjacent sequences. J. Virol 67, 3931–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins LK, Wold WS, 1995. The E3–20.5K membrane protein of subgroup B human adenoviruses contains O-linked and complex N-linked oligosaccharides. Virology 210, 335–344. 10.1006/viro.1995.1350. [DOI] [PubMed] [Google Scholar]
- Hay RT, 1985. Origin of adenovirus DNA replication. Role of the nuclear factor I binding site in vivo. J. Mol. Biol 186, 129–136. 10.1016/0022-2836(85)90263-3. [DOI] [PubMed] [Google Scholar]
- Hayes BW, Telling GC, Myat MM, Williams JF, Flint SJ, 1990. The adenovirus L4 100-Kilodalton protein is necessary for efficient translation of viral late messenger-Rna species. J. Virol 64, 2732–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SS, Szolajska E, Schoehn G, Franqueville L, Myhre S, Lindholm L, Ruigrok RW, Boulanger P, Chroboczek J, 2005. The 100K-chaperone protein from adenovirus serotype 2 (Subgroup C) assists in trimerization and nuclear localization of hexons from subgroups C and B adenoviruses. J. Mol. Biol 352, 125–138. 10.1016/j.jmb.2005.06.070. [DOI] [PubMed] [Google Scholar]
- Houng HS, Clavio S, Graham K, Kuschner R, Sun W, Russell KL, Binn LN, 2006. Emergence of a new human adenovirus type 4 (Ad4) genotype: identification of a novel inverted terminal repeated (ITR) sequence from majority of Ad4 isolates from US military recruits. J. Clin. Virol 35, 381–387. 10.1016/j.jcv.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Iacovides DC, O’Shea CC, Oses-Prieto J, Burlingame A, McCormick F, 2007. Critical role for arginine methylation in adenovirus-infected cells. J. Virol 81, 13209–13217. 10.1128/JVI.01415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SC, Davison AJ, Carr S, Bennett AM, Phillpotts R, Wilkinson GW, 2004. Characterization and manipulation of the human adenovirus 4 genome. J. Gen. Virol 85, 3361–3366. 10.1099/vir.0.80386-0. [DOI] [PubMed] [Google Scholar]
- Kajan GL, Lipiec A, Bartha D, Allard A, Arnberg N, 2018. A multigene typing system for human adenoviruses reveals a new genotype in a collection of Swedish clinical isolates. PLoS One 13, e0209038 10.1371/journal.pone.0209038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajon AE, Moseley JM, Metzgar D, Huong HS, Wadleigh A, Ryan MAK, Russell KL, 2007. Molecular epidemiology of adenovirus type 4 infections in US military recruits in the postvaccination era (1997–2003). J. Infect. Dis 196, 67–75. 10.1086/518442. [DOI] [PubMed] [Google Scholar]
- Kajon AE, Lamson DM, Bair CR, Lu XY, Landry ML, Menegus M, Erdman DD, St George K, 2018. Adenovirus type 4 respiratory infections among civilian adults, Northeastern United States, 2011–2015. Emerg. Infect. Dis 24, 201–209. 10.3201/eid2407.180137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalimuddin S, Chan YFZ, Wu IQ, Tan QL, Murthee KG, Tan BH, Oon LLE, Yang Y, Lin RTP, Joseph U, Sessions OM, Smith GJD, Ooi EE, Low JGH, 2017. A report of adult human adenovirus infections in a tertiary hospital. Open Forum Infect Dis 4 10.1093/ofid/ofx053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel R, Srinivasan A, D’Agata EMC, Lu XY, Erdman D, Jhung M, 2010. Outbreak of adenovirus type 4 infection in a long-term care facility for the Elderly. Infect. Control Hosp. Epidemiol 31, 755–757. 10.1086/653612. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM, 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd AH, Garwicz D, Oberg M, 1995. Human and simian adenoviruses: phylogenetic inferences from analysis of VA RNA genes. Virology 207, 32–45. 10.1006/viro.1995.1049. [DOI] [PubMed] [Google Scholar]
- Koyuncu OO, Dobner T, 2009. Arginine methylation of human adenovirus type 5 L4 100-Kilodalton protein is required for efficient virus production. J. Virol 83, 4778–4790. 10.1128/JVI.02493-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG, 2007. Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Li QG, Wadell G, 1988. The degree of genetic variability among adenovirus type 4 strains isolated from man and chimpanzee. Arch. Virol 101, 65–77. 10.1007/bf01314652. [DOI] [PubMed] [Google Scholar]
- Li Y, Wold WS, 2000. Identification and characterization of a 30K protein (Ad4E3–30K) encoded by the E3 region of human adenovirus type 4. Virology 273, 127–138. 10.1006/viro.2000.0384. [DOI] [PubMed] [Google Scholar]
- Lichtenstein DL, Toth K, Doronin K, Tollefson AE, Wold WS, 2004a. Functions and mechanisms of action of the adenovirus E3 proteins. Int. Rev. Immunol 23, 75–111 [DOI] [PubMed] [Google Scholar]
- Lichtenstein DL, Doronin K, Toth K, Kuppuswamy M, Wold WSM, Tollefson AE, 2004b. Adenovirus E3–6.7K protein is required in conjunction with the E3-RID protein complex for the internalization and degradation of TRAIL receptor 2. J. Virol 78, 12297–12307. 10.1128/JVI.78.22.12297-12307.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC, 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol 73, 152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Martin N, Ramani SR, Hackney JA, Tom I, Wranik BJ, Chan M, Wu J, Paluch MT, Takeda K, Hass PE, Clark H, Gonzalez LC, 2016. The extracellular interactome of the human adenovirus family reveals diverse strategies for immunomodulation. Nat. Commun 7, 11473 10.1038/ncomms11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölsa M, Hemmila H, Ronkko E, Virkki M, Nikkari S, Ziegler T, 2016. Molecular characterization of adenoviruses among Finnish military conscripts. J. Med. Virol 88, 571–577. 10.1002/jmv.24364. [DOI] [PubMed] [Google Scholar]
- Montell C, Courtois G, Eng C, Berk A, 1984. Complete Transformation by adenovirus-2 requires both E1a-proteins. Cell 36, 951–961. 10.1016/0092-8674(84)90045-x. [DOI] [PubMed] [Google Scholar]
- Muhire BM, Varsani A, Martin DP, 2014. SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9, e108277 10.1371/journal.pone.0108277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mul YM, Verrijzer CP, van der Vliet PC, 1990. Transcription factors NFI and NFIII/oct-1 function independently, employing different mechanisms to enhance adenovirus DNA replication. J. Virol 64, 5510–5518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzzi A, Rocchi G, Lumbroso B, Tosato G, Barbieri F, 1975. Acute haemorrhagic conjunctivitis during an epidemic outbreak of adenovirus-type-4 injection. Lancet 2, 822–823. 10.1016/s0140-6736(75)80061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narra R, Bono P, Zoccoli A, Orlandi A, Piconi S, Grasselli G, Crotti S, Girello A, Piralla A, Baldanti F, Lunghi G, 2016. Acute respiratory distress syndrome in adenovirus type 4 pneumonia: a case report. J. Clin. Virol 81, 78–81. 10.1016/j.jcv.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Pruijn GJM, Vandriel W, Vandervliet PC, 1986. Nuclear factor-Iii, a novel sequence-specific DNA-binding protein from Hela-cells stimulating adenovirus DNA-replication. Nature 322, 656–659. 10.1038/322656a0. [DOI] [PubMed] [Google Scholar]
- Purkayastha A, Ditty SE, Su J, McGraw J, Hadfield TL, Tibbetts C, Seto D, 2005. Genomic and bioinformatics analysis of HAdV-4, a human adenovirus causing acute respiratory disease: implications for gene therapy and vaccine vector development. J. Virol 79, 2559–2572. 10.1128/JVI.79.4.2559-2572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Lam TT, Carvalho LM, Pybus OG, 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren CS, Nakazono N, Ishida S, Fujii S, Yoshii T, Yamazaki S, Ishii K, Fujinaga K, 1985. Genome type Analysis of adenovirus type-4 isolates, recently obtained from clinically different syndromes in some areas in Japan. Jpn. J. Med. Sci. Biol 38, 195–199 [DOI] [PubMed] [Google Scholar]
- Robinson CM, Rajaiya J, Zhou XH, Singh G, Dyer DW, Chodosh J, 2011. The E3 CR1-gamma gene in human adenoviruses associated with epidemic keratoconjunctivitis. Virus Res 160, 120–127. 10.1016/j.virusres.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AE, Lu X, Killerby M, Campbell E, Gallus L, Kamau E, Froh IB, Nowak G, Erdman DD, Sakthivel SK, Gerber SI, Schneider E, Watson JT, Johnson LA, 2019. Outbreak of acute respiratory illness associated with adenovirus type 4 at the U.S. Naval academy. 2016. MSMR 26, 21–27 [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP, 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol 61, 539–542. 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld PJ, Oneill EA, Wides RJ, Kelly TJ, 1987. Sequence-specific interactions between cellular DNA-binding proteins and the adenovirus origin of DNA-replication. Mol. Cell. Biol 7, 875–886. 10.1128/mcb.7.2.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Vandenberghe LH, Kryazhimskiy S, Grant R, Calcedo R, Yuan X, Keough M, Sandhu A, Wang Q, Medina-Jaszek CA, Plotkin JB, Wilson JM, 2009. Isolation and characterization of adenoviruses persistently shed from the gastrointestinal Tract of non-human primates. PLoS Pathog 5 10.1371/journal.ppat.1000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Gonzalez G, Wada Y, Setiyono A, Handharyani E, Rahmadani I, Taha S, Adiani S, Latief M, Kholilullah ZA, 2016. Divergent bufavirus harboured in megabats represents a new lineage of parvoviruses. Sci. Rep 6, 24257 10.1038/srep24257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepetiuk SK, Norton R, Kok T, Irving LG, 1993. Outbreak of adenovirus type 4 conjunctivitis in South Australia. J. Med. Virol 41, 316–318. 10.1002/jmv.1890410411. [DOI] [PubMed] [Google Scholar]
- Team RC, 2013. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria: Available online at: https://www.R-project.org/. [Google Scholar]
- Thandapani P, O’Connor TR, Bailey TL, Richard S, 2013. Defining the RGG/RG motif. Mol. Cell 50, 613–623. 10.1016/j.molcel.2013.05.021. [DOI] [PubMed] [Google Scholar]
- Tian X, Qiu H, Zhou Z, Wang S, Fan Y, Li X, Chu R, Li H, Zhou R, Wang H, 2018. Identification of a critical and conformational neutralizing epitope in human adenovirus type 4 hexon. J. Virol 92 10.1128/JVI.01643-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollefson AE, Scaria A, Hermiston T, Ryerse JS, Wold LJ, Wold WSM, 1996. The adenovirus death protein (E3–11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol 70, 2296–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki-Wang L, Aoki K, Isobe K, Shiao S, Toba K, Kobayashi N, Noguchi Y, Ohno S, 1997. Genome analysis of adenovirus type 4 strains isolated from acute conjunctivitis in Japan. Jpn. J. Ophthalmol 41, 308–311 [DOI] [PubMed] [Google Scholar]
- Tullo AB, Higgins PG, 1980. An outbreak of adenovirus type-4 conjunctivitis. Br. J. Ophthalmol 64, 489–493. 10.1002/jmv.1890410411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon VK, Conn GL, 2016. Adenovirus VA RNA: an essential pro-viral non-coding RNA. Virus Res 212, 39–52. 10.1016/j.virusres.2015.06.018. [DOI] [PubMed] [Google Scholar]
- Walsh MP, Seto J, Liu EB, Dehghan S, Hudson NR, Lukashev AN, Ivanova O, Chodosh J, Dyer DW, Jones MS, Seto D, 2011. Computational analysis of two species C human adenoviruses provides evidence of a novel virus. J. Clin. Microbiol 49, 3482–3490. 10.1128/JCM.00156-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HGH, Moran E, Yaciuk P, 1995. E1a promotes association between P300 and prb in multimeric complexes required for normal biological-activity. J. Virol 69, 7917–7924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winberg G, Shenk T, 1984. Dissection of overlapping functions within the adenovirus type-5 E1a gene. EMBO J 3, 1907–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windheim M, Hilgendorf A, Burgert HG, 2004. Immune evasion by adenovirus E3 proteins: exploitation of intracellular trafficking pathways. Curr. Top. Microbiol. Immunol 273, 29–85. 10.1007/978-3-662-05599-1_2. [DOI] [PubMed] [Google Scholar]
- Wold WS, Hermiston TW, Tollefson AE, 1994. Adenovirus proteins that subvert host defenses. Trends Microbiol 2, 437–443. 10.1016/0966-842X(94)90801-X. [DOI] [PubMed] [Google Scholar]
- Wold WS, Tollefson AE, Hermiston TW, 1995. E3 transcription unit of adenovirus. Curr. Top. Microbiol. Immunol 199 (Pt 1), 237–274. 10.1007/978-3-642-79496-4_13. [DOI] [PubMed] [Google Scholar]
- Wunderlich K, van der Helm E, Spek D, Vermeulen M, Gecgel A, Pau MG, Vellinga J, Custers J, 2014. An alternative to the adenovirus inverted terminal repeat sequence increases the viral genome replication rate and provides a selective advantage in vitro. J. Gen. Virol 95, 1574–1584. 10.1099/vir.0.064840-0. [DOI] [PubMed] [Google Scholar]
- Xi Q, Cuesta R, Schneider RJ, 2004. Tethering of eIF4G to adenoviral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev 18, 1997–2009. 10.1101/gad.1212504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitomi H, Sera N, Gonzalez G, Hanaoka N, Fujimoto T, 2017. First isolation of a new type of human adenovirus (genotype 79), species Human mastadenovirus B (B2) from sewage water in Japan. J. Med. Virol 89, 1192–1200. 10.1002/jmv.24749. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Vrancken B, Feng Y, Dellicour S, Yang Q, Yang W, Zhang Y, Dong L, Pybus OG, Zhang H, Tian H, 2017. Cross-border spread, lineage displacement and evolutionary rate estimation of rabies virus in Yunnan Province, China. Virol. J 14, 102 10.1186/s12985-017-0769-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kang JE, Dehghan S, Sridhar S, Lau SKP, Ou JX, Woo PCY, Zhang QW, Seto D, 2019. A survey of recent adenoviral respiratory pathogens in Hong Kong reveals emergent and recombinant human adenovirus type 4 (HAdV-E4) circulating in civilian populations. Viruses-Basel 11 10.3390/v11020129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.