

ABSTRACT
Major efforts are made to characterize the presence of microRNA (miRNA) and messenger RNA in blood plasma to discover novel disease-associated biomarkers. MiRNAs in plasma are associated to several types of macromolecular structures, including extracellular vesicles (EV), lipoprotein particles (LPP) and ribonucleoprotein particles (RNP). RNAs in these complexes are recovered at variable efficiency by commonly used EV- and RNA isolation methods, which causes biases and inconsistencies in miRNA quantitation. Besides miRNAs, various other non-coding RNA species are contained in EV and present within the pool of plasma extracellular RNA. Members of the Y-RNA family have been detected in EV from various cell types and are among the most abundant non-coding RNA types in plasma. We previously showed that shuttling of full-length Y-RNA into EV released by immune cells is modulated by microbial stimulation. This indicated that Y-RNAs could contribute to the functional properties of EV in immune cell communication and that EV-associated Y-RNAs could have biomarker potential in immune-related diseases. Here, we investigated which macromolecular structures in plasma contain full length Y-RNA and whether the levels of three Y-RNA subtypes in plasma (Y1, Y3 and Y4) change during systemic inflammation. Our data indicate that the majority of full length Y-RNA in plasma is stably associated to EV. Moreover, we discovered that EV from different blood-related cell types contain cell-type-specific Y-RNA subtype ratios. Using a human model for systemic inflammation, we show that the neutrophil-specific Y4/Y3 ratios and PBMC-specific Y3/Y1 ratios were significantly altered after induction of inflammation. The plasma Y-RNA ratios strongly correlated with the number and type of immune cells during systemic inflammation. Cell-type-specific “Y-RNA signatures” in plasma EV can be determined without prior enrichment for EV, and may be further explored as simple and fast test for diagnosis of inflammatory responses or other immune-related diseases.
KEYWORDS: Y-RNA, biomarker, endotoxemia, inflammation, sepsis, lipoprotein particles, extracellular vesicles
Introduction
Body fluids contain a plethora of RNA molecules that have been released by cells. Macromolecular structures in plasma that contain extracellular RNAs (exRNAs) include extracellular vesicles (EV), lipoprotein particles (LPP) and ribonucleoprotein complexes (RNPs) [1–3]. In recent years, the major focus has been on characterizing the RNA content of EV. EV are nano-sized membrane vesicles that are involved in transferring transmembrane and cytosolic proteins, lipids, and RNA between different cells [4,5]. The release and molecular composition of EV changes depending on the activation or differentiation status of the EV-producing cell. EVs have therefore not only been implicated as important mediators of intercellular communication, but also as biomarkers for disease [6–12]. Numerous studies addressed the cell-type-specific profiles of EV-associated RNA and how such profiles change due to cell signalling, oncogenic transformation, or other disease-related processes [13–19]. Most of this research has focused on the microRNA (miRNA) and messenger RNA (mRNA) content of EV because of their known functions in gene expression. Changes in EV-associated levels of these RNA types in plasma or serum have been demonstrated for diseases such as cancer, rheumatoid arthritis and neurodegenerative diseases [10,20,21]. Since recent years, there has been growing awareness that separation of EV, LPP and RNP is technically challenging due to their overlap in size and buoyant density. The levels of contamination with LPP and RNP in EV preparations obtained with the commonly used EV-isolation methods are variable. This introduces biases in EV-miRNA biomarker studies and reduces inter-study reproducibility of data [22,23].
Our previous work indicated that EV also contain less well-known types of non-coding RNA [19,24]. One of these RNA types, Y-RNA, has been abundantly detected in EV from several different cell types (reviewed in [25]). Y-RNAs are highly conserved non-coding RNAs with sizes of ~ 100 nt. Inside cells, these RNAs are involved in various basic cellular processes such as DNA replication [26,27] and RNA quality control [28,29]. The human genome encodes four Y-RNA subtypes [30], which are hairpin-like structures with homologous stem regions and subtype-specific loop sequences [31]. Although several research groups proposed that EV mainly contain specific fragments of Y-RNA, our recent data demonstrate that this is a common artefact in small RNA sequencing procedures [19]. Difficulties to ligate the adapters to the 5ʹ-triphosphate group of full length Y-RNAs and the strict size selection applied during small RNA library preparation strongly bias towards detection of fragmented Y-RNAs [32]. Using Northern blotting, we provided evidence that most of the Y-RNA in EV from cultured primary dendritic cells was in the full-length form [19]. Moreover, we showed that the dendritic cells could regulate the amount of full length Y-RNA sorted into EV in response to immune-activating or immune-suppressing stimuli [19], similar to the regulated incorporation of specific miRNAs [13,33,34]. This suggests that Y-RNAs can contribute to the functional properties of EV in immune cell communication and that EV-associated Y-RNAs could have biomarker potential in immune-related diseases.
Y-RNA is not only present at high levels in EV from several cell lines, but is also among the most abundant RNA types in plasma of healthy individuals [35,36]. The levels of circulating Y-RNA have been proposed to correlate with cancer and coronary artery disease [37,38], but these studies were biased towards detection of Y-RNA fragments. It is currently unknown whether full length Y-RNAs in plasma are associated to both EV, LPP, and RNP, similar to what has been shown for miRNAs [1–3]. It is also unknown which blood-related cell types can release Y-RNA containing EV into the circulation and whether the levels of full length Y-RNA in plasma are regulated by immune-related stimuli.
We here show that the majority of full length Y-RNA in plasma is stably associated to EV. Interestingly, we found that EV released by different blood-related cell types contain unique quantitative ratios of different Y-RNA subtypes. In response to TLR-mediated stimulation, several of these cell types showed increased release of Y-RNA containing EV, but the cell-type-specific Y-RNA-subtype ratios remained stable under these conditions. In a human endotoxemia model for systemic inflammation, we observed significant alterations in the Y-RNA content within the total pool of plasma EV. A substantial increase was observed in the neutrophil-specific Y-RNA-subtype ratio and this correlated tightly with the increase in number of neutrophils that occurred during inflammation. Detection of Y-RNA-subtype ratios in plasma EV can be determined without prior enrichment for EV and may be further explored as simple and fast test for diagnosis of inflammatory responses or other immune-related diseases.
Materials and methods
Clinical samples
Plasma samples of the human endotoxemia study were obtained under approval of the Academic Medical Centre Medical Ethical Committee and are according to the Declaration of Helsinki, including Good Clinical Practice [39,40]. This study has been registered at the Dutch Trial Register (NTR4455). All enrolled volunteers provided written informed consent before enrolment. Two volunteers displayed aberrant cytokine levels at either t = 0 or t = 2 and were excluded from all further analyses.
Blood and plasma from healthy volunteers was obtained following approval of the Medical Ethical Committees of Utrecht Medical Centre, Amsterdam Medical Centre and Sanquin Research. All volunteers provided written informed consent, the experiments abide by the Declaration of Helsinki principles for human research ethics.
Plasma collection and fractionation
During the human endotoxemia study, plasma samples were collected as described previously [39,40]. In brief, arterial blood samples were collected in two tubes with 0.11 M sodium citrate (Vacutainer, Becton Dickinson). Samples were collected directly before infusion of LPS and before infusion of the transfusion product and every 2 h thereafter until 6 h after transfusion. Tubes were centrifuged at 1,500 g for 10 min at 20°C, the supernatant was centrifuged again at 1,550 g for 20 min, plasma was frozen at −80°C until analysis. Parallel blood samples were drawn for determining blood cell counts and cytokine levels.
For preparation of all other plasma samples from healthy donors, blood was collected in the morning by venepuncture with a 21 G needle into a citrate tube (Greiner Vacuette 9NC NaC 3,2%), and was processed within 30 minutes after collection. Tubes were centrifuged at 2,500 g for 15 min at RT, supernatant was pipetted off using a plastic Pasteur pipette. Supernatant was centrifuged again 3,000 g for 15 min, supernatant was collected and frozen directly at −80°C in 0.5 mL aliquots in Eppendorf LoBind Tubes.
For fractionation of plasma (Figure 1(b)), 0.5 mL plasma was thawn at RT and fractionated on a qEV Classic size exclusion column (Izon Science, Christchurch, New Zealand) eluted with 1x PBS (Gibco, Paisley, UK). 0.5 mL fractions were collected manually. Fractions 7–12 (“early”) and fractions 17–24 (“late”) were pooled into two SW40 tubes and were centrifuged for 65 min at 100,000 g (k-factor: 381.5). A stricter separation between large and small structures present in plasma was achieved by omitting the intermittent fractions 13–16 from further analysis. 90% of the supernatant (“sup”) was removed by pipetting and stored at 4°C, and the last 10% was decanted, after which the pellets were resuspended in 50 µl PBS + 0.2% EV-depleted BSA (which was cleared of aggregates by overnight ultracentrifugation at 100,000 g). Resuspended pellets were overlaid with sucrose density gradients (2.5 M–0.4 M) and centrifuged for 15–18 h at 192,000 g in a SW40 rotor (k-factor 144.5). High-density (1.25 g/mL, “hi dens”) and intermediate density (1.11–1.18 g/mL, “int dens”) fractions were diluted four times in PBS + 0.2% EV-depleted BSA and ultracentrifuged for 65 min at 192,000 g in a SW40 rotor (k-factor 144.5). Pellets were resuspended in 60 µl PBS, divided into three aliquots which were subjected to different enzymatic treatments. Stored 100,000 g supernatants were concentrated on PBS-washed Amicon Ultra 100kDa spin filters (15 min 3,000 g) before being subjected to enzymatic treatment.
Figure 1.
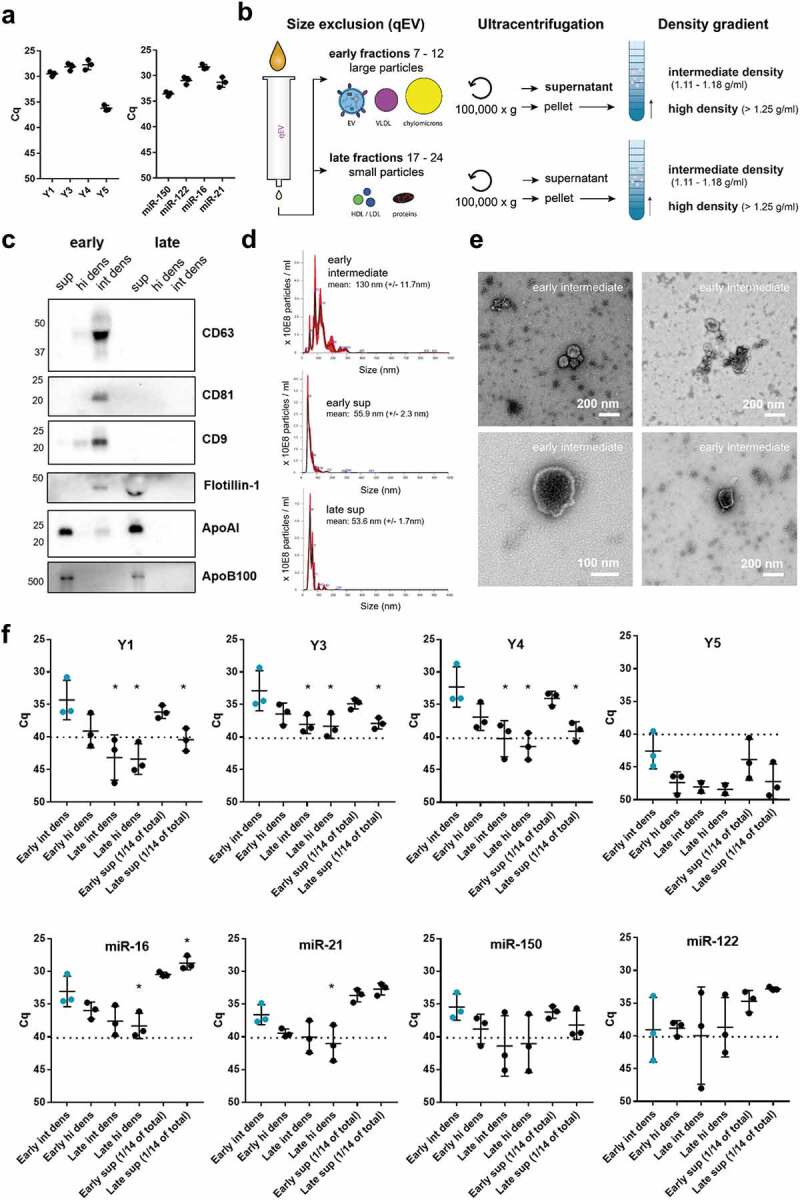
Distribution of full-length Y-RNA subtypes over RNA carriers in plasma with different sizes and densities.
Protease, RNase and detergent treatments
Each of the plasma fractions was subjected to treatment with combinations of detergent, protease and RNase according to Table 1.
Table 1.
Overview of enzymatic treatments on plasma fractions.
| Treatment | Untreated | Protease + RNase | Protease + RNase + detergent |
|---|---|---|---|
| NP-40 | - | - | + |
| Proteinase K | - | + | + |
| RNase A | - | + | + |
First, 1% NP-40 detergent or an equal volume of PBS was added and samples were incubated 15 min on ice. Next, 45 ng/mL Proteinase K (Roche, Basel, Switzerland) or an equivalent volume of PBS was added and incubated 30 min at 37°C, after which the reaction was stopped by adding 5 mM PMSF (Roche, Basel, Switzerland) an incubation on ice for 10 min. Finally, 8 U/mL RNase A (Roche, Basel, Switzerland) or an equivalent volume of PBS was added, incubated at 37°C for 30 min. Immediately hereafter, the RNA isolation procedure was started by adding 700 µl Qiazol (Qiagen, Hilden, Germany).
Blood cell culture and medium preparation
Preparation of EV-depleted media
EV were depleted from foetal calf serum (FCS) for preparation of EV-depleted culture medium [41]. In brief, FCS was diluted to 30% and centrifuged overnight at 100,000 g in a SW28 rotor (k-factor 334.2). Supernatants were recovered by pipetting, filtered through a 0.45 µm bottle top filter (Millipore, Leiden, NL). EGM-2 medium contained only 2% of serum and was thus ultracentrifuged as complete medium overnight at 100,000 g in a SW28 rotor (k-factor 334.2). Supernatants were recovered by pipetting, filtered through a 0.45 µm bottle top filter (Millipore, Leiden, NL) before use.
PBMC
Blood was drawn with a 21 G needle into a 10 ml Lithium Hep Green Vacutainer (Beckton Dickinson, Franklin Lakes, NJ), layered onto a 15 mL FicollPaque (GE, Piscataway, NJ) cushion and centrifuged for 20 minutes at 560 g. The plasma layer was discarded, after which the PBMC interphase was removed with a plastic Pasteur pipette and mixed with 20 mL RPMI-1640 (Gibco, Paisley, UK). Cells were washed 3× by centrifugation for 10 minutes at 560 g. PBMC were seeded in RPMI-1640 with 10% EV-depleted FCS, 100 IU/mL penicillin and 100 µg streptomycin (Gibco, Paisley, UK) at 1E6 cells/ml for EV-production. PBMC were checked for neutrophil contamination by flow cytometry (see below) which was <3% for all donors. Where indicated, cells were stimulated with 1 µg/mL LPS (O111-B4, cat L2630, Sigma-Aldrich, St. Louis, MO). EV were produced in 4 h with 12–20×10E6 PBMC, after which medium was collected for EV isolation. Cell viability was checked by Trypan blue exclusion and was >95% for all cultures.
Neutrophils
Blood was drawn with a Vacutainer Eclipse needle into a Vacuette sodium heparine 9 ml tube (Greiner Bio-One, Alphen aan de Rijn, Netherlands). Blood was diluted with HBSS (Sigma Aldrich H6136) after which granulocytes and erythrocytes were separated from PBMC by density gradient centrifugation on 20 ml Lymphoprep (STEMcell Technologies, Köln, Germany). The granulocyte/erythrocyte pellet was subsequently transferred to a new 50 mL tube followed by erythrocyte lysis in ice-cold erythrocyte lysis buffer (0.155 M NH4 Cl (Sigma), 1 mM KHCO3 (Merck), 80 µM EDTA (Merck) in Milli-Q) in 2 consecutive rounds. The final cell pellet was washed in PBS, centrifuged 8 min at 400 g and seeded in IMDM + ultraglutamine (Gibco) supplemented with 1% EV-depleted FCS (Sigma-Aldrich, F7524), and 86 µg/mL gentamycin (Duchefa, DUC586714) at 1E6 cells/mL for EV production. Purity of isolated cells was determined on flow cytometry (see below). Neutrophils were positive for CD15 and contained less than 3% PBMC (determined by FCS/SSC detection). Where indicated, cells were stimulated with 100 ng/mL LPS (O111-B4, cat L2360, Sigma-Aldrich, St. Louis, MO). EV were produced in 2 h with 9x10E6 neutrophils, after which medium was collected for EV isolation. Cell viability after culture was checked by PI or DAPI staining, which was >90% for all cultures.
Platelets
Whole blood was collected in 3.2% trisodium citrate Vacuette tubes (Greiner, Kremsmünster, Austria) and was centrifuged at 160 g for 15 minutes at RT without a brake. Platelet-rich plasma (PRP) was transferred into a new tube containing 1/10 volume of Acid-Citrate-Dextrose (ACD; 38 mM citric acid, 75 mM sodium citrate, 135 mM dextrose). PRP was then centrifuged at 400 g for 15 min at room temperature with brake level 1. Afterwards, the supernatant was discarded and the pellet was resuspended in modified Tyrode’s buffer (10 mM HEPES, 137 mM NaCl, 2.8 mM KCl, 12 mM NaHCO3) with 5.55 mM D-glucose, pH 6.5. Then, prostaglandin I2 (PGI2, Cayman Chemical Company) was prediluted into modified Tyrode’s buffer (pH 6.5)) and added to a final concentration of 0.5 µM, after which the previous centrifugation step was repeated immediately. Finally, the platelets were resuspended in modified Tyrode’s, supplemented with 5.55 mM D-glucose, pH 7.4. Platelet suspensions were counted using the CELL-DYN Emerald System (Abbott, Green Oaks, IL) and adjusted to 200x10E6 platelets/mL in 1 mL for EV production. Where indicated, platelets were stimulated with 1 µg/ml LPS (O111-B4, cat L2360, Sigma-Aldrich, St. Louis). After 2 h of culture, 1 ml platelet suspension was collected, topped up with 10 mL PBS + 1% EV-depleted BSA and subjected to EV isolation.
HUVEC
Three pools of Human Umbilical Vein Endothelial cells (HUVEC) were obtained from Lonza (Verviers, Belgium). Cells were thawn and cultured for 5 passages before being seeded at 0.55E6 cells/cm2 in EV-depleted EGM-2 complete medium with 2 mM L-glutamine, 100 U/ml pen/strep in porcine skin-gelatin-coated culture dishes for EV production from 9×10E6 cells. Where indicated, HUVEC were stimulated with 1 µg/ml LPS (O55-B5, cat L2880, Sigma-Aldrich, St. Louis). After 4 h, cell viability was checked by LIVE/DEAD (>95% for all cultures) (ThermoScientific), medium was collected and subjected to EV isolation.
Red blood cells
9 mL of blood was collected into a Vacuette Natrium Heparine tube (Greiner Bio-One). Red blood cells were isolated by the following centrifugation steps [42]: 15 min at 1000 rpm, 5 min at 2500 rpm, 5 min at 2500 rpm. After isolation, red blood cells were incubated for 2 h in HEPES buffer (132 mM NaCl, 20 mM HEPES, 6 mM KCl, 1 mM MgSO4, 1.2 mM K2HPO4, (Sigma-Aldrich, Spruce, USA) supplemented with 1 mM Ca2+ and 1 mg/ml glucose), EV were produced from 2×10E9 red blood cells in 1 mL. Where indicated, RBC were stimulated with 1 µg/ml LPS (O111-B4, cat L2360, Sigma-Aldrich, St. Louis). After 2 h, 10 mL PBS + 1% EV-depleted BSA was added to the supernatants which were then subjected to EV isolation.
Flow cytometry
5x10E4 to 1x10E5 cells were labelled for 30 minutes in 18 µl to 100 µl PBS + 1% BSA containing one or combinations of the following antibodies: anti-CD16-PECy7 (Biolegend; 1:1000; 3G8), anti-CD62 L-APCCy7 (Biolegend; 1:25; Greg-56), anti-CD63-APC (Biolegend; 1:100; H5 C6), anti-CD66b-PE (Biolegend; 1:100; G10F5), DAPI (Sigma-Aldrich), anti-CD3-PacificBlue (Beckman Coulter, 1: 50, UCHT-1), anti-CD14-FITC (Miltenyi, 1:100, Tük 4), anti-ICAM-1-AlexaFluor-405 (SantaCruz, 1:100, sc-107-af405), an in-house VHH against P-selectin (1:125, B10.6 [43],), or Annexin-V (250 ug/ml, VPS diagnostics, cat. nr. A705). Neutrophil activation was assessed by measuring the increase in CD62L expression [44] and the simultaneous decrease in CD16 and increase in CD63 expression [45,46]; HUVEC activation was assessed by evaluating the increase in ICAM-1 expression [47]; red blood cell activation was assessed by evaluating phosphatidylserine exposure detected using annexin-V staining [48]; platelet activation was assessed by investigating P-selectin expression [49]. Surface labelling of cells was measured on a FACSCanto (BD Biosciences) or a LSR-II (BD Biosciences) flow cytometer relative to unlabelled controls.
EV isolation and fluorescent labelling
Cells were incubated in their respective media for 2–4 h, after which EV were isolated as described before [19]. Supernatants were centrifuged at 4°C, twice for 10 min at 200 g, twice for 10 min at 500 g, and 30 min at 10,000 g in a SW40 rotor (k-factor 2773.9). Supernatant was carefully pipetted off and transferred to new SW40 tubes, EV were pelleted at 100,000 g for 65 min (k-factor 381.5). EV pellets were resuspended in 20 µl PBS + 0.2% EV-depleted BSA and labelled with 1.5 µl PKH67 (Sigma-Aldrich) in 180 µl Diluent C, the reaction was stopped by adding 100 µl EV-depleted medium. Labelled EV were overlaid with a sucrose density gradient as described above and centrifuged for 15–18 h at 192,000 g (k-factor 144.5). Equal volumes of non-conditioned culture medium were processed in parallel to quantify background levels of residual medium-derived RNA [41].
High resolution flow cytometry
High-resolution flow cytometric analysis of PKH67-labelled EV was performed on a BD Influx flow cytometer (BD Biosciences, San Jose, CA) with an optimized configuration for small particle analysis as previously described [50,51]. We applied fluorescence threshold triggering to discriminate PKH67 labelled EV from non-fluorescent noise signals. Forward scatter (FSC) was detected at a 15–25 degree collection angle. Fluorescent polystyrene 100 and 200 nm beads (FluoSpheres, Invitrogen, Carlsbad, CA) were used to calibrate the fluorescence and reduced width-FSC settings before each measurement. Sucrose gradient fractions were diluted 10–20 times in PBS and vortexed just before measurement. Samples were measured at maximally 10,000 events per second, which is far below the electronic pulse processing limit of the BD Influx [52]. Serial dilutions of peak fractions were included to control for potential “invisible swarm” effects [53].
Nanoparticle tracking analysis (NTA)
Particles in the early intermediate density fraction were pelleted at 192,000 g. Pellets were resuspended in 50 µl PBS and diluted 50x before measurement on a Nanosight NS500 instrument (Malvern, Worchestershire, UK). Early supernatant and late supernatant fractions were diluted 100× before measurement by NTA. Data acquisition and processing were performed using NTA software 3.3. Each sample was recorded for three times 30 seconds, 25 frames per second, camera level 14, detection threshold 4.
Electron microscopy
Particles in the early intermediate density fraction were pelleted at 192,000 g. Pellets were resuspended in 7 µl PBS and were added on top of carbon-coated 300 mesh, copper grids and incubated for 3 min at room temperature. The grids were then washed two times in 50 µl water and stained with 2% uranyl acetate. Imaging was performed on a T20 electron microscope (FEI) operated at 200 keV. Images were recorded on a CCD Eagle camera (FEI).
Immunoblotting
EV produced by equal numbers of stimulated and unstimulated cells were purified as described before, and resuspended in 30 µl 2× SDS sample buffer. Cell lysates were prepared in PBS + 1% NP-40 and Complete Mini Protease inhibitor cocktail tables (Roche, Basel, Switzerland) and incubation for 15 min on ice. Nuclei were spun down at 16,000 g for 15 min. Protein content of cleared cell lysates was measured by BCA assay (ThermoScientific), and equal amounts of cell lysate proteins were used in immunoblotting. Equal volumes of fractionated plasma samples were mixed with 2× SDS sample buffer. Samples were incubated at 100°C for 3 min for detection of CD9, for 10 min for detection of ApoAI and ApoB100, and were not heated for detection of CD63 and CD81. Proteins were separated on a 10% SDS-PAGE gel (6% gel for ApoB100), after which proteins were transferred onto an Immobilon-P 0.45 µm PVDF membrane (Millipore, Cork, Ireland). After blocking for 1–2 h in blocking buffer (0.5% Cold Fish Skin Gelatin in PBS + 0.05% Tween-20), blots were incubated overnight at 4°C with one of the following primary antibodies: anti-human-CD9 (BioLegend, 1: 1000, clone HI9a), anti-human-CD63 (BD Bioscience, 1: 500, clone H5C6), anti-human-CD81 (SantaCruz, 1: 1000, clone B-11), anti-human-flotillin-1 (BD Transduction Labs, 1: 1000, clone 18), anti-human-ApoAI (SantaCruz, 1: 1000, clone sc376818), anti-human-ApoB100 (Biotechne, 1: 1000, clone AF3260), or anti-human-Calnexin (Transduction Labs, 1: 1000, clone 37) in blocking buffer, washed three times in PBS-Tween, and incubated for 1–2 h with HRP-coupled secondary antibodies (Dako, 1: 5000–1: 10 000). Blots were washed again three times in PBS-Tween, followed by two washes in plain PBS and incubation with ECL solution (ThermoScientific, SuperSignal West Dura Extended Duration Substrate, cat. 34,075). Blots were analysed using a BioRad Chemidoc imager (BioRad, Hercules, CA) and Image Lab software.
RNA analysis
RNA was isolated from cell culture EV isolates using the miRNeasy micro kit according to the manufacturer’s guidelines (Qiagen, Hilden, Germany). RNA was isolated from the 100 µl plasma aliquots according to the miRNeasy micro serum/plasma guidelines. RNA was eluted in 15 µl RNase-free water supplied with the kit. The Bioanalyzer Pico 6000 kit (Agilent, Waldbronn, Germany) was used to quantify the RNA. cDNA was prepared using the miScript RT II kit (Qiagen, Hilden, Germany) using equal input volumes of RNA (12 µl per reaction) and HiFlex buffer. Quantification of specific RNA molecules by qPCR was done in 8 µl reactions, containing 2 µl of 10× diluted cDNA template, 4 µl of SYBR Green SensiMix PCR master mix (BioLine Reagents Ltd., UK) and 100 nM primers (IDT, Leuven, Belgium). Cycling conditions were 95°C for 10 min followed by 50 cycles of 95°C for 10 s, 57°C for 30 s and 72°C for 20 s, run on a BioRad CFX384 machine (BioRad, Hercules, CA). Cq values were determined at a threshold of 300 fluorescent units, data analysis was done in BioRad CFX Manager.
PBMC activation was determined by measuring mRNA levels of the cytokines TNFα and IL-6 [54]. PBMC mRNA was isolated using the MN Nucleospin RNA kit (Macherey-Nagel, Düren, Germany). RNA was eluted in 60 µl RNase-free water and quantified by Nanodrop 2000 (ThermoScientific). cDNA was prepared using the RevertAid cDNA kit with random hexamer primers (ThermoScientific), qPCR reactions were set up as described previously using primer pairs for TNFα, IL-6, and using beta-actin and GAPDH as reference genes. Cycling conditions were 95°C for 10 min followed by 40 cycles of 95°C for 10 s, 60°C for 30 s and 72°C for 20 s, run on a BioRad CFX96 machine (BioRad, Hercules, CA). Data analysis was done in BioRad CFX Manager.
Primer sequences can be found in Table 2 (1–8 were combined with miScript Universal reverse primer, primers 9–16 were matched forward and reverse primers):
Table 2.
qPCR primers.
| No | Primer name | Sequence (5ʹ–3ʹ) |
|---|---|---|
| 1 | Y1_loop-F | GATCGAACTCCTTGTTCTACTC |
| 2 | Y3_loop-F | AGATTTCTTTGTTCCTTCTCCACTC |
| 3 | Y4_loop-F | GTGTCACTAAAGTTGGTATACAAC |
| 4 | Y5_loop-F | GTTAAGTTGATTTAACATTGTCTC |
| 5 | hsa-miR-150-5p | TCTCCCAACCCTTGTACCAGTG |
| 6 | hsa-miR-21-5p | TAGCTTATCAGACTGATGTTGA |
| 7 | hsa-miR-122-5p | TGGAGTGTGACAATGGTGTTTG |
| 8 | hsa-miR-16-5p | TAGCAGCACGTAAATATTGGCG |
| 9 | hGAPDH-F | TGCACCACCAACTGCTTAGC |
| 10 | hGAPDH-R | GGCATGGACTGTGGTCATGAG |
| 11 | hActin-F | CCTTCCTGGGCATGGAGTCCTG |
| 12 | hActin-R | GGAGCAATGATCTTGATCTTC |
| 13 | hIL-6-F | AACCTGAACCTTCCAAAGATGG |
| 14 | hIL-6-R | TCTGGCTTGTTCCTCACTACT |
| 15 | hTNFa-F | ATGAGCACTGAAAGCATGATCC |
| 16 | hTNFa-R | GAGGGCTGATTAGAGAGAGGTC |
Statistics
Statistical analyses were done in SPSS (v24, IBM) or R Studio (v 3.5.2) with ggplot2 and ggally. The Kolmogorov-Smirnov test was used to assess the normality of the data. Various statistical tests were used which are indicated in the respective figure legends. p values <0.05 were considered statistically significant. Y-RNA abundance ratios were calculated from the differences in Cq value (dCq) between individual Y-RNA subtypes. For example: Y4/Y3 = dCqY3-Y4 = CqY3 – CqY4. The resultant values represent the relative abundance between two Y-RNA subtypes, for example: a Y4/Y3 ratio of zero means that both Y-RNA subtypes are present in equal amounts; a ratio of 1 means Y4 is twice as abundant as Y3; a ratio of −1 means Y4 is two times less abundant as Y3. Receiver-operator characteristics curve was plotted using the pROC plugin for R [55].
Availability of protocols
Written details on experimental procedures have been submitted to the EV-TRACK knowledgebase (EV-TRACK ID: 190044) [56].
Results
Full length Y-RNA in human plasma is predominantly associated to EV
Previously, it was shown that miRNAs in plasma are associated to both EV, RNP and LPP [1–3]. We here investigated whether full length Y-RNAs in human plasma also associate to these three types of macromolecular structures. We first assessed whether all four Y-RNA subtypes could be detected in unfractionated plasma. To reliably distinguish between the different Y-RNA subtypes, we designed primers complementary to the loop sequences, which are unique to each of the four Y-RNA subtypes. RNA was isolated directly from 100 µl human plasma, followed by RT-qPCR-based quantification of all Y-RNA subtypes. For comparison, we analysed a selected set of miRNAs previously shown to be abundantly present in plasma [57] and to be associated to different RNA carriers [2]. These included miR-21-5p and miR-16-5p (detected in both EV and LPP), miR-150-5p (mainly detected in EV), and miR-122-5p (mainly detected in LPP) (Figure 1(a)). Y1, Y3 and Y4 were as abundant as the highly abundant miRNAs, whereas the levels of Y5 were relatively low. Next, we separated EV, RNP and LPP based on differences in size and buoyant density using a combination of isolation methods (Figure 1(b)). We used size-exclusion chromatography to separate large EV and (lipo)protein particles (“early fractions”) from small (lipo)protein particles and soluble proteins (“late fractions”). Fractions 13–16 were omitted from further analysis because they contained low numbers of mixed large EV and small lipoparticles. The “early” and “late” fractions were subjected to ultracentrifugation at 100,000 g to separate pelletable from non-pelletable structures. Non-pelletable structures (“sup”) were concentrated by ultrafiltration, and the pelleted particles were separated into particles with densities characteristic for EV (intermediate density, “int dens”) and particles with a higher density (“hi dens”), by using density gradient ultracentrifugation. The resulting six fractions were assessed for the presence of the common EV markers CD9, CD63, CD81 and flotillin-1, and the lipoprotein markers ApoAI and ApoB100 (Figure 1(c)). Consistent with their reported association with differently sized lipoproteins, ApoAI (present in 75–1200 nm chylomicrons and 5 − 12 nm HDL) and ApoB100 (present in 30–80 nm VLDL, 25–35 nm ILDL and 18 − 25 nm LDL) were detected in the early and late supernatant fractions, and were not efficiently pelleted in line with their low density [58,59]. Common EV-associated proteins were predominantly detected on larger particles (present in the early SEC fractions) that sedimented at 100,000 g and displayed intermediate buoyant densities. This is in accordance with the typical size (50–300 nm) and buoyant density (1.11–1.18 g/ml) of EV. Besides a specific band for flotillin-1 in the early intermediate density fraction, a non-specific band with lower molecular weight was observed in the late sup fraction. Additionally, a faint signal for CD9 was detected in the early supernatant fraction (Figure 1(c), Supplementary Figure 1), suggesting the presence of an EV-subset that did not sediment efficiently during ultracentrifugation. Nanoparticle tracking analysis showed that the early intermediate density fraction contained a heterogeneous mixture of particles in the size range characteristic for EV (50–200 nm), whereas the supernatant fractions contained smaller particles (Figure 1(d)). The size and morphology of EV in the early intermediate density fraction was further confirmed by transmission electron microscopy (Figure 1(e)). Next, we compared the six fractions for the presence of full length Y-RNA subtypes and selected miRNAs by RT-qPCR (Figure 1(f)). Transcripts for which Cq values >40 were obtained were considered to be absent from the fractions. All Y-RNA subtypes were comparably distributed over the different plasma fractions. Y-RNAs were mainly detected in the early intermediate “EV” fraction and early supernatant fractions, while they were almost absent in the late supernatant fraction. The distribution of miRNAs, on the contrary, varied per miRNA species. MiR-16, and miR-21 were most abundantly detected in early and late “LPP” fractions, and to a lesser extent in the early “EV” fraction. MiR-122 was exclusively detected in the “LPP” supernatant fractions. The distribution of miR-150 was similar to Y-RNAs, with high levels present in the early intermediate “EV” fraction. These data show that full length Y-RNAs are predominantly detected in plasma fractions that contain EV.
Due to the overlap in size and density of different extracellular RNA carriers, the enrichment methods used above did not allow for strict separation of EV from all LPP subsets and RNP. Additional methods were needed to further substantiate the presence of Y-RNA in plasma EV. We therefore combined our method for plasma fractionation with protease/RNase treatments. MiRNAs and miRNA-protein complexes inside EV are thought to be protected against degradation by these enzymes by the EV membrane and will only become sensitive to these enzymes upon detergent lysis. In contrast, degradation of the protein scaffold of RNP and LPP by proteases will render the RNAs associated to these structures sensitive to RNases [2]. We selected the two plasma fractions containing the highest levels of Y-RNA (“early intermediate density” and “early sup”) and an additional fraction in which high levels of miRNAs were detected (“late sup”) for treatment with different combinations of RNase, protease, and detergent (Figure 2). As expected, all Y-RNA subtypes in the early intermediate (EV) fractions were resistant to protease + RNase treatment, while addition of detergent led to complete degradation of the enclosed Y-RNAs (Figure 2(a)). The low amounts of miR-16, miR-21 and miR-122 present in these early intermediate fractions were also protected unless detergent was added (Figure 2(b)). These data strengthen the conclusion that the “early intermediate density” fractions contain a pure population of EV that protect enclosed miRNAs and Y-RNAs against degradation. In contrast, the majority of miR-16, miR-21 and miR-122 in the lipoprotein-enriched late SEC fractions (“late sup”) were sensitive to enzymatic degradation. This corroborates previous findings that miRNAs in late SEC fractions are associated to LPP [2]. Only very low levels of miR-150 were detected in these late SEC fractions. In the lipoprotein-enriched early SEC fractions (“early sup”), miR-16, miR-21 and miR-122 were degraded by protease/RNase treatment, while Y-RNA and miR-150 were protected against degradation unless detergent was added (Figure 2(a,b)). A likely explanation of these data is that this LPP-enriched fraction contains a low number of miRNA/Y-RNA containing EV that did not pellet efficiently during the 100,000 g centrifugation step. This is supported by the detection of residual amounts of CD9 in this fraction (Figure 1(c) and Supplementary Figure 1). However, we cannot fully exclude the presence of a yet unknown type of Y-RNA containing LPP/RNP that protects RNA against protease/RNase treatment. Most importantly, our data indicate that full-length Y-RNAs in plasma are highly stable and are predominantly associated with EV.
Figure 2.
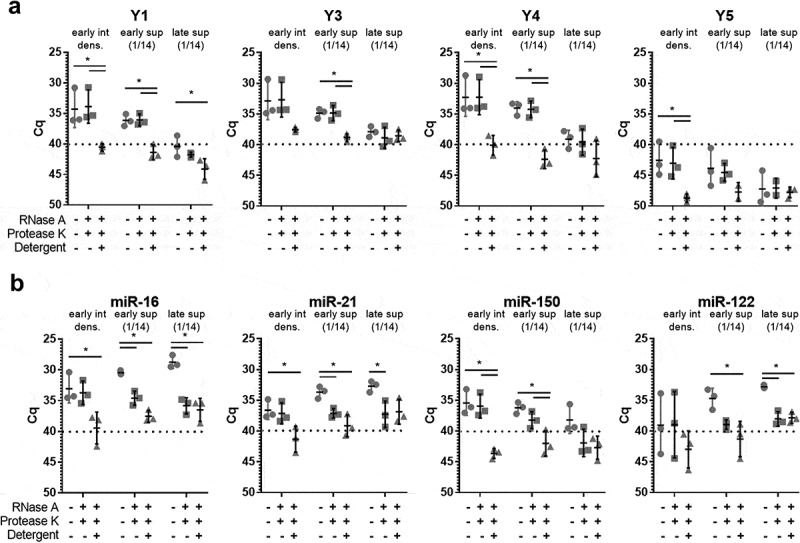
Full-length Y-RNA in plasma is protected from enzymatic degradation.
Blood-associated cell types release EV with cell-type-specific Y-RNA signatures
EV in plasma are likely derived from multiple cell types. It is currently unknown whether a broad range of blood-related cell types has the capacity to release EV containing full length Y-RNA into the extracellular space. To investigate this, we isolated EV from in vitro cultures of human red blood cells (RBC), neutrophils, peripheral blood mononuclear cells (PBMC, which consist of T cells, B cells, and monocytes), and endothelial cells (HUVEC). We additionally prepared EV from platelets since these are known to be abundantly present in blood plasma [60]. We aimed to compare the number of EV released by these different cell types within a similar time frame. Because primary neutrophils are short-lived in culture [61], EV from all different blood-related cell types were collected after a short culturing period of 2–4 h. EV were purified by differential centrifugation followed by density gradient ultracentrifugation. By using high-resolution flow cytometric quantification of EV [50,51], we observed that all cells released EV within the given culture period, although the number of released EV per cell strongly differed between cell types (Figure 3(a)). Irrespective of these differences in released EV, we used RT-qPCR to assess whether full length Y-RNAs could be detected in EV from the selected blood cells (Figure 3(b)). Y-RNA subtypes Y1, Y3 and Y4 were abundantly detected in EV from all cell types. Y5 levels were substantially lower and were excluded in subsequent computational analyses because of the low reliability with which such low-abundance transcripts can be quantified. Across the different cell types, we found a statistically significant log-linear relationship between Y-RNA abundance and EV number, which indicates that cells that release more EV also release more Y-RNA (Figure 3(c)). Furthermore, we observed remarkable differences in the relative abundance of Y-RNA subtypes in EV from various cell types. Notably, neutrophil-derived EV were unique in their high levels of Y4 and low levels of Y3. In contrast, PBMC derived EV and platelet-derived EV contained high levels of both Y4 and Y3, but low levels of Y1. To further explore these cell-specific Y-RNA profiles, we calculated abundance-ratios between each of the Y-RNA subtypes, which we defined as the log2fold difference between individual Y-RNA subtypes (Figure 3(d)). Based on these calculations, we conclude that EV from neutrophils uniquely displayed high Y4/Y3 and negative Y3/Y1 ratios. PBMC- and platelet-EV were characterized by high Y3/Y1 ratios but Y4/Y3 ratios around zero. In EV from HUVEC and RBC, on the contrary, none of the Y-RNA-subtype ratios deviated significantly from zero. Overall, our results indicate that blood-related cell types differ in the amount of EV-associated Y-RNA they release. In addition, our data illustrate that EV released by different blood cells vary in Y-RNA subtype composition and therefore have a specific “Y-RNA signature”.
Figure 3.
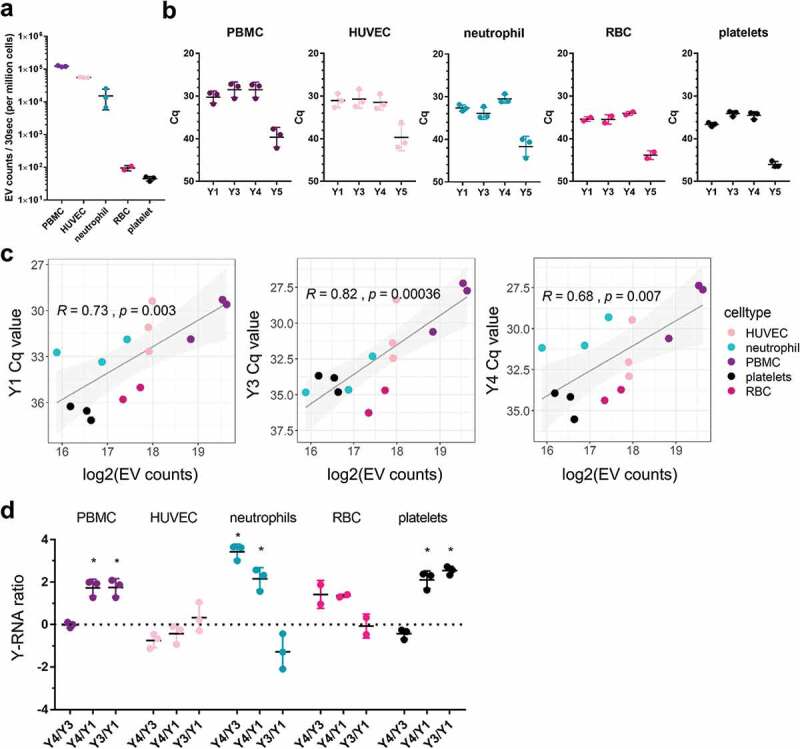
Blood-associated cell types release EV with cell-type-specific Y-RNA signatures.
LPS stimulation of neutrophils affects the number, but not the characteristic Y-RNA-subtype ratios of EV
Next, we investigated whether the Y-RNA load and signature of EV released by the different cell types changed after microbial stimulation. To this end, the different blood cells were cultured for 2–4 h in the presence or absence of LPS. Cellular activation was assessed by flow cytometric analysis of early activation markers (for details, see Materials and Methods), or by measuring early cytokine mRNAs by RT-qPCR (Figure 4(a-b)). Within this short time frame, neutrophils from all donors responded to LPS exposure as indicated by a clear downregulation of CD62L, but not all donors showed the phenotype of fully activated neutrophils (CD16low CD63high). PBMC showed increased expression of TNFα and IL-6 mRNA (Figure 4(b)). HUVEC and platelets showed a modest upregulation of activation markers, whereas RBC did not respond to LPS stimulation (Figure 4(a)). In response to LPS, neutrophils from all donors showed a rapid and strong, but variable increase in the number of released EV, as quantified by high-resolution flow cytometry (Figure 4(c)) and western blot analysis of the common EV proteins CD9 and CD63 (Figure 4(d)). The increase in EV-release correlated with the neutrophil activation status (%CD16low CD63high) (Supplementary Figure 2A). The expected increase in EV release by PBMC due to LPS stimulation [62] was also observed, but differed by only 1.2-fold at this early time point (Figure 4(c)). Under the selected conditions, no increase in the number of released EV was observed for the other cell types. For LPS-stimulated neutrophils, but not for PBMC, we additionally observed a significant increase in the total amount of Y-RNA released via EV (Figure 4(e)), which is in line with the previously observed correlations between EV counts and Y-RNA levels (Figure 3(c)). Furthermore, we found that Y-RNA release directly correlated with neutrophil activation (%CD16low CD63high) (Supplementary Figure 2B). Importantly, our data indicated that the cell-type-specific Y-RNA-subtype ratios (Y4/Y3, Y4/Y1 and Y3/Y1) did not change in response to LPS stimulation (Figure 4(f)). Together, the data indicated that LPS stimulation of specific blood-related cell types can increase the total amount of EV-associated Y-RNA by increased sorting of Y-RNA into EV and/or by increased release of EV. However, the cell-type-specific Y-RNA signatures remain stable under these conditions.
Figure 4.
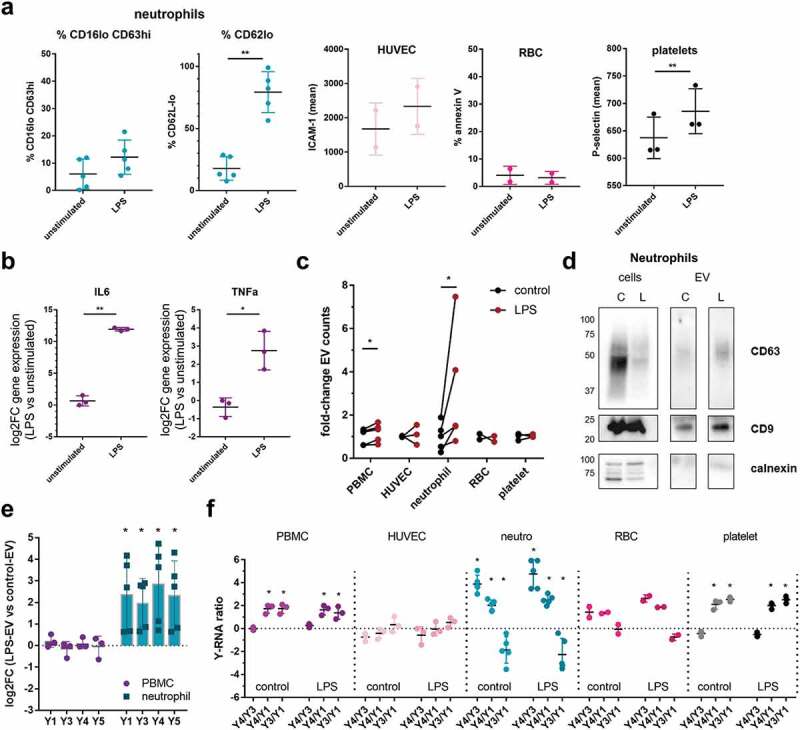
LPS stimulation changes the quantity of EV-associated Y-RNA by specific blood-related cell types, while cell-type-specific Y-RNA signatures remain stable.
Systemic inflammation induces alterations in the Y-RNA-subtype ratios in the total pool of plasma EV
Next, we investigated whether the Y-RNA signatures could also be identified in complex biofluids such as plasma. We hypothesized that the Y-RNA-subtype ratios in blood are influenced by the presence of different cell types in the circulation that produce EV with characteristic Y-RNA signatures. To test this hypothesis, we investigated changes in human plasma Y-RNA ratios after injection of healthy volunteers with LPS, which induces a rapid change in neutrophil and lymphocyte numbers [63]. This experimental human endotoxemia model is often used to study systemic inflammatory processes, such as those occurring during sepsis [64,65]. We analysed plasma samples of an earlier conducted study, in which plasma was collected from volunteers before and 2 or 6 hours after infusion with LPS [39,40]. Clinical responses in these volunteers were characterized by an increase in body temperature and heart rate, and increased levels of circulating inflammatory cytokines [39,40]. In addition, induction of systemic endotoxemia led to increased numbers of neutrophils and decreased numbers of lymphocytes and monocytes in the circulation, similar to what has been observed during sepsis [39,40,63,66]. The preferential localization of circulating Y-RNAs in EV (Figures 1 and 2) allowed us to determine Y-RNA-subtype ratios in plasma without prior isolation of EV. We isolated total RNA from plasma obtained before and 2 h after infusion of LPS. A slight increase in average RNA concentration could be observed at t = 2, with considerable variability between individuals (Figure 5(a)). We subsequently quantified the Y-RNA subtypes by RT-qPCR and calculated the different Y-RNA ratios before (black dots) and 2 h after (red dots) infusion of LPS (Figure 5(b)). The overall Y-RNA-subtype ratios in plasma are sum-ups of the Y-RNA ratios of all the individual EV present in plasma. Interestingly, substantial changes were detected in Y-RNA ratios in whole plasma 2 h after induction of inflammation. We observed a highly significant increase in the Y4/Y3 ratio (p = 0.001) and a significant decrease in the Y3/Y1 ratio (p = 0.03), while the Y4/Y1 ratio did not change (p = 0.078). The increase in Y4/Y3 ratio, which is specific for neutrophil-derived EV (Figure 3(e)) accompanied the previously observed increase in number of neutrophils (2.2-fold increase, p = 0.001) in the t = 2 blood samples [39]. Similarly, the decrease in Y3/Y1 ratio, which may indicate reduced numbers of EV released by PBMC (Figure 3(e)) accompanied the observed decrease in numbers of lymphocytes (2.2-fold decrease, p = 4.2E-9) and/or monocytes (19-fold decrease, p = 2.3E-10) [39]. To further investigate this, we determined pairwise Pearson’s correlations of the Y-RNA-subtype ratios with the different types of immune cells or with the different cytokines in the combined t = 0 and t = 2 datasets (Figure 5(c), see Supplementary Figure 3 for a full overview of these correlations). Importantly, we found a strong correlation between the neutrophil specific Y4/Y3 ratios and the blood neutrophil counts (R = 0.52, p = 0.0024) (Figure 5(d)). The decrease in Y3/Y1 ratios correlated with a decrease in monocyte numbers, but these data did not reach statistical significance (Figure 5(e)). Besides changes in immune cell counts, the levels of pro- and anti-inflammatory cytokines are highly increased during systemic endotoxemia and sepsis [63,67]. We observed that the neutrophil-specific Y4/Y3 ratio in plasma strongly correlated with levels of the pro-inflammatory cytokines IL-6 (R = 0.51, p = 0.0028), IL-8 (R = 0.43 p = 0.013) and also with TNFα (R = 0.35, p = 0.05), whereas the Y3/Y1 ratio negatively correlated with levels of the anti-inflammatory cytokine IL-10 (R = −0.39, p = 0.029) (Figure 5(f)). This raised the question whether the observed changes in Y-RNA-subtype ratios were mostly determined by the increased number of circulating immune cells or by the increased levels of cytokines that may influence the release of EV and/or their molecular composition. We therefore also analysed plasma samples collected 6 hrs after LPS infusion (blue dots). At this time point, body temperature, heart rate and cytokine levels had returned to baseline levels [39]. Additionally, while monocyte counts were increased to normal levels, neutrophil counts remained elevated on t = 6. We isolated RNA from total plasma collected at t = 6 and observed that the RNA concentrations at this time point had returned to values that were slightly lower than those observed on t = 0 (Figure 5(g)). Importantly, we observed that the Y4/Y3 ratio at t = 6 was still highly increased relative to t = 0 (Figure 5(h)). Moreover, the Y4/Y3 levels still correlated strongly with the neutrophil counts at this time point (Figure 5(i–j)). In contrast, the other correlations observed at t = 2 were not observed at t = 6 (Figure 5(i) and Supplementary Figure 4). The Y3/Y1 levels, which were decreased on t = 2, returned to control levels at t = 6 (Figure 5(h)), thereby following the pattern for monocyte counts (Figure 5(k)). This indicated that the observed alterations in Y-RNA-subtype ratios predominantly correlated with the numbers of circulating immune cells, but not with inflammatory cytokine levels.
Figure 5.

Y-RNA-subtype ratios in total plasma change during systemic endotoxemia.
We performed receiver-operator characteristics (ROC) analysis [55] of the Y4/Y3 ratios determined in this endotoxemia model to explore whether plasma Y4/Y3 ratio values could discriminate individuals in the early and late (resolving) phases of inflammation from healthy individuals (Figure 6). The Y4/Y3 ratio data of both t = 0 versus t = 2, and t = 0 versus t = 6 yielded excellent AUC values of 0.82 and 0.88, respectively. This suggests that increased Y4/Y3 ratio values accurately mark the past or present occurrence of systemic inflammatory processes.
Figure 6.

ROC curves of Y-RNA ratios.
Taken together, we have shown that Y-RNA in plasma is predominantly localized in EV. Furthermore, we discovered that EV from different blood cells contain cell-type-specific Y-RNA-subtype ratios. Systemic inflammation induced significant alterations in neutrophil- and PBMC-specific Y-RNA ratios observed in plasma. The detected changes in Y-RNA-subtype ratios strongly correlated with the inflammation-induced changes in the number of circulating neutrophils and monocytes. These immune cell-type-specific “Y-RNA signatures” in plasma EV can be determined without prior enrichment for EV, and may be further explored as biomarkers for inflammatory responses or other immune-related diseases
Discussion
Several studies have reported the abundant presence of Y-RNA in cell culture EV and body fluids [24,32,35,36,41,68–72]. Yet, the function and biomarker potential of extracellular Y-RNAs is an understudied research area. We therefore have limited knowledge on the presence of full length versus fragmented versions of Y-RNA in the extracellular space, the type of macromolecular structures to which extracellular Y-RNA is associated, and how this varies between different Y-RNA subtypes. Here, we performed extensive fractionation and characterization of macromolecular structures in plasma to show that the majority of full length Y-RNA is present in EV. This is importantly different from most of the studied miRNAs in plasma, which are known to distribute over both EV, LPP and RNP [1–3]. In addition, we show that EV released by various blood-related cell types differed in Y-RNA-subtype composition. This cell-type-specific “Y-RNA signature” remained stable upon LPS-stimulation of these cells. In a human endotoxemia model for systemic inflammation, we obtained proof-of-concept that the number of circulating immune cells correlates with the abundance of cell-type-specific Y-RNA-subtype ratios in plasma. The abundance of full length Y-RNAs in body fluids, their stability due to association with EV, and the immune cell-type-specific Y-RNA signatures may render these non-coding RNAs interesting candidate biomarkers for immune-related diseases.
In the field of exRNA research there is an increasing awareness that extracellular miRNAs are distributed over different macromolecular complexes, including EV, LPP and RNP [5,23,59,73]. These different RNA carriers overlap in size and buoyant density [59], which complicates strict separation of the different particles. The different types of EV isolation methods that are currently in use vary largely in the level of contamination with LPP and RNP [22,74–76]. Moreover, the currently used RNA isolation kits exhibit variable recovery efficiencies for RNAs enclosed in each of the different macromolecular complexes (EV, LPP and RNP) [22]. All of these factors complicate the accurate characterization of carrier-specific RNA profiles and cause inter-study and inter-laboratory variation in exRNA analysis. In this study, plasma was fractionated by consecutive application of size-exclusion chromatography and density gradient centrifugation. This is currently considered the most optimal method for separation of EV and LPP/RNP [59]. Yet, our data indicate that this approach did not achieve complete separation of EV and LPP/RNP. This is illustrated by some residual ApoAI (found in chylomicrons and HDL [58]) in the EV fractions. This may be due to incomplete separation of EV and LPP using this approach, or because LPP can physically interact with EV [77]. In addition, the non-pelletable fractions that were most enriched in the LPP proteins ApoAI and ApoB100 contained some residual CD9, which may point towards a subset of EV that are not efficiently pelleted at 100,000 g. Another approach to distinguish between EV- or LPP/RNP-associated RNA is to test for sensitivity to Protease/RNase treatment. Arroyo et al. applied enzymatic treatment of unfractionated plasma to demonstrate that plasma contained both miRNAs that are insensitive to protease/RNase treatment, likely due to enclosure in EV, and miRNAs that are sensitive to these enzymes [2]. In our current study, we built on this approach by performing size- and density-based enrichment for EV and LPP/RNP prior to applying the enzymatic treatment. Our data illustrate that the early SEC fractions not only contained EV-associated miRNAs that were resistant to enzymatic degradation, but also LPP/RNP-associated miRNAs that were sensitive to degradation. All of the full-length Y-RNA in plasma, on the contrary, was protected from degradation, supporting a predominant localization inside EV. In previous publication it was reported that Y-RNA in plasma and in culture medium of glioblastoma cells was associated to RNP complexes [69,73,78]. However, the technologies used in the glioblastoma studies preferentially detected Y-RNA fragments and the data indicated that such fragments were more abundant in RNP than in EV [69,73]. The study that identified RNP-associated Y-RNA in plasma employed plasma samples collected from individuals with autoimmune diseases [78,79]. It is possible that the distribution of full length Y-RNA over EV and other RNA carriers differs between disease processes. It is known that plasma sample of patients with autoimmune diseases contain more Y-RNA containing RNP and higher levels of anti-Ro60 antibodies compared to healthy controls [80]. Our current data show that in healthy plasma the full-length Y-RNA molecules are predominantly associated to EV.
The human genome encodes four different Y-RNAs, but little is known about functional differentiation of these Y-RNA subtypes inside cells. In many exRNA sequencing studies, the data were biased towards detection of Y-RNA fragments derived from the Y-RNA stem regions that are highly conserved between the Y-RNA subtypes [25]. The use of loop-specific primers, employed in our current study, allowed discrimination of the full length forms of the different Y-RNA subtypes. The cell-type-specific Y-RNA ratio signatures were most pronounced for EV from neutrophils (high Y4/Y3 – low Y3/Y1) and PBMC (high Y3/Y1 – high Y4/Y1). We used a human systemic endotoxemia model to provide proof-of-concept that Y-RNA-subtype ratios in plasma can change during systemic inflammation. This model system is commonly used to mimic the inflammatory response during sepsis [39,64,65]. The inflammatory responses induced by LPS infusion recapitulate those during sepsis, including major changes in circulating immune cells and inflammatory cytokines [39,63]. Our data suggest that the abundant presence of specific immune cell types, such as neutrophils, can cause changes in the plasma Y-RNA composition. The neutrophil-specific Y4/Y3 ratio was increased at 2 and 6 h after induction of systemic inflammation and correlated well with the number of neutrophils in the circulation. An earlier study reported a strong increase in neutrophil-derived large microparticles in plasma of sepsis patients [81]. In our current study, we focused on small EV and demonstrate in primary neutrophil cultures that in vitro LPS stimulation induces a large and rapid increase in the number of small EV released by these cells, which correlated with their activation status. Neutrophils are the most abundant immune cells in blood and are among the first to migrate and respond to infection [82,83]. The increased Y4/Y3 ratio we observed during systemic inflammation may therefore be caused by both the larger number of neutrophils that release EV and the activation-induced increase in EV release per neutrophil. Besides the changes in neutrophil-specific Y-RNA signatures, we observed a decrease in the Y3/Y1 ratio during inflammation. Our in vitro experiments indicated that EV from both PBMC and platelets were characterized by high Y3/Y1 ratios. Although we cannot exclude that EV from platelets contribute to the changes in Y3/Y1 ratios during inflammation, we observed that the Y3/Y1 levels in the endotoxemia plasma samples correlated with the monocyte counts at t = 2 and t = 6. Future research is needed to further establish the monocyte-specificity of Y3/Y1 ratios in EV and whether plasma Y3/Y1 ratios can be used as indicators of monocyte-driven disease processes.
Based on our data, we argue that Y-RNA-subtype ratios represent an interesting set of biomarkers to be further explored as biomarkers in the context of infection and inflammation. Detection of EV-associated full-length Y-RNAs may provide several advantages over detection of miRNAs, which have until now been a primary focus in EV-RNA biomarker research [22,84–86]. First, our data indicate that full-length Y-RNAs are predominantly present in EV. As a result, they are stable and resistant to enzymes that degrade proteins and RNAs, which reduces variability in their detection. Second, quantification of EV-associated full length Y-RNA in plasma does not require initial enrichment or purification of EV. Quantification of Y-RNA ratios can therefore be achieved by simply isolating total RNA from unprocessed plasma. This circumvents introduction of variation caused by variability in efficiency or purity of EV isolation. Third, Y-RNA-subtype ratios are based on the relative abundance of two transcripts in the same sample, which circumvents the need for RT-qPCR normalization or spike-in controls, which may further increase its detection accuracy.
Assessment of Y-RNA-subtype ratios could potentially be used in diagnosis and monitoring of inflammatory conditions such as sepsis syndrome, for which early biomarkers are an unmet need [87–89]. Assessment of Y-RNA-subtype ratios such as the Y4/Y3 ratio could be useful in diagnosis of sepsis because these ratios showed sufficient discriminatory potential between inflammatory and healthy plasma at the early activation of systemic inflammation and in the later resolution phase. At the later timepoint in our endotoxemia model, neutrophil counts and Y4/Y3 ratios were still high whereas the level of pro-inflammatory cytokines had returned to normal levels. This suggests that neutrophils and their EV may play a role in both the induction and the resolution of inflammation. In support of this, EV have been shown to exert both pro- and anti-inflammatory roles (reviewed in [90]). EV from activated neutrophils can cause tissue damage in the lungs [91], whereas anti-inflammatory effects include the reduction of phagocytosis and increasing LPS-resistance in dendritic cells [92]. Compared to the volunteers in the endotoxemia study, sepsis patients are much more heterogeneous with regard to the initial site of infection, causative organisms, and the overall health status of the patient [93]. We propose that combined assessment of Y-RNA-subtype ratios and other clinical parameters may provide novel opportunities for stratification of sepsis patient populations, or give additional information on the disease stadium. In case of local instead of systemic inflammation, neutrophils migrate into the inflamed tissues. Neutrophil-derived EV levels in the circulation may remain high under these conditions based on the idea that EV can cross epithelial and endothelial barriers [94,95]. Future research is needed to evaluate the applicability of Y-RNA-subtype-based diagnosis in diseases involving local tissue inflammation, such as in rheumatoid arthritis.
In conclusion, our data demonstrate that full length Y-RNAs in plasma are stably associated to EV. Moreover, we provide proof-of-concept that that Y-RNA ratios in pools of plasma EV are indicative for the number and type of immune-related cells during systemic inflammation. Hence, EV-associated Y-RNAs provide an abundant and stable source of candidate biomarkers for infectious and inflammatory diseases.
Supplementary Material
Acknowledgments
The authors would like to thank Dr G.J.A. Arkesteijn for the assistance with high resolution flow cytometric analysis of EV, E.G.F. Borg for assistance with the flow cytometric analyses, and Dr T. Zeev Ben Mordehai for assistance with electron microscopy.
Disclosure of interest
The authors report no conflicts of interest
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Vickers KC, Palmisano BT, Shoucri BM, et al. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol [Internet] 2011;13:423–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arroyo JD, Chevillet JR, Kroh EM, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A [Internet] 2011;108: 5003–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Turchinovich A, Weiz L, Langheinz A, et al. Characterization of extracellular circulating microRNA. Nucleic Acids Res [Internet] 2011. [cited 2014 July13];39:7223–7233. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3167594&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].van Niel G, D’Angelo G, Raposo G.. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol [Internet] 2018;19:213–228. [DOI] [PubMed] [Google Scholar]
- [5].Mateescu B, Kowal EJK, van Balkom BWM, et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA – an ISEV position paper. J Extracell Vesicles [Internet] 2017;6:1286095 Available from: https://www.tandfonline.com/doi/full/10.1080/20013078.2017.1286095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Redzic JS, Balaj L, van der Vos KE, et al. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol [Internet]2014;28:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Revenfeld ALS, Bæk R, Nielsen MH, et al. Diagnostic and prognostic potential of extracellular vesicles in peripheral blood. Clin Ther [Internet]2014;36:830–846. [DOI] [PubMed] [Google Scholar]
- [8].De Toro J, Herschlik L, Waldner C, et al. Emerging roles of exosomes in normal and pathological conditions: new insights for diagnosis and therapeutic applications. Front Immunol. 2015;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tofaris GK. A critical assessment of exosomes in the pathogenesis and stratification of Parkinson’s disease. J Parkinsons Dis. 2017;7:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Buzas EI, György B, Nagy G, et al. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol [Internet] 2014;10: 356–364. [DOI] [PubMed] [Google Scholar]
- [11].Zijlstra C, Stoorvogel W. Prostasomes as a source of diagnostic biomarkers for prostate cancer. J Clin Invest. 2016;126:1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Quek C, Hill AF. The role of extracellular vesicles in neurodegenerative diseases. Biochem Biophys Res Commun [Internet] 2017;483:1178–1186. [DOI] [PubMed] [Google Scholar]
- [13].Montecalvo A, Larregina AT, Shufesky WJ, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood [Internet] 2012;119: 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rak J. Extracellular vesicles - biomarkers and effectors of the cellular interactome in cancer. Front Pharmacol. 2013;4(MAR):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cha DJ, Franklin JL, Dou Y, et al. KRAS-dependent sorting of miRNA to exosomes. Elife. 2015;4:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Thompson AG, Gray E, Heman-Ackah SM, et al. Extracellular vesicles in neurodegenerative disease-pathogenesis to biomarkers. Nat Rev Neurol [Internet]2016;12:346–357. [DOI] [PubMed] [Google Scholar]
- [17].Nabet BY, Qiu Y, Shabason JE, et al. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell [Internet]2017;170:352–366.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Datta Chaudhuri A, Dastgheyb RM, Yoo SW, et al. TNFα and IL-1β modify the miRNA cargo of astrocyte shed extracellular vesicles to regulate neurotrophic signaling in neurons. Cell Death Dis [Internet] 2018;9 Available from: http://dx.doi.10.1038/s41419-018-0369-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Driedonks TAP, van der Grein SG, Ariyurek Y, et al. Immune stimuli shape the small non‑coding transcriptome of extracellular vesicles released by dendritic cells. Cell Mol Life Sci [Internet] 2018;3857–3875. Available from: https://doi.10.1007/s00018-018-2842-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Katsuda T, Kosaka N, Ochiya T. The roles of extracellular vesicles in cancer biology: toward the development of novel cancer biomarkers. Proteomics. 2014;14:412–425. [DOI] [PubMed] [Google Scholar]
- [21].Cheng L, Doecke JD, Sharples RA, et al. Prognostic serum miRNA biomarkers associated with alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol Psychiatry [Internet] 2014. [cited 2015 May8]: 1–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25349172 [DOI] [PubMed] [Google Scholar]
- [22].Buschmann D, Kirchner B, Hermann S, et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J Extracell Vesicles Internet 2018;7:1481321 Available from: https://doi.10.1080/20013078.2018.1481321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Srinivasan S, Yeri A, Cheah PS, et al. Small RNA sequencing across diverse biofluids identifies optimal methods for exRNA isolation. Cell [Internet] 2019;177: 446–462.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nolte-’t Hoen ENM, Buermans HPJ, Waasdorp M, et al. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res [Internet] 2012. [cited 2014 October21];40:9272–9285. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3467056&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Driedonks TAP, Nolte-’t Hoen ENM. Circulating Y-RNAs in extracellular vesicles and ribonucleoprotein complexes; implications for the immune system. Front Immunol [Internet] 2019;9:1–15. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2018.03164/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Christov CP, Gardiner TJ, Szüts D, et al. Functional requirement of noncoding Y RNAs for human chromosomal DNA replication. Mol Cell Biol [Internet] 2006;26: 6993–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gardiner TJ, Christov CP, Langley AR, et al. A conserved motif of vertebrate Y RNAs essential for chromosomal DNA replication. RNA. 2009;15:1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sim S, Weinberg DE, Fuchs G, et al. The subcellular distribution of an RNA quality control protein, the Ro autoantigen, is regulated by noncoding Y RNA binding. Mol Biol Cell. 2009;20:1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fuchs G, Stein AJ, Fu C, et al. Structural and biochemical basis for misfolded RNA recognition by the Ro autoantigen. Nat Struct Mol Biol. 2006;13:1002–1009. [DOI] [PubMed] [Google Scholar]
- [30].Perreault J, Noël JF, Brière F, et al. Retropseudogenes derived from the human Ro/SS-A autoantigen-associated hY RNAs. Nucleic Acids Res. 2005;33:2032–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rutjes SA, van der Heijden A, Utz PJ, et al. Rapid nucleolytic degradation of the small cytoplasmic Y RNAs during apoptosis. J Biol Chem [Internet] 1999;274: 24799–24807. [DOI] [PubMed] [Google Scholar]
- [32].Vojtech L, Woo S, Hughes S, et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res [Internet] 2014;42:7290–7304. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gku347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Alexander M, Hu R, Runtsch MC, et al. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun [Internet] 2015;6:7321 Available from: http://www.nature.com/doifinder/10.1038/ncomms8321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Y, Liu D, Chen X, et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell. 2010;39:133–144. [DOI] [PubMed] [Google Scholar]
- [35].Yeri A, Courtright A, Reiman R, et al. Total extracellular small RNA profiles from plasma, saliva, and urine of healthy subjects. Sci Rep Internet 2017;7:44061 Available from: http://www.nature.com/articles/srep44061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Godoy PM, Bhakta NR, Barczak AJ, et al. Large differences in small RNA composition between human biofluids. Cell Rep [Internet]2018;25:1346–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dhahbi JM, Spindler SR, Atamna H, et al. Deep sequencing of serum small RNAs identifies patterns of 5ʹ tRNA half and yrna fragment expression associated with breast cancer. Biomark Cancer 2014;6:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Repetto E, Lichtenstein L, Hizir Z, et al. RNY-derived small RNAs as a signature of coronary artery disease. BMC Med [Internet] 2015;13:259 Available from:: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4599655&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peters AL, van Hezel ME, Cortjens B, et al. Transfusion of 35-day stored RBCs in the presence of endotoxemia does not result in lung injury in humans*. Crit Care Med Internet 2016;44:e412–e419. Available from: http://insights.ovid.com/crossref?an=00003246-201606000-00055 [DOI] [PubMed] [Google Scholar]
- [40].Peters AL, van Hezel ME, Klanderman RB, et al. Transfusion of 35-day-stored red blood cells does not alter lipopolysaccharide tolerance during human endotoxemia. Transfusion. 2017;57:1359–1368. [DOI] [PubMed] [Google Scholar]
- [41].Driedonks TAP, Nijen-Twilhaar MK, Nolte-’t Hoen ENM. Technical approaches to reduce interference of fetal calf serum derived RNA in the analysis of extracellular vesicle RNA from cultured cells. J Extracell Vesicles [Internet] 2018;8 Available from: https://doi.10.1080/20013078.2018.1552059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Burger P, Hilarius-Stokman P, De Korte D, et al. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood. 2012;119:5512–5522. [DOI] [PubMed] [Google Scholar]
- [43].Schmitt C, Abt M, Ciorciaro C, et al. First-in-man study with inclacumab, a human monoclonal antibody against P-selectin. J Cardiovasc Pharmacol. 2015;65:611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Monteseirín J, Chacón P, Vega A, et al. L-selectin expression on neutrophils from allergic patients. Clin Exp Allergy. 2005;35:1204–1213. [DOI] [PubMed] [Google Scholar]
- [45].Amulic B, Cazalet C, Hayes GL, et al. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459–489. [DOI] [PubMed] [Google Scholar]
- [46].Lacy P. Mechanisms of degranulation in neutrophils. Allergy, Asthma Clin Immunol. 2006;2:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Meager A. Cytokine regulation of cellular adhesion molecule expression in inflammation. Cytokine Growth Factor Rev. 1999;10:27–39. [DOI] [PubMed] [Google Scholar]
- [48].Daleke DL. Regulation of phospholipid asymmetry in the erythrocyte membrane. Curr Opin Hematol. 2008;15:191–195. [DOI] [PubMed] [Google Scholar]
- [49].Stenberg PE, McEver RP, Shuman MA, et al. A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J Cell Biol. 1985;101:880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].van der Vlist EJ, Nolte-’t Hoen ENM, Stoorvogel W, et al. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protoc [Internet] 2012;7:1311–1326. Available from: http://www.nature.com/doifinder/10.1038/nprot.2012.065 [DOI] [PubMed] [Google Scholar]
- [51].Nolte-’t Hoen ENM, van der Vlist EJ, Aalberts M, et al. Quantitative and qualitative flow cytometric analysis of nanosized cell-derived membrane vesicles. Nanomedicine [Internet] 2012. [cited 2014 November5];8:712–720. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22024193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Groot Kormelink T, Arkesteijn GJA, Nauwelaers FA, et al. Prerequisites for the analysis and sorting of extracellular vesicle subpopulations by high-resolution flow cytometry. Cytom Part A. 2016;89:135–147. [DOI] [PubMed] [Google Scholar]
- [53].Libregts SFWM, Arkesteijn GJA, Németh A, et al. Flow cytometric analysis of extracellular vesicle subsets in plasma: impact of swarm by particles of non-interest. J Thromb Haemost. 2018;16:1423–1436. [DOI] [PubMed] [Google Scholar]
- [54].De Groote D, Zangerle PF, Gevaert Y, et al. Direct stimulation of cytokines (IL-1β, TNF-α, IL-6, IL-2, IFN-γ and GM-CSF) in whole blood. I. Comparison with isolated PBMC stimulation. Cytokine. 1992;4:239–248. [DOI] [PubMed] [Google Scholar]
- [55].Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics [Internet] 2011;12 Available from:: http://link.springer.com/10.1007/s00134-009-1641-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Van Deun J, Mestdagh P, Agostinis P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods [Internet] 2017;14:228–232. Available from: http://www.nature.com/doifinder/10.1038/nmeth.4185 [DOI] [PubMed] [Google Scholar]
- [57].Williams Z, Ben-Dov IZ, Elias R, et al. Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc Natl Acad Sci [Internet] 2013. [cited 2015 March17];110:4255–4260. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1214046110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Feingold K, Grunfeld C. Introduction to lipids and lipoproteins In: Feingold K, Anawalt B, Boyce A, editors. Endotext. [Internet] MDtext.com, Inc, South Dartmouth (MA), 2015. Available from: https://europepmc.org/books/NBK305896;jsessionid=FC86076A4DE6F06B7F048A4DFF4C7F05%0A [Google Scholar]
- [59].Karimi N, Cvjetkovic A, Jang SC, et al. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell Mol Life Sci [Internet] 2018;1–14. Available from: https://doi.10.1007/s00018-018-2773-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Arraud N, Linares R, Tan S, et al. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost. 2014;12:614–627. [DOI] [PubMed] [Google Scholar]
- [61].Sabroe I, Prince LR, Jones EC, et al. Selective roles for toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol. 2014;170:5268–5275. [DOI] [PubMed] [Google Scholar]
- [62].Wang J, Williams JC, Davis BK, et al. Monocytic microparticles activate endothelial cells in an IL-1b dependent manner. Blood. 2011;118:2366–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lowry SF. Human endotoxemia: A model for mechanistic insight and therapeutic targeting. Shock. 2005;24:94–100. [DOI] [PubMed] [Google Scholar]
- [64].Andreasen A, Krabbe K, Krogh-Madsen R, et al. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. [DOI] [PubMed] [Google Scholar]
- [65].van Lier D, Geven C, Leijte GP, et al. Experimental human endotoxemia as a model of systemic inflammation. Biochimie [Internet] 2018;1–8. Available from: https://doi.10.1016/j.biochi.2018.06.014 [DOI] [PubMed] [Google Scholar]
- [66].Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol [Internet] 2013;13:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Netea MG, Van Der Meer JWM, Van Deuren M, et al. Proinflammatory cytokines and sepsis syndrome: not enough, or too much of a good thing? Trends Immunol. 2003;24:254–258. [DOI] [PubMed] [Google Scholar]
- [68].Bellingham SA, Coleman BM, Hill AF. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res [Internet] 2012;40:10937–10949. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gks832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wei Z, Batagov AO, Schinelli S, et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat Commun [Internet] 2017;8:1145 Available from: http://www.nature.com/articles/s41467-017-01196-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tosar JP, Gambaro F, Sanguinetti J, et al. Assessment of small RNA sorting into different extracellular fractions revealed by high-throughput sequencing of breast cell lines. Nucleic Acids Res [Internet] 2015;1–16. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gkv432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shurtleff MJ, Yao J, Qin Y, et al. Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc Natl Acad Sci [Internet] 2017;114:E8987–E8995. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1712108114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lässer C, Shelke GV, Yeri A, et al. Two distinct extracellular RNA signatures released by a single cell type identified by microarray and next-generation sequencing. RNA Biol [Internet] 2017;14:58–72. Available from: https://www.tandfonline.com/doi/full/10.1080/15476286.2016.1249092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jeppesen DK, Fenix AM, Franklin JL, et al. Reassessment of exosome composition. Cell [Internet] 2019;177:428–445.e18. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867419302120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Van Deun J, Mestdagh P, Sormunen R, et al. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J Extracell Vesicles. 2014;3:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Karttunen J, Heiskanen M, Navarro-Ferrandis V, et al. Precipitation-based extracellular vesicle isolation from rat plasma co-precipitate vesicle-free microRNAs. J Extracell Vesicles [Internet] 2018;8 Available from: http://www.tandfonline.com/action/journalInformation?journalCode=zjev20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles [Internet] 2018;8:1535750 Available from: https://www.tandfonline.com/doi/full/10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sódar BW, Kittel Á, Pálóczi K, et al. Low-density lipoprotein mimics blood plasma-derived exosomes and microvesicles during isolation and detection. Sci Rep [Internet] 2016;6:24316 Available from: http://www.nature.com/articles/srep24316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hendrick JP, Wolin SL, Rinke J, et al. Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins: further characterization of the Ro and La small ribonucleoproteins from uninfected mammalian cells. Mol Cell Biol. 1981;1:1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Clancy RM, Alvarez D, Komissarova E, et al. Ro60-associated single-stranded RNA links inflammation with fetal cardiac fibrosis via ligation of TLRs: a novel pathway to autoimmune-associated heart block. J Immunol [Internet] 2010;184:2148–2155. Available from: http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.0902248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Garberg H, Jonsson R, Brokstad KA. The serological pattern of autoantibodies to the Ro52, Ro60, and La48 autoantigens in primary Sjögren’s syndrome patients and healthy controls. Scand J Rheumatol. 2005;34:49–55. [DOI] [PubMed] [Google Scholar]
- [81].Nieuwland R, Berckmans RJ, McGregor S, et al. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood [Internet] 2000;95:930–935. Available from: http://bloodjournal.hematologylibrary.org/content/95/3/930.full.pdf [PubMed] [Google Scholar]
- [82].Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol [Internet] 2019;4531200:255–265. [DOI] [PubMed] [Google Scholar]
- [83].Rosales C. Neutrophil: A cell with many roles in inflammation or several cell types? Front Physiol. 2018;9:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Vasilescu C, Rossi S, Shimizu M, et al. MicroRNA fingerprints identify miR-150 as a plasma prognostic marker in patients with sepsis. PLoS One. 2009;4:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ma Y, Liu Y, Hou H, et al. MiR-150 predicts survival in patients with sepsis and inhibits LPS-induced inflammatory factors and apoptosis by targeting NF-κB1 in human umbilical vein endothelial cells. Biochem Biophys Res Commun [Internet]2018;500:828–837. [DOI] [PubMed] [Google Scholar]
- [86].Benz F, Roy S, Trautwein C, et al. Circulating MicroRNAs as biomarkers for sepsis. Int J Mol Sci. 2016;17(1),78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Reinhart K, Bauer M, Riedemann NC, et al. New approaches to sepsis: molecular diagnostics and biomarkers. Clin Microbiol Rev. 2012;25:609–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Terrasini N, Lionetti V. Exosomes in critical illness. Crit Care Med. 2017;45:1054–1060. [DOI] [PubMed] [Google Scholar]
- [89].Raeven P, Zipperle J, Drechsler S. Extracellular vesicles as markers and mediators in sepsis. Theranostics. 2018;8:3348–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Groot Kormelink T, Mol S, de Jong EC et al. The role of extracellular vesicles when innate meets adaptive. Semin Immunopathol.2018; 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Genschmer KR, Russell DW, Lal C, et al. Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell [Internet]2019;176:113–126.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Eken C, Gasser O, Zenhaeusern G, et al. Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol [Internet] 2008;180:817–824. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18178820 [DOI] [PubMed] [Google Scholar]
- [93].Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:703–707. [DOI] [PubMed] [Google Scholar]
- [94].Alvarez-Erviti L, Seow Y, Yin H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol [Internet]2011;29:306–309. [DOI] [PubMed] [Google Scholar]
- [95].EL Andaloussi S, Mäger I, Breakefield XO, et al. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov [Internet] 2013;12:347–357. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23584393 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.