

Abstract
Peptides are an important class of molecules with diverse biological activities. Many endogenous peptides, especially neuropeptides and peptide hormones, play critical roles in development and regulating homeostasis. Furthermore, as drug candidates their high receptor selectivity and potent binding leads to reduced off-target interactions and potential negative side effects. However, the therapeutic potential of peptides is severely hampered by their poor stability in vivo and low permeability across biological membranes. Several strategies have been successfully employed over the decades to address these concerns, and one of the most promising strategies is glycosylation. It has been demonstrated in numerous cases that glycosylation is an effective synthetic approach to improve the pharmacokinetic profiles and membrane permeability of peptides. The effects of glycosylation on peptide stability and peptide-membrane interactions in the context of blood-brain barrier penetration will be explored. Numerous examples of glycosylated analogues of endogenous peptides targeting class A and B G-protein coupled receptors (GPCRs) with an emphasis on O-linked glycopeptides will be reviewed. Notable examples of N-, S-, and C-linked glycopeptides will also be discussed. A small section is devoted to synthetic methods for the preparation of glycopeptides and requisite amino acid glycoside building blocks.
Keywords: Glycopeptide, Hormone, Opioid peptide, CNS, Blood-brain barrier, GPCR
1. Introduction
Signaling and neuromodulatory peptides are a major class of biomolecules that play a myriad of important physiological roles. The ability of peptides to modulate diverse processes necessary for life has drawn the attention of a large number of research groups who aim to develop peptides for potential therapeutics to address a variety of medical disorders. Endogenous peptides act as neurotransmitters and signaling molecules that regulate and modulate intracellular communication, sometimes between adjacent cells and sometimes between remote organ systems [1,2]. Peptides exhibit high receptor affinity and low toxicity compared to small molecule drugs. Despite their immense therapeutic potential peptides suffer from low bioavailability and poor pharmacokinetic properties (PK/PD), as they are often metabolized by numerous proteases and peptidases. Various strategies have been employed to make peptides more effective drug candidates, including the use of non-natural amino acids, D-amino acids β-amino acids, cyclization, N-methylation, conjugation to drug delivery vectors, lipidation, and PEGylation [3,4,5,6,7]. Although these modifications have shown success in improving stability, many of the resulting stable peptide-based drugs are still incapable of crossing cellular barriers, such as ocular compartments, the blood nerve barrier (BNB), and the blood brain barrier (BBB). Cyclization and lipidation have shown enhancement of BBB penetration for some peptides by increasing their lipophilicity. [7] However, lipidation may pose a disadvantage because the introduction of highly lipophilic moieties can decrease water solubility, resulting in the reduction of overall bioavailability. Glycosylation provides an attractive alternative strategy for improving penetration of the BBB and enhancing stability while also increasing water solubility. Glycosylation of proteins in vivo is an essential post-translational modification for membrane proteins. Endogenous glycoproteins and glycopeptide hormones play critical roles in cell-cell adhesion, recognition processes, immune responses, ovulation, and more [8,9,10]. As interest in endogenous glycopeptides and glycoproteins has grown over the decades, novel methods detailing their isolation and characterization by nanoflow liquid chromatography coupled to mass spectrometry have been extensively developed. [11]. Glycosylation of endogenous neuropeptides has resulted in enhanced resistance to proteolytic degradation and, perhaps most importantly for our purposes, can modulate interactions with biological membranes, ultimately inducing penetration of the BBB [12, 13, 14].
In the late 1980s several research groups began the investigation of glycosylated analogues of various endogenous peptides and their therapeutic potential, particularly those with a role in the CNS. Several of the initial studies on therapeutic glycopeptides focused on the different subgroups of endogenous opioid peptides. Both the in vitro stability and in vivo biological activity of many of these analogues were improved significantly compared to the native un-glycosylated peptides. Given the success of these pioneering studies, other endogenous peptides targeting both class A and class B G-protein coupled receptors (GPCRs) have been subjected to glycosylation and subsequent in vitro and in vivo studies. In this review we will survey past and current examples of potentially therapeutic glycopeptides that target both class A and class B GPCRs. Some examples of non-therapeutic glycopeptides will also be discussed. The focus of this review will primarily be on O-linked glycopeptides, but notable examples of glycopeptides containing N-, S-, and C-linked carbohydrate moieties will be briefly discussed. Additionally, a small section will be devoted to the synthetic methodology employed for convenient introduction of carbohydrate moieties into peptide backbones using Fmoc-based solid phase peptide synthesis (SPPS).
1.1. General Review of Approaches to Make Peptides “Druggable”
When performing structure-activity relationship (SAR) studies on a peptide it is essential to determine what changes to the amino acid sequence will be produce an ideal drug candidate. It is important to identify regions within the peptide sequence that are susceptible to enzymatic cleavage and amino acid residues which are responsible for pharmacological efficacy. Residues that do not greatly affect the overall biological activity of the peptide but are enzymatically labile can be replaced by more stable moieties. Additionally, amino acid motifs that are pharmacophoric elements can be appropriately substituted by an amino acid residue that can modify the receptor binding and biological activity of the peptide, and simultaneously improve stability. One notable example of this strategy is the replacement of Tyr in opioid peptides with 2,5-dimethyltyrosine (χ-space) [15]. Peptide cyclization offers several advantages including stabilizing the peptide’s bioactive conformation and improving penetration of biological membranes. This is especially true for the hydrocarbon-stapled class of cyclic peptides that can penetrate cell membranes. A second notable example comes from Sawyer and coworkers at Merck. They designed and synthesized a number of novel cyclic peptides with an olefin bridge that exhibit potent and selective anticancer activity in vitro and in vivo [16]. Peptide lipidation has also been extensively explored as a method for enhanced membrane permeability, which is rationalized by the hypothesis that increasing the lipophilicity of the peptide will promote interactions with the lipid bilayer. Typically, a lipoamino acid moiety containing a linear alkyl side chain is introduced during solid phase synthesis [17]. This approach has been applied to the development of potent glucagon analogues and to human neutrophil elastase inhibitors with improved transport across the human epidermis in vitro [18,19]. PEGylation of the peptide backbone has also been widely explored as a means of improving the water solubility of the peptide, increasing its plasma half-life, and reducing renal clearance. However, further investigation of these proteins has indicated potential health concerns from introducing PEGylated drug conjugates into the human body, including pronounced cellular vacuolization, making this strategy a less attractive alternative [20,21]. Glycosylation is a strategy that improves membrane permeability and stability, increases water solubility, and causes minimal side effects. Glycosylation of peptides and the resultant effects on physical and chemical properties of peptides will be discussed in the next section.
1.2. Advantages of Peptide Backbone Glycosylation: Improved Stability and BBB Penetration
Glycosylation of the peptide backbone increases steric bulk around the amide bond, providing a shield from certain proteases and peptidases (Figure 1A). Additionally, the steric bulk helps prevent aggregation of individual peptide monomers and the hydroxyl groups of the sugar moiety improve water solubility. This translates into increased in vitro and in vivo half-lives and improved stability. It has also been postulated that the presence of the carbohydrate moiety influences the interactions a peptide has with biological membranes. Polt and coworkers have suggested that glycopeptides have biousian character, literally meaning “two essences.” These “essences” refer to the two different conformational states glycopeptides can adopt in H2O (bulk) and when bound to a membrane. Glycopeptides have a relatively lipophilic backbone that interacts with the membrane surface in an α-helical conformation, while the carbohydrate moiety increases the water solubility and allows the glycopeptide to interact with the aqueous compartment in an ensemble of random coil conformations. This allows the peptide to interact with both the membrane surface and aqueous compartment in a “hopping” motion (Figure 1B), improving bioavailability by reducing an inefficient three-dimensional search for its target receptor to a simpler two-dimensional search, ultimately reducing the amount of space the peptide must survey.
Figure 1.
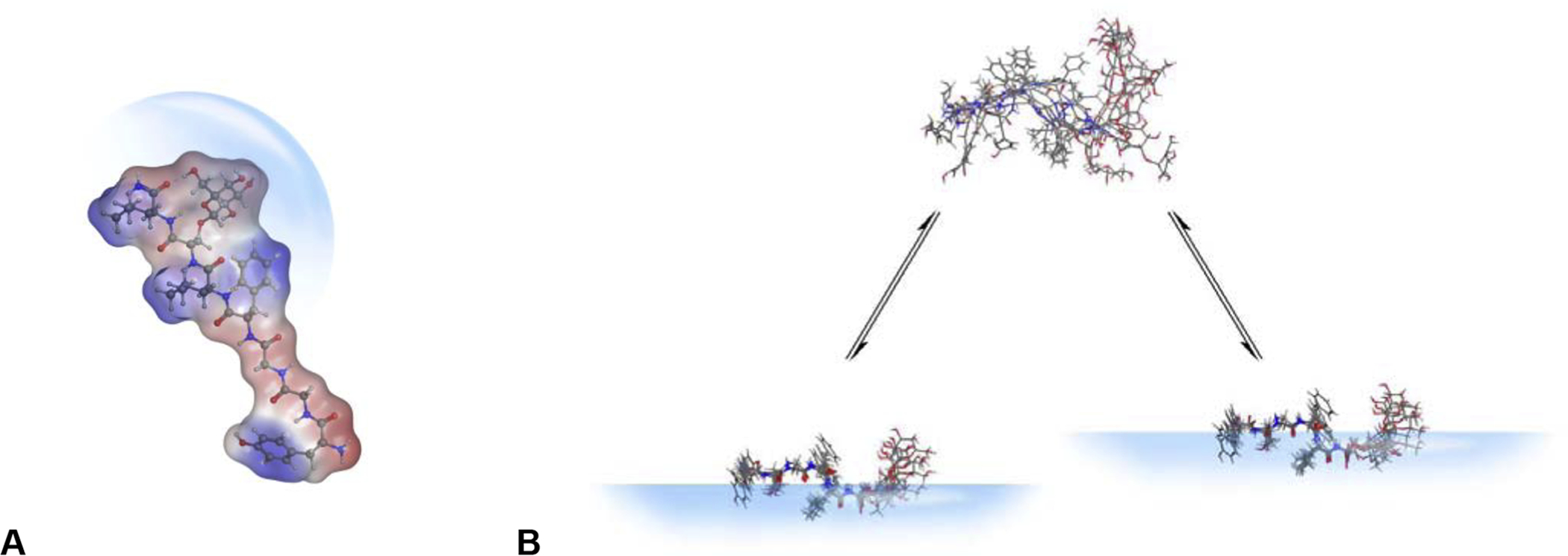
A. Glycosylation provides a steric shield towards proteolysis. B. Introduction of a carbohydrate moiety into the peptide backbone gives the peptide “biousian character.” A glycopeptide lacking a carbohydrate will strongly interact with the lipid bilayer and adopt an amphipathic α-helical conformation. The introduction of a carbohydrate will increase the water solubility of the peptide message and allow it to “hop” into the aqueous phase where it can adopt an ensemble of random coil conformations. This “hopping” behavior allows glycopeptides to assess a greater amount of surface area compared to their un-glycosylated counterparts.
Another important characteristic of glycopeptides is their ability to penetrate the BBB. The BBB consists primarily of tightly packed endothelial cells that act as a gatekeeper to the brain, only allowing in key nutrients for brain function (Figure 2). This includes small ions, water, and biomolecules like glucose and essential amino acids. Many of these species pass through the BBB either by passive diffusion or via a transporter protein. Although the exact mechanism for BBB penetration by glycopeptides is not fully understood, several mechanisms have been proposed, and several lines of evidence suggest that the glycopeptides are active in the CNS. One of the proposed mechanisms is adsorptive endocytosis. The “hopping” motions of the glycopeptides induce negative membrane curvature, and once a particular glycopeptide concentration is reached, the membrane will collapse in on itself and form a vesicle that transports the glycopeptide across the BBB.
Figure 2.
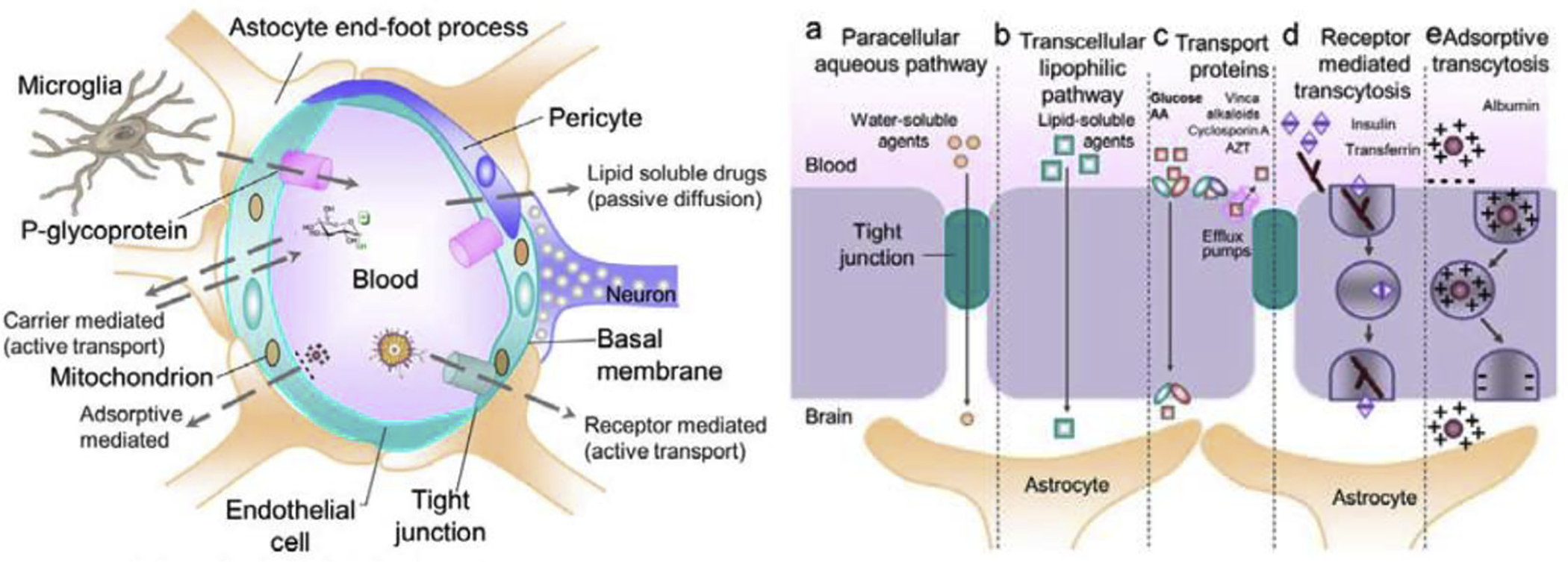
The BBB is comprised of tightly packed endothelial cells that act as a barrier preventing foreign substances from entering the brain. Essential nutrients can pass through the BBB via passive transport, passage through specific proteins or channels, receptor mediated transport, and adsorptive transcytosis. Adapted from Nanotechnology-based Targeted Drug Delivery Systems for Brain Tumors https://doi.org/10.1016/B978-0-12-812218-1.00003-8.
2. Chemical Methods for O-Linked Glycosidation and Glycopeptide Synthesis
In glycopeptide synthesis the carbohydrate moiety can be introduced in a number of ways. The focus of this review is not on synthetic methods for glycopeptide synthesis, so only a few notable examples will be discussed. The most straightforward way to incorporate carbohydrates into peptides is by synthesizing amino acid glycoside building blocks. Historically, this was achieved by the Koenigs-Knorr glycosylation [22]. Protected glycosyl bromides are reacted with the desired amino acid (generally serine or threonine) in the presence of a metal salt, usually silver or mercury, to afford the desired carbohydrate-amino acid linkage. Over the last few decades more selective and milder glycosylation procedures have been developed by various groups utilizing novel glycosyl donors including trichloroacetimidates and thioglycosides [23,24]. Other methods are reviewed elsewhere [25]. One method developed by Polt and coworkers utilizes Schiff base chemistry to enhance the nucleophilicity and reactivity of a serine glycosyl acceptor [26]. The nitrogen atom of the Schiff base forms hydrogen bonds with the hydroxyl group in the serine side chain. This interaction increases the nucleophilicity of the alcohol oxygen atom. In the case where an amine is present, it acts as a hydrogen bond donor to the alcohol side chain, diminishing its nucleophilicity and reducing its reactivity. Another method developed in the Polt laboratory uses InBr3 as a promoter. InBr3 is typically thought to be a very poor Lewis acid in glycosylation chemistry but is reactive enough in refluxing solvent to promote the glycosylation reaction. Additionally, it was found that the InBr3 can be used in catalytic amounts, which is advantageous over other methods requiring stoichiometric quantities of promoter [27]. Synthetic methodologies and considerations for the synthesis of glycopeptides on the solid phase are extensively reviewed elsewhere [28,29,30,31].
In addition to simple amino acids and their Schiff base analogues, α azido amino acids have also been explored to a small extent as glycosyl acceptors. Due to their relatively small size, azides are a useful alternative to the bulky Fmoc and Boc protecting groups, which can decrease reactivity in some cases. In the context of glycosylation, this strategy has been especially useful in the preparation of sterically hindered α-methylserine analogues containing a glucoside. [32]. This approach has also been successful in the synthesis of glycosylated amino acid building blocks for an in vitro translational system to make glycoproteins [33]. Interestingly, α azido amino acids were also studied as glycosyl acceptors using enzymatic catalysis, and it was found that they undergo glycosylation selectively and in good yields [34]. The azido group is also a useful functional handle that can be converted into a variety of triazole-containing scaffolds via copper catalyzed click reactions with alkynes [35]. O’Flaherty and coworkers utilized this approach to synthesize several galactosylated α azido amides containing a triazole linkage with a lipid tail [36]. It has also been demonstrated that α azido amino acids and their glycosylated analogues can be successfully implemented into SPPS protocols to yield biologically relevant peptides [37,38,39]. Due to the broad synthetic utility of azides, their ability to tolerate both chemical and enyzymatic glycosylation reaction conditions, and their successful incorporation in SPPS methodologies demonstrates their emerging importance in the synthesis of novel neoglycopeptides.
3. Examples of N-, S-, and C-linked Glycopeptides
Although the focus of this review is O-linked glycopeptides, notable examples of N-linked, S-linked, and C-linked require some discussion. N-linked carbohydrates are typically appended to an asparagine side chain via an amide bond, and the S-linked glycopeptides contain carbohydrates moieties bonded through a cysteine side chain. C-linked glycopeptides are a special case that occur naturally in some antifreeze glycoproteins found in various fish and in human RNase Us [40,41,42]. The carbohydrate is linked through a carbon atom in the indole ring of a tryptophan side chain. Synthetic methodologies have been extensively explored to afford novel C-linked glycosyl amino acid building blocks that mimic serine O-linked glycosyl amino acids, which are covered elsewhere [43,44,45,46,47,48]. S- and C-linked glycopeptides are especially stable towards proteolysis as compared to O- and N-linked carbohydrates because of the non-natural linkage through sulfur or carbon, which is not recognized by many enzymes.
3.1. N-Linked Glycopeptides
One of the most common post translational modifications in proteins is N-linked glycosylation through an Asn amide nitrogen. This is done at the specific consensus sequence Asn-X-Thr/Ser, where X is any amino acid except for proline [49]. N-linked glycopeptides are commonly synthesized on solid supports using traditional solid phase peptide synthesis protocols via the introduction of sugar amino acid building blocks; enzymatic methods have also been exploited [50,51,52,53,54]. One of the earliest examples of the synthesis of N-linked glycopeptides in solution was performed by Rocchi and coworkers. They introduced a 2-amino-2-deoxy-β-D-glucopyranose moiety into the C-terminus of the peptide by either reductive glucosamination or C-terminal amidation [55]. Guillemin and coworkers synthesized several glycosylated analogues related to somatostatin containing either glucose or N-acetylglucosamine linked through the amide nitrogen of Asn5 [56]. This is one of the earliest examples of a glycopeptide targeting a class B GPCR. Furthermore, the glycosylated analogues exhibited similar in vitro and in vivo profiles to native somatostatin. Albert and coworkers synthesized several orally bioavailable glycopeptide analogues of octreotide, a more stable derivative of somatostatin [57]. The carbohydrate moieties were introduced at the N-terminal amino group via the Amadori reaction [58]. The aldehyde of the aldose reacts with the free amine at the N-terminus to form a Schiff base that undergoes an irreversible Amadori rearrangement, affording the stable N-linked carbohydrate motif. Interestingly, this reaction can proceed in the absence of enzymes [59]. The glycosylated analogues exhibited similar receptor binding affinities to that of octreotide. In vivo studies examining inhibition of growth hormone secretion showed that the glycosylated analogues were more potent than octreotide following subcutaneous administration in anaesthetized rats [57]. The maltoside-containing analog was studied further in rhesus monkeys [57]. Following oral administration of the maltoside analogue at a dose of 10 μg/kg plasma concentrations of growth hormone were reduced by 50% for 8 hours. This is remarkable considering that octreotide exhibited similar effects for only 4 hours at a dose of 100 μg/kg. Another interesting example is the synthesis of pramlintide glycopeptides that act as amylin receptor agonists by a combined solid phase and enzymatic approach [60]. In this work, Fairbanks and colleagues improved the physiochemical properties of pramlintide, a synthetic analogue of the naturally occurring peptide amylin prepared for the treatment of type I and II diabetes, by introducing GlcNAc moieties N-linked through specific Asn residues. The GlcNAc residues were then elaborated using a variety of endo-β-N-acetylglucosaminidases to afford highly diverse glycan structures. Select pramlintide glycopeptides were evaluated for their ability to attenuate blood glucose levels in vivo, and it was found that they control glucose levels as well as, if not better than, native pramlintide.
3.2. S-Linked Glycopeptides
The synthesis of S-linked glycopeptides is generally achieved in either solution or in the solid phase and utilizes sugar amino acid building blocks [61,62]. The carbohydrate moiety is typically linked through a cysteine or cysteine derivative side chain [31,63]. S-linked glycopeptides have been sought after due to their improved stability towards enzymatic degradation as compared to their O-glycosylated glycopeptide counterparts. In a recent study, Pratt et al. reported a robust synthetic method to afford S-β-D-GlcNAc building blocks for SPPS and their incorporation into model peptides and proteins [64]. NMR and computational studies on these S-linked glycopeptides indicated the sulfur linkage as a good structural mimic for an oxygen linkage, and in vitro stability studies on a S-β-D-GlcNAc analogue of α-synuclein showed that the S-linked analogue is stable towards human O-GlcNAc transferase.
3.3. C-Linked Glycopeptides
Similarly to S-linked glycopeptides, C-linked glycopeptides have been pursued due to their superior enzymatic stability as compared to native glycopeptide linkages. Since the syntheses of C-linked amino acid building blocks are difficult, much attention has been focused on improved synthetic methodologies to afford SPPS-friendly sugar amino acid building blocks. Furthermore, due to these synthetic challenges the conformational dynamics of C-linked glycopeptides as compared to N- and O-linked glycopeptides has not been investigated in-depth. Early work performed by Bertozzi addressed many of the synthetic challenges, and in one particular study the conformational effects of a C-linked glycoside in a model peptide was studied [65]. Circular Dichroism studies revealed that both C- and O- linked glycopeptide analogues destabilized the helical structure of the native peptide to a similar extent, and the CD data obtained for both glycopeptides had similar profiles. As of late, the synthesis of biologically active C-linked glycopeptides is still limited due to the difficult syntheses of the peptide building blocks. However, many novel asymmetric methods have been explored to synthesize these building blocks in a reasonable quantity for peptide synthesis [66,67,68].
4. O-linked Glycopeptides Targeting Class A G-Protein Coupled Receptors (GPCRs)
GPCRs comprise the largest class of membrane receptors in humans [69]. They are involved in the regulation of many important physiological functions, ranging from neuroprotection to pain relief. Because of their significance, many small molecule and peptide drugs have been developed to target various GPCRs. Roughly 40% of drugs currently on the market target a GPCR. [70]. GPCRs can be broken down into several classes based on their overall structure. The largest of these is the class A family of GPCRs, which are commonly referred to as Rhodopsin-like. The class B GPCRs, or the secretin family of GPCRs, are unique from class A in that they contain a large extracellular N-terminal domain required for binding their relatively large endogenous peptide ligands. Class C GPCRs will not be discussed in this review. Our discussion of class A GPCR glycopeptide ligands is divided into two parts: opioid peptides and other nonopioid peptides. Class B glycopeptide ligands discussed include GLP-1, VIP, and calcitonin.
4.1. O-linked Glycopeptides Targeting Class A GPCRs: Opioid Peptides
Opioid glycopeptides comprise one of the most extensively studied groups of glycosylated neuropeptides due to their importance in regulating responses to painful stimuli. One of the major drawbacks preventing analogues of endogenous opioid peptides from being used as treatments for neuropathic pain is their inability to cross the BBB efficiently. This prompted investigations into the synthesis and biological evaluation of opioid glycopeptides in the 1980s and 1990s. The following sections will cover notable examples of such opioid glycopeptides based on their corresponding endogenous structure. Specifically, glycopeptides related to enkephalin, endomorphin, β-endorphin, dermorphin, deltorphin, DAMGO, morphiceptin, and DALDA will be discussed.
4.1.1. Glycopeptides Targeting Class A GPCRs: Enkephalin-Inspired Glycopeptides
Enkephalins represent a class of endogenous opioid peptides that are selective for the δ opioid receptor. Following the discovery and characterization of the enkephalins by Hughes and coworkers, there were a large number of research groups designing and synthesizing more stable and efficacious analogues [71]. Many of these groups explored the effects of glycosylation on the biological properties of the enkephalins.
Peter Varga of the Schiller lab synthesized several glycosylated enkephalin analogues (Figure 3) containing O-linked β-D-glucosides appended through the phenolic oxygen of the N-terminal tyrosine residues [72]. Specifically, glucosylated analogues related to enkephalin and [Leu5] enkephalin were synthesized and evaluated for their in vitro biological activity in the standard guinea pig ileum (GPI) and mouse vas deferens (MVD) assays. It was found that glycosylation of the N-terminal tyrosine side chain resulted in analogues that were either inactive at concentrations up to 3×10−5 M or were extremely weak agonists. These studies further confirmed the importance of the tyrosine hydroxyl side chain group in the activity of opioid peptides.
Figure 3. Phenolic Leu-Enkephalin β-D-Glucoside.
Glycosylation of the phenolic oxygen of Leucine Enkephalin strongly reduced activity in the MVD and GPI assays. (Varga and Schiller, 1987) [72].
Following the work on the glucosylated enkephalins at the N-terminus, Horvat and Schiller examined enkephalin glycopeptides containing a glycoside at the C-terminus (Figure 4) [73]. They introduced β-D-Glucose into the C-terminal residue via an ester linkage through either C-1 or C-6 of the glucose residue. Like the previous series of compounds, the C-terminal glycopeptides were examined for their in vitro activity in the GPI and MVD assays. In particular, the 1-O glucosylated analogues exhibited somewhat higher and lower potency as compared to [Leu5] enkephalin in the GPI and MVD assays, respectively. The 6-O glucosylated analogues were less potent in both assays. Interestingly, the introduction of the carbohydrate units into the C-terminus through the ester linkage resulted in a slight increase in δ opioid receptor selectivity.
Figure 4. C-Terminal Ester Enkephalin Glycopeptides.
Enkephalin glycopeptides containing a β-D-glucose moiety at the C-terminus exhibited similar activity to that of [Leu5] enkephalin (Horvat and Schiller, 1988) [73].
One important N-linked glycopeptide worth mentioning in this section is the N-linked glucosyl analogue of [D-Met2, Pro5] enkephalin by Torres and coworkers (Figure 5) [74]. The glucose moiety was linked through the C-terminal amide nitrogen, and the peptide was synthesized in solution phase using Boc chemistry. The in vitro potency of this compound was relatively low compared to morphine in the GPI assay, but it exhibited significantly higher activity in vivo in the tail immersion assay following intraperitoneal (i.p.) administration. Specifically, this compound was 2000 times more active in rats and 200 times more active in mice than morphine.
Figure 5. C-Terminally N-Glycosyl Enkephalin Glycopeptides.
An N-linked glucosylated enkephalin glycopeptide exhibited significantly greater analgesic activity compared to morphine in vivo. (Torres 1988) [74].
Following previous studies on the antinociceptive activity of novel C-terminally glycosylated enkephalin amide analogues, Rodriguez and coworkers synthesized enkephalin analogues containing hydroxyproline β-D-glucosides or galactosides (Figure 6) [75,76]. These compounds were examined for the antinociceptive activity in vivo through tail immersion and paw pressure behavioral studies. The compounds were injected by either intrathecal (i.t.) or intracerebroventricular (i.c.v.) administration. The results from these studies showed that the glycosylated enkephalin analogues were much more potent than morphine and the un-glycosylated enkephalin analogues. Notably, in the paw pressure assay the galactosylated analogue was approximately 5500 times more potent than morphine, 1000 times more potent than the un-glycosylated control, and 160 times more potent than the glucosylated analogue.
Figure 6. Hydroxyproline (Hyp)-Containing Glycopeptides Related to Enkephalin.
Hydroxyproline glycopeptides related to enkephalin were roughly 5500 times more potent than morphine in vivo. (Rodriguez 1989) [75].
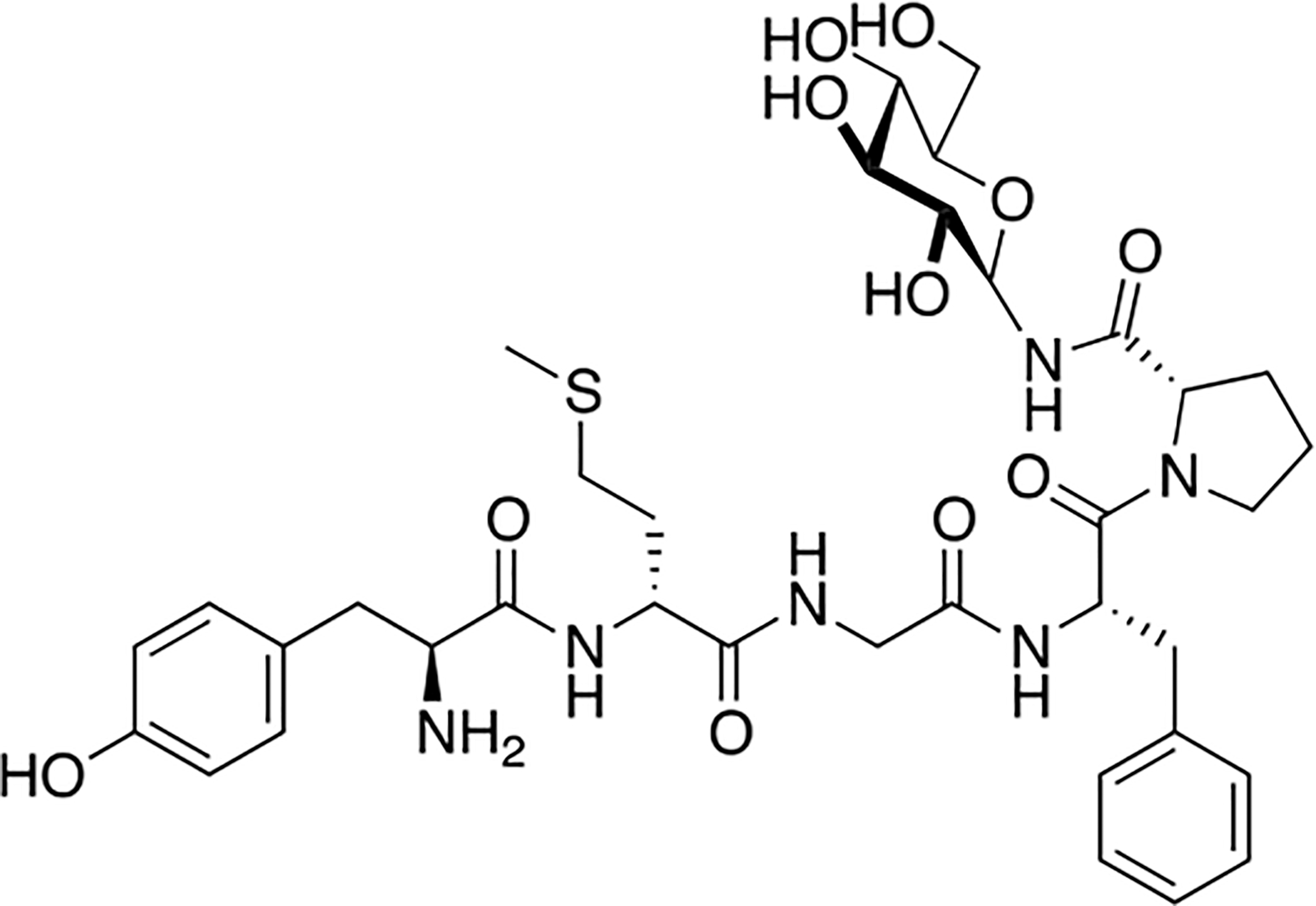
Polt and coworkers synthesized a series of glycopeptides related to [Met5] Enkephalinamide and the δ opioid receptor selective peptide, DPDPE (Figure 7) [77,78]. The carbohydrate moiety was introduced via coupling of a serine β-D-glucoside building block during solid phase peptide synthesis. One notable example from this work is the C-terminally glycosylated DPDPE analogue, which retained opioid receptor binding and was more δ opioid receptor selective than its un-glycosylated analogue. The Glycopeptide was then examined in vivo using both tail flick and hot plate assays. Following i.p. administration, this glycopeptide exhibited potent analgesia that could be antagonized by naloxone administered i.c.v. but not i.p., suggesting that the glycopeptide was acting centrally. This indicates that the glycopeptides are able to penetrate the blood brain barrier to a significant extent. Williams and colleagues further studied one particular DPDPE glycopeptide for its stability in vitro and BBB penetration in vivo [79]. Specifically, they performed stability studies both in mouse brain homogenate and serum and examined penetration properties in bovine brain microvessel endothelial cells. Additional studies on CNS uptake were performed via in situ brain perfusion. In vitro stability studies showed that the glycosylated and un-glycosylated analogues had similar half-lives in brain homogenate (>8 hours) and serum (~5 hours). Interestingly, the glycosylated analogue did not exhibit significantly different BBB penetration rates from the un-glycosylated analogue in vitro, but the permeability of the glycosylated analogue was much greater than that of the un-glycosylated analogue in the in situ penetration experiments.
Figure 7. Opioid Glycopeptide Derived from DPDPE.
Glycopeptides related to the [Met5] enkephalinamide and DPDPE were synthesized and studied for antinociception in vivo. One particular glucosylated analogue of DPDPE produced analgesia following i.p. administration. Naloxone administration antagonized the effects of this glycopeptide following i.c.v. administration but not i.p. administration, suggesting this glycopeptide is able to penetrate the BBB and act in the CNS. (Polt 1994) [77].
An Enkephalin glycopeptide produced by Polt and coworkers containing a C-terminal serine glucoside was examined for its binding and functional activity in vitro and then evaluated for its ability to produce analgesia in vivo (Figure 8) [80]. In situ perfusion experiments revealed enhanced brain uptake of the glycosylated analogue compared to the un-glycosylated peptide. GPI and MVD functional assay results showed that the glycopeptide was more active than morphine as it exhibited a much lower IC50 value. The results from the in vivo studies revealed that the glycopeptide produced significant analgesia comparable to morphine, and the un-glycosylated analogue was not as potent as the glycosylated analogue. However, when administered peripherally (i.p.) there was a reduction in potency. This reduction was small, however, which indicates that the glycopeptide drug successfully penetrated the BBB.
Figure 8. An Analogue (Roque’s Enkephalin) Containing A Serine Glucoside at the C-Terminus.
Enkephalin glycopeptide containing a C-terminal serine glucoside produced potent analgesia following both i.v. and i.p. administration. (Polt 2000) [80].
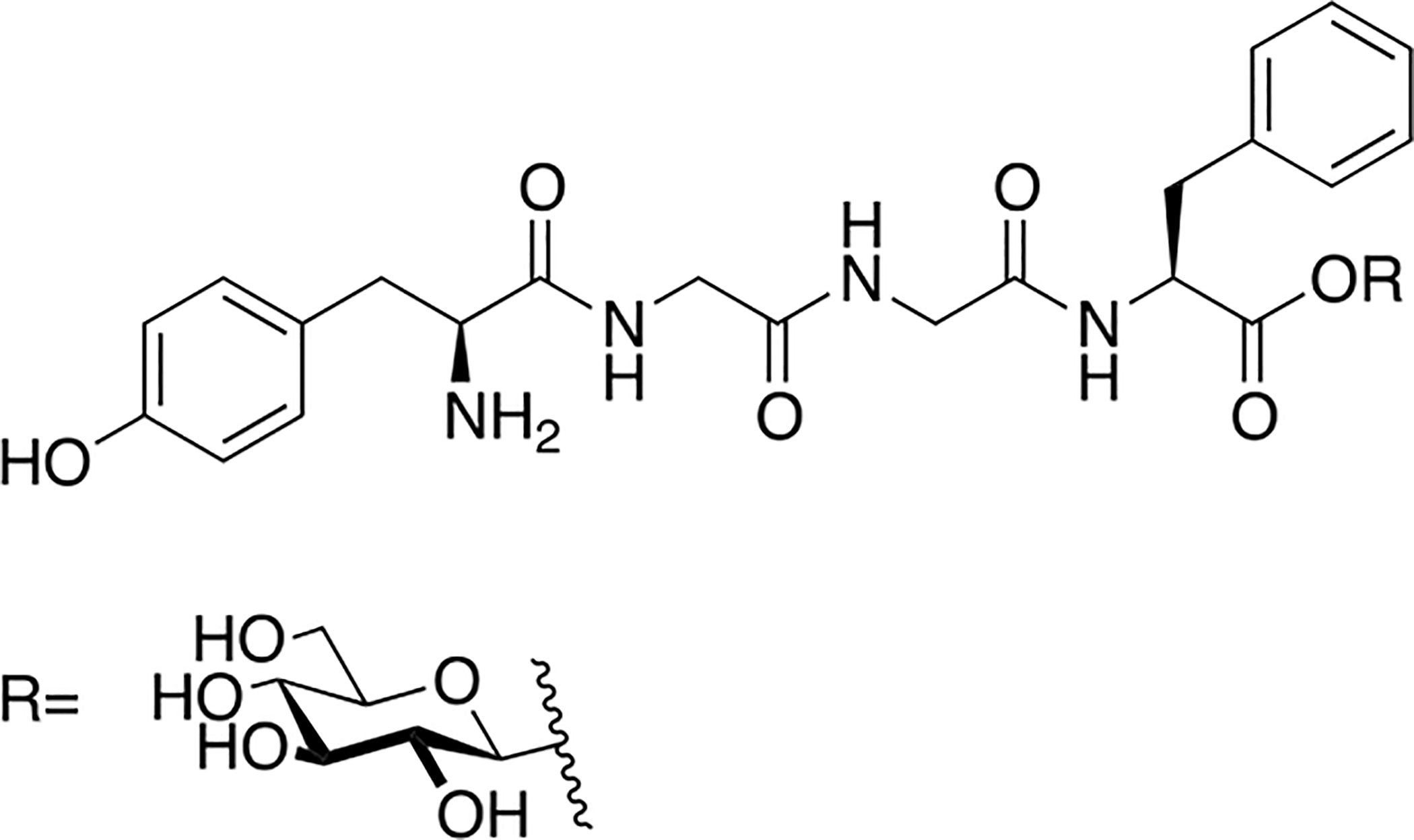
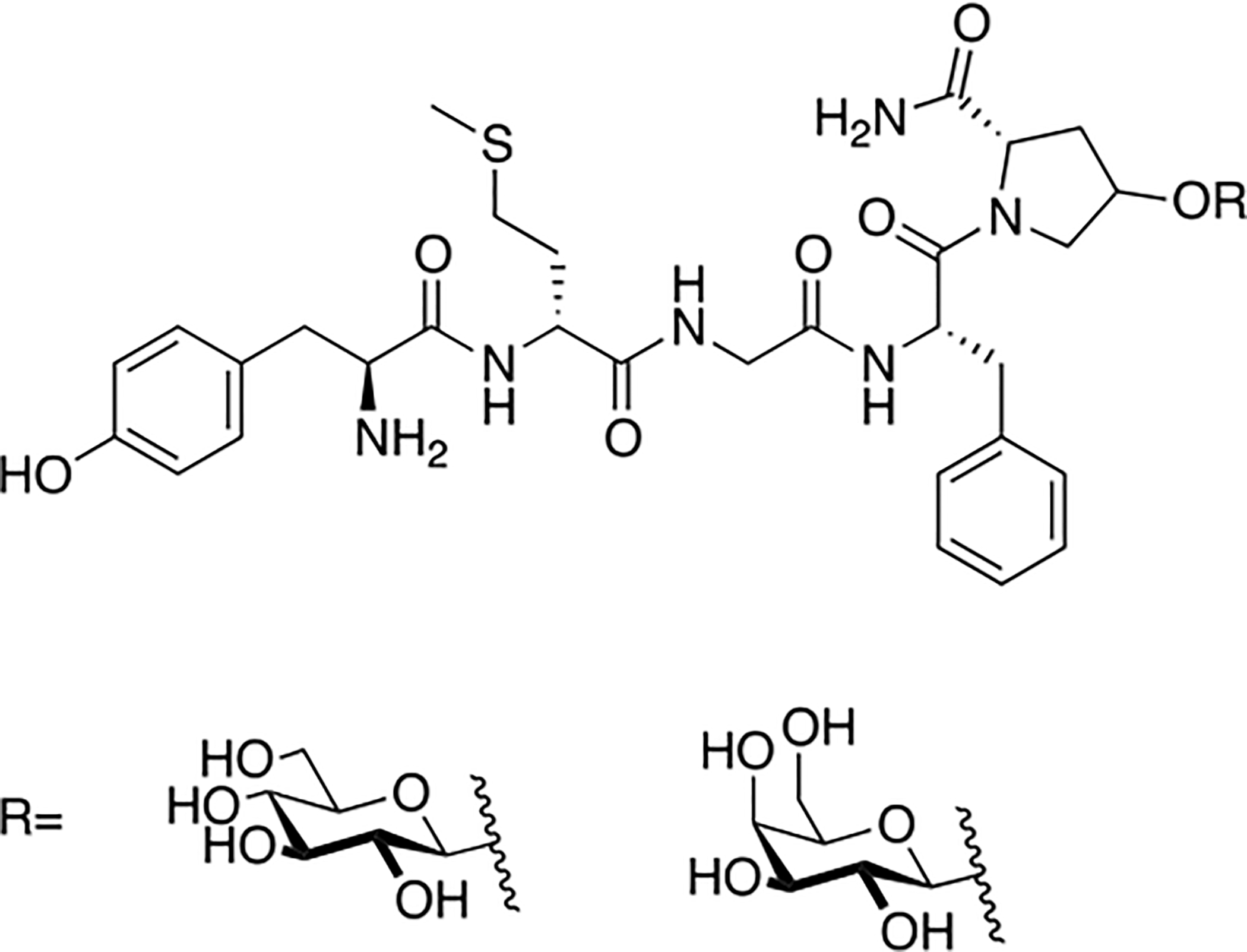
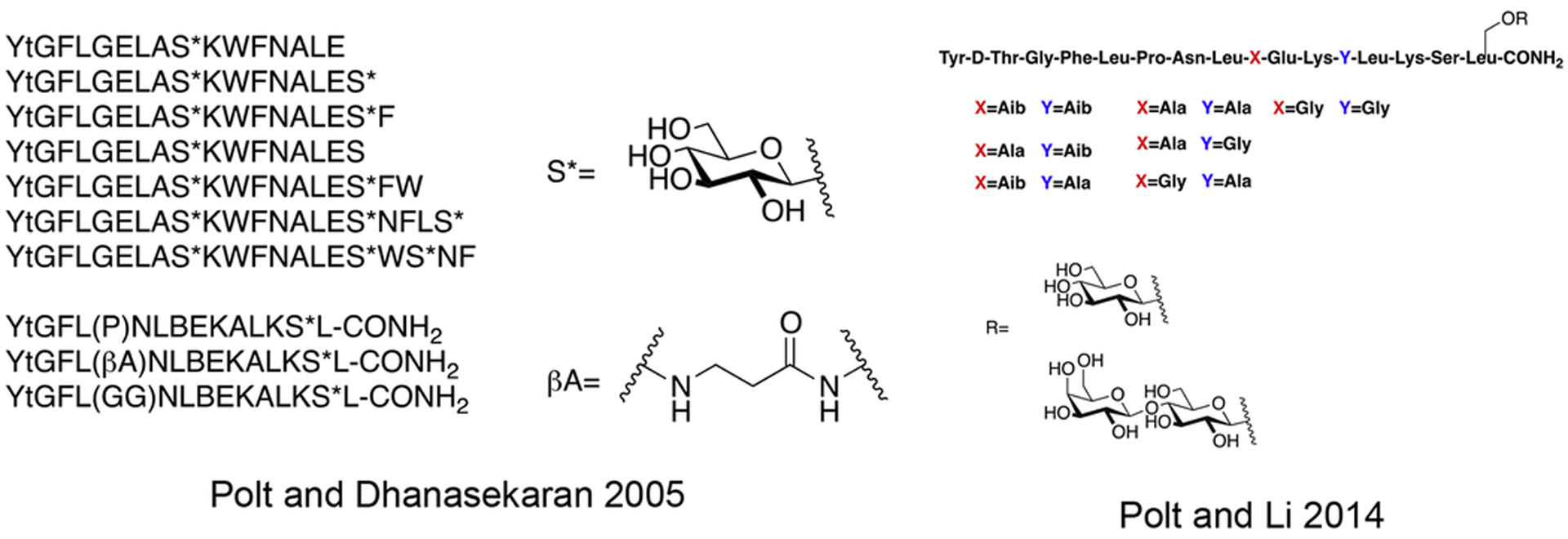
Polt and coworkers continued their studies of opioid glycopeptides by synthesizing a series of glycosylated enkephalin analogues containing a C-terminal carbohydrate moiety linked through either a serine or a threonine side chain (Figure 9) [81]. The carbohydrate moieties appended to the peptide backbone included β-D-xylose, β-D-glucose, α-D-mannose, β-lactose, β-maltose, β-maltotriose, and β-melibiose. Stabilities (t1/2) of the glycopeptide analogues in mouse brain and serum were significantly greater than the parent un-glycosylated peptide. It is important to note that improvements in stability correlated with increased degree of glycosylation. The glycosylated analogues exhibited low nanomolar binding at the μ and δ opioid receptors, which was consistent with functional activity data. Interestingly, replacing either β-D-xylose or β-D-glucose with α-D-mannose resulted in decreased affinity at the target receptors and decreased δ opioid receptor selectivity. Increased potency was observed as a result of using a disaccharide rather than a monosaccharide. It was hypothesized that the improved in vivo profiles were a result of increased hydrodynamic volume. To test this, a trisaccharide-containing glycopeptide was synthesized and evaluated in vitro and in vivo, but this compound was less potent than the disaccharide compounds.
Figure 9. Enkephalin Inspired Glycopeptides Containing Mono-, Di-, and Trisaccharide Motifs.
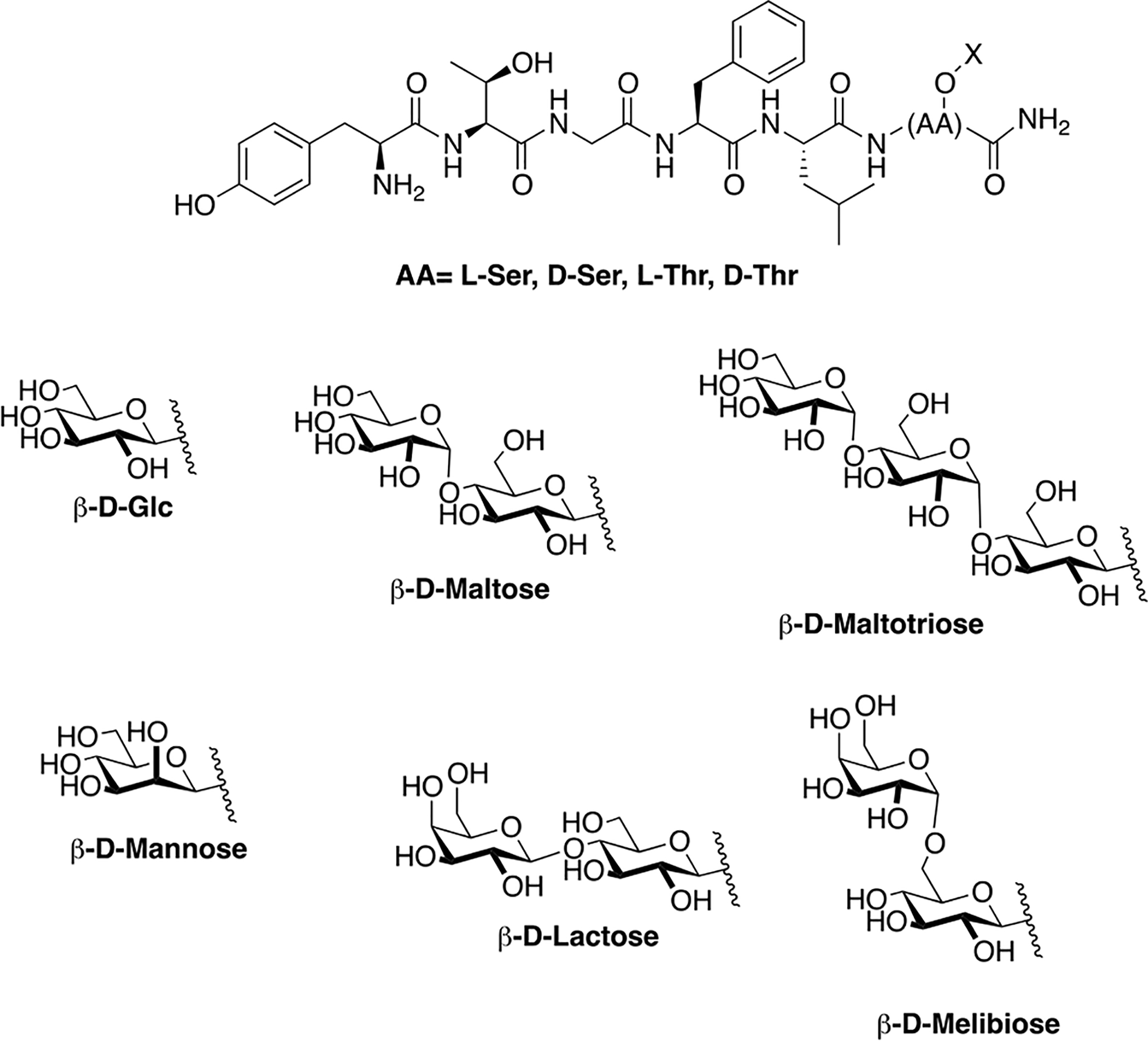
Enkephalin glycopeptides containing a variety of mono-, di-, and trisaccharides were synthesized and evaluated for their ability to produce analgesia in vivo by simultaneous agonism of both the mu and delta receptor. The lactoside (MMP2200, or Lactomorphin) was manufactured commercially at 400 g scale in a cGMP compliant fashion by PolyPeptide Labs, and by Lonza, which has since sold its peptide manufacturing facility to PPL. It is still being studied, and is used as a laboratory standard. (Polt 2004) [81].
Lactoside MMP2200, an enkephalin analogue containing a β-lactose moiety, was chosen to be extensively studied in vivo for its ability to penetrate into the CNS (Figure 10). Mabrouk and coworkers measured the concentration of MMP2200 entering the CNS, specifically in the dorsolateral striatum, following i.p. administration [82]. MMP2200 entering the CNS was measured using in vivo microdialysis coupled to mass spectrometry using live and fully conscious rats. It was determined that low nanomolar concentrations initially entered the dorsolateral striatum. 80 minutes after administration MMP2200 could still be detected at a concentration of approximately 250 pM.
Figure 10. MMP2200: A Novel Opioid Glycopeptide with Excellent BBB Penetration Properties.
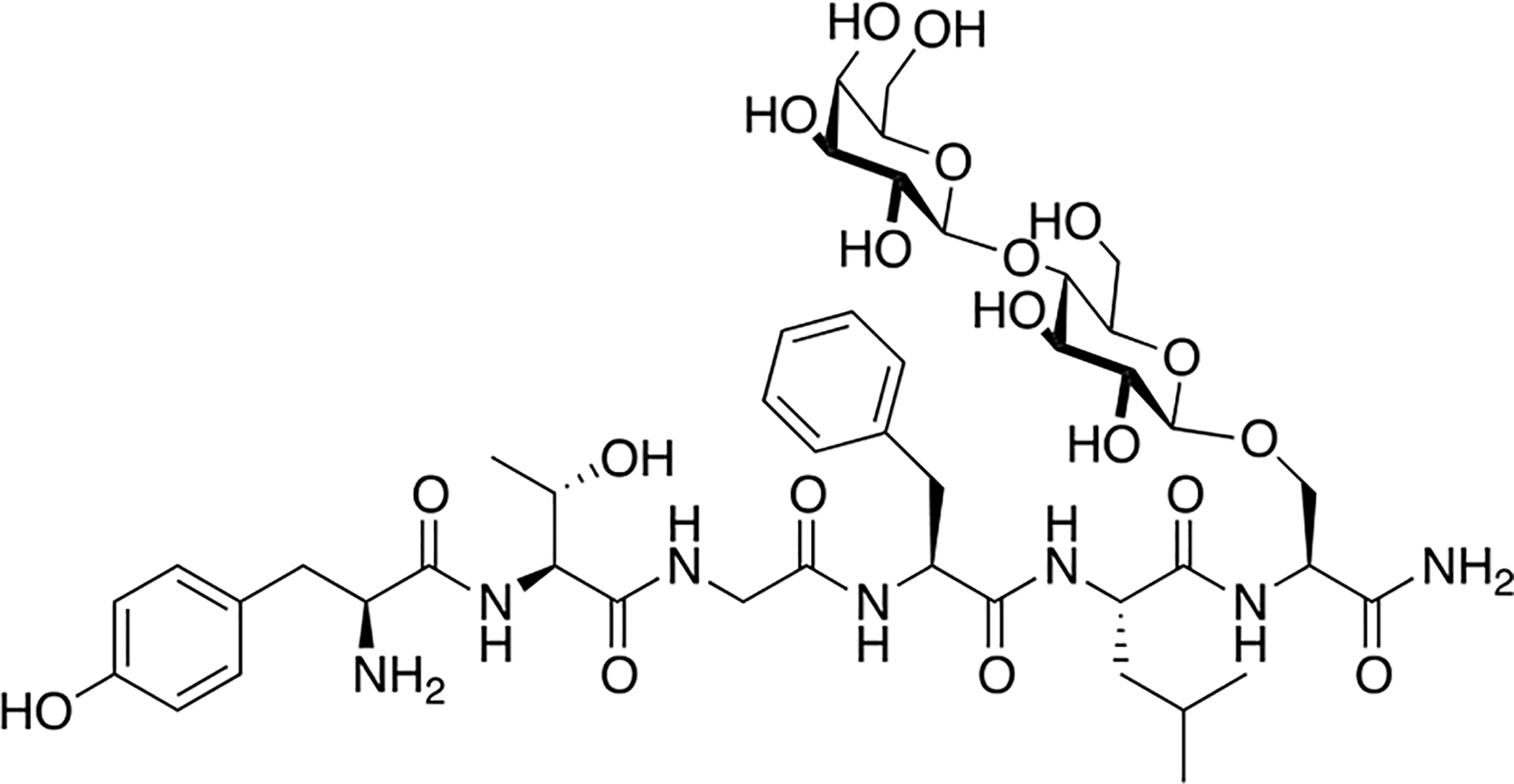
The lactoside MMP2200 produced in Polt’s laboratory was studied by in vivo microdialysis coupled with LC-MS to evaluate the extent of its BBB penetration. The results from these studies showed that MMP2200 entered the brain at low nanomolar concentrations and was still detected in the brain 80 minutes after administration. (Polt 2005) [82].
Valencia and coworkers revisited their previously synthesized enkephalin β-D-glucosides and β-D-galactosides and re-examined their receptor binding and antinociceptive properties (Figure 11) [83]. They also synthesized several new analogues containing α-D-mannose, β-D-lactose, and β-D-cellobiose to further investigate the effects of the carbohydrate unit on biological activity and conformation. In receptor binding studies, it was found that introducing disaccharides increased selectivity at the μ opioid receptor, while monosaccharides showed no shift in selectivity. However, the glucose analogue was slightly more δ opioid receptor selective compared to the other compounds. The mannosylated glycopeptide was selected for in vivo studies and produced significant analgesia in the tail-flick assay following i.p. administration, indicating that it had readily penetrated the BBB. The mannosylated glycopeptide was selected for in vivo study because of previous work by Valencia’s group, in which a mannosylated morphine analogue was found to be significantly more potent and longer lasting than native morphine in rats following i.p. administration [84].
Figure 11. Enkephalin Glycopeptides with Carbohydrate Moieties O-Linked to Hydroxyproline.
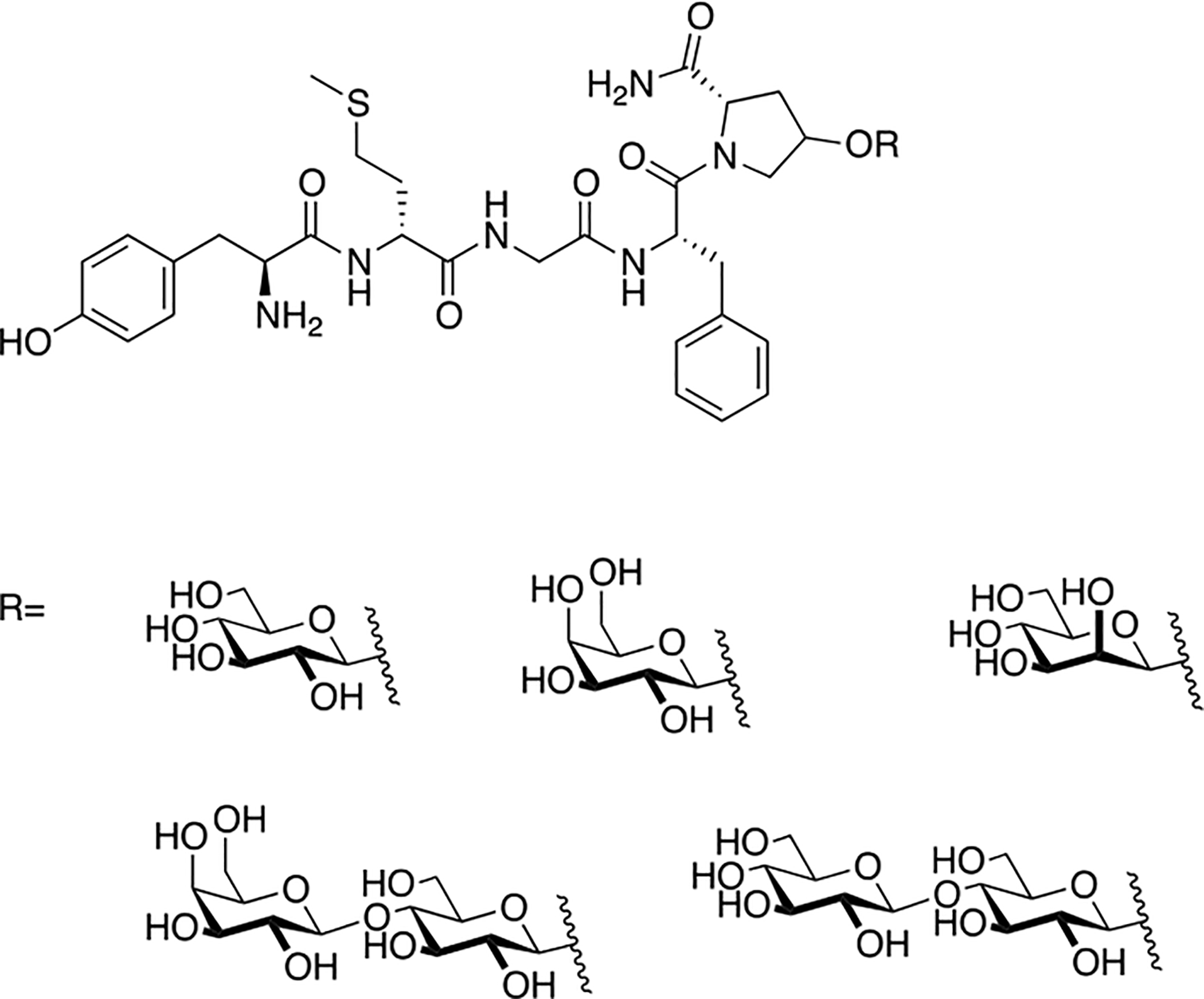
Enkephalin glycopeptides studied by Valencia and Rodriguez in the 1980s were later synthesized and evaluated for their activity in vitro. Additional analogues containing different carbohydrate moieties were also examined. The mannosylated analogue in particular exhibited desirable transport properties and had a better in vivo profile than morphine. (Valencia 2017) [83].
4.1.2. Glycopeptides Targeting Class A GPCRs: Endomorphin Glycopeptides
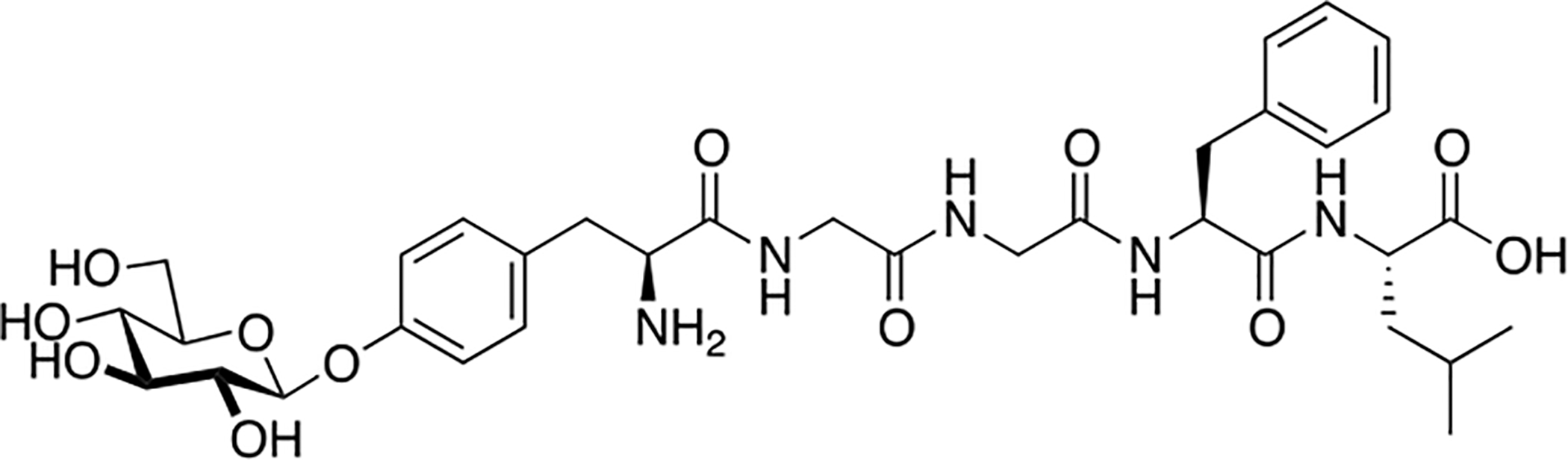
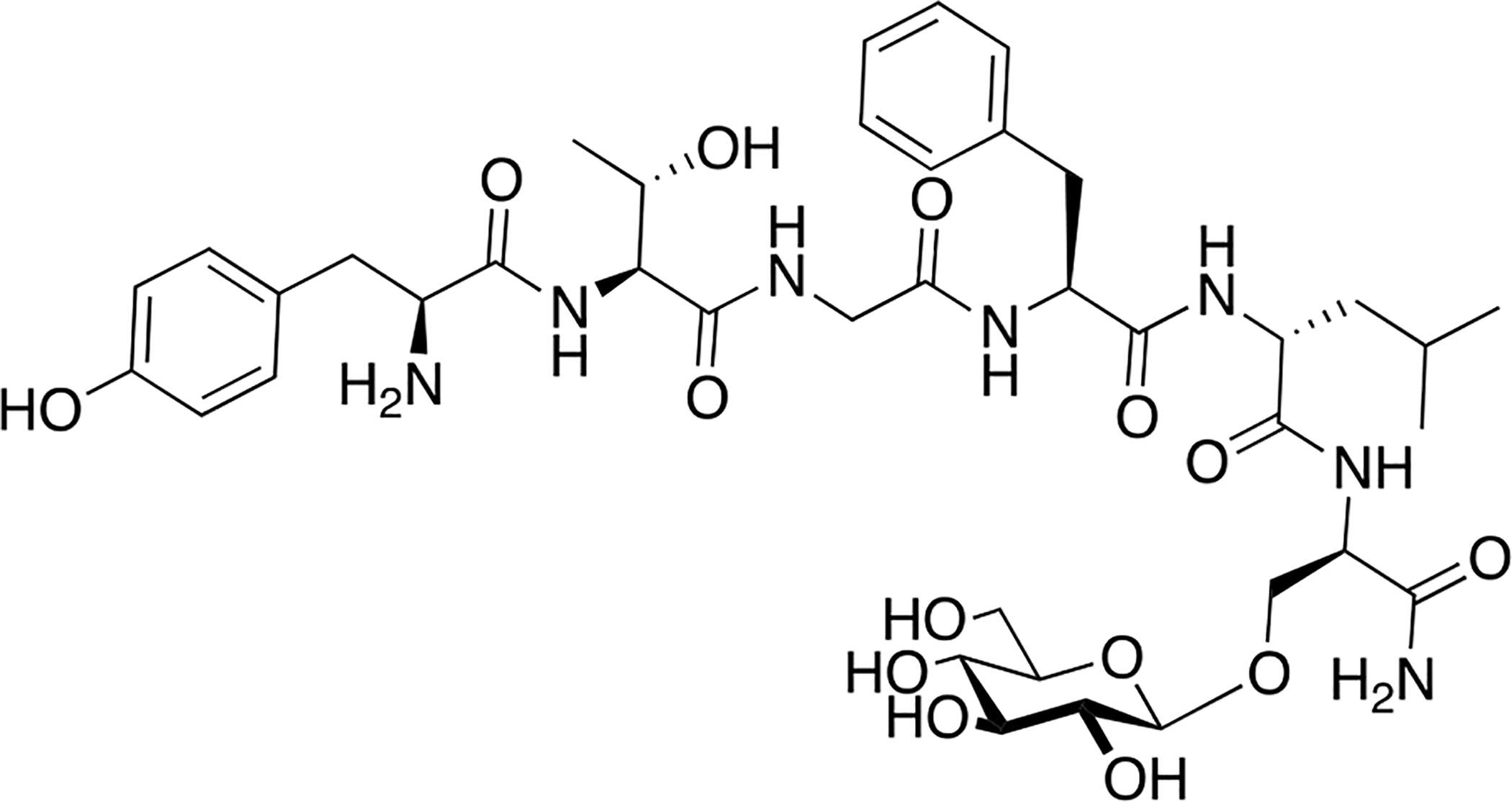
Varamini and coworkers of the Toth lab synthesized a novel endomorphin-1 analogue containing a β-D-lactose moiety linked to the N-terminal tyrosine amino group via a succinamide acid spacer (Figure 12) [85]. Modifications to the N-terminus of opioid peptides have been established to typically reduce receptor affinity and functional activity, but this particular analogue exhibited only slight reductions in its μ opioid receptor affinity and agonist activity in comparison to its un-glycosylated analogue. Furthermore, this analogue exhibited exceptional stability both in vitro and in vivo. Most notably, it was found to be orally active following oral administration at a dosage of 10 μmol/kg to CCI rats. Significant concentrations in the micromolar range of this analogue were present in the rat’s blood plasma up to 90 minutes after administration. Additionally, the glycosylated analogue exhibited higher permeability compared to the un-glycosylated compound in Caco-2 cell monolayers. Due to this analogue’s improved stability, minimal reductions in pharmacological efficacy, and oral bioavailability, it represents a potential lead candidate for the development of future endomorphin-1 analogues that are orally active and successfully alleviate pain.
Figure 12. An Orally Active Endomporphin-1 Glycopeptide.
A novel endomorphin glycopeptide containing a lactose moiety at the N-terminus interestingly showed only slight reductions in biological activity in both receptor binding and functional activity experiments. This analogue was highly stable both in vitro and in vivo and was found to be orally active. (Varamini and Toth 2013) [85].
Olczak and coworkers synthesized several novel endomorpin-2 glycopeptides containing a β-D-glucose residue appended to the Tyr3 side chain phenolic oxygen (Figure 13) [86]. The glucose residue was either fully acetylated or fully de-acetylated. Initial receptor binding studies at the μ opioid receptor using the GPI assay showed that the glycopeptide containing the fully acetylated glycoside did not bind to the receptor. However, the fully de-acetylated analogue exhibited binding in the low nanomolar range with a 70-fold decrease in binding compared to the native un-glycosylated compound. The de-acetylated compound was studied further in vivo and exhibited potent antinociception at a dose of 3 mg/kg following i.v. administration. To examine whether or not the antinociceptive effects were mediated centrally or peripherally, naloxone methiodide was administered along with the de-acetylated compound. The naloxone methiodide did not block the effects of the de-acetylated compound, suggesting central activity. These results also indicate that the glycosylated endomorphin-2 analogue can penetrate the BBB.
Figure 13. Phenolic Endomorphin Glycopeptides.
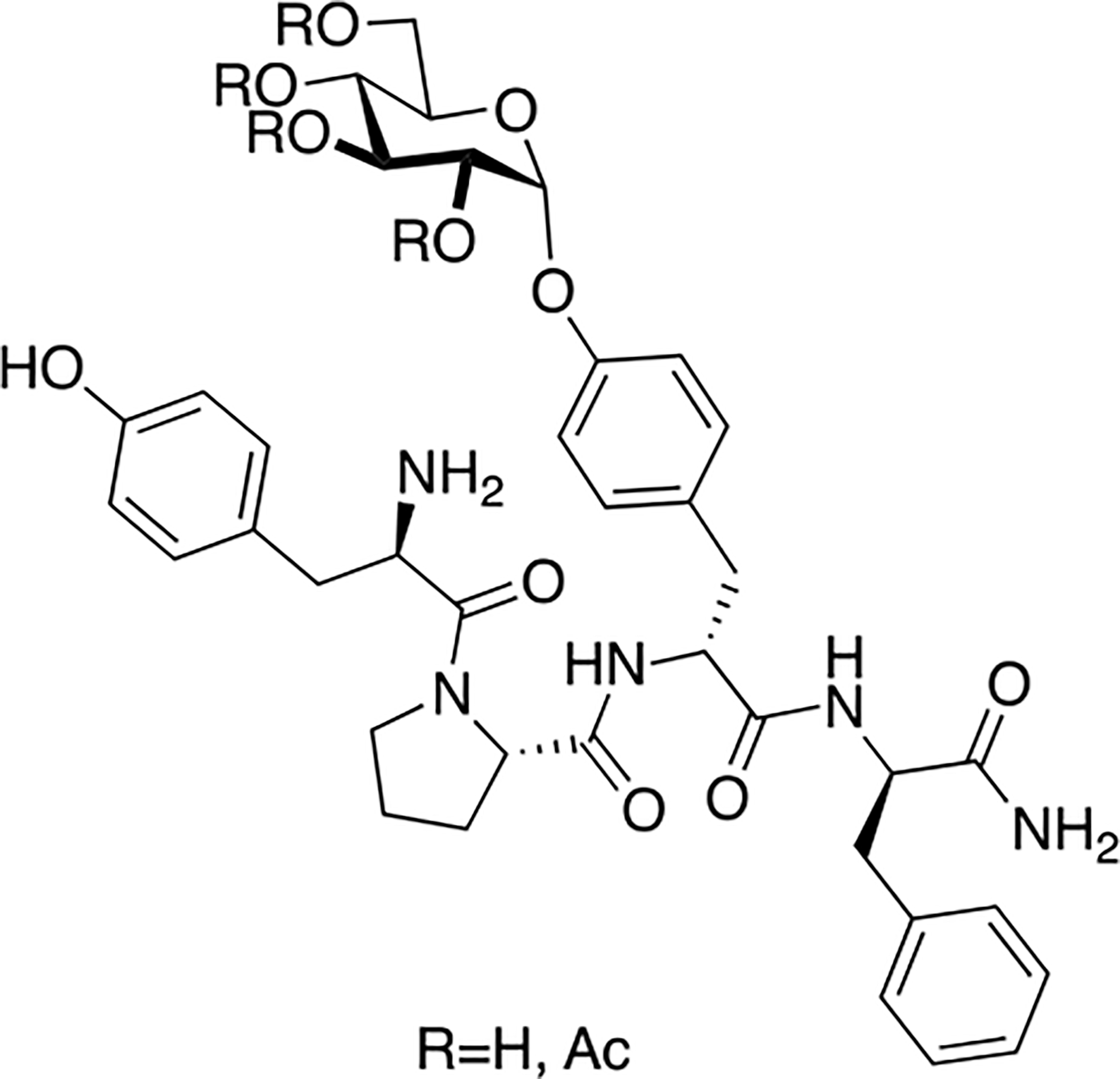
Endomorphin glycopeptides containing carbohydrate moieties linked through the phenolic oxygen of a tyrosine residue was found to act centrally in vivo and penetrate the BBB. (Olczack 2013) [86].
4.1.3. Glycopeptides Targeting Class A GPCRs: Endorphin Glycopeptides
It has been established from many in vivo studies that glycopeptides can penetrate the BBB, but the exact mechanism has yet to be elucidated. To this end, Dhanasekaran of the Polt laboratory synthesized a series of opioid glycopeptides related to β-endorphin and studied their conformations using 2D NMR and circular dichroism in water, trifluoroethanol, and SDS micelles (Figure 14) [87]. The sequences of the peptides were based on work previously performed by Kaiser, who proposed that artificially constructed C-terminal regions mimicking β-endorphin would exhibit similar binding properties to that of native β-endorphin, as long as the appropriate linker between the message and address regions was used, the N-terminus was preserved, and the C-terminal region retained amphipathic character [88]. NMR and CD studies confirmed that the glycopeptides adopted highly helical conformations in the presence of SDS micelles. Further experiments using plasmon-waveguide resonance (PWR) in the presence of phosphatidyl choline bilayers revealed that the glycopeptides adopted an α-helical conformation in the presence of the bilayers and that they were able to dissociate from them and adopt a random coil conformation.
Figure 14. Amphipathic Glycopeptides Related to β-Endorphin.
Dhanasekaran of the Polt’s lab investigated the conformation of a number of glycosylated endorphin analogues by 2D NMR and circular dichroism in various media. (Polt 2005) [87].
Polt and coworkers synthesized a series of amphipathic glycopeptides related to β-endorphin (Figure 15) [89]. Their goal was to examine how varying the amphipathicity of different glycopeptides would affect their binding, in vivo activity, and BBB penetration. These particular glycopeptides are thought to have biousian character; they rapidly interconvert between a more water-soluble random coil conformation and a lipophilic (α-helix preferring) conformation in the presence of a lipid bilayer or cell membrane. It has been hypothesized that the biousian character of these glycopeptides promotes BBB penetration. Receptor binding studies showed that glycosylation did not significantly alter binding to the opioid receptor subtypes, as the values were similar to that of their un-glycosylated analogues. In vivo studies revealed that the glycosylated analogues exhibited good activity in the tail flick assay following i.c.v. administration. The un-glycosylated analogues did not perform as well as the glycosylated analogues in the tail flick assay following i.v. administration. This is likely due to their low water solubility relative to glycopeptide analogues. It is possible that the un-glycosylated analogues were too lipophilic and remained bound to the lipid bilayer rather than passing through the BBB.
Figure 15. β-Endorphin Inspired Amphipathic Glycopeptides.
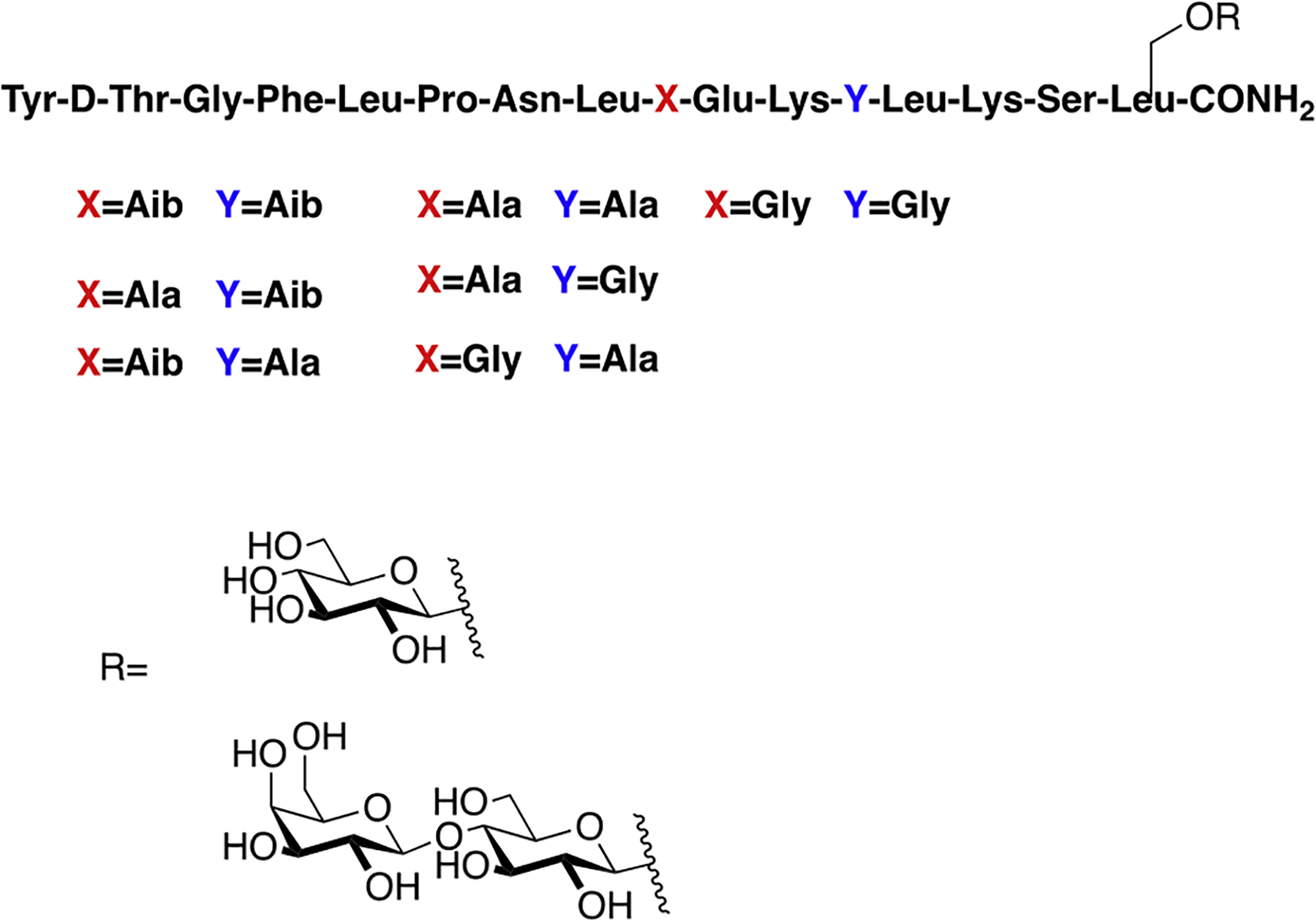
Glycopeptides related to β-endorphin were synthesized and studied for their biological activity and transport properties. One of the key features of these particular glycopeptides is their varying degrees of amphipathicity. Glycopeptides performed significantly better than their un-glycosylated analogues in both in vitro functional assays and in vivo studies (Polt 2014) [89].
4.1.4. DAMGO-Derived Glycopeptides
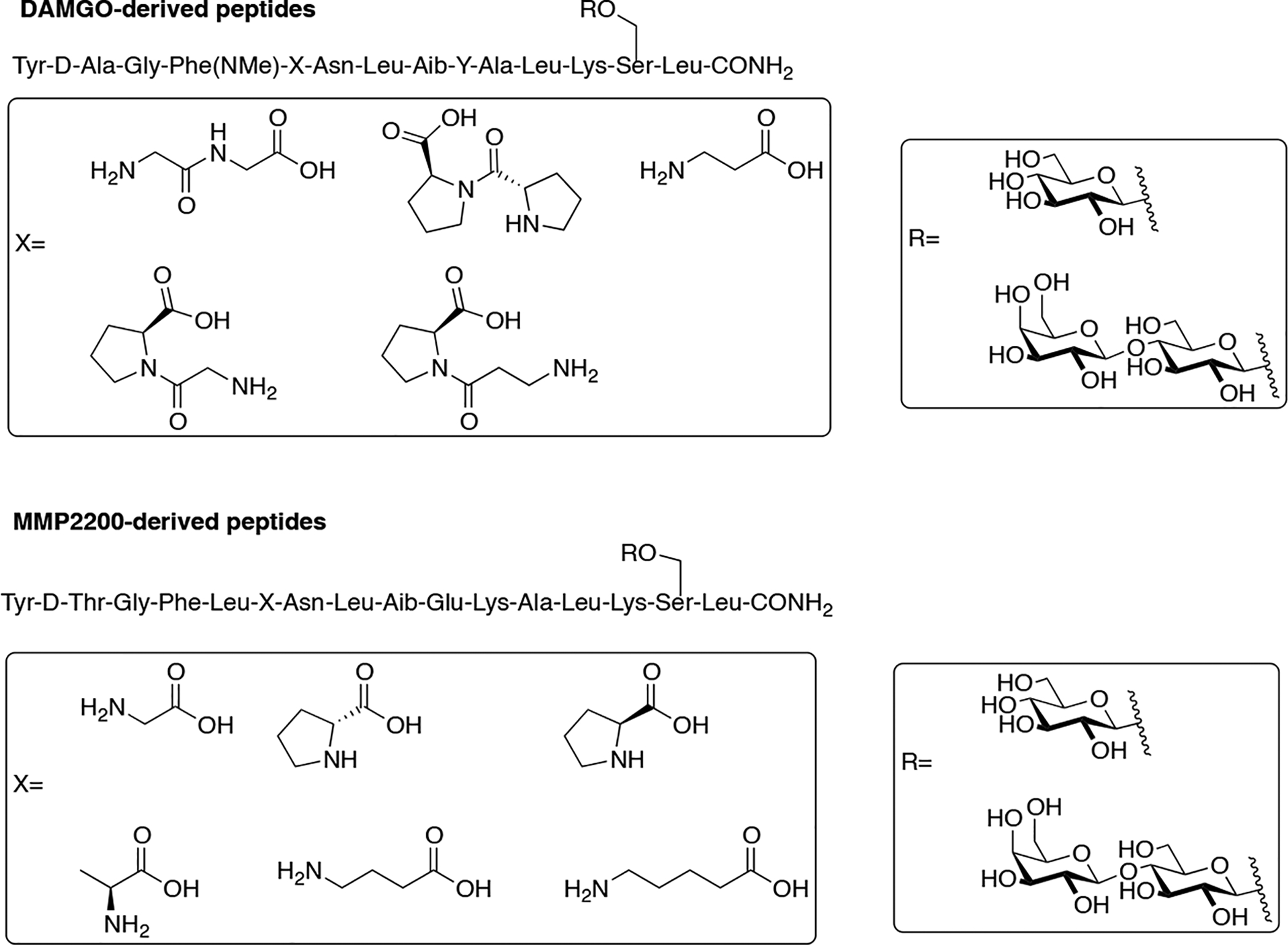
Given the previous successes of enkephalin-based glycopeptides, Polt and colleagues explored other opioid peptides and evaluated the effect of glycosylation on their stability and activity in vivo (Figure 16) [90]. Native DAMGO was modified in this study by replacing the glycinol moiety at the C-terminus with various serine glycosides including a β-D-xyloside, β-D-glucoside, and β-lactoside. These compounds were evaluated for binding at the opioid receptors and their activity in the classic tail-flick assay in rats. The results from the binding experiments showed that all of the glycopeptides retained μ opioid receptor selectivity. Following i.c.v and i.v. administration, it was found that the glycopeptide analogues were significantly more potent than native DAMGO and morphine. The lactosylated glycopeptide was slightly less potent compared to the glycopeptides containing a monosaccharide.
Figure 16. Glycopeptides Related to the μ-Selective Opioid Agonist DAMGO.

DAMGO-derived glycopeptides were evaluated for their activity in vivo. Variation in the amphipathic character of the glycopeptide shifted its biological activity in a predictable manner. (Polt 2007) [90].
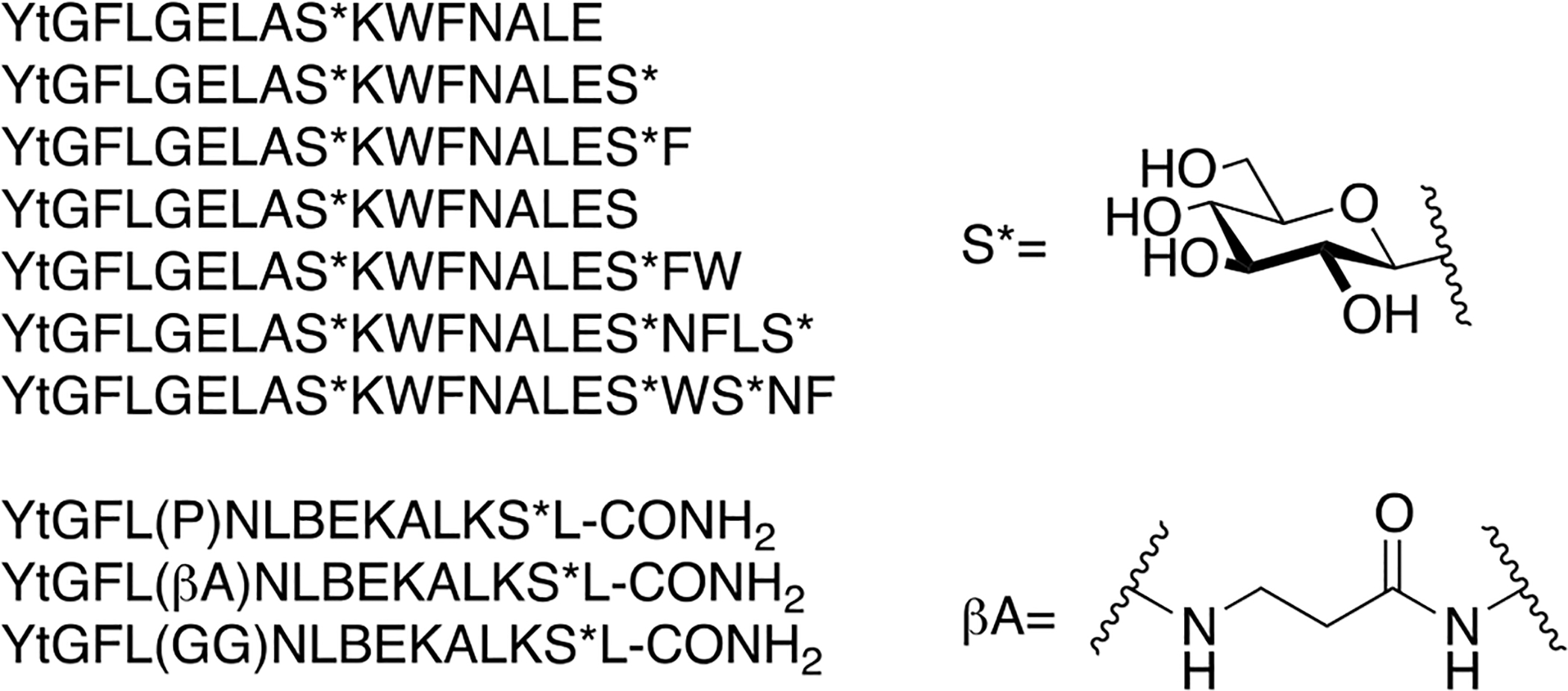
Based on the previous successes with glycopeptides related to β-endorphin, Polt and collogues designed and synthesized several highly amphipathic glycopeptides related to the opioid peptides DAMGO and DTLET in order to further validate the biousian hypothesis (Figure 17) [91]. A DAMGO or DTLET “message” segment was covalently linked to an “address” region responsible for receptor binding via either flexible (β-alanine, GABA, DAVA etc.) or rigid (Pro, Pro-Pro, D-Pro, etc.) linkers. It was found that more flexible linkers were required to maintain binding and antinociceptive activity. Shorter, more rigid linkers resulted in compounds that exhibited relatively lower binding and decreased antinociceptive activity compared to the compounds with long, flexible linkers. This is presumably due to restricted conformational flexibility, preventing the message segment from inserting into the opioid receptor active site. Based on in vivo studies it was determined that the glycosylated analogues could penetrate the BBB and induce antinociception, while the un-glycosylated analogues were less potent, suggesting that they could not penetrate the BBB. This finding further highlights the importance of glycosylation on the transport properties of peptide-based drugs.
Figure 17. Amphipathic Glycopeptides Based DAMGO and DTLET.
Highly amphipathic glycopeptides related to DAMGO and DTLET were synthesized and studied for their in vivo antinociception. These glycopeptides contained various flexible and rigid linkers separating the message and address segments. As seen with the previous study on amphipathic β-endorphin glycopeptides, the un-glycosylated analogues did not perform as well as their glycosylated counterparts. (Polt 2015) [91].
4.1.5. Dermorphin and Deltorphin Glycopeptides
The dermorphins and deltorphins represent two classes of opioid peptides secreted from the skin glands of phyllomedusa amphibians. The dermorphins are μ selective agonists while the deltorphins are δ selective agonists. Exhaustive SAR studies have been performed on these two classes of peptides, but up until the late 1990s, glycosylation had not been explored. Salvadori and coworkers set out to introduce O-linked β-D-glucose residues into the backbones of the dermorphin and deltorphin skeletons (Figure 18) [92]. They achieved this via an O-linkage through either a Thr4 sidechain or Thr7 sidechain. Glycopeptides containing a glucose residue appended to Thr4 exhibited significant reductions in opioid receptor binding affinity, whereas the compounds glycosylated at Thr7 only showed only a modest loss in binding, mostly retaining low nanomolar activity similar to that of native dermorphin and deltorphin. In vitro functional activity obtained in the GPI and rabbit jejunum assays was consistent with the receptor binding affinity data. These results indicate, as with many other opioid glycopeptides, that modifications at the C-terminus are much less detrimental than modifications towards the N-terminus. The glycosylated deltorphins and dermorphins exhibited potent analgesia in vivo in the tail-flick assay following both i.c.v. and subcutaneous administration and were more active than their un-glycosylated counterparts and morphine. The fact these glycopeptides induced analgesia in both i.c.v. and subcutaneous routes supports their ability to penetrate the BBB.
Figure 18. Dermorphin and Deltorphin Inspired Glycopeptides.

Dermorphin and deltorphin glycopeptides were active analgesic agents in vivo following both i.v and subcutaneous administration. This suggests these glycopeptides can successfully penetrate the BBB (Salvadori 1995) [92].
Rocchi and colleges synthesized a series of glycopeptides related to the μ-selective opioid peptide dermorphin (Figure 19) [93, 94]. In one case they introduced a β-D-glucose residue linked through the Ser7 side chain oxygen. In another case they substituted Ser7 with Ala and introduced a C-linked α-D-galactose residue through the methyl side chain of Ala. In both cases the carbohydrate residues were either fully acetylated or de-acetylated. The de-acetylated glycopeptides exhibited slightly lower binding affinities compared to the un-glycosylated compounds, and the acetylated glycopeptides showed further reductions in binding affinity. Although slight reductions in binding were observed, the glycosylated analogues were roughly twice as potent as native dermorphin in vivo in the tail-flick assay. The compounds were administered to rats either i.c.v. or subcutaneously. Naloxone was also administered to determine whether the antinociceptive effects were mediated peripherally or centrally. Naloxone administered i.c.v antagonized glycodermorphin-induced antinociception to a significant degree, while administration subcutaneously did not antagonize the antinociceptive effects, suggesting centrally-mediated antinociception. BBB penetration was quantified as the ratio of AD50 values following i.c.v. and subcutaneous administration. It was determined that the de-acetylated glycopeptides penetrated the BBB more efficiently than the acetylated glycopeptides and the non-glycosylated analogues. Interestingly, the C-linked glycopeptide exhibited the highest penetration followed by the O-linked analogue. Stability studies in rat brain and liver homogenates showed that the glycopeptide analogues were more enzymatically stable than their non-glycosylated counterparts.
Figure 19. Novel O- and C-Linked Dermorphin Glycopeptides.
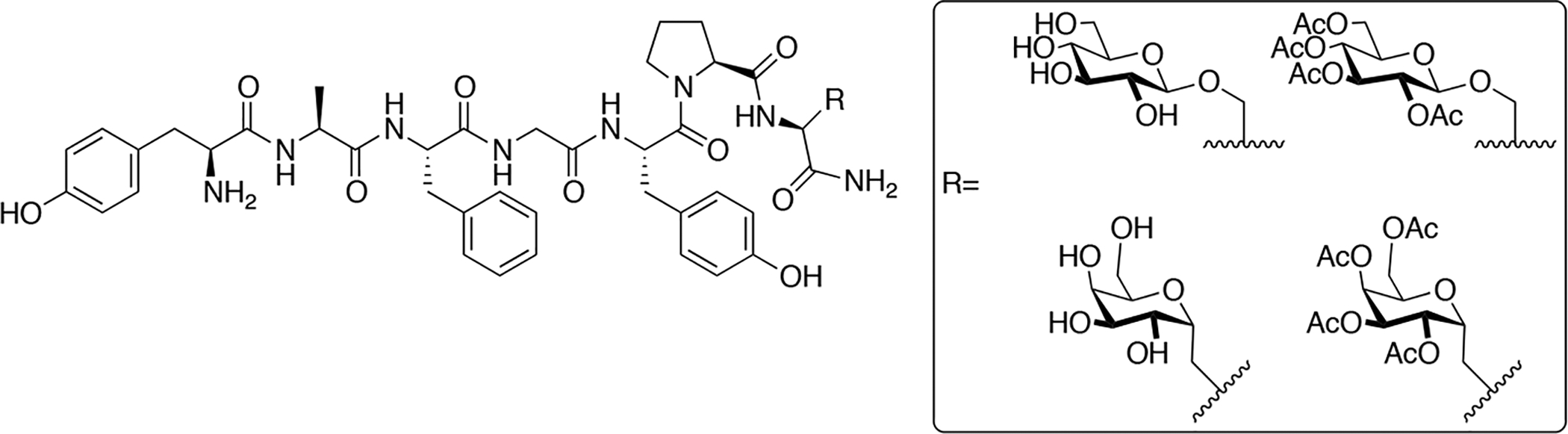
Dermorphin glycopeptides containing both O- and C-linked carbohydrate moieties were evaluated in vitro and in vivo. Stability studies showed that the glycopeptide analogues were more stable compared to the un-glycosylated control compounds. Penetration studies revealed that the C-linked galactosylated glycopeptide exhibited the most efficient BBB penetration. (Rocchi 1999) [93, 94].
Drug candidate BBI-11008 is an opioid glycopeptide related to the dermorphins and deltorphins (Figure 20) [95]. It exhibits mixed μ/δ (δ-selective) activity with no agonist activity at the κ opioid receptor. It was recently demonstrated to be antinociceptive in vivo in models of inflammatory pain and acute thermal pain following i.p administration, and was less potent in these models than morphine. The side effect profiles of BBI-11008 were evaluated by in vivo respiration and drug self-administration assays. In the respiration assay, the effects of BBI-11008 on the volume of gas inhaled or exhaled by the rats (the respiratory minute volume) were compared to that of morphine. It was found that BBI-11008 led to a slight increase in respiratory minute volume followed by a subsequent decrease, whereas morphine exhibited a dose dependent decrease in respiratory minute volume indicative of respiratory depression. Studies revealed that rats readily self-administered fentanyl, while this behavior was not observed with BBI-11008 indicating a lower likelihood of addiction potential. Additionally, oral administration of BBI-11008 led to efficacious antinociception in vivo, despite what is typically low oral bioavailability in peptides. Taken altogether, the evidence presented in this study suggests that BBI-11008 may be a useful alternative to standard opioid therapy.
Figure 20. A Novel Dermorphin/Deltorphin Inspired Glycopeptide with Reduced Side Effects.
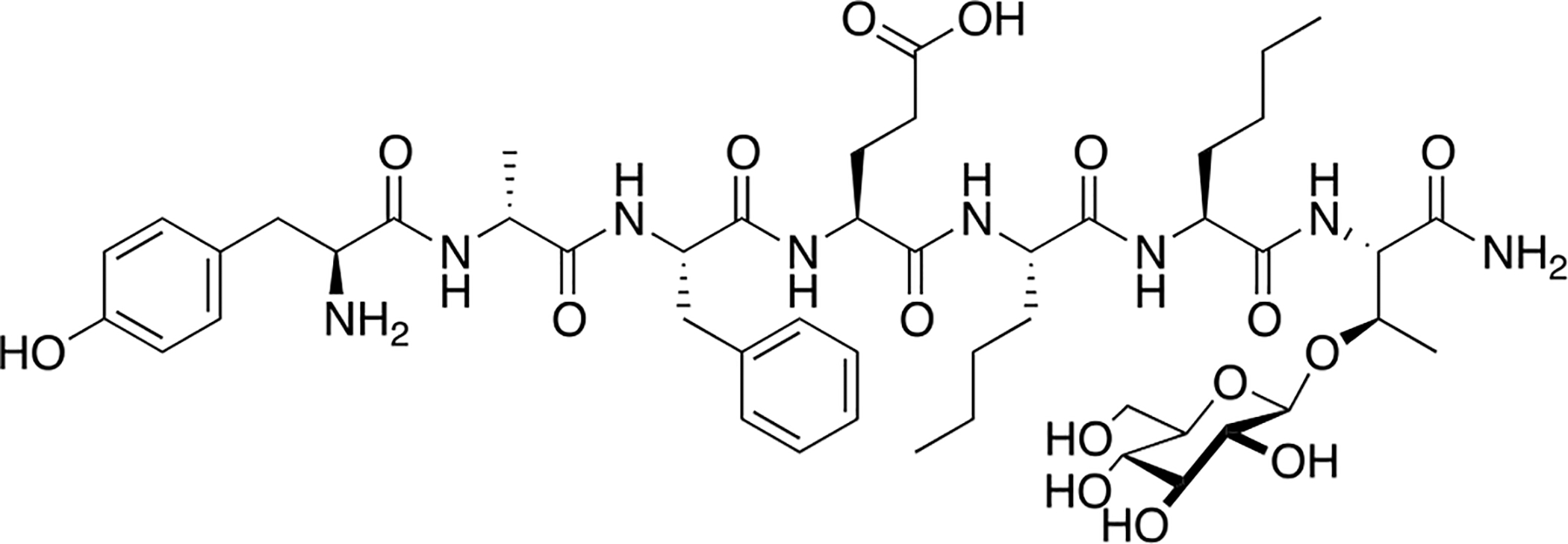
BBI-11008 is a δ-selective agonist with antinociceptive effects in models of acute and chronic pain and shows an improved side effect profile compared to morphine and fentanyl. BBI-11008 was also able to induce antinociception in a model of acute thermal pain following oral administration. (Bilsky and Polt 2020) [95]
4.2. Neurotensin glycopeptides
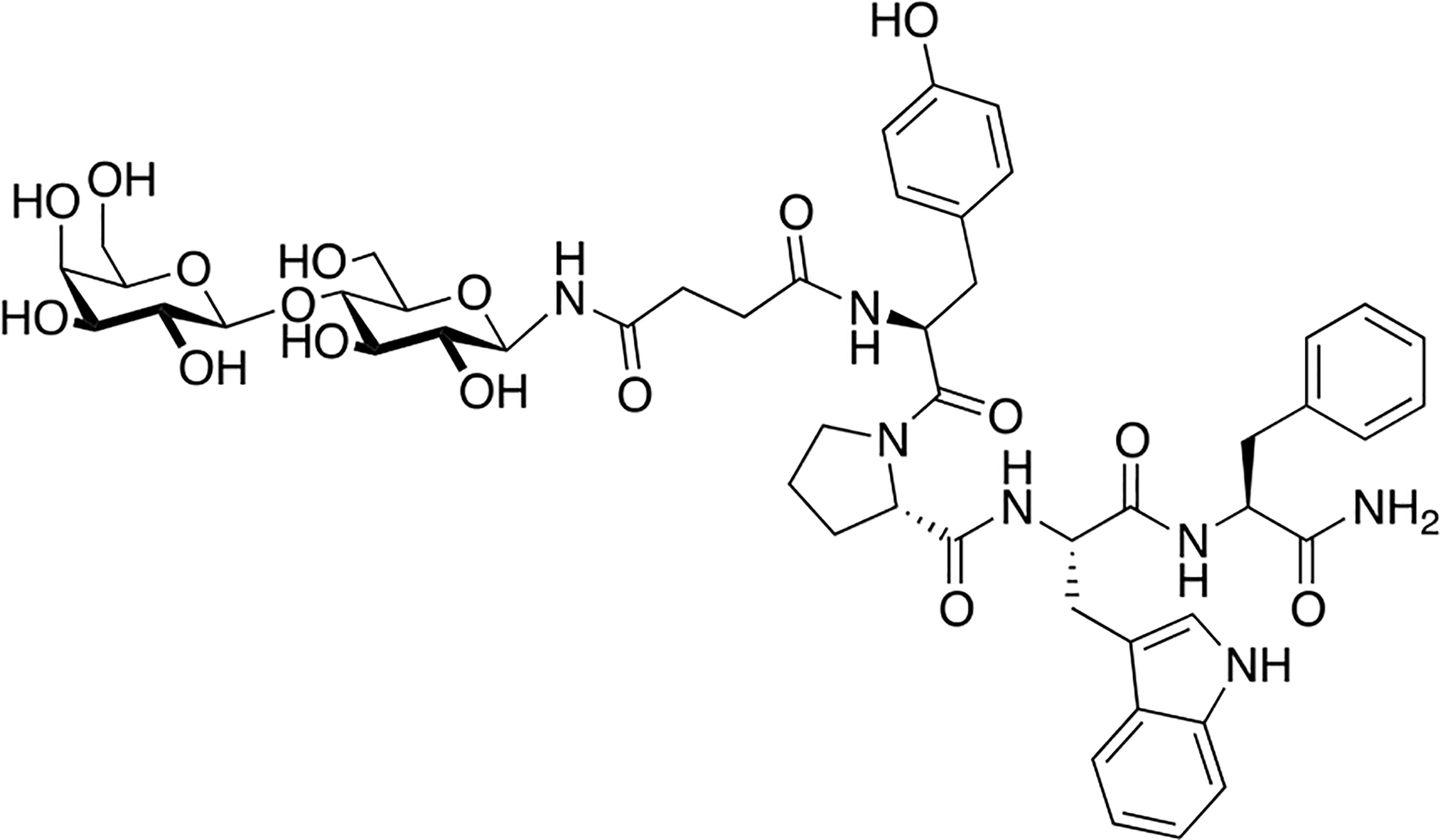
Lee and coworkers prepared three glycosylated analogues of neurotensin, an endogenous neuropeptide. Neurotensin exerts antinociceptive and antiepileptic activity [96,97,98], and has been implicated in the treatment of various psychiatric disorders, including schizophrenia [99]. They introduced a variety of glycosides in position 7 of native neurotensin, including O-linked serine melibioside, O-linked Thr α-TF antigen, and a lactosylated PEG3 conjugate appended through the carboxyl sidechain of Glu (Figure 21). The glycosylated analogues exhibited low nanomolar binding affinities and were able to promote Ca2+ production in vitro. The compound’s anticonvulsant activity was evaluated in vivo, showing that only sub-picomolar concentrations were required to produce anticonvulsant effects. These results demonstrate the potential for glycosylated neurotensin analogues as new and exciting candidates for anticonvulsant and antiepileptic therapies.
Figure 21. Novel Neurotensin Glycopeptides with Anticonvulsant Activity.
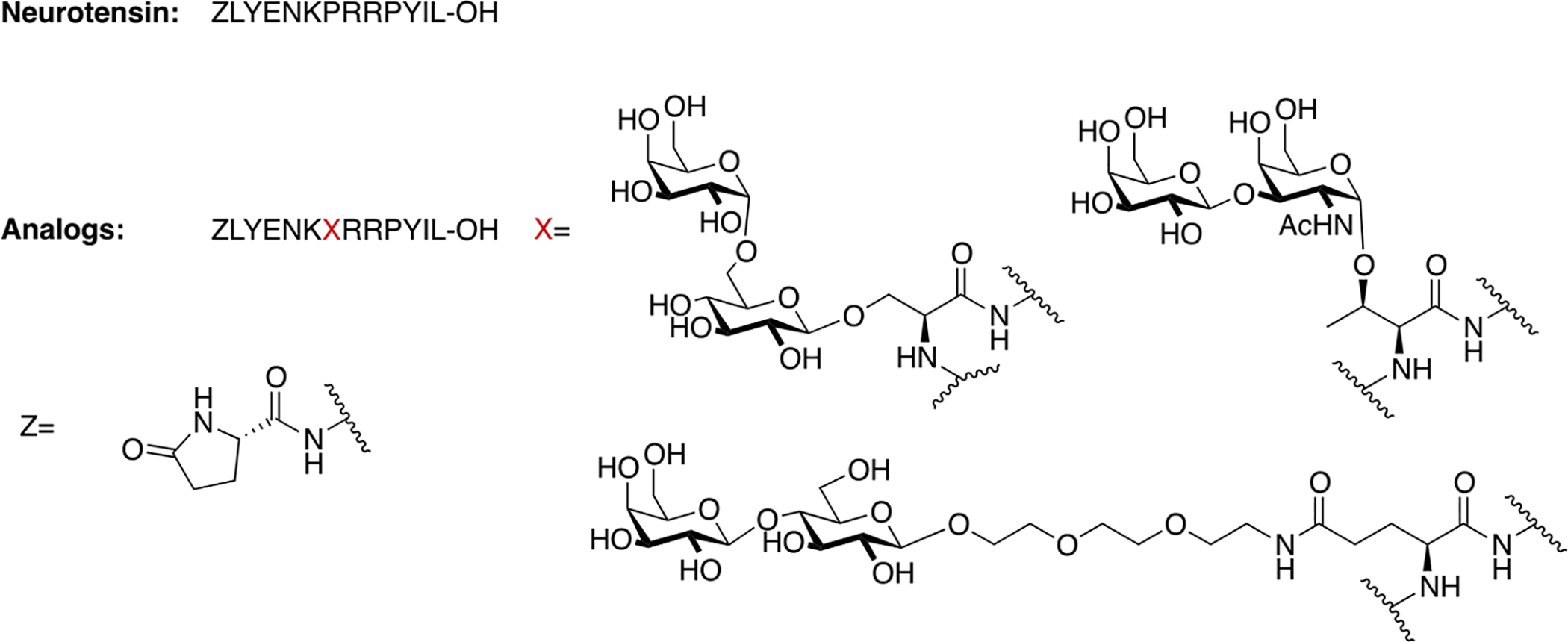
Neurotensin glycopeptides were evaluated for their anticonvulsant activity in vivo. Satisfyingly, the glycopeptide analogues were active anticonvulsant agents at sub-picomolar concentrations. (Lee 2009) [96].
Contulakin G is a naturally occurring O-linked glycopeptide isolated from the cone snail, Conus geographus, that exhibits antinociceptive activity (Figure 22) [100,101]. It is glycosylated at Thr10 with a β-D-Gal-(1→3)-α-GalNAc moiety. This compound was synthesized and evaluated for its ability to bind and agonize the Neurotensin1 receptor compared to the receptor’s native peptide ligand neurotensin. It was found that Contulakin G was a weak agonist at the neurotensin1 receptor and was not as potent as native neurotensin in producing an antinociceptive response. Computational studies indicated that the carbohydrate makes the peptide more water soluble, allowing for dissociation from the Neurotensin1 receptor and a subsequent decrease in receptor desensitization.
Figure 22. Naturally Occurring Glycopeptide Contulakin G.
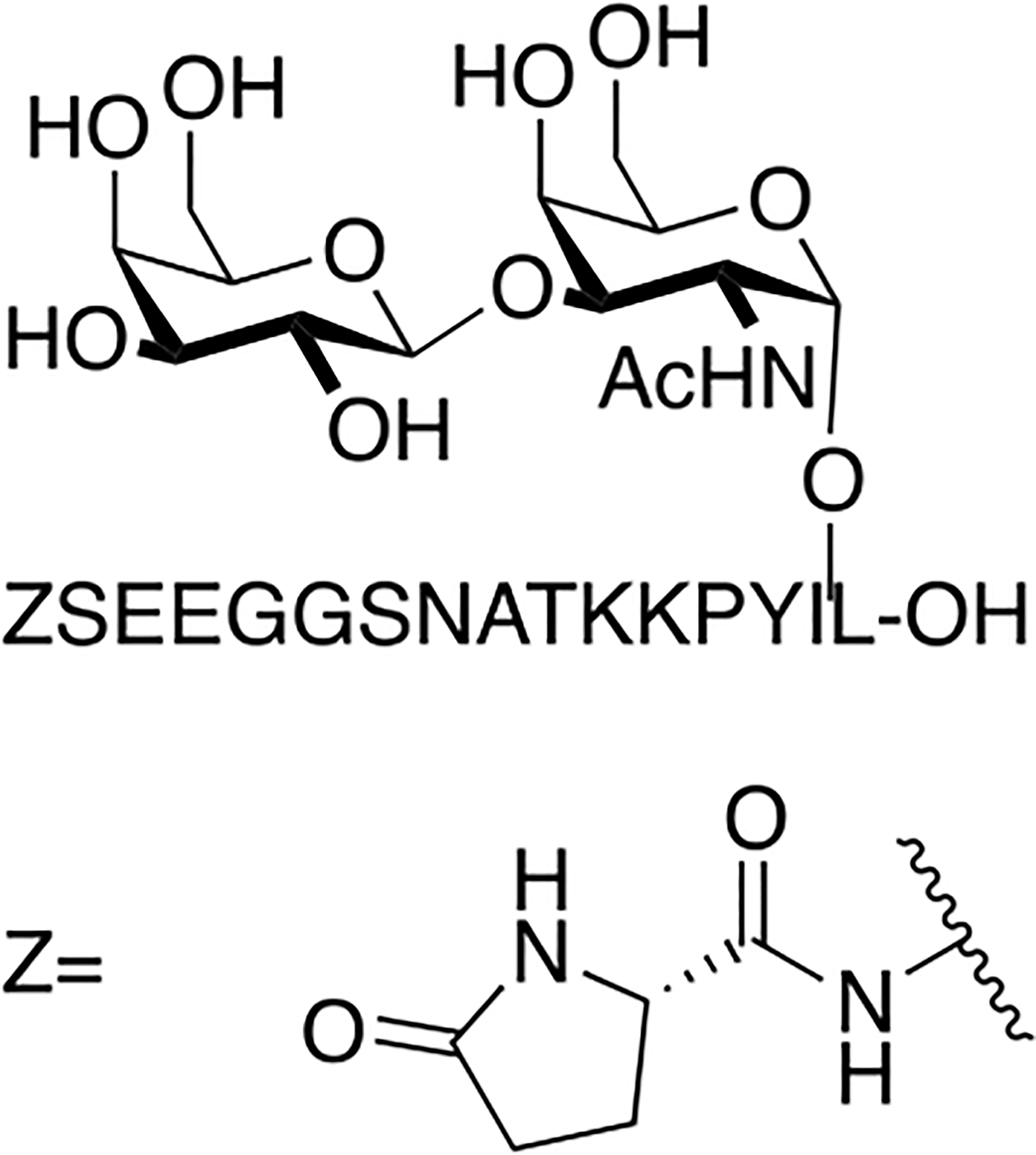
Contulakin G was synthesized using standard Fmoc-based SPPS. It was evaluated for its activity at the neurotensin1 receptor and compared to the receptor’s native ligand, neurotensin. Contulakin G was a weak antagonist at the neurotensin1 receptor and was less potent than neurotensin. (Lee 2015) [100].
4.3. Angiotensin-1–7 (Ang-1–7)-derived Glycopeptides
PNA5 is a glycopeptide derived from angiotensin 1–7 (Ang-1–7), an endogenous peptide metabolite of angiotensin II (Figure 23) [102]. Glycosylation of Angiotensin 1–7 (DRVYIHP), replaces the seventh residue (proline) with a serine, and has a glucose sugar moiety attached to the serine and is amidated on the C-terminus resulting in Ang-1–6-Ser-O-Glu-NH2 (PNA5). PNA5 has outstanding brain penetration and enhanced stability as compared to the native Ang-(1–7) [103, 12, 104, 105]. Further, none of the other published studies with oral formulations of Ang-(1–7) related peptides or small molecules [106, 107, 108] have exhibited the excellent brain penetration that has observed with our glycosylated Ang-(1–7) peptides, which will be key for developing an effective CNS anti-inflammatory, cognitive protective therapeutic. Recent results from an in vivo swine pilot study showed PNA5 has 110% greater t1/2, 146% greater CMax and 187% greater AUC in serum than the native Ang-(1–7) following a 230 microgram/kg s.c. injection (n=3). Rat brain dialysate levels of both PNA5 and Ang-(1–7) following 10 mg/kg injection in healthy rats showed that PNA5 exhibited an average CMax and AUC log fold increase of 2.7 ± 0.2 and 2.3 ± 0.1 over Ang-(1–7),respectively (n=8).
Figure 23. PNA5:
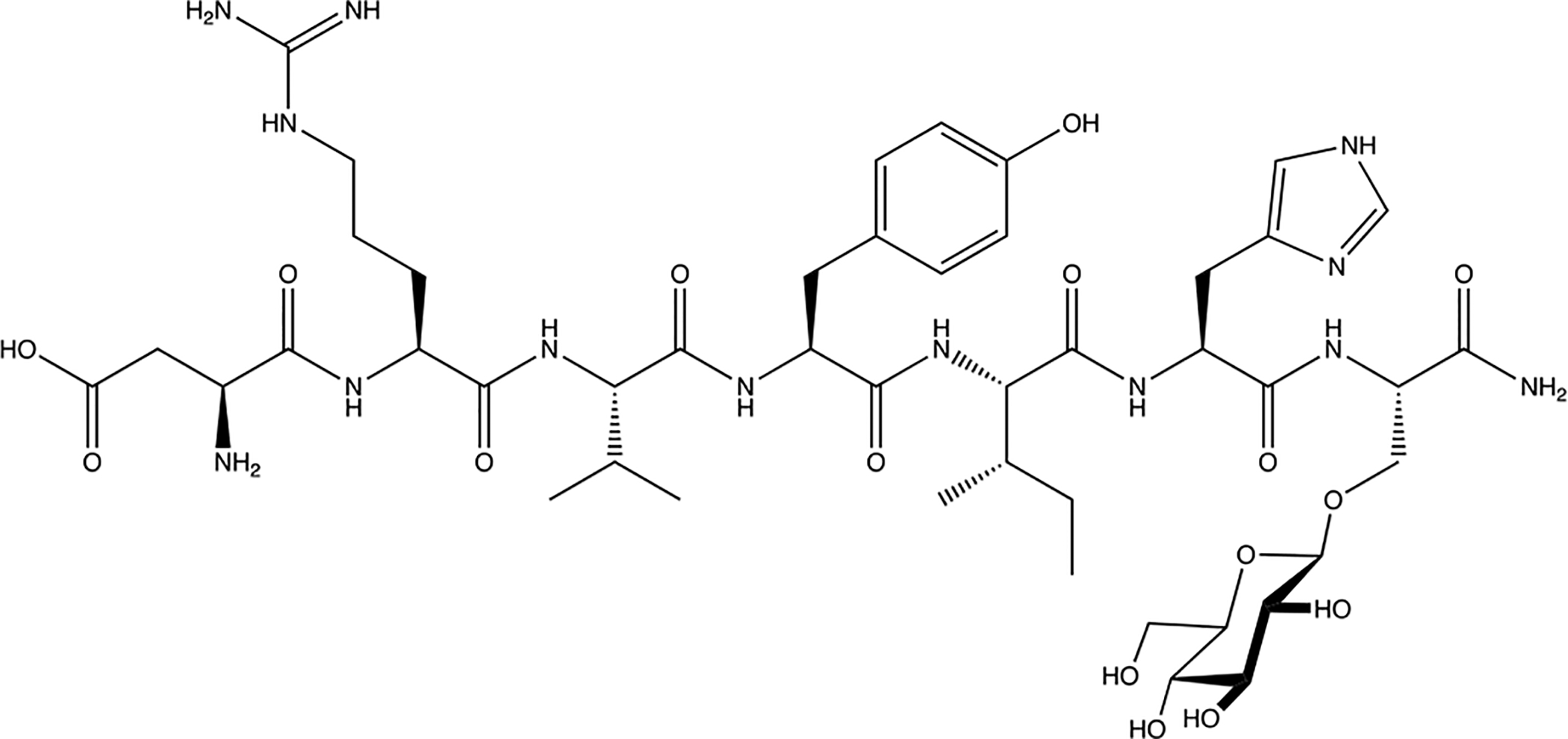
PN5 is a glycosylated Ang-1–7 analogue with protective effects in vascular cognitive impairment, enhanced PK properties in vivo, and improved BBB permeability (Hay et al 2019) [103]
Angiotensin-(1–7) and PNA5 bind to the G-protein linked Mas receptor 109, 103, 110, 111, 112, 113] which is expressed in both peripheral tissues and in the brain. Within the brain, the Mas receptor is expressed on neurons, glia and vascular endothelial cells [114]. Activation of Mas decreases ROS and brain inflammation, increases cerebral circulation via increases in endothelial NO release 115, 116, 117] increases induction of neuroprotective cytokines and inhibits hypoxia-inducing factor-1alpha [115, 118, 119]. Because the Mas receptor is found in high quantities within the hippocampus and perirhinal cortex as well as in vascular endothelial cells, PNA5 will be particularly effective in targeting memory impairments associated with hypoxia and inflammation-related neurodegenerative disease [109]. The hippocampus and perirhinal cortex, two areas important in memory, express high levels of the MasR [120, 114]. Recently, the Ang-(1–7)/Mas pathway was found to be essential for normal object recognition memory [121], suggesting that it is an important modulator of learning and memory. Hay and colleagues have shown that PNA5 rescues VCID induced cognitive impairment in a preclinical model of vascular dementia (VCID) [103]. In these studies, cognitive impairment, as measured by object recognition tests and the Morris Water Maze, was reversed following a 3-week treatment with subcutaneous PNA5 [103]. Activation of MasR has been shown to decrease ROS and brain inflammation, increases cerebral circulation via increases in endothelial NO release, increase induction of neuroprotective cytokine and inhibits hypoxia-inducing factor-1alpha [122, 115, 118, 119]. Given that the inflammatory response in VCID is known to involve increases in circulating and brain inflammatory factors, it is believed that PNA5 our glycosylated Ang-(1–7) derivatives will be better drugs than either the native Ang-(1–7) or related small molecules due to the ability of our glycosylated derivatives to access the brain MasR and inhibit inflammation and ROS formation within the blood-brain-barrier.
4.4. Miscellaneous Glycopeptides
Gregorio Valencia and coworkers synthesized several glycosylated analogues of the opioid tetrapeptide Morphiceptin (Tyr-Pro-Phe-Pro), which is derived from bovine β-casein (Figure 24) [123]. In their work, Pro4 was mutated to Hyp in order to introduce either a glucose or galactose residue through its hydroxyl side chain. The glycosylated compounds did not exhibit the desired antinociceptive properties. Analysis in vitro using the GPI assay revealed that the glycosylated analogues and the analogue containing a Hyp residue in the 4th position were less potent than native morphiceptin. In vivo studies using tail-immersion and paw-pressure assays confirmed that introducing either a glycosylated or an un-glycosylated Hyp residue in the 4th position results in less active compounds than morphine. This suggests that the Hyp-containing morphiceptin analogue and the glycosylated compounds adopt different bioactive conformations than native morphiceptin. This is an interesting result in consideration of prior studies showing opioid glycopeptides mainly retained activity in vitro and in vivo when they contained carbohydrate moieties at the C-terminus.
Figure 24. Glycopeptides Related to Morphiceptin.
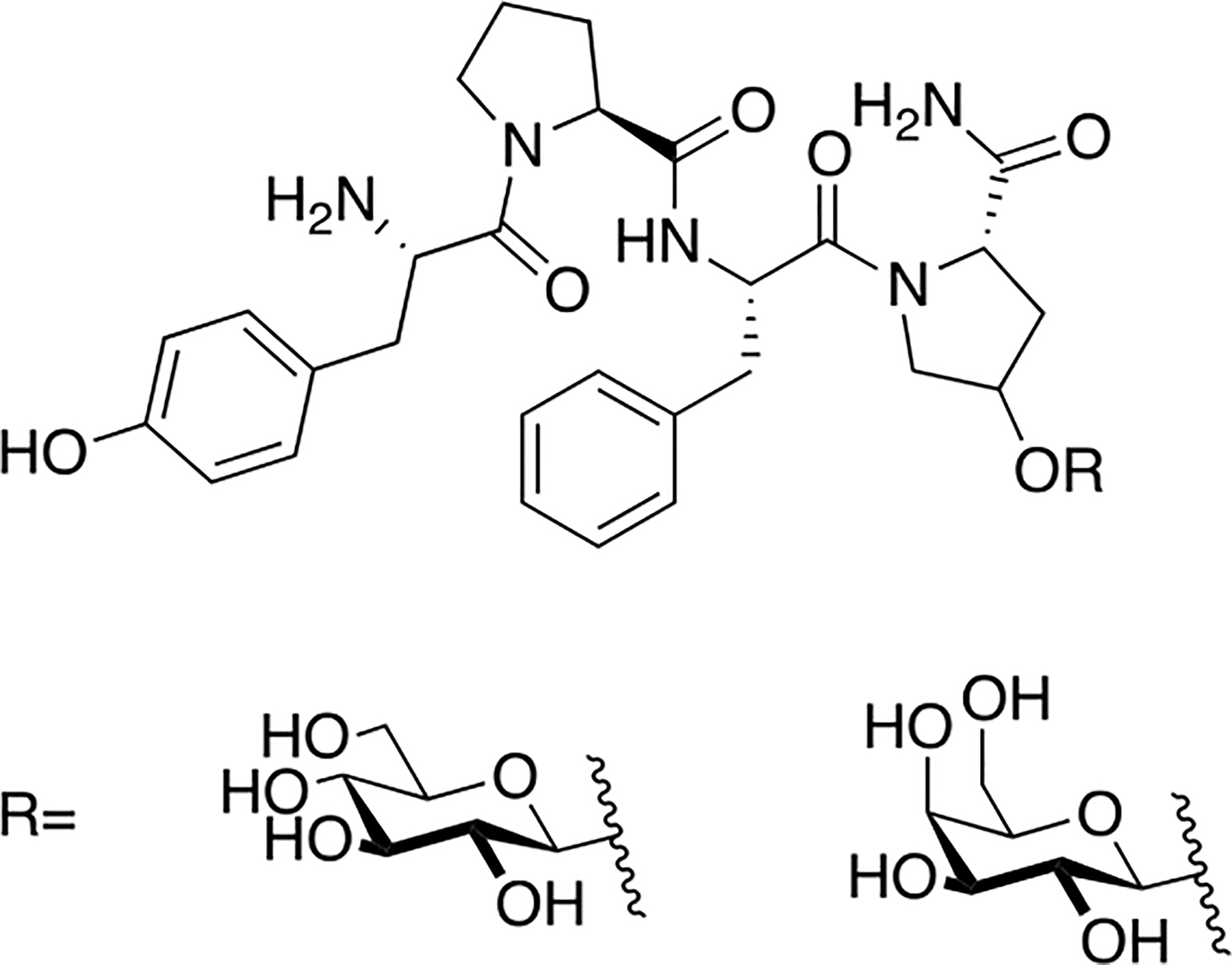
Morphiceptin glycopeptides containing either glucosylated or galactosylated hydroxyproline moieties were less potent than morphine in vivo. Interestingly, this is the opposite result of incorporating a carbohydrate moiety into opioid peptides. This indicates that morphiceptin adopts a different bioactive conformation than opioid peptides do. (Valencia 1990) [123].
Khilberg and coworkers synthesized several glycosylated analogues of [1-desamino,8-D-Arg]vasopressin containing a serine galactoside in place of Gln4 (Figure 25) [124]. The carbohydrate was either fully acetylated or fully de-acetylated. Their overall aim was to develop vasopressin analogues as antidiuretic agents with improved stability and potency. In enzyme stability studies, the galactosylated analogue was shown to be more stable than native vasopressin and exhibited improved intraintestinal transport. However, the analogues were weak antagonists at the vasopressin receptors and did not exhibit any antidiuretic activity. NMR studies in aqueous solution were conducted to determine the effect of glycosylation on the analogues’ conformation. It was found that the conformations of the glycosylated and un-glycosylated analogues were similar in aqueous solution, so it was hypothesized that the steric bulk of the sugar prevented interactions between the peptide ligand and the receptor’s active site. However, further 2D NMR studies simulating membrane-like environments are necessary to provide more insight and to fully elucidate the bioactive conformation of the glycosylated analogues.
Figure 25. Vasopressin Glycopeptides Containing Serine Glucosides and Galactosides.

Vasopressin glycopeptides were synthesized and evaluated for their antidiuretic activity in vivo. The glycopeptide analogues did not produce the desired activity, which was believed to be the result of unfavorable conformations induced by the carbohydrate moiety. (Khilberg 1996) [124].
Following previous work on opioid glycopeptides, Valencia and coworkers explored glycopeptides related to the N/OFQ peptide [125]. N/OFQ is an endogenous peptide that selectively targets the NOP receptor, which is widely expressed in the CNS and spinal cord [126]. This peptide has been shown to induce antinociception in vivo. The glycosylated analogues were designed with the carbohydrate moiety positioned in the address segment of the peptide to reduce interference with the message region, which is responsible for activation of the receptor (Figure 26). The glycosylation sites that were chosen include Thr5 and Ser7. Thr5 was glycosylated with α-D-GalNAc, and Ser10 was glycosylated with either α- or β-D-GlcNAc. The analogues glucosylated at Ser10 only exhibited a slight decrease in receptor binding compared to native N/OFQ, while the galactosylated analogue showed improved binding. The galactosylated analogue at Thr5 displayed a much more pronounced decrease in binding affinity compared to the other glycopeptides, indicating glycosylation too close to the message region can hinder interactions with the receptor’s active site. This was confirmed by conformational studies using both CD and NMR in the presence of SDS micelles.
Figure 26. N/OFQ-Derived Glycopeptides.
Glycopeptides related to the N/OFQ peptide were studied for the antinociceptive properties in vivo. The analogues containing a carbohydrate closer to the C-terminus exhibited similar binding and functional activity to native N/OFQ, whereas glycopeptides with a carbohydrate closer to the N-terminus showed reductions in biological activity. (Valencia 2011) [125].
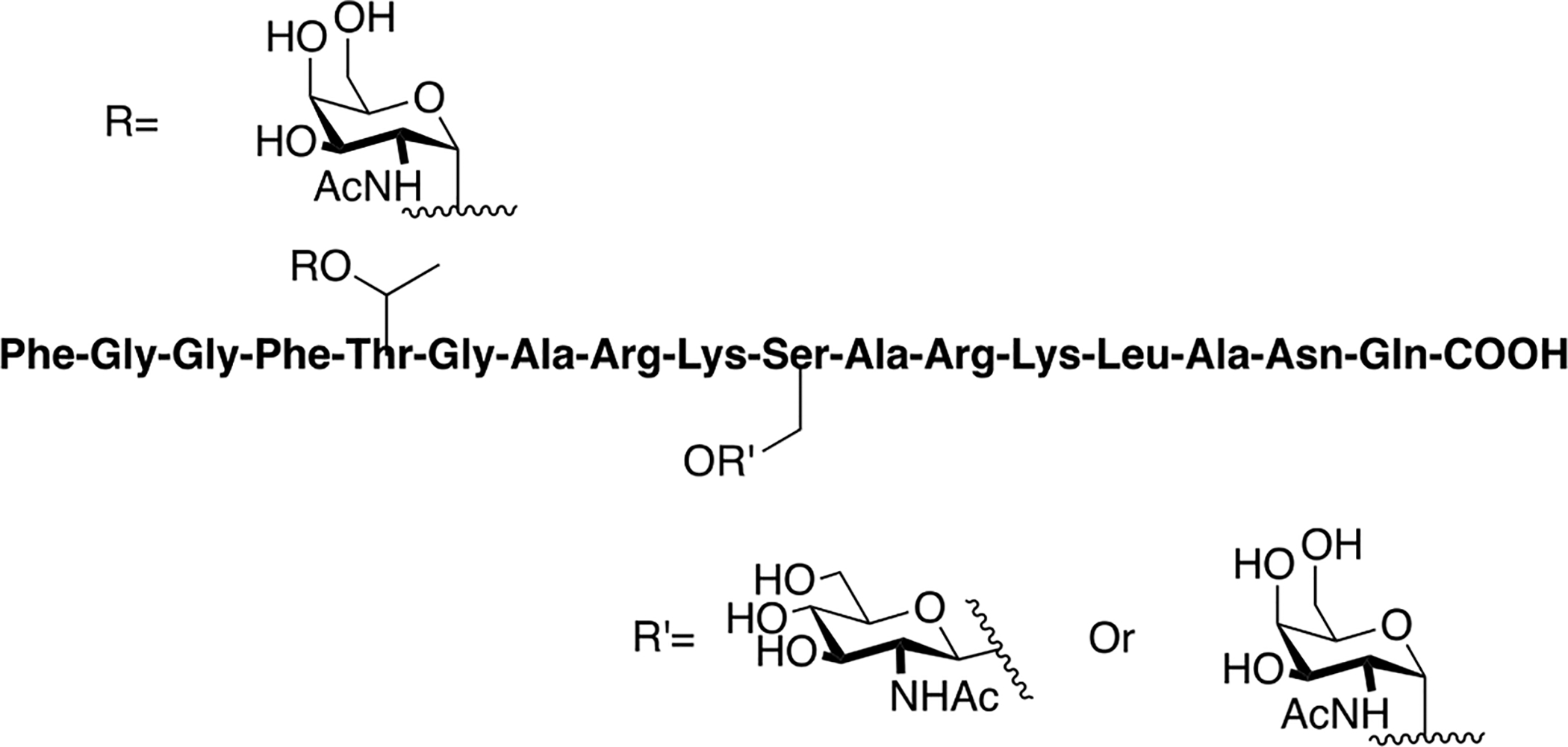
Jansson and coworkers synthesized several medium length peptides (25 residues) related to glycogenin [127]. Although this fragment does not actually bind to a GPCR, it is still an interesting O-linked glycopeptide worthy of discussion. Glycogenin is a primer glycoprotein involved in glycogen biosynthesis that aquires a glucose residue through the phenolic side chain of Tyr194 via a reaction with uridine 5”-diphospho(UDP)-glucose (Figure 27) [128,129]. Following the addition of the first glucose residue, seven more glucose residues are added autocatalytically [130.] The purpose of this work was to explore how the anomeric configuration of the carbohydrate linkage at Tyr194 influenced the ability of the fragments to undergo glucosyl transfer by recombinant glycogenin. Analogues containing no carbohydrate, β- and α-maltoside linkages, β and α glucoside linkages were prepared using Fmoc-based SPPS. The extent of glucosyl transfer of these model glycopeptides was quantified using labelled UDP-glucose. The β- and α-maltosides were better acceptors compared to the β- and α- glucosides, and the un-glycosylated analogue did not undergo any glucosyl transfer, highlighting the importance of the first glucose residue.
Figure 27. Glycopeptides Derived from a Fragment of Glycogenin.
Synthetic glycogenin fragments were prepared to examine the substrate acceptor specificity of native glycogenin. (Jansson 1995) [127].
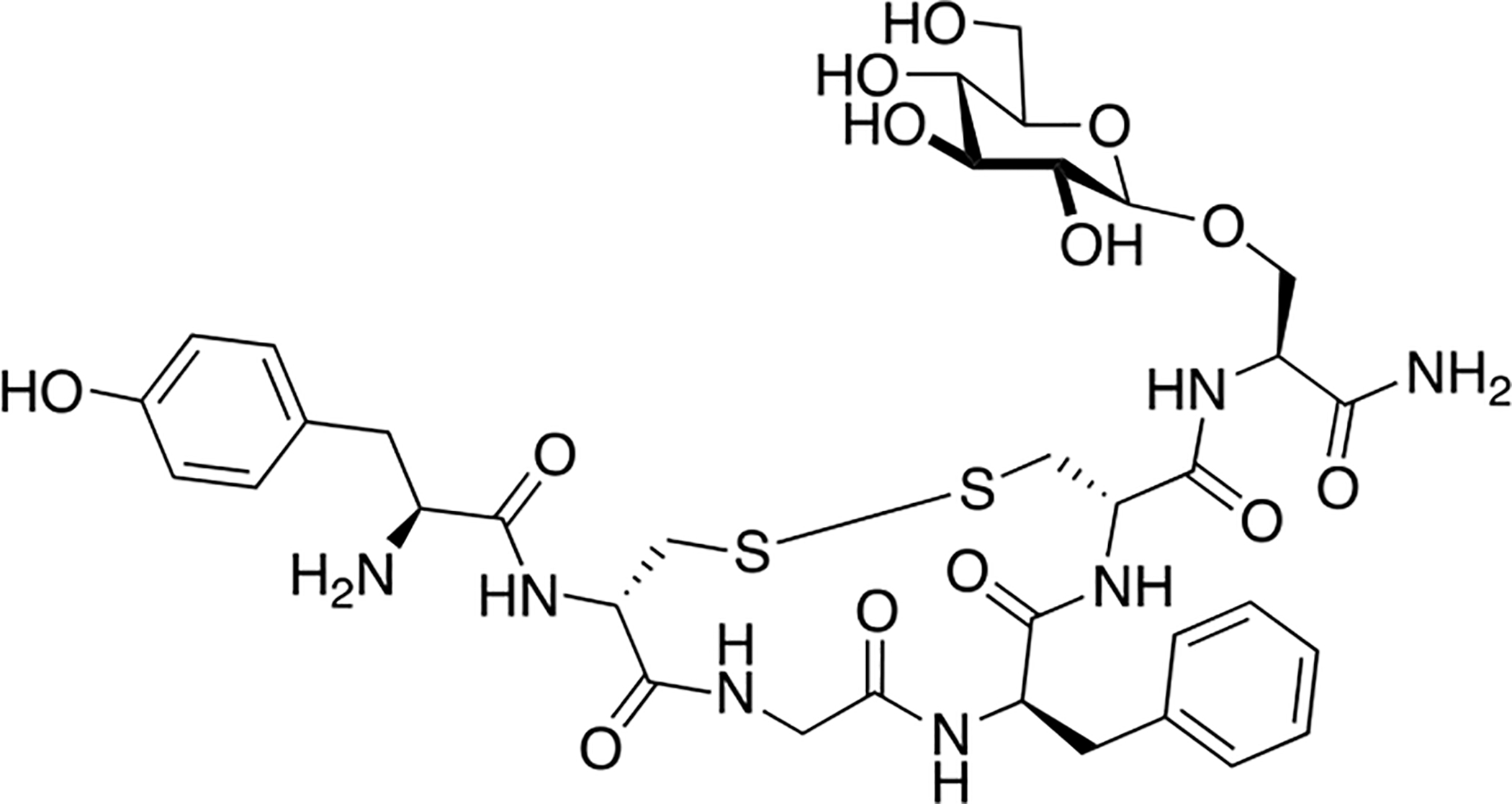
Schiller and colleagues synthesized a series of glycopeptides related to the μ opioid receptor selective peptides DALDA and Dmt1-DALADA [131]. These glycopeptides contained novel linkages with the carbohydrate was anchored to the glycopeptide through triazole linkage (Figure 28). Husigen cycloaddition chemistry was used to introduce a glycosyl azide donor to an alkyne acceptor, resulting in a triazole linkage. Initial studies involved glycosylation of the N-terminus, which resulted in analogues with low efficacy in vivo, prompting investigation into the C-terminus as a potential carbohydrate linkage site. Glycosylation at the C-terminus proved to be successful; the C-terminally modified glycopeptides showed low nanomolar binding in vitro. The glycopeptides exhibited significant antinociception in the hot plate and tail-flick assays following i.v. administration. The galactosylated analogue (X=CH2, Y=β-D-galactose from Figure 27) was evaluated for its BBB permeability in caco-2 cells and compared to transport rates exhibited by native Dmt1-DALDA. It was found that the glycopeptide analogue exhibited comparable transport rates to native Dmt1-DALDA, albeit with a slight reduction.
Figure 28. Triazole-Containing Opioid Glycopeptides Related to DALDA and Dmt1-DALDA.
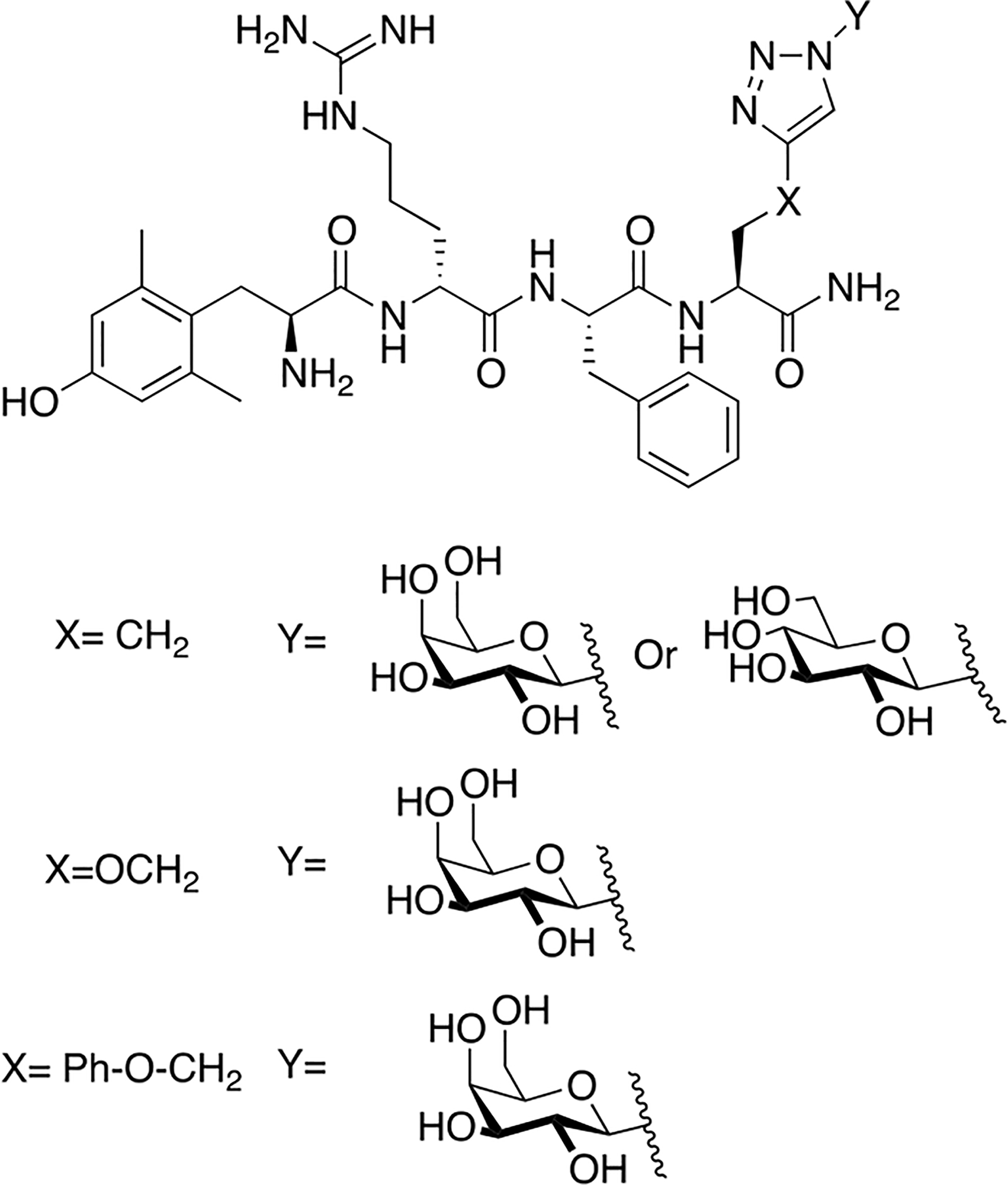
Several novel glycopeptides related to DALDA and Dmt1-DALADA containing carbohydrates linked through triazole moieties were synthesized and studied for the antinociceptive activity in vivo. The glycopeptide analogues exhibited antinociceptive activity, and the galactoside was specifically selected to be studied for its transport properties in caco-2 cells. It showed similar transport properties to native Dmt1-DALADA (Schiller 2014) [131].
A novel bifunctional peptide with μ/δ opioid receptor agonist activity and neurokinin-1 receptor antagonist activity (TY027) with the structure Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-3’,5’-Bzl(CF3)2) was glycosylated at various positions using a serine β-D-glucoside building block by Hruby and coworkers (Figure 29) [132, 133,134]. The optimal position was determined to be at the C-terminal region of the opioid pharmacophore just preceding the NK-1 receptor antagonist pharmacophore. This glucosylated TY027 analogue exhibited exceptional stability in rat plasma with roughly 70% of the compound present after 24 hours. Further, it exhibited similar receptor binding and functional activity profiles to native TY027. Interestingly, the glucosylated analogue displayed a well-defined conformation in the presence of dodecylphosphocholine (DPC) micelles. These results seem to indicate that the carbohydrate does not interfere substantially with binding and activation of the opioid and NK-1 receptors.
Figure 29. Glycopeptides Related to the Bifunctional Opioid Peptide TY027.
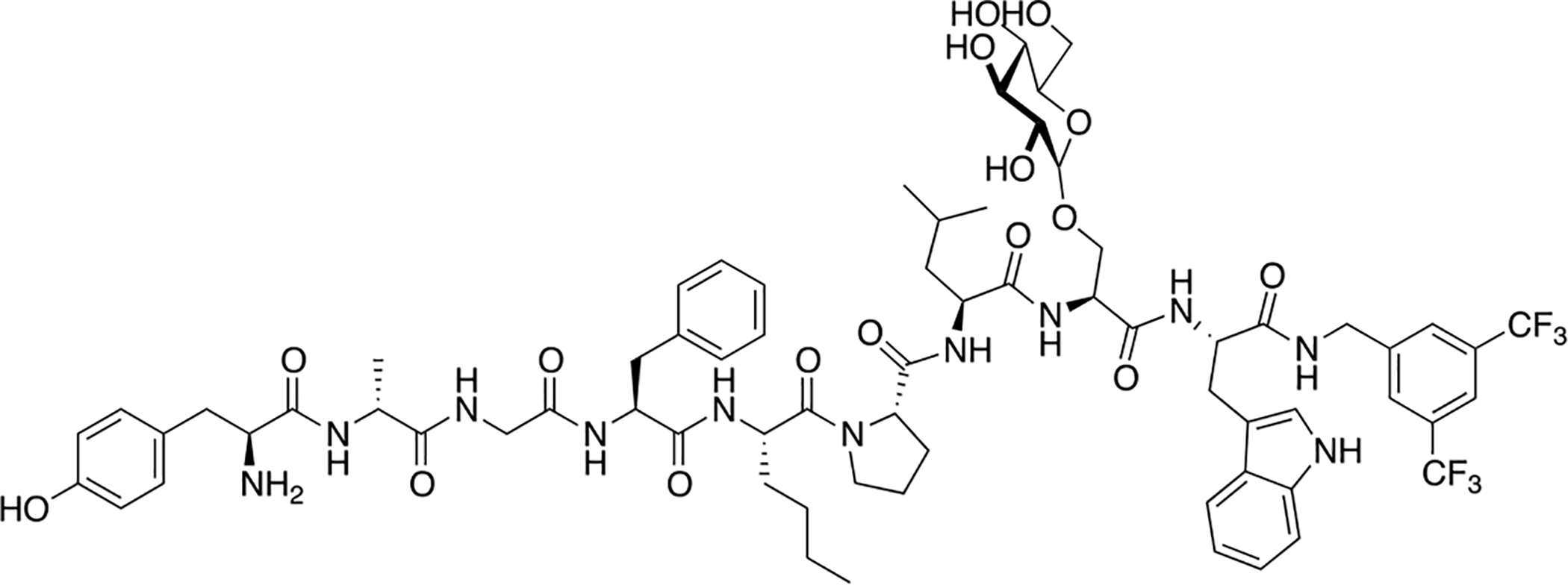
A bifunctional glycopeptide with agonist activity at the opioid receptors and antagonist activity at the NK-1 receptor exhibited exceptional stability in rat plasma but performed slightly better than the un-glycosylated control in vitro. (Hruby 2009) [132].
5. O-Linked and N-Linked Glycopeptides Targeting Class B GPCRs
Class B GPCR peptide ligands are much greater in size than their class A GPCR ligand counterparts and are generally more difficult to synthesize. Due to the difficulty of their synthesis, this class of peptide ligands has not been extensively studied for glycosylation. However, there are a few recent examples including glycosylated vasoactive intestinal peptide (VIP) analogues containing both O- and N-linked carbohydrates, O-glycosylated calcitonin analogues, and GLP-1 analogues containing an N-linked carbohydrate moiety. Although the focus of this review is predominantly on O-linked glycopeptides, N-linked glycopeptides targeting class B GPCRs should be included in this discussion. The structures of the various glycopeptides targeting class B GPCRs are summarized in Table 8.
Table 8.
Representative Structures of Glycopeptides Targeting Class B GPRs
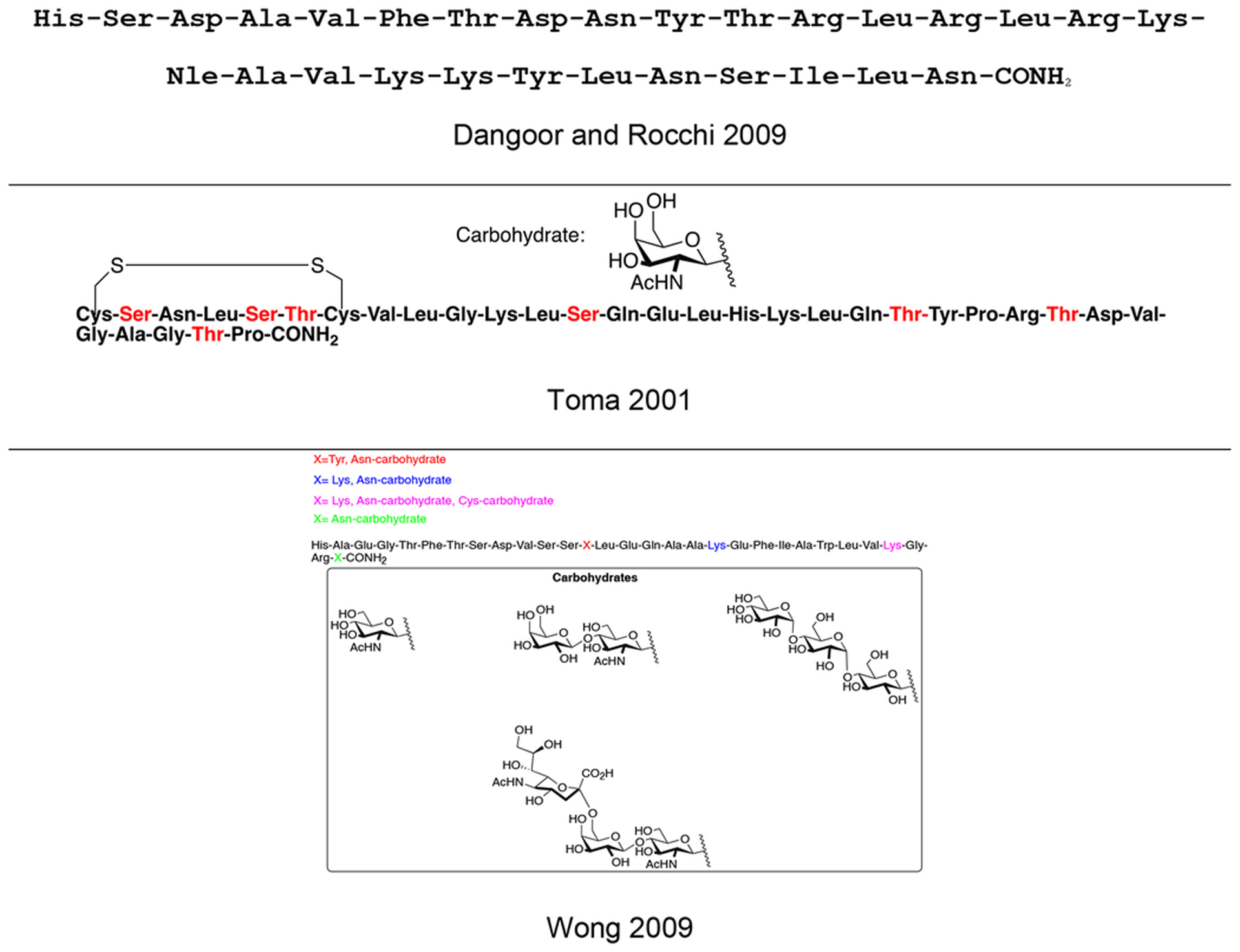
|
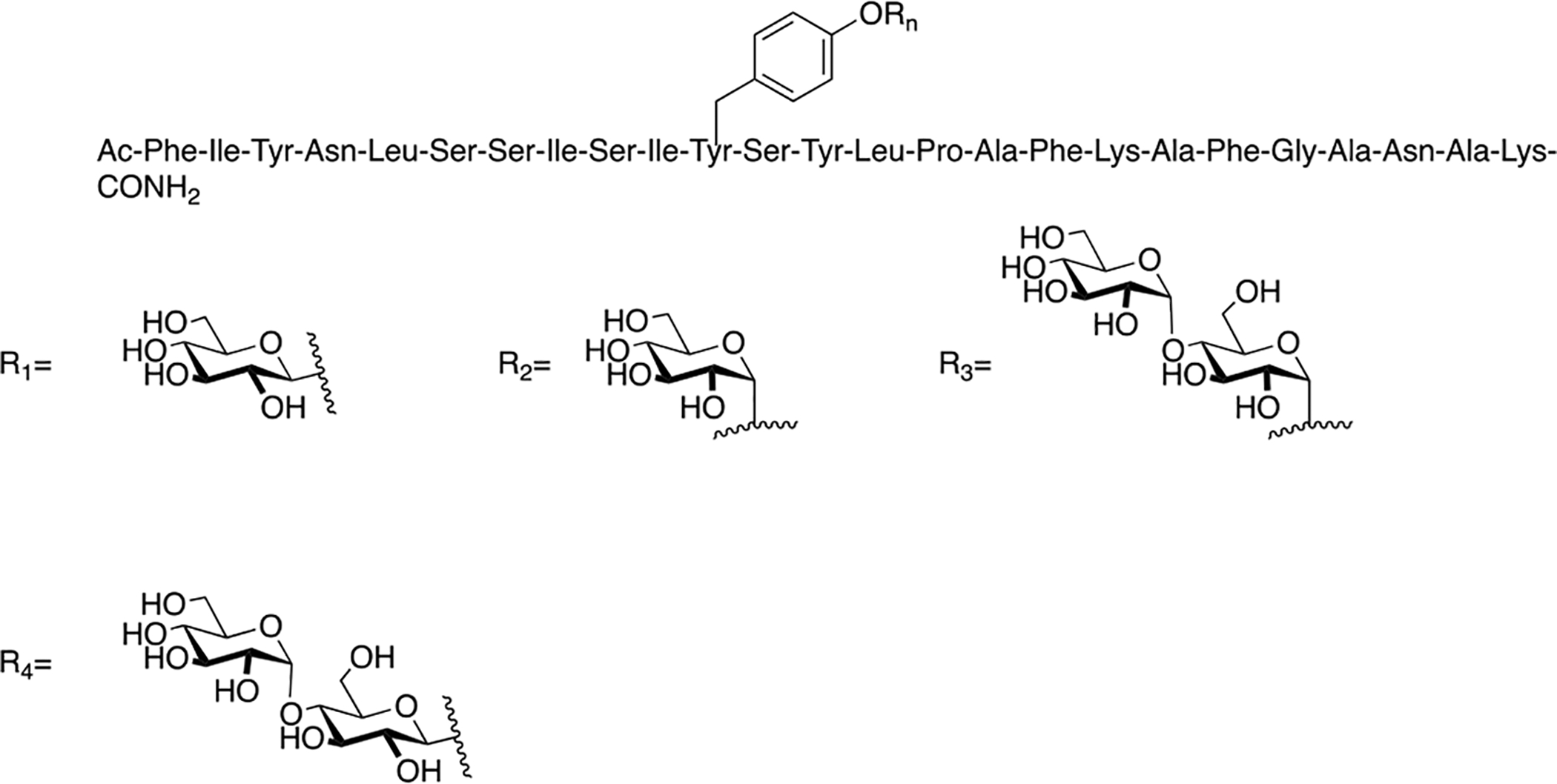
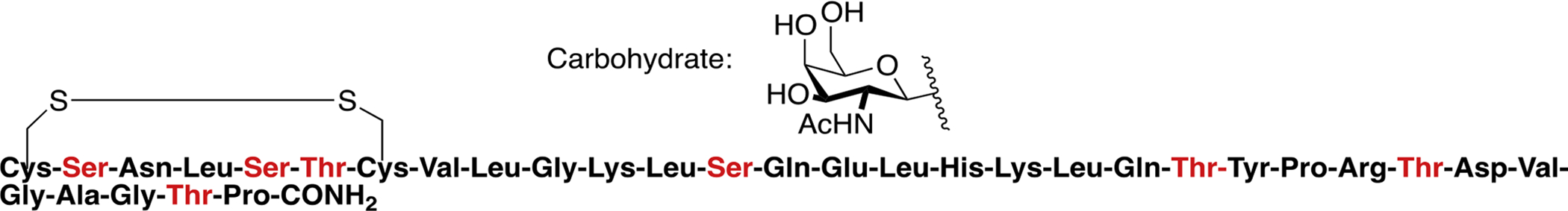
Dangoor and coworkers synthesized a library of glycopeptides related to the vasoactive intestinal peptide (VIP) [135]. They introduced N-linked carbohydrate moieties at Asn9, Asn24, and Asn28 and O-linked carbohydrate moieties were introduced in Ser2, Thr7, Thr11, Tyr22, and Ser25 (Figure 30). The structures of the specific carbohydrates used at their corresponding positions are summarized in Table 3. Receptor binding experiments revealed that introducing a carbohydrate moiety into the C-terminus only resulted in small decreases in binding, while the effect a carbohydrate inserted closer to the N-terminus was detrimental. Despite these decreases in binding, it was found that all glycopeptide analogues were full agonists at the VPAC1 receptor. This was especially true for analogues glycosylated at Thr7 and Thr11. However, glycosylation at Thr11 afforded an analogue with exceptional stability towards degradation by trypsin compared to native VIP and the other glycopeptide analogues.
Figure 30. VIP Glycopeptides.
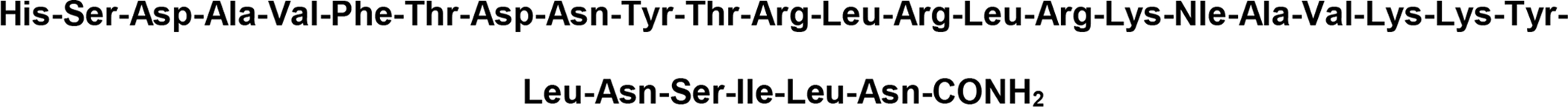
Glycopeptides related to the vasoactive intestinal peptide (VIP) were glycosylated at several different positions to determine the sequence-dependent effect of glycosylation on biological activity. (Dangoor 2009) [135].
Table 3.
Summary of Endorphin Glycopeptides
|
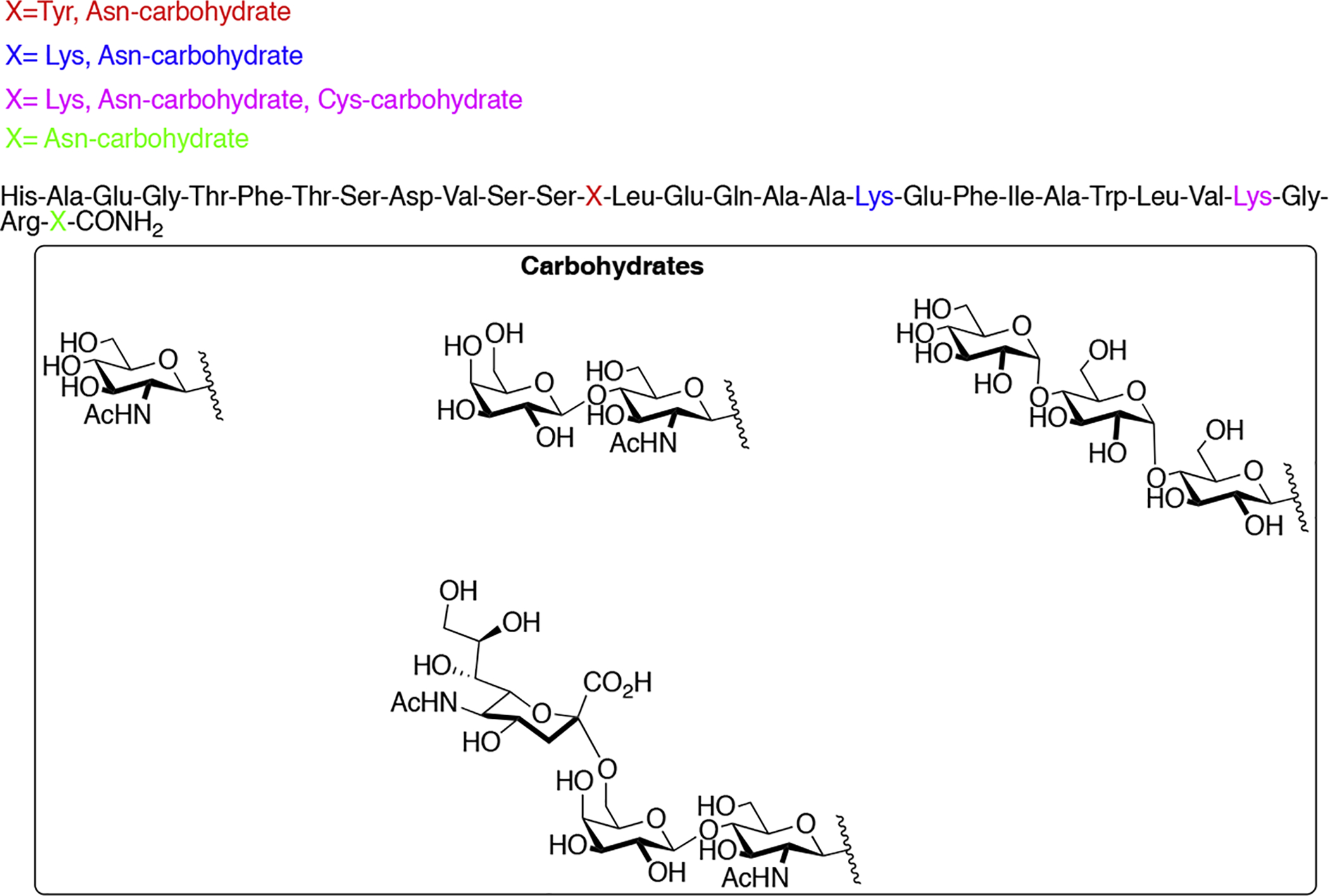
Toma and coworkers synthesized a series of O-glycosylated calcitonin analogues containing GalNAc moieties appended to various Ser and Thr residues (Figure 31) [136]. Calcitonin is primarily involved in regulating calcium homeostasis and is used to treat diseases affecting bones and bone metabolism, such as osteoporosis and Paget’s Disease [137]. Toma’s study aimed to determine how glycosylation affected the 3D structure of calcitonin, its biodistribution, and biological activity in vitro and in vivo. CD studies revealed that glycosylation at Ser2 and Ser21 reduced the α-helical content of the peptide, but other glycosylated analogues exhibited similar α-helical content. Interestingly, it was determined in receptor binding studies that the glycosylated analogues at Ser2 and Ser21 exhibited similar binding affinities to native calcitonin, whereas the other analogues showed significant reductions in binding. In vivo studies evaluating the hypocalcemic activity of the glycosylated analogues showed that the compounds glycosylated at Ser2 and Ser21 exhibited slightly greater activity compared to the other analogues. Biodistribution studies revealed important glycosylation-site dependent trends. The analogues glycosylated at Ser2 and Ser21 accumulated primarily in bone and in the blood at lower concentrations. Surprisingly, the glycosylated Ser2 analogue was found at low concentrations in the kidney while the kidney concentration of the Ser21 glycosylated analogue was significantly higher. The analogues glycosylated at Thr25 and Thr31 were detected predominantly in the liver.
Figure 31. O-linked Calcitonin Glycopeptides.
Glycosylated calcitonin analogues were synthesized and evaluated for their hypocalcemic activity in vivo. (Toma 2001) [136].
Additional examples of novel N-linked glycopeptides for the treatment of diabetes and regulation of glucose levels includes work from the Kondo group. In one study, they explored the chemoenzymatic synthesis of glycosylated GLP-1 analogues (Figure 32) [138]. They introduced GlcNAc, LacNAc, and sialyl LacNAc residues via an amide linkage through an Asn residue. LacNAc moieties were introduced with glycotransferases, while the other carbohydrates were introduced by traditional solid phase peptide synthesis methods. Other analogues containing S-linked glucose, maltose, and maltotriose through cysteine side chains were also synthesized. It was found in studies of functional activity and binding that glycosylation in the C-terminal region of the peptide resulted in analogues with retained agonist activity close to that of native GLP-1. The LacNAc analogue exhibited slightly lower activity compared to the other compounds. Glycosylation closer towards the N-terminus or in the middle of the peptide afforded analogues with decreased agonist activity. Enzymatic stability studies using DPP-IV showed that glycosylation vastly improved the half-life of the newly synthesized analogues. The LacNAc analogue in particular exhibited the greatest stability compared to the other glycopeptides. In vivo experiments in which glucose levels were measured following subcutaneous administration showed that all of the glycopeptide analogues lowered blood glucose levels with the LacNAc analogue reducing most effectively. This study is consistent with many of the results obtained for glycopeptides targeting class A GPCRs.
Figure 32. Glycopeptides Related to GLP-1.
N-linked glycopeptide analogues of GLP-1 were synthesized using a combined SPPS and enzymatic approach. They exhibited improved binding activity, functional activity, and stability in vitro. (Kondo 2009) [138].
Watanabe and coworkers synthesized the LacNAc analogue originally synthesized by Wong and coworkers in 2009 [139] to examine the biodistribution of the LacNAc-containing GLP-1 glycopeptide with non-invasive imaging studies in vivo. Additionally, ex vivo studies were conducted by administering an I25-labeled LacNAc analogue to rats. Their organs were collected following injection and the compound was quantified using a gamma counter. Ultimately, they found the concentration of the LacNAc analogue decreased in the liver while plasma concentrations significantly increased. Further, the LacNAc analogue was found in higher concentrations in the kidney as compared to native GLP-1. They hypothesized that the increased kidney clearance was due to the increased hydrophilicity of this glycopeptide analogue.
6. Considerations for Large Scale Manufacture of Glycopeptide Drugs
Considering the vast success of various classes of glycopeptides in pre-clinical studies, it is imperative to develop efficient methods for their synthesis on an industrial scale. There are significant challenges in the manufacture of peptide drugs– indeed, as there are with all classes of drugs. Chemists in both academia and industry have devised creative and effective solutions for the synthesis and purification of peptides. One of the world’s leading drugs to treat hypertension is lisinopril (Zestril®), an angiotensin-converting enzyme (ACE) inhibitor designed from a peptide substrate for ACE. By “inverting” the phenylalanine residue and extending the phenylalanine sidechain by one carbon, a highly effective drug is produced that is metabolically stable and excreted unchanged in urine [140]. Several other “blockbuster” peptide drugs have also been produced on an industrial scale, including oxytocin, various vasopressin analogues, parathyroid hormone, luteinizing hormone-releasing hormone, somatostatin, and octreotide, among others [141]. Syntheses of this magnitude are typically accomplished using classical solution phase, solid phase, or hybrid solution/solid phase synthesis strategies. Some current limitations that have yet to be fully addressed in large-scale peptide production include reproducible preparation of SPPS resins, general solutions for the synthesis of difficult peptide sequences, and convenient ways of monitoring coupling and deprotection reaction progress during synthesis [142]. In the case of glycopeptides, some additional complications include the cost of large-scale preparation of the desired carbohydrate bearing amino acid building blocks and the deprotection of carbohydrate hydroxyl protecting groups. For example, hydrazine hydrate can effectively cleave acetyl protection of the hydroxyl groups while the glycopeptide is still bound to the resin, but on an industrial scale this is not ideal due to toxicity and safety concerns. Another strategy is to cleave the glycopeptide from the resin, followed by a Zemplén de-acetylation reaction in solution. This can become problematic if the solubility of the glycopeptide is low, but more importantly the basic conditions of the reaction could potentially lead to β-elimination of the carbohydrate moiety and other degradations of the peptide chain. Thus, the development of less toxic, scalable, and mild methods for carbohydrate protecting group removal should be pursued. Alternatively, new protecting groups meeting these criteria can be prepared. Despite these limitations, several examples of the preparation of complex glycopeptides and glycoproteins using novel chemoenzymatic glycosylation methods and native chemical ligation coupled with SPPS have been reported [143–145]. Furthermore, the development of protecting group-free chemoenzymatic peptide ligation reactions in aqueous solution have been developed and applied to bioactive peptides [146,147]. Current advances in peptide and glycoprotein synthesis make the development of methods for kilogram to metric-ton syntheses of therapeutic glycopeptides certainly feasible.
7. Clinical Implications for the Future of Glycopeptide Drugs
Peptides have high affinities for their binding sites, possess immense chemical and biological diversity, and offer great solutions to a variety of diseases and metabolic syndromes. Over 60 peptide drugs have now been approved for marketing, and more than 150 are now in active clinical development [148]. Glycosylation has proven to be an extremely effective way to improve the pharmacodynamic and pharmacokinetic profiles of biologically active peptides, which are not limited to the peptides produced from the human genome [149]. It offers several advantages over traditional methods for improving the drug-like properties of peptides including improving BBB penetration, increasing water solubility and bioavailability, and significantly reducing toxicity. Furthermore, there are well-established chemical and enzymatic methods to introduce a wide variety of different carbohydrate linkages into the peptide backbone. Early studies on opioid glycopeptides in the 1980s and 1990s set the stage for a paradigm shift in peptide synthesis. It was originally believed that increasing the lipophilicity of a drug allows it to efficiently penetrate the BBB. Since this is true in a vast number of cases, it had not been predicted that the introduction of a water-soluble carbohydrate into a drug would improve its BBB penetration and biological activity. This discovery prompted several more syntheses of novel glycopeptides targeting the CNS. Glycopeptides targeting class A GPCRs have dominated the field of glycopeptide drug development, leaving class B GPCR peptide ligands largely untouched with the exception of VIP, GLP-1, and calcitonin. More class B GPCR peptide ligands need to be examined due to their prevalence in neuroprotection and regulation of inflammatory responses. However, the positive results obtained from the limited number of studies on class B GPCR peptide ligands are promising and suggest that glycosylation can improve the stability and BBB penetration of peptides of this size and molecular weight. There are challenges associated with industrial production of glycopeptide drug candidates, including high throughput parallel synthesis, but advances in large-scale manufacturing of peptide drugs [150] including both hybrid SPPS/solution phase methods and enzyme-mediated reactions can lead to new avenues for their preparation. The future is bright for this field and, given the successes and promising in vivo results from past glycopeptide studies, the number of glycopeptide drug candidates is sure to grow significantly in the coming years.
Table 1.
Summary of Enkephalin Glycopeptides.
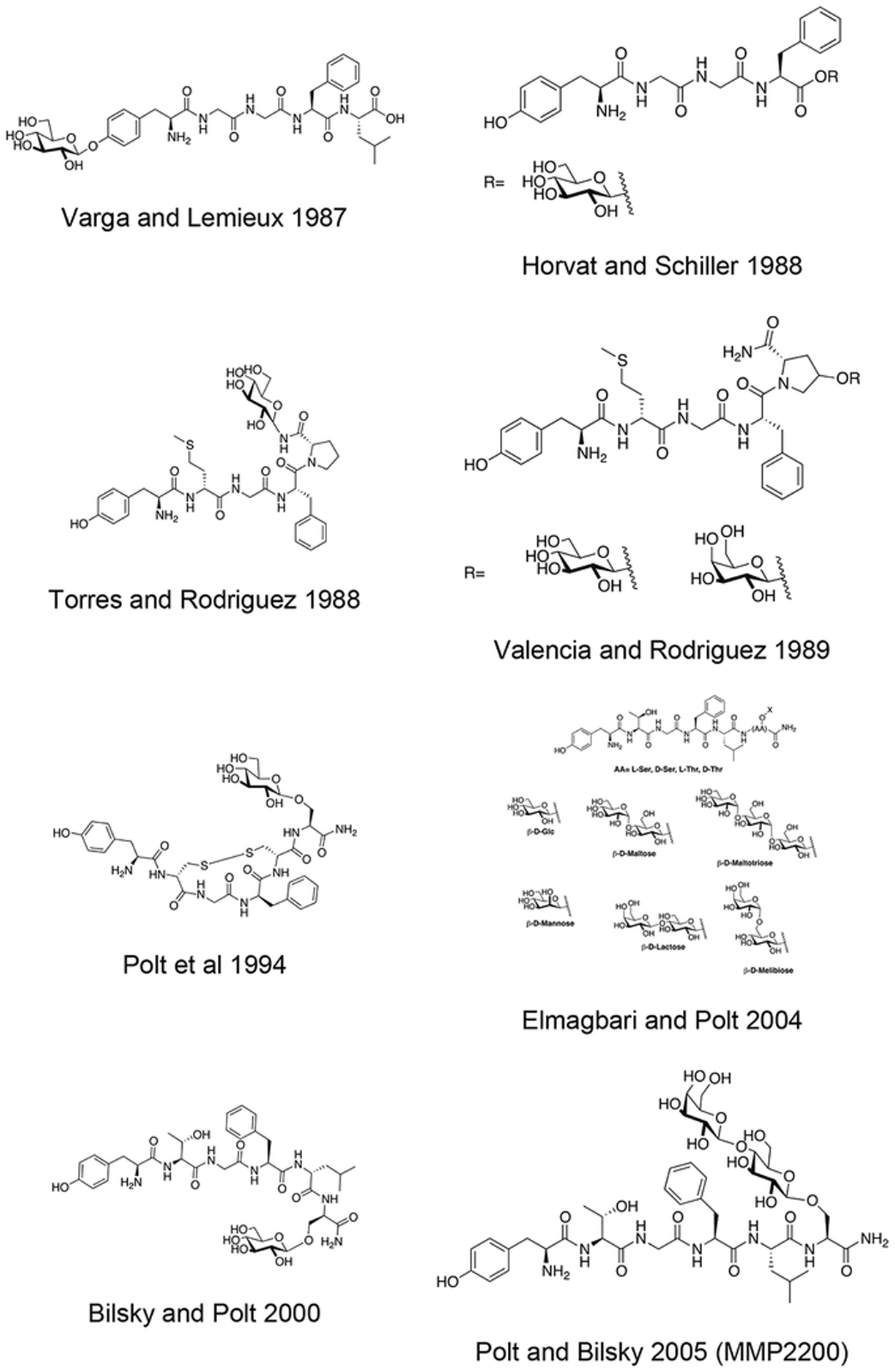
|
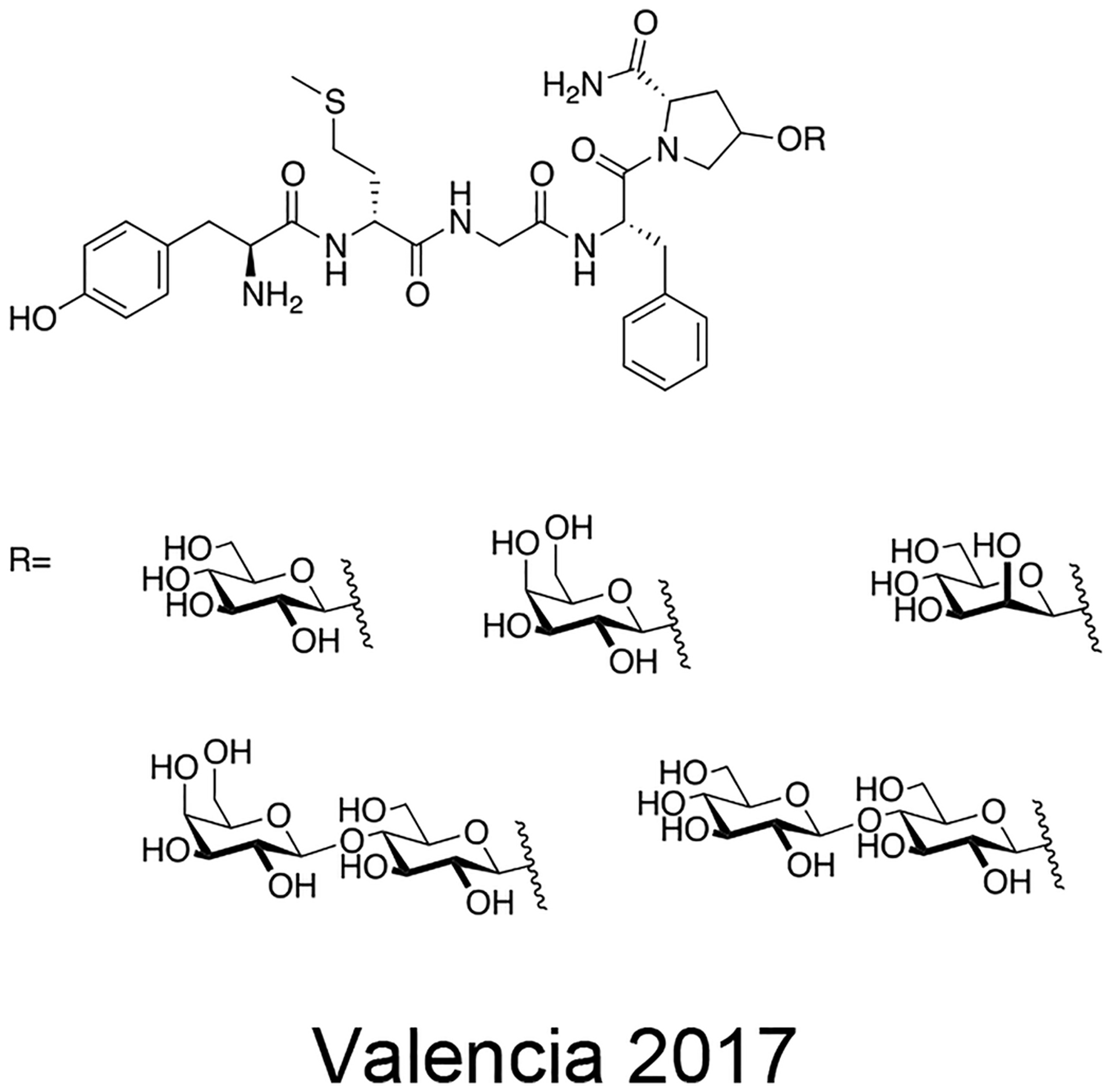
|
Table 2.
Summary of Endomorphin Glycopeptides
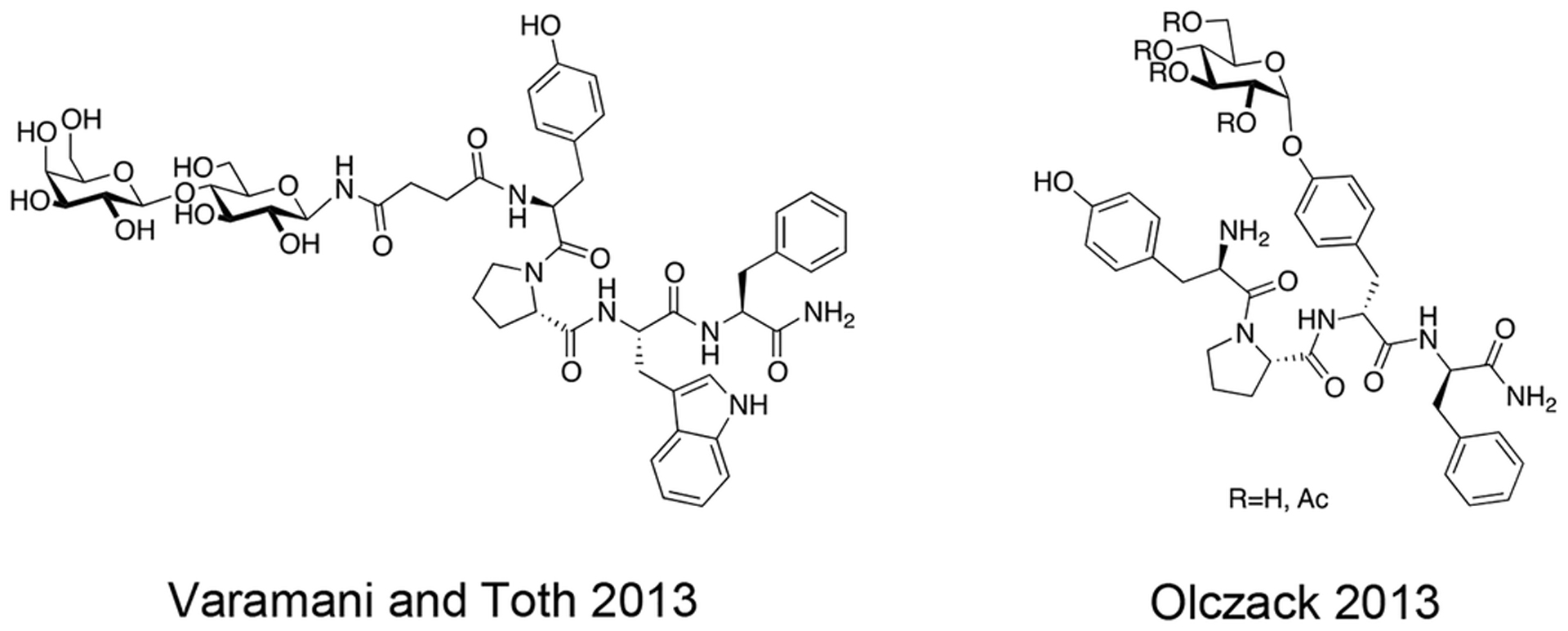
|
Table 4.
Summary of DAMGO Glycopeptides
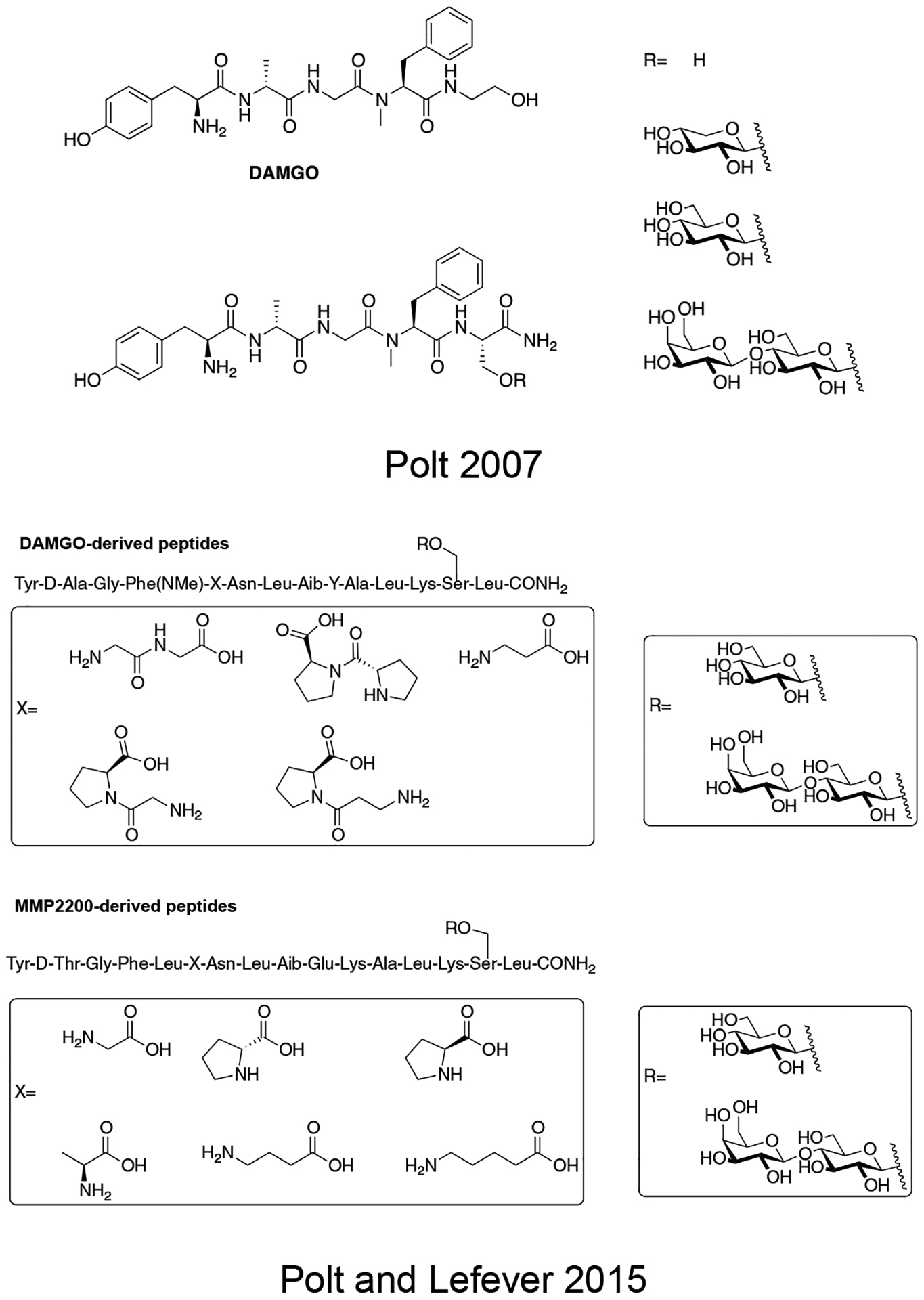
|
Table 5.
Summary of Dermorphin and Deltorphin Glycopeptides
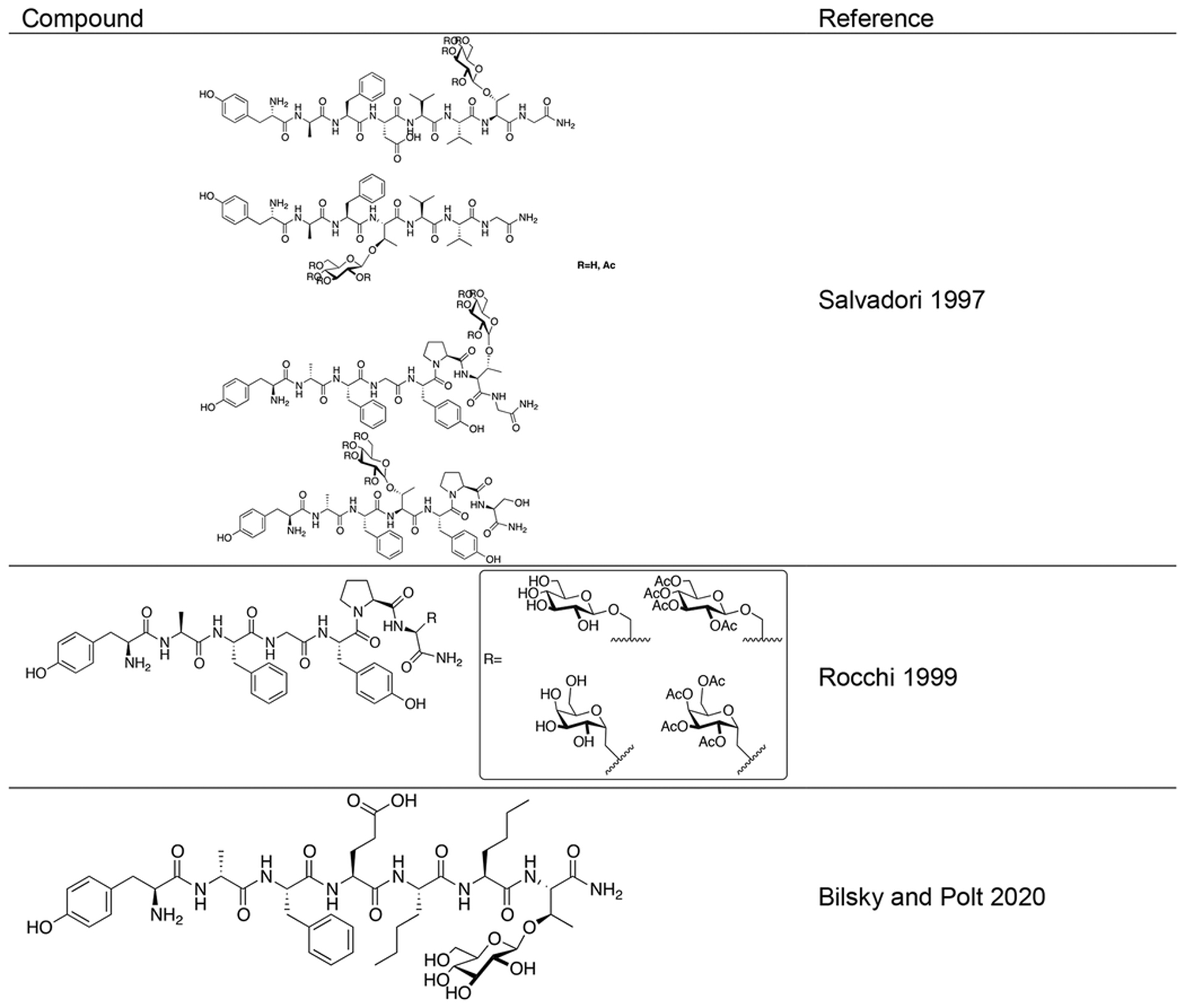
|
Table 6.
Summary of Glycopeptides Targeting Neurotensin-1 Receptor
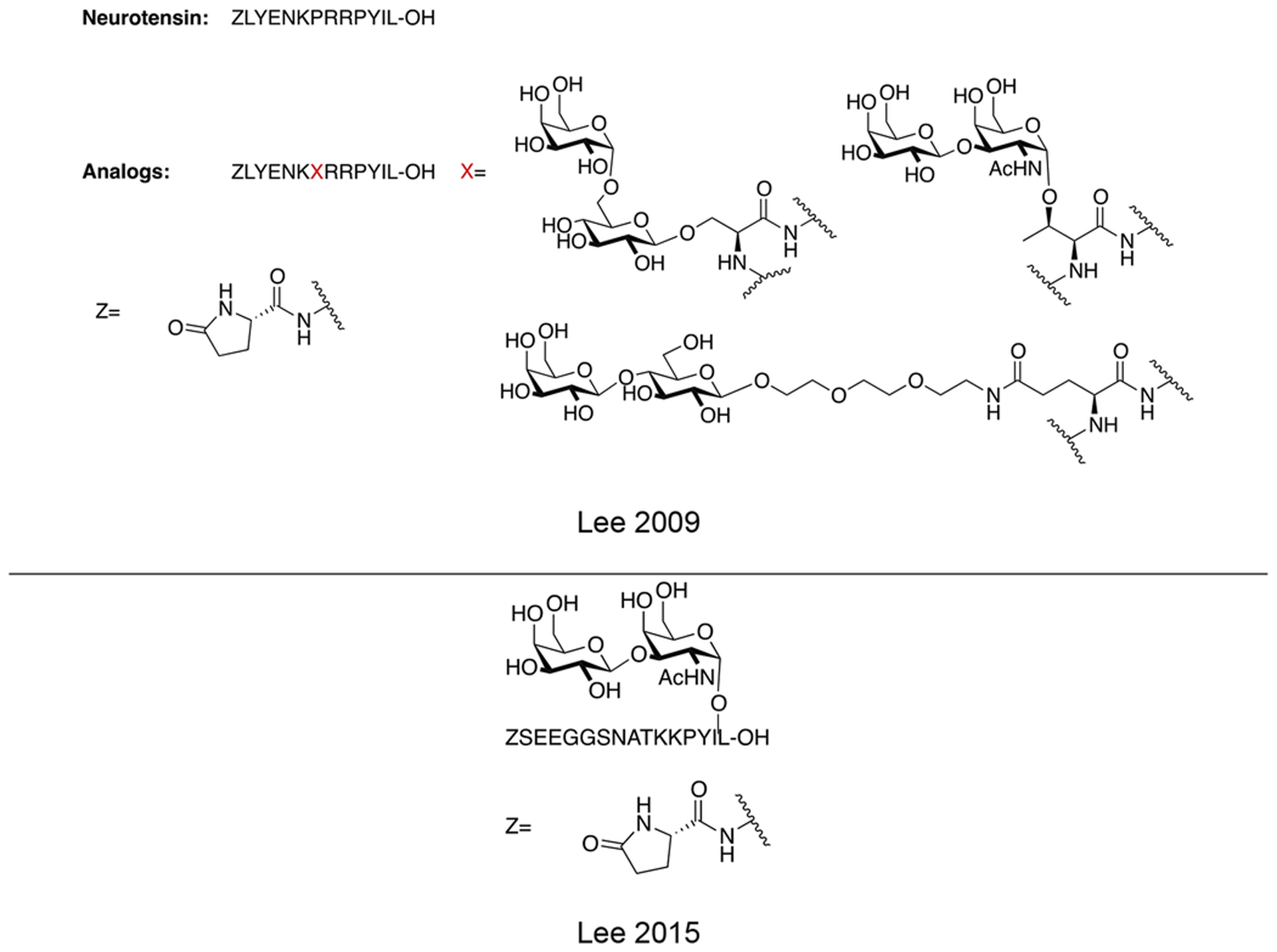
|
Table 7.
Summary of Miscellaneous Glycopeptides
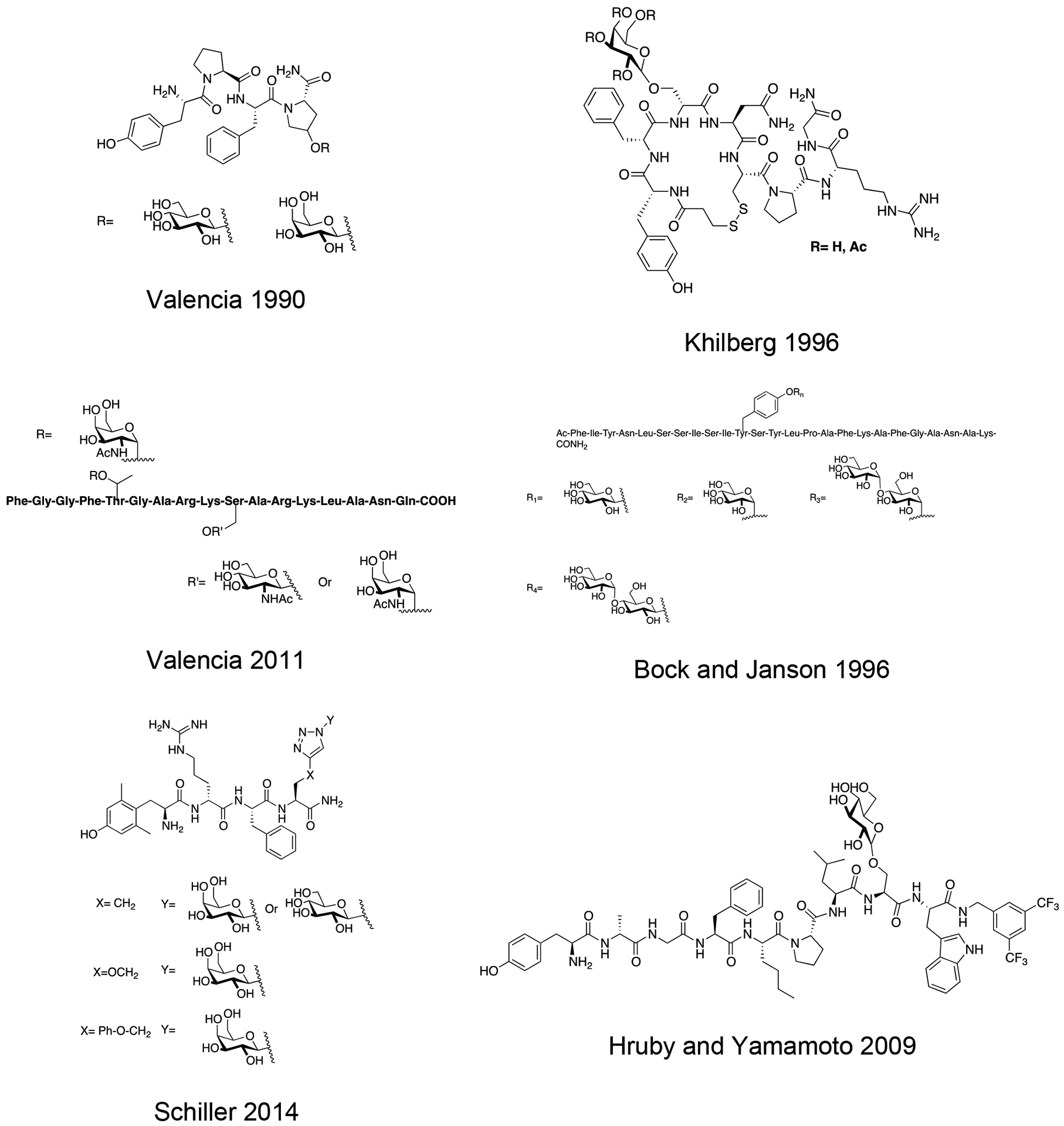
|
Table 9.
Summary of Carbohydrate Substitutions in VIP Glycopeptide Analogues (Dangoor, et al)
| Native Residue | Glycosylated substitution |
|---|---|
| Ser2 | 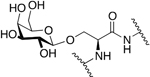 |
| Thr7 |  |
| Asn9 | 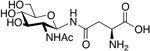 |
| Thr11 | 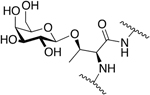 |
| Tyr22 | 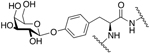 |
| Asn24 | 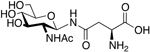 |
| Ser25 | 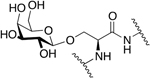 |
| Asn28 | 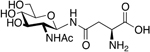 |
Highlights.
Glycosylation has been shown to dramatically improve stability of peptides
Glycopeptides are able to penetrate the blood-brain barrier (BBB) to a greater extent than their non-glycosylated counterparts
Glycopeptide analogues of their native neuropeptides generally retain potency and efficacy at their target receptors
Glycosylation can be successfully applied to peptides of varying size
Acknowledgements.
The authors thank the Office of Naval Research, the National Institutes of Health, the Michael J. Fox Foundation and the Migraine Research Foundation for the generous support of our studies. The authors would also like to thank Alexander Marciniak and Dillon Hanrahan for their help in figure preparation and Hannah Goodman for help in the editing process.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
References
- (1).Hruby VJ; Cai M Design of Peptide and Peptidomimetic Ligands with Novel Pharmacological Activity Profiles. Annu. Rev. Pharmacol. Toxicol, 2013, 53, 557–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fosgerau K; Hoffmann T Peptide Therapeutics: Current Status and Future Directions. Drug Discov. Today, 2015, 20 (1), 122–128. [DOI] [PubMed] [Google Scholar]
- (3).Hruby VJ Designing Peptide Receptor Agonists and Antagonists. Nat. Rev. Drug Discov, 2002, 1 (11), 847–858. [DOI] [PubMed] [Google Scholar]
- (4).Werner HM; Horne WS Folding and Function in α/β-Peptides: Targets and Therapeutic Applications. Curr. Opin. Chem. Biol, 2015, 28, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cromm PM; Spiegel J; Grossmann TN Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem. Biol, 2015, 10 (6), 1362–1375. [DOI] [PubMed] [Google Scholar]
- (6).Chatterjee J; Gilon C; Hoffman A; Kessler H N-Methylayion of Peptides: A New Perspective in Medicinal Chemistry. Acc. Chem. Res, 2008, 41 (10), 1331–1342. [DOI] [PubMed] [Google Scholar]
- (7).Varamini P; Toth I Lipid- and Sugar-Modified Endomorphins: Novel Targets for the Treatment of Neuropathic Pain. Front. Pharmacol, 2013, 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Buskas T; Ingale S; Boons GJ Glycopeptides as Versatile Tools for Glycobiology. Glycobiology, 2006, 16 (8), 113–136. [DOI] [PubMed] [Google Scholar]
- (9).Bertozzi CR; Kiessling LL Chemical Glycobiology. Science. 2001, 291 (5512), 2357–2364. [DOI] [PubMed] [Google Scholar]
- (10).Dwek RA Glycobiology. Ann. Rev. Biochem, 1988, 57, 785–838. [DOI] [PubMed] [Google Scholar]
- (11).Liu Y; Cao Q; Li L Isolation and Characterization of Glycosylated Neuropeptides, 1st ed.; Elsevier Inc., 2019; Vol. 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jones EM; Polt R CNS Active O-Linked Glycopeptides. Front. Chem,. 2015, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Polt R; Dhanasekaran M; Keyari CM Glycosylated Neuropeptides: A New Vista for Neuropsychopharmacology? Med. Res. Rev, 2005, 25 (5), 557–585. [DOI] [PubMed] [Google Scholar]
- (14).Moradi SV; Hussein WM; Varamini P; Simerska P; Toth I Glycosylation, an Effective Synthetic Strategy to Improve the Bioavailability of Therapeutic Peptides. Chem. Sci,. 2016, 7 (4), 2492–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bryant SD; Jinsmaa Y; Salvadori S; Okada Y; Lazarus LH Dmt and Opioid Peptides : A Potent Alliance. 2003, Biopolymers (Pept Sci), 71, 86–102. [DOI] [PubMed] [Google Scholar]
- (16).Chang YS; Graves B; Guerlavais V; Tovar C; Packman K; To K-H; Olson KA; Kesavan K; Gangurde P; Mukherjee A; Baker T; Darlak K; Elkin C; Filipovic Z; Qureshi FZ; Cai H; Berry P; Feyfant E; Shi XE; Horstick J; Annis DA; Maning AM; Fotouhi N; Nash H; Vassilev LT; Sawyer TK Stapled α−helical Peptide Drug Development: A Potent Dual Inhibitor of MDM2 and MDMX for P53-Dependent Cancer Therapy. Proc. Natl. Acad. Sci, 2013, 110 (36), E3445–E3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Simerska P; Moyle PM; Toth I Moderin Lipid-, Carbohydrate-, and Peptide-Based Delivery Systems for Peptide, Vaccine, and Gene Products. Med. Res. Rev, 2011, 31 (4), 520–547. [DOI] [PubMed] [Google Scholar]
- (18).Caccetta R; Blanchfield JT; Harrison J; Toth I; Benson HAE Epidermal Penetration of a Therapeutic Peptide by Lipid Conjugation; Stereo-Selective Peptide Availability of a Topical Diastereomeric Lipopeptide. Int. J. Pept. Res. Ther, 2006, 12 (3), 327–333. [Google Scholar]
- (19).Ward BP; Ottaway NL; Perez-Tilve D; Ma D; Gelfanov VM; Tschöp MH; DiMarchi RD Peptide Lipidation Stabilizes Structure to Enhance Biological Function. Mol. Metab, 2013, 2 (4), 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).European Medicines Agency: EMA/CHMP/SWP/647258/2012 – CHMP Safety Working Party’s response to the PDCO regarding the use of PEGylated drug products in the paediatric population. July 2019.
- (21).Shubin AV; Demidyuk IV; Komissarov AA; Rafieva LM; Kostrov SV; Shubin AV; Demidyuk IV; Komissarov AA; Rafieva LM; Kostrov SV Cytoplasmic Vacuolization in Cell Death and Survival. Oncotarget, 2016, 7 (34), 55863–55889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Koenigs W; Knorr E Ueber einige Derivate des Traubenzuckers und der Galactose. Ber. Dtsch. Chem. Ges 1901, 34, 957–981. [Google Scholar]
- (23).Schmidt RR; Michel J Facile Synthesis of α- And β- O-Glycosyl Imidates; Preparation of Glycosides and Disaccharides. Angew. Chemie Int. Ed. English, 1980, 19 (9), 731–732. [Google Scholar]
- (24).Lian G; Zhang X; Yu B Thioglycosides in Carbohydrate Research. Carbohydr. Res, 2015, 403, 13–22. [DOI] [PubMed] [Google Scholar]
- (25).Toshima K; Tatsuta K Recent Progress in O-Glycosylation Methods and Its Application to Natural Products Synthesis. Chem. Rev, 1993, 93 (4), 1503–1531. [Google Scholar]
- (26).Mitchell SA; Pratt MR; Hruby VJ; Polt R Solid-Phase Synthesis of O-Linked Glycopeptide Analogues of Enkephalin. J. Org. Chem, 2001, 66, 2327–2342. [DOI] [PubMed] [Google Scholar]
- (27).Lefever MR; Szabò LZ; Anglin B; Ferracane M; Hogan J; Cooney L; Polt R Glycosylation of α-Amino Acids by Sugar Acetate Donors with InBr3. Minimally Competent Lewis Acids. Carbohydr. Res, 2012, 351, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Seitz O Glycopeptide Synthesis and the Effects of Glycosylation on Protein Structure and Activity. Chembiochem, 2000, 1 (4), 214–246. [DOI] [PubMed] [Google Scholar]
- (29).Buskas T; Ingale S; Boons GJ Glycopeptides as Versatile Tools for Glycobiology. Glycobiology, 2006, 16 (8), 113R–136R. [DOI] [PubMed] [Google Scholar]
- (30).Herzner H; Reipen T; Schultz M; Kunz H Synthesis of Glycopeptides Containing Carbohydrate and Peptide Recognition Motifs. Chem. Rev, 2000, 100 (12), 4495–4537. [DOI] [PubMed] [Google Scholar]
- (31).Taylor CM Glycopeptides and Glycoproteins: Focus on the Glycosidic Linkage. Tetrahedron, 1998, 54 (38), 11317–11362. [Google Scholar]
- (32).Avenoza A; Peregrina JM; Martin ES Synthesis of a new conformationally constrained glycoamino acid building block Tetrahedron Lett, 2003, 44 (33), 6413–6416. [Google Scholar]
- (33).Manabe S; Sakamoto K; Nakahara Y; Sisido M; Hohsaka T; Ito Y Preparation of Glycosylated Amino Acid Derivatives for Glycoprotein Synthesis by In Vitro Translation System. Bioor. Med. Chem, 2002, 10 (3), 573–581. [DOI] [PubMed] [Google Scholar]
- (34).Baker A; Turner NJ; Webberley MC Improved Strategy for the Stereoselective Synthesis of Glycosides Using Glycosidases as Catalysts. Tetrahedron: Asymmetry, 1994, 5 (12), 2517–2522. [Google Scholar]
- (35).Liang L; Astruc D The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coordination Chemistry Reviews, 2011, 255 (23–24), 2933–2945. [Google Scholar]
- (36).O’Flaherty R; Velasco-Torrijos T Glycosylated α-Azido Amino Acids: Versatile Intermediates in the Synthesis of Neoglycoconjugates. Synlett, 2018, 29, 904–907. [Google Scholar]
- (37).Paulsen H; Peters S; Bielfeldt T; Meldal M; Bock K Synthesis of the glycosyl amino acids Nα-Fmoc-Ser[Ac4-β-D-Galp-(1→3)-Ac2-α-D-GalN3p]-OPfp and Nα-Fmoc-Thr[Ac4-β-D-Galp-(1→3)-Ac2-α-D-GalN3p]-Opfp and the application in the solid-phase peptide synthesis fo multiply glycosylated mucin peptides with Tn and T antigenic structures. Carbohydrate Research, 1995, 268 (1), 17–34. [DOI] [PubMed] [Google Scholar]
- (38).TornØe CW; Davis P; Porreca F; Meldal M α-Azido Acids for Direct Use in Solid-phase Peptide Synthesis. J. Peptide. Sci, 2000, 6 (12), 594–602. [DOI] [PubMed] [Google Scholar]
- (39).Halkes KM; St. Hilaire PM; Jansson AM; Gotfredsen CH; Meldal M Synthesis and application of sialic acid-containing building blocks for glycopeptide libraries.1 Establishing glycosylation conditions. J. Chem. Soc., Perkin Trans, 2000, 1 (13), 2127–2133. [Google Scholar]
- (40).Hofsteenge J; Müller DR; de Beer T; Löffler A; Richter WJ; Vliegenthart JFG New Type of Linkage between a Carbohydrate and a Protein: C-Glycosylation of a Specific Tryptophan Residue in Human RNase Us. Biochemistry, 1994, 33 (46), 13524–13530 [DOI] [PubMed] [Google Scholar]
- (41).Feeny RE; Burcham TS; Yeh Y Antifreeze Glycoproteins From Polar Fish Blood. Annu. Rev. Biophys. Biophys. Chem, 1986, 15, 59–78. [DOI] [PubMed] [Google Scholar]
- (42).Harding MM; Anderberg PI; Haymet ADJ “Antifreeze” Glycoproteins from Polar Fish. Eur. J. Biochem, 2003, 270 (7), 1381–1392. [DOI] [PubMed] [Google Scholar]
- (43).Kobertz WR; Bertozzi CR; Bednarski MD An Efficient Method for the Synthesis of α- and β-C-Glycosyl Aldehydes. Tetrahedron Lett, 1992, 33 (6), 737–740. [Google Scholar]
- (44).Marcaurelle LA; Bertozzi CR New Directions in the Synthesis of Glycopeptide Mimetics. Chem. Eur. J, 1999, 5 (5), 1384–1390. [Google Scholar]
- (45).Bertozzi CR; Bednarski MD The Synthesis of 2-Azido C-Glycosyl Sugars. Tetrahedron Lett, 1992, 33 (22), 3109–3112. [Google Scholar]
- (46).Nolen EG; Kurish AJ; Potter JM; Donahue LA; Orlando MD Stereoselective Synthesis of α-C-Glucosyl Serine and Alanine via a Cross-Metathesis/Cyclization Strategy. Org. Lett, 2005, 7 (15), 3383–3386. [DOI] [PubMed] [Google Scholar]
- (47).Norsikian S; Zeitouni J; Rat S; Gérard S; Lubineau A New and General Synthesis of β-C-Glycosylformaldehydes from Easily Available β-C-Glycosylpropanones. Carbohydr. Res, 2007, 342 (18), 2716–2728. [DOI] [PubMed] [Google Scholar]
- (48).Dondoni A; Marra A Methods for Anomeric Carbon-Linked and Fused Sugar Amino Acid Synthesis: The Gateway to Artificial Glycopeptides. Chem. Rev, 2000, 100 (12), 4395–4421. [DOI] [PubMed] [Google Scholar]
- (49).Imperiali B; O’Connor SE Effect of N-Linked Glycosylatian on Glycopeptide and Glycoprotein Structure. Curr. Opin. Chem. Biol, 1999, 3 (6), 643–649. [DOI] [PubMed] [Google Scholar]
- (50).Urge L; Otvos L; Lang E; Wroblewski K; Laczko I; Hollosi M Fmoc-Protected, Glycosylated Asparagines Potentially Useful as Reagents in the Solid-Phase Synthesis of N-Glycopeptides. Carbohydr. Res, 1992, 235 (C), 83–93. [DOI] [PubMed] [Google Scholar]
- (51).Rákosi K; Szolomájer-Csikós O; Kalmár L; Szurmai Z; Kerékgyártó J; Tóth GK Synthesis of N-Glycopeptides Applying Glycoamino Acid Building Blocks with a Combined Fmoc/Boc Strategy. Protein Pept. Lett, 2011, 18 (7), 679–683. [DOI] [PubMed] [Google Scholar]
- (52).Lomino JV; Naegeli A; Orwenyo J; Amin MN; Aebi M; Wang LX A Two-Step Enzymatic Glycosylation of Polypeptides with Complex N-Glycans. Bioorganic Med. Chem, 2013, 21 (8), 2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Li B; Zeng Y; Hauser S; Song H; Wang LX Highly Efficient Endoglycosidase-Catalyzed Synthesis of Glycopeptides Using Oligosaccharide Oxazolines as Donor Substrates. J. Am. Chem. Soc, 2005, 127 (27), 9692–9693. [DOI] [PubMed] [Google Scholar]
- (54).Gamblin DP; Scanlan EM; Davis BG Glycoprotein Synthesis: An Update. Chem. Rev, 2009, 109 (1), 131–163. [DOI] [PubMed] [Google Scholar]
- (55).Filippi B; Biondi L; Filira F; Rocchi R; Bellini C; Medica P Synthesis and Biological Activity of (D) Ala2, Leu5- Enkephalins Containing Hydrophilic or Hydrophobic Moieties. Biopolymers. 1983, 22, 575–578. [DOI] [PubMed] [Google Scholar]
- (56).Ling N; Braseau P; Benoit R; Wasada T; Harris D; Unger R; Guillemin R Synthesis and Biological Activity of Glycosylated Analogs of Somatostatin. Biochemical and Biophysical Research Communications., 1979, 91 (2), 614–622. [DOI] [PubMed] [Google Scholar]
- (57).Albert R; Marbach P; Bauer W; Briner U; Fricker G; Brums C; Pless J SDZ CO 611: A Highly Potent Glycated Analog of Somatostatin with Improved Oral Activity. Life Sci, 1993, 53 (6), 517–525. [DOI] [PubMed] [Google Scholar]
- (58).Yaylayan VA; Huyqhues-Despointes A Chemistry of Amadori Rearrangement Products: Analysis, Synthesis, Kinetics, Reactions, and Spectroscopic Properties. Critical Reviews in Food Science and Nutrition, 1994, 34 (4), 321–369. [DOI] [PubMed] [Google Scholar]
- (59).Schleicher E; Deufel T; Wieland H Non-Enzymatic Glycosylations of Human Serum Lipoproteins. Elevated ε-Lysine Glycosylated Low Density Lipoprotein in Diabetic Patients. FEBS Letters, 1981, 129 (1), 1–4. [DOI] [PubMed] [Google Scholar]
- (60).Tomabechi Y; Krippner G; Rendle PM; Squire MA; Fairbanks AJ Glycosylation of Pramlintide: Synthetic Glycopeptides That Display in Vitro and in Vivo Activities as Amylin Receptor Agonists. Chem. Eur. J, 2013, 19 (45), 15084–15088. [DOI] [PubMed] [Google Scholar]
- (61).Katayama H; Asahina Y; Hojo H Chemical Synthesis of the S-Linked Glycopeptide, Sublancin. J. Pept. Sci, 2011, 17 (12), 818–821. [DOI] [PubMed] [Google Scholar]
- (62).Zhu X; Pachamuthu K; Schmidt RR Synthesis of S -Linked Glycopeptides in Aqueous Solution. J. Org. Chem, 2003, 68 (14), 5641–5651. [DOI] [PubMed] [Google Scholar]
- (63).Elofsson M; Walse B; Kihlberg J Building Blocks for Glycopeptide Synthesis: Glycosylation of 3-Mercaptopropionic Acid and Fmoc Amino Acids with Unprotected Carboxyl Groups. Tetrahedron Lett, 1991, 32 (51), 7613–7616. [Google Scholar]
- (64).De Leon CA; Levine PM; Craven TW; Pratt MR The Sulfur-Linked Analogue of O-GlcNAc (S-GlcNAc) Is an Enzymatically Stable and Reasonable Structural Surrogate for O-GlcNAc at the Peptide and Protein Levels. Biochemistry, 2017, 56 (27), 3507–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Bertozzi CR; Hoeprich PD;Bednarski MD Synthesis of Carbon-Linked Glycopeptides as Stable Glycopeptide Models. J. Org. Chem,1992, 57 (23), 6092–6094. [Google Scholar]
- (66).Debenham SD; Debenham JS; Burk MJ; Toone EJ Synthesis of Carbon-Linked Glycopeptides through Catalytic Asymmetric Hydrogenation. J. Am. Chem. Soc, 1997, 119 (41), 9897–9898. [Google Scholar]
- (67).Westermann B; Walter A; Florke U; Altenbach HJ Chiral Auxiliary Based Approach toward the Synthesis of C-Glycosylated Amino Acids. Org Lett, 2001, 3 (9), 1375–1378. [DOI] [PubMed] [Google Scholar]
- (68).Palomo C; Oiarbide M; Landa A; González-Rego MC; García JM; González A; Odriozola JM; Martín-Pastor M; Linden A Design and Synthesis of a Novel Class of Sugar-Peptide Hybrids: C-Linked Glyco β-Amino Acids through a Stereoselective “Acetate” Mannich Reaction as the Key Strategic Element. J. Am. Chem. Soc, 2002, 124 (29), 8637–8643. [DOI] [PubMed] [Google Scholar]
- (69).Kobilka BK G Protein Coupled Receptor Structure and Activation. Biochim. Biophys. Acta, 2007, 1768 (4), 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Stevens RC; Cherezov V; Katritch V; Abagyan R; Kuhn P; Rosen H; Wüthrich K The GPCR Network: A Large-Scale Collaboration to Determine Human GPCR Structure and Function. Nat. Rev. Drug Discov, 2013, 12 (1), 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hughes J; Smith T; Morgan B; Fothergill L. Purification and Properties of Enkephalin – The Possible Endogenous Ligand for the Morphine Receptor. Life Sci., 1975, 16 (12), 1753–1758. [DOI] [PubMed] [Google Scholar]
- (72).Varga L; Horvat S; Lemieux C; Schiller PW Synthesis and Biological Activity of some Glucose-enkephalin Conjugates. Int. J. Peptide Protein Res, 1987, 30, 371–378. [DOI] [PubMed] [Google Scholar]
- (73).Horvat J; Horvat S; Lemieux C; Schiller PW Synthesis and Biological Activity of [Leu5]Enkephalin Derivatives Containing D-glucose. Int. J. Pept. Protein Res, 1988, 31 (5), 499–507. [DOI] [PubMed] [Google Scholar]
- (74).Torres JL; Reig F; Valencia G; Rodríguez RE; García-Antón JM [D-Met2, Pro5] Enkephalin [N1.5-β-d-glucopyranosyl] Amide: A Glycosylpeptide with High Antinociceptive Activity. Int. J. Pept. Protein Res, 1988, 31 (5), 474–480. [DOI] [PubMed] [Google Scholar]
- (75).Rodriguez RE; Rodriguez FD; Sacristán MP; Torres JL; Valencia G; Garcia Antón JM New Glycosylpeptides with High Antinociceptive Activity. Neurosci. Lett, 1989, 101 (1), 89–94. [DOI] [PubMed] [Google Scholar]
- (76).Torres J; Pepermans H; Valencia G; Reig F; Binst G Van. Synthesis and Conformational Analysis of a Series of Galactosyl Enkephalin Analogues Showing High Analgesic Activity.The EMBO Journal., 1989, 8 (10), 2925–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Polt R; Porreca F; Szabó LZ; Bilsky EJ; Davis P; Abbruscato TJ; Davis TP; Harvath R; Yamamura HI; Hruby VJ Glycopeptide Enkephalin Analogues Produce Analgesia in Mice: Evidence for Penetration of the Blood-Brain Barrier. Proc. Natl. Acad. Sci, 1994, 91 (15), 7114–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Mosberg HI; Omnaas JR; Medzihradsky F; Smith CB Cyclic, Disulfide- and Dithioether-Containing Opioid Tetrapeptides: Development of a Ligard with High Delta Opioid Receptor Selectivity and Affinity. Life Sci, 1988, 43 (12), 1013–1020. [DOI] [PubMed] [Google Scholar]
- (79).Williams SA; Abbruscato TJ; Szabó LZ; Polt R; Hruby VJ; Davis TP The Effect of Glycosylation on the Uptake of an Enkephalin Analogue into the Central Nervous System Biol. Physiol. Blood-Brain Barrier, 46; Couraud PP; Scherman D Springer, Boston, MA, 1996; 69–77. [Google Scholar]
- (80).Bilsky EJ; Egleton RD; Mitchell SA; Palian MM; Davis P; Huber JD; Jones H; Yamamura HI; Janders J; Davis TP; et al. Enkephalin Glycopeptide Analogues Produce Analgesia with Reduced Dependence Liability. J. Med. Chem, 2000, 43 (13), 2586–2590. [DOI] [PubMed] [Google Scholar]
- (81).Elmagbari NO; Egleton RD; Palian MM; Lowery JJ; Schmid WR; Davis P; Navratilova E; Dhanasekaran M; Keyari CM; Yamamura HI; et al. Antinociceptive Structure-Activity Studies with Enkephalin- Based Opioid Glycopeptides. Pharmacology, 2004, 311 (1), 290–297. [DOI] [PubMed] [Google Scholar]
- (82).Mabrouk OS; Falk T; Sherman SJ; Kennedy RT; Polt R CNS Penetration of the Opioid Glycopeptide MMP-2200: A Microdialysis Study. Neurosci. Lett, 2012, 531 (2), 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Rosa M; Gonzalez-Nunez V; Barreto-Valer K; Marcelo F; Sánchez-Sánchez J; Calle LP; Arévalo JC; Rodríguez RE; Jiménez-Barbero J; Arsequell G; Valencia G Role of the Sugar Moiety on the Opioid Receptor Binding and Conformation of a Series of Enkephalin Neoglycopeptides. Bioorganic Med. Chem, 2017, 25 (7), 2260–2265. [DOI] [PubMed] [Google Scholar]
- (84).Arsequell G; Salvatella M; Valencia G; Fernández-Mayoralas; Fontanella M; Venturi C; Jimenéz-Barbero J; Marrón E; Rodríguez RE Synthesis, Conformation, and Biological Characterization of a Sugar Derivative of Morphine that Is a Potent, Long-Lasting, and Nontolerant Antinociceptive. J. Med. Chem, 2009, 52, 2656–2666. [DOI] [PubMed] [Google Scholar]
- (85).Varamini P; Mansfeld FM; Blanchfield JT; Wyse BD; Smith MT; Toth I Synthesis and Biological Evaluation of an Orally Active Glycosylated Endomorphin-1. J. Med. Chem, 2012, 55 (12), 5859–5867. [DOI] [PubMed] [Google Scholar]
- (86).Fichna J; Mazur M; Grzywacz D; Kamysz W; Perlikowska R; Piekielna J; Sobczak M; Sałaga M; Toth G; Janecka A; et al. Novel Glycosylated Endomorphin-2 Analog Produces Potent Centrally-Mediated Antinociception in Mice after Peripheral Administration. Bioorganic Med. Chem. Lett, 2013, 23 (24), 6673–6676. [DOI] [PubMed] [Google Scholar]
- (87).Dhanasekaran M; Palian MM; Alves I; Yeomans L; Keyari CM; Davis P; Bilsky EJ; Egleton RD; Yamamura HI; Jacobsen NE; et al. Glycopeptides Related to β-Endorphin Adopt Helical Amphipathic Conformations in the Presence of Lipid Bilayers. J. Am. Chem. Soc, 2005, 127 (15), 5435–5448. [DOI] [PubMed] [Google Scholar]
- (88).Taylor JW; Kaiser ET The Structural Characterization of Beta-Endorphin and Related Peptide Hormones and Neurotransmitters. Pharmacol. Rev, 1986, 38 (4), 291–319. [PubMed] [Google Scholar]
- (89).Li Y; St. Louis L; Knapp BI; Muthu D; Anglin B; Giuvelis D; Bidlack JM; Bilsky EJ; Polt R Can Amphipathic Helices Influence the CNS Antinociceptive Activity of Glycopeptides Related to β-Endorphin? J. Med. Chem, 2014, 57 (6), 2237–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Lowery JJ; Yeomans L; Keyari CM; Davis P; Porreca F; Knapp BI; Bidlack JM; Bilsky EJ; Polt R Glycosylation Improves the Central Effects of DAMGO. Chem. Biol. Drug Des, 2007, 69 (1), 41–47. [DOI] [PubMed] [Google Scholar]
- (91).Lefever M; Li Y; Anglin B; Muthu D; Giuvelis D; Lowery JJ; Knapp BI; Bidlack JM; Bilsky EJ; Polt R Structural Requirements for CNS Active Opioid Glycopeptides. J. Med. Chem, 2015, 58 (15), 5728–5741. [DOI] [PubMed] [Google Scholar]
- (92).Tomatis R; Marastoni M; Balboni G; Guerrini R; Capasso A; Sorrentino L; Santagada V; Caliendo G; Lazarus LH; Salvadori S Synthesis and Pharmacological Activity of Deltorphin and Dermorphin-Related Glycopeptides. J. Med. Chem, 1997, 40, 2948–2952. [DOI] [PubMed] [Google Scholar]
- (93).Negri L; Lattanzi R; Tabacco F; Orrù L; Severini C; Scolaro B; Rocchi R Dermorphin and Deltorphin Glycosylated Analogues: Synthesis and Antinociceptive Activity after Systemic Administration. J. Med. Chem, 1999, 42 (3), 400–404. [DOI] [PubMed] [Google Scholar]
- (94).Negri L; Lattanzi R; Tabacco F; Scolaro B; Rocchi R Glycodermorphins: Opioid Peptides with Potent and Prolonged Analgesic Activity and Enhanced Blood-Brain Barrier Penetration. Br. J. Pharmacol, 1998, 124, 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Stevenson GW; Giuvelis D; Cormier J; Cone K; Atherton P; Krivitsky R; Warner E; St. Laurent B; Dutra J; Bidlack JM; Szabó L; Polt R; Bilsky EJ Behavioral pharmacology of the mixed-action delta-selective opioid receptor agonist BBI-11008: studies on acute, inflammatory and neuropathic pain, respiration, and drug self-administration. Psychopharmacology, 2020, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Lee HK; Zhang L; Smith MD; White HS; Bulaj G Glycosylated Neurotensin Analogues Exhibit Sub-Picomolar Anticonvulsant Potency in a Pharmacoresistant Model of Epilepsy. ChemMedChem, 2009, 4 (3), 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Al-Rodhan NRF; Richelson E; Gilbert JA; McCormick DJ; Kanba KS; Pfenning MA; Nelson A; Larson EW; Yaksh TL Structure-Antinociceptive Activity of Neurotensin and Some Novel Analogues in the Periaqueductal Gray Region of the Brainstem. Brain Res, 1991, 557 (1–2), 227–235. [DOI] [PubMed] [Google Scholar]
- (98).Dobner PR Neurotensin and Pain Modulation. Peptides, 2006, 27 (10), 2405–2414. [DOI] [PubMed] [Google Scholar]
- (99).Boules M; Li Z; Smith K; Fredrickson P; Richelson E Diverse Roles of Neurotensin Agonists in the Central Nervous System. Front. Endocrinol. (Lausanne), 2013, 4, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Lee HK; Zhang L; Smith MD; Walewska A; Vellore NA; Baron R; McIntosh JM; White HS; Olivera BM; Bulaj G A Marine Analgesic Peptide, Contulakin-G, and Neurotensin Are Distinct Agonists for Neurotensin Receptors: Uncovering Structural Determinants of Desensitization Properties. Front. Pharmacol, 2015, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Craig a G.; Norberg T; Griffin D; Hoeger C; Akhtar M; Schmidt K; Low W; Dykert J; Richelson E; Navarro V; Mazella J; Watkins M; Hillyard D; Imperial J; Cruz LJ; Olivera BM Contulakin-G, an O -Glycosylated Invertebrate Neurotensin. J. Biol. Chem, 1999, 274 (20), 13752–13759. [DOI] [PubMed] [Google Scholar]
- (102).Passos-Silva DG; Verano-Braga T; Santos RAS Angiotensin-(1–7): Beyond the Cardio-Renal Actions. Clin. Sci 2013, 124 (7), 443–456. [DOI] [PubMed] [Google Scholar]
- (103).Hay M; Polt R; Heien ML; Vanderah TW; Largent-Milnes TM; Rodgers K; Falk T; Bartlett MJ; Doyle KP; Konhilas JP A Novel Angiotensin-(1–7) Glycosylated MAs Receptor Agonist for Treating Vascular Cognitive Impairment and Inflammation-Related Memory Dysfunction. J. Pharmacol. Exp. Ther 2019, 369 (1), 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Malakoutikhah M; Teixidó M; Giralt E Toward an Optimal Blood-Brain Barrier Shuttle by Synthesis and Evaluation of Peptide Libraries. J. Med. Chem 2008, 51 (16), 4881–4889. [DOI] [PubMed] [Google Scholar]
- (105).Dhanasekaran M; Polt R New Prospects for Glycopeptide Based Analgesia: Glycoside-Induced Penetration of the Blood-Brain Barrier. Curr Drug Deliv. 2005, 2 (1), 59–73. [DOI] [PubMed] [Google Scholar]
- (106).Gaffney K; Weinberg M; Soto M; Louie S; Rodgers K Developmentof Angiotensin II (1–7) Analog as an Oral Therapeutic for the Treatment of Chemotherapy-Induced Myelosuppression. Haematologica. 2018, 103 (12), 567–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Bennion DM; Jones CH; Donnangelo LL; Graham JT; Isenberg JD; Dang AN; Rodriguez V; Sinisterra RDM; Sousa FB; Santos RAS; Sumners C Neuroprotection by Post-Stroke Administration of an Oral Formulation of Angiotensin-(1–7) in Ischaemic Stroke. Exp. Physiol 2018, 103 (6), 916–923. [DOI] [PubMed] [Google Scholar]
- (108).Wang Y; Qian C; Roks AJM; Westermann D; Schumacher SM; Escher F; Schoemaker RG; Reudelhuber TL; Van Gilst WH; Schultheiss HP; Tschöpe C; Walther T Circulating Rather than Cardiac Angiotensin-(1–7) Stimulates Cardioprotection after Myocardial Infarction. Circ. Hear. Fail 2010, 3 (2), 286–293. [DOI] [PubMed] [Google Scholar]
- (109).Hay M; Vanderah TW; Samareh-Jahani F; Constantopoulos E; Uprety AR; Barnes CA; Konhilas J Cognitive Impairment in Heart Failure: A Protective Role for Angiotensin-(1–7). Behavioral Neuroscience. 2017, 131 (1), 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Forte BL; Slosky LM; Zhang H; Arnold MR; Staatz WD; Hay M; Largent-Milnes TM; Vanderah TW Angiotensin-(1–7)/Mas Receptor as an Antinociceptive Agent in Cancer-Induced Bone Pain. Pain 2016, 157 (12), 2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Xue B; Zhang Z; Johnson RF; Guo F; Hay M; Johnson AK Central Endogenous Angiotensin-(1–7) Protects against Aldosterone/NaCl-Induced Hypertension in Female Rats. Am. J. Physiol. - Hear. Circ. Physiol 2013, 305 (5), 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Mordwinkin NM; Russell JR; Burke AS; Dizerega GS; Louie SG; Rodgers KE Toxicological and Toxicokinetic Analysis of Angiotensin (1–7) in Two Species. J. Pharm. Sci 2012, 101 (1), 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Rodgers KE; Oliver J; DiZerega GS Phase I/II Dose Escalation Study of Angiotensin 1–7 [A(1–7)] Administered before and after Chemotherapy in Patients with Newly Diagnosed Breast Cancer. Cancer Chemother. Pharmacol 2006, 57 (5), 559–568. [DOI] [PubMed] [Google Scholar]
- (114).Bunnemann B; Fuxe K; Metzger R; Mullins J; Jackson TR; Hanley MR; Ganten D Autoradiographic Localization of Mas Proto-Oncogene MRNA in Adult Rat Brain Using in Situ Hybridization. Neurosci. Lett 1990, 114 (2), 147–153. [DOI] [PubMed] [Google Scholar]
- (115).Zheng J; Li G; Chen S; Bihl J; Buck J; Zhu Y; Xia H; Lazartigues E; Chen Y; Olson JE Activation of the ACE2/Ang-(1–7)/Mas Pathway Reduces Oxygen-Glucose Deprivation-Induced Tissue Swelling, ROS Production, and Cell Death in Mouse Brain with Angiotensin II Overproduction. Neuroscience 2014, 273, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Regenhardt RW; Desland F; Mecca AP; Pioquinto DJ; Afzal A; Mocco J; Sumners C Anti-Inflammatory Effects of Angiotensin-(1–7) in Ischemic Stroke. Neuropharmacology 2013, 71, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Jiang T; Gao L; Guo J; Lu J; Wang Y; Zhang Y Suppressing Inflammation by Inhibiting the NF-IB Pathway Contributes to the Neuroprotective Effect of Angiotensin-(1–7) in Rats with Permanent Cerebral Ischaemia. Br. J. Pharmacol 2012, 167 (7), 1520–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Chang RL; Lin JW; Kuo WW; Hsieh DJ; Yeh YL; Shen CY; Day CH; Ho TJ; Viswanadha VP; Huang CY Angiotensin-(1–7) Attenuated Long-Term Hypoxia-Stimulated Cardiomyocyte Apoptosis by Inhibiting HIF-1 α Nuclear Translocation via Mas Receptor Regulation. Growth Factors, 2016, 34 (1–2), 11–18. [DOI] [PubMed] [Google Scholar]
- (119).Ferrario CM Angiotensin-Converting Enzyme 2 and Angiotensin-(1–7): An Evolving Story in Cardiovascular Regulation. Hypertension 2006, 47 (3 II), 515–521. [DOI] [PubMed] [Google Scholar]
- (120).Doobay MF; Talman LS; Obr TD; Tian X; Davisson RL; Lazartigues E Differential Expression of Neuronal ACE2 in Transgenic Mice with Overexpression of the Brain Renin-Angiotensin System. Am. J. Physiol. - Regul. Integr. Comp. Physiol 2007, 292 (1), 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Lazaroni TLN; Raslan ACS; Fontes WRP; de Oliveira ML; Bader M; Alenina N; Moraes MFD; dos Santos RA; Pereira GS Angiotensin-(1–7)/Mas Axis Integrity Is Required for the Expression of Object Recognition Memory. Neurobiol. Learn. Mem 2012, 97 (1), 113–123. [DOI] [PubMed] [Google Scholar]
- (122).Sumners C; Horiuchi M; Widdop RE; Mccarthy C; Unger T; Steckelings UM Protective Arms of the Renin-Angiotensin-System in Neurological Disease. Clin. Exp. Pharmacol. Physiol 2013, 40 (8), 580–588. [DOI] [PubMed] [Google Scholar]
- (123).Bardají E; Torres JL; Clapés P; Albericio F; Barany G; Rodríguez RE; Sacristán MP; Valencia G Synthesis and Biological Activity of O-Glycosylated Morphiceptin Analogues. J. Chem. Soc. Perkin Trans, 1991, 1 (7), 1755–1759. [Google Scholar]
- (124).Kihlberg J; Åhman J; Walse B; Drakenberg T; Nilsson A; Soderberg-ahlm C; Olsson H Glycosylated Peptide Hormones: Pharmacological Properties and Conformational Studies of Analogues of [1-Desamino,8-D-Arginine] Vasopressin. J. Med. Chem, 1995, 38, 161–169. [DOI] [PubMed] [Google Scholar]
- (125).Arsequell G; Rosa M; Mayato C; Dorta RL; Gonzalez-Nunez V; Barreto-Valer K; Marcelo F; Calle LP; Vázquez JT; Rodríguez RE; et al. Synthesis, Biological Evaluation and Structural Characterization of Novel Glycopeptide Analogues of Nociceptin N/OFQ. Org. Biomol. Chem, 2011, 9 (17), 6133–6142. [DOI] [PubMed] [Google Scholar]
- (126).Reinscheid RK; Nothacker H; Bourson A; Ardati A; Henningsen RA; Bunzow JR; Grandy DK; Langen H; Monsma FJ Jr..; Civelli O Orphain FQ : A Neuropeptide That Activates an Opioidlike G Protein-Coupled Receptor. Science., 1995, 270 (3), 792–794. [DOI] [PubMed] [Google Scholar]
- (127).Jansson AM; Jensen KJ; Meldal M; Lomako J; Lomako WM; Olsen CE; Bock K Solid-Phase Glycopeptide Synthesis of Tyrosine-Glycosylated Glycogenin Fragments as Substrates for Glucosylation by Glycogenin. J. Chem. Soc. - Perkin Trans, 1996, 1 (10), 1001–1006. [Google Scholar]
- (128).Rodriguez IR; Whelan WJ A Novel Glycosyl-Amino Acid Linkage: Rabbit-Muscle Glycogen is Covalently Linked to a Protein via Tyrosine. Biochemical and Biophysical Research Communications, 1985, 132 (2), 829–836 [DOI] [PubMed] [Google Scholar]
- (129).Campbell DG; Cohen P The amino acid sequence of rabbit skeletal muscle glycogenin. Eur. J. Biochem, 1989, 185 (1), 119–125. [DOI] [PubMed] [Google Scholar]
- (130).Alonso MD; Lomako J; Lomako WM; Whelan WJ Tyrosine-194 of glycogenin undergoes autocatalytic glucosylation but is not essential for catalytic function and activity. FEBS Letters, 1994, 342, 38–42. [DOI] [PubMed] [Google Scholar]
- (131).Ballet S; Betti C; Novoa A; Tömböly C; Uhd Nielsen C; Helms HC; Lesniak A; Kleczkowska P; Chung NN; Lipkowski AW; Brodin B; Tourwé D; Schiller PW In Vitro Membrane Permeation Studies and in Vivo Antinociception of Glycosylated Dmt1-DALDA Analogues. ACS Med. Chem. Lett, 2014, 5 (4), 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Yamamoto T; Nair P; Jacobsen NE; Vagner J; Kulkarni V; Davis P; Ma SW; Navratilova E; Yamamura HI; Vanderah TW; Porreca F; Lai J; Hruby VJ Improving Metabolic Stability by Glycosylation: Bifunctional Peptide Derivatives That Are Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J. Med. Chem, 2009, 52 (16), 5164–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Yamamoto T; Nair P; Davis P; Ma S; Navratilova E; Moye S; Tumati S; Lai J; Vanderah TW; Yamamura HI; Porreca F; Hruby VJ Design, Synthesis, and Biological Evaluation of Novel Bifunctional C-Terminal-Modified Peptides for δ/μ Opioid Receptor Agonists and Neurokinin-1 Receptor Antagonists. J Med. Chem, 2007, 50 (12), 2779–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Yamamoto T; Nair P; Jacobsen NE; Davis P; Ma SW; Navratilova E; Moye S; Lai J; Yamamura HI; Vanderah TW; Porreca F; Hruby VJ. The Importance of Micelle-Bound States for the Bioactivities of Bifunctional Peptide Derivatives for δ/μ Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J. Med. Chem, 2008, 51 (20), 6334–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Dangoor D; Biondi B; Gobbo M; Vachutinski Y; Fridkin M; Gozes I; Rocchi R Novel Glycosylated VIP Analogs: Synthesis, Biological Activity, and Metabolic Stability. J. Pept. Sci, 2008, 14 (10), 321–328. [DOI] [PubMed] [Google Scholar]
- (136).Tagashira M; Iijima H; Isogai Y; Hori M; Takamatsu S; Fujibayashi Y; Yoshizawa-Kumagaye K; Isaka S; Nakajima K; Yamamoto T; Teshima T; Toma K Site-Dependent Effect of O-Glycosylation on the Conformation and Biological Activity of Calcitonin. Biochemistry, 2001, 40 (37), 11090–11095. [DOI] [PubMed] [Google Scholar]
- (137).Azria M; Copp DH; Zanelli JM 25 Years of Salmon Calcitonin: From Synthesis to Therapeutic Use. Calcif. Tissue Int, 1995, 57 (6), 405–408. [DOI] [PubMed] [Google Scholar]
- (138).Ueda T; Tomita K; Notsu Y; Ito T; Fumoto M; Takakura T; Nagatome H; Takimoto A; Mihara SI; Togame H; Kawamoto K; Iwasaki T; Asakura K; Oshima T; Hanasaki K; Nishimura S; Kondo H Chemoenzymatic Synthesis of Glycosylated Glucagon-like Peptide 1: Effect of Glycosylation on Proteolytic Resistance and in Vivo Blood Glucose-Lowering Activity. J. Am. Chem. Soc, 2009, 131 (17), 6237–6245. [DOI] [PubMed] [Google Scholar]
- (139).Watanabe A; Nishijima KI; Zhao S; Tanaka Y; Itoh T; Takemoto H; Tamaki N; Kuge Y Effect of Glycosylation on Biodistribution of Radiolabeled Glucagon-like Peptide 1. Ann. Nucl. Med, 2012, 26 (2), 184–191. [DOI] [PubMed] [Google Scholar]
- (140).Goa KL; Balfour JA; Zuanetti G Lisinopril. Drugs 1996, 52 (4), 564–588. [DOI] [PubMed] [Google Scholar]
- (141).Andersson L; Blomberg L; Flegel M; Lepsa L; Nilsson B; Verlander M Large-Scale Synthesis of Peptides. Biopolymers. 2000, 55 (3), 227–250. [DOI] [PubMed] [Google Scholar]
- (142).Verlander M Industrial Applications of Solid-Phase Peptide Synthesis - A Status Report. Int. J. Pept. Res. Ther 2007, 13 (1–2), 75–82. [Google Scholar]
- (143).Wang LX; Amin MN Chemical and Chemoenzymatic Synthesis of Glycoproteins for Deciphering Functions. Chem. Biol 2014, 21 (1), 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Dawson PE; Muir TW; Kent SBH Synthesis of Proteins by Native Chemical Ligation. Science. 1994, 266 (November), 776–779. [DOI] [PubMed] [Google Scholar]
- (145).Haase C, Seitz O (2006) Chemical Synthesis of Glycopeptides. In: Wittmann V. (eds) Glycopeptides and Glycoproteins Topics in Current Chemistry, vol 267 Springer, Berlin, Heidelberg [Google Scholar]
- (146).Schmidt M; Toplak A; Quaedflieg PJLM; Ippel H; Richelle GJJ; Hackeng TM; van Maarseveen JH; Nuijens T Omniligase-1: A Powerful Tool for Peptide Head-to-Tail Cyclization. Adv. Synth. Catal 2017, 359 (12), 2050–2055. [Google Scholar]
- (147).Toplak A; Nuijens T; Quaedflieg PJLM; Wu B; Janssen DB Peptiligase, an Enzyme for Efficient Chemoenzymatic Peptide Synthesis and Cyclization in Water. Adv. Synth. Catal 2016, 358 (13), 2140–2147. [Google Scholar]
- (148).Lau JL, Dunn MK, Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem 2018, 26, 2700–2707. [DOI] [PubMed] [Google Scholar]
- (149).Baptista L,C; Sun Y; Carter CS; Buford TW Crosstalk Between the Gut Microbiome and Bioactive Lipids: Therapeutic Targets in Cognitive Frailty. Front. Nutr 2020, 7 (17), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Gyros Protein Technologies White Paper: Meeting the Challenge of Synthesizing Complex Therapeutic Peptides https://page.gyrosproteintechnologies.com/wp-meeting-the-challenge-of-synthesizing-complex-therapeutic-peptides. Web page accessed on 6/10/2020.