

Abstract
Acyclic cucurbit[n]uril molecular containers 1 and 2C3 have previously been shown to strongly bind to the neuromuscular blocking agents rocuronium, vecuronium, pancuronium, and cisatracurium in vitro by optical methods and to reverse neuromuscular block in vivo in rats. In this paper we study the in vitro binding of a panel of acyclic CB[n]-type receptors toward the four neuromuscular blocking agents and acetylcholine to develop structure-binding affinity relationships. The selected variants include those with different aromatic sidewalls (e.g. 1Me4 with dimethyl o-xylylene walls; 3 with 1,8-linked naphthalene walls), with different glycoluril oligomer lengths (e.g. 4 and 5 based on glycoluril trimer), and with different linker lengths between aromatic wall and SO3− solubilizing group (e.g. 2C2 – 2C4). Based on the analysis of complexation induced changes in 1H NMR chemical shift we conclude that the hydrophobic regions of the guests bind in the hydrophobic cavity of the hosts with the cationic moieties of the guest binding at the ureidyl C=O portals by ion-dipole and ion-ion interactions. The thermodynamic parameters of binding were determined by direct and competition isothermal titration calorimetry experiments. We find that hosts 4 and 5 based on glycoluril trimer form significantly weaker complexes with the streroidal NMBAs than with the analogues hosts based on glycoluril tetramer (1 and 2C3). Similarly, hosts 1Me4 and 3 with different length and height aromatic walls do not exhibit the extreme binding constants displayed by 2C3 but rather behave similarly to 1. Finally, we find that hosts 2C2 and 2C4 bind only slightly more weakly to the NMBAs than 2C3, but retain the ability to discriminate against acetylcholine, and possess higher inherent water solubility than 2C3. Host 2C4, in particular, holds potential for future in vivo applications.
Keywords: Cucurbituril, neuromuscular blocking agent, reversal agent, molecular container, drugs
Graphical Abstract

Introduction.
The administration of neuromuscular blocking agents (NMBAs) is an essential element of care given to over 400 million patients per year during surgical procedures in operating rooms, intensive care units, and emergency medicine departments.1 Specifically, NMBAs are used during general anesthesia to block neuromuscular transmission which prevents movement during surgery and optimizes surgical conditions. The NMBAs exert their function by binding to the Acetyl choline receptor (AChR) at the neuromuscular junction.1–2 The most widely used NMBAs in clinical practice are rocuronium (roc), vecuronium (vec), and cisatracurium (cis) (Figure 1).1 Unfortunately, an estimated 20–50% of patients that receive NMBAs experience postoperative respiratory disfunction that can lead to airway obstruction, hypoxia, and longer stays in the post-anesthesia care unit all of which increase the risk of morality and healthcare costs.3 Accordingly, strategies to reverse neuromuscular block at the end of surgery are important in clinical practice to control costs and improve patient outcomes. Classical reversal strategies involve the administration of small molecules like neostigmine and edrophonium that bind to and inhibit the activity of Acetylcholine esterase which increases the concentration of acetyl choline (ACh) at the neuromuscular junction to more effectively displace the NMBA from the AChR.4 An alternative strategy is to directly compete with the AChR for binding to the NMBA. This strategy was first implemented in the form of the γ-cyclodextrin derivative Sugammadex which displays high affinity binding to rocuronium (Ka = 1.05 × 107 M-1) in water in vitro.5 In vivo, Sugammadex reverses neuromuscular block by sequestering rocuronium and vecuronium in the bloodstream, thereby depleting their concentration at the neuromuscular junction, and promoting the clearance of the Sugammadex•rocuronium complex in the urine.6 Sugammadex has been having a major impact on the clinical practice of anesthesia in Europe since its approval in 2008, but was only approved by the US FDA in December 2015 after 4 concerns about allergic reactions and hemorragic side effects had been addressed. Worldwide sales of Sugammadex under the tradename BridionTM by Merck amounted to $704 million in 2017.7 Accordingly, when we started our work in this area in 2010 we saw a need to develop new classes of molecular containers that could act as broad spectrum reversal agents (e.g. roc, vec, and cis).8
Figure 1.
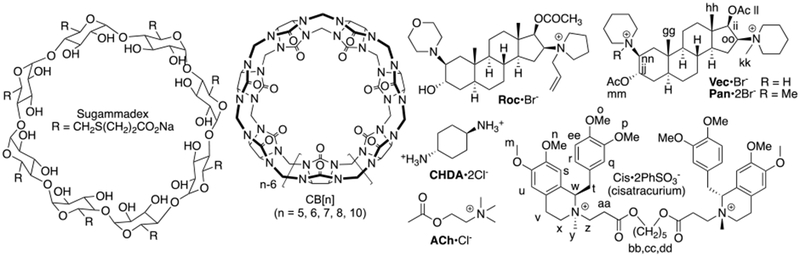
Chemical structures of Sugammadex, CB[n], and guest.
Our group has a long-standing interest in the synthesis and molecular recognition properties of macrocyclic cucurbit[n]uril (CB[n]) molecular containers (Figure 1).9 CB[n] are pumpkin shaped hosts that comprise n glycoluril units connected by 2n methylene brides that feature a hydrophobic cavity guarded by two symmetry equivalent electrostatically negative ureidyl C=O portals.10 Guest compounds that feature a hydrophobic region flanked by two ammonium moieties can form extraordinarily tight complexes (Ka up to 1017 M-1) with CB[n] hosts.11 Accordingly, we and others, have considered the use of CB[n]-type receptors as potential reversal agents for NMBAs.8a,12 Unfortunately, the CB[n] that are large enough to encapsulate the steroidal nucleus of roc and vec (CB[8] and CB[10]) display low μM solubility in water10b,13 which greatly limits their potential in vivo applications. CB[7] which does have good water solubility (20 mM)10b was shown by Macartney to bind to the ammonium end groups of steroidal NMBAs rather than engulf the steroid ring system.12b Over the years, our group has sought to understand the mechanism of CB[n] formation and use that knowledge to prepare new CB[n] type receptors with enhanced solubility and clickable functional groups.9b,14 For example, we prepared acyclic CB[n]-type receptors (e.g. 1 and 2, Figure 2) that comprise a central glycoluril tetramer, two aromatic sidewalls, and four sodium sulfonate groups to greatly improve aqueous solubility.15 Related receptors that lack the SO3 - solubilizing groups have also been studied by Sindelar and co-workers.16 Initially, we studied the ability of 1 and 2 to function as solubilizing agents for insoluble drugs and carbon nanotubes and as components of sensing arrays for pharmaceutical agents.15,17 As part of follow-up studies of the use of 1 and 2 to function as solubilizing excipients, we had cause to prepare numerous structural variants containing different numbers of glycoluril rings (1, 2, 3, 4),17d different aromatic walls,17b and different solubilizing group linker lengths18 including compounds 1 – 5 shown in Figure 2. In 2012, we reported that 1 and 2 bind to roc, vec, and cis in vitro with μM to nM binding affinity.8a In vivo experiments (rat) showed that 1 or 2 reverse the effects of roc, vec, and cis in a dose dependent manner and restores the train-of-four (TOF) ratio faster than placebo and neostigmine and comparable to or faster than Sugammadex.8b,8c As part of our efforts to further develop 2 as a broad spectrum reversal agent for NMBA reversal we studied the binding of 2 toward a panel of 27 drugs commonly used during or after surgery by experiment and simulation to assess the potential for displacement interactions that could lead to undesired recurarization.8d Most recently, we have investigated the ability of 1 and 2 to act as in vivo sequestration agents for drugs of abuse (e.g. methamphetamine).19 In a complementary line of inquiry, the group of Ruibing Wang has pursued the use of macrocyclic cucurbiturils as reversal agents.20 In this paper, we measure the binding affinity of a panel of previously prepared acyclic CB[n] type receptors (1 – 5) toward the panel of NMBAs (roc, vec, pan, cis) and acetylcholine (ACh) to more fully define the structure– binding affinity relationships.
Figure 2.
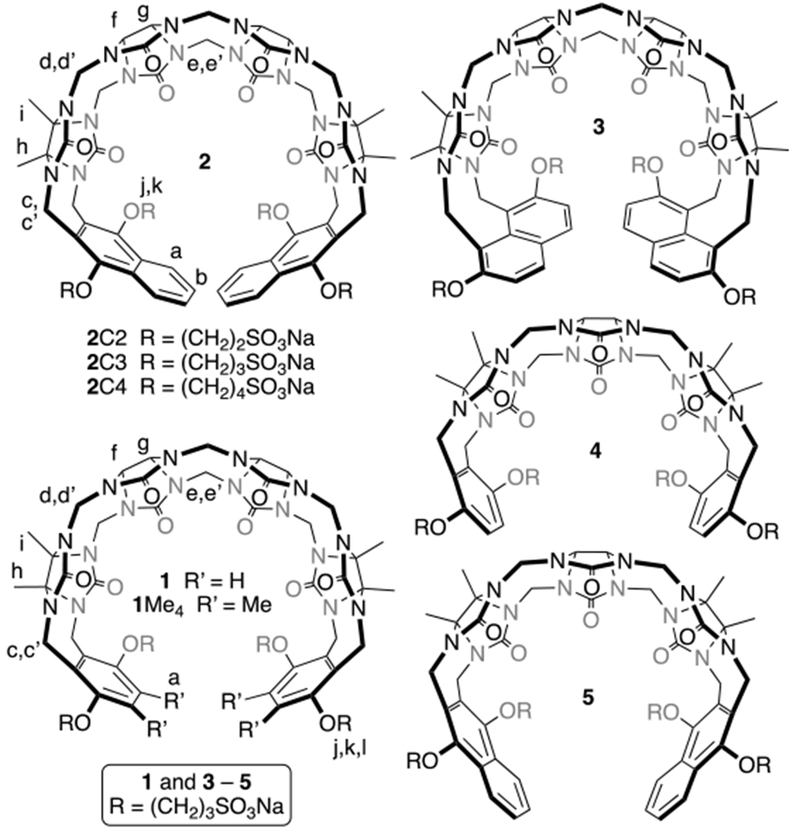
Chemical structures of acyclic CB[n]-type receptors used in this study.
Results and Discussion.
This results and discussion section is organized as follows. First, we describe the rationale for our selection of the eight different hosts used in this study. Next, we present the results of 1H NMR experiments that provide qualitative information on the geometry of the host•guest complexes. Subsequently, we present the results of isothermal titration calorimetry experiments used to measure the thermodynamic parameters for the various host•guest complexes. Finally, we discuss the trends in the Ka values as a function of host structure.
Selection of Hosts 1 – 5.
As described above, our investigations into the use of acyclic CB[n]-type compounds as reversal agents have focussed on lead compounds 1 and 2 due to their high binding affinity and their excellent biocompatibility in vitro and in vivo (e.g. high maximum tolerated dose, no hERG activity, no genotoxicity).15,18,21 In addition to 1 and 2, we also selected compounds 1Me4 and 3 that feature dimethyl o-xylylene and 2,7-dialkoxynaphthalene sidewalls. These structural changes influence the depth (e.g. 3 is deeper), the size (e.g. 1Me4 is intermediate in size between 1 and 2), and the nature (e.g. the aliphatic Me groups of 1 become part of the walls that define the molecular recognition surface) of the cavity. Finally, we selected compounds 4 and 5 that differ from 1 and 2 in that they are based on a central glycoluril trimer but possess common aromatic sidewalls.17d The cavities of hosts 4 and 5 are smaller than those of 1 and 2 and may be more complementary to the narrower diammonium region of cis.17d Finally, we selected compounds 2C2 and 2C4 which differ in the length of the alkylene linker between the aromatic sidewall and the solubilizing sulfonate groups.18 Previously, 2C2 (68 mM) and 2C4 (196 mM) have been shown to have higher aqueous solubility than 2 (14 mM) which would be advantageous for formulation if the binding constants of 2C2 or 2C4 were comparable to those of 2 toward the NMBAs.
1H NMR Investigations of the Host-Guest Complexes.
Initially, we performed qualitative 1H NMR investigations of the interactions between containers 1 – 5 and guests (roc, vec, pan, cis, ACh, and CHDA) to glean information about complex geometry and stoichiometry. It is well known that the ureidyl C=O region of macrocyclic CB[n] and acyclic CB[n]-type receptors constitute ammonium ion binding regions due to ion-dipole interactions.10a,22 Conversely, the cavity of macrocyclic CB[n] and acyclic CB[n]-type receptors constitutes a hydrophobic binding region; the cavity is also a magnetic shielding region by virtue of the π-systems of the glycoluril units and the aromatic sidewalls.9b,23 In accord with these expectations, the 1H NMR spectra of complexes between hosts 1 – 5 and guests ACh and CHDA (Supporting Information) feature significant upfield shifting for the methylene protons of ACh and CHDA which indicates they are located in the cavity of the host. Somewhat surprisingly, the NMe3 protons of ACh also undergo an upfield shifting which probably reflects an out-of-plane geometrical helical distortion of the hosts which allows the NMe3 + group to engage in cation-π interactions with the aromatic sidewalls of the hosts. Figure 3a–c show the 1H NMR spectra recorded for cis, 2C2, and an equimolar mixture of cis and 2C2. Upon formation of the 2C2•cis complex we observe a large upfield change in chemical shift for Hcc and Hdd which means that host resides in large part on the central (CH2)5 linker between the benzyl isoquinoline units as expected. Conversely, we observe a significant downfield change in chemical shift of Ha and Hb on the naphthalene sidewalls of 2C2 upon complex formation. We attribute this change to the presence of intramolecular edge-to-face π−π interactions between the naphthalene walls of uncomplexed 2C2 which are disrupted upon complex formation.17b Related downfield changes in chemical shift of the protons on the aromatic sidewalls are also seen for hosts 1, 2, and 3.17b For other host-guest combinations, multiple resonances are observed for the aromatic walls upon complexation. For example, upon formation of the 3•pan complex (Figure S54) we observe that the two symmetry equivalent aromatic protons (Ha and Hb) of C2v-3 split into 8 resonances which reflects the overall C1-symmetry of the 3•pan complex due to enantiomerically pure pan guest and slow guest exchange kinetics. For the 5•vec and 5•pan complexes (Figures S57 and S60) we observe splitting of the two aromatic resonances of C2v-5 (Ha and Hb) into four resonances (rather than eight) because fast guest exchange kinetics averages the two possible orientations (e.g. up or down) of vec or pan within the cavity of 5. Related increases in the number of resonances are observed for the CH2-groups of the glycoluril backbone of C2v-5 (e.g. two doublets from 5.0 – 5.5 ppm; Figure S57) upon formation of the 5•vec complex (e.g. four doublets from 5.0 – 5.6 ppm). Figure 3d–g show the 1H NMR spectra recorded for vec, 1Me4, and 1:1 and 1:2 mixtures of 1Me4 and vec. As expected, at a 1:1 ratio of 1Me4:vec, we observe significant upfield shifts for the axial steroidal CH3-groups (Hgg, Hhh) which indicates that the steroidal nucleus of vec is bound inside the cavity of 1Me4 which allows the pendant ammonium groups to engage in ion-ion and ion-dipole interactions with the C=O portals and SO3 - solubilizing groups of the host. At a 1:2 ratio of 1Me4:vec the resonances for the axial steroidal Me groups are broadened into the baseline which indicates that exchange processes with intermediate kinetics occur on the 1H NMR timescale. Figure 4 shows a cross-eyed stereoview of an MMFF minimized geometry of the 1Me4•vec complex which illustrates the three dimensional arrangement of vec inside the cavity of the host. Interestingly, the axial Me-groups point into the concavity shaped by the glycoluril tetramer whereas the axial steroidal C-H bonds on the α-face are oriented toward the aromatic walls of the host. Overall, our analysis of the changes in 1H NMR chemical shifts upon complexation are consistent with the complexation of the hydrophobic portions of the guests within the hydrophobic cavity of the acyclic CB[n]-type receptors with electrostatic interactions at the portals as expected based on the literature precedents.9b,22
Figure 3.
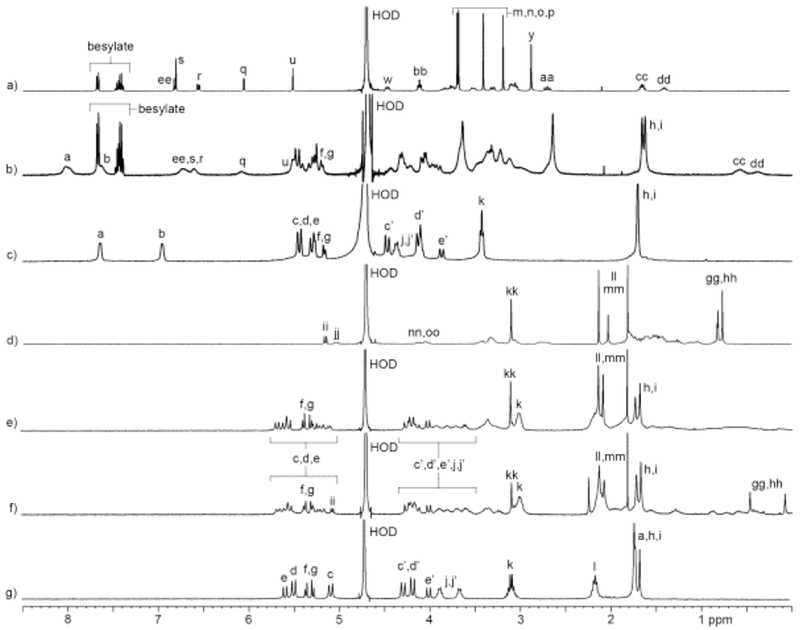
1H NMR spectra recorded (400 MHz, RT, D2O) for solutions containing: a) cis (1 mM), b) cis (1 mM) and 2C2 (1 mM), c) 2C2 (1 mM), d) vec (1 mM), e) 1Me4 (1 mM) and vec (2 mM), f) 1Me4 (1 mM) and vec (2 mM), and g) 1Me4 (1 mM).
Figure 4.
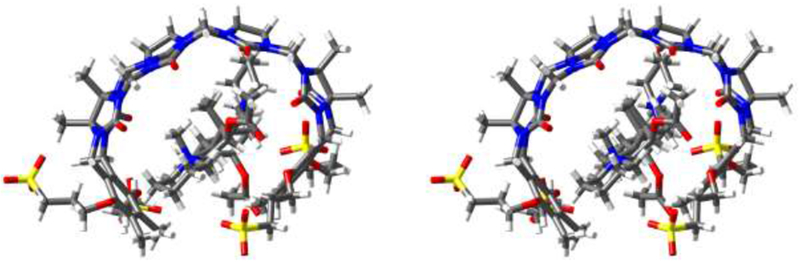
Cross-eyed stereoview of the MMFF minimized geometry of 1Me4•vec. Color code: C, grey; H, white; N, blue; O, red; S, yellow.
Determination of the Thermodynamics of Host•Guest Complexes by Isothermal Titration Calorimetry.
After having observed clear cavity binding of the NMBA guests within the cavity of hosts 1 – 5 by 1H NMR spectroscopy, we set out to determine the strength and thermodynamic parameters of the complexes. Given that the tight binding previously observed with 28a and the complexity and broadening observed in the 1H NMR of many of the complexes would complicate Ka determination by 1H NMR spectroscopy we decided to use ITC which would deliver the full thermodynamic parameters. Table 1 presents the Ka, Δ rG, ΔrH, and TΔrS values for the various host•guest complexes. For the weaker complexes (e.g. Ka ≤ 5 × 106 M-1) we were able to perform direct titrations of host with guest. For example, Figure 5a shows the thermogram obtained when a solution of 2C4 (70 μM) in the cell was titrated with a solution of CHDA (1 mM) in the syringe. Fitting of the data to a 1:1 binding model implemented within the PEAQ-ITC analysis software allowed a determination of Ka = 1.00 × 106 M-1 and ΔrH = −3.59 kcal mol-1; complexation is also entropically favourable with - TΔrS = −4.59 kcal mol-1. For complexes with higher Ka values, direct titrations are not reliable because the c-values are larger than 100024 Accordingly, to measure Ka > 107 M-1 we performed competitive ITC titrations. In competitive ITC titrations, a solution of host and weak binding guest (Ka and ΔrH previously determined) in the cell is typically titrated with a solution of the tighter binding guest.25 As the guest exchange process occurs, the difference in ΔrH between the two competing complexes is evolved so it is important to select the competitor wisely. We have found that CHDA is a suitable competitor in many cases. Figure 5c shows the thermogram obtained during the titration of a solution of 2C4 (70 μM) and CHDA (0.5 mM) with a solution of roc (1.08 mM) in the syringe. Figure 5d shows the fitting of the data to a competitive binding model using the known thermodynamic parameters for the 2C4•CHDA complex (Figure 5b) as inputs which allowed us to determine Ka = 1.32 × 109 M-1 and ΔrH = −17.1 kcal mol-1 for the 2C4•roc complex. The thermodynamic parameters for the remaining host•guest complexes (Table 1) were measured by analogous direct or competitive ITC titrations as documented in the Supporting Information.
Table 1.
Thermodynamic parameters (Ka (M−1), ΔrH, ΔrG, and - TΔrS (kcal mol−1) and determined for the host•guest complexes by ITC.
| Ka / 106 (M−1); ΔrH kcal mol−1; ΔrG (kcal mol−1); - TΔrS (kcal mol−1) | ||||||
|---|---|---|---|---|---|---|
| CHDA | ACh | roc | vec | pan | cis | |
| 1 Ka | 2.30 | 0.0229 | 3.51 | 4.2 | 0.758 | 0.132 |
| ΔrH | −5.69 | −6.87 | −8.64 | −5.56 | −4.35 | −16.4 |
| ΔrG | −8.68 | −5.95 | −8.93 | −9.05 | −8.02 | −6.99 |
| −2.99 | 0.917 | −0.291 | −3.49 | −3.67 | 9.40 | |
| − T ΔrS | ||||||
| 1Me4 Ka | 0.730 | 0.0302 | 2.51 | 5.41 | 1.41 | 0.0914 |
| ΔrH | −6.91 | −6.31 | −14.9 | −6.68 | −5.46 | −10.1 |
| ΔrG | −8.00 | −6.41 | −8.73 | −9.19 | −8.39 | −6.77 |
| −1.09 | −1.17 | 6.21 | −2.51 | −2.93 | 3.30 | |
| − T ΔrS | ||||||
| 2C2 Ka | 2.00 | 0.158 | 1090[a] | 862[a] | 148[a] | 0.13 |
| ΔrH | −6.64 | −8.32 | −16.1 | −9.29 | −8.69 | −13.2 |
| ΔrG | −8.60 | −7.09 | −12.3 | −12.2 | −11.1 | −6.83 |
| −1.96 | 1.23 | 3.74 | −2.90 | −2.46 | 6.35 | |
| − T ΔrS | ||||||
| 2C3 Ka | 2.49 | 0.179 | 5780[a] | 4020[a] | 800[a] | 0.128 |
| ΔrH | −5.05 | −7.27 | −13.5 | −9.33 | −9.31 | −13.3 |
| ΔrG | −8.73 | −7.17 | −13.3 | −13.1 | −12.1 | −6.97 |
| −3.68 | 0.102 | 0.201 | −3.77 | −2.84 | 6.34 | |
| − T ΔrS | ||||||
| 2C4 Ka | 1.00 | 0.0588 | 1320[a] | 231[a] | 200[a] | 0.194 |
| ΔrH | −3.59 | −4.57 | −17.1 | −6.26 | −6.11 | −10.9 |
| ΔrG | −8.19 | −6.51 | −12.4 | −11.4 | −11.3 | −7.21 |
| −4.59 | −1.94 | 4.65 | −5.15 | −5.21 | 3.64 | |
| − T ΔrS | ||||||
| 3 Ka | 0.14 | n.b. | 0.847 | 0.971 | 0.195 | 0.0971 |
| ΔrH | −4.79 | −9.05 | −4.15 | −4.68 | −8.30 | |
| ΔrG | −7.02 | −8.09 | −8.17 | −7.22 | −6.80 | |
| −2.24 | 0.967 | −4.02 | −2.53 | 1.49 | ||
| − T ΔrS | ||||||
| 4 Ka | n.d. | 0.000704 | 0.0214 | 0.0348 | 0.0382 | 0.0225 |
| ΔrH | −4.99 | −6.08 | −3.67 | −4.01 | −6.69 | |
| ΔrG | −3.88 | −5.91 | −6.20 | −6.25 | −5.94 | |
| 1.11 | 0.173 | −2.53 | −2.24 | 0.754 | ||
| − T ΔrS | ||||||
| 5 Ka | n.d. | 0.0015 | 1.31 | 0.98 | 0.588 | 0.0562 |
| ΔrH | −3.78 | −7.21 | −4.89 | −3.63 | −7.06 | |
| ΔrG | −4.33 | −8.35 | −8.18 | −7.87 | −6.48 | |
| −0.555 | −1.14 | −3.28 | −4.24 | 0.582 | ||
| − T ΔrS | ||||||
n.d.: not determined because competition experiments were not necessary. n.b.: no binding detected.
competition ITC titration using CHDA as competitor.
Figure 5.
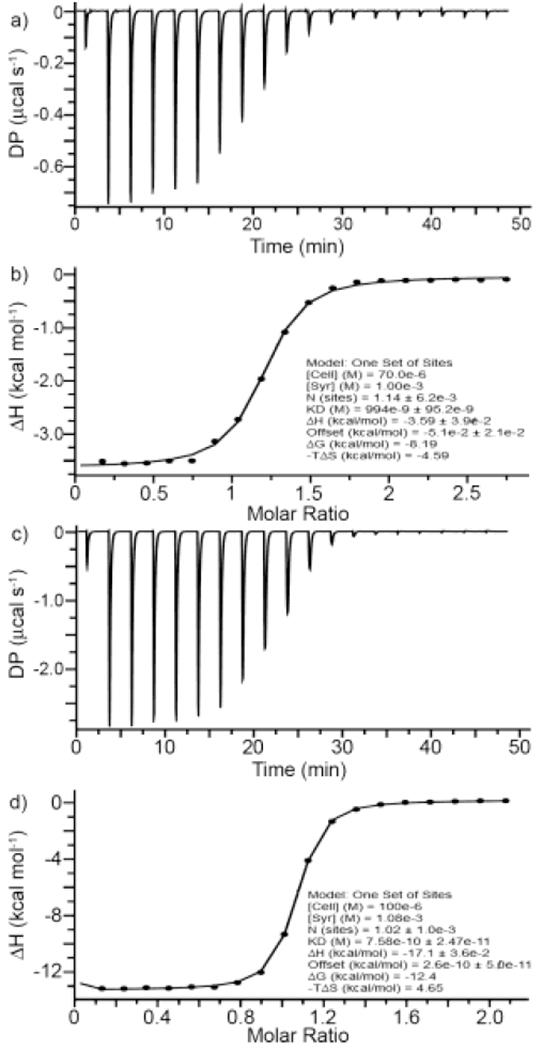
Thermograms recorded (298.0 K, 20 mM sodium phosphate buffered H2O, pH 7.4) during the titration of: a) a solution of 2C4 (70 μM) with a solution of CHDA (1 mM) in the syringe, and c) a solution of 2C4 (100 μM) and CHDA (500 μM) in the cell with a solution of Roc (1.08 mM) in the syringe. The data was fitted using the PEAQ-ITC analysis software to: b) a one-set-of-sites binding model to extract the thermodynamic parameters for 2C4•CHDA, and d) to a competitive binding model using the thermodynamic parameters for 2C4•CHDA as inputs to determine the thermodynamic parameters for 2C4•Roc.
Discussion of the Thermodynamic Parameters.
There are a number of interesting trends observed in the thermodynamic parameters of binding that are worthy of discussion. First, and most relevant toward the proposed use of 2C3 as an in vivo reversal agent for steroidal neuromuscular blocking agents is that 2C3 displays nanomolar affinity toward roc, vec, and pan and displays 4 – 20-fold higher affinity than either 2C2 or 2C4. Figure 6 presents the thermodynamic data in bar chart form to allow straightforward comparisons. Host 2C3 has previously been found to be a more efficient solubilizing agent for insoluble drugs which can be traced to its higher binding constants than 2C2 and 2C4 in accord with present results.18 Hosts 1, 1Me4, and 3 – 5 display lower affinity (2 × 104 – 5 × 106 M-1) toward roc, vec, and pan. Amongst these three steroidal NMBA’s pan usually forms the weakest complex which is surprising given that pan contains two permanent quaternary ammonium groups which would be expected to enhance binding.11d Host 2C3 binds roc 32000-fold stronger than Ach which is important because the sequestration of ACh from the neuromuscular junction is undesirable. In this regard, it is worth noting that 2C4 (2C2) is 22000-fold (6900-fold) selective for roc over ACh while maintaining sub nM affinity toward roc. Host 2C4 has higher inherent solubility (196 mM) than 2C3 (14 mM) and 2C2 (68 mM) which suggests that 2C4 may be more easily formulated for potential in vivo use.18
Figure 6.
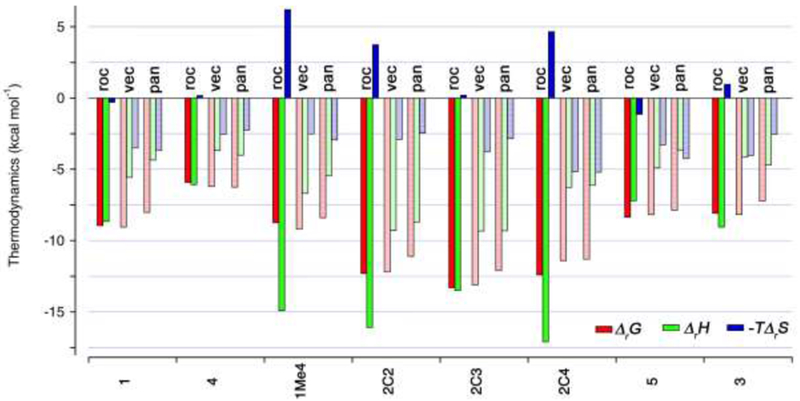
Bar chart depicting the thermodynamic parameters of binding (Δ rG, red bars; ΔrH, green bars; -TΔrS, blue bars) in kcal mol-1 for complexes of roc, vec, and pan toward hosts 1, 4, 1Me4, 2C2, 2C3, 2C4, 5, and 3.
Part of the impetus to perform this structure–binding affinity study was to determine whether superior acyclic CB[n]-type receptors could be discovered for the benylisoquinoline NMBA cis which is the third most popular NMBA in clinical use. We hoped that the narrow (CH2)5 chain of the guest would thread more efficiently through the narrower hosts 4 and 5 created from glycoluril trimer or host 3 with taller sidewalls. Experimentally, we found that 1 – 3 bound cis with Ka values in the 105 M-1 range and that 4 and 5 do so even more weakly with Ka values from 22500 – 56200 M-1. Host 2C4 displays highest affinity toward cis and possesses excellent aqueous solubility. Interestingly, the host•guest complexes with cis are all entirely driven by favourable enthalpic changes and feature entropically unfavourable contributions to ΔrG. We believe that the conformational restriction of the long oligomethylene chain of cis needed to optimize binding is responsible for this thermodynamic signature.
Comparison between structurally related hosts toward common guests is also worthwhile. For example, hosts 1 and 2C3 are homologues of 4 and 5 containing one more glycoluril ring. This additional glycoluril ring was expected to increase the size (volume) of the host cavity and thereby influence binding affinity. Experimentally, we find that 1 is a significantly better host than 4 toward roc (164-fold), vec (121-fold), pan (20-fold), cis (6-fold), and ACh (32-fold). Similarly, we find that 2C3 is a significantly better host than 5 toward roc (4400-fold),vec (4100-fold), pan (1360-fold), cis (2.3-fold), and ACh (119-fold). We believe the superior affinity and selectivity displayed by 1 and 2C3 reflects the larger cavity size / volume which probably contains a larger number of high energy waters23 that provide an enhanced driving force for complexation. Selectivity is lowest for the narrow cis guest which probably reflects a better complementarity between the narrow host with the narrower guest. A related comparison can be made between the behaviour of hosts 2C3 and 3 which are isomers of one another. Host 2C3 with its 2,3-linked naphthalene sidewalls possesses a wider cavity, but host 3 with its 1,8-linked naphthalene sidewalls possesses a narrower but deeper cavity. The SO3 - solubilizing groups of 3 are also farther from the cavity and ureidyl C=O portals of the host. Experimentally, we find that 2C3 is a better host than 3 for roc (6800-fold), vec (4100-fold), pan (4100-fold), and cis (1.3). Once again, the selectivity is largest for the larger steroidal guests that can properly fill the larger cavity of 2C3 without requiring conformational changes of the host and lowest for cis whose (CH2)5 chain is more complementary to the narrower cavity of 3. Finally, one can consider the binding trends for hosts 1, 1Me4, and 2C3 which differ systematically in the length of their aromatic sidewalls. For the smaller guests CHDA, ACh, and cis only small differences (1.4 – 7.8-fold) are observed in the binding constants toward 1, 1Me4, and 2C3 whereas for the larger steroidal guests roc, vec, and pan the differences are much larger (434 – 1055-fold). Our interpretation is that the cavity of 2C3 is particularly poorly solvated (e.g. high energy waters) and is able to adjust its conformation (e.g. helical twist) to fully complement the larger steroidal guest in ways that are not possible for small hosts and not needed for the smaller guests. As found in previous studies,17b,17d,18 hosts 2C2 – 2C4 are the most potent hosts known to date in the acyclic CB[n] series.
Conclusion.
summary, we have studied the interaction of a series of acyclic cucurbit[n]uril-type receptors (1 – 5) toward four NMBAs, ACh, and CHDA to further define the structure–binding affinity relationships relevant for their development as in vivo reversal agents. Based on the result of 1H NMR experiments, we conclude that the NMBAs bind with their hydrophobic residues located in the hydrophobic cavity of 1 – 5 which places their cationic moieties at the ureidyl C=O portals of the hosts. Isothermal titration calorimetry was used to measure the thermodynamic parameters for binding between hosts 1 – 5 and the NMBAs and ACh. We find that host 2C3 and its relatives 2C2 and 2C4 display nanomolar affinity toward the steroidal NMBAs and maintain a high selectivity against Ach as needed for the proposed in vivo use. Hosts 2C4 and 2C2 with their higher aqueous solubility compared to 2C3 are viable alternative reversal agents for steroidal NMBAs. All eight hosts displayed affinities toward cis in the 2 × 104 – 2 × 105 M-1 range according to ITC. No binding improvement relative to compound 1 (Ka = 1.32 × 105 M-1 toward cis) which was previously used to reverse cis in vivo in rats8b was found. Finally, hosts 4 and 5 which are based on glycoluril trimer units rather than tetramer units (e.g. 1 and 2) display 20 – 4400-fold weaker binding to the steroidal NMBAs which we attribute to the smaller host cavity and a small number of high energy waters23a which are known to provide a driving force in CB[n] complexation. Amongst the acyclic CB[n] tested, we conclude that 2C3 and its analogues 2C4 and 2C2 remain the most promising agents for further development as in vivo reversal agents for neuromuscular blocking agents.
Experimental Details.
General. Hosts 1 – 5 were prepared by the literature procedures.15,17a,17b,17d The neuromuscular blocking agents (roc, vec, pan, and cis) and Ach were purchased from commercial suppliers and used without further purification. 1H NMR spectra were measured on commercial NMR spectrometers operating at 400 MHz. ITC data was collected on a Malvern Microcal PEAQ-ITC instrument with a 200 μL cell volume. We used an injection syringe of 40 μL capacity. In each case, the host and guest solutions were prepared in a 20 mM NaH2PO4 buffer (pH 7.4). The sample cell was filled to capacity (200 μL) with the host solution and the guest solution was titrated (first injection = 0.4 μL, subsequent 18 injections = 2 μL) into the cell. Competition (displacement) titrations were performed for container•drug complexes with binding constants exceeding Ka = 5 × 106 M-1 using CHDA as a weaker binding ligand included with the host in the ITC cell. Data was fitted, as appropriate, with either the single set of sites model or the competitive binding model within the MicroCal PEAQ-ITC analysis software.
Supplementary Material
Acknowledgement.
We thank the National Science Foundation (CHE-1404911 and CHE-1807486 to L.I.) and the National Institutes of Health (CA-168365 to L.I.) for financial support. D.S. thanks the University of Maryland for a Howard Hughes Medical Institute undergraduate research fellowship.
Footnotes
Dedicated to Professor Kata Mlinaric-Majerski on the occasion of her 70th birthday.
Disclosure statement.
The University of Maryland holds patents on the use of acyclic CB[n]-type receptors in biomedical applications where L.I. is named as an inventor.
References.
- 1).Raghavendra T, Soc JR. Med. 2002, 95, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Bowman WC, Br. J. Pharmacol. 2006, 147, S277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).a) Butterly A; Bittner EA; George E; Sandberg WS; Eikermann M; Schmidt U, Br. J. Anaesth. 2010, 105, 304. [DOI] [PubMed] [Google Scholar]; b) Maybauer DM; Geldner G; Blobner M; Puehringer F; Hofmockel R; Rex C; Wulf HF; Eberhart L; Arndt C; Eikermann M, Anaesthesia 2007, 62, 12. [DOI] [PubMed] [Google Scholar]
- 4).Eikermann M; Zaremba S; Malhotra A; Jordan AS; Rosow C; Chamberlin NL, Br. J. Anaesth. 2008, 101, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).) Bom A; Bradley M; Cameron K; Clark JK; Van Egmond J; Feilden H; MacLean EJ; Muir AW; Palin R; Rees DC; Zhang M-Q, Angew. Chem., Int. Ed. 2002, 41, 265. [DOI] [PubMed] [Google Scholar]
- 6).) Naguib M, Anesth. Analg. 2007, 104, 575. [DOI] [PubMed] [Google Scholar]
- 7).) https://www.pharmalive.com/merck-2018-the-6-billion-surprise/. Accessed by L. Isaacs, March 25, 2019. [Google Scholar]
- 8).a) Ma D; Zhang B; Hoffmann U; Sundrup MG; Eikermann M; Isaacs L, Angew. Chem., Int. Ed. 2012, 51, 11358. [DOI] [PubMed] [Google Scholar]; b) Hoffmann U; Grosse-Sundrup M; Eikermann-Haerter K; Zaremba S; Ayata C; Zhang B; Ma D; Isaacs L; Eikermann M, Anesthesiology 2013, 119, 317. [DOI] [PubMed] [Google Scholar]; c) Haerter F; Simons JC; Foerster U; Duarte IM; Diaz-Gil D; Ganapati S; Eikermann-Haerter K; Ayata C; Zhang B; Blobner M; Isaacs L; Eikermann M, Anesthesiology 2015, 123, 1337–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ganapati S; Zavalij PY; Eikermann M; Isaacs L, Org. Biomol. Chem. 2016, 14, 1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).a) Lagona J; Mukhopadhyay P; Chakrabarti S; Isaacs L, Angew. Chem., Int. Ed. 2005, 44, 4844. [DOI] [PubMed] [Google Scholar]; b) Isaacs L, Acc. Chem. Res. 2014, 47, 2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).a) Mock WL; Shih N-Y, J. Org. Chem. 1986, 51, 4440 [Google Scholar]; b) Lee JW; Samal S; Selvapalam N; Kim H-J; Kim K, Acc. Chem. Res. 2003, 36, 621. [DOI] [PubMed] [Google Scholar]
- 11).a) Rekharsky MV; Mori T; Yang C; Ko YH; Selvapalam N; Kim H; Sobransingh D; Kaifer AE; Liu S; Isaacs L; Chen W; Moghaddam S; Gilson MK; Kim K; Inoue Y, Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 20737. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moghaddam S; Yang C; Rekharsky MV; Ko YH; Kim K; Inoue Y; Gilson MK, J. Am. Chem. Soc. 2011, 133, 3570. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu S; Ruspic C; Mukhopadhyay P; Chakrabarti S; Zavalij PY; Isaacs L, J. Am. Chem. Soc. 2005, 127, 15959. [DOI] [PubMed] [Google Scholar]; d) Cao L; Sekutor M; Zavalij PY; Mlinaric-Majerski K; Glaser R; Isaacs L, Angew. Chem., Int. Ed. 2014, 53, 988. [DOI] [PubMed] [Google Scholar]; e) Assaf KI; Nau WM, Chem. Soc. Rev. 2015, 44, 394. [DOI] [PubMed] [Google Scholar]
- 12).a) Macartney DH, Future Med. Chem 2013, 5, 2075. [DOI] [PubMed] [Google Scholar]; b) Gamal-Eldin MA; Macartney DH, Can. J. Chem. 2014, 92, 243 [Google Scholar]; c) Lazar AI; Biedermann F; Mustafina KR; Assaf KI; Hennig A; Nau WM, J. Am. Chem. Soc. 2016, 138, 13022. [DOI] [PubMed] [Google Scholar]
- 13).) Liu S; Zavalij PY; Isaacs L, J. Am. Chem. Soc. 2005, 127, 16798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).) Isaacs L, Chem. Commun. 2009, 619. [DOI] [PubMed] [Google Scholar]
- 15).) Ma D; Hettiarachchi G; Nguyen D; Zhang B; Wittenberg JB; Zavalij PY; Briken V; Isaacs L, Nat. Chem. 2012, 4, 503. [DOI] [PubMed] [Google Scholar]
- 16).a) Stancl M; Hodan M; Sindelar V, Org. Lett. 2009, 11, 4184. [DOI] [PubMed] [Google Scholar]; b) Stancl M; Gilberg L; Ustrnul L; Necas M; Sindelar V, Supramol. Chem. 2014, 26, 168 [Google Scholar]; c) Stancl M; Gargulakova Z; Sindelar V, J. Org. Chem. 2012, 77, 10945. [DOI] [PubMed] [Google Scholar]
- 17).a) Ma D; Zavalij PY; Isaacs L, J. Org. Chem. 2010, 75, 4786. [DOI] [PubMed] [Google Scholar]; b) Zhang B; Isaacs L, J. Med. Chem. 2014, 57, 9554. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang B; Zavalij PY; Isaacs L, Org. Biomol. Chem. 2014, 12, 2413. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gilberg L; Zhang B; Zavalij PY; Sindelar V; Isaacs L, Org. Biomol. Chem. 2015, 13, 4041. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Minami T; Esipenko NA; Zhang B; Isaacs L; Nishiyabu R; Kubo Y; Anzenbacher P, J. Am. Chem. Soc. 2012, 134, 20021. [DOI] [PubMed] [Google Scholar]; f) Minami T; Esipenko N; Akdeniz A; Zhang B; Isaacs L; Anzenbacher P, J. Am. Chem. Soc. 2013, 135, 15238. [DOI] [PubMed] [Google Scholar]; g) Shcherbakova EG; Zhang B; Gozem S; Minami T; Zavalij Peter Y; Pushina M; Isaacs L; Anzenbacher PJ, J. Am. Chem. Soc. 2017, 139, 14954. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Shen C; Ma D; Meany B; Isaacs L; Wang Y, J. Am. Chem. Soc. 2012, 134, 7254. [DOI] [PubMed] [Google Scholar]
- 18).) Sigwalt D; Moncelet D; Falcinelli S; Mandadapu V; Zavalij PY; Day A; Briken V; Isaacs L, ChemMedChem 2016, 11, 980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).) Ganapati S; Grabitz SD; Murkli S; Scheffenbichler F; Rudolph MI; Zavalij PY; Eikermann M; Isaacs L, ChemBioChem 2017, 18, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).a) Chen H; Y-W. CJ; Li S; Liu JJ; Wyman I; Lee SM-Y; Macartney DH; Wang R, RSC Adv. 2015, 5, 63745 [Google Scholar]; b) Huang Q; Kuok KI; Zhang X; Yue L; Lee SMY; Zhang J; Wang R, Nanoscale 2018, 10, 10333. [DOI] [PubMed] [Google Scholar]; c) Yang X; Li S; Wang Z; Lee SMY; Wang L-H; Wang R, Chem. - Asian J. 2018, 13, 41; [DOI] [PubMed] [Google Scholar]; d) Yin H; Wang R, Isr. J. Chem. 2018, 58, 188. [Google Scholar]
- 21).a) Diaz-Gil D; Haerter F; Falcinelli S; Ganapati S; Hettiarachchi GK; Simons JCP; Zhang B; Grabitz SD; Duarte IM; Cotten JF; Eikermann-Haerter K; Deng H; Chamberlin NL; Isaacs L; Briken V; Eikermann M, Anesthesiology 2016, 125, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hettiarachchi G; Samanta SK; Falcinelli S; Zhang B; Moncelet D; Isaacs L; Briken V, Mol. Pharmaceutics 2016, 13, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).) Masson E; Ling X; Joseph R; Kyeremeh-Mensah L; Lu X, RSC Adv. 2012, 2, 1213. [Google Scholar]
- 23).a) Nau WM; Florea M; Assaf KI, Isr. J. Chem. 2011, 51, 559 [Google Scholar]; b) Biedermann F; Uzunova VD; Scherman OA; Nau WM; De Simone A, J. Am. Chem. Soc. 2012, 134, 15318. [DOI] [PubMed] [Google Scholar]
- 24).a) Broecker J; Vargas C; Keller S, Anal. Biochem. 2011, 418, 307. [DOI] [PubMed] [Google Scholar]; b) Wiseman T; Williston S; Brandts JF; Lin L-N, Anal. Biochem. 1989, 179, 131. [DOI] [PubMed] [Google Scholar]
- 25).) Krainer G; Keller S, Methods 2015, 76, 116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.