

Abstract
Cell–cell fusion is indispensable for creating life and building syncytial tissues and organs. Ever since the discovery of cell–cell fusion, how cells join together to form zygotes and multinucleated syncytia has remained a fundamental question in cell and developmental biology. In the past two decades, Drosophila myoblast fusion has been used as a powerful genetic model to unravel mechanisms underlying cell–cell fusion in vivo. Many evolutionarily conserved fusion-promoting factors have been identified and so has a surprising and conserved cellular mechanism. In this review, we revisit key findings in Drosophila myoblast fusion and highlight the critical roles of cellular invasion and resistance in driving cell membrane fusion.
Keywords: myoblast fusion, cell–cell fusion, Drosophila genetics, asymmetric fusogenic synapse, actin-propelled membrane protrusions, mechanosensory response, mechanical tension
INTRODUCTION
Cell–cell fusion is a fascinating process underlying fertilization, skeletal muscle development and regeneration, bone remodeling, immune response, and placenta formation (2, 28, 126). Failure in cell–cell fusion leads to defects such as infertility, congenital myopathy, osteopetrosis, immune deficiency, and pre-eclampsia. Despite the diversity of cell types that undergo fusion, all cell–cell fusion events commence from the recognition and adhesion of two fusion partners and end with the merging of their plasma membranes and union of their cytoplasm.
As with any membrane fusion event, the rate-limiting step of cell–cell fusion is bringing the two membranes destined for fusion into close proximity of one another, thus allowing lipid mixing and fusion pore formation (68). Cell adhesion molecules (CAMs) are obvious facilitators for cell–cell fusion, given their function as velcro between cell membranes. However, numerous cell–cell junctions exist in multicellular organisms between cells that do not fuse, suggesting that cell fusion is a tightly regulated process beyond cell adhesion and that additional cellular machineries must be involved to promote membrane juxtaposition and merger.
For the past two decades, Drosophila myoblast fusion has been used as a powerful genetic model to study cell–cell fusion in vivo (1, 33, 77, 90, 109). Unbiased genetic screens have led to the identification of CAMs, adaptor proteins, actin cytoskeletal regulators, and vesicle trafficking proteins with roles in myoblast fusion (Table 1). Although CAMs are expected components in myoblast fusion, the requirement for the intracellular actin cytoskeleton in promoting cell membrane fusion of myoblasts was initially puzzling. Because the actin cytoskeleton is involved in many cellular processes, such as cell migration, division, adhesion, contraction, protrusion formation, and shape change (95), it was unclear at the time if the actin cytoskeleton had a general function in maintaining the cellular homeostasis of fusion partners or if it played a specific role at sites of fusion.
Table 1.
Molecular components of myoblast fusion
| Fusion regulator | Protein type | Cell type | Localization at fusogenic synapse | Function in myoblast fusion | Reference(s) |
|---|---|---|---|---|---|
| Duf/Kirre | Ig domain–containing CAM | FC | Ring-like structure | Promotes FC–FCM adhesion and binds to Sns and Hbs in tram to establish fusogenic synapse | 104, 121 |
| Rst | Ig domain–containing CAM | FC | ND | Promotes FC–FCM adhesion and binds to Sns and Hbs in tram to establish fusogenic synapse | 121 |
| Sns | Ig domain–containing CAM | FCM | Ring-like structure | Promotes FC–FCM adhesion and binds to Duf and Rst in tram to establish fusogenic synapse | 18, 106 |
| Hbs | Ig domain–containing CAM | FCM | ND | Promotes FC–FCM adhesion and binds to Duf and Rst in tram to establish fusogenic synapse | 5, 48 |
| Sing | Multipass transmembrane protein | ND | ND | Potentially involved in vesicle trafficking | 51 |
| Rols7/Ants | Ankyrin repeat-, tetratricopeptide repeat-, and coiled-coil domain–containing protein | FC | Ring-like structure | Replenishes Duf at the fusogenic synapse by vesicle trafficking | 27, 85, 99 |
| Dock | SH2 and SH3 domain–containing adaptor protein | ND | ND | Links CAMs and actin cytoskeletal regulators | 74 |
| Drk | SH2 and SH3 domain–containing adaptor protein | ND | ND | Links CAMs and actin cytoskeletal regulators | 74 |
| Crk | SH2 and SH3 domain–containing adaptor protein | ND | ND | Links CAMs and actin cytoskeletal regulators | 7, 73, 79 |
| Sltr/WIP | WASP-binding protein | FCM | Actin focus | Recruits WASP to the fusogenic synapse | 79, 84 |
| WASP | Actin NPF | FCM | Actin focus | Promotes branched actin polymerization; required for actin foci formation and membrane protrusion generation | 12, 59, 79, 84, 108, 114 |
| Blow | PH domain–containing protein | FCM | Actin focus | Competes with WASP for WIP binding to destabilize the WASP-WIP complex | 38, 73 |
| Mbc | Bipartite Rac GEF | FCM | Actin focus | Activates Rac proteins together with Elmo | 50, 66, 106 |
| Elmo | Bipartite Rac GEF | FCM | Actin focus | Activates Rac proteins together with Mbc | 58 |
| Rac1 | Small G protein | FCM | Actin focus | Activates Scar complex and group I Pak together with Rac2 | 63, 82 |
| Rac2 | Small G protein | FCM | Actin focus | Activates Scar complex and group I Pak together with Rac1 | 63, 82 |
| Scar/WAVE | Actin NPF | FCM, FC | Actin focus | Promotes branched actin polymerization; required for actin foci formation in FCMs and actin sheath formation in FCs | 12, 59, 100, 114 |
| Kette | Component of the SCAR complex | FCM, FC | Actin focus | Stabilizes the Scar complex; not required for membrane protrusion generation | 65, 73, 111, 114 |
| Arp3 | Component of the Arp2/3 actin nucleator | FCM, FC | Actin focus | Promotes nucleation of branched actin filament | 12, 100 |
| ArpC1 | Component of the Arp2/3 actin nucleator | FCM, FC | Actin focus | Promotes nucleation of branched actin filament | 84 |
| DPak3 | Serine/threonine kinase | FCM | Actin focus | Promotes invasive protrusions with DPak1 | 44 |
| DPak1 | Serine/threonine kinase | FCM | Actin focus | Promotes invasive protrusions with DPak3 | 44 |
| Loner/Schizo | Arf GEF | FCM, FC | Actin focus | Activates Arf proteins | 22, 29 |
| Arf1 | Small G protein | ND | ND | Regulates N-Cad | 42 |
| Arf6 | Small G protein | ND | ND | Regulates Rac localization | 29, 42 |
| Rho1 | Small G protein | FC | Actin sheath | Activates Rok | 78 |
| Rok | Serine/threonine kinase | FC | Actin sheath | Phosphoactivates and activates MyoII | 78 |
| Nonmuscle MyoII | Actin motor | FC | Actin sheath | Mechanosensor; increases cortical tension via actomyosin contraction | 78 |
| βH-Spectrin | Spectrin cytoskeleton subunit | FC | Actin sheath | Mechanoresponsive as a heterotetramer with α-spectrin; restricts Duf at the fusogenic synapse; constricts FCM protrusions | 45 |
| α-Spectrin | Spectrin cytoskeleton subunit | FC | Actin sheath | Mechanoresponsive as a heterotetramer with βH-spectrin; restricts Duf at the fusogenic synapse; constricts FCM protrusions | 45 |
| PIP2 | Phospholipid | FCM, FC | Enriched on membrane | Controls localization of actin regulators at the fusogenic synapse | 17 |
| Dia | Actin NPF | FCM? | Actin focus | ND | 34 |
| D-Titin | Giant filamentous protein | FCM? | Actin focus | ND | 83, 85, 131 |
| WHAMY | Actin NPF | ND | ND | ND | 20 |
| Rab11 | Small G protein | ND | ND | ND | 13 |
Abbreviations: βH-spectrin, βHeavy-spectrin; Ants, Antisocial; Arf1, ADP-ribosylation factor 1; Arf6, ADP-ribosylation factor 6; Arp3, Actin-related protein 3; ArpC1, Actin-related protein C1; Blow, Blown fuse; CAM, cell adhesion molecule; Crk, Crk oncogene; Dia, Diaphanous; Dock, Dreadlocks; DPak1, Drosophila p21-activated kinase 1; DPak3, Drosophila p21-activated kinase 3; Drk, Downstream of receptor kinase; Duf, Dumbfounded; Elmo, Engulfment and cell motility protein; FC, founder cell; FCM, fusion-competent myoblast; GEF, guanine nucleotide exchange factor; Hbs, Hibris; Ig, immunoglobulin; Mbc, Myoblast city; MyoII, myosin II; ND, not determined; NPF, nucleation-promoting factor; PH, pleckstrin homology; PIP2, phosphatidylinositol 4,5-bisphosphate; Rok, Rho kinase; Rols7, Rolling pebbles 7; Rst, Roughest; Scar, Suppresser of cAMP receptor; Sing, Singles bar; Sltr, Solitary; Sns, Sticks and stones;WASP,Wiskott–Aldrich syndrome protein;WAVE,WASP family verprolin homologs; WIP,WASP-interacting protein.
The discovery of actin-enriched structures at sites of fusion opened up a new chapter in studying the cell biology of myoblast fusion (76, 79, 100). Surprisingly, these actin-enriched structures are asymmetric and invasive, drilling fingerlike protrusions from one fusion partner into another (114). The receiving fusion partners, conversely, build stiffer cortices to resist the invasive forces (78). The mechanical interactions between the two fusion partners push the two cell membranes closer than the distance achieved by CAMs alone to promote cell membrane fusion (77). Interestingly, similar actin-propelled invasive protrusions have been found at sites of mammalian cell fusion (98, 119). Indeed, many of the molecular components identified in Drosophila myoblast fusion are also found in mammals (1, 33, 77), demonstrating conserved cellular and molecular mechanisms underlying cell–cell fusion in higher eukaryotes. Here, we review the key findings in Drosophila myoblast fusion over the past two decades and highlight the critical function of the actin cytoskeleton in cell membrane fusion.
EMBRYONIC MYOBLAST FUSION
The musculature of Drosophila is generated de novo twice during its life span. The larval musculature forms during embryogenesis and is then disintegrated during metamorphosis when the adult musculature is generated (112). Both larval and adult musculatures are composed of multinucleated muscle fibers created by the fusion of mononucleated muscle cells. To date, much of the mechanistic understanding of Drosophila myoblast fusion has come from studies of the formation of larval body wall muscles during embryogenesis. We therefore focus our review largely on embryonic myoblast fusion.
RENDEZVOUS OF FUSION PARTNERS: THE INITIAL ATTRACTION AND ENGAGEMENT
Two Types of Embryonic Muscle Cells in Drosophila: Origin and Specification
The embryonic muscle cells in Drosophila arise from the somatic mesoderm (40, 123). Clusters of mesodermal cells become promuscle groups by expressing the transcription factor Lethal of scute (23). Within each group, one cell with higher Ras and Delta expression is specified to be the muscle progenitor cell via Notch-mediated lateral inhibition (24). Activated Notch inhibits muscle progenitor cell formation, whereas activated Ras promotes the progenitor cell fate (4). The muscle progenitor cells then undergo cell division and give rise to either two muscle founder cells or a founder cell and an adult muscle progenitor. The remaining cells in the promuscle cluster become fusion-competent myoblasts (FCMs) (25, 103).
A typical abdominal hemisegment of a Drosophila embryo contains 30 muscle founder cells. These founder cells are further divided into different subsets based on their expression of distinct transcription factors (40), such as the homeodomain proteins S59 (41), Even-skipped (122), and Ladybird (71); the LIM domain protein Apterous (19); the basic helix-loop-helix proteins Vestigial (10) and Nautilus (8); the COE family protein Collier (31); and the zinc-finger protein Krüppel (105). In addition, the chromatin remodeling factor Sin3A is involved in founder cell fate determination (39). Each founder cell resides at a specific location of a hemisegment, and together they prefigure the stereotypical pattern of the multinucleated muscle fibers.
In contrast to founder cells, all the FCMs are specified by a single transcription factor, Lame duck (Lmd) (43). Lmd is a member of the Gli superfamily of zinc-finger-containing transcription factors and activates the expression of FCM-specific genes. In lmd mutant embryos, although founder cells are properly specified, all FCMs are absent, leading to an absence of multinucleated muscle fibers (43). Another zinc-finger transcription factor, Tramtrack 69, is activated by Lmd in FCMs to repress the expression of founder cell–specific genes and to stabilize the FCM cell fate (30).
Fusion Occurs Between Muscle Founder Cells and Fusion-Competent Myoblasts
Fusion between embryonic muscle cells in Drosophila was first suggested by transmission electron microscopy (TEM) analysis that displayed disintegrated plasma membranes between muscle cells (115), and it was later visualized by immunohistochemical studies (9) (Figure 1a). The first genetic evidence for the presence of founder cells in Drosophila embryos came from the study of the myoblast city (mbc) mutant embryos, in which a subset of mononucleated muscle cells elongate to form miniature muscle fibers in the place of the wild-type multinucleated muscle fibers (106) (Figure 1b). These elongated miniature muscle cells were named muscle founder cells because they act as founding seeds to attract the surrounding FCMs and determine the position, orientation, size, and pattern of neuronal innervation of future muscle fibers. FCMs function as building blocks by fusing with neighboring founder cells to generate multinucleated muscle fibers. Upon the completion of each fusion event, the nucleus of the fused FCM adopts the transcription profile of the founder cell, such that the multinucleated myotube behaves as a larger founder cell and proceeds with additional rounds of fusion.
Figure 1.
Milestones in the study of Drosophila myoblast fusion. (a) Multinucleated muscle cells visualized in Drosophila embryo (9). (b) Founder cells identified (106). (c) Cell adhesion molecules discovered (18, 104). (d) Discovery of actin-enriched structure (actin focus) at the site of myoblast fusion (76, 79, 100). (e) Discovery of the asymmetric fusogenic synapse (114). (f) Discovery of the role of mechanical tension in promoting myoblast fusion (78).
Cell Adhesion Molecules: Mediators of Muscle Cell Recognition and Adhesion
Once the muscle cell fates are specified, the distinct transcription factors in founder cells and FCMs begin to activate different sets of target genes to regulate myoblast fusion. Both types of cells express immunoglobulin (Ig) domain–containing CAMs to mediate the recognition and adhesion of founder cells and FCMs (Figure 2a).
Figure 2.
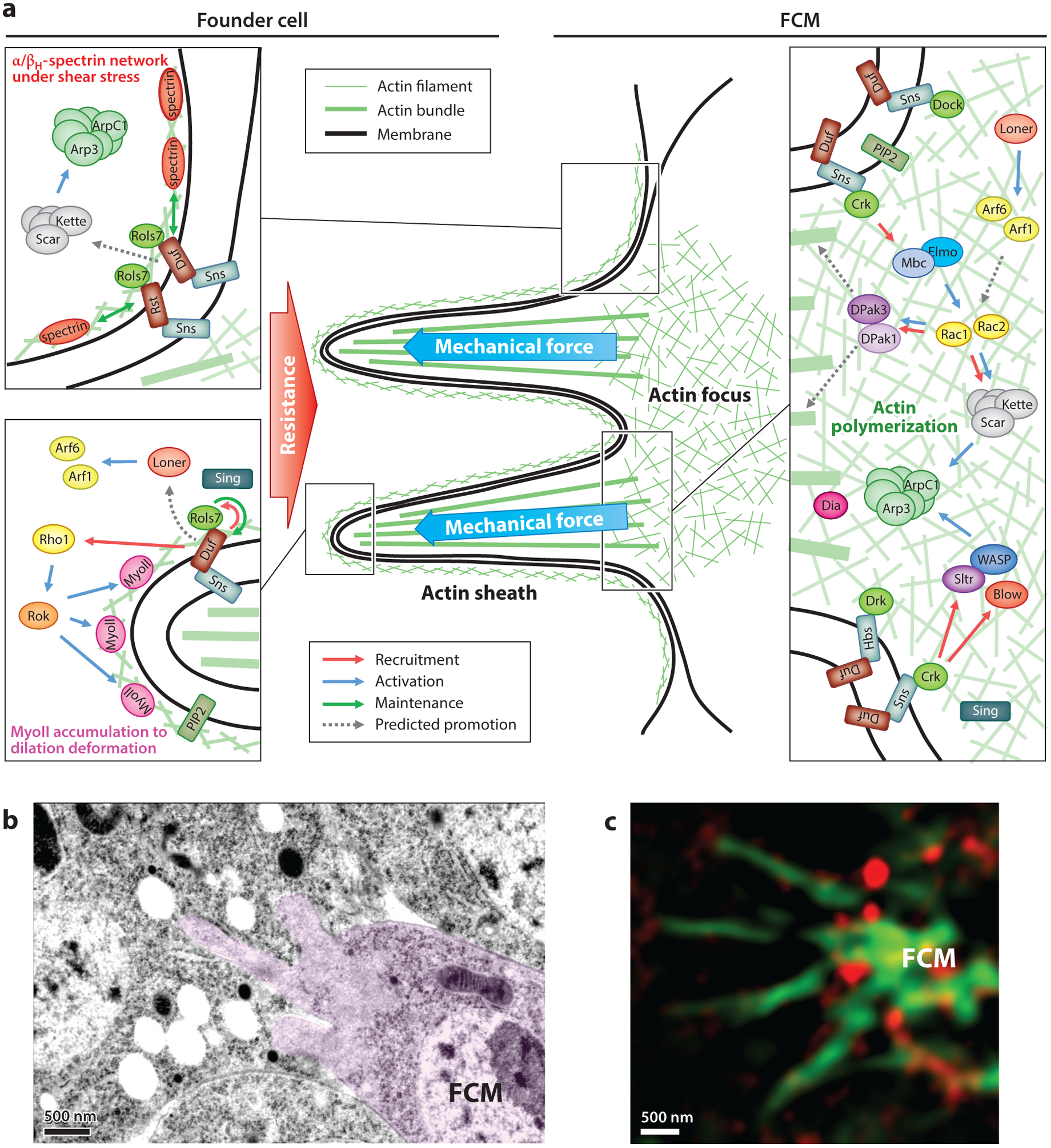
The asymmetric fusogenic synapse. (a) Molecular components and signaling pathways at the fusogenic synapse. Scar, Arp2/3 complex, Loner, Arf, Sing, and PIP2 are shown once in the two founder cell boxes for the sake of simplicity. (b) Invasive protrusions at the fusogenic synapse visualized by transmission electron microscopy. A mononucleated FCM (pink) is projecting membrane protrusions into the binucleated myotube. Note the exclusion of ribosomes and intracellular organelles in the protrusive area, which is filled with actin filaments. Panel b adapted from Reference 114 with permission. (c) The spectrin network constricts the invasive protrusions, visualized by superresolution microscopy. Actin-propelled protrusions (green) from the FCM penetrate through microdomains free of accumulated βH-spectrin (red) in the apposing founder cell. Panel c adapted from Reference 45 with permission. Abbreviations: Arf, ADP-ribosylation factor; Arp2/3, actin-related protein 2/3; βH, βHeavy; Blow, Blown fuse; Crk, Crk oncogene; Dia, Diaphanous; Dock, Dreadlocks; DPak, Drosophila p21-activated kinase; Drk, Downstream of receptor kinase; Duf, Dumbfounded; Elmo, Engulfment and cell motility protein; FCM, fusion-competent myoblast; Hbs, Hibris; Mbc, Myoblast city; MyoII, Myosin II; PIP2, phosphatidylinositol 4,5-bisphosphate; Rok, Rho kinase; Rols, Rolling pebbles; Rst, Roughest; Scar, Suppresser of cAMP receptor/WAVE; Sing, Singles bar; Sns, Sticks and stones; Sltr, Solitary/WIP; WASP, Wiskott–Aldrich syndrome protein; WAVE, WASP family verprolin homologs; WIP, WASP-interacting protein.
Duf and Rst: founder cell–specific adhesion molecules.
Dumbfounded (Duf), also known as Kirre, was the first CAM identified in myoblast fusion via a founder cell–specific enhancer trap, rP298-lacZ, in which lacZ was inserted in the duf promoter (104) (Figure 1c). Overexpressing Duf in epithelial cells attracts FCMs to ectopic locations, demonstrating Duf’s role as a myoblast attractant (104). Loss of Duf together with its paralog Roughest (Rst) causes a complete disruption of myoblast fusion, whereas either single mutant has wild-type musculature, demonstrating the redundant functions of Duf and Rst in the fusion process (104, 121). Indeed, expressing either Duf or Rst in embryonic muscle cells completely rescues the fusion defect in duf,rst double mutant embryos (104, 121).
Sns and Hbs: fusion-competent myoblast–specific adhesion molecules.
Sticks and stones (Sns) was identified based on its fusion-defective mutant phenotype (18) (Figure 1d). Sns and its paralog Hibris (Hbs) are specifically expressed in FCMs (5, 18). Although the hbs mutant exhibits little myoblast fusion defect (5, 48), the sns,hbs double mutant showed a complete lack-of-fusion phenotype that is more severe than the sns single mutant, suggesting that Sns and Hbs are partially redundant in the fusion process with Sns playing a major role (116). Indeed, expressing Sns in FCMs fully rescues the sns,hbs double mutant phenotype, whereas Hbs expression only partially rescues the fusion defect (116). Interestingly, Hbs appears to have a dominant-negative effect on Sns, since overexpression of Hbs in wild-type embryos resulted in a myoblast fusion defect (5), likely due to increased Sns/Hbs heterodimerization (116).
Transinteractions between immunoglobin domain–containing cell adhesion molecules.
Two lines of genetic evidence suggest potential transinteractions between the CAMs in founder cells and FCMs. First, ectopic expression of Duf or Rst in epithelial cells attracts FCMs to these locations (104, 121). Second, in duf,rst double mutant embryos, Sns is no longer enriched at the founder cell and FCM contact sites, but instead becomes evenly distributed at the cell cortex (56). The specific affinities between different CAMs in trans have been revealed by S2 cell aggregation assays, in which two groups of normally nonadherent S2 cells expressing distinct CAMs are tested for intergroup aggregation. Duf and Rst show both homophilic interactions with themselves and heterophilic interactions with Sns (48, 56, 116) and Hbs (48, 116), whereas no homophilic interactions are detected for Sns and Hbs (56). The transheterophilic interaction between Duf and Sns is confirmed by coimmunoprecipitation of these proteins (56). Despite their transheterophilic interactions, overexpressing these founder cell– and FCM-specific CAMs in two populations of S2 cells, respectively, does not induce cell fusion (29, 118). Consistent with this observation, crystallographic studies of Caenorhabditis elegans orthologs of Duf/Rst (SYG-1) and Sns/Hbs (SYG-2) showed that SYG-1 and SYG-2 interact through their most N-terminal Ig domains and form an L-shaped rigid structure that props the cell membranes of two adherent cells apart at a distance of approximately 45 nm (91). This distance is too large for membrane fusion to occur because that would require much closer proximity of 1–2 nm. Thus, CAMs can bring two cells to a certain proximity, and the rigid CAM structures formed in trans may then block further membrane juxtaposition and fusion, consistent with the nonfusogenic nature of most cell–cell junctions in multicellular organisms.
Adaptor Proteins: Relaying the Fusion Signal
The engagement of cell type–specific CAMs establishes future sites of fusion and initiates a cascade of signaling events in both founder cells and FCMs. Adaptor proteins play a critical role in relaying signals from the cell membrane to intracellular components (Figure 2a).
Rols7/Ants: a founder cell–specific adaptor protein.
Three independent studies uncovered a role for rolling pebbles 7 (rols7), also known as antisocial (ants), in myoblast fusion (27, 85, 99). Rols7 encodes a founder cell–specific protein containing ankyrin repeats, tetratricopeptide repeats, and a RING finger (27, 85, 99). Rols7 is localized in a Duf- and Rst-dependent manner at the site of fusion (27, 85). In the duf,rst double mutant embryos, Rols7 becomes evenly distributed throughout the founder cell cytoplasm (27, 85, 86). Ectopic expression of Duf in salivary gland epithelia cells or cultured S2 cells recruits Rols7 to the cell cortex (86). The recruitment of Rols7 by Duf is likely to occur through biochemical interactions of these two proteins (27) mediated by the ankyrin repeats of Rols7 (86). Reciprocally, Rols7 maintains Duf localization at the founder cell membrane (86). In the rols7 mutant, Duf is not detected at the fusion sites and is present at a lower level in the founder cell cytoplasm (86). It is proposed that Rols7 promotes the enrichment of Duf and self-enrichment at the fusion sites by cotranslocation with Duf in exocytic vesicles to the plasma membrane.
Regulation of vesicle trafficking during myoblast fusion remains unclear. To date, two vesicle trafficking-related proteins have been implicated in myoblast fusion, Singles bar (Sing) (51) and Rab11 (13). Sing is a multipass transmembrane protein that belongs to the family of MARVEL domain–containing proteins, which have been implicated in the biogenesis of exocytic vesicles (107). Sing is required for myoblast fusion and is expressed in both founder cells and FCMs (51). Although there is an accumulation of intracellular vesicles in sing mutant embryos, the translocation of Duf and Rols7 is not affected, leaving open the question whether Sing is involved in vesicle trafficking during myoblast fusion (51). Rab11 is another vesicle trafficking-related protein implicated in myoblast fusion. Overexpression of the constitutively active or dominant-negative form of Rab11 caused a mild myoblast fusion defect (13). However, it is unclear whether endogenous Rab11 and/or other Rab GTPases contribute to myoblast fusion.
SH2 and SH3 domain–containing adaptor proteins: Dock, Drk, and Crk.
A potential role for the Src Homology 2 (SH2) and Src Homology 3 (SH3) domain–containing proteins dreadlocks (Dock), downstream of receptor kinase (Drk), and Crk oncogene (Crk) in myoblast fusion was first implicated by biochemical studies. Both Crk and Dock have been shown to bind the FCM-specific CAMs Sns and Hbs in addition to downstream actin cytoskeletal regulators (7, 73, 74, 79), which suggests that these proteins function as adaptors linking the CAMs and the actin cytoskeleton. In addition, Dock also binds Duf in founder cells (74). dock and drk single mutants and the dock,drk double mutant do not show any myoblast fusion defect, likely due to maternal contributions and potential functional redundancy with crk (74). The localization of crk on the small fourth chromo-some, of which few genetic tools are available, has hampered the generation of a crk single mutant and a dock,drk;crk triple mutant. Nevertheless, the dock mutant shows genetic interactions with mutations in sns, hbs, and genes regulating the actin cytoskeleton, which indicates its functional significance in myoblast fusion (74).
INTERACTIONS BETWEEN FUSION PARTNERS: INCREASED INTIMACY DRIVEN BY ASYMMETRIC ACTIN POLYMERIZATION AND ACTOMYOSIN DYNAMICS
Although the Ig domain–containing CAMs are essential for myoblast recognition and adhesion, they are not sufficient to bring cell membranes into close enough contact for fusion. Genetic analyses have led to the identification of many actin polymerization and actomyosin contractility regulators essential for myoblast fusion. Subsequent cell biological and ultrastructural analyses have revealed that a major function of the actin cytoskeleton is pushing cell membranes into closer proximity to one another at the site of fusion.
Actin Polymerization–Mediated Mechanical Force Generation by Fusion-Competent Myoblasts
Of the two muscle cell types, FCMs are the more aggressive fusion partners. They utilize the actin polymerization machinery to propel invasive protrusions into the apposing founder cells. In this section, we review how actin polymerization regulators were discovered and how they function together to control the invasive behavior of the FCMs (Figure 2a).
Actin polymerization regulators: essential roles revealed by genetic analyses.
Drosophila genetics has been instrumental in identifying actin polymerization regulators in myoblast fusion. These discoveries have led to exciting directions of investigation and a deep understanding of the cellular mechanisms underlying cell–cell fusion.
Mbc, Elmo, and Rac.
myoblast city (mbc) was the very first myoblast fusion mutant identified in Drosophila (106). Its human homolog DOCK180 was shown to alter cell morphology at the time of discovery (67), suggesting a link between myoblast fusion and the actin cytoskeleton (50). DOCK180 forms a complex with Engulfment and cell motility protein (ELMO), and the DOCK180–ELMO complex functions as a bipartite guanine nucleotide exchange factor (GEF) for the small GTPase Rac (21). Indeed, the Drosophila elmo maternal/zygotic mutant also exhibits a severe myoblast fusion defect, and Elmo can enhance Mbc-dependent activation of Rac proteins (58). The first evidence linking Rac with myoblast fusion came from genetic experiments in Drosophila expressing constitutively active Rac1V12 or dominant-negative Rac1N17 in embryonic muscle cells, both of which resulted in severe myoblast fusion defects (82). Subsequent loss-of-function analyses revealed normal somatic musculature in the rac1 and rac2 single mutants, but severe fusion defects in the rac1,rac2 double mutant (63), demonstrating that Rac1 and Rac2 function redundantly in myoblast fusion.
Kette, Scar, and Arp2/3.
Rac regulates the actin cytoskeleton by activating the suppresser of cAMP receptor (Scar)/WASP family verprolin homologs (WAVE) complex, which is a five-subunit protein complex consisting of Scar/WAVE, Sra1/PIR, Kette/Nap1/Hem, Abi, and HSPC300 (96). Scar is a member of the WASP (Wiskott–Aldrich syndrome protein) family of actin nucleation-promoting factors (NPFs) for the actin-related protein (Arp)2/3 complex, a seven-subunit protein complex that nucleates actin monomers to form branched actin filaments (120). A role for kette in myoblast fusion was originally identified by an unbiased screen of ethyl methanesulfonate (EMS)-induced mutant alleles (111). The kette mutant exhibits a strong myoblast fusion defect despite undergoing a low level of fusion (111), further implicating the actin cytoskeleton in myoblast fusion. Later, scar was also identified as a molecular component in myoblast fusion. Although the scar zygotic mutant shows a mild fusion defect, embryos with reduced maternal and zygotic scar contributions exhibit a severe fusion defect (100). Consistent with the requirement for the Scar complex in myoblast fusion, mutants of arp3 and arpC1, which encode two components of the Arp2/3 complex, are also defective in myoblast fusion (12, 84, 100).
WASP and WIP.
WASP is another NPF for the Arp2/3 complex (96). WASP is stabilized in a tight complex by WASP-interacting protein (WIP) (57). A role for WASP in myoblast fusion was uncovered through both forward and reverse genetic approaches (79, 84, 108). While the zygotic wasp null mutant has largely normal musculature, the maternal/zygotic wasp mutant shows severe defects in myoblast fusion, suggesting that the maternally contributed WASP masks the effects of zygotic mutations (79, 84). Indeed, a zygotic wasp mutant carrying a truncated form of the protein that misses the Arp2/3-binding domain functions as a dominant-negative form by interfering with the maternally contributed WASP (84, 108). A role for Drosophila WIP (D-WIP), also known as Solitary (Sltr), in myoblast fusion was revealed by an EMS mutagenesis screen (79) and a reverse genetic analysis on the basis of its interaction with WASP (84). sltr mutant displays a severe myoblast fusion defect despite a low level of fusion (79, 84). Interestingly, Sltr expression is restricted to FCMs prior to fusion, because no Sltr expression is detected in the lmd mutant embryos (79, 84). This is the first actin cytoskeletal regulator found to be expressed in only one of the two muscle cell types.
F-actin focus: zooming in to the site of fusion.
The identification of many actin cytoskeletal regulators through genetic analyses highlights the essential function of the actin cytoskeleton in myoblast fusion. Subsequently, striking actin cytoskeletal rearrangements at sites of fusion were revealed by confocal microscopy (76, 79, 100). Prior to each fusion event, there is a burst of actin polymerization at the founder cell and FCM contact site, resulting in the formation of a dense oval-shaped F-actin focus (76, 79, 100) (Figure 1d). Time-lapse imaging confirmed that these actin foci indeed correspond to sites of fusion and that they assemble and dissolve within a 5.7–29.5-min time frame with an average life span of approximately 11.9 min (100). Many of the actin regulators implicated in myoblast fusion colocalize with the actin foci, such as WASP (108), Sltr (79), Mbc, and Kette (100). These actin foci appear to be surrounded by ring-like structures formed by CAMs and adaptor proteins, such as Duf, Sns, and Rols7 (76, 114). Initially, however, the cellular origin of the actin foci was unclear. One study suggested that a larger portion of each actin focus resided in the FCM (79), whereas other studies concluded that each actin focus was evenly divided between the adherent founder cell and the FCM (76, 100).
Breaking symmetry: discovery of the invasive podosome-like structure and the asymmetric fusogenic synapse.
Confusion over the cellular origin of the actin foci was resolved by cell type– specific expression of green fluorescent protein (GFP)-tagged actin (114). While GFP-actin expressed in FCMs colocalizes with the oval-shaped dense actin foci, GFP-actin expressed in founder cells does not accumulate at the sites of fusion, demonstrating that the actin foci are generated exclusively in FCMs (114) (Figure 1e). Although founder cells do not accumulate dense actin foci, there is a thin sheath of actin underlying the founder cell membranes, which is barely visible in wild-type embryos (114). The FCM-specific actin focus (with an average size of 1.7 μm2) dynamically changes its shape and protrudes toward the founder cell, causing an inward curvature on the founder cell membrane (114). Electron microscopy (EM) analyses revealed multiple actin-propelled fingerlike protrusions, each with an average diameter of 250 nm projecting as long as 1.9 μm into the founder cell (114) (Figure 2b). Each actin focus contains an average of 4.3 fingerlike protrusions (114). When viewed along the protrusive axis, the dense actin focus is seen surrounded by a ring of CAMs (76, 114). The characteristic organization of the actin focus surrounded by adhesion molecules, together with its dynamic and protrusive nature, resembles that of a podosome, which has been extensively studied in the migration and adhesion of cultured cell (26). This actin-enriched protrusive structure in the FCM is therefore termed a podosome-like structure (PLS) (114). Such an actin-propelled invasive structure has also been observed in cultured Drosophila primary muscle cells (66). The closely juxtaposed cell membrane contact zone mediated by invasive protrusions encircled by CAMs at the site of fusion has since been referred to as the fusogenic synapse (114).
WASP and Scar complexes: distinct requirement, recruitment, and function.
Although both WASP and Scar are members of the WASP family and activate the actin nucleation activity of Arp2/3, they have distinct functions in myoblast fusion due to their differential expression and modes of recruitment.
Differential requirement in founder cells and fusion-competent myoblasts.
The WASP and Scar complexes are differentially required in the two types of muscle cells. Several lines of evidence support an FCM-specific role for the WASP–Sltr complex. First, Sltr is specifically expressed in FCMs (79, 84). Second, no fusion defect is induced when dominant-negative forms of WASP or Sltr are expressed in founder cells (84). And third, expressing WASP in founder cells alone does not rescue the fusion defect in wasp mutant embryos as does WASP expression in all muscle cells (108). Cell type–specific rescue of sltr with an UAS–Sltr transgene is technically challenging, since the UAS–Sltr transgene has leaky expression in all muscle cells even without a GAL4 driver (P. Jin & E. Chen, unpublished material). Conversely, the Scar complex is required in both founder cells and FCMs, because expressing Scar (or Kette) in all muscle cells—but not in either founder cells or FCMs alone—fully rescues the scar (or kette) mutant phenotype (65, 114). Despite the requirement for the Scar complex in both muscle cell types, its upstream regulators, Mbc and Rac, have been shown to function specifically in FCMs (66). FCM-specific expression of Mbc (or Rac1) is sufficient to fully rescue the mbc (or rac1,rac2) mutant phenotype (66). This raises the intriguing question of how the Scar complex is regulated in founder cells.
The requirement for both WASP and Scar complexes in FCMs and for Scar complex alone in founder cells correlates with the distinct actin polymerization patterns in these two cells—the formation of oval-shaped dense actin foci in FCMs and thin actin sheaths in founder cells. In single mutants of either NPF (or their interacting proteins), the FCM-generated actin remains enriched at the asymmetric fusogenic synapse due to the presence of the remaining NPF (59, 66, 79, 100, 114). Only when the activities of both WASP and Scar complexes are eliminated in double mutants, such as sltr,scar and sltr;kette, do the actin foci no longer form at the fusogenic synapse (34, 114), which leads to a complete block of myoblast fusion (12, 114). Thus, WASP and Scar complexes have redundant functions in actin foci formation.
Different modes of recruitment to the fusogenic synapse.
All of the above-mentioned actin cytoskeletal regulators colocalize with the actin foci at the fusogenic synapse (59, 66, 79, 100, 108, 114). The FCM-specific CAM Sns recruits Sltr, likely through the SH2 and SH3 domain– containing adaptor protein Crk, and Sltr in turn recruits WASP to the fusogenic synapse via biochemical interactions (79, 84). Sns may also recruit Mbc though Crk and other unknown factors (7). The small GTPase Rac, downstream of Mbc, recruits the Scar complex to the fusogenic synapse (59). The recruitment of the two NPF complexes is independent of each other. For example, the Sltr enrichment at the fusogenic synapse is unaffected in the mbc mutant and the rac1,rac2 double mutant (79), and the Scar enrichment is undisrupted in sltr mutants (59).
Distinct cellular functions.
Although both WASP and Scar have similar biochemical functions in activating Arp2/3, their cellular functions are not interchangeable. Expression of Scar in the sltr mutant does not rescue the fusion defect caused by the absence of Sltr (114). Moreover, WASP is required for the invasiveness of the PLS, because in sltr mutant embryos actin-filled fingers are either wide and tubby or folded upon each other and unable to project into the apposing founder cell (114). In contrast, kette mutant embryos show invasive protrusions similar to those in the wild type embryos, suggesting that the Scar complex is not required for the invasiveness of these protrusions (65, 114). Normal PLS invasion in the kette mutant raises the question of how the loss of the Scar complex disrupts myoblast fusion. Since Scar is also required in founder cells to generate the thin sheath of actin, it is conceivable that the loss of Scar may weaken the response of founder cells to the invasive protrusions from the FCMs. Indeed, expressing WASP or Sltr in founder cells to activate Arp2/3 in the kette mutant partially rescues the fusion defect (65), likely by replenishing the actin sheath. An additional function of Kette has been proposed in which Kette may be involved in dissolving the fusion-inhibitory cellular junctions formed by the CAM N-cadherin (N-Cad), although the mechanism underlying the Kette and N-Cad interaction is unknown (65).
Regulation of podosome-like structure dynamics and organization: Blow and Pak.
When the actin foci were initially characterized, it was thought that the size of the foci was a major determining factor for their function (100). Subsequent analyses revealed that actin polymerization dynamics and actin filament organization within the actin foci are the key determinants for PLS function.
Blown fuse.
blown fuse (blow) was first identified as an axon guidance mutant (125), but later studies found that the primary defect in the blow mutant embryos was in myoblast fusion (38). Blow is another FCM-specific protein colocalizing with the actin foci, like WASP and Sltr (73). Its recruitment to the fusogenic synapse is mediated by Sns and Crk but is independent of Mbc, Kette, and Sltr (73). Biochemically, Blow competes with WASP for Sltr binding, thus modulating the stability of the WASP–Sltr complex (73). Because the WASP–Sltr complex binds to the barbed ends of actin filaments, Blow-mediated WASP–Sltr destabilization facilitates the dissociation of WASP from the actin filaments, which results in filament end capping and initiation of new branched actin filaments. Indeed, fluorescent recovery after photobleaching analyses demonstrate rapid and full recovery of GFP-actin at the fusogenic synapse in wild-type embryos but slow and partial recovery in the blow mutant (73). EM studies reveal compromised protrusions in blow mutant, suggesting that the dynamic branched actin polymerization is required for generating short and mechanically stiff actin filaments suitable for propelling invasive protrusions (73).
Group I p21-activated kinases.
A Drosophila group I p21-activated kinase (Pak), DPak3, was identified from a deficiency screen as a myoblast fusion-promoting protein (44). Paks are serine/threonine kinases known to regulate actin cytoskeletal organization (3, 16). Two Drosophila group I Paks, DPak3 and DPak1, have partially redundant functions in myoblast fusion, with DPak3 playing a major role (44). DPak3 is specifically required in FCMs, because FCM-specific DPak3 expression fully rescues the dpak3 mutant phenotype (44). DPak3 is enriched at the fusogenic synapse, colocalizing with the F-actin focus within the PLS, and this enrichment is dependent on Rac but independent of Sltr or Kette (44). EM analysis of dpak3 mutant embryos showed compromised invasive protrusions that contain ribosomes and intracellular organelles, demonstrating a role for DPak3 in organizing the actin filaments within the PLS into a densely packed network, which in turn generates sufficient mechanical force to promote PLS invasion and fusion pore formation (44).
Other fusion-promoting factors with unclear functions in myoblast fusion.
Besides the actin polymerization regulators described above, several other proteins have actin cytoskeleton-associated functions during myoblast fusion. However, their precise roles in the fusion process have not been clearly demonstrated.
Loner.
loner, also known as schizo, was identified in an EMS mutagenesis screen for myoblast fusion mutants (29). Loner is a member of the BRAG family of ADP-ribosylation factor (Arf) GEFs, which are known to activate the Arf GTPases at plasma membranes and endosomes (32). A function of Loner in founder cells is supported by its expanded expression in Notch mutant embryos that contain more founder cells, its localization at the vicinity of the founder cell nuclear marker rP298-lacZ, and its recruitment by Duf to cell contact sites in cultured Drosophila cells (29). However, because the FCM-specific transcription factor Lmd, the PLS at the fusogenic synapse, and muscle type–specific GAL4 drivers were neither identified nor available at the time of the initial characterization of Loner, its potential role in FCMs could not be assessed. Later studies demonstrated a role for Loner in both founder cells and FCMs, based on its expression in both cell populations and the partial rescue of the loner mutant phenotype by either founder cell– or FCM-specific expression of the gene (42, 100). The function of Loner in the fusion process, however, remains unclear. One study showed that Loner controls the localization of Rac (29), whereas another showed that Loner does not play a role in actin foci formation at the fusogenic synapse (100). Protein interaction studies revealed that Loner interacts with two CAMs, Duf (22) and N-Cad (42). Although N-Cad itself is not required for myoblast fusion, it exhibits genetic interactions with loner. Removing N-cad suppresses the fusion defect of the loner mutant (42). A similar genetic interaction was observed between N-cad and kette, suggesting that both Loner and Kette may be involved in dissolving N-Cad-containing cellular junctions that may inhibit myoblast fusion (65). However, it remains unclear how these N-Cad junctions are related to the fusogenic synapse.
Arf.
Structure–function analysis of Loner suggests that its GEF activity is essential for myoblast fusion (29). In vitro GDP release assays showed that the GEF domain of Loner has specific activity toward Arf6 but not Arf1. Founder cell–specific expression of a dominant-negative form of Arf6 results in a mild fusion defect (29). In addition, a role for Arf6 in the fusion of cultured mammalian muscle cells has been demonstrated (6). However, arf6 null mutant is homozygous viable (49, 70) without exhibiting any myoblast fusion defects (42), suggesting that Arf6 either is not required for myoblast fusion in vivo or has a redundant function with other Arfs in the fusion process. Similar redundant functions among the Rac GTPases have been demonstrated in myoblast fusion (63). For example, expression of dominant-negative Rac1 in muscle cells causes a severe myoblast fusion defect, whereas the rac1 single mutant has normal musculature due to the redundant function of rac2 (63, 82). A later study showed that Loner binds a dominant-negative form of Arf1 (Arf1DN) with higher affinity than that of Arf6 (42). Moreover, the expression of Arf1DN in all muscle cells results in a mild fusion defect in some embryos and the expression of constitutively active Arf1 in all muscle cells partially suppresses the fusion defects in loner mutant embryos (42). Taken together, these results suggest that Arf1 and Arf6 may have redundant functions in myoblast fusion, with Arf1 playing a major role. Further investigations are required to resolve the Arf conundrum.
D-Titin.
D-Titin is a giant filamentous protein best known for its role in sarcomere assembly and function (124). A role for D-Titin in myoblast fusion has been identified based on a partial loss-of-fusion phenotype in D-Titin mutant embryos (83, 131). D-Titin is expressed in the somatic mesoderm and accumulates at the myoblast–myotube contact sites (85, 131). Furthermore, D-Titin colocalizes with Sltr at the fusogenic synapse (79), suggesting that it may be involved in regulating actin filament organization within the PLS. Despite these observations, the precise function of D-Titin in the fusion process requires further investigation.
Diaphanous.
Diaphanous (Dia) is one of the Drosophila formins that promote linear actin nucleation (61). Dia colocalizes with the actin foci at the fusogenic synapse in FCMs, and its recruitment is dependent on the FCM-specific CAM Sns but independent of all the upstream regulators of Arp2/3 (34). dia mutant embryos and embryos expressing a dominant-negative form of Dia in all muscle cells exhibit thinner muscle fibers with reduced number of nuclei, indicating a potential block of myoblast fusion. However, it is unclear whether this effect is caused by a specific defect in the fusion process or an earlier defect in FCM maturation, because most of the mononucleated cells marked by diaDN-GFP do not appear to express the muscle structural protein myosin heavy chain (MHC), indicating that they are not fully differentiated (34). In addition, it is unclear whether Dia polymerizes actin at the fusogenic synapse because Dia remains enriched at these sites in sltr;kette mutant embryos, where F-actin foci fail to form (34). Further experiments are necessary to clarify the endogenous function of Dia in myoblast fusion.
Whamy.
WHAMY is another member of the WASP family of Arp2/3 NPFs (101). The whamy single mutant has normal musculature, but together with a mutation in wasp, the whamy,wasp double mutant exhibits a mild fusion defect (20). Overexpressed WHAMY in somatic mesoderm does not localize to the fusogenic synapse but rather to muscle attachment sites (20), raising the question of how WHAMY coordinates with WASP to regulate myoblast fusion.
Mechanosensitive Response-Mediated Tension Generation by Founder Cells
The invasive protrusions from the FCM at the fusogenic synapse create a hand-in-glove type of interaction between the two fusion partners (Figure 2a). Such an interaction significantly increases the cell surface contact area between the two cell membranes and allows intimate apposition of the two lipid bilayers beyond that brought about by CAMs. Recent studies revealed that founder cells are not passive fusion partners. Instead, they mount mechanosensitive responses to the invasive protrusions from FCMs to push back the FCM (78) (Figure 1f), restrict the boundary of the fusogenic synapse (45), and sculpt the invasive protrusions to facilitate cell membrane fusion (45) (Figure 2a).
The mechanosensitive actomyosin network: building up the resistance.
A function for the small GTPase Rho1 was uncovered in myoblast fusion when expressing a dominant-negative form of Rho1 in muscle cells caused a fusion defect (78). It is well known that Rho1 activates Rho kinase (Rok), which in turn phosphorylates the regulatory light chain (RLC) of the nonmuscle myosin II (MyoII) to activate MyoII (15). Rho1, Rok, and MyoII are all enriched at the fusogenic synapse, and, strikingly, only on the side of the founder cell (78). Despite the normal musculature in the rho1 and rok single mutant embryos due to maternal contribution, the rok;rho1 double mutant embryos exhibit a myoblast fusion defect (78). Founder cell–specific expression of phosphomimetic, but not nonphosphorylatable, MyoII RLC partially rescues the rok;rho1 mutant phenotype, demonstrating that the function of Rho1–Rok is to activate MyoII in founder cells (78).
The enrichment of MyoII at the fusogenic synapse can be mechanically triggered, because MyoII still accumulates in the absence of Duf-mediated Rho1–Rok accumulation in Drosophila embryos (78). In support of this, mechanosensitive accumulation of MyoII has been demonstrated by two complementary biophysical approaches, micropipette aspiration (MPA) and atomic force microscopy (AFM), which apply pulling and pushing forces to cells, respectively. Under both conditions, MyoII accumulates in response to applied mechanical force prior to Rho1–Rok accumulation in a motor domain–dependent manner (78). These experiments highlight a role of MyoII as a mechanosensor during myoblast fusion.
The accumulation of MyoII at the fusogenic synapse increases cortical stiffness and provides resistance to the invasive protrusions. This has been demonstrated by the presence of abnormally long invasive protrusions from FCMs in embryos where MyoII activity is decreased in founder cells (78). Moreover, when MyoII is knocked down in the receiving fusion partner in a cell-fusion culture system (118), cortical stiffness decreases as measured by MPA and AFM accompanied by a defect in cell–cell fusion (78). However, overexpression of the actin crosslinker Fimbrin in MyoII knockdown cells restores the cortical tension and significantly rescues the cell fusion defect (78). Thus, MyoII elevates cortical stiffness in the receiving fusion partner to resist the PLS invasion and keep the apposing cell membranes at a close distance for fusion.
The mechanosensitive spectrin network: restricting the boundary of the fusogenic synapse and sculpting the invasive protrusions.
Spectrin was identified by a deficiency screen for myoblast fusion mutants (45). Prior to the functional studies of spectrin in myoblast fusion, it was best known as a membrane skeleton scaffold protein critical for maintaining cell shape and providing mechanical support for the plasma membrane (11, 129). The basic unit of spectrin is a flexible, chain-like α/β heterotetramer with actin-binding domains localized at the two ends. Of the two β-spectrins in Drosophila, βHeavy-spectrin (βH-spectrin), also known as Karst (Kst), and β-spectrin, only the former plays a role in myoblast fusion (45). Cell type–specific rescue experiments demonstrate a founder cell–specific role for the α/βH-spectrin heterotetramers in myoblast fusion (45). Correspondingly, α/βH-spectrin are enriched at the fusogenic synapse in the founder cell, closely abutting the actin focus in the FCM (45).
In contrast to its well-established scaffolding function, βH-spectrin is dynamically recruited to the fusogenic synapse and dissolves together with the actin focus upon fusion pore formation (45). Recruitment of βH-spectrin to the fusogenic synapse still occurs in the absence of Duf-mediated chemical signaling, suggesting that βH-spectrin may accumulate in response to mechanical stimuli. Indeed, biophysical analyses using MPA and AFM demonstrate a mechanosensitive accumulation of βH-spectrin, which requires its actin-binding and tetramerization activities (45). Moreover, MPA experiments and mathematical modeling demonstrate that βH-spectrin responds to shear deformation, which corresponds to the base areas of invasive protrusions (45). Accumulated α/βH-spectrin, in turn, forms a physical barrier for future protrusions from the FCM, such that new protrusions can penetrate only through spectrin-free domains and trigger additional spectrin accumulation in these areas. Eventually, an uneven α/βH-spectrin network forms with a few small spectrin-free microdomains that allow the penetrance of narrow invasive protrusions (45). Such a spectrin network at the fusogenic synapse has at least two functions. First, it serves as a cellular fence to restrict CAMs to the fusogenic synapse via biochemical interactions and steric hindrance. Second, it serves as a cellular sieve to constrict the diameter of the invasive protrusions (45) (Figure 2c). The increased mechanical tension generated by the narrow protrusions helps overcome energy barriers for membrane apposition and drives cell membrane fusion.
LIPID BILAYER MERGER: FORMATION OF FUSION PORES
The ultimate goal of CAM engagement and actin cytoskeletal rearrangement/contraction at the fusogenic synapse is to bring the two lipid bilayers into close proximity and prime them for fusion. To date, fusogenic proteins directly involved in fusion pore formation in Drosophila muscle cells have not be identified. However, EM studies have revealed the morphology of fusion pores between fusing muscle cells, and the functions of membrane lipids in the fusion process have begun to be uncovered.
Ultrastructural analyses of the fusogenic synapse.
The first comprehensive EM study of embryonic myoblast fusion identified several characteristic structures along the muscle cell contact zone: paired electron-dense vesicles (termed prefusion complexes); relatively rare electron-dense plaques; and multiple membrane discontinuities (MMDs) with diameters between 50 and 250 nm (38). It was proposed that the vesicles release electron-dense materials to form the plaques, which induce the formation of multiple fusion pores along the muscle cell contact zone (38). Subsequent studies over the next ten years reproduced these structures using the same conventional room temperature chemical fixation method, making this a prevailing model describing the distinct steps of myoblast fusion. However, additional EM studies using the same method revealed similar MMDs between muscle cells in fusion mutants, as well as between cells that do not normally fuse (114), raising the question whether these MMDs on the plasma membrane are indeed fusion pores. Because the conventional fixation method may not allow ultrafast penetration of fixatives into tissues underneath the epithelial cell layer, it is prone to generating artifacts. Thus, the high-pressure freezing and freeze substitution (HPF/FS) method was applied to Drosophila embryos in order to achieve optimal preservation, especially of plasma membranes in mesodermal cells.
With the HPF/FS method, MMDs are no longer observed between muscle cells in wild-type and fusion mutant embryos (114), indicating that these may be artifacts generated by the conventional method due to insufficient fixation and the subsequent extraction of the membranous materials by osmium treatment. HPF/FS EM analyses revealed fusion pores as single-channel openings connecting the two fusing cells (114). Because of the ultrafast expansion of fusion pores under 200 nm demonstrated by biophysical analysis (75, 93, 94), the smallest fusion pores observed by EM so far have diameters of approximately 300 nm (114).
The FCM-projected fingerlike protrusions at the fusogenic synapse were first revealed by HPF/FS EM analyses and confirmed by conventional EM studies (114). The invasive fingerlike protrusions provide a clear morphological marker for the fusogenic synapse at the ultrastructural level. No electron-dense vesicles or plaques are associated with the invasive protrusions at the fusogenic synapses, suggesting that these structures either may correspond to early events in the fusion process prior to PLS formation or are irrelevant to myoblast fusion. The former possibility is supported by the presence of electron-dense vesicles associated with micro-tubules pointing toward muscle cell contact sites without actin enrichment, suggesting that these vesicles may undergo exocytosis and may be involved in CAM trafficking (79). The function of electron-dense plaques is completely unknown. A recent study found an N-Cad-dependent increase of electron-dense plaques in the kette mutant embryos, suggesting that the plaques may block myoblast fusion (65). However, it is unclear how these plaques are related to the fusogenic synapse.
Phosphatidylinositol 4,5-bisphosphate: involvement in myoblast fusion.
Fusion pore formation requires the destabilization of the two lipid bilayers, leading to the formation of a membranous opening that connects the two cells. Thus, investigating the function of lipids is important for understanding myoblast fusion. To date, only one lipid has been implicated in Drosophila myoblast fusion, which is phosphatidylinositol 4,5-bisphosphate (PIP2) (17). PIP2 is enriched at the fusogenic synapse, and its enrichment is dependent on CAMs but not actin regulators (17). It has been shown that overexpressing PIP2-binding pleckstrin homology (PH) domain of phospholipase C gamma (PLCγ) in muscle cells severely blocks myoblast fusion presumably by sequestering PIP2 (17). In addition, Rac, Scar, and WASP are no longer enriched at the fusogenic synapse in PLCγPH-overexpressing embryos, a finding that supports a potential contribution of PIP2 signaling in actin foci formation (17). However, most of the unfused cells in these fusion-defective embryos were labeled by PLCγPH-GFP expression and were MHC negative (17), suggesting that these mononucleated cells may not have fully differentiated. Therefore, the specific function of PIP2 at the fusogenic synapse needs to be assessed in embryos with decreased PIP2 levels in properly differentiated muscle cells.
PUPAL MYOBLAST FUSION
Drosophila adult musculature accommodates diverse types of movements, including flying, walking, mating, and feeding. Adult muscles form by myoblast fusion during pupal development either by building on a larval muscle scaffold or through a de novo process (62). Specifically, the larval oblique muscles escape histolysis and become the template for the dorsal longitudinal muscles (DLMs) (52, 54). On the other hand, the dorsoventral muscles (DVMs) are generated de novo without a pre-existing template (54). DLMs and DVMs constitute the indirect flight muscles (IFM).
Relatively little is known about the mechanisms underlying pupal myoblast fusion, partly because most loss-of-function alleles of genes promoting pupal myoblast fusion have early lethal phases (112). The application of RNA interference (RNAi) (37, 89) has made pupal musculature a genetically amenable system to study the molecular mechanisms of myoblast fusion (88, 110).
A Repeated Fate Determination of Myoblasts—Similar Fusion Partners and Adhesion Molecules
The adult muscle progenitors are first set aside during embryogenesis and then proliferate to generate myoblast precursors during metamorphosis (55). Similar to embryonic myoblast fusion, adult myoblast precursors are also specified into two populations, founder cells and FCMs (40). Most of the precursors differentiate into FCMs, whereas a small population takes on the founder cell fate. Interestingly, adult founder cell specification does not require Notch-mediated lateral inhibition (46) but requires fibroblast growth factor signaling (47). The founder cells, together with the persisting larval muscle scaffold for the DLMs, specifically express Duf (46, 60) and seed future adult muscle fibers (102). Most studies of pupal myoblast fusion have been performed with the DLMs, within which the larval muscle templates can be clearly identified.
A unique aspect of pupal myoblast fusion is the requirement for long-range cell migration prior to the fusion process. As myoblasts reach the vicinity of founder cells or the larval muscle scaffold, their Notch signaling is switched off such that they can differentiate into mature FCMs expressing Sns and Hbs (60). Subsequent local migration of FCMs is mediated by CAMs (60). Ex vivo culture of isolated IFMs identified the presence of long filopodia emanating from the template myotubes, which facilitate heterotypic adhesion between myotubes and FCMs (113). Formation of the filopodia requires a processive actin polymerase, Enabled, and a BAR-domain protein, IRSp53 (113). Founder cell– and FCM-specific CAMs accumulate at the sites of contact between myotube filopodia and FCMs (113). Sns and Hbs function more equally in the pupal FCMs than in embryos, and double knockdown of Sns and Hbs, but not single knockdowns, results in pupal myoblast fusion defects (60). EM analysis of sns and hbs double knockdown muscles showed looser adhesion between FCMs and template myotubes, where most of the contact sites are more than 50 nm apart, compared to <22 nm apart in wild type (35), consistent with a function of Sns and Hbs in mediating muscle cell adhesion.
Another Round of Mechanical Interactions Between Muscle Cells
Pupal and embryonic myoblast fusion appear to share similar molecular and cellular mechanisms of bringing membranes into close proximity following cell adhesion. The first genetic clue indicating a function for the actin cytoskeleton came from myoblast-specific expression of dominant-negative Rac1, which causes fusion defects in DLM and DVM (46, 53). Subsequently, a role for wasp in pupal myoblast fusion was uncovered (88). In wasp hypomorphic mutants, muscle fibers are underdeveloped with differentiated but unfused myoblasts clumping around them, and somatic muscle expression of dominant-negative WASP results in a similar phenotype (88). RNAi knockdown of WASP, Scar, and Arp2/3 also leads to compromised fusion, demonstrating a critical role for Arp2/3-mediated branched actin polymerization in pupal myoblast fusion (88). Consistent with the similar molecular requirement, actin enrichment is observed at the contact sites between pupal myoblasts and myotubes (88), as is observed at the fusogenic synapses in embryos (76, 79, 100). Moreover, cell type–specific labeling of F-actin with Moesin-GFP showed FCM-specific actin foci and a myotube-specific actin sheath, recapitulating the asymmetric fusogenic synapse in embryonic myoblast fusion (88, 114). Not surprisingly, the Arp2/3 complex and its NPFs are enriched within the actin foci (88).
Ultrastructural analysis of the pupal fusogenic synapse also revealed fingerlike protrusions (35) similar to those in embryos (114). These protrusions are relatively rare in the wild type as compared to the fusion-defective wasp mutant or the kette or sing knockdown pupae (35), consistent with fewer actin foci in wild-type compared to fusion-mutant embryos. The distances between adherent muscle cells were measured in the pupae, and the wild-type cells were found to have the closest distance, followed by the intermediate distance between the Arp2/3 knockdown cells and the largest distance between sns and hbs double knockdown cells (35). However, these measurements were not taken in the areas of invasive protrusions. Therefore, it is unclear how these measurements are related to the fusogenic synapse. Additional EM analyses showed multiple closely abutting points of membrane along the muscle cell contact zone, as well as multiple membranous openings (35). Because these EM studies used a hybrid method of both conventional room temperature chemical fixation and HPF/FS, it is unclear whether the small openings (<50 nm) resulted from insufficient chemical fixation as shown in embryos (38, 114).
COMMON MECHANISMS AND MAJOR UNANSWERED QUESTIONS
Common mechanisms of cell–cell fusion have emerged from studies of both embryonic and pupal myoblast fusion in Drosophila. Both systems use actin-propelled invasive membrane protrusions to bring two cell membranes into close proximity following cell adhesion (Figure 2a). Similar protrusions have also been observed in cultured Drosophila S2R+ cells induced to fuse (118), in fusing mammalian myoblasts and osteoclasts (98, 119), and in C. elegans epithelial seam cell–hyp7 cell fusion (127). These findings suggest that different cell–cell fusion events utilize a common cellular strategy to promote cell–cell fusion.
Despite these exciting discoveries, several major questions remain unanswered in Drosophila myoblast fusion. First, transmembrane fusogenic proteins have yet to be identified. Recent studies of mouse myoblast fusion uncovered a bipartite myoblast-specific fusogen, Myomaker and Myomixer/Myomerger/Minion (14, 87, 97, 128). Although these two proteins are conserved in vertebrate myoblast fusion (36, 80, 117, 130), neither has a Drosophila homolog, suggesting that fusogens are likely to be species- and tissue-specific. Second, the functions of various lipids in Drosophila myoblast fusion remain unclear. Mammalian studies implicated phosphatidylserine (PS) (72, 81) and PS receptors BAI1, BAI3, and Stabilin-2 in myoblast fusion (64, 69, 92). The potential functions of PS and many other lipids in Drosophila myoblast fusion remain elusive. Third, calcium signaling has been implicated in myoblast fusion for decades, but its potential function at the fusogenic synapse is completely unknown. Fourth, although there have been significant insights into the process of actin polymerization at the fusogenic synapse, we do not yet understand how the actin foci are depolymerized upon fusion pore formation.
CONCLUDING REMARKS
The past two decades have witnessed significant progress in our understanding of conserved mechanisms underlying myoblast fusion. The discovery of the asymmetric fusogenic synapse involving invasive protrusions and the corresponding mechanosensitive response led to a biophysical framework of cell–cell fusion. This new conceptual framework highlights the interplay of pushing and resisting forces between the two fusion partners, which brings apposing cell membranes into close proximity and facilitates fusogen engagement and membrane fusion (Figure 2a). These findings have fundamentally changed our view of how cells fuse and provide a solid foundation upon which future studies can be built. With a sophisticated toolbox leveraging new methods from genetics, cell biology, biochemistry, and biophysics, the next decade will undoubtedly bring about many new discoveries in this exciting field.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health grants R01 AR053173 and R01 GM098816, an American Heart Association Established Investigator Award, and a Howard Hughes Medical Institute Faculty Scholar Award to E.H.C. D.M.L. is supported by a Canadian Institute of Health Research postdoctoral fellowship.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abmayr SM, Pavlath GK. 2012. Myoblast fusion: lessons from flies and mice. Development 139:641–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar PS, Baylies MK, Fleissner A, Helming L, Inoue N, et al. 2013. Genetic basis of cell–cell fusion mechanisms. Trends Genet. 29:427–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arias-Romero LE, Chernoff J. 2008. A tale of two Paks. Biol. Cell 100:97–108 [DOI] [PubMed] [Google Scholar]
- 4.Artero R, Furlong EE, Beckett K, Scott MP, Baylies M. 2003. Notch and Ras signaling pathway effector genes expressed in fusion competent and founder cells during Drosophila myogenesis. Development 130:6257–72 [DOI] [PubMed] [Google Scholar]
- 5.Artero RD, Castanon I, Baylies MK. 2001. The immunoglobulin-like protein Hibris functions as a dose-dependent regulator of myoblast fusion and is differentially controlled by Ras and Notch signaling. Development 128:4251–64 [DOI] [PubMed] [Google Scholar]
- 6.Bach A-S, Enjalbert S, Comunale F, Bodin S, Vitale N, et al. 2010. ADP-ribosylation factor 6 regulates mammalian myoblast fusion through phospholipase D1 and phosphatidylinositol 4,5-bisphosphate signaling pathways. Mol. Biol. Cell 21:2412–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balagopalan L, Chen M-H, Geisbrecht ER, Abmayr SM. 2006. The CDM superfamily protein MBC directs myoblast fusion through a mechanism that requires phosphatidylinositol 3,4,5-triphosphate binding but is independent of direct interaction with DCrk. Mol. Cell. Biol 26:9442–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balagopalan L, Keller CA, Abmayr SM. 2001. Loss-of-function mutations reveal that the Drosophila nautilus gene is not essential for embryonic myogenesis or viability. Dev. Biol 231:374–82 [DOI] [PubMed] [Google Scholar]
- 9.Bate M 1990. The embryonic development of larval muscles in Drosophila. Development 110:791–804 [DOI] [PubMed] [Google Scholar]
- 10.Bate M, Rushton E, Frasch M. 1993. A dual requirement for neurogenic genes in Drosophila myogenesis. Dev. 119(Suppl.):149–61 [PubMed] [Google Scholar]
- 11.Bennett V, Lorenzo DN. 2013. Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr. Top. Membr 72:1–37 [DOI] [PubMed] [Google Scholar]
- 12.Berger S, Schäfer G, Kesper DA, Holz A, Eriksson T, et al. 2008. WASP and SCAR have distinct roles in activating the Arp2/3 complex during myoblast fusion. J. Cell Sci 121:1303–13 [DOI] [PubMed] [Google Scholar]
- 13.Bhuin T, Roy JK. 2009. Rab11 is required for myoblast fusion in Drosophila. Cell Tissue Res 336:489–99 [DOI] [PubMed] [Google Scholar]
- 14.Bi P, Ramirez-Martinez A, Li H, Cannavino J, McAnally JR, et al. 2017. Control of muscle formation by the fusogenic micropeptide myomixer. Science 356:323–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop AL, Hall A. 2000. Rho GTPases and their effector proteins. Biochem. J 348:241–55 [PMC free article] [PubMed] [Google Scholar]
- 16.Bokoch GM. 2003. Biology of the p21-activated kinases. Annu. Rev. Biochem 72:743–81 [DOI] [PubMed] [Google Scholar]
- 17.Bothe I, Deng S, Baylies M. 2014. PI(4,5)P2 regulates myoblast fusion through Arp2/3 regulator localization at the fusion site. Development 141:2289–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bour BA, Chakravarti M, West JM, Abmayr SM. 2000. Drosophila SNS, a member of the immunoglobulin superfamily that is essential for myoblast fusion. Genes Dev 14:1498–511 [PMC free article] [PubMed] [Google Scholar]
- 19.Bourgouin C, Lundgren SE, Thomas JB. 1992. apterous is a Drosophila LIM domain gene required for the development of a subset of embryonic muscles. Neuron 9:549–61 [DOI] [PubMed] [Google Scholar]
- 20.Brinkmann K, Winterhoff M, Önel S-F, Schultz J, Faix J, Bogdan S. 2016. WHAMY is a novel actin polymerase promoting myoblast fusion, macrophage cell motility and sensory organ development in Drosophila. J. Cell Sci 129:604–20 [DOI] [PubMed] [Google Scholar]
- 21.Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, et al. 2002. Unconventional Rac–GEF activity is mediated through the Dock180–ELMO complex. Nat. Cell Biol 4:574–82 [DOI] [PubMed] [Google Scholar]
- 22.Bulchand S, Menon SD, George SE, Chia W. 2010. The intracellular domain of Dumbfounded affects myoblast fusion efficiency and interacts with Rolling pebbles and Loner. PLOS ONE 5:e9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carmena A, Bate M, Jiménez F. 1995. lethal of scute, a proneural gene, participates in the specification of muscle progenitors during Drosophila embryogenesis. Genes Dev 9:2373–83 [DOI] [PubMed] [Google Scholar]
- 24.Carmena A, Buff E, Halfon MS, Gisselbrecht S, Jiménez F, et al. 2002. Reciprocal regulatory interactions between the Notch and Ras signaling pathways in the Drosophila embryonic mesoderm. Dev. Biol 244:226–42 [DOI] [PubMed] [Google Scholar]
- 25.Carmena A, Gisselbrecht S, Harrison J, Jiménez F, Michelson AM. 1998. Combinatorial signaling codes for the progressive determination of cell fates in the Drosophila embryonic mesoderm. Genes Dev 12:3910–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen EH. 2011. Invasive podosomes and myoblast fusion. Curr. Top. Membr 68:235–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen EH, Olson EN. 2001. Antisocial, an intracellular adaptor protein, is required for myoblast fusion in Drosophila. Dev. Cell 1:705–15 [DOI] [PubMed] [Google Scholar]
- 28.Chen EH, Olson EN. 2005. Unveiling the mechanisms of cell-cell fusion. Science 308:369–73 [DOI] [PubMed] [Google Scholar]
- 29.Chen EH, Pryce BA, Tzeng JA, Gonzalez GA, Olson EN. 2003. Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector ARF6. Cell 114:751–62 [DOI] [PubMed] [Google Scholar]
- 30.Ciglar L, Girardot C, Wilczyński B, Braun M, Furlong EEM, et al. 2014. Coordinated repression and activation of two transcriptional programs stabilizes cell fate during myogenesis. Development 141:2633–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crozatier M, Vincent A. 1999. Requirement for the Drosophila COE transcription factor Collier in formation of an embryonic muscle: transcriptional response to notch signalling. Development 126:1495–504 [DOI] [PubMed] [Google Scholar]
- 32.D’Souza RS, Casanova JE. 2016. The BRAG/IQSec family of Arf GEFs. Small GTPases 7:257–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng S, Azevedo M, Baylies M. 2017. Acting on identity: myoblast fusion and the formation of the syncytial muscle fiber. Semin. Cell Dev. Biol 72:45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng S, Bothe I, Baylies MK. 2015. The formin Diaphanous regulates myoblast fusion through actin polymerization and Arp2/3 regulation. PLOS Genet 11:e1005381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhanyasi N, Segal D, Shimoni E, Shinder V, Shilo B-Z, et al. 2015. Surface apposition and multiple cell contacts promote myoblast fusion in Drosophila flight muscles. J. Cell Biol 211:191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Gioia SA, Connors S, Matsunami N, Cannavino J, Rose MF, et al. 2017. A defect in myoblast fusion underlies Carey–Fineman–Ziter syndrome. Nat. Commun 8:16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dietzl G, Chen D, Schnorrer F, Su K-C, Barinova Y, et al. 2007. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448:151–56 [DOI] [PubMed] [Google Scholar]
- 38.Doberstein SK, Fetter RD, Mehta AY, Goodman CS. 1997. Genetic analysis of myoblast fusion: blown fuse is required for progression beyond the prefusion complex. J. Cell Biol 136:1249–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobi KC, Halfon MS, Baylies MK. 2014. Whole-genome analysis of muscle founder cells implicates the chromatin regulator Sin3A in muscle identity. Cell Rep 8:858–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobi KC, Schulman VK, Baylies MK. 2015. Specification of the somatic musculature in Drosophila. Wiley Interdiscip. Rev. Dev. Biol 4:357–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dohrmann C, Azpiazu N, Frasch M. 1990. A new Drosophila homeo box gene is expressed in mesodermal precursor cells of distinct muscles during embryogenesis. Genes Dev 4:2098–111 [DOI] [PubMed] [Google Scholar]
- 42.Dottermusch-Heidel C, Groth V, Beck L, Önel S-F. 2012. The Arf-GEF Schizo/Loner regulates N-cadherin to induce fusion competence of Drosophila myoblasts. Dev. Biol 368:18–27 [DOI] [PubMed] [Google Scholar]
- 43.Duan H, Skeath JB, Nguyen HT. 2001. Drosophila Lame duck, a novel member of the Gli superfamily, acts as a key regulator of myogenesis by controlling fusion-competent myoblast development. Development 128:4489–500 [DOI] [PubMed] [Google Scholar]
- 44.Duan R, Jin P, Luo F, Zhang G, Anderson N, Chen EH. 2012. Group I PAKs function downstream of Rac to promote podosome invasion during myoblast fusion in vivo. J. Cell Biol 199:169–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duan R, Kim JH, Shilagardi K, Schiffhauer E, Lee D, et al. 2018. Spectrin is a mechanoresponsive protein shaping fusogenic synapse architecture during myoblast fusion. Nat. Cell Biol 20:688–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dutta D, Anant S, Ruiz-Gomez M, Bate M, VijayRaghavan K. 2004. Founder myoblasts and fibre number during adult myogenesis in Drosophila. Development 131:3761–72 [DOI] [PubMed] [Google Scholar]
- 47.Dutta D, Shaw S, Maqbool T, Pandya H, VijayRaghavan K. 2005. Drosophila Heartless acts with Heart-broken/Dof in muscle founder differentiation. PLOS Biol 3:e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dworak HA, Charles MA, Pellerano LB, Sink H. 2001. Characterization of Drosophila hibris, a gene related to human nephrin. Development 128:4265–76 [DOI] [PubMed] [Google Scholar]
- 49.Dyer N, Rebollo E, Domínguez P, Elkhatib N, Chavrier P, et al. 2007. Spermatocyte cytokinesis requires rapid membrane addition mediated by ARF6 on central spindle recycling endosomes. Development 134:4437–47 [DOI] [PubMed] [Google Scholar]
- 50.Erickson MRS, Galletta BJ, Abmayr SM. 1997. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J. Cell Biol 138:589–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Estrada B, Maeland AD, Gisselbrecht SS, Bloor JW, Brown NH, Michelson AM. 2007. The MARVEL domain protein, Singles Bar, is required for progression past the pre-fusion complex stage of myoblast fusion. Dev. Biol 307:328–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandes J, Bate M, Vijayraghavan K. 1991. Development of the indirect flight muscles of Drosophila. Development 113:67–77 [DOI] [PubMed] [Google Scholar]
- 53.Fernandes JJ, Atreya KB, Desai KM, Hall RE, Patel MD, et al. 2005. A dominant negative form of Rac1 affects myogenesis of adult thoracic muscles in Drosophila. Dev. Biol 285:11–27 [DOI] [PubMed] [Google Scholar]
- 54.Fernandes JJ, Keshishian H. 1996. Patterning the dorsal longitudinal flight muscles (DLM) of Drosophila: insights from the ablation of larval scaffolds. Development 122:3755–63 [DOI] [PubMed] [Google Scholar]
- 55.Figeac N, Jagla T, Aradhya R, Da Ponte JP, Jagla K. 2010. Drosophila adult muscle precursors form a network of interconnected cells and are specified by the rhomboid-triggered EGF pathway. Development 137:1965–73 [DOI] [PubMed] [Google Scholar]
- 56.Galletta BJ, Chakravarti M, Banerjee R, Abmayr SM. 2004. SNS: adhesive properties, localization requirements and ectodomain dependence in S2 cells and embryonic myoblasts. Mech. Dev 121:1455–68 [DOI] [PubMed] [Google Scholar]
- 57.García E, Jones GE, Machesky LM, Antón IM. 2012. WIP: WASP-interacting proteins at invadopodia and podosomes. Eur. J. Cell Biol 91:869–77 [DOI] [PubMed] [Google Scholar]
- 58.Geisbrecht ER, Haralalka S, Swanson SK, Florens L, Washburn MP, Abmayr SM. 2008. Drosophila ELMO/CED-12 interacts with Myoblast city to direct myoblast fusion and ommatidial organization. Dev. Biol 314:137–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gildor B, Massarwa R, Shilo B-Z, Schejter ED. 2009. The SCAR and WASp nucleation-promoting factors act sequentially to mediate Drosophila myoblast fusion. EMBO Rep 10:1043–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gildor B, Schejter ED, Shilo B-Z. 2012. Bidirectional Notch activation represses fusion competence in swarming adult Drosophila myoblasts. Development 139:4040–50 [DOI] [PubMed] [Google Scholar]
- 61.Goode BL, Eck MJ. 2007. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem 76:593–627 [DOI] [PubMed] [Google Scholar]
- 62.Gunage RD, Dhanyasi N, Reichert H, VijayRaghavan K. 2017. Drosophila adult muscle development and regeneration. Semin. Cell Dev. Biol 72:56–66 [DOI] [PubMed] [Google Scholar]
- 63.Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, et al. 2002. Rac function and regulation during Drosophila development. Nature 416:438–42 [DOI] [PubMed] [Google Scholar]
- 64.Hamoud N, Tran V, Croteau L-P, Kania A, Côté J-F. 2014. G-protein coupled receptor BAI3 promotes myoblast fusion in vertebrates. PNAS 111:3745–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hamp J, Löwer A, Dottermusch-Heidel C, Beck L, Moussian B, et al. 2016. Drosophila Kette coordinates myoblast junction dissolution and the ratio of Scar-to-WASp during myoblast fusion. J. Cell Sci 129:3426–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haralalka S, Shelton C, Cartwright HN, Katzfey E, Janzen E, Abmayr SM. 2011. Asymmetric Mbc, active Rac1 and F-actin foci in the fusion-competent myoblasts during myoblast fusion in Drosophila. Development 138:1551–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, et al. 1996. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol. Cell. Biol 16:1770–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hernández JM, Podbilewicz B. 2017. The hallmarks of cell–cell fusion. Development 144:4481–95 [DOI] [PubMed] [Google Scholar]
- 69.Hochreiter-Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, et al. 2013. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497:263–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang J, Zhou W, Dong W, Watson AM, Hong Y. 2009. Directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. PNAS 106:8284–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jagla T, Bellard F, Lutz Y, Dretzen G, Bellard M, Jagla K. 1998. ladybird determines cell fate decisions during diversification of Drosophila somatic muscles. Development 125:3699–708 [DOI] [PubMed] [Google Scholar]
- 72.Jeong J, Conboy IM. 2011. Phosphatidylserine directly and positively regulates fusion of myoblasts into myotubes. Biochem. Biophys. Res. Commun 414:9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin P, Duan R, Luo F, Zhang G, Hong SN, Chen EH. 2011. Competition between Blown fuse and WASP for WIP binding regulates the dynamics of WASP-dependent actin polymerization in vivo. Dev. Cell 20:623–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaipa BR, Shao H, Schäfer G, Trinkewitz T, Groth V, et al. 2013. Dock mediates Scar- and WASp-dependent actin polymerization through interaction with cell adhesion molecules in founder cells and fusion-competent myoblasts. J. Cell Sci 126:360–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaplan D, Zimmerberg J, Puri A, Sarkar DP, Blumenthal R. 1991. Single cell fusion events induced by influenza hemagglutinin: studies with rapid-flow, quantitative fluorescence microscopy. Exp. Cell Res 195:137–44 [DOI] [PubMed] [Google Scholar]
- 76.Kesper DA, Stute C, Buttgereit D, Kreisköther N, Vishnu S, et al. 2007. Myoblast fusion in Drosophila melanogaster is mediated through a fusion-restricted myogenic-adhesive structure (FuRMAS). Dev. Dyn 236:404–15 [DOI] [PubMed] [Google Scholar]
- 77.Kim JH, Jin P, Duan R, Chen EH. 2015. Mechanisms of myoblast fusion during muscle development. Curr. Opin. Genet. Dev 32:162–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim JH, Ren Y, Ng WP, Li S, Son S, et al. 2015. Mechanical tension drives cell membrane fusion. Dev. Cell 32:561–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim S, Shilagardi K, Zhang S, Hong SN, Sens KL, et al. 2007. A critical function for the actin cytoskeleton in targeted exocytosis of prefusion vesicles during myoblast fusion. Dev. Cell 12:571–86 [DOI] [PubMed] [Google Scholar]
- 80.Landemaine A, Rescan PY, Gabillard JC. 2014. Myomaker mediates fusion of fast myocytes in zebrafish embryos. Biochem. Biophys. Res. Commun 451:480–84 [DOI] [PubMed] [Google Scholar]
- 81.Leikina E, Melikov K, Sanyal S, Verma SK, Eun B, et al. 2013. Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J. Cell Biol 200:109–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luo L, Liao YJ, Jan LY, Jan YN. 1994. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev 8:1787–802 [DOI] [PubMed] [Google Scholar]
- 83.Machado C, Andrew DJ. 2000. D-Titin: a giant protein with dual roles in chromosomes and muscles. J. Cell Biol 151:639–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Massarwa R, Carmon S, Shilo B-Z, Schejter ED. 2007. WIP/WASp-based actin-polymerization machinery is essential for myoblast fusion in Drosophila. Dev. Cell 12:557–69 [DOI] [PubMed] [Google Scholar]
- 85.Menon SD, Chia W. 2001. Drosophila Rolling pebbles: a multidomain protein required for myoblast fusion that recruits D-Titin in response to the myoblast attractant Dumbfounded. Dev. Cell 1:691–703 [DOI] [PubMed] [Google Scholar]
- 86.Menon SD, Osman Z, Chenchill K, Chia W. 2005. A positive feedback loop between Dumbfounded and Rolling pebbles leads to myotube enlargement in Drosophila. J. Cell Biol 169:909–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Millay DP, O’Rourke JR, Sutherland LB, Bezprozvannaya S, Shelton JM, et al. 2013. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 499:301–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mukherjee P, Gildor B, Shilo B-Z, VijayRaghavan K, Schejter ED. 2011. The actin nucleator WASp is required for myoblast fusion during adult Drosophila myogenesis. Development 138:2347–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ni J-Q, Zhou R, Czech B, Liu L-P, Holderbaum L, et al. 2011. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8:405–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Önel SF, Rust MB, Jacob R, Renkawitz-Pohl R. 2014. Tethering membrane fusion: common and different players in myoblasts and at the synapse. J. Neurogenet 28:302–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Özkan E, Chia PH, Wang RR, Goriatcheva N, Borek D, et al. 2014. Extracellular architecture of the SYG-1/SYG-2 adhesion complex instructs synaptogenesis. Cell 156:482–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park S-Y, Yun Y, Lim J-S, Kim M-J, Kim S-Y, et al. 2016. Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat. Commun 7:10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Plonsky I, Cho M-S, Oomens AGP, Blissard G, Zimmerberg J. 1999. An analysis of the role of the target membrane on the Gp64-induced fusion pore. Virology 253:65–76 [DOI] [PubMed] [Google Scholar]
- 94.Plonsky I, Zimmerberg J. 1996. The initial fusion pore induced by baculovirus GP64 is large and forms quickly. J. Cell Biol 135:1831–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pollard TD, Cooper JA. 2009. Actin, a central player in cell shape and movement. Science 326:1208–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pollitt AY, Insall RH. 2009. WASP and SCAR/WAVE proteins: the drivers of actin assembly. J. Cell Sci 122:2575–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Quinn ME, Goh Q, Kurosaka M, Gamage DG, Petrany MJ, et al. 2017. Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat. Commun 8:15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Randrianarison-Huetz V, Papaefthymiou A, Herledan G, Noviello C, Faradova U, et al. 2018. Srf controls satellite cell fusion through the maintenance of actin architecture. J. Cell Biol 217:685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rau A, Buttgereit D, Holz A, Fetter R, Doberstein SK, et al. 2001. rolling pebbles (rols) is required in Drosophila muscle precursors for recruitment of myoblasts for fusion. Development 128:5061–73 [DOI] [PubMed] [Google Scholar]
- 100.Richardson BE, Beckett K, Nowak SJ, Baylies MK. 2007. SCAR/WAVE and Arp2/3 are crucial for cytoskeletal remodeling at the site of myoblast fusion. Development 134:4357–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rottner K, Hänisch J, Campellone KG. 2010. WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol 20:650–61 [DOI] [PubMed] [Google Scholar]
- 102.Roy S, VijayRaghavan K. 1998. Patterning muscles using organizers: Larval muscle templates and adult myoblasts actively interact to pattern the dorsal longitudinal flight muscles of Drosophila. J. Cell Biol 141:1135–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ruiz-Gómez M, Bate M. 1997. Segregation of myogenic lineages in Drosophila requires Numb. Development 124:4857–66 [DOI] [PubMed] [Google Scholar]
- 104.Ruiz-Gómez M, Coutts N, Price A, Taylor MV, Bate M. 2000. Drosophila dumbfounded: a myoblast attractant essential for fusion. Cell 102:189–98 [DOI] [PubMed] [Google Scholar]
- 105.Ruiz-Gómez M, Romani S, Hartmann C, Jäckle H, Bate M. 1997. Specific muscle identities are regulated by Krüppel during Drosophila embryogenesis. Development 124:3407–14 [DOI] [PubMed] [Google Scholar]
- 106.Rushton E, Drysdale R, Abmayr SM, Michelson AM, Bate M. 1995. Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophila muscle development. Development 121:1979–88 [DOI] [PubMed] [Google Scholar]
- 107.Sánchez-Pulido L, Martıín-Belmonte F, Valencia A, Alonso MA. 2002. MARVEL: a conserved domain involved in membrane apposition events. Trends Biochem. Sci 27:599–601 [DOI] [PubMed] [Google Scholar]
- 108.Schäfer G, Weber S, Holz A, Bogdan S, Schumacher S, et al. 2007. The Wiskott-Aldrich syndrome protein (WASP) is essential for myoblast fusion in Drosophila. Dev. Biol 304:664–74 [DOI] [PubMed] [Google Scholar]
- 109.Schejter ED. 2016. Myoblast fusion: experimental systems and cellular mechanisms. Semin. Cell Dev.Biol 60:112–20 [DOI] [PubMed] [Google Scholar]
- 110.Schnorrer F, Schönbauer C, Langer CCH, Dietzl G, Novatchkova M, et al. 2010. Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464:287–91 [DOI] [PubMed] [Google Scholar]
- 111.Schröter RH, Lier S, Holz A, Bogdan S, Klämbt C, et al. 2004. kette and blown fuse interact genetically during the second fusion step of myogenesis in Drosophila. Development 131:4501–9 [DOI] [PubMed] [Google Scholar]
- 112.Schulman VK, Dobi KC, Baylies MK. 2015. Morphogenesis of the somatic musculature in Drosophila melanogaster. Wiley Interdiscip. Rev. Dev. Biol 4:313–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Segal D, Dhanyasi N, Schejter ED, Shilo B-Z. 2016. Adhesion and fusion of muscle cells are promoted by filopodia. Dev. Cell 38:291–304 [DOI] [PubMed] [Google Scholar]
- 114.Sens KL, Zhang S, Jin P, Duan R, Zhang G, et al. 2010. An invasive podosome-like structure promotes fusion pore formation during myoblast fusion. J. Cell Biol 191:1013–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shafiq SA. 1963. Electron microscopic studies on the indirect flight muscles of Drosophila melanogaster. II. Differentiation of myofibrils. J. Cell Biol 17:363–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shelton C, Kocherlakota KS, Zhuang S, Abmayr SM. 2009. The immunoglobulin superfamily member Hbs functions redundantly with Sns in interactions between founder and fusion-competent myoblasts. Development 136:1159–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shi J, Bi P, Pei J, Li H, Grishin NV, et al. 2017. Requirement of the fusogenic micropeptide myomixer for muscle formation in zebrafish. PNAS 114:11950–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shilagardi K, Li S, Luo F, Marikar F, Duan R, et al. 2013. Actin-propelled invasive membrane protrusions promote fusogenic protein engagement during cell–cell fusion. Science 340:359–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shin N-Y, Choi H, Neff L, Wu Y, Saito H, et al. 2014. Dynamin and endocytosis are required for the fusion of osteoclasts and myoblasts. J. Cell Biol 207:73–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stradal TE, Scita G. 2006. Protein complexes regulating Arp2/3-mediated actin assembly. Curr. Opin. Cell Biol 18:4–10 [DOI] [PubMed] [Google Scholar]
- 121.Strünkelnberg M, Bonengel B, Moda LM, Hertenstein A, de Couet HG, et al. 2001. rst and its paralogue kirre act redundantly during embryonic muscle development in Drosophila. Development 128:4229–39 [DOI] [PubMed] [Google Scholar]
- 122.Su M-T, Fujioka M, Goto T, Bodmer R. 1999. The Drosophila homeobox genes zfh-1 and even-skipped are required for cardiac-specific differentiation of a numb-dependent lineage decision. Development 126:3241–51 [DOI] [PubMed] [Google Scholar]
- 123.Tixier V, Bataillé L, Jagla K. 2010. Diversification of muscle types: recent insights from Drosophila. Exp. Cell Res 316:3019–27 [DOI] [PubMed] [Google Scholar]
- 124.Tskhovrebova L, Trinick J. 2010. Roles of titin in the structure and elasticity of the sarcomere. J. Biomed.Biotechnol 2010:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Van Vactor D, Sink H, Fambrough D, Tsoo R, Goodman CS. 1993. Genes that control neuromuscular specificity in Drosophila. Cell 73:1137–53 [DOI] [PubMed] [Google Scholar]
- 126.Willkomm L, Bloch W. 2015. State of the art in cell–cell fusion In Cell Fusion, ed. Pfannkuche K, pp. 1–19. Methods Mol. Biol. 1313. Clifton, NJ: Humana; [DOI] [PubMed] [Google Scholar]
- 127.Yang Y, Zhang Y, Li WJ, Jiang Y, Zhu Z, et al. 2017. Spectraplakin induces positive feedback between fusogens and the actin cytoskeleton to promote cell-cell fusion. Dev. Cell 41:107–20.e4 [DOI] [PubMed] [Google Scholar]
- 128.Zhang Q, Vashisht AA, O’Rourke J, Corbel SY, Moran R, et al. 2017. The microprotein Minion controls cell fusion and muscle formation. Nat. Commun 8:15664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang R, Zhang CY, Zhao Q, Li DH. 2013. Spectrin: structure, function and disease. Sci. China Life Sci 56:1076–85 [DOI] [PubMed] [Google Scholar]
- 130.Zhang W, Roy S. 2017. Myomaker is required for the fusion of fast-twitch myocytes in the zebrafish embryo. Dev. Biol 423:24–33 [DOI] [PubMed] [Google Scholar]
- 131.Zhang Y, Featherstone D, Davis W, Rushton E, Broadie K. 2000. Drosophila D-Titin is required for myoblast fusion and skeletal muscle striation. J. Cell Sci 113:3103–15 [DOI] [PubMed] [Google Scholar]