

SUMMARY
Programmable RNA editing is gaining momentum as an approach to repair mutations, but its efficiency in repairing endogenous mutant RNA in complex tissue is unknown. Here we apply this approach to the brain and successfully repair a guanosine-to-adenosine mutation in methyl CpG binding protein 2 RNA that causes the neurodevelopmental disease Rett syndrome. Repair is mediated by hippocampal injections of juvenile Mecp2317G>A mice with an adeno-associated virus expressing the hyperactive catalytic domain of adenosine deaminase acting on RNA 2 and Mecp2 guide. After 1 month, 50% of Mecp2 RNA is recoded in three different hippocampal neuronal populations. MeCP2 protein localization to heterochromatin is restored in neurons to 50% of wild-type levels. Whole-transcriptome RNA analysis of one neuronal population indicates that the majority of off-target editing sites exhibit rates of 30% or less. This study demonstrates that programmable RNA editing can be utilized to repair mutations in mouse models of neurological disease.
Graphical Abstract
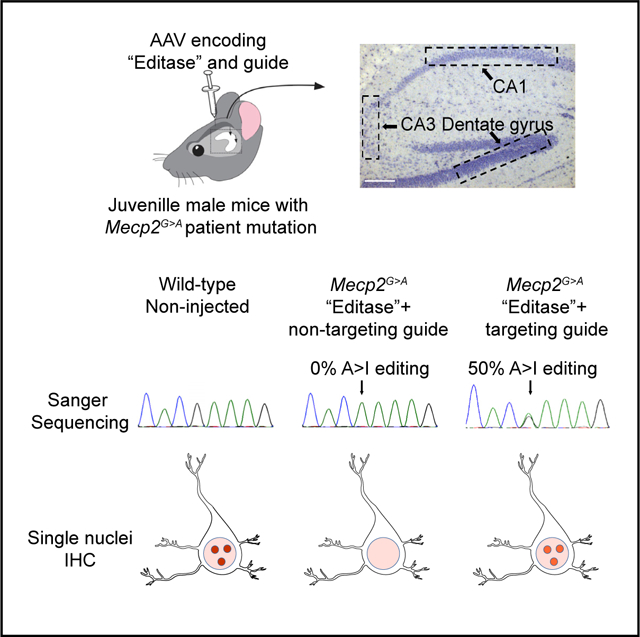
In Brief
Sinnamon et al. show that the Mecp2 guide-targeted “editase,” introduced by an adeno-associated virus into the hippocampus of a Rett syndrome mouse model, repairs a large fraction of pathological Mecp2G>A RNA. MeCP2 function, visualized by chromatin association in neurons, is repaired to similar levels. The results point toward a potential therapy for Rett syndrome.
INTRODUCTION
Adenosine deaminase enzymes catalyze the hydrolytic deamination of adenosine to inosine in RNA (A-to-I RNA editing) (Bass and Weintraub, 1987, 1988; Kim et al., 1994; Melcher et al., 1996; O’Connell et al., 1998). Inosine is generally treated as guanosine by the translational machinery (Basilio et al., 1962; Hoernes et al., 2018; Licht et al., 2019), resulting in codon changes with important consequences for protein function, particularly in the nervous system (Bhalla et al., 2004; Burns et al., 1997; Sommer et al., 1991). Two catalytically active adenosine deaminase acting on RNA enzymes, ADAR1 and ADAR2, are expressed at high levels in the CNS (Tan et al., 2017). The native proteins are recruited to their natural target RNAs, which can be overlapping or unique to each molecule (Burns et al., 1997; Maas et al., 1996; Melcher et al., 1996; Yang et al., 1997), by intrinsic domains that recognize double-stranded RNA (Kim et al., 1994; Melcher et al., 1996). Several pioneering studies have paved the way for programmable A-to-I editing of exogenous RNAs by harnessing the deaminase domains of ADAR1 or ADAR2 (Montiel-Gonzalez et al., 2013; Stafforst and Schneider, 2012; Woolf et al., 1995). Many of these studies utilized fusions of the ADAR deaminase domain with a heterologous RNA binding protein (for example, Cas13 or the bacteriophage λN peptide) with a RNA guide specific for the target RNA substrate (Cox et al., 2017; Katrekar et al., 2019; Montiel-Gonzalez et al., 2013, 2019; Montiel-González et al., 2016; Schneider et al., 2014; Sinnamon et al., 2017; Vogel et al., 2018). Endogenous RNAs have been repaired by programmable RNA editing in cell lines and primary cells (Cox et al., 2017; Merkle et al., 2019; Sinnamon et al., 2017; Vogel et al., 2018).
A recent study has demonstrated repair in mouse models of Duchenne muscular dystrophy and ornithine transcarbamylase deficiency (Katrekar et al., 2019), establishing the utility of this approach for muscle and the liver. However, both of these tissues have the advantage of relative cellular homogeneity. The large cellular complexity in the CNS raises the question of whether RNA structure and RNA binding proteins in different cell types will create a more or less accessible transcript for RNA repair by programmable editing. Therefore, direct tests in a mouse model of neurological disease are clearly warranted.
Mouse models of Rett syndrome are ideally suited to test the efficacy of programmable RNA editing in vivo. Rett syndrome is caused by de novo loss-of-function mutations in the gene encoding the X-linked transcriptional regulator MECP2 (Amir et al., 1999). MeCP2 pathological mutations result primarily in a neurological phenotype characterized in females by regression of speech and purposeful hand motions and the appearance of seizures and respiratory abnormalities (Neul et al., 2010). MeCP2 is expressed in most, if not all, neurons and glia (Ballas et al., 2009; Lioy et al., 2011; Rakela et al., 2018; Shahbazian et al., 2002; Skene et al., 2010), and previous studies have indicated that these cells can be targeted throughout the brain and spinal cord by adeno-associated virus 9 (AAV9) for facile analyses (Gadalla et al., 2013, 2017; Garg et al., 2013; Mendell et al., 2017; Sinnett et al., 2017). Importantly, a database of mutations causing Rett syndrome (Fyfe et al., 2003) indicates that 36% are caused by G>A mutations or C>T mutations that create opal stop codons, raising the possibility that adenosine deamination in these contexts may repair MeCP2 protein function. We previously generated a mouse line in which the mouse Mecp2 gene contained the human patient mutation MECP2317G>A (R106Q) (Sinnamon et al., 2017). This mutation, resulting in Rett syndrome, is located in the DNA binding domain. Consequently, MeCP2 protein becomes unstable and has a greatly reduced ability to bind to chromatin (Kudo et al., 2003; Sinnamon et al., 2017; Yang et al., 2016), easily quantifiable metrics for recovery of protein function.
We previously showed repair of Mecp2317G>A RNA and stabilization and chromatin binding of MeCP2 protein in neurons cultured from these mice. The neurons were infected with an AAV bearing a fusion between a mutated hyperactive ADAR2 deaminase domain and the bacteriophage λN peptide (“editase”; Montiel-Gonzalez et al., 2013). A nuclear localization signal had also been added to the editase to increase nuclear editing efficiency (Sinnamon et al., 2017; Vallecillo-Viejo et al., 2018). The editase was targeted to Mecp2 RNA with a guide that also included a sequence recognized by the bacteriophage λN peptide (Sinnamon et al., 2017). Despite efficient editing in primary neurons, whether the virus could deliver sufficient amounts of editase in vivo, whether different neuronal types would be equally accessible to directed RNA editing, and the extent of off-target editing remained open questions.
Here, using a Rett syndrome mouse model, we determine whether mutant Mecp2 RNA, in distinct neuronal subpopulations in the postnatal mouse brain, is accessible to programmable RNA repair and whether MeCP2 protein function is also restored. We also determine the on-and off-target editing landscape in one of these populations of neurons by whole-transcriptome analysis.
RESULTS
On-and Off-Target Editing Analyses of Mecp2 RNA
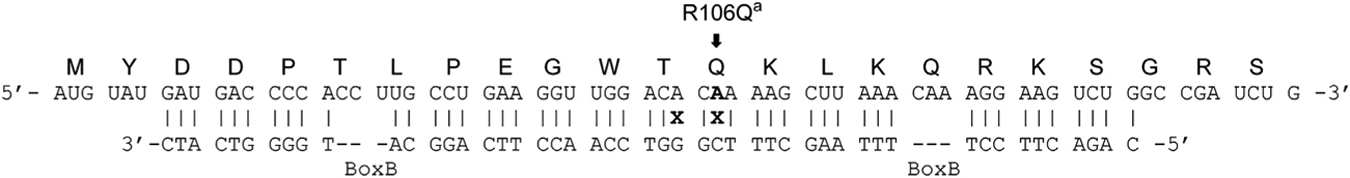
Six copies of a Mecp2 targeting guide or a non-targeting guide (Table S1) were cloned downstream of the U6 small nuclear RNA polymerase III promoter and upstream of the editase. The Mecp2 guide (Table 1) is complementary to Mecp2 RNA and contains two copies of the BoxB hairpin recognized by the λN RNA binding domain as well as an A-C mismatch at the target adenosine to increase editing efficiency (Källman et al., 2003; Schneider et al., 2014; Sinnamon et al., 2017; Wong et al., 2001). The guide also contains an additional A-G mismatch located 3 nt upstream of the target adenosine to diminish off-target editing at this site (Schneider et al., 2014; Sinnamon et al., 2017).
Table 1.
Editing of Adenosines within Mecp2 RNA Identified by Sanger Sequencing Analysis
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Target | ||||||||
| DG | Replicate | Injection Condition | E102G | E102E | T105A | T105T | R106Qa | K107R |
| 1 | 6xU6-Mecp2 targeting guide-hSynI editase | ND | 44.5% | ND | ND | 39.4% | ND | |
| 2 | 6xU6-Mecp2 targeting guide-hSynI editase | 10.2% | 41.3% | 15.3% | 9.4% | 57.9% | 9.6% | |
| 3 | 6xU6-Mecp2 targeting guide-hSynI editase | 7.8% | 39.2% | 8.3% | ND | 49.7% | ND | |
| 1 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| 2 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| 3 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| CA1 | Replicate | Injection Condition | E102G | E102E | T105A | T105T | R106Qa | K107R |
| 1 | 6xU6-Mecp2 targeting guide-hSynI editase | 5.8% | 64.7% | ND | ND | 63.9% | 64.7% | |
| 2 | 6xU6-Mecp2 targeting guide-hSynI editase | ND | 24.4% | 6% | ND | 39.6% | ND | |
| 3 | 6xU6-Mecp2 targeting guide-hSynI editase | ND | 18.3% | ND | ND | 52.4% | ND | |
| 1 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| 2 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| 3 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| CA3 | Replicate | Injection Condition | E102G | E102E | T105A | T105T | R106Qa | K107R |
| 1 | 6xU6-Mecp2 targeting guide-hSynI editase | 5.9% | 38% | 16.3% | 8.4% | 41% | 6.2% | |
| 2 | 6xU6-Mecp2 targeting guide-hSynI editase | 9.3% | 39.6% | 18.9% | 12.1% | 54.9% | 12.9% | |
| 3 | 6xU6-Mecp2 targeting guide-hSynI editase | 7.5% | 26.8% | 12.7% | 9.8% | 49.7% | ND% | |
| 1 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| 2 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
| 3 | 1xU6-non-targeting guide-hSynI editase | ND | ND | ND | ND | ND | ND | |
Top: Mecp2 mRNA and the deduced primary amino acid sequence (top row) relative to the guide RNA (bottom row). x, mismatched adenosines. Bottom: rates of editing at the adenosines located within the guide region for each biological replicate. The detection limit for this assay was determined previously to be 5% editing (Sinnamon et al., 2017). Any sites that had 5% or less editing are listed as having no detectable editing (ND). There was no detectable editing in Mecp2 RNA outside of the guide region.
The target adenosine, Mecp2317 in the RNA strand (MeCP2R106Q).
Because the target adenosine in Mecp2317G>A is not in an ideal sequence context for ADAR2 editing (Eggington et al., 2011), we utilized a version of the editase that contains a mutation in the deaminase domain, E488Q, that results in hyperactivity (Kuttan and Bass, 2012; Sinnamon et al., 2017). The non-targeting guide lacks the BoxB RNA hairpins as well as any Mecp2 sequences. The hemagglutinin (HA) epitope-tagged editaseE488Q was placed under control of the human Synapsin I promoter to direct expression to neurons after stereotactic injection of the viruses (Figure 1A). We produced AAV vectors expressing each component by packaging these expression cassettes into the AAV PHP.B capsid.
Figure 1. Hippocampal Expression of RNA Editing Components following Stereotaxic Injection of Mecp2317G>A Mice.
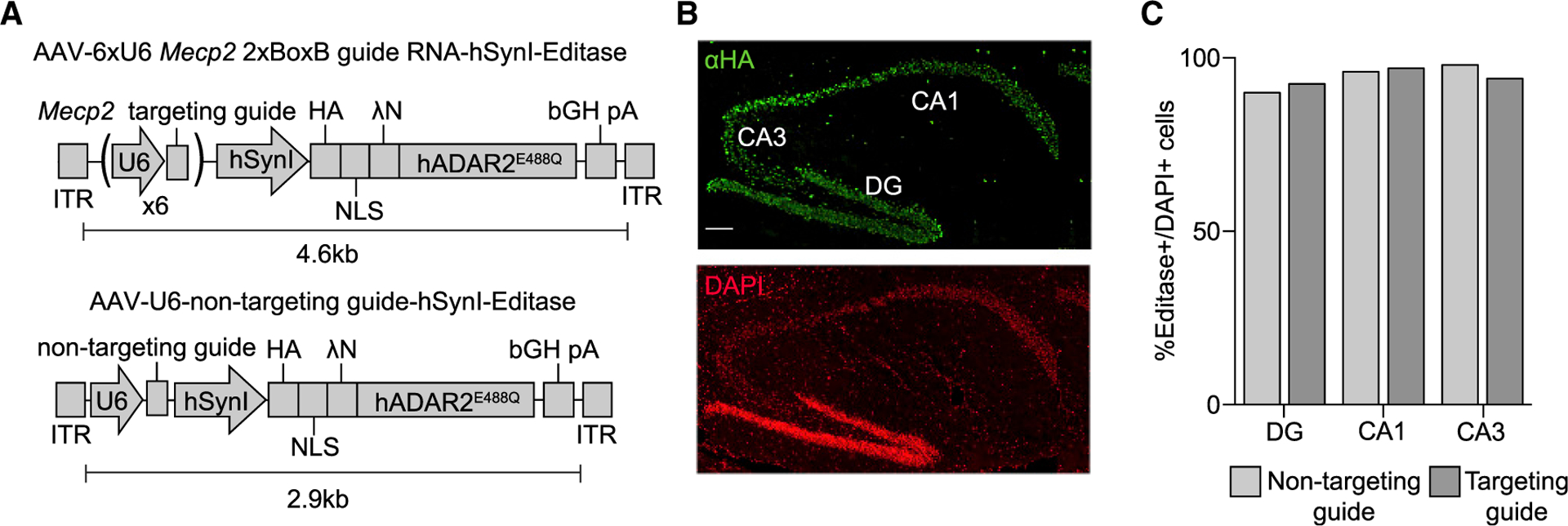
(A) Schematic of AAV editase expression vectors. Each construct contains the human Synapsin I promoter for neuronal editase expression and either six individual U6 promoters, each driving expression of one copy of the Mecp2 2xBoxB targeting guide (top) or a single human U6 promoter driving expression of a small non-targeting RNA (bottom).
(B) Confocal images of a Mecp2317G>A mouse 3 weeks after hippocampal injection of the AAV PHP.B vector. HA immunostaining identifies the editase in the dentate gyrus (DG) and CA1 and CA3 pyramidal neuronal layers. Scale bar, 100 μm.
(C) Quantification of HA-editase-positive cells for each virus relative to the total number of cells in each region (mean, n = 2 mice per condition). More than 100 cells were counted per hippocampal region per replicate.
We first established expression of the editase by immunohistochemistry for the HA epitope tag after stereotaxic injection of both vectors into the hippocampus. We observed editase expression from both vectors in the major neuronal populations of the hippocampus (Figures 1B and 1C). We then determined on-and off-target A-to-I editing rates within Mecp2 by Sanger sequencing analysis. RNA was isolated from the intact hippocampus as well as from different subpopulations of hippocampal neurons (CA1, CA3, and dentate gyrus) isolated by laser capture micro-dissection from each of three mice. In the isolated intact hippocampus, we measured 35% ± 7% A-to-I editing at the target adenosine with the Mecp2 guide, with negligible editing observed with the non-targeting guide (Figure 2A).
Figure 2. Efficient Editing of Mecp2 RNA following Hippocampal Injection of Mecp2317G>A Male Mice (Post-natal Day 28 [P28]).
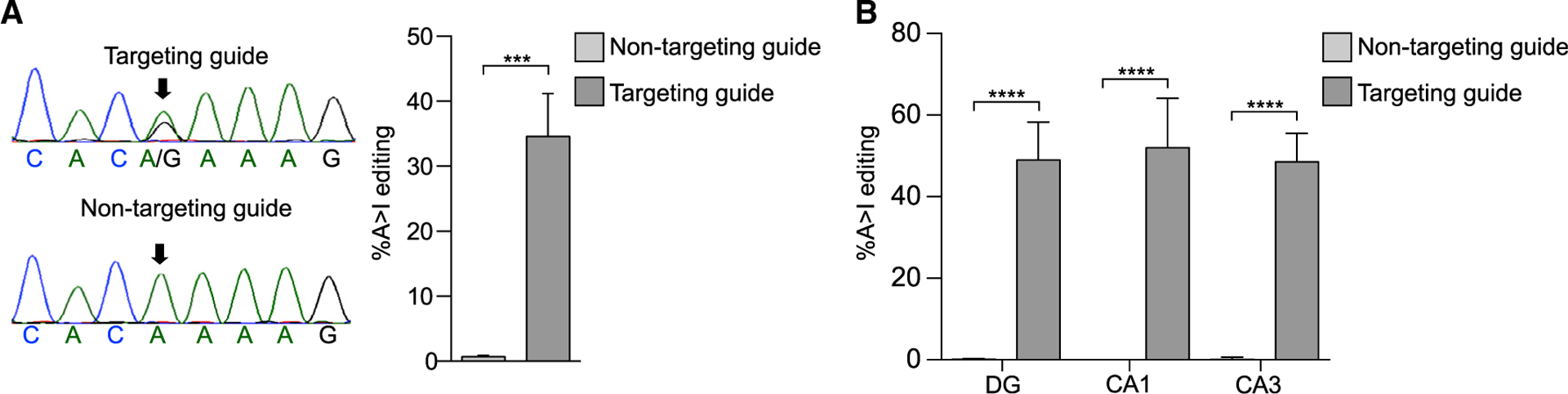
(A) Left: sequencing chromatograms of cDNA from an intact hippocampus injected with the editase and the indicated guides 3 weeks after viral injection. An arrow denotes the on-target base. Right: quantification of editing (mean ± SD, n = 3 mice per condition). ***p < 0.01 unpaired two-tailed t test.
(B) Quantification of editing in hippocampal neurons following laser capture micro-dissection. Mean ± SD, n = 3 mice/condition 3 weeks after viral injection. ****p < 0.001, one-way ANOVA and Tukey’s multiple comparisons test.
Remarkably, when cDNA was prepared from the individual hippocampal neuronal populations, the mean editing rates at the target adenosine varied only minimally, between 49% and 52% (dentate gyrus [DG], 49% ± 9%; CA1, 52% ± 12%; CA3, 49% ± 7%). Editing was again negligible with the non-targeting guide (Figure 2B). External to the guide region, no off-target sites were detected within the Mecp2 transcript. Within the guide region, we detected five bystander off-target editing sites, only one of which (E102E) was consistently edited at rates above background across neuronal populations and individual mice (18%–65%; Table 1). The bystander site that contained an A-G mismatch in the guide, T105T, based on our previous work, showed only minimal editing over our 5% background (DG, 6% ± 4%; CA1, 4% ± 3%; CA3, 10% ± 2%). These results demonstrate the efficacy of adding mismatches to diminish bystander editing efficiency in vivo as well as in vitro (Schneider et al., 2014; Sinnamon et al., 2017). We addressed the question of off-target editing rates in the wild-type allele because female Rett syndrome models are mosaic for wild-type and mutant cells. For this purpose, we injected the hippocampus of a wild-type mouse, where all cells have the wild-type allele, with the same virus used for the mutant mice (Table S2). We found very similar bystander sites and rates in these injected mice, with no other off-target editing within the rest of the Mecp2 transcript. This result suggests that off-target editing will be similar between the wild-type and mutant Mecp2 alleles in females, but it needs to be tested formally in female Rett syndrome mice in future studies.
Because the editing rates were so similar among the different neuronal populations, we next performed a whole-transcriptome RNA sequencing (RNA-seq) analysis on one of the neuronal populations, DG, to determine A-to-I on-and off-target editing rates (Figure 3). Whole-exome sequencing was also performed on the same samples. Single-nucleotide polymorphisms and edited sites in the non-injected control samples, presumably reflecting endogenous ADAR activity, were removed from the results presented here. Importantly, the on-target and off-target editing rates within the guide region matched those from our Sanger sequencing analysis using the same RNA. Additionally, editing sites were not detected anywhere in the Mecp2 RNA in mice infected with the non-targeting guide virus. These results support our RNA-seq bioinformatics pipeline.
Figure 3. Off-Target Editing Rates Determined by Whole-Transcriptome Analysis in the DG Are Graded and Depend on the Level of Editase Expression.
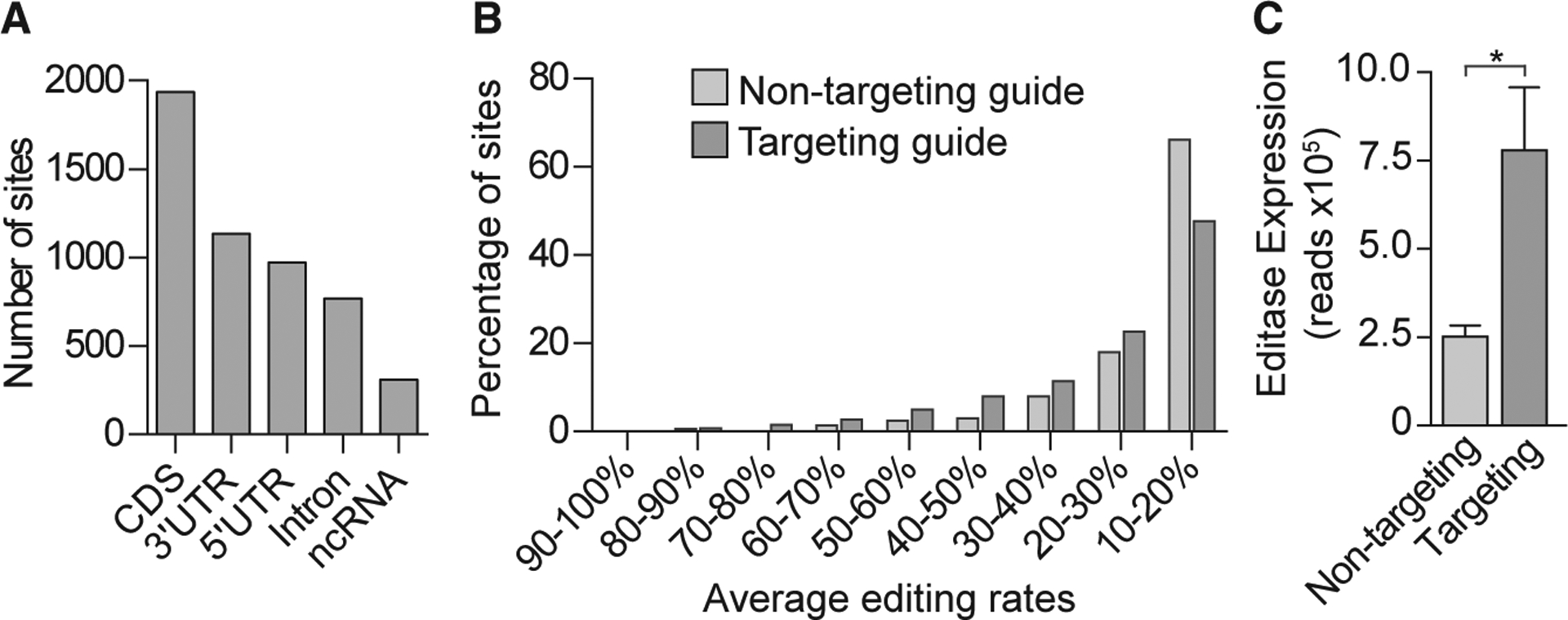
(A) Histogram showing the number of total off-target sites, independent of injection condition, located in coding sequences (CDSs), 3’ untranslated regions (UTRs), 5’ UTRs, and non-coding RNA (ncRNA).
(B) Histogram showing the percentage of transcriptome-wide RNA editing sites, binned according to the average editing rates (n = 3 biological replicates.
(C) Editase RNA-seq reads (mean ± SD, n = 3 mice/condition) that aligned to the editase CDS for each injection condition. *p < 0.05, two-tailed unpaired t test.
Global Off-Target Analysis
We identified 2,984 off-target sites from mice injected with the Mecp2 targeting guide virus and 909 off-target sites from mice injected with the non-targeting guide virus. The off-target sites under both conditions were distributed throughout the primary transcript (Figure 3A). When the number of sites and the percentages of A-to-I editing were considered for targeting and non-targeting guides, a majority (70% and 84%, respectively) represented editing at rates of 30% or less (Figure 3B). Consistent with a previous study (Cox et al., 2017), we found that nearly all (97%) of the off-target sites under the non-targeting condition were included in the sites under the Mecp2 targeting condition and that off-target editing was influenced by editase levels (Figures 3B and 3C). To confirm that on-target editing of Mecp2 RNA was guide dependent and independent of editase levels, we performed a new RNA-seq analysis on the same RNA from our previous neuronal culture study (Sinnamon et al., 2017), where, fortuitously, the editase level was higher in cells infected with the non-targeting virus (Figure S1A). Despite the higher editase levels under the non-targeting condition, there was no on-target editing without the Mecp2 targeting guide (Figure S1B). Further, off-target editing was influenced by the editase expression level (Figure S1C). The U6 promoter number did not influence editase protein levels in the transfection analysis, suggesting that variability in editase levels in neuronal cultures and in vivo reflect variability in viral infection parameters (Figure S2).
Repair of MeCP2 Protein Function
The Mecp2317G>A mutation destabilizes MeCP2 protein in vitro (Sinnamon et al., 2017; Yang et al., 2016) and in vivo (Figures 4A–4C) and results in greatly diminished binding to methylated DNA (Kudo et al., 2003; Sinnamon et al., 2017; Yang et al., 2016). We wanted to find out, at the single-cell level, whether the broadly distributed off-target editing sites we detected would prevent MeCP2 protein function; for example, by causing further destabilization, altering chromatin, or preventing nuclear entry. Because MeCP2 binds to methylated DNA that is enriched in mouse satellite sequences in the pericentromeric heterochromatin, enrichment in heterochromatin has been an in vivo proxy for MeCP2 DNA binding ability (Brown et al., 2016; Heckman et al., 2014). Therefore, we used confocal microscopy to quantitate the enrichment of MeCP2 within regions of interest in neuronal heterochromatin foci in the hippocampus of two Mecp2317G>A mice for each viral condition (STAR Methods). Two non-injected wild-type mice were used as controls. We analyzed the same fields—CA1, CA3, and DG—that were used for Sanger sequencing analyses (Figure 2B). Neither the number of DAPI- labeled heterochromatin puncta per nucleus nor the average size of heterochromatic puncta was altered between virus-infected and wild-type nuclei. In all fields, for CA3 and DG sections, both mice infected with a virus expressing the Mecp2 targeting guide showed heterochromatic enrichment of MeCP2 protein (see Figures 4A and 4D and 4C and 4F). Further, the amount of MeCP2 within heterochromatin relative to wild-type MeCP2 was nearly the same as the amount of on-target editing by Sanger and whole transcriptomic sequencing (compare Figures 4D and 4E and 2B). In contrast, in all neurons infected with the non-targeting virus, MeCP2 protein was destabilized and not enriched in heterochromatin (Figures 4A–4C).
Figure 4. Editase Programmable RNA Editing Restores the Ability of MeCP2 to Associate with Heterochromatin in Mecp2317G>A Mice.
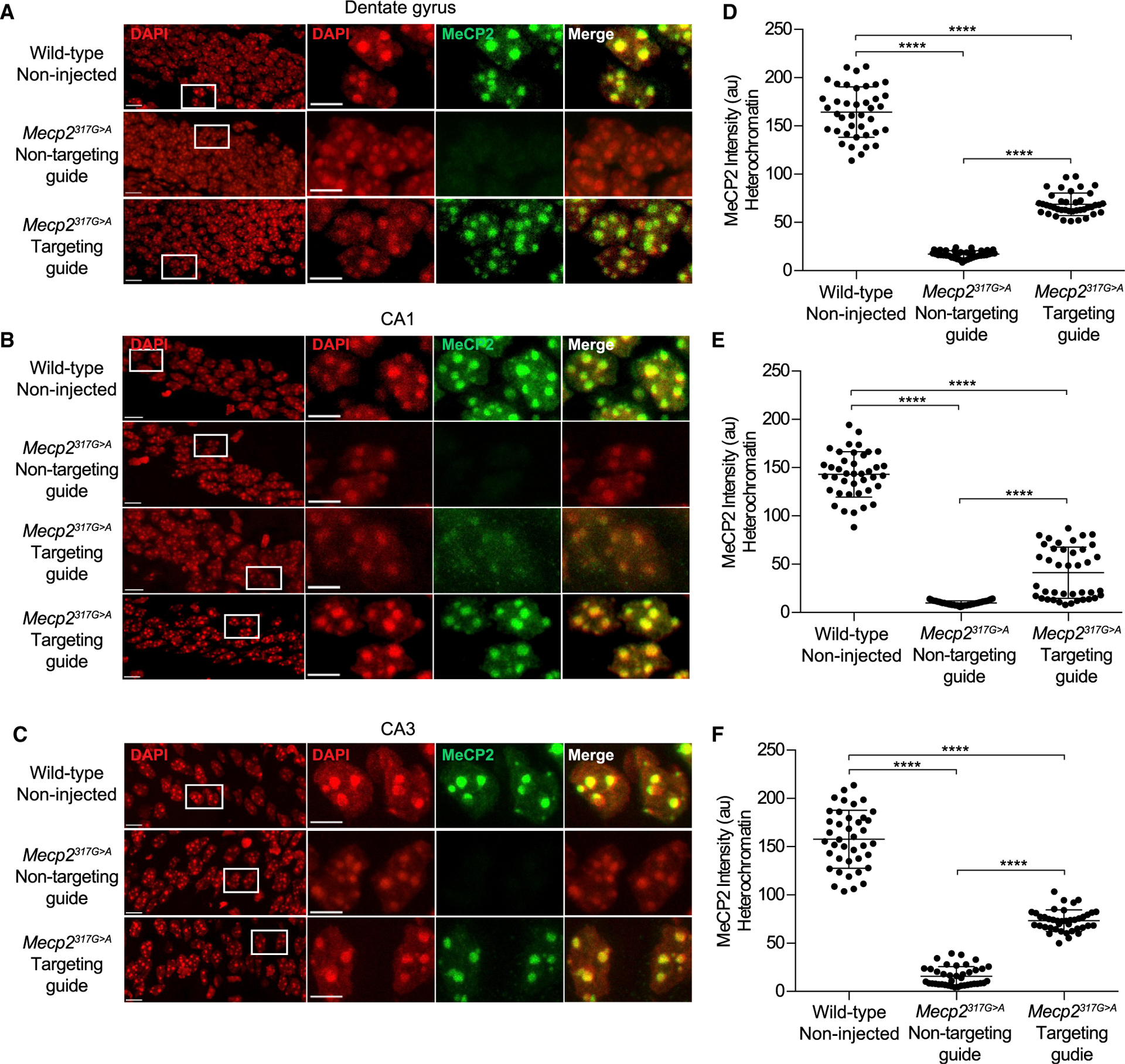
(A–C) Confocal images of hippocampal neuronal nuclei immunolabeled for MeCP2. DAPI staining defines the nucleus and heterochromatic foci. Boxes enclose regions of higher magnification. Scale bars, 10 μm for higher-power images and 5 mm for lower-power images. All images were acquired at the same intensity measurements.
(D–F) Quantification of immunolabeled MeCP2 associated with heterochromatic foci (STAR Methods). Each dot represents a single cell (40 cells total from two mice).
(A) and (D) show dentate granule neurons. (B) and (E) show CA1 pyramidal neurons. (C) and (F) show CA3 pyramidal neurons. a.u., arbitrary units. ****p < 0.0001 by Kruskal-Wallis test and Dunn’s multiple comparisons test.
Interestingly, one mouse injected with the Mecp2 targeting virus did not show MeCP2 enrichment within the heterochromatin of CA1 neurons, although the editase was expressed and enrichment was robust in the CA3 and DG in the same mouse. This result is reflected in the bimodal distribution of heterochromatin-associated MeCP2 in the Mecp2 targeting guide condition of CA1 neurons (Figures 4B and 4E). We cannot measure RNA repair and immunohistochemistry in the same mouse for technical reasons. However, the lack of functional MeCP2 in this mouse may reflect the presence of a CA1-specific off-target site that was detected in one of the three additional injected mice that were tested for RNA editing by Sanger analysis (Table 1). The bystander off-target site was located 3 nt 3’ of the target adenosine and recoded the codon from lysine to arginine. Because this change occurred in the DNA binding domain, it could potentially prevent binding of MeCP2 to chromatin, although it is not a mutation causing Rett syndrome (Fyfe et al., 2003). Perhaps the off-target editing event in CA1 detected by Sanger sequencing and the lack of MeCP2 enrichment in the heterochromatin of CA1 neurons were consequences of more virus being delivered to the CA1 region of those mice because of inconsistencies in injection placement. Regardless, because the off-target site was within the guide region, in future constructs the aberrant editing can be eliminated by adding an A:G mismatch at this site in the guide RNA (Sinnamon et al., 2017; Schneider et al., 2014), which we showed to be effective at the bystander off-target T105T in this study (Table 1).
DISCUSSION
Genomic and RNA base editing are potential therapeutic approaches for treating human disease (Gaudelli et al., 2017; Komor et al., 2016; Montiel-Gonzalez et al., 2019). Genomic editing has been utilized to successfully repair a gene responsible for hearing loss in mice (Yeh et al., 2018). We focused on RNA base editing because it does not have the same sequence constraints as DNA base editing. Additionally, as shown here, rates of off-target editing are graded with programmable RNA editing (Figure 3; Cox et al., 2017; Vallecillo-Viejo et al., 2018). Earlier work has demonstrated the potential of site-directed RNA editing for treating human disease. For example, heterologous expression in Xenopus oocytes of an editase system repaired a chloride channel mutation underlying cystic fibrosis (Montiel-Gonzalez et al., 2013). More recently, recoding of a pathogenic mutation in PINK1, associated with Parkinson’s disease, was achieved by full-length ADAR2-mediated editing in HeLa cells (Wettengel et al., 2017), and A-to-I and C-to-U recoding of 11 endogenous genes was achieved using an ADAR2-Cas13-guided system in HEK293 cells (Abudayyeh et al., 2019; Cox et al., 2017). A demonstration of successful in vivo RNA editing was published recently using G>A mutant mouse models of Duchenne muscular dystrophy and ornithine in vivo transcarbamylase deficiency (Katrekar et al., 2019). Our study demonstrates that in vivo programmable RNA editing can also efficiently edit target RNA in heterogeneous nervous tissue, resulting in functional repair of a patient mutation in mice representing a human neurological disease.
Importantly, and in contrast to the only other in vivo study of programmable editing, we also examine on- and off-target editing efficiencies in vivo in a tissue with cellular heterogeneity. Although on-target editing was efficient and fairly uniform among the three different neuronal populations, which differ anatomically and functionally, two types of off-target editing occurred: bystander, within the guide region, and transcriptome-wide. Within the guide region, we identified only one bystander off-target site that was edited consistently across the neuronal populations, but editing did not alter the amino acid (E102E). Given that an A:G mismatch at another bystander off-target site, T105T, significantly reduced the editing rate in vivo, subsequent guides should include a mismatch at this site as well. Even though neither bystander off-target editing changed the amino acid, mismatches at these sites will have the benefit of preventing promiscuous inosine decoding (Hoernes et al., 2018; Licht et al., 2019), in the case that decoding also occurs at a reasonable frequency in non-dividing neurons, as reported for dividing cells. Regarding off-target editing within the whole transcriptome, as described previously (Cox et al., 2017; Katrekar et al., 2019; Vallecillo-Viejo et al., 2018), the hyperactive editase resulted in global off-target editing within neurons in the brain. Importantly, only off-target editing was dependent on the level of editase expression, consistent with a previous in vitro study (Cox et al., 2017). An advantage of this finding is that future experiments to evaluate rescue of Rett syndrome-like phenotypes in mice will result in lower levels of editase per cell because of peripheral virus delivery, which is necessary to infect the entire brain. Additionally, several groups are already beginning to identify editase molecules with higher specificity and efficiency (Cox et al., 2017; Monteleone et al., 2019) for in vivo testing.
Despite the off-target editing, the efficient on-target editing rates determined by Sanger and whole-transcriptome sequencing in neurons in this study were consistent with the amount of heterochromatin-associated MeCP2 estimated from our imaging experiments (compare Figures 4D–4F and 2B). Although the presence of immunolabeled MeCP2 in heterochromatin is an indirect measurement of MeCP2 DNA binding ability, many previous studies have indicated that it is an excellent proxy based on comparisons with in vitro binding studies (Brown et al., 2016; Goffin et al., 2011; Heckman et al., 2014). We noted that the variability of MeCP2 heterochromatin association in mutant neurons in mice infected with the Mecp2 targeting guide was significantly less than that of native MeCP2 in wild-type neurons (DG, p < 0.0001; CA1, p = 0.0025; CA3 p < 0.0001). This result could reflect a still unidentified compensatory mechanism in the heterochromatin in mutants from loss of MeCP2 from inception, which reduces the accessible number of MeCP2 binding sites. Further studies examining programmable editase:guide editing at earlier time points may shed light on this intriguing observation.
Our results showed a uniformity of 50% editing and a comparable association of MeCP2 protein with heterochromatin across several hippocampal neuronal subtypes. This result suggests that, using peripheral injections, neuronal populations across the brain should share a similar repair rate. With peripheral injections, comprehensive behavioral testing combined with quantitative measurements of MeCP2 protein function and gene expression are possible and will need to be performed in male and female Rett syndrome mouse models. How much repaired MeCP2 per cell is necessary and how many neuronal and glial cells need to be repaired to reverse Rett syndrome phenotypes in mice is not known. Previous reports indicate that the mouse and human nervous systems are very sensitive to the levels of MeCP2 expression (Kerr et al., 2008; Samaco et al., 2008), and even a 2-fold increase in MeCP2 levels results in a neurological phenotype (Collins et al., 2004; Van Esch et al., 2005). Thus, although it is unlikely that 50% repair per cell will result in a wild-type mouse, as evaluated by the above metrics, this level of repair may be reasonably expected to result in significant improvement in Rett syndrome-like phenotypes in treated mice, and, importantly, programmable editing will never lead to overexpression of MeCP2.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the lead contact, Gail Mandel (mandelg@ohsu. edu)
Materials Availability
Reagents generated in this study are available from the Lead Contact with the completion of a Materials Transfer Agreement.
Data and Code Availability
RNA-seq and exome sequencing data files have been uploaded to the Sequencing Read Archive (SUB7012760).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies
All animal procedures were approved by the Oregon Health and Science University Institutional Animal Care and Use Committee. Mice were housed with littermates on a 12:12 light/dark cycle. The generation of Mecp2317G > A mice and genotyping protocols have been described previously (Sinnamon et al., 2017).
Cell lines
Human embryonic kidney HEK293 cells (AAV-293, Agilent, cat# 240073; RRID: CVCL_6871) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Lonza, cat# BE12–614F) supplemented with 10% fetal bovine serum (FBS), L-glutamine and penicillin-streptomycin. Mouse neuro-2A cells (ATCC, cat# CCL-131; RRID: CVLCL-0470, were grown in DMEM (Thermo Scientific, cat # 11965092) supplemented with 10% FBS. All cell lines were kept at 37°C in a 5% CO2 humidified incubator.
METHOD DETAILS
Plasmid constructs
The creation of the plasmids containing the AAV vector genomes that were used for AAV vector production (pGM1258, pGM1186) was described previously (Sinnamon et al., 2017). Plasmid pGM1267 was generated by removing the six copies of the human U6 promoter and Mecp2 guide sequences from pGM1258 by restriction digest (NdeI/ApaI) and adding a single copy of the human U6 promoter and Mecp2 guide between these two sites. The single human U6-Mecp2 guide sequence was generated by PCR amplification from pGM1108 (Sinnamon et al., 2017) using primers to add NdeI/ApaI restriction sites. The guide sequences and the cloning primers are shown in Table S1. Plasmid DNA to generate viral vectors was prepared using the NucleoBond Xtra Maxi endotoxin free kit (Takara Bio, cat # 740424.10) prior to production. The AAV helper plasmid expressing the AAV2 Rep proteins and the AAV-PHP.B capsid protein (i.e., the AAV-PHP.B helper plasmid) is a derivative of the AAV9 helper plasmid, p5E18-VD2/9 (Gao et al., 2002) and was constructed by inserting a PHP.B 7-mer peptide-coding DNA sequence (Deverman et al., 2016) into the wild-type AAV9 capsid protein open-reading frame between the amino acid positions 588 and 589.
AAV vectors
AAV vectors used in the study were produced in human embryonic kidney HEK293 cells (AAV-293, Agilent, RRID: CVCL_6871) by an adenovirus-free plasmid transfection method and purified by two rounds of cesium chloride (CsCl) density-gradient ultracentrifugation followed by dialysis as described elsewhere (Earley et al., 2017). To package AAV vector genome in the AAV.PHP.B capsids, we used the AAV-PHP.B helper plasmid as described above. The purified AAV vectors were in PBS with 5% sorbitol (w/v) and 0.001% Pluronic F-68 (v/v). The titer of each AAV vector was determined using quantitative dot blot using a probe generated against the Editase-coding sequence.
Stereotaxic injections
P28-P35 Mecp2317G>A and wild-type male mice were deeply anesthetized with 4% isoflurane (v/v) and stabilized in a custom stereotaxic apparatus, modified from a David Kopf system. After being placed in the apparatus, mice were kept under 2% isoflurane (v/v) for the remainder of the surgery. A dental drill was used to make holes in the skull and each hippocampal hemisphere was injected using a pulled glass micropipette (diameter 10–15 mm) backfilled with AAV. Injections were made at the following coordinates relative to Bregma: medial-lateral (ML): 1.40 mm, anterior-posterior (AP): 1.50 mm at depths of 1.50 and 1.65 mm; ML: 1.75 mm and AP: 2.25 mm at depths of 1.75 mm and 2.00 mm. At each location 2.75 3 109 viral genomes of virus were delivered. Following injections animals were allowed to recover on a heated pad prior to being returned to their home cage.
Laser capture microdissection
Three weeks after stereotaxic injection, Mecp2317G>A mice were anesthetized by intraperitoneal injection of 2,2,2-tribromoethanol (Sigma Aldrich, cat# T48402) and sacrificed by decapitation. Whole brains were washed in ice cold phosphate buffered saline (PBS), embedded in Tissue Freezing Medium (Electron Microscopy Sciences, cat: #72592) and stored at −80°C. Sagittal sections (12 μm) were cut at −25°C using a cryostat and loaded on poly (L) lysine (Sigma Aldrich, cat# P2636) coated PEN 1.0 membrane slides (Zeiss, cat #415190-9041-000). Immediately after sectioning, slides were fixed in 70% ethanol, stained with an abbreviated hematoxylin staining protocol, and stored at −80°C. Pyramidal cells from the CA1 and CA3 regions of the hippocampus along with dentate granule neurons were isolated for RNA analysis and cerebellar tissue was isolated for whole-exome sequencing using the Zeiss Palm Microbeam system.
Sanger sequencing analysis
Whole hippocampal tissue and laser captured hippocampal fields were isolated from male mice three weeks post stereotaxic injection. Total RNA from intact hippocampal tissue was isolated using Trizol reagent (Thermo Fisher Scientific, cat#15596026) according to the manufacturer’s instructions. RNA was isolated from laser captured cell populations using the RNeasy Micro kit (QIAGEN, Cat# 74004) according to the manufacturer’s instructions. All samples were tested for RNA purity using a Bioanalyzer 2100 and the Agilent RNA 6000 pico kit (Agilent, cat# 5067–1513). All samples had integrity scores of > 8.5. RNA was reverse transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen, cat# 18080051) and primed using oligo dT. Endogenous Mecp2 cDNA was amplified for analysis using a forward primer in the 5’ untranslated region and a reverse primer located in the 3’ untranslated region PCR products were fractionated on a 1% agarose gel and purified using the QIAquick gel extraction kit (QIAGEN, cat# 28706) before being submitted for Sanger sequence analysis. Sanger sequencing was performed using an Applied Biosystems 3730xl 96-capillary DNA analyzer. All primers are listed in Table S1. The C/T peak heights of the antisense strand were quantified from the resulting four-dye-trace sequences using the Bioedit software package (www.mbio.ncsu.edu/BioEdit/bioedit.html; RRID: SCR_007361) as previously described (Eggington et al., 2011; Sinnamon et al., 2017).
Quantification of editing rates was performed using the antisense strand because A/G peaks have more inconsistent heights (Nurpeisov et al., 2003). All chromatographs in the figures are shown as the reverse complement to show the mixed peaks at the target adenosine.
Whole transcriptomic analysis
cDNA libraries were made by the OHSU Massively Parallel Sequencing Shared Resource using the SMARTer RNA kit (Clontech, cat# 63490). Library quality was assessed using a TapeStation 220 and libraries were quantified by qPCR using a KAPA Library Quantification kit (Roche, cat# 7960140001). Libraries were sequenced using 100-cycle paired-end runs on a HiSeq 2500. Whole genomic DNA was isolated from laser captured samples using a PureLink genomic DNA mini kit (Thermo Fisher Scientific, cat# K182001) and quantified using a TapeStation 220. Libraries were made from 50 ng of genomic DNA using a Seq-Cap Exome Plus capture kit (Roche, cat# 06740189001) and quantified by qPCR using a KAPA Library Quantification kit. Exome libraries were then sequenced using 100-cycle single-read runs on a HiSeq 2500.
Whole exome DNA sequencing results were aligned to the C57BL/6J reference genome using Bwa-mem 0.717 (RRID: SCR_010910). The RNA-seq results were aligned to the mm10 reference genome using Bowtie 1.2.2 (RRID: SCR_005476). Single nucleotide polymorphisms (SNPs) were defined as DNA sample calls, which had < 99% of the reads aligning to the reference nucleotide across all sequenced samples and were excluded from downstream analysis.
RNA editing events were identified by comparing the adenosine or thymine nucleotides from the reference DNA sequence to the RNA sequencing results using the REDItoolDnaRna workflow (Picardi and Pesole, 2013, RRID: SCR_012133) with the following parameters:
e,E exclude multihits for RNA-Seq, DNA-Seq
d, D exclude duplicates for RNA-Seq, DNA-Seq
p User pair concordant reads only (for RNA-Seq only)
u, U Consider mapping quality for RNA-Seq, DNA-Seq
m 20,20 Minimum mapping quality score for RNA-Seq, DNA-Seq
a,A6–0 Trim 6 bases up and 0 bases down per read for RNA-Seq, DNA-Seq
I,L Remove substitutions in homopolymeric regions
v1 Minimum number of reads supporting variation
n 0.0 Minimum editing frequency for RNA-Seq, DNA-Seq
To be considered for further analysis, an editing event had to be present in all three biological replicates from each sample type (non-targeting guide and targeting guide) but not present in the non-injected controls.
Immunostaining
Mice were anesthetized by intraperitoneal injection of 2,2,2-tribromoethanol (Sigma Aldrich, cat# T48402) and sacrificed by transcardial perfusion of PBS, followed by 4% depolymerized paraformaldehyde. Brains were removed and equilibrated in 30% sucrose overnight at 4°C before being embedded in Tissue Freezing Medium (Electron Microscopy Sciences, cat #72592) and stored at −80°C. Sagittal sections (14 μm) were cut at −20°C using a cryostat and stored at −20°C. Sections underwent antigen retrieval with −20°C acetone for 8 minutes followed by washes with water and PBS before treatment with boiling citrate buffer (10 mM sodium citrate, 0.1% Tween-20, pH 6.0) for 10 minutes. Sections were cooled to room temperature (RT) and incubated in blocking buffer containing PBST (0.01% Triton X-100 in PBS, pH 7.4) and 10% normal donkey serum (Jackson Immunoresearch labs, cat# 017-00-001) for 30 minutes at RT. Sections were incubated overnight at 4°C with rabbit anti-MeCP2 (rabbit mab D4F3, Cell Signaling, cat# 34456, 1:500) and rat anti-HA (rat mab 3F10; Roche cat# 1867432991, 1:200) antibodies diluted in blocking buffer.
Sections were washed 3x with PBST and incubated for 1 hour at RT with Alexa Fluor secondary antibodies (Thermo Fisher Scientific, donkey anti-rat 488 (cat# A-31573) and donkey anti-rabbit 647 (cat# A-31573), 1:500) diluted in blocking buffer. Sections were washed 3x with PBST, then washed again with PBS before being incubated with 300 nM 4’,6-diamidino-2-phenylindole (DAPI, Thermo Scientific, cat# D1306) in PBS for 20 minutes. After a final wash in PBS, sections were mounted using ProLong Gold antifade mountant (Thermo Fisher Scientific, cat# 936934), which was allowed to cure overnight.
Image Acquisition and Analysis
A Zeiss 710 laser scanning confocal microscope equipped with a 63x Plan-Apo objective and Zen Digital Imaging Softare (Zeiss, RRID:SCR_013672) was used to acquire sequential 1 μm optical sections for creation of Z stack images. The field size corresponded to 18211.5 mm2 with a resolution of 1024×1024 pixels. Fluorescence images corresponding to HA label (488 laser), MeCP2 label (633 nm laser) and DAPI (405 nm laser) were sequentially acquired. For each, the laser strength was set to sub-saturating levels corresponding to 0 to 255. These acquisition settings were then applied to all samples. The fraction of HA immuno-labeled cells was determined by counting the fraction of DAPI positive nuclei that were also HA positive. Cells were determined to be HA positive if the fluorescence intensity was above non-injected controls. At least 100 cells were counted in each region of the hippocampus (CA1, CA3 and dentate gyrus) for each mouse using the ImageJ cell counter plugin (National Institutes of Health; imagej.nih.gov/ij/, version 1.60_65 (32bit), RRID: SCR_003070) and two Mecp2317G>A mice were analyzed for each viral condition. As a proxy for the ability of MeCP2 to bind DNA, the MeCP2 intensity in pericentromeric heterochromatic foci was determined. Once again, the laser strengths for MeCP2 and DAPI were adjusted individually to fall within a non-saturating 0–255 range and the acquisition parameters were held constant for all subsequent measurements. Regions of heterochromatin selected for measurement were based on both size (R0.3 mm2) and average intensity (R80 on a scale of 0 to 255). Using ImageJ, the corresponding MeCP2 fluorescence intensity for 4 to 6 heterochromatic foci per cell was determined on the basis of 4 × 4 pixel ROIs. These values were then used to generate the average intensity value for each cell. For each of two animals, 20 cell averages were generated for each of 3 regions of the hippocampus (CA1, CA3 and dentate gyrus). This process was repeated for both viral conditions (wild-type non-injected, Mecp2317G>A targeting guide, Mecp2317G>A non-targeting guide). The resultant amplitude distributions (n = 40 cells per condition) were subject to statistical comparison using a Kruskal-Wallis test followed by a Dunn’s multiple comparisons test. Further, for each hippocampal region the variance of MeCP2 fluorescence intensity for each condition was subject to statistical testing using an F-test for equality of variances.
Transient Transfections and Cell Culture
Neuro2A cells (ATCC CCL-131) were maintained in DMEM (Thermo Fisher Technologies, cat # 11965092) in 10% FBS at 37°C in a 5% CO2 humidified incubator. For analysis of Editase protein expression, cells were seeded at a density of 1.25×105 cells per well of a 12-well plate. After 24 h, cells were transfected with 1 mg of plasmid DNA containing the human Synapsin I promoter expressing Editase and six copies of the Mecp2 targeting guide (pGM1258), one copy of the Mecp2 targeting guide (pGM1267) or one copy of the non-targeting guide (previously referred to as Editase alone, pGM1186). Plasmid DNA was transfected using a 2:1 ratio of Lipofectamine 2000 transfection regent (Thermo Fisher Scientific, cat#11668019) and DNA in Opti-MEM reduced serum media (Thermo Fisher Scientific, cat# 31985062).
Western Blotting
Transfected Neuro2A cells were lysed 72 hr after transfection using 100 μL of whole-cell lysis buffer (25 mM Tris, pH 7.6, 150 mM NaCl, 1% Igepal CA-630 (Sigma, cat# I8896), 1% deoxycholate, 0.1% SDS, protease inhibitor (Complete EDTA-free; Roche, cat # 11836170001), 1 mM beta-mercaptoethanol, and 250 units per mL Benzonase (Sigma-Aldrich, cat# E1014). Lysates were centrifuged at 16,000 × g for 10 min at 4°C and the soluble fraction isolated. Protein concentrations from the soluble fraction were measured using the Pierce BCA protein assay (Thermo Fisher Scientific, cat# 23225). 20 mg of protein lysate per sample was separated on NuPage 4%–12% Bis–Tris gels (Thermo Fisher Scientific, cat# NP0335BOX) in NuPAGE MES-SDS running buffer (Thermo Fisher Scientific, cat# NP002), and transferred onto a nitrocellulose membrane (GE Healthcare Life Sciences, cat# 45004001). Membranes were blocked with 3% bovine serum albumin in 1 × TBST (TBS with 0.05% Tween 20) for 1 h, then incubated with mouse anti-HA (1:000, Biolegend, cat# 901514, RRID: AB_2565336) and rabbit anti–Histone H3 (1:5000, Abcam, Cat# Ab1791; RRID: AB_302613) overnight at 4°C. After washing three times with 1 × TBST, blots were incubated with goat anti-mouse IgG DyLight 680 (1:10,000 dilution; Thermo Scientific, cat# 33518) and goat anti-rabbit IgG Dylight 800 (1:10,000 dilution; Thermo Scientific, cat# 33571) diluted in 3% BSA in 1xTBST for 1 h. Blots were imaged and quantified using the Odyssey Imaging System (LI-COR Biosciences).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests with the exception of the whole transcriptome analysis were performed using GraphPad Prism version 6.0e software (RRID: SCR_002798). The rate of A-to-I editing from whole hippocampal samples was compared between viral conditions using an unpaired t test and within isolated neuronal populations using one-way ANOVA followed by Tukey’s post hoc multiple comparisons tests. Western blots comparing the level of Editase protein and the number of RNA-seq reads aligning to the Editase coding sequence were compared using unpaired two-tailed t tests. The level of MeCP2 fluorescence in heterochromatic foci was compared within hippocampal regions using a Kruskal-Wallis test followed by a Dunn’s post hoc multiple comparisons test. The variance of MeCP2 fluorescence intensity was subjected to testing using an F-test for equality of variances. All experimental results are expressed as the mean ± the standard deviation.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-HA clone 3F10 | Roche | Cat# 11867432001; RRID: AB_2314622 |
| Rabbit monoclonal anti-MeCP2 | Cell Signaling | Cat# 34456; RRID: AB_2143849 |
| Mouse monoclonal Anti-HA | Biolegend | Cat# 901514; RRID: AB_2565336 |
| Rabbit polyclonal Anti-Histone H3 | Abcam | Cat# Ab1791; RRID: AB_302613 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21208; RRID: AB_2535794 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross- Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573; RRID: AB_2536183 |
| Goat anti-mouse IgG DyLight IR 680 | Thermo Fisher Scientific | Cat# 35518; RRID: AB_614942 |
| Goat anti-rabbit IgG Dylight IR 800 | Thermo Fisher Scientific | Cat#SA5–10036; RRID:AB_2556616 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TRIzol | Thermo Fisher Scientific | Cat# 15596026 |
| Poly(L) lysine hydrobromide | Sigma Aldrich | Cat# P2636 |
| 2,2,2-tribromoethanol | Sigma Aldrich | Cat# T48402 |
| 4’,6’-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | Cat#D1306 |
| Critical Commercial Assays | ||
| SMARTer RNA kit | Clonetech | Cat# 634940 |
| Seq-Cap Exome Plus capture kit | Roche | Cat# 06740189001 |
| KAPA Library Quantification kit | Roche | Cat# 7960140001 |
| NucleoBond Xtra Maxi Endotoxin Free Kit | Takara Bio | Cat #740424.10 |
| RNeasy Micro kit | QIAGEN | Cat# 74004 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Invitrogen Superscript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat# 18080051 |
| QIAquick gel extraction kit | QIAGEN | Cat# 28706 |
| Agilent RNA 6000 Pico Kit | Agilent | Cat#5067–1513 |
| PureLink Genomic DNA Isolation kit | Thermo Fisher Scientific | Cat#K182001 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668019 |
| Deposited Data | ||
| Whole transcriptome RNA-seq | This paper | SUB7012760 |
| Whole exome sequencing | This paper | SUB7012760 |
| Experimental Models: Cell Lines | ||
| HEK293 Cells (AAV-293) | Agilent | cat# 240073; RRID: CVCL_6871) |
| Neuro-2A Cells (N2A) | ATCC | Cat#CCL-131; RRID: CVLCL-0470 |
| Experimental Models: Organisms/Strains | ||
| Mecp2317G > A; C57BL/6J mice | Sinnamon et al., 2017 | NA |
| C57BL/6J | The Jackson Laboratory | JAX Stock 000664 |
| Oligonucleotides | ||
| Guide sequences; see Table S1 | This paper | NA |
| PCR and sequencing primers; see Table S1 | This paper | NA |
| Primers for cloning of pGM1267; see Table S1 | This paper | NA |
| Recombinant DNA | ||
| Plasmid: P5E18-VD2/9 | Gao et al., 2002 | NA |
| Plasmid: PHP.B 7-mer peptide coding DNA sequence | Deverman et al., 2016 | NA |
| Plasmid: pGM1258 | Sinnamon et al., 2017 | NA |
| Plasmid: pGM1186 | Sinnamon et al., 2017 | NA |
| Plasmid: pGM1108 | Sinnamon et al., 2017 | NA |
| Plasmid: pGM1267 | This paper | NA |
| Software and Algorithms | ||
| ImageJ, Version 1.60_65 (32bit) | https://imagej.nih.gov/ij/ | RRID: SCR_003070 |
| Bioedit Software Package | https://bioedit.software.informer.com/7.2/ | RRID: SCR_007361 |
| Zen Digital Imaging Software | Zeiss | RRID: SCR_013672 |
| Bwa-mem 0.717 | http://bio-bwa.sourceforge.net | RRID: SCR_010910 |
| Bowtie 1.2.2. | https://sourceforge.net/projects/bowtie-bio/files/bowtie/1.2.2 | RRID: SCR_005476 |
| REDItoolDNARNA | Picardi and Pesole 2013 | RRID: SCR_012133 |
| Graph Pad Prism 6.0e | Graph Pad | RRID: SCR_002798 |
| Other | ||
| Tissue Freezing Medium | Electron Microscopy Sciences | Cat# 72592 |
| Pen 1.0 membrane slides | Zeiss | Cat# 415190-9041-000 |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | Cat# P36934 |
| Normal Donkey Serum | Jackson Immunoresearch Labs | Cat# 017-00-001 |
Highlights.
Programmable RNA editing to repair neurological disease lacks in vivo evidence
A guide-programmed “editase” tests RNA editing in mice with a human MECP2G>A mutation
Editing repairs the Mecp2 RNA mutation and protein function across neuronal types
Off-target but not on-target editing increases with increased editase levels
ACKNOWLEDGMENTS
We thank Christine Schmidt-Weber and Justine Nguyen for mouse husbandry, Joshua Rosenthal (Marine Biological Laboratories, MA) for help and advice when applying the editase system to Rett syndrome, and James M. Wilson (University of Pennsylvania) for the p5E18-VD2/9 plasmid. Discussions and encouragement from people in the laboratories of Adrian Bird (Wellcome Trust Centre for Cell Biology, University of Edinburgh), Michael E. Greenberg (Harvard Medical School), and Paul Brehm (Vollum Institute, Oregon Health and Science University) are greatly appreciated. An NIH Director’s Transformative Research award (NS087726) to G.M., Joshua Rosenthal, Paul Brehm and John Adelman, NIH R01 NS088399 (to H.N.), and grants from the Rett Syndrome Research Trust (to G.M. and J.R.S.) funded this work.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107878.
DECLARATION OF INTERESTS
G.M. is a scientific co-founder of Vico Therapeutics. She holds equity but is not an employee, and her laboratory receives no Vico funding. G.M. and J.R.S. are co-inventors on provisional patent application PCT/US18/55029 filed by Oregon Health and Science University.
REFERENCES
- Abudayyeh OO, Gootenberg JS, Franklin B, Koob J, Kellner MJ, Ladha A, Joung J, Kirchgatterer P, Cox DBT, and Zhang F (2019). A cytosine deaminase for programmable single-base RNA editing. Science 365, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, and Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, and Mandel G (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci 12, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilio C, Wahba AJ, Lengyel P, Speyer JF, and Ochoa S (1962). Synthetic polynucleotides and the amino acid code. V. Proc. Natl. Acad. Sci. USA 48, 613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, and Weintraub H (1987). A developmentally regulated activity that unwinds RNA duplexes. Cell 48, 607–613. [DOI] [PubMed] [Google Scholar]
- Bass BL, and Weintraub H (1988). An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098. [DOI] [PubMed] [Google Scholar]
- Bhalla T, Rosenthal JJ, Holmgren M, and Reenan R (2004). Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat. Struct. Mol. Biol 11, 950–956. [DOI] [PubMed] [Google Scholar]
- Brown K, Selfridge J, Lagger S, Connelly J, De Sousa D, Kerr A, Webb S, Guy J, Merusi C, Koerner MV, and Bird A (2016). The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum. Mol. Genet 25, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, and Emeson RB (1997). Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387, 303–308. [DOI] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, and Zoghbi HY (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet 13, 2679–2689. [DOI] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, and Zhang F (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Bane-rjee A, Wu WL, Yang B, Huber N, Pasca SP, and Gradinaru V (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol 34, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley LF, Powers JM, Adachi K, Baumgart JT, Meyer NL, Xie Q, Chapman MS, and Nakai H (2017). Adeno-associated Virus (AAV) Assembly-Activating Protein Is Not an Essential Requirement for Capsid Assembly of AAV Serotypes 4, 5, and 11. J. Virol 91, e01980–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, and Bass BL (2011). Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun 2, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe S, Cream A, de Klerk N, Christodoulou J, and Leonard H (2003). InterRett and RettBASE: International Rett Syndrome Association databases for Rett syndrome. J. Child Neurol 18, 709–713. [DOI] [PubMed] [Google Scholar]
- Gadalla KK, Bailey ME, Spike RC, Ross PD, Woodard KT, Kalburgi SN, Bachaboina L, Deng JV, West AE, Samulski RJ, et al. (2013). Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol. Ther 21, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla KKE, Vudhironarit T, Hector RD, Sinnett S, Bahey NG, Bailey MES, Gray SJ, and Cobb SR (2017). Development of a Novel AAV Gene Therapy Cassette with Improved Safety Features and Efficacy in a Mouse Model of Rett Syndrome. Mol. Ther. Methods Clin. Dev 5, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, and Wilson JM (2002). Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 99, 11854–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, Foust KD, Kaspar BK, Bird A, and Mandel G (2013). Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci 33, 13612–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017). Programmable base editing of A●T to G●C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Allen M, Zhang L, Amorim M, Wang IT, Reyes AR, Mercado-Berton A, Ong C, Cohen S, Hu L, et al. (2011). Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci 15, 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman LD, Chahrour MH, and Zoghbi HY (2014). Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. eLife 3, e02676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoernes TP, Faserl K, Juen MA, Kremser J, Gasser C, Fuchs E, Shi X, Siewert A, Lindner H, Kreutz C, et al. (2018). Translation of non-standard codon nucleotides reveals minimal requirements for codon-anticodon interactions. Nat. Commun 9, 4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Källman AM, Sahlin M, and Ohman M (2003). ADAR2 A–>I editing: site selectivity and editing efficiency are separate events. Nucleic Acids Res. 31, 4874–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrekar D, Chen G, Meluzzi D, Ganesh A, Worlikar A, Shih YR, Varghese S, and Mali P (2019). In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 16, 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr B, Alvarez-Saavedra M, Sáez MA, Saona A, and Young JI (2008). Defective body-weight regulation, motor control and abnormal social interactions in Mecp2 hypomorphic mice. Hum. Mol. Genet 17, 1707–1717. [DOI] [PubMed] [Google Scholar]
- Kim U, Wang Y, Sanford T, Zeng Y, and Nishikura K (1994). Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. USA 91, 11457–11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, and Liu DR (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo S, Nomura Y, Segawa M, Fujita N, Nakao M, Schanen C, and Tamura M (2003). Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J. Med. Genet 40, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttan A, and Bass BL (2012). Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. USA 109, E3295–E3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht K, Hartl M, Amman F, Anrather D, Janisiw MP, and Jantsch MF (2019). Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res. 47, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, and Mandel G (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Melcher T, Herb A, Seeburg PH, Keller W, Krause S, Higuchi M, and O’Connell MA (1996). Structural requirements for RNA editing in glutamate receptor pre-mRNAs by recombinant double-stranded RNA adenosine deaminase. J. Biol. Chem 271, 12221–12226. [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, and Higuchi M (1996). A mammalian RNA editing enzyme. Nature 379, 460–464. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, et al. (2017). Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Merkle T, Merz S, Reautschnig P, Blaha A, Li Q, Vogel P, Wettengel J, Li JB, and Stafforst T (2019). Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol 37, 133–138. [DOI] [PubMed] [Google Scholar]
- Monteleone LR, Matthews MM, Palumbo CM, Thomas JM, Zheng Y, Chiang Y, Fisher AJ, and Beal PA (2019). A Bump-Hole Approach for Directed RNA Editing. Cell Chem. Biol 26, 269–277.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Gonzalez MF, Vallecillo-Viejo I, Yudowski GA, and Rosenthal JJ (2013). Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. USA 110, 18285–18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-González MF, Vallecillo-Viejo IC, and Rosenthal JJ (2016). An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 44, e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Gonzalez MF, Diaz Quiroz JF, and Rosenthal JJC (2019). Current strategies for Site-Directed RNA Editing using ADARs. Methods 156, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey ME, Schanen NC, Zappella M, et al. ; RettSearch Consortium (2010). Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol 68, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurpeisov V, Hurwitz SJ, and Sharma PL (2003). Fluorescent dye terminator sequencing methods for quantitative determination of replication fitness of human immunodeficiency virus type 1 containing the codon 74 and 184 mutations in reverse transcriptase. J. Clin. Microbiol 41, 3306–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MA, Gerber A, and Keegan LP (1998). Purification of native and recombinant double-stranded RNA-specific adenosine deaminases. Methods 15, 51–62. [DOI] [PubMed] [Google Scholar]
- Picardi E, and Pesole G (2013). REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814. [DOI] [PubMed] [Google Scholar]
- Rakela B, Brehm P, and Mandel G (2018). Astrocytic modulation of excitatory synaptic signaling in a mouse model of Rett syndrome. eLife 7, e31629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Fryer JD, Ren J, Fyffe S, Chao HT, Sun Y, Greer JJ, Zoghbi HY, and Neul JL (2008). A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum. Mol. Genet 17, 1718–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MF, Wettengel J, Hoffmann PC, and Stafforst T (2014). Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res. 42, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, and Zoghbi HY (2002). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet 11, 115–124. [DOI] [PubMed] [Google Scholar]
- Sinnamon JR, Kim SY, Corson GM, Song Z, Nakai H, Adelman JP, and Mandel G (2017). Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc. Natl. Acad. Sci. USA 114, E9395–E9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett SE, Hector RD, Gadalla KKE, Heindel C, Chen D, Zaric V, Bailey MES, Cobb SR, and Gray SJ (2017). Improved MECP2 Gene Therapy Extends the Survival of MeCP2-Null Mice without Apparent Toxicity after Intracisternal Delivery. Mol. Ther. Methods Clin. Dev 5, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, and Bird AP (2010). Neuronal MeCP2 is expressed at near his-tone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B, Köhler M, Sprengel R, and Seeburg PH (1991). RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19. [DOI] [PubMed] [Google Scholar]
- Stafforst T, and Schneider MF (2012). An RNA-deaminase conjugate selectively repairs point mutations. Angew. Chem. Int. Engl 51, 11166–11169. [DOI] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. ; GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration &Visualization—EBI; Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz (2017). Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallecillo-Viejo IC, Liscovitch-Brauer N, Montiel-Gonzalez MF, Eisenberg E, and Rosenthal JJC (2018). Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 15, 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. (2005). Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet 77, 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel P, Moschref M, Li Q, Merkle T, Selvasaravanan KD, Li JB, and Stafforst T (2018). Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 15, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettengel J, Reautschnig P, Geisler S, Kahle PJ, and Stafforst T (2017). Harnessing human ADAR2 for RNA repair -Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 45, 2797–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SK, Sato S, and Lazinski DW (2001). Substrate recognition by ADAR1 and ADAR2. RNA 7, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf TM, Chase JM, and Stinchcomb DT (1995). Toward the therapeutic editing of mutated RNA sequences. Proc. Natl. Acad. Sci. USA 92, 8298–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Sklar P, Axel R, and Maniatis T (1997). Purification and characterization of a human RNA adenosine deaminase for glutamate receptor B premRNA editing. Proc. Natl. Acad. Sci. USA 94, 4354–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kucukkal TG, Li J, Alexov E, and Cao W (2016). Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. ACS Chem. Biol 11, 2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WH, Chiang H, Rees HA, Edge ASB, and Liu DR (2018). In vivo base editing of post-mitotic sensory cells. Nat. Commun 9, 2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and exome sequencing data files have been uploaded to the Sequencing Read Archive (SUB7012760).