

Abstract
Coronaviridae is a peculiar viral family, with a very large RNA genome and characteristic appearance, endowed with remarkable tendency to transfer from animals to humans. Since the beginning of the 21st century, three highly transmissible and pathogenic coronaviruses have crossed the species barrier and caused deadly pneumonia, inflicting severe outbreaks and causing human health emergencies of inconceivable magnitude. Indeed, in the past two decades, two human coronaviruses emerged causing serious respiratory illness: severe acute respiratory syndrome coronavirus (SARS-CoV-1) and Middle Eastern respiratory syndrome coronavirus (MERS-CoV), causing more than 10,000 cumulative cases, with mortality rates of 10 % for SARS-CoV-1 and 34.4 % for MERS-CoV. More recently, the severe acute respiratory syndrome coronavirus virus 2 (SARS-CoV-2) has emerged in China and has been identified as the etiological agent of the recent COVID-19 pandemic outbreak. It has rapidly spread throughout the world, causing nearly 22 million cases and ∼ 770,000 deaths worldwide, with an estimated mortality rate of ∼3.6 %, hence posing serious challenges for adequate and effective prevention and treatment. Currently, with the exception of the nucleotide analogue prodrug remdesivir, and despite several efforts, there is no known specific, proven, pharmacological treatment capable of efficiently and rapidly inducing viral containment and clearance of SARS-CoV-2 infection as well as no broad-spectrum drug for other human pathogenic coronaviruses. Another confounding factor is the paucity of molecular information regarding the tendency of coronaviruses to acquire drug resistance, a gap that should be filled in order to optimize the efficacy of antiviral drugs.
In this light, the present review provides a systematic update on the current knowledge of the marked global efforts towards the development of antiviral strategies aimed at coping with the infection sustained by SARS-CoV-2 and other human pathogenic coronaviruses, displaying drug resistance profiles. The attention has been focused on antiviral drugs mainly targeting viral protease, RNA polymerase and spike glycoprotein, that have been tested in vitro and/or in clinical trials as well as on promising compounds proven to be active against coronaviruses by an in silico drug repurposing approach. In this respect, novel insights on compounds, identified by structure-based virtual screening on the DrugBank database endowed by multi-targeting profile, are also reported. We specifically identified 14 promising compounds characterized by a good in silico binding affinity towards, at least, two of the four studied targets (viral and host proteins). Among which, ceftolozane and NADH showed the best multi-targeting profile, thus potentially reducing the emergence of resistant virus strains. We also focused on potentially novel pharmacological targets for the development of compounds with anti-pan coronavirus activity. Through the analysis of a large set of viral genomic sequences, the current review provides a comprehensive and specific map of conserved regions across human coronavirus proteins which are essential for virus replication and thus with no or very limited tendency to mutate. Hence, these represent key druggable targets for novel compounds against this virus family. In this respect, the identification of highly effective and innovative pharmacological strategies is of paramount importance for the treatment and/or prophylaxis of the current pandemic but potentially also for future and unavoidable outbreaks of human pathogenic coronaviruses.
Keywords: SARS-CoV-2, Coronavirus, Outbreaks, Antiviral agents, Antiviral resistance, Conservation, RNA polymerase, Protease, Spike, Nucleoside analogs, Protease inhibitors, Entry inhibitors
Introduction
Emerging infectious diseases are occurring at an increasing frequency worldwide, having a profound impact on public health. In the past two decades, two highly pathogenic and transmissible coronaviruses emerged causing serious respiratory illness: severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1, in 2002) and Middle East respiratory syndrome coronavirus (MERS-CoV, in 2012) (Cui et al., 2019). These two viruses had caused more than 10,000 cumulative cases, with mortality rates of 10 % for SARS-CoV-1 and 34.4 % for MERS-CoV, representing the first global threat of the 21st century. The latter continues to cause sporadic cases of severe respiratory illness (https://www.who.int/emergencies/mers-cov/en).
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported on December 2019 from Wuhan City, Hubei, China (Zhu et al., 2020; Lu et al., 2020) and is currently causing major concern in the medical community, as it exhibits faster human-to-human transmission often combined with asymptomatic, or minimally symptomatic course of the infection (Drożdżal et al., 2020). These two factors are perhaps the major contribution of the rapid propagation of the virus. Indeed, SARS-CoV-2 has been declared pandemic, and has been responsible for nearly 22 million cases, with ∼ 770,000 deaths worldwide (GISAID, 16th August 2020), causing a global health emergency of inconceivable magnitude.
Coronaviruses are large (with the largest genomes of all RNA viruses), enveloped, with a very characteristic appearance, positive-sense single-stranded RNA viruses classified into 4 genera: α-, β-, δ- and γ-coronaviruses. Members of the subfamily Coronavirinae are widespread among mammals, often causing only mild respiratory or enteric infections. Over 60 coronaviruses have been isolated from bats (BatCoV) and most of these are in the genus β-coronavirus. The β-coronavirus genus comprises the human SARS-CoV-1 and -2 (belonging to the subgroup 2b), MERS-CoV (subgroup 2c), and HCoV-OC43 and HCoV-HKU1 (subgroup 2a). Conversely, HCoV-229E and HCoV-NL63 belong to the genus α-coronavirus, (subgroup 1b) (Yang and Leibowitz, 2015; Cui et al., 2019; Song et al., 2019).
SARS-CoV-2 is characterized by a large genome with a length of 29,891 nucleotides, encoding 9,860 amino acids for a total of 4 structural proteins and 16 non-structural proteins (NSP) with regulatory functions.
Among these proteins, the 3-chymotrypsin-like protease (3CL-PR), the RNA-dependent-RNA polymerase (RdRp) and the spike protein, have been proposed as druggable targets (Chan et al., 2020; Huang et al., 2020; Liu et al., 2020).
In particular, the 3CL-PR is involved in the polyproteins cleavage, giving rise to viral proteins essential for the life cycle of the virus (Zhang et al., 2020a). This enzyme, sharing structural similarity among coronaviruses, is a homodimer with a catalytic dyad involving a His residue at position 41 and a Cys at position 145 (Huang et al., 2004; Shan et al., 2004). 3CL-PR is composed of two domains I and II (residues 10–99 and 100–182, respectively) forming the substrate-binding site, whereas domain III (residues 198–303) is involved in regulating the dimerization of the two subunits (Shi and Song, 2006; Goyal and Goyal, 2020; Zhang et al., 2020a).
NSP12 is characterized by an N-terminal nidovirus RdRp-associated nucleotidyltransferase (NiRAN) domain, an interface domain, and a C-terminal domain acting as RdRp. This enzyme is crucial for both the replication of the viral genome as well as for the synthesis of viral mRNAs (Gao et al., 2020; Romano et al., 2020; Wu et al., 2020).
RdRp is characterized by the classical architecture of the viral polymerases (Mcdonald, 2013) with three subdomains: a fingers subdomain (residues Leu366 to Ala581 and Lys621 to Gly679), a palm subdomain (residues Thr582 to Pro620 and Thr680 to Gln815), and a thumb subdomain (residues His816 to Phe920) (Gao et al., 2020). The active site resides in the palm subdomain and is composed of at least five conserved motifs defined as A–E (Gao et al., 2020; Hillen et al., 2020). In particular, motif A contains the classic divalent-cation–binding residue Asp618, while motif C contains the catalytic residues Ser759, Asp760 and Asp761.
The Spike is a homotrimeric viral surface glycoprotein, which is critical for virus binding and entry into the target host cell. The spike glycoprotein undergoes a cleavage by host proteases, giving rise to two distinct subunits. The former is the S1 subunit (residues 1–685) containing the receptor binding domain (RBD, residues 319–541), while the latter is the S2 subunit (residues 686–1273) including the fusion peptide (residues 788–806), two heptad repeats (HR1 and HR2, residues 912–984 and 1163–1213), a transmembrane domain (residues 1214–1237), and a cytoplasmic domain (residues 1238–1273, promoting fusion between the viral envelope and plasma membrane of the host cell (Lan et al., 2020; Xia and Liu et al., 2020). SARS‐CoVs use the angiotensin‐converting‐enzyme 2 (ACE2) receptor to bind to the host cell, expressed on diverse respiratory epithelial cells, alveolar macrophages, enterocytes of the small intestine and monocytes (Hamming et al., 2004; Guo et al., 2008; Qi et al., 2020). Following the binding to the cellular receptor, the HR1 and HR2 of the S2 subunit undergo hydrophobic interactions with each other, resulting in the formation of a six-helix bundle (6-HB) fusion core, hence allowing for viral and cellular membranes to be in close proximity for fusion and infection to occur (Bosch et al., 2004). So far, with the exception of the nucleotide analogue prodrug remdesivir, an RdRp inhibitor, there is no other known specific, effective, proven pharmacological treatment for SARS-CoV-2 infection. Researchers all over the world are exploring a large variety of therapeutic approaches in order to discover potential candidates capable of coping with both the current and potential future outbreaks sustained by coronaviruses.
This review provides a comprehensive overview of the current and innovative antiviral strategies targeting viral protease, RdRp and spike glycoprotein, that have been proposed to cope with the infection sustained by SARS-CoV-2 and other pathogenic human coronaviruses. Thanks to the analysis of a large sequence datasets, a detailed information of the conserved regions in 3CL-PR, RdRp and spike across human coronaviruses is also presented. These regions can represent novel druggable targets for compounds with pan anti-coronavirus activity.
Current status on antivirals against SARS Cov-2 and other human pathogenic coronaviruses
Currently, no approved therapies are available against SARS-CoV-2 and other human pathogenic coronaviruses, with the exception of remdesivir which was recently approved on July 2020 by the European Medicine Agency (EMA) in the context of severe COVID-19 (European Medicines Agency, 2020a). Several options can be considered to control or prevent the spread of SARS-CoV-2 infection, including vaccines, convalescent plasma, interferon-based therapies, small-molecule drugs, repurposing of approved drugs, cell-based therapies, monoclonal antibodies, as well as combinations of pharmacologic and non-pharmacologic interventions (Bloch et al., 2020; Cohen, 2020; Li and De Clercq, 2020). However, such interventions are likely to require years of research and development. Given the urgency of blocking the SARS-CoV-2 outbreak at global level, there has been considerable interest in both repurposing of approved antiviral agents and in drug development for the treatment of other infections caused by human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), filoviruses and influenza, based on therapeutic experience with the previous SARS-CoV-1 and MERS-CoV infections (Li and De Clercq, 2020). Several highly conserved regions between different human pathogenic coronaviruses (including SARS-CoV-1 and MERS-CoV) that are of interest for therapeutic interventions have been already identified thanks to the rapid genomic sequencing of SARS-CoV-2. These regions involve key viral enzymes, essential for virus replication such as the 3CL-PR, papain-like protease, and RdRp. Importantly, structural analyses of viral proteins suggest that key drug-binding pockets in these viral enzymes are conserved across SARS-CoV-2, SARS-CoV-1 and MERS-CoV (Morse et al., 2020).
Based on the mentioned considerations, some protease inhibitors and nucleoside analogues have been considered antiviral agents against SARS-CoV-2 (Table 1 ).
Table 1.
Antiviral agents against SARS-CoV-2 under investigation.
| Inhibitor class/Target/Mechanism of action | Drug | Chemical structure | Clinical trials evaluating agents for treatment against SARS-CoV-2a | Indications for the use | Human coronaviruses involved | In vitro activity against coronaviruses | Log P values | pKa values | References |
|---|---|---|---|---|---|---|---|---|---|
| Protease inhibitor | Darunavir | 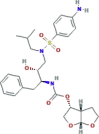 |
Ongoing clinical phase III trials investigating darunavir/ritonavir and darunavir/cobicistat | Approved for treatment against HIV NIH recommends against the use of HIV protease inhibitors for the treatment of COVID-19, except in a clinical trial |
SARS-CoV-2 | It showed no activity against SARS-CoV-2 at clinically relevant concentrations (EC50 >100 μM). These data do not support the use of darunavir for treatment of SARS-CoV-2 | 1.76b 1.82c 1.89d |
13.59e 2.39f |
(Bhimraj et al., 2020; Johnson and Johnson Services, 2020; NIH, 2020) |
| Protease inhibitor | Danoprevir | 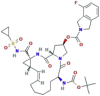 |
Completed two phase IV clinical trials | Approved for treatment of HCV in China | SARS-CoV-2 | No information, | 2.37b 2.55c |
3.77e −3.5f |
(Lythgoe and Middleton, 2020) |
| Protease inhibitor | Lopinavir | 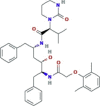 |
Ongoing several phase III trials | Approved for treatment against HIV NIH recommends against the use of Lopinavir/ritonavir for the treatment of COVID-19, except in a clinical trial |
SARS-CoV-2, SARS-CoV-1, MERS-CoV |
In vitro activity against SARS-CoV-1, MERS-CoV and SARS-CoV-2 (EC50 = 26.63 μM) | 3.91b 4.69c |
13.39e −1.5f |
(Wu et al., 2004; Chu et al., 2004; de Wilde et al., 2014; Chan et al., 2015a; Kim et al., 2015; Bin et al., 2016; Totura and Bavari, 2019; Cao et al., 2020; Chen et al., 2020b; Choy et al., 2020; Lythgoe and Middleton, 2020; NIH, 2020) |
| Protease inhibitor In MERS, it inhibits spike-mediated membrane fusion |
Nafamostat | 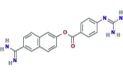 |
Currently undergoing three phase II clinical trials | Approved for anticoagulant therapy in Asian countries | SARS-CoV-2, MERS-CoV, HCoV-229E |
In vitro activity against SARS-CoV-2 (EC50 = 22.50 μM) Reduced titers of HCoV-229E |
1.91b 2.52c |
11.32f | (Wang et al., 2020a; Yamaya et al., 2020) |
| Protease inhibitor | Ritonavir | 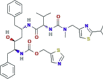 |
Ongoing several phase III trials | Approved for treatment against HIV NIH recommends against the use of Lopinavir/ritonavir for the treatment of COVID-19, except in a clinical trial |
SARS-CoV-2, SARS-CoV-1 |
In vitro activity against SARS-CoV-1, while the antiviral activity is absent against SARS-CoV-2 | 4.24b 5.22c 3.9d |
13.68e 2.84f |
(Wu et al., 2004; Chu et al., 2004; de Wilde et al., 2014; Chan et al., 2015a; Kim et al., 2015; Bin et al., 2016; Totura and Bavari, 2019; Cao et al., 2020; Chen et al., 2020b; Choy et al., 2020; Lythgoe and Middleton, 2020; NIH, 2020) |
| Protease inhibitor | TMC-310911 (ASC-09) |
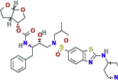 |
Two ongoing clinical trials investigating ASC-09 in combination with lopinavir/ritonavir or ritonavir | It is a novel investigational protease inhibitor that is structurally similar to the currently available darunavir. It is being investigated for use in HIV-1 infections | SARS-CoV-2 | No information | 4.5b 5.32c |
13.46e 9.02f |
(Stellbrink et al., 2014; Lythgoe and Middleton, 2020) |
| Nucleoside reverse transcriptase inhibitor | Azvudine (RO-0622) |
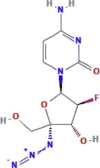 |
Ongoing clinical trials | It is an experimental drug with antiviral activity against HIV, HBV and HCV | SARS-CoV-2 | No information | – | – | (Lythgoe and Middleton, 2020); |
| Nucleoside reverse transcriptase inhibitor | Emtricitabine | 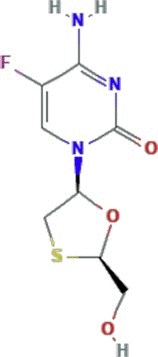 |
Two ongoing clinical trials investigating emtricitabine in combination with tenofovir as an option to combat SARS-CoV-2 | Emtricitabine and tenofovir are approved as treatment combination against HIV | SARS-CoV-2, HCoV-229E |
In vitro activity against HCoV-229E | −0.8b−0.9 c− 0.43d |
2.65d 14.29e −3.1f |
(Lythgoe and Middleton, 2020; Parang et al., 2020) |
| RNA polymerase inhibitor It acts as a prodrug and undergoes ribosylation and phosphorylation intracellularly to become the active favipiravir-RTP. |
Favipiravir (T-705, Avigan) |
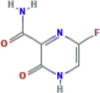 |
Currently undergoing several phase II & III clinical trials | Approved in Japan against influenza | SARS-CoV-2 | Limited in vitro activity against SARS-CoV-2. | 0.49b 0.25c |
9.39e −3.7f |
(Furuta et al., 2017; Choy et al., 2020; Li et al., 2020; Wang et al., 2020a) |
| RNA polymerase inhibitor | Galidesivir (BCX4430) |
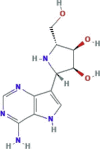 |
One ongoing phase I clinical trial | It has been investigated for use against Zaire Ebolavirus | SARS-CoV-2, SARS-CoV-1, MERS-CoV |
In vitro activity against SARS-CoV-1 and MERS-CoV , but not in SARS-CoV-2 |
−1.2b− 2.1c |
12.95e 8.46f |
(Choy et al., 2020; Li and De Clercq, 2020) |
| RNA polymerase inhibitor It inhibits the action of RNA polymerase: by incorporating into RNA, additional nucleotides cannot be added, terminating RNA transcription |
Remdesivir (GS-5734) |
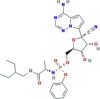 |
Currently undergoing several phase III trials Compassionate use protocol Expanded access |
Approved for emergency use in treating patients infected with SARS-CoV-2 by FDA, in India, Taiwan and Singapore EMA approved |
SARS-CoV-2, SARS-CoV-1, MERS-CoV, HCoV-NL63 |
In vitro activity against SARS-CoV-2, SARS-CoV-1, MERS-CoV. In general, potential effective pan-CoV antiviral It potently blocks SARS-CoV-2 at low-micromolar concentration (EC50 = 0.77 μM) and shows high safety index (>129.87) |
2.2b 2.0c |
10.23e 0.65f |
(Warren et al., 2016; Sheahan et al., 2017; Lo et al., 2017; Agostini et al., 2018; Beigel et al., 2020; Wang et al., 2020a; NIH, 2020; Parang et al., 2020; Wang et al., 2020c; Choy et al., 2020; European Medicines Agency, 2020a, 2020b; Goldman et al., 2020; Grein et al., 2020; Holshue et al., 2020; Li and De Clercq, 2020) |
| Guanosine analogue inhibitor It inhibits viral RNA synthesis as well as mRNA capping, and induces RNA mutations |
Ribavirin | 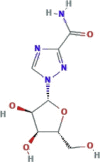 |
Currently undergoing phase III clinical trials evaluation in combination a pegylated interferon | Approved for treatment against HCV and RSV | SARS-CoV-2, SARS-CoV-1, MERS-CoV, HCoV-OC43 | Limited in vitro activity (if any) against SARS-CoV-1, MERS-CoV and SARS-CoV-2 |
−1.9b−2.8 c− 1.85d |
5.1d 11.88e −1.2f |
(Ferron et al., 2018; Totura and Bavari, 2019; Choy et al., 2020; Li and De Clercq, 2020; Wang et al., 2020a; NIH, 2020) |
| Nucleotide reverse transcriptase inhibitor | Tenofovir | 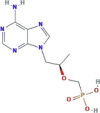 |
Two ongoing clinical trials investigating tenofovir in combination with emtricitabine | Approved for treatment against HIV and HBV | SARS-CoV-2, HCoV-229E |
In vitro activity against HCoV-229E | −1.5b−3.4 c− 1.6d |
3.8&6.7d 1.35e 3.75f |
(Lythgoe and Middleton, 2020; Parang et al., 2020) |
| Polymerase acidic endonuclease inhibitor It blocks the transcription of mRNA by inhibiting the activity of endonuclease |
Baloxavir marboxil (Xofluza) | 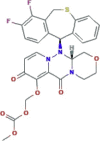 |
Ongoing clinical trials approved by the Chinese Clinical Trial Registry (ChiCTR) | Approved for influenza | SARS-CoV-2 | Limited in vitro activity against SARS-CoV-2 (EC50 > 50 μM) | 2.12b 3.38c 2.24d |
−0.6f | (Lythgoe and Middleton, 2020; Wang et al., 2020b) |
| Ribonucleoside analogue inducing mutations in RNA virions It is phosphorylated in tissue to the active 5’-triphosphate form, which is incorporated into the genome of new virions, resulting in the accumulation of inactivating mutations, known as viral error catastrophe |
β-d-N4-hydroxycytidine (NHC, EIDD-1931); EID 2801 prodrug |
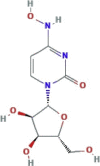 |
No clinical trials available | In experimental phase Therapeutic administration of EIDD-2801, an orally bioavailable NHC prodrug (β-d-N4- hydroxycytidine-5′-isopropyl ester) |
SARS-CoV-2, SARS-CoV-1, MERS-CoV, zoonotic group 2b or 2c Bat-CoVs |
In vitro activity against SARS-CoV-2, other human coronaviruses and bat coronaviruses | −2b− 2.7c |
12.55e 2.39f |
(Hampton, 2020; Sheahan et al., 2020) |
| Neuraminidase inhibitor | Oseltamivir (Tamiflu) | 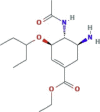 |
Ongoing randomized clinical trials | Approved for influenza A/B (both for treatment and prophylaxis) | SARS-CoV-2, MERS-CoV |
No in vitro activity against SARS-CoV-2 | 1.3b 1.16c 1 C |
14.03e 9.31f |
(Choy et al., 2020; Lythgoe and Middleton, 2020; NIH, 2020) |
| S protein/ACE2 membrane fusion inhibitor It prevents the fusion of the virus with the target membrane and blocks the entry of the virus into the target cell |
Umifenovir (Arbidol) |
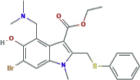 |
Ongoing randomized clinical trials | Available in Russia and China against influenza | SARS-CoV-2, SARS-CoV-1 |
It efficiently inhibits infection with SARS-CoV-2 in vitro (EC50 = 4.11 μM) |
4.97b 3.75c |
6.01e 9.87f |
(Blaising et al., 2014; Lu, 2020; Lythgoe and Middleton, 2020; Wang et al., 2020b, d) |
| Viral entry inhibitor It inhibits terminal glycosylation of ACE2. ACE2 that is not in the glycosylated state may less efficiently interact with the SARS-CoV-2 spike protein, further inhibiting viral entry |
Chloroquine phosphate (Aralen/ generic) |
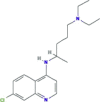 |
Ongoing randomized clinical trials | Approved for the treatment of malaria It was granted an FDA Emergency Use Authorization against SARS-CoV-2 (see Cloroquine). |
SARS-CoV-2, SARS-CoV-1, MERS-CoV, HCoV-229E, HCoV-OC43 |
In vitro activity against several human coronaviruses It potently blocks SARS-CoV-2 at low-micromolar concentration (EC50 = 1.13; μM 5.47 μM) with high safety index (>88.50) |
5.28b 3.93c 4.63d |
10.1d −4.3e 10.32f |
(Vincent et al., 2005; Plantone and Koudriavtseva, 2018; Wang et al., 2020a; NIH, 2020; WHO. COVID-NMA project. Cochrane, 2020)(Yao et al., 2020); |
| Viral entry inhibitor Same mechanism of action of chloroquine |
Hydroxychloroquine sulfate (Plaquenil/ generic) |
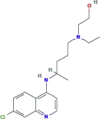 |
Ongoing randomized clinical trials | Approved for the treatment of malaria It was granted an FDA Emergency Use Authorization against SARS-CoV-2 (see Cloroquine). |
SARS-CoV-2 | It potently blocks SARS-CoV-2 at low-micromolar concentration (EC50 = 0.72 μM) | 3.87b 2.89c |
9.67d 15.59e 9.76f |
(Plantone and Koudriavtseva, 2018; Boulware et al., 2020; Gautret et al., 2020; NIH, 2020; WHO. COVID-NMA project. Cochrane, 2020; Yao et al., 2020) |
| Viral entry inhibitor An algae-derived lectin, a potent viral entry inhibitor. A carbohydrate-binding protein made of 121 amino acids, 12.7 kDa It binds to the SARS-CoV spike glycoprotein, thus inhibiting viral entry PDB code: 2GTY Ref: DOI: 10.1016/j.str.2006.05.017 |
Griffithsin | SLTHRKFGGSGGSP FSGLSSIAVRSGSYL DXIIIDGVHHGGSG GNLSPTFTFGSGEYI SNMTIRSGDYIDNIS FETNMGRRFGPYG GSGGSANTLSNVK VIQINGSAGDYLDS LDIYYEQY 
|
No clinical trials available | Phase 1 studies for the prevention of HIV transmission In experimental phase |
SARS-CoV-2, SARS-CoV-1 |
In vitro activity against SARS-CoVs | – | – | (Imai et al., 2005; O’Keefe et al., 2010; Li and De Clercq, 2020; Lusvarghi and Bewley, 2016) |
| Viral entry inhibitor It inhibits the low pH cleavage of the viral spike protein and the continuation of virus replication cycle |
Teicoplanin | 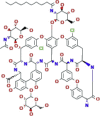 |
An ongoing phase III clinical trial approved by the Iranian Registry of Clinical Trials (IRCT) | Antibiotic commonly used to treat Gram‐positive bacterial infections. It showed efficacy against various viruses such as Ebola virus, influenza virus, flavivirus, HCV, HIV, as well as coronaviruses such as MERS-CoV and SARS-CoV-1 | SARS-CoV-2, SARS-CoV-1, MERS-CoV |
It potently inhibits SARS-CoV-1, MERS-CoV |
– | – | (Zhou et al., 2016; Baron et al., 2020; Li and De Clercq, 2020) |
| Viral entry inhibitor EK1C4 is one of a series of lipopeptides derived from EK1 It is a potent fusion inhibitor against the protein-mediated membrane fusion of SARS-CoV-2 and other coronaviruses |
EK1C4 | SLDQINVTFLDLEY EMKKLEEAIKKLEE SYIDLKEL-GSGSG- PEG4-Chol 3D structure not available |
No clinical trials available | In experimental phase | SARS-CoV-2, SARS-CoV-1, MERS-CoV, HCoV-OC43, HCoV-NL63, HCoV-229E |
It potently inhibits SARS-CoV-2 (IC50 = 1.3 nM), SARS-CoV-1, MERS-CoV and other human coronaviruses |
– | – | (Xia et al., 2019, 2020) |
Abbreviations: ACE2 = angiotensin-converting enzyme 2 gene; EC50 = half-maximal effective concentration; EMA = European Medicines Agency; FDA = US Food & Drug Administration; HBV = hepatitis B virus; HCV = hepatitis C virus; HIV = human immunodeficiency virus; MERS-CoV = Middle Eastern respiratory syndrome coronavirus; NIH = National Institutes of Health; RdRp = RNA-dependent RNA polymerase; RNA = ribonucleic acid; RSV = respiratory syncytial virus; 3CL-PR = 3CL-protease; SARS-CoV-1 = severe acute respiratory syndrome coronavirus; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Clinical trials focused of drugs against SARS-CoV-2 infected patients available at https://clinicaltrials.gov/ (accessed on 12 August 2020).
Predicted properties by ALOGPS (available at https://www.drugbank.ca/drugs; accessed on 12 August 2020).
Predicted properties by ChemAxon (available at https://www.drugbank.ca/drugs; accessed on 12 August 2020).
Exprimental properties (available at https://www.drugbank.ca/drugs; accessed on 12 August 2020).
Strongest acidic and f basic pKa value predicted by ALOGPS (available at https://www.drugbank.ca/drugs; accessed on 12 August 2020).
Protease inhibitors
Several clinical trials are ongoing to test whether protease inhibitors such as lopinavir, ritonavir and darunavir used for HIV treatment, or danoprevir used for HCV treatment, are also effective against SARS-CoV-2 (Table 1) (Lythgoe and Middleton, 2020; https://clinicaltrials.gov/; accessed on 12 August 2020).
The combination of lopinavir/ritonavir is the most common exploratory antiviral regimen against SARS-CoV-2, appearing in 81 investigational studies (https://clinicaltrials.gov/; accessed on 12 August 2020). Lopinavir is a potent antiretroviral protease inhibitor used in combination with ritonavir to treat patients with HIV infection (Croxtall and Perry, 2010). Although ritonavir was initially developed as an HIV protease inhibitor, it is now almost exclusively used at a low dose as a pharmacokinetic booster to increase levels of other HIV protease inhibitors, including lopinavir, through inhibition of the Cytochrome P450 3A4 pathway (Hull and Montaner, 2011). In vitro studies showed activity of lopinavir/ritonavir against the 3CL-PR of SARS-CoV-1 (Wu et al., 2004). During the first SARS epidemic, the administration of lopinavir/ritonavir in combination with ribavirin in SARS-CoV-1-infected individuals, was associated with decreased viral load, decreased adverse clinical outcomes, acute respiratory distress syndrome, or death, when compared with historical control cases (Chu et al., 2004). The in vitro activity of lopinavir/ritonavir was also demonstrated against MERS-CoV (de Wilde et al., 2014). In the marmoset model of MERS-CoV infection, the oral administration of lopinavir/ritonavir resulted in a modest improvement of MERS disease symptoms including decreased pulmonary infiltrates, decreased interstitial pneumonia, and decreased weight loss (Chan et al., 2015a). In MERS-CoV-infected patients treated with regimens containing lopinavir/ritonavir, positive disease outcomes including defervescence, viral clearance from serum and sputum, and survival, were observed (Kim et al., 2015; Bin et al., 2016). Lopinavir/ritonavir has been also proposed as a possible treatment against SARS-CoV-2. However, the results of a trial performed on 199 hospitalized adult patients with confirmed SARS-CoV-2 infection in China showed that the use of lopinavir/ritonavir had no significant benefit beyond standard-care in either reduction of viral load or overall mortality (Cao et al., 2020). The authors reported several limitations, including lack of treatment blinding, with study participants and investigators being aware of treatment assignments, thus reducing study objectivity. Additional studies are certainly required to determine whether or not lopinavir/ritonavir treatment given at a certain stage of illness can reduce some COVID-19 complications. While already multiple other ongoing studies exploring lopinavir/ritonavir in SARS-CoV-2 infection are ongoing, none utilizes a double-blind methodology to address the mentioned above limitation.
Currently, the “COVID-19 Treatment Guidelines Panel” recommended the use of lopinavir/ritonavir or other protease inhibitors for the treatment of SARS-CoV-2 infections only in the context of clinical trials, because of unfavorable pharmacodynamics and negative clinical trial data (NIH, 2020).
RNA polymerase inhibitors
Concerning nucleoside analogues, those in the form of adenine or guanine analogues, target the RdRp and block viral RNA synthesis in a broad spectrum of RNA viruses, including human coronaviruses (Li and De Clercq, 2020). This is the reason why several nucleos(t)ide analogues such as remdesivir, ribavirin and favipiravir have been considered potential drugs against SARS-CoV-2.
Remdesivir (GS-5734) was initially developed for the management of the Ebola and Marburg viruses (Warren et al., 2016; Wang et al., 2020a). This molecule is a small nucleotide analogue with a similar chemical structure of tenofovir alafenamide, a reverse transcriptase inhibitor approved for the treatment of HIV infection. Remdesivir has a broad antiviral activity in vitro against several RNA virus families including Filoviridae, Pneumoviridae, Paramyxoviridae, and Coronaviridae (Warren et al., 2016; Lo et al., 2017; Sheahan et al., 2017; Totura and Bavari, 2019). Several in vitro and in vivo data on remdesivir against a panel of coronaviruses, including highly pathogenic coronaviruses and potentially emergent BatCoVs (BatCoV-HKU5, BatCoV-HKU3, BatCoV-SHC014, and BatCoV-WIV1), support the development of this drug as an important potential pan-coronavirus antiviral agent (Sheahan et al., 2017; Agostini et al., 2018; Totura and Bavari, 2019). In particular, studies demonstrated that remdesivir decreases viral titers and viral RNA in in vitro models of both SARS-CoV-1 and MERS-CoV infection of primary human airway epithelial (HAE) cell cultures (Sheahan et al., 2017; Totura and Bavari, 2019), and a recent study reported that this molecule inhibits SARS-CoV-2 (EC50 = 0.77 μM in Vero E6 cells) (Wang et al., 2020a). Moreover, in vivo activity against coronaviruses was supported by improved disease signs (weight loss, lung viral titers) in MA15 SARS-CoV-infected mice treated prophylactically or therapeutically with remdesivir (Sheahan et al., 2017; Totura and Bavari, 2019). There has been much interest in remdesivir, following treatment of the first SARS-CoV-2 case, and subsequent recovery (Holshue et al., 2020). Currently, 44 trials taking place globally to investigate the efficacy of remdesivir for SARS-CoV-2 infection have been registered (https://clinicaltrials.gov/; accessed on 12 August 2020).
Potentially important clinical benefit of using remdesivir for COVID-19 patients emerged from an expanded access program (Grein et al., 2020). More recently however, data published from a first randomized clinical trial from China in adults with severe SARS-CoV-2 infection, did not show an association of remdesivir with statistically significant clinical benefits (Wang et al., 2020c). Of note, the study was prematurely terminated due to low patient enrolment, which would limit its power. In the Adaptive COVID-19 Treatment Trial (ACTT), including 1063 adult hospitalized patients with SARS-CoV-2 infection and evidence of lower respiratory tract involvement, a lower median time of recovery was found in patients treated with remdesivir compared to those who received placebo (11 [95 % C.I: 9–12] versus 15 [95 % C.I: 13–19] days, p < 0.001) (Beigel et al., 2020). Moreover, results from the SIMPLE trial showed that in patients with severe SARS-CoV-2 infection not requiring mechanical ventilation, a five-day dosing duration of remdesivir led to "similar improvement in clinical status" as the 10-day treatment course (Goldman et al., 2020). This study was not placebo-controlled; therefore the magnitude of benefit could not be determined. Recently, an expansion phase of the study has been added and will enroll a large cohort of 5600 additional patients, including those on mechanical ventilation.
On the basis of preliminary clinical trial data, the American COVID-19 Treatment Guidelines Panel recommended the administration of the investigational antiviral agent remdesivir to hospitalized patients infected with SARS-CoV-2 displaying a severe disease (NIH, 2020). However, remdesivir is not approved by the Food and Drug Administration (FDA). It is available only through an FDA emergency use authorization, in clinical trials, or through an emergency access program for children and adult patients hospitalized with severe COVID-19 (Updated 7/30/2020). On 25 June 2020, the European Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion recommending the granting of a conditional marketing authorization for remdesivir. It is currently indicated for the treatment of COVID-19 in adults and adolescents (aged 12 years and older with body weight at least 40 kg) with pneumonia requiring supplemental oxygen (European Medicines Agency, 2020b).
Ribavirin is a guanosine analogue previously approved and largely used for the treatment of HCV and respiratory syncytial virus (RSV). This drug shows in vitro activity against a large number of highly lethal emerging viruses. Ribavirin inhibits RNA synthesis by interfering with viral RdRp as well as inhibiting mRNA capping. However, while studies in vitro demonstrated that SARS-CoV-1, MERS-CoV, and HCoV-OC43 were sensitive to ribavirin, the doses that significantly inhibited the viral replication exceeded ribavirin blood concentrations attainable by typical human regimens (Totura and Bavari, 2019). Coronaviruses are one of few RNA viruses with a genomic proofreading mechanism, and therefore, a decreased in vitro activity of ribarivin than expected, was observed. This decrease is related to the excision of ribavirin and other nucleoside analogues by conserved coronavirus proofreading mechanisms (Ferron et al., 2018). Moreover, a limited activity against MA15 SARS-CoV by ribavirin alone was found in mouse models, thus suggesting that ribavirin treatment exacerbated SARS disease symptoms (Day et al., 2009). However, combination treatment of ribavirin and type I Interferons in primate models, improved MERS disease signs (Falzarano et al., 2013b). Ribavirin was used as part of treatment regimens for SARS and MERS patients; but, its efficacy was very limited in patients with highly pathogenic coronavirus respiratory syndromes (Totura and Bavari, 2019). Whether ribavirin exerts sufficient potency against SARS-CoV-2 remains to be determined.
Favipiravir (T-705), a guanine analogue approved for influenza treatment, can effectively inhibit the RdRp of RNA viruses such as influenza, Ebola, yellow fever, chikungunya, norovirus and enterovirus (Oestereich et al., 2014; Li and De Clercq, 2020). This compound showed in vitro activity against SARS-CoV-2 as well, albeit at a high concentration (EC50 = 61.88 μM in Vero E6 cells) (Wang et al., 2020a). Further in vivo studies are required to evaluate this antiviral nucleoside analogue.
Patients with SARS-CoV-2 are being recruited in randomized trials to evaluate the efficacy of favipiravir in combination with interferon-α and favipiravir in combination with baloxavir marboxil (an approved influenza inhibitor targeting the cap-dependent endonuclease).
Viral entry inhibitors
Further potential drug candidates for the treatment of SARS-CoV-2 infections are chloroquine phosphate and hydroxychloroquine sulfate. Chloroquine is an aminoquinolone derivative. Since its development in the 1940s, it was the drug of choice in the treatment of malaria until the development of newer antimalarials such as pyrimethamine, artemisinin, and mefloquine (Plantone and Koudriavtseva, 2018). Chloroquine and its derivative hydroxychloroquine have since been repurposed for the treatment of a number of other conditions including HIV, drug-resistant HCV-infection, systemic lupus erythematosus, and rheumatoid arthritis (Plantone and Koudriavtseva, 2018). These two drugs are being investigated for the treatment of SARS-CoV-2 (Devaux et al., 2020; Wang et al., 2020a; Yao et al., 2020). They inhibit terminal glycosylation of ACE2, the receptor that both SARS-CoVs target for cell entry. ACE2 not fully glycosylated may less efficiently interact with the SARS-CoV spike glycoproteins, thus facilitating the inhibition of viral entry (Vincent et al., 2005). In vitro, they inhibit several human pathogenic coronaviruses (Vincent et al., 2005; Devaux et al., 2020; Wang et al., 2020a; Yao et al., 2020). In particular, both chloroquine and hydroxychloroquine potently inhibit SARS-CoV-2 with a low micromolar concentration (Wang et al., 2020a; Yao et al., 2020) (Table 1).
Several clinical trials are undergoing to investigate the efficacy of these two drugs against SARS-CoV-2 (https://clinicaltrials.gov/; accessed on 12 August 2020). Several critical scientific questions have been raised about data reported in the paper by Mandeep Mehra et al., -Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis1- published in The Lancet on May 22, 2020, that was later retracted (The Lancet Editors, 2020). A recent randomized, placebo-controlled trial on 821 asymptomatic participants found hydroxychloroquine was no better than a placebo in preventing infection of the coronavirus (Boulware et al., 2020). At the same time, a targeted update on the 11th of June 2020 of the systematic review and meta-analyses about the efficacy of hydroxychloroquine or chloroquine for treatment of COVID-19, highlighted that so far there is very low certainty evidence from randomized clinical trials and quasi-experimental studies that hydroxychloroquine results in little or no benefit over standard care for the treatment of COVID-19 (WHO. COVID-NMA project. Cochrane, 2020). Overall, potential prevention and/or clinical benefits of hydroxychloroquine use remain to be determined.
Another drug that can interfere with viral entry, by targeting the viral spike glycoprotein, is griffithsin. This drug is a red-alga-derived lectin that binds to oligosaccharides on the surface of various viral glycoproteins, including HIV glycoprotein 120, and SARS-CoV spike glycoprotein (Lusvarghi and Bewley, 2016; Li and De Clercq, 2020). Griffithsin has been tested in phase I studies for HIV prevention, but studies are required to investigate the potency and delivery systems of spike inhibitors for the treatment or prevention of SARS-CoV-2.
It has been demonstrated that the binding of the coronavirus spike protein to ACE2 leads to ACE2 downregulation, thus resulting in excessive production of angiotensin. This in turn contributes to lung injury, as angiotensin-stimulated a type I angiotensin II receptor (AT1R) resulting in increased pulmonary vascular permeability, thereby mediating increased lung pathology (Imai et al., 2005). Therefore, although it could seem paradoxical, higher ACE2 expression due to the treatment of SARS-CoV-2 infected patients with AT1R blockers may protect them against acute lung injury rather than exposing them at higher risk to develop SARS-CoV infection.
Recently, Shuai Xia and colleagues found that the lipopeptide EK1C4 acts as a potent fusion inhibitor against SARS-CoV-2 spike protein-mediated membrane fusion and pseudovirus infection with IC50 values of 1.3 and 15.8 nM, respectively (Xia, Liu et al., 2020). This lipopeptide is about 241- and 149-fold more potent than the original pan-coronavirus fusion inhibitor, EK1, which targeted the HR1 domain (Xia et al., 2019). A high activity of EK1C4 was also shown against membrane fusion and infection of other human coronavirus pseudoviruses, including SARS-CoV-1 and MERS-CoV; moreover, EK1C4 potently inhibited the replication of five live human coronaviruses, including SARS-CoV-2 (Xia, Liu et al., 2020). In mice, a protection against HCoV-OC43 infection was found by using intranasal application of EK1C4 before or after challenge with HCoV-OC43, suggesting that EK1C4 could be used for prevention and treatment of infection by the currently circulating SARS-CoV-2 and other emerging SARS-CoVs (Xia, Liu et al., 2020).
Current status of antiviral resistance of coronaviruses
Drug resistance development is influenced by several parameters including drug potency and genetic barrier, host factors as well as viral factors such as viral fitness and intrinsic biology.
Considering viral factors, RNA viruses are generally characterized by a higher degree of genetic variability (Lauring and Andino, 2010). This is due to lack of proofreading activity by RdRp leading to high error rates and low replicative fidelity. Coronaviruses represent an exception to this rule, since they are the only RNA viruses encoding an exoribonuclease activity in the nonstructural protein 14 (ExoN) (Denison et al., 2011; Pruijssers and Denison, 2019). The presence of ExoN represents an obstacle to the development of nucleos(t)ide analogues, a broad-spectrum class of viral RdRp inhibitors largely used in treating multiple viral infections (Agostini et al., 2018). Indeed, ExoN acts by removing incorporated nucleos(t)ide analogues, conferring a sort of “innate” resistance to the majority of these compounds (Pruijssers and Denison, 2019; Shannon et al., 2020) and introducing a unique concept in mechanisms modulating drug resistance emergence. In line with this concept, in vitro studies showed that murine hepatitis virus (MHV) and SARS-CoV strains, lacking the proofreading activity of ExoN, were more susceptible to 5-fluorouracil and ribavirin than wild type (Smith et al., 2013). Similarly, a previous study has shown that the RdRp mutations V553I and M611 F capable to affect the fidelity of RdRp can confer resistance to 5-fluorouracil and/or to 5-azacytidine only when the activity of ExoN was abrogated, supporting that ExoN proofreading activity exerts an epistatic effect to the nucleotide selectivity of RdRp (Table 2 ) (Sexton et al., 2016). Overall, this supports a cooperation between RdRp and ExoN to optimize both fidelity and replication kinetics (Sexton et al., 2016).
Table 2.
Amino acid substitutions associated with reduced susceptibility to drugs with anti-coronavirus activity in in vitro model and by homology modeling.
| Antiviral drug | Drug-resistance mutationa | Corresponding residue in SARS-CoV-2b | Fold Change in EC50c | References |
|---|---|---|---|---|
| RdRp | ||||
| Remdesivir | F476LMHV | F480 | 2.4 | Agostini et al. (2018) |
| V553LMHV | V557 | 5 | ||
| F476LMHV+V553LMHV | F480+V553 | 5.6 | ||
| F480LSARS-CoV-1+V557LSARS-CoV-1 | F480+V557 | 6 | ||
| 5-FU | V553IMHV | V557 | n.a. | Sexton et al. (2016) |
| M611FMHV | M615 | n.a. | ||
| 5-AZC | V553IMHV | V557 | n.a | |
| Ribavirin | G64SPV | N459 | n.a. | Neogi et al. (2020) |
| L420APV | D865 | n.a. | ||
| Favipiravir | K159RCVB3 | K545 | n.a. | |
| 3CL-PR | ||||
| GRL-001 | T26IMHV | T26 | 3.06c | Deng et al. (2014) |
| D65GMHV | N65 | 2.56c | ||
| T26IMHV + D65GMHV | T26+N65 | >6c | ||
| T26IMHV + A298DMHV | T26+S301 | >6c | ||
| GC376 | N25SFel-Cov | T25 | 1.38 | Perera et al. (2019) |
| A252SFel-Cov | A255 | 1.15 | ||
| K260NFel-Cov | D263 | 1.05 | ||
| N25SFel-Cov + K260NFel-Cov | T25+D263 | 1.53 | ||
| N25SFelCov + A252SFelCov + K260NFel-Cov | T25+A255+D263 | 1.68 | ||
Abbreviations: RdRp, RNA-dependent RNA polymerase; 5-FU, 5-fluorouracil; 5-AZC, 5-azacytidine; 3CL-PR, 3CL protease; MHV, murine hepatitis virus; SARS-CoV-1, severe acute respiratory syndrome coronavirus; PV, poliovirus; CVB3, Coxsackievirus B3; FelCov, feline coronavirus.
The column reports the drug-resistance mutations identified for each antiviral drug. The virus in which the mutation has been identified is reported as subscript.
For each drug-resistance mutation, the corresponding residue in SARS-CoV-2 RdRp and 3CL-PR has been determined basing on homology of SARS-CoV-2 sequences with MHV and SARS-CoV-1 sequences.
The fold change in EC50 was calculated basing on the EC50 value of the mutant and EC50 value of wild-type reported in Reference #4.
So far, only two nucleos(t)ide analogues potently inhibit the coronavirus replication even in presence of ExoN. This is the case of remdesivir and the ribonucleoside analogue β-D-N4-hydroxycytidine, for whom an additional mechanism of action (beyond acting as chain terminator) has been hypothesized (Agostini et al., 2018, 2019). Both compounds are characterized by a high barrier towards resistance-acquisition in in vitro studies. For remdesivir, drug resistance was observed after several passages in MHV at residues F476 L and V553L (Table 2). These mutations correspond to F480 L and V557 L in SARS-CoV-1 RdRp and can confer resistance also in SARS-CoV-1 despite affecting viral fitness and virulence (Table 2) (Agostini et al., 2018). Regarding β-D-N4-hydroxycytidine, a low level resistance was detected only after the appearance of several mutations in MHV and MERS-CoV (Agostini et al., 2019).
Few resistance data were reported on 3CL-PR inhibitors. Using MHV in vitro model, single mutation (T26I and D65 G) and double mutations (T26I + D65 G and T26I + A298D) (Table 2) were associated with drug resistance to the broad-spectrum 3CL-PR inhibitor GRL-001 (Deng et al., 2014). Drug resistance mutations emerged after only four passages in cell culture suggesting a low barrier to resistance of this compound. Again, the emergence of drug resistance was associated with a reduced viral replication capacity and virulence supporting a high cost in term of viral fitness (Deng et al., 2014).
Recent reports described the effects of specific 3CL-PR mutations (N25S, A252S and K260 N) emerging in a patient with acquired feline infectious peritonitis receiving prolonged treatment with the 3CL-PR inhibitor GC376 (Pedersen et al., 2018; Perera et al., 2019). Considering single (N25S, A252S or K260 N), double (N25S + K260 N) or triple (N25S + A252S + K260 N) amino acid changes, only those containing N25S where associated with a marginal reduction in susceptibility to GC376 (Perera et al., 2019). Conversely, drug resistance profiles to lopinavir, used in clinical practice for the treatment of SARS-CoV-2 infected patients have not yet been defined.
Overall, limited information is available on the emergence of drug resistance against RdRp and 3CL-PR inhibitors. While drug resistance against the above-mentioned active RNA polymerase inhibitors was difficult to select also due to the presence of ExoN, mutations conferring resistance to 3CL-PR inhibitors tend to emerge rapidly. However, for both drug-classes, the resistance phenotype impaired viral fitness in vitro and attenuated virulence in in vivo models. Identifying and understanding drug resistance against anti-coronavirus agents will be a crucial aspect that deserves to be finely elucidated for optimized antiviral strategies.
Degree of genetic conservation among the three main pharmacological targets
A comprehensive characterization of conserved regions in 3CL-PR, RdRp and spike from SARS-CoV-2, and other human coronaviruses, is of crucial importance for the best definition of targets for the design and development of novel compounds with anti-pan-coronavirus activity. This is critical since coronaviruses are endowed by high tendency to spillover from animals to humans, and thus future cross-species transmission events leading to severe outbreaks in humans are not unexpected. Overall, this reinforces the need to set up an armamentarium of effective drugs to be used as treatment and/or prophylaxis to cope with potential future coronavirus infections. In particular, the identification of conserved regions, essential for viral replication and thus with no/very limited tendency to mutate, may offers the basis for the identification of pharmacological targets associated with a limited natural drug resistance and potential pan-activity versus all human coronaviruses.
The degree of conservation was defined on a large number of SARS-CoV-2 sequences retrieved from the start of the epidemic, from GISAID public database (https://www.gisaid.org/), with a total of 11,918 3CL-PR, 11,185 RdRp and 9,111 Spike glycoprotein sequences. The degree of conservation in SARS-CoV-2 was compared to that observed in sequences from SARS-CoV-1 (N = 40), MERS-CoV (N = 242) and other human coronaviruses (N = 55 for HCoV-NL63, 21 for HCoV-229E, 15 for HCoV-HKU and 126 for HCoV-OC43).
3. CL protease
The analysis of the 3CL-PR sequences from SARS-CoV-2 infected patients revealed a very high degree of genetic conservation. Indeed, 98.0 % (300/306) amino acid residues showed < 0.1 % variability (Table 3 ). Among them, 179 residues were found never mutated. Even more, in the remaining 6 residues, the degree of variability never exceeds 5%. The analysis of specific domains revealed that, the highest degree of genetic conservation was observed in domains II and III with 100 % and 100 % of residues showing ≤ 0.1 % variability. A slight decrease in the extent of genetic conservation was found for domain I with 96.7 % of residues with ≤ 0.1 % variability.
Table 3.
Extent of genetic conservation in 3CL-Protease, RNA-dependent RNA polymerase and in the spike protein in the different human coronaviruses.
| Coronavirus species | % of conserved residues (N of conserved residues/Total N of residues in a protein)a |
|||
|---|---|---|---|---|
| 3CL-PR | RdRp | Spike subunit 1 |
Spike subunit 2 |
|
| Group 2b | ||||
| SARS-CoV-2 | 98.0 (300/306) | 98.8 (560/567) | 98.4 (674/685) | 98.6 (580/588) |
| SARS-CoV-1 | 98.7 (302/306) | 97.5 (553/567) | 98.1 (654/667) | 99.1 (583/588) |
| Group 2c | ||||
| MERS-CoV | 95.4 (292/303) | 99.8 (566/567) | 96.9 (728/751) | 98.0 (590/602) |
| Group 2a | ||||
| HCoV-OC43 | 93.4 (283/303) | 99.6 (565/567) | 85.9 (663/772) | 96.0 (571/595) |
| HCoV-HKU1 | 98.7 (299/303) | 99.5 (564/567) | 97.4 (740/760) | 99.7 (594/596) |
| Group 1b | ||||
| HCoV-NL63 | 99.0 (300/303) | 95.9 (544/567) | 87.8 (657/748) | 96.4 (586/608) |
| HCoV-229E | 97.4 (294/302) | 98.1 (556/567) | 91.2 (515/565) | 98.2 (595/606) |
For SARS-CoV-2, conserved residues are defined as those in which aa substitutions are observed with a frequency <0.1 %, while for all other coronaviruses, conserved residues were defined as those in which no aa substitutions were observed.
Abbreviations: 3CL-PR, 3CL protease; RdRp, RNA-dependent RNA polymerase.
Number of sequences analyzed are: 11,918 for SARS-CoV-2 3CL-PR, 11,185 for SARS-CoV-2 RdRp, and 9,111 for SARS-CoV-2 Spike, 40 for SARS-CoV-1 3CL-PR, RdRP and Spike, 126 for HCoV-OC43 3CL-PR, RdRP and Spike, 55 for HCoV-NL63 3CL-PR, RdRP and Spike, 20 for HCoV-229E 3CL-PR, RdRP and Spike, 15 for HCoV-HKU1 3CL-PR, RdRP and Spike, and 242 for MERS-CoV 3CL-PR, RdRP and Spike.
A high degree of genetic conservation was also observed for other species of human coronaviruses with a percentage of conserved residues ranging from 93.4 % for HCoV-OC43 up to 99.0 % for HCoV-NL63. Overall findings support a limited 3CL-PR genetic diversification within each species of human coronaviruses (Table 3).
A completely different scenario is observed when 3CL-PR sequences from all human coronaviruses were compared (Table 4 ). Here, only 85 residues were found conserved across the different species of human coronaviruses. In particular, these invariant residues were scattered throughout the individual sequence or forming conserved regions composed of at most 5 residues (Fig. 1 ). Expectedly, the two residues involved in the catalytic dyad were fully conserved: The His residue at position 41 resides in the largest conserved region encompassing amino acids 38–42, whereas the Cys residue at position 145 is located within a conserved triplet encompassing positions 145–147 (Fig. 1). Beyond the catalytic dyad, other 15 residues comprise the substrate-binding cleft as evident from the crystal structure of SARS-CoV-1/2 3CL-PR (Hsu et al., 2005; Muramatsu et al., 2016; Goyal and Goyal, 2020; Zhang et al., 2020a) Among them, only 5 residues (at positions 147, 163, 166, 187 and 192) were fully conserved (Zhang et al., 2020a), while the remaining tend to have a group-specific pattern of amino acids.
Table 4.
Degree of amino acid identity in 3CL-Protease, RNA-dependent RNA polymerase and in the spike protein across the different human coronaviruses compared to SARS-CoV-2.
| Coronavirus species | % of similarity (N of identical amino acid residue compared to SARS-CoV-2)a |
|||
|---|---|---|---|---|
| 3CL-PR | RdRp | Spike subunit 1 |
Spike subunit 2 |
|
| Group 2b | ||||
| SARS-CoV-1 | 96.1 (294) | 98.2 (557) | 63.9 (438) | 90.0 (529) |
| Group 2a | ||||
| HCoV-OC43 | 48.4 (148) | 71.6 (406) | 21.3 (146) | 41.0 (241) |
| HCoV-HKU1 | 49.0 (150) | 71.8 (407) | 20.6 (141) | 38.6 (227) |
| Group 1b | ||||
| HCoV- NL63 | 43.5 (133) | 60.1 (341) | 17.7 (121) | 32.8 (193) |
| HCoV-229E | 40.8 (125) | 61.7 (344) | 15.3 (105) | 33.8 (199) |
| Group 2c | ||||
| MERS-CoV | 50.3 (154) | 75.3 (427) | 19.3 (132) | 43.0 (253) |
The degree of identity is calculated as the % of identical amino acid residues in a specific protein between SARS-CoV-2 and each of the other human coronaviruses. The denominator used to calculate the % is the number of residues in SARS-CoV-2 proteins: 306 for 3CL-PR, 567 for RdRp, 685 for the spike subunit 1 and 588 for the spike subunit 2.
Abbreviations: 3CL-PR, 3CL protease; RdRp, RNA-dependent RNA polymerase.
Number of sequences analyzed are: 11,918 for SARS-CoV-2 3CL-PR, 11,185 for SARS-CoV-2 RdRp, and 9,111 for SARS-CoV-2 Spike), 40 for SARS-CoV-1 3CL-PR, RdRP and Spike, 126 for HCoV-OC43 3CL-PR, RdRP and Spike, 55 for HCoV-NL63 3CL-PR, RdRP and Spike, 20 for HCoV-229E 3CL-PR, RdRP and Spike, 15 for HCoV-HKU1 3CL-PR, RdRP and Spike, and 242 for MERS-CoV 3CL-PR, RdRP and Spike.
Fig. 1.

Amino acid sequence alignment of 3CL-PR across SARS-CoV-2, SARS-CoV-1, HCoV-NL63, HCoV-229E, HCoV-HKU-1, HCoV-OC43 and MERS-CoV. Conserved amino acids shared across human coronaviruses are indicated by dots and highlighted in cyan. Amino acid residues of the catalytic dyad are highlighted in dark red, residues involved in dimerization interface are in light blue according to Goyal and Goyal (2020), Zhang et al. (2020), while residues composing the substrate-binding cleft are in dark blue according to Muramatsu et al. (2016), Hsu et al. (2005), Zhang et al. (2020), Goyal and Goyal (2020). The domains of 3CL-PR are reported according to Zhang et al. (2020). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Particular attention should be given to the Ser residues at positions 139 and 147 located near the active site. Indeed, mutations at these positions can profoundly abrogate protease activity suggesting that targeting this site can serve as the basis for broad-spectrum therapeutic agents against 3CL-PR (Bacha et al., 2004; Barrila et al., 2010; Goyal and Goyal, 2020).
Recently, peptidomimetic α-ketoamides have been proposed as potential inhibitors of SARS-CoV-2 3CL-PR (Zhang et al., 2020b, a). These compounds can interact with residues at positions 1, 40, and 166 in the core of the substrate-binding cleft (Zhang et al., 2020a). The high degree of conservation across all human coronaviruses of the residues involved in the substrate binding cleft supports a potential anti pan-coronavirus activity of these compounds that deserves further investigation.
Among the 10 residues involved in the dimerization (Goyal and Goyal, 2020; Zhang et al., 2020a), 6 residues at positions 14, 28, 139, 140, 290 and 299, were conserved across the different species. The remaining residues at positions 4, 10, 11, 298, were characterized by a group-specific pattern of genetic diversification (Fig. 1).
RNA-dependent RNA polymerase
The analysis of the RdRp sequences from SARS-CoV-2 isolated from infected patients revealed a very high degree of genetic conservation. Strikingly, 98.8 % of the amino acid residues (560/567) displayed < 0.1 % variability (Table 3). Among them, 420 residues were found to be never mutated and the remaining 7 residues displayed a very low degree of variability never exceeding 1%. As evident form the analysis of specific domains, the thumb domain showed the highest degree of genetic conservation with 100 % of residues exhibiting ≤ 0.1 % variability (105/105), whereas the finger and palm domains exhibited 98.2 % (270/275) and 98.9 % (173/175) amino acid conservation, respectively.
As observed for 3CL-PR too, the other species of human coronaviruses showed a high degree of genetic conservation in RdRp with a residues conservation ranging from 95.9 % for HCoV-NL63 up to 99.8 % for MERS-CoV, re-indicating a limited intra-species genetic variability (Table 3).
However, unlike 3CL-PR, a higher degree of RdRp genetic conservation across all human coronaviruses was observed with nearly 50 % of the amino acid residues fully conserved (271/567) (Table 4, Fig. 2 ). This corroborates the crucial role of this enzyme for viral replication and life cycle and its role as a druggable target for a pan-coronavirus pharmacological approach. In particular, a large fraction (45.4 %) of conserved residues (123/27) clustered into 21 regions composed of 4–12 consecutive invariant residues, while the remaining invariant residues were scattered throughout the sequence, either individually or either pairs or triplets (Fig. 2).
Fig. 2.

Amino acid sequence alignment of RdRp across SARS-CoV-2, SARS-CoV-1, HCoV-NL63, HCoV-229E, HCoV-HKU-1, HCoV-OC43 and HCoV-MERS. The right hand RdRp domain (residues 366-920) is reported.
Conserved amino acids across human coronaviruses are indicated by dots and highlighted in cyan. Residues encompassing motifs A–E are highlighted in light blue. The catalytic residues S759, D760 and D761 and the classic divalent-cation–binding residue D618 are highlighted in dark red. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The start and end of each RdRp functional domains (fingers, palm and thumb) are also indicated. The numbering of RdRp domains and motifs is according to Gao et al., Science 2020.
In motif A (encompassing aa 611–627), 14 out of 17 residues were conserved including the classic divalent cation-binding residue Asp618 (Fig. 2). The RdRp Asp618 is known to be conserved in most viral polymerases including that of hepatitis C virus (corresponding to Asp220) and of poliovirus (corresponding to residue Asp233) (Gong and Peersen, 2010; Appleby et al., 2015). A superimposable scenario is observed for motif C (aa:753−767) with 13/15 residues fully conserved including the catalytic residues Ser759, Asp760 and Asp761 crucial for RNA-dependent RNA synthesis (Subissi et al., 2014; Gordon et al., 2020). Notably, in motif C, the variant residue at position 766 was a Tyr in all species with the exception of SARS-CoV-2 in which a Phe was observed (Fig. 2). Further studies are necessary to determine whether the presence of a Phe can confer higher processivity to SARS-CoV-2 RdRp. Similarly, the variant residue at position 762 was a Gly in all species with the exception of the group 2b coronaviruses including SARS-CoV-1 and SARS-CoV-2 in which an Ala was present, suggesting a group-specific genetic adaptation (Fig. 2).
The positively charged residues at position 545, 553 and 555, located in motif F and involved nucleotide triphosphate (NTP) entry channel are also fully conserved across the different species of human coronaviruses, as well as residues Asp623, Asn691 and Ser682 capable of recognizing the 2′−OH group of the NTP, thus allowing the RdRp to be specific for the synthesis of RNA rather than DNA (Fig. 2) (Hillen et al., 2020).
Recently, residues involved in the interaction with remdesivir have been characterized (Gao et al., 2020; Hillen et al., 2020). In particular, it has been shown that residue Thr680 forms hydrogen bonds not only with the incoming endogenous NTP but also with the 2′ OH of remdesivir, while residue Val557 stabilizes the interaction with the incoming triphosphate remdesivir (Gao et al., 2020). Other residues (Arg555, Ser682, Asp623 and Asn691) are located close to the region involved in remdesivir interaction and Asn691 is also potentially involved in the interaction with sofosbuvir (Gao et al., 2020). All of them are were fully conserved across human coronaviruses (Fig. 2).
Spike glycoprotein
The analysis of the spike glycoprotein sequences too, within each species, showed a high degree of genetic conservation despite being a surface glycoprotein. Overall, in SARS-CoV-2, 98.5 % of the amino acid residues (1254/1273) showed < 0.1 % variability with no difference between the S1 and S2 subunits (98.4 % [674/685] in subunit S1 and 98.6 % [580/588] in subunit S2) (Table 3). Among them, 897 residues were never found mutated (476 in S1 and 421 in S2). Notably, only the Asp residue at position 614, mapping in the junction between subunit S1 and S2, showed a high degree of genetic variability exceeding 60 %. The main mutation observed at this residue was D614 G which has been proposed as a novel serine protease cleavage site capable of significantly enhancing the fusion with cell membrane in vitro (Bhattacharyya et al., 2020).
By analyzing specific domains, the highest degree of genetic conservation was observed in the receptor binding domain (99.1 % of residues with ≤ 0.1 % variability), fusion peptide (100 %), Heptad repeat region 2 (100 %), and in the transmembrane domain (100 %). A slight reduction in the extent of genetic conservation was found for the heptad repeat region 1 and for the cytoplasmic domain with 97.4 % and 97.2 % of residues with ≤ 0.1 % variability, respectively.
High degree of genetic conservation was also observed for other species of human coronaviruses ranging from 85.9 % in HCoV-OC43 up to 98.1 % in SARS-CoV-1 for subunit 1, and from 96.0 % in HCoV-OC43 up to 99.7 % in HCoV-HKU1 for subunit 2. The higher degree of conservation in subunit S2 than in S1 can be explained by the fact that the S1 subunit is exposed to the environment and can thus be subject to immunological pressure (Table 3, Fig. 3, Fig. 5C).
Fig. 3.

Amino acid sequence alignment of the Spike subunit S2 across SARS-CoV-2, SARS-CoV-1, HCoV-NL63, HCoV-229E, HCoV-HKU-1, HCoV-OC43 and MERS-CoV. Conserved amino acids across human coronaviruses are indicated by dots and highlighted in cyan. The figures report only the functional domain of the spike subunit S2 according to Xia et al., 2020. FP, fusion peptide; HR, heptad repeats; TMD, transmembrane domain; Cyt-D, cytoplasmic domain.
Fig. 5.

Surface 3D representation of the conserved regions of SARS-CoV-2. In panels A) 3CL-PR, B) RdRp and C) spike subunit S2, optimized structures are shown. Amino acid residues that are conserved in all coronaviruses, those conserved in SARS-CoV-2, SARS-CoV-1 and MERS-CoV, those conserved in SARS-CoV-2 and SARS-CoV-1, those conserved in SARS-CoV-2 and at least another CoV and those that are present only in SARS-CoV-2 are indicated, respectively, in blue, light blue, pale cyan, salmon and red. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The inter-species analysis of the spike subunits compared to SARS-CoV-2, confirmed the higher degree of conservation in subunit S2 than in S1 across human coronaviruses, ranging from 15.3 % in HCoV-229E up to 63.9 % in SARS-CoV-1 for subunit 1, and from 32.8 % in HCoV-NL63 up to 90.0 % in SARS-CoV-1 for subunit 2 (Table 4). In particular, the inter-species analysis of subunit S2 revealed that 109/240 residues were conserved across human coronaviruses (Fig. 3 ). Most of them (70/109) were found to be clustered in regions composed of at most 4 residues, while the remaining 39 residues were scattered individually throughout the S2 subunit.
The heptad repeat 1 was the most conserved domain with 32.8 % (24/73) of invariant residues. In group 1b (HCoV-NL63 and HCoV-229E), this region (encompassing positions 939–940) was characterized by a long insertion of 14 residues, mainly identical (10/14). In the heptad repeat 1, between residues 922–943, we found 9 specific amino acids in SARS-CoV-2 which were different from SARS-CoV-1; furthermore, three amino acids were also different from all other human coronaviruses (Fig. 3). This is in line with a recent study suggesting that these unilateral replacements in the heptad repeat domain 1 of SARS-CoV-2 could enhance the interactions between heptad repeat 1 and 2, thus further stabilizing the 6-helices bundle and in turn increasing viral infectivity (Xia, Liu et al., 2020). In particular, this study showed that the presence of a Ser residue at position 929 can determine the establishment of a novel hydrogen bond (not observed in SARS-CoV-1) with the Asn at position 1192 (Xia, Liu et al., 2020). In line with a higher degree of conservation, the heptad repeats 1 has been proposed as a target for the development of viral fusion inhibitors with group-specific (Lu et al., 2014; Channappanavar et al., 2015) or with anti-pan-coronavirus activity (Xia, Liu et al., 2020; Xia, Zhu et al., 2020). Among them, the compound EK1C4 showed a broad-spectrum inhibitory activity against infection by SARS-CoV-2, SARS-CoV-1, MERS-CoV and other HCoVs. Furthermore, EK1C4 was characterized by long-lasting prophylactic effect against HCoV−OC43 in mice, supporting its use against SARS-CoV-2 infection (Xia, Liu et al., 2020).
The other domains of subunit 2 showed a degree of conservation ranging from 7.8 % for the heptad repeat 2 up to 16 % for the transmembrane domain, while no conserved residues were detected in the fusion peptide.
Completely different scenario is observed for subunit S1 which displays remarkable tendency towards group-specific amino acids profile (Fig. 4 ). For this reason, the analysis of genetic conservation was carried out by comparing subunit S1 sequences within each group of human coronaviruses separately, and was focused on the receptor binding domain (RBD), the key functional component of subunit S1.
Fig. 4.

Receptor binding domain (RBD) sequence alignment of the spike subunit S1 within different groups of human coronaviruses. Amino acid sequences of SARS-CoV-2 and SARS-CoV-1 (group 2b), HCoV-HKU-1 and HCoV-OC43 (group 2a), HCoV-NL63, and HCoV-229E (group 1b), and MERS-CoV are shown. Conserved amino acids within each group are denoted as dots and highlighted in cyan.
In group 2b (comprising SARS-CoV-1 and SARS-CoV-2, both of which use the ACE2 receptor for their entry), 73.5 % (164/223) showed a full genetic concordance (Fig. 4). These include the 8 Cys residues at positions 336, 361, 379, 432, 391, 480, 488 and 525, crucial for the establishment of disulfide bonds that stabilize the RBD structure (Lan et al., 2020). A recent study has identified 14 SARS-CoV-2 residues involved in the interaction with the ACE2 receptor (Lan et al., 2020). Among them, only 8 were shared and conserved among both SARS-CoVs: Tyr449, Tyr453, Asn487, Tyr489, Gly496, Thr500, Gly502 and Tyr505 of SARS-CoV-2 (corresponding to Tyr436, Tyr440, Asn473, Tyr475, Gly482, Thr486, Gly488 and Tyr491 in SARS-CoV-1, respectively) (Lan et al., 2020). Among them, Tyr residues at positions 449, 489 and 505 in SARS-CoV-2 were critical for the establishment of hydrogen bonds with polar hydroxyl groups in ACE2. Conversely, 5 positions were characterized by different amino acids with superimposable biochemical properties: Leu455/Tyr442, Phe456/Leu443, Phe486/Leu472, Gln493/Asn479 and Asn501/Thr487 (corresponding to the respective Tyr442, Leu443, Leu472, Asn479 and Thr487 in SARS-CoV-1) (Lan et al., 2020). The remaining positions corresponds to Gln498 and Tyr484 in SARS-CoV-2 and SARS-CoV-1 (Lan et al., 2020).
Previous studies have shown that SARS-CoV-2 RBD is endowed with significantly higher binding affinity to ACE2 than SARS-CoV-1. Thus, it is conceivable to hypothesize that the amino acids changes have been evolved in order to optimize such interaction.
Focusing on the other human coronaviruses, a lower degree of genetic conservation is observed for group 2a comprising HCoV-OC43 and HCoV-HKU1 sharing 54.4 % of their amino acid residues (148/272). Both viruses employ sialoglycan-based receptors with 9-O-acetylated sialic acid (9-O-Ac-Sia) as a key component to enter the target cell. Nevertheless, a previous study showed that HCoV-OC43 and HCoV-HKU1 are characterized by a different adaptation to human sialome that can explain the evolutionary genetic divergence observed in the RBD (Hulswit et al., 2019). Finally, a peculiar scenario emerges for group 1b comprising HCoV-229E and HCoV-NL63, which share only 59/144 (41.0 %) amino acid residues in the RBD, all showing no amino acid substitutions. Interestingly, this finding can be explained by the fact that, while HCoV-NL63 interacts with ACE2, HCoV-229E specifically binds to the aminopeptidase N (APN or CD13). Thus, the different receptor usage can pose a different selective pressure favoring a substantial degree of genetic diversification.
With the aim to represent the 3D localization of all the conserved regions, we report the surface optimized structures of SARS-CoV-2 3CL-PR (Fig. 5 A), RdRp (Fig. 5B) and spike S2 subunit (Fig. 5C) by coloring the residues that are conserved in all coronaviruses, those conserved in SARS-CoV-2, SARS-CoV-1 and MERS-CoV, those conserved in SARS-CoV-2 and SARS-CoV-1, those conserved in SARS-CoV-2 and at least another coronavirus, as well as those that are present solely in SARS-CoV-2 respectively, in blue, light blue, pale cyan, salmon and red. Interestingly, we could observe some highly conserved putative additional binding pockets in both 3CL-PR and RdRp that will be further investigated in future studies.
Glycosylation profiling in the Spike protein
The glycosylation of proteins present in the viral envelope, plays a critical role in viral pathogenesis including mediating protein folding and stability and shaping viral tropism (Watanabe et al., 2019, 2020). Thus, we analyzed the profiles of consensus N-glycosylation sites (Asn-X-Ser/Thr, where X is any amino acid excluding proline) in the overall spike glycoprotein and their level of conservation by using the “N-Glycosite” algorithm available at: http://www.hiv.lanl.gov/. Interestingly, a heavy enrichment of consensus N-linked glycosylation sites in the spike glycoprotein characterizes human coronaviruses. Indeed, the highest number of N -linked glycosylation sites was observed in group 1b (33 for HCoV-229E and 39 for HCoV-NL63) followed by group 2a (24 for HCoV-OC43 and 28 HCoV-HKU1), group 2c (23 for MERS-CoV) and group 2b (22 for both SARS-CoV-2 and SARS-CoV-1). No mutations abrogating the N-linked glycosylation sites were observed in highly pathogenic coronaviruses SARS-CoV-2, SARS-CoV-1 and MERS-CoV, suggesting high degree of genetic conservation. Conversely, mutations abrogating the N-linked glycosylation site were observed in HCoV-OC43 (at positions 152, 214, 484, 728, and 899), followed by HCoV-NL63 (at positions 24, 98, 178, 626), HCoV-229E (at positions 20, 98) and HCoV-HKU1 (only at position 58). The high degree of conservation of the N-linked glycosylation sites, particularly in highly pathogenic coronaviruses, suggests their role as a target of the so-called carbohydrate-binding agents (CBA), an intriguing class of antiviral compounds capable to prevent viral entry into the target cell. Interestingly, previous studies have shown that long-term pharmacological pressure with CBA can result in the selection of resistant viral strains with mutations abrogating the N-linked glycans (François and Balzarini, 2012). Since the shield of carbohydrates can mask viral epitopes from neutralizing antibodies, CBAs could also enhance the capability of the immune system in blocking viral infection (François and Balzarini, 2012). A previous study has shown that CBA can efficiently inhibit SARS-CoV-1 replication (Keyaerts et al., 2007), supporting their role, not only for treatment but also for prevention of coronavirus infection.
Potential new antiviral agents against SARS-CoV-2 and other human pathogenic coronaviruses based on structural modeling
As previously discussed, albeit their high species diversity, coronaviruses share key genomic elements that are crucial for drug design process. Two viral proteases, the papain-like protease (PLpro) and the 3CL-PR, are involved in cleaving the large replicase polyprotein 1a (pp1a) and pp1ab to produce NSPs, such as RdRp and helicase, responsible for the replication and transcription of the virus (Boheemen et al., 2012; Chan et al., 2015b). The surface structural spike glycoprotein, composed of the amino-terminal receptor-binding S1 and carboxy-terminal membrane fusion S2 subunits, is of particular interest for antiviral development because of its critical role in the virus-host cell receptor interaction. Binding of the S1 subunit RBD to the host receptor triggers conformational changes in the S2 subunit (the stalk region of S) to enable fusion (Lu et al., 2014). ACE2 (used by SARS-CoV-1 and −2 as well as HCoV-NL63), dipeptidyl peptidase 4 (DPP4; used by MERS-CoV), aminopeptidase N (used by HCoV-229E), and O-acetylated sialic acid (used by HCoV-OC43 and HCoVHKU1) represent the key functional host cell receptors utilized by human pathogenic coronaviruses (Vlasak et al., 1988; Yeager et al., 1992; Li et al., 2003; Hofmann et al., 2005; Raj et al., 2013; Huang et al., 2015). In order to promote the process of cell surface non-endosomal virus entry at the plasma membrane, other host proteases, such as transmembrane protease serine 2 (TMPRSS2) and TMPRSS11D (also known as airway trypsin-like protease), cleave the spike into the S1 and S2 subunits (Shirato et al., 2013). The MERS-CoV spike is also activated by furin, a serine endoprotease implicated in the processing of fusion proteins and cell entry of other RNA viruses. Furin is also involved in MERS-CoV S1/S2 cleavage during egress from the infected cell (Mille and Whittaker, 2014).
Before the SARS epidemic, only two human pathogenic coronaviruses (HCoV-229E and HCoVOC43 were known, as usually associated with self-limited upper respiratory tract infections (Chan et al., 2012). Therefore, when the high pathogenic SARS-CoV-1 suddenly emerged in the late 2002, the researchers involved in antiviral development were underprepared, and applied three general approaches to discover potential anti-coronavirus treatment options for human-pathogenic coronaviruses, especially for SARS and later MERS coronaviruses (Barnard and Kumaki, 2011; Kilianski and Baker, 2014).
The first approach to drug discovery was based on testing existing broad-spectrum antiviral drugs that have been used to treat other viral infections by using standard assays that measure the effects of these drugs on the cytopathic effect, virus yield and plaque formation of live and/or pseudotyped coronaviruses. Examples of drugs identified using this approach include interferon α, interferon β, interferon ɤ, ribavirin and inhibitors of cyclophilin (Cinatl et al., 2003; So et al., 2003; Pfefferle et al., 2011; Chan et al., 2013; de Wilde et al., 2013; Falzarano et al., 2013a; Tanaka et al., 2013). The advantage of using these drugs is related to their availability with known pharmacokinetic and pharmacodynamic properties, side effects and dosing regimens. However, they do not have specific anti-coronaviruses effects and may be associated with severe adverse effects.
The second anti-coronavirus drug discovery approach was represented by a drug repurposing screening method that involves chemical libraries comprising large numbers of existing compounds or databases (de Wilde et al., 2014; Elshabrawy et al., 2014). This approach allows to rapidly identify many readily available compounds that can then be further characterized by antiviral assays. Albeit many of the identified drugs exhibit anti-coronavirus activities in vitro, most are not clinically useful because they are either associated with immunosuppressive effects or have therapeutic dosage limitations.
The third approach for anti-coronavirus drug discovery was based on the de novo development of novel, specific agents thanks to the genomic and biophysical understanding of the individual coronaviruses, such as siRNA molecules or inhibitors that target specific viral enzymes involved in the viral replication cycle. Although most of these drugs have potent in vitro and/or in vivo anti-coronavirus activity, their pharmacokinetic and pharmacodynamics properties and side effect profiles have yet to be evaluated in animal and human trials. Furthermore, the development of these candidate drugs into clinically useful therapeutic options with reliable delivery modes for patients usually takes years (Zumla et al., 2016). In this perspective, in the drug development process, several computational methods, and in particular virtual screening (vs) techniques, proved to be important tools to speed up the process. During the early 1980s, the ability to rationally design drugs using protein structures was an unrealistic goal for many structural biologists. The first projects were underway in the mid-80 s, and by the early 1990s the first success stories were published (Erickson et al., 1990; Roberts et al., 1990; Dorsey et al., 1994). Today, even though there is still quite a bit of fine-tuning necessary to perfect the process, structure-based drug design is an integral part of most industrial drug discovery programs (Mountain, 2003) and is the major subject of research for many academic research laboratories. The improvement of both proteomics and structural genomics and the development in information technology played a key role for the success story in the discovery of new lead drugs. Excellent drug targets are identified at an increasing pace using developments in bioinformatics. High-throughput crystallography, as well as nuclear magnetic resonance (NMR), have seen a number of advances in the past years, thus shortening the timeline for determining structures. Faster computers and the availability of relatively inexpensive clusters of computers have increased the speed at which drug leads can be identified and evaluated in silico.
By exploiting these increasingly advanced technologies, it has been possible to design very promising molecules able to recognize different targets of the β-coronaviruses.
Specifically, the SARS‐CoV-1 main protease has been comprehensively explored as a drug target, and many potent enzyme inhibitors have been identified (Bacha et al., 2004; Grum-Tokars et al., 2008). Interestingly, as discussed above, 3CL-PR enzymes from different coronaviruses are known to share significant sequence and 3D structural homology providing a strong structural basis for designing wide‐spectrum anti‐coronavirus inhibitors (Fig. 5A) (Yang et al., 2006; Wang et al., 2016). For example, in 2017 Abuhammad and collaborators applied Quantitative Structure‐Activity Relationship (QSAR)‐guided modeling together with docking simulations in order to carry out the first pharmacophore modeling study of a set of peptidomimetic inhibitors of the bat HKU4‐CoV 3CL-PR. Specifically, the structural features pivotal in ligand recognition, as well as the most important 3CL-PR binding pocket regions, were highlighted by the obtained pharmacophore models, that were further used to screen the National Cancer Institute database for novel non-peptidomimetic 3CL-PR inhibitors. The identified hits were tested as potential HKU4‐CoV and MERS‐CoV 3CL-PR inhibitors. Among them, two phenylsulfonamide derivatives displayed moderate inhibitory activity against the MERS‐CoV 3CL-PR, thus representing a potential starting point for the development of novel anti‐MERS agents (Abuhammad et al., 2017).
The same target of the six human coronaviruses HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, SARS-CoV-1 and MERS-CoV was also investigated by Berry and coworkers, who screened the ZINC drugs-now library by applying both high-throughput pharmacophore modeling and molecular docking experiments. The results obtained from the consensus virtual screening approach, performed by using Vina, Glide, GOLD and MM-GBSA, were further validated by means of molecular dynamics simulations. The authors identified 19 best hits and characterized them in terms of shape and features, thus highlighting 15 significantly dissimilar clusters. Interestingly, in the perspective of a future lead optimization, the incorporation of a lactam ring was strongly suggested since it was found to enhance the interactions of the natural substrate and is also effective against the SARS-CoV-1 3CL-PR (Berry et al., 2015).
Another crucial target largely investigated by means of computational techniques is represented by the ACE2 receptor, considered an important therapeutic target for controlling cardiovascular diseases and SARS outbreaks. Specifically, in a study published in 2004, about 140,000 small molecules were screened by in silico molecular docking, and those with the best in silico binding affinity were tested for their ability to inhibit both ACE2 catalytic activity and SARS-CoV-1 spike protein-mediated cell fusion. The N-(2-aminoethyl)-1-aziridine-ethanamine was identified as a novel ACE2 inhibitor that is also effective in blocking the SARS-CoV-1 spike protein-mediated cell fusion (Huentelman et al., 2004).
Repurposed drugs anti SARS-CoV-2 identified by computational techniques already published
More recently, by applying in silico docking models, a library of known bioactive compounds against several sites on the spike protein and the catalytic site of the SARS-CoV-2 3CL-PR has been screened. Many ligands were identified as promising inhibitors for SARS-CoV-2, such as zanamivir, indinavir, saquinavir and remdesivir, able to well recognize the viral main protease (Hall and Ji, 2020).
In a drug repurposing perspective, the SARS-CoV-2 RdRp was modeled and targeted using different anti-polymerase drugs already approved against various viruses, such as HIV, HCV and Ebola. The authors showed the potential effectiveness of ribavirin, remdesivir, sofosbuvir, galidesivir and tenofovir as potent inhibitors of SARS-CoV-2 RdRp, and also suggested a guanosine derivative (IDX-184), setrobuvir and YAK as the most promising antiviral leads to specifically combat the SARS-CoV-2 strain (Elfiky, 2020). Recently, by structurally superposing the HCV polymerase bound to sofosbuvir with the SARS-CoV RdRp, Jácome and coworkers demonstrated that the residues of the drug-binding pocket are present also in RdRp. Moreover, these residues were found to be conserved in several SARS and MERS-related coronaviruses polymerases, thus suggesting the possibility to use sofosbuvir against these highly infectious pathogens (Jácome et al., 2020).
Another in silico drug repurposing study was carried out against the main protease of SARS-CoV-2. The authors combined docking experiments with molecular dynamics simulations (MDs) in order to screen 1,615 FDA-approved against the enzyme active site. Specifically, the predicted binding modes of the top scoring hits were fully characterized by MDs and, in agreement with other recent in silico drug repurposing studies, aliskiren, capreomycin and isovuconazonium, as well as ceftolozane, cobicistat and carfilzomib emerged as novel potential inhibitors (Qiang Wang et al., 2020). Saquinavir was also well ranked, but MDs revealed an unstable binding mode. In addition, the protocol favorably ranked dronedarone, a molecule recently reported as an active inhibitor of SARS-CoV-2 virus (Jeon et al., 2020), suggesting that its target could be the viral main protease (Cesar and Alejandro, 2020). This enzyme was also investigated by Abhithaj J. and coworkers, who applied pharmacophore modeling followed by docking simulations, by screening the DrugBank database and thus identifying cobicistat, larotrectinib and simeprevir as potential candidates for repurposing (Abhithaj et al., 2020). Other very promising compounds selected by means of another recent structure-based vs were two directly-acting antiviral drugs, that are velpatasvir and ledipasvir, both of which are inhibitors of the HCV NS5A protein and marketed as approved drugs in combination with sofosbuvir (Chen et al., 2020a). Whereas, by means of a blind molecular docking approach against the SARS-CoV-2 3CL-PR, Das and collaborators identified 33 molecules, which included natural products, anti-virals, anti-fungals, anti-nematodes and anti-protozoals, able to well recognize the viral enzyme, with rutin showing the highest inhibitor efficacy (Das et al., 2020). All published repurposed drugs identified by computational techniques are depicted in Table S1.
New repurposed drugs anti SARS-CoV-2, identified by computational techniques
In this context, our research group also applied an in silico drug repurposing approach by investigating the molecular recognition of the DrugBank database, thus including about 15,000 small molecules (approved drugs, experimental and investigational compounds, as well as nutraceuticals) against: a) the crystal structure of the complex resulting from the interaction between SARS-CoV-2 main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) (PDB code 6Y2G); b) the structure of the chimeric receptor-binding domain of spike of SARS-CoV-2 complexed with its receptor human ACE2 (PDB code 6VW1), considering both ACE2 alone and in complex with spike, as well as c) the nsp12-nsp7-nsp8 complex bound to the template-primer RNA and triphosphate form of remdesivir (PDB code 7BV2). All the experimental details are reported in the Supporting Information.
Based on their theoretical binding affinity, expressed as Glide-score (G-score), we filtered the best molecules obtained by our structure-based vs, selecting only those with a score within 2 kcal/mol above the minimum energy. Thereby, we selected ∼100 compounds for each analyzed target, and by visual inspection and chemical diversity, we identified the most promising ones characterized by a good theoretical binding affinity towards at least two of the four studied targets. Specifically, as reported in Table 5 , we found 4 cephalosporins, 2 flavone compounds, 3 purine analogues (including remdesivir), 2 peptide derivatives, 2 triazoles and a benzeneacetamide, identified as the best 14 candidates that deserve further investigation in future in vitro studies to test their potential antiviral activity.
Table 5.
The best 14 compounds identified by the structure-based virtual screening approach against the SARS-CoV-2.
| DrugBank ID | 3CLpro (-8.83)a | ACE2 (-10.29)a | ACE2/spike (-7.25)a | RdRp (-11.54)a | 2D Structure | DRUG NAME |
|---|---|---|---|---|---|---|
| DB03632 | −7.29 | −9.50 | −6.97 | −10.65 |  |
Argifin |
| DB01329 | −7.82 | −6.31 | −4.77 | −11.54 |  |
Cefoperazone |
| DB00430 | −7.66 | −8.84 | −4.81 | −11.07 |  |
Cefpiramide |
| DB01415 | −6.92 | −8.88 | −4.03 | −10.07 | 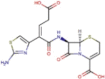 |
Ceftibuten |
| DB09050 | −7.91 | −9.15 | −6.53 | −10.74 | 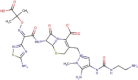 |
Ceftolozane |
| DB08995 | −6.85 | −6.86 | −6.12 | −8.07 | 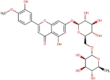 |
Diosmin |
| DB11633 | −6.85 | −5.56 | −5.30 | −7.18 | 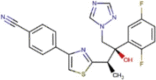 |
Isavuconazole |
| DB00722 | −6.85 | −6.68 | −5.30 | −9.88 | 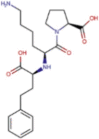 |
Lisinopril |
| DB00157 | −7.79 | −8.64 | −6.79 | −9.69 | 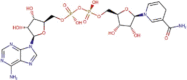 |
NADH |
| DB11871 | −7.72 | −8.57 | −6.41 | −8.23 | 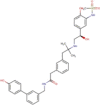 |
PF-00610355 (investigational) |
| DB121383 | −7.20 | −7.24 | −5.25 | −10.72 | 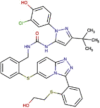 |
PF-03715455 (investigational) |
| DB14761 | −6.96 | −7.47 | −3.98 | −9.91 |  |
Remdesivir |
| DB12846 | −7.31 | −8.65 | −5.96 | −8.14 |  |
Reproterol |
| DB01698 | −7.81 | −6.76 | −4.54 | −9.58 |  |
Rutin |
2D representation, DrugBank code, drug name and Glide score (G-Score) values of the best 14 compounds identified by the structure-based virtual screening approach against the SARS-CoV-2 main protease (3CL-PR), polymerase (RdRp) and the host ACE2 enzyme, alone and complexed to the viral spike glycoprotein. The compounds are in alphabetical order.
This value indicates the absolute best G-score value for each analyzed target and is expressed in kcal/mol.
As shown in the Supporting Information (Figs. S1–S5), most of the identified compounds were involved in several hydrogen bonds (HBs) with some crucial residues of the viral main protease, such as Asn142, Gly143, Cys145, Glu166 and Thr190, as well as in many Van der Waals contacts and a pivotal π-π stacking interaction with His41 (the residue involved in the catalytic dyad together with Cys145). The other four 3CL-PR residues are part of the substrate-binding cleft and are fully conserved among SARS-CoVs (Fig. 1).
Regarding the RdRp, Lys545, Ser549, Lys551 and Arg553 were found to establish a strong hydrogen bonding network with most of the selected molecules, further stabilized by means of additional HBs with some RNA nucleobases, such as uracil at position 18, adenine at position 19 and uracil at position 20, also involved in salt bridges and π-cation interactions (Supporting information, Figs. S6–S10). All 4 residues are located in the RdRp Motif F and are fully conserved among all human coronaviruses (Fig. 2). We observed that all the studied cephalosporins were able to interact with the magnesium cations, both by means of coordination and π-cation interactions, thus rationalizing their better theoretical affinities towards the viral enzyme.
In the molecular recognition of the ACE2 host protein, many residues of the catalytic domain were involved in productive interactions with several analyzed drugs, such as Asp206, Asn394, Arg514 and Lys562, able to establish HBs together with salt bridges and π-cation interactions (Supporting information, Figs. S11–S15). Moreover, argifin and three of the four cephalosporins were able to coordinate the divalent zinc cation present in the catalytic site, thus increasing their stabilization within the enzyme binding pocket.
Finally, by analyzing the ACE2-spike interface region, we observed that most of the identified compounds were involved in HBs with Glu35, Asp38, Lys68 and Glu75 residues of ACE2 enzyme. Moreover, Lys31 and Lys68 were found to establish crucial salt bridges and π-cation interactions with several ligands, as reported in the Supporting Information (Figs. S16–S20).
Among the most promising drugs, ceftolozane, a semi-synthetic broad-spectrum fifth generation cephalosporin approved by the FDA for use in combination with tazobactam for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia, and the coenzyme NADH, useful in treating Parkinson’s disease, chronic fatigue syndrome, Alzheimer’s disease and cardiovascular pathologies, were able to well recognize all the investigated targets, thus providing the best potential multi-targeting profile.
As shown in Fig. 6 A, ceftolozane was involved in 7 HBs and a salt bridge within the 3CL-PR binding pocket, while in the RdRp best pose, the cephalosporin was well stabilized through several interactions among its heterocyclic rings and both RNA nucleobases and lysine residues at positions 545 and 551 (Fig. 6B). Within the ACE2 catalytic site, as well as at the interface with the spike glycoprotein, the anti-bacterial antibiotic was involved in 4 HBs and a salt bridge (Fig. 6D and C).
Fig. 6.

3D representation of the lowest energy pose of ceftolozane. Ceftolozane is docked into A) 3CL-PR, B) RdRp, C) ACE2/spike interface and D) ACE2 proteins. The ligand is depicted in green carbon sticks, whereas the targets are shown, respectively, as salmon, slate, yellow and orange cartoon and the zinc cations are represented as light magenta spheres. Salt bridges, HBs and π-cation interactions are reported as magenta, yellow and dark green dashed lines, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The interesting results related to ceftolozane and to the other identified cephalosporins could rationalize the suggestion of Berry and coworkers (Berry et al., 2015) to incorporate a lactam ring in the lead optimization process of potential SARS-CoV 3CL-PR inhibitors.
As evident from Fig. 7 A, NADH was well stabilized within 3CL-PR binding pocket by means of 8 HBs, while after the molecular recognition of the viral RdRp, the coenzyme was involved in 3 HBs, 2 salt bridges and one π-cation interaction (Fig. 7B). Within the ACE2 catalytic site and at the interface with the spike glycoprotein, NADH was found to establish, respectively, 8 and 6 HBs and a salt bridge (Fig. 7D and C).
Fig. 7.

3D representation of the lowest energy pose of NADH. NADH is docked into: A) 3CL-PR, B) RdRp, C) ACE2/spike interface and D) ACE2 proteins. The ligand is depicted as green carbon sticks, the targets are shown, respectively, as salmon, slate, yellow and orange cartoon and the zinc cations are represented as light magenta spheres. Salt bridges, HBs and π-cation interactions are reported as magenta, yellow and darkgreen dashed lines, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Our results, which are in accord with some published data, suggest a potential multi-targeting behavior of the screened compounds, thus potentially reducing the generation of drug resistant viral strains. We are interested in seeing these compounds move to drug testing as antivirals against SARS-CoV-2, and in vitro studies are underway in our laboratory in this respect.
Conclusions and future directions
The urgent identification of effective interventions against SARS-CoV-2 infection is a major challenge. The development of new effective broad-spectrum drugs targeting coronaviruses will increase the number of therapeutic options for patients with SARS-CoV-2 and other pathogenic coronaviruses infection. In this review the current antiviral strategies and global efforts towards the development of novel compounds aimed at coping with the infection sustained by SARS-CoV-2 and other pathogenic human coronaviruses have been discussed, with a particular focus on antiviral drugs already tested in clinical trials and able to inhibit viral protease, polymerase and spike glycoprotein.
So far, despite the huge number of clinical trials evaluating the efficacy of several drugs against SARS-CoV-2, the RdRp inhibitor remdesivir remains the only antiviral drug currently authorized for the treatment of SARS-CoV-2 infection. Even if it was associated with an improvement in clinical recovery and a reduction in the risk of mortality compared with standard of care, the results require confirmation in future prospective ongoing clinical trials.
With the overall increased numbers of treated patients with new antivirals, it will be important to increase our understanding of efficacy, safety in the context of the COVID 2019 disease, and the molecular mechanisms by which this virus acquires drug resistance. This review also provided insight on the peculiar coronavirus mechanisms underlying resistance, highlighting a sort of innate drug resistance to RdRp nucleoside analogues, mediated by the exonuclease genomic proofreading mechanism. Conversely, paucity of information is available on the profiles of drug resistance mutations against anti-coronavirus agents, so filling this gap is crucial in optimizing antiviral strategies.
Moreover, a large dataset of coronaviruses genomic sequences has been evaluated, thus providing a comprehensive map of conserved regions in the abovementioned viral proteins across human coronaviruses. In particular, the analysis of 3CL-PR, RdRp and spike glycoprotein sequences from SARS-CoV-2 infected patients revealed a very high degree of genetic conservation, with 98.0 %, 98.8 % and 98.5 % amino acid residues showing < 0.1 % variability, respectively. Interestingly, these highly conserved protein regions could represent novel pharmacological targets for compounds with antiSARS-CoV-2 activity to be thoroughly investigated in future studies. Furthermore, by applying an in silico drug repurposing approach, we analyzed the molecular recognition of the DrugBank database against the viral protease, polymerase and the human ACE2 enzyme, alone and in complex with the receptor-binding domain of the spike glycoprotein, thus identifying the most promising compounds displaying a good theoretical binding affinity towards at least two of the four studied targets. In particular, we identified 4 cephalosporins, 2 flavone compounds, 3 purine analogues (including remdesivir), 2 peptide derivatives, 2 triazoles and a benzeneacetamide. Most of the identified compounds were well recognized by the target receptors through several productive interactions with crucial residues of both viral and host proteins, thus increasing their in silico binding affinity. Among which, the 5th generation antibacterial cephalosporin ceftolozane and the coenzyme NADH exhibited the best multi-targeting profile. Interestingly, we observed that all the studied cephalosporins were well ranked against all the targets studied, thus confirming the key role of the β-lactam ring in the drug discovery process as already proposed for SARS-CoV-1 inhibitors. Therefore, a very promising future direction in the research of novel anti-coronavirus agents could be represented by the lead optimization of such a chemical scaffold. Further in vitro studies are necessary in order to verify the antiviral activity of the top rating molecules against SARS-CoV-2 and hopefully their ability to alleviate the emergence as well as surmount drug resistant viral strains.
As abovementioned, the current review provides not just classical evidence, but also highlights innovative antiviral drugs. Other future directions could be developed to also antagonize other nonstructural and structural viral proteins, or modulate essential host elements of viral infection. For instance, other potential targets could be based on the design of drugs targeting cell machinery, such as the stimulation of the autophagy process. Indeed, it is known that SARS-CoV-2 (like other human coronaviruses) can hamper the unfolded protein response (UPR) and consequently the induction of autophagy in order to improve its replicative capacity. In this perspective, a very intriguing therapeutic strategy could be to unlock the virus induced autophagy blockage, thus decreasing viral replication capacity (Boga and Coto-Montes, 2020).
Overall, this review corroborates the urgent need to define highly effective and innovative broad-spectrum antiviral agents aimed at coping the current and the future unavoidable outbreaks of coronaviruses.
Acknowledgement
The authors acknowledge the Mu.Ta.Lig. COST Action (CA15135) for the scientific cooperation and Fondazione Vironet C for financial support to the study.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.drup.2020.100721.
Appendix A. Supplementary data
The following is Supplementary data to this article:
References
- Abhithaj J., et al. 2020. Repurposing Simeprevir, Calpain Inhibitor IV and a Cathepsin F Inhibitor Against SARS-CoV-2: A Study Using in Silico Pharmacophore Modeling and Docking Methods. chemRxiv. [DOI] [Google Scholar]
- Abuhammad A., et al. Computational modeling of the bat HKU4 coronavirus 3CL pro inhibitors as a tool for the development of antivirals against the emerging Middle East respiratory syndrome (MERS) coronavirus. J. Mol. Recognit. 2017;30(11):e2644. doi: 10.1002/jmr.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini M.L., et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio. 2018;9(2):1–15. doi: 10.1128/mBio.00221-18. Edited by K. Subbarao. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini M.L., et al. Small-Molecule Antiviral -D-N4-Hydroxycytidine Inhibits a Proofreading-Intact Coronavirus with a High Genetic Barrier to Resistance. J. Virol. 2019;(October):1–14. doi: 10.1128/JVI.01348-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby T.C., et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science. 2015;347(6223):771–775. doi: 10.1126/science.1259210. [DOI] [PubMed] [Google Scholar]
- Bacha U., et al. Identification of Novel Inhibitors of the SARS Coronavirus Main Protease 3CL pro. Biochemistry. 2004;43(17):4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Kumaki Y. Recent developments in anti-severe acute respiratory syndrome coronavirus chemotherapy. Future Virol. 2011;6(5):615–631. doi: 10.2217/fvl.11.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron S.A., et al. Teicoplanin: an alternative drug for the treatment of COVID-19? Int. J. Antimicrob. Agents. 2020;55(4):105944. doi: 10.1016/j.ijantimicag.2020.105944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrila J., et al. Mutation of Asn28 Disrupts the Dimerization and Enzymatic Activity of SARS 3CL pro. Biochemistry. 2010;49(20):4308–4317. doi: 10.1021/bi1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J.H., et al. Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2007764. NEJMoa2007764. [DOI] [PubMed] [Google Scholar]
- Berry M., Fielding B., Gamieldien J. Potential broad Spectrum inhibitors of the coronavirus 3CLpro: a virtual screening and structure-based drug design study. Viruses. 2015;7(12):6642–6660. doi: 10.3390/v7122963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya C., et al. 2020. Global Spread of SARS-CoV-2 Subtype With Spike Protein Mutation D614G Is Shaped by Human Genomic Variations That Regulate Expression of TMPRSS2 and MX1 Genes. bioRxiv, p. 2020.05.04.075911. [DOI] [Google Scholar]
- Bhimraj A., et al. Infectious diseases society of America guidelines on the treatment and management of patients with COVID-19. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin S.Y., et al. Environmental contamination and viral shedding in MERS patients during MERS-CoV outbreak in South Korea. Clin. Infect. Dis. 2016;62(6):755–760. doi: 10.1093/cid/civ1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaising J., Polyak S.J., Pécheur E.-I. Arbidol as a broad-spectrum antiviral: An update. Antiviral Res. 2014;107:84–94. doi: 10.1016/j.antiviral.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch E.M., et al. Deployment of convalescent plasma for the prevention and treatment of COVID-19. J. Clin. Invest. 2020;130(6):2757–2765. doi: 10.1172/JCI138745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boga J.A., Coto-Montes A. ER stress and autophagy induced by SARS-CoV-2: the targets for melatonin treatment. Melatonin Research. 2020;3(3):346–361. doi: 10.32794/mr11250067. [DOI] [Google Scholar]
- Boheemen S.Van, et al. Genomic characterization of a newly discovered coronavirus. mBio. 2012;3(6):1–9. doi: 10.1128/mBio.00473-12.Editor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., et al. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. U. S. A. 2004;101(22):8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulware D.R., et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Engl. J. Med. 2020:1–9. doi: 10.1056/NEJMoa2016638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B., et al. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. N. Engl. J. Med. 2020;382(19):1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesar M.-M., Alejandro R.-L. 2020. Identification of Potential Inhibitors of SARS-CoV-2 Main Protease Via a Rapid In-silico Drug Repurposing Approach. chemRxiv. [DOI] [Google Scholar]
- Chan J.F.W., et al. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 2012;65(6):477–489. doi: 10.1016/j.jinf.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.W., et al. Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J. Infect. 2013;67(6):606–616. doi: 10.1016/j.jinf.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.-W., et al. Treatment with lopinavir/ritonavir or Interferon-β1b improves outcome of MERS-CoV infection in a nonhuman primate model of common marmoset. J. Infect. Dis. 2015;212(12):1904–1913. doi: 10.1093/infdis/jiv392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.W., et al. Middle east respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015;28(2):465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.-W., et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., et al. Protective effect of intranasal regimens containing peptidic middle east respiratory syndrome coronavirus fusion inhibitor against MERS-CoV infection. J. Infect. Dis. 2015;212(12):1894–1903. doi: 10.1093/infdis/jiv325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.W., Yiu C.P.B., Wong K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000 Research. 2020;9:1–9. doi: 10.12688/f1000research.22457.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.M., et al. Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus. World J. Pediatr. 2020;16:240–246. doi: 10.1007/s12519-020-00345-5. Springer Singapore. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy K.-T., et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res. 2020;178:104786. doi: 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.M., et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59(3):252–256. doi: 10.1136/thorax.2003.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinatl J., et al. Treatment of SARS with human interferons. Lancet. 2003;362(9380):293–294. doi: 10.1016/S0140-6736(03)13973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M.S. Hydroxychloroquine for the prevention of Covid-19 — searching for evidence. N. Engl. J. Med. 2020;383:585–586. doi: 10.1056/NEJMe2020388. p. NEJMe2020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxtall J.D., Perry C.M. Lopinavir/Ritonavir: a review of its use in the management of HIV-1 infection. Drugs. 2010;70(14):1885–1915. doi: 10.2165/11204950-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17(3):181–192. doi: 10.1038/s41579-018-0118-9. Springer US. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., et al. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020;0(0):1–11. doi: 10.1080/07391102.2020.1763201. Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day C.W., et al. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology. 2009;395(2):210–222. doi: 10.1016/j.virol.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H., et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J. Gen. Virol. 2013;94(Part 8):1749–1760. doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H., et al. Creening of an FDA-approved compound library identifies four small-molecule inhibitors of middle east respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014;58(8):4875–4884. doi: 10.1128/AAC.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., et al. Coronaviruses resistant to a 3C-Like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. J. Virol. 2014;88(20):11886–11898. doi: 10.1128/jvi.01528-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M.R., et al. Coronaviruses. RNA Biol. 2011;8(2):270–279. doi: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux C.A., et al. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int. J. Antimicrob. Agents. 2020;55(5):105938. doi: 10.1016/j.ijantimicag.2020.105938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey B.D., et al. L-735,524: The Design of a Potent and Orally Bioavailable HIV Protease Inhibitor. J. Med. Chem. 1994;37(21):3443–3451. doi: 10.1021/jm00047a001. [DOI] [PubMed] [Google Scholar]
- Drożdżal S., et al. FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist. Updates. 2020;53:100719. doi: 10.1016/j.drup.2020.100719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A.A. Ribavirin, remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. 2020;253(January):117592. doi: 10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshabrawy H.A., et al. Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and ebola, Hendra, and nipah viruses by using a novel high-throughput screening assay. J. Virol. 2014;88(8):4353–4365. doi: 10.1128/jvi.03050-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson J., et al. Design, activity, and 2.8 A crystal structure of a C2 symmetric inhibitor complexed to HIV-1 protease. Science. 1990;249(4968):527–533. doi: 10.1126/science.2200122. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency . 2020. First COVID-19 Treatment Recommended for EU Authorization. Available at: https://www.ema.europa.eu/en/documents/press-release/first-covid-19-treatment-recommended-eu-authorisation_en.pdf (Accessed: 3 July 2020) [Google Scholar]
- European Medicines Agency . 2020. Product Information As Approved by the CHMP on 25 June 2020, Pending Endorsement by the European Commission. Available at: https://www.ema.europa.eu/en/medicines/human/summaries-opinion/veklury (Accessed: 3 July 2020) [Google Scholar]
- Falzarano D., De Wit E., Martellaro C., et al. Inhibition of novel β coronavirus replication by a combination of interferon-α2b and ribavirin. Sci. Rep. 2013;3:1–6. doi: 10.1038/srep01686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzarano D., De Wit E., Rasmussen A.L., et al. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV–infected rhesus macaques. Nat. Med. 2013;19(10):1313–1317. doi: 10.1038/nm.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F., et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. 2018;115(2):E162–E171. doi: 10.1073/pnas.1718806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François K.O., Balzarini J. Potential of carbohydrate-binding agents as therapeutics against enveloped viruses. Med. Res. Rev. 2012;32(2):349–387. doi: 10.1002/med.20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y., Komeno T., Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 2017;93(7):449–463. doi: 10.2183/pjab.93.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautret P., et al. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: a pilot observational study. Travel Med. Infect. Dis. 2020;34:101663. doi: 10.1016/j.tmaid.2020.101663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GISAID, 2020. Available at: https://www.gisaid.org/.
- Goldman J.D., et al. Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2015301. p. NEJMoa2015301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P., Peersen O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. 2010;107(52):22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C.J., et al. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020;295(15):4773–4779. doi: 10.1074/jbc.AC120.013056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal B., Goyal D. Targeting the dimerization of main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020;22:297–305. doi: 10.1021/acscombsci.0c00058. [DOI] [PubMed] [Google Scholar]
- Grein J., et al. Compassionate use of remdesivir for patients with severe Covid-19. N. Engl. J. Med. 2020:1–10. doi: 10.1056/nejmoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grum-Tokars V., et al. Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133(1):63–73. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., et al. Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008;133(1):4–12. doi: 10.1016/j.virusres.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D.C., Ji H. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020;(January):101646. doi: 10.1016/j.tmaid.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming I., et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203(2):631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton T. New flu antiviral candidate may thwart drug resistance. JAMA. 2020;323(1):17. doi: 10.1001/jama.2019.20225. [DOI] [PubMed] [Google Scholar]
- Hillen H.S., et al. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;(May) doi: 10.1038/s41586-020-2368-8. Springer US, p. 2020.04.27.063180. [DOI] [PubMed] [Google Scholar]
- Hofmann H., et al. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U.S.A. 2005;102(22):7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holshue M.L., et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020;382(10):929–936. doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M.-F., et al. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280(35):31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., et al. 3C-like Proteinase from SARS Coronavirus Catalyzes Substrate Hydrolysis by a General Base Mechanism. Biochemistry. 2004;43(15):4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- Huang X., et al. Human Coronavirus HKU1 Spike Protein Uses O -Acetylated Sialic Acid as an Attachment Receptor Determinant and Employs Hemagglutinin-Esterase Protein as a Receptor-Destroying Enzyme. J. Virol. 2015;89(14):7202–7213. doi: 10.1128/jvi.00854-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., et al. Pharmacological Therapeutics Targeting RNA-Dependent RNA Polymerase, Proteinase and Spike Protein: From Mechanistic Studies to Clinical Trials for COVID-19. J. Clin. Med. 2020;9(4):1131. doi: 10.3390/jcm9041131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huentelman M.J., et al. Structure-Based Discovery of a Novel Angiotensin-Converting Enzyme 2 Inhibitor. Hypertension. 2004;44(6):903–906. doi: 10.1161/01.HYP.0000146120.29648.36. [DOI] [PubMed] [Google Scholar]
- Hull M.W., Montaner J.S.G. Ritonavir-boosted protease inhibitors in HIV therapy. Ann. Med. 2011;43(5):375–388. doi: 10.3109/07853890.2011.572905. [DOI] [PubMed] [Google Scholar]
- Hulswit R.J.G., et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. U.S.A. 2019;116(7):2681–2690. doi: 10.1073/pnas.1809667116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y., et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jácome R., et al. Sofosbuvir as a potential alternative to treat the SARS-CoV-2 epidemic. Sci. Rep. 2020;10(1):9294. doi: 10.1038/s41598-020-66440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S., et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020:64. doi: 10.1128/AAC.00819-20. e0081920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson & Johnson Services . 2020. Lack of Evidence to Support Use of Darunavir-based Treatments for SARS-CoV-2.https://www.jnj.com/lack-of-evidence-to-support-darunavir-based-hiv-treatments-for-coronavirus Available at: [Google Scholar]
- Keyaerts E., et al. Plant lectins are potent inhibitors of coronaviruses by interfering with two targets in the viral replication cycle. Antiviral Res. 2007;75(3):179–187. doi: 10.1016/j.antiviral.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilianski A., Baker S.C. Cell-based antiviral screening against coronaviruses: developing virus-specific and broad-spectrum inhibitors. Antiviral Res. 2014;101(January):105–112. doi: 10.1016/j.antiviral.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U.J., et al. Combination therapy with lopinavir/ritonavir, ribavirin and interferon-alpha for Middle East respiratory syndrome: a case report. Antivir. Ther. (Lond.) 2015;21(5):455–459. doi: 10.3851/IMP3002. [DOI] [PubMed] [Google Scholar]
- Lan J., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. Springer US. [DOI] [PubMed] [Google Scholar]
- Lauring A.S., Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6(7):1–8. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV) Nat. Rev. Drug Discov. 2020;19(3):149–150. doi: 10.1038/d41573-020-00016-0. Springer US. [DOI] [PubMed] [Google Scholar]
- Li W., et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., et al. Coronavirus infections and immune responses. J. Med. Virol. 2020;92(4):424–432. doi: 10.1002/jmv.25685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6(3):315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M.K., et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep. 2017;7(1):43395. doi: 10.1038/srep43395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Drug treatment options for the 2019-new coronavirus (2019-nCoV) Biosci. Trends. 2020;14(1):69–71. doi: 10.5582/bst.2020.01020. [DOI] [PubMed] [Google Scholar]
- Lu L., et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014;5 doi: 10.1038/ncomms4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. Elsevier Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusvarghi S., Bewley C. Griffithsin: an antiviral lectin with outstanding therapeutic potential. Viruses. 2016;8(10):296. doi: 10.3390/v8100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lythgoe M.P., Middleton P. Ongoing clinical trials for the management of the COVID-19 pandemic. Trends Pharmacol. Sci. 2020;41(6):363–382. doi: 10.1016/j.tips.2020.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcdonald S.M. RNA synthetic mechanisms employed by diverse families of RNA viruses. Wiley Interdiscip. Rev. RNA. 2013;4(4):351–367. doi: 10.1002/wrna.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mille J.K., Whittaker G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. U.S.A. 2014;111(42):15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse J.S., et al. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019‐nCoV. ChemBioChem. 2020;21(5):730–738. doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountain V. Astex, Structural Genomix, and Syrrx. I can see clearly now: structural biology and drug discovery. Chem. Biol. 2003;10(2):95–98. doi: 10.1016/s1074-5521(03)00030-9. [DOI] [PubMed] [Google Scholar]
- Muramatsu T., et al. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. 2016;113(46):12997–13002. doi: 10.1073/pnas.1601327113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neogi U., et al. Feasibility of known RNA polymerase inhibitors as Anti-SARS-CoV-2 drugs. Pathogens. 2020;9(5):320. doi: 10.3390/pathogens9050320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH . National Institutes of Health, NIH; 2020. COVID-19 Treatment Guidelines Panel. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines.https://www.covid19treatmentguidelines.nih.gov/ Available at: [PubMed] [Google Scholar]
- O’Keefe B.R., et al. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family coronaviridae. J. Virol. 2010;84(5):2511–2521. doi: 10.1128/JVI.02322-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestereich L., et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res. 2014;105:17–21. doi: 10.1016/j.antiviral.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Parang K., et al. Comparative antiviral activity of remdesivir and Anti-HIV nucleoside analogs against human coronavirus 229E (HCoV-229E) Molecules. 2020;25(10):2343. doi: 10.3390/molecules25102343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N.C., et al. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018;20(4):378–392. doi: 10.1177/1098612X17729626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera K.D., et al. Characterization of amino acid substitutions in feline coronavirus 3C-like protease from a cat with feline infectious peritonitis treated with a protease inhibitor. Vet. Microbiol. 2019;237(January):108398. doi: 10.1016/j.vetmic.2019.108398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle S., et al. The SARS-Coronavirus-host interactome: identification of cyclophilins as target for pan-Coronavirus inhibitors. PLoS Pathog. 2011;7(10) doi: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantone D., Koudriavtseva T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin. Drug Investig. 2018;38(8):653–671. doi: 10.1007/s40261-018-0656-y. [DOI] [PubMed] [Google Scholar]
- Pruijssers A.J., Denison M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019;35(January):57–62. doi: 10.1016/j.coviro.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F., et al. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020;526(1):135–140. doi: 10.1016/j.bbrc.2020.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang Wang, et al. 2020. Virtual Screening of Approved Clinic Drugs With Main Protease (3CL’, Preprints, (March)https://www.preprints.org/manuscript/202003.0144/v1 Available at: [Google Scholar]
- Raj V.S., et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495(7440):251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts N., et al. Rational design of peptide-based HIV proteinase inhibitors. Science. 1990;248(4953):358–361. doi: 10.1126/science.2183354. [DOI] [PubMed] [Google Scholar]
- Romano M., et al. A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells. 2020;9(5):1267. doi: 10.3390/cells9051267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton N.R., et al. Homology-based identification of a mutation in the coronavirus RNA-Dependent RNA polymerase that confers resistance to multiple mutagens. J. Virol. 2016;90(16):7415–7428. doi: 10.1128/jvi.00080-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y.-F., Li S.-F., Xu G.-J. A novel auto-cleavage assay for studying mutational effects on the active site of severe acute respiratory syndrome coronavirus 3C-like protease. Biochem. Biophys. Res. Commun. 2004;324(2):579–583. doi: 10.1016/j.bbrc.2004.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon A., et al. Remdesivir and SARS-CoV-2: structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antiviral Res. 2020;178(March) doi: 10.1016/j.antiviral.2020.104793. Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T.P., et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017;9(396) doi: 10.1126/scitranslmed.aal3653. p. eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T.P., et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020;12(541):1–21. doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Song J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006;273(5):1035–1045. doi: 10.1111/j.1742-4658.2006.05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K., Kawase M., Matsuyama S. Middle east respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 2013;87(23):12552–12561. doi: 10.1128/jvi.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.C., et al. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 2013;9(8) doi: 10.1371/journal.ppat.1003565. Edited by M. S. Diamond, p. e1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So L.K.-Y., et al. Development of a standard treatment protocol for severe acute respiratory syndrome. Lancet. 2003;361(9369):1615–1617. doi: 10.1016/S0140-6736(03)13265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., et al. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. 2019;11(1) doi: 10.3390/v11010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellbrink H.-J., et al. Antiviral Activity, Pharmacokinetics, and Safety of the HIV-1 Protease Inhibitor TMC310911, Coadministered With Ritonavir, in Treatment-Naive HIV-1–Infected Patients. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2014;65(3):283–289. doi: 10.1097/QAI.0000000000000003. [DOI] [PubMed] [Google Scholar]
- Subissi L., et al. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. 2014;111(37):E3900–E3909. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y., Sato Y., Sasaki T. Suppression of coronavirus replication by cyclophilin inhibitors. Viruses. 2013;5(5):1250–1260. doi: 10.3390/v5051250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Lancet Editors Expression of concern: hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis. Lancet. 2020;395(10240):e102. doi: 10.1016/S0140-6736(20)31290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totura A.L., Bavari S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin. Drug Discov. 2019;14(4):397–412. doi: 10.1080/17460441.2019.1581171. Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent M.J., et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005;2:1–10. doi: 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasak R., et al. Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc. Natl. Acad. Sci. U.S.A. 1988;85(12):4526–4529. doi: 10.1073/pnas.85.12.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., et al. Structure of main protease from human coronavirus NL63: insights for wide Spectrum anti-Coronavirus drug design. Sci. Rep. 2016;6(December 2015):1–12. doi: 10.1038/srep22677. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020;6(1):28. doi: 10.1038/s41421-020-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., et al. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin. Infect. Dis. 2020;71:769–777. doi: 10.1093/cid/ciaa272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren T.K., et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., et al. Exploitation of glycosylation in enveloped virus pathobiology. Biochimica et Biophysica Acta - General Subjects. 2019;1863:1480–1497. doi: 10.1016/j.bbagen.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., et al. Site-specific glycan analysis of the SARS-CoV-2 spike. Sci. 2020;369(May):330–333. doi: 10.1126/science.abb9983. p. eabb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. COVID-NMA project. Cochrane . 2020. Targeted Update: Safety and Efficacy of Hydroxychloroquine or Chloroquine for Treatment of COVID-19. Updated 17 June 2020. Available at: https://www.who.int/publications/m/item/targeted-update-safety-and-efficacy-of-hydroxychloroquine-or-chloroquine-for-treatment-of-covid-19 (Accessed: 5 July 2020) [Google Scholar]
- Wu C.-Y., et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. 2004;101(27):10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Liu M., et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30(4):343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019;5(4) doi: 10.1126/sciadv.aav4580. p. eaav4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Zhu Y., et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020;(February):3–5. doi: 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaya M., et al. Protease inhibitors: candidate drugs to inhibit severe acute respiratory syndrome coronavirus 2 replication. Tohoku J. Exp. Med. 2020;251(1):27–30. doi: 10.1620/tjem.251.27. [DOI] [PubMed] [Google Scholar]
- Yang D., Leibowitz J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends’. Virus Res. 2015;206:120–133. doi: 10.1016/j.virusres.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Bartlam M., Rao Z. Drug design targeting the main protease, the achilles heel of coronaviruses. Curr. Pharm. Des. 2006;12(35):4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- Yao X., et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Clin. Infect. Dis. 2020;71:732–739. doi: 10.1093/cid/ciaa237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager C.L., et al. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature. 1992;357(6377):420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Kusov Y., et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020;63(9):4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- Zhou N., et al. Glycopeptide antibiotics potently inhibit cathepsin l in the late Endosome/Lysosome and block the entry of ebola virus, middle east respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV) J. Biol. Chem. 2016;291(17):9218–9232. doi: 10.1074/jbc.M116.716100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., et al. A novel coronavirus from patients with pneumonia in China 2019. N. Engl. J. Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., et al. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016;15(5):327–347. doi: 10.1038/nrd.2015.37. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.