

Antimicrobial peptides (AMPs) have seen limited clinical use as antimicrobial agents, largely due to issues relating to toxicity, short biological half-life, and lack of efficacy against Gram-negative bacteria. However, the development of novel AMP-nanomedicines, i.e., AMPs entrapped in nanoparticles, has the potential to ameliorate these clinical problems. The authors investigated two novel nanomedicines based on AA139, an AMP currently in development for the treatment of multidrug-resistant Gram-negative infections.
KEYWORDS: Gram-negative bacteria, Klebsiella pneumoniae, antimicrobial peptides, biodistribution, experimental therapeutics, laboratory animals, micelles, nanomedicines, polymeric nanoparticles
ABSTRACT
Antimicrobial peptides (AMPs) have seen limited clinical use as antimicrobial agents, largely due to issues relating to toxicity, short biological half-life, and lack of efficacy against Gram-negative bacteria. However, the development of novel AMP-nanomedicines, i.e., AMPs entrapped in nanoparticles, has the potential to ameliorate these clinical problems. The authors investigated two novel nanomedicines based on AA139, an AMP currently in development for the treatment of multidrug-resistant Gram-negative infections. AA139 was entrapped in polymeric nanoparticles (PNPs) or lipid-core micelles (MCLs). The antimicrobial activity of AA139-PNP and AA139-MCL was determined in vitro. The biodistribution and limiting doses of AA139-nanomedicines were determined in uninfected rats via endotracheal aerosolization. The early bacterial killing activity of the AA139-nanomedicines in infected lungs was assessed in a rat model of pneumonia-septicemia caused by extended-spectrum β-lactamase-producing Klebsiella pneumoniae. In this model, the therapeutic efficacy was determined by once-daily (q24h) administration over 10 days. Both AA139-nanomedicines showed equivalent in vitro antimicrobial activities (similar to free AA139). In uninfected rats, they exhibited longer residence times in the lungs than free AA139 (∼20% longer for AA139-PNP and ∼80% longer for AA139-MCL), as well as reduced toxicity, enabling a higher limiting dose. In rats with pneumonia-septicemia, both AA139-nanomedicines showed significantly improved therapeutic efficacy in terms of an extended rat survival time, although survival of all rats was not achieved. These results demonstrate potential advantages that can be achieved using AMP-nanomedicines. AA139-PNP and AA139-MCL may be promising novel therapeutic agents for the treatment of patients suffering from multidrug-resistant Gram-negative pneumonia-septicemia.
INTRODUCTION
Pneumonia is responsible for over 230,000 deaths and €10 billion in economic costs across the European Union every year (1, 2), with Gram-negative bacteria being the primary causative pathogens in ∼70% of health care-associated pneumonia cases (3). Therefore, the availability of effective antimicrobial drugs is crucial if clinicians are to adequately treat patients suffering from Gram-negative bacterial pneumonia (4). Unfortunately, the current crisis of growing antimicrobial resistance and fewer antimicrobials reaching the market has resulted in a lack of new antimicrobials that are effective against multidrug-resistant Gram-negative bacteria (5, 6). In response to this growing health care crisis, researchers have focused on the development and improvement of known but underutilized antibiotic classes to generate new approaches for treating bacterial pneumonia.
One class of antibiotics under investigation is the antimicrobial peptides (AMPs), a broad family of antibiotics that are produced in nature by organisms as a natural defense against microbes (7). Although AMPs have been known since the 1950s, this family of antibiotics has seen only limited use for the treatment of pneumonia, largely due to a number of hurdles that have limited their clinical use, including toxic side effects (8), short biological half-life due to degradation by proteases (8, 9), and limited efficacy against Gram-negative bacteria (10).
Some of these issues may be offset by the concurrent development of new AMPs and new delivery systems for these antimicrobials. For example, entrapment of AMPs into nanoparticles to produce nanomedicines may generate several solutions to the hurdles traditionally associated with the clinical utility of AMPs in the treatment of multidrug-resistant infections (11, 12). Notably, nanomedicine formulations could potentially reduce toxic side effects and extend the biological half-life of AMPs (13). Furthermore, the direct delivery of antibiotics to the site of infection represents an additional therapeutic improvement (14), which could facilitate increased antibiotic activity at the actual site of infection (15, 16) and lead to reduced systemic toxicity and “collateral damage” to patients (17, 18).
As such, the direct delivery of AMP nanomedicines to the lungs is a promising research strategy at a time when traditional antibiotics and treatment for respiratory tract infections are becoming ineffective (19, 20). In this respect, the European Union’s Seventh Framework Programme provided funding to the PneumoNP research consortium, with the goal of developing and investigating novel AMP nanomedicines in the treatment of pneumonia (21).
One of the AMPs investigated in the PneumoNP project was AA139, which was originally isolated from the marine lugworm Arenicola marina as “Arenicin-3” and was subsequently further developed to decrease the original AMP’s plasma protein-binding properties, cytotoxicity, and hemolytic activity (22, 23). AA139 is a cationic AMP with a 21-residue amphipathic hairpin structure that appears to have a dual mode of action involving directly binding the AMP to membrane phospholipids and interrupting phospholipid transportation pathways, resulting in membrane dysregulation and bacterial cell death (22, 24). AA139 has previously demonstrated potent in vitro and in vivo activity against multidrug-resistant Gram-negative bacteria (25, 26) and is not readily absorbed into systemic circulation from the lungs following inhalation in mice (Adenium Biotech ApS, personal communication).
With respect to the development of nanomedicines, PneumoNP utilized several different nanomedicine formulations, including polymeric nanoparticles (PNPs) and lipid-core micelles (MCLs). PNPs are hydrophilic dextran-based single-chain polymer nanoparticles to which cationic drugs like AA139 can be attached by electrostatic interaction for increased drug delivery (27). MCLs are self-assembling colloidal nanoparticles with a hydrophilic surface and a hydrophobic core in which drugs like AA139 can be entrapped for drug delivery (28). Both PNPs (29) and MCLs (30) are suitable for nanomedicine formulation due to their favorable biocompatibility, nontoxicity, biodistribution, and ease of modification. PNPs and MCLs are promising nanocarriers for pulmonary drug delivery, as the relatively small size of PNPs (1 to 20 nm) may allow for improved penetration of the respiratory mucus (31), and MCLs have been shown to facilitate sustained drug release in the lungs (32).
In the present study, the following two novel AA139 nanomedicines were developed using either PNPs or MCLs: AA139-PNP, in which AA139 has been attached to PNPs by electrostatic interactions; and AA139-MCL, in which AA139 has been enclosed in the hydrophobic core of MCLs. Their antimicrobial activity was assessed first via in vitro concentration- and time-dependent bactericidal activity studies against extended-spectrum β-lactamase (ESBL)-producing Klebsiella pneumoniae. In vivo studies were then performed involving the administration of the nanomedicines by endotracheal aerosolization as a means of direct delivery to the lungs, the primary site of infection in pneumonia. First, the biodistribution and residence times in the lungs of the nanomedicines were determined, and the maximum tolerated dose (MTD) was assessed in uninfected rats. Next, the early bacterial killing activity of the nanomedicines was assessed in a rat model of pneumonia-septicemia caused by ESBL-producing K. pneumoniae. Finally, the therapeutic efficacy of the nanomedicines was determined in the pneumonia-septicemia rat model by once-daily (q24h) administration over 10 days.
RESULTS
Concentration- and time-dependent antimicrobial activity of AA139, AA139-PNP, and AA139-MCL in vitro.
Time-kill kinetics (TKK) assays were used to determine the antimicrobial activity of free AA139, AA139-PNP, and AA139-MCL against K. pneumoniae ESBL over 24 hours. The bacterial populations rapidly increased in the absence of antibiotics within 24 hours of incubation (Fig. 1). Free AA139 showed bactericidal activity, resulting in bacterial growth inhibition at 2 mg/liter AA139, a 100-fold reduction in bacterial numbers within 2 hours at 4 mg/liter AA139, and near-complete bacterial killing after 24 hours at 8 mg/liter AA139. AA139-PNP and AA139-MCL both showed similar bactericidal activity compared with free AA139, but the bacterial growth inhibition observed at 2 mg/liter AA139 (P = 0.0461) was not observed for 2 mg/liter AA139-PNP and 2 mg/liter AA139-MCL. Unlike free AA139 and AA139-MCL, AA139-PNP did not lead to complete bacterial killing after 24 hours at 8 mg/liter AA139-PNP. Unlike free AA139 and AA139-PNP, AA139-MCL did not result in a 100-fold reduction in bacterial numbers within 2 hours at 4 mg/liter AA139-MCL but did at 8 mg/liter AA139-MCL. The rebound after 24 hours observed for 4 mg/liter AA139-PNP and 4 mg/liter AA139-MCL is likely the result of degradation of AA139 (25; Adenium Biotech ApS, personal communication). Unloaded MCLs and unloaded PNPs did not show any antimicrobial activity.
FIG 1.
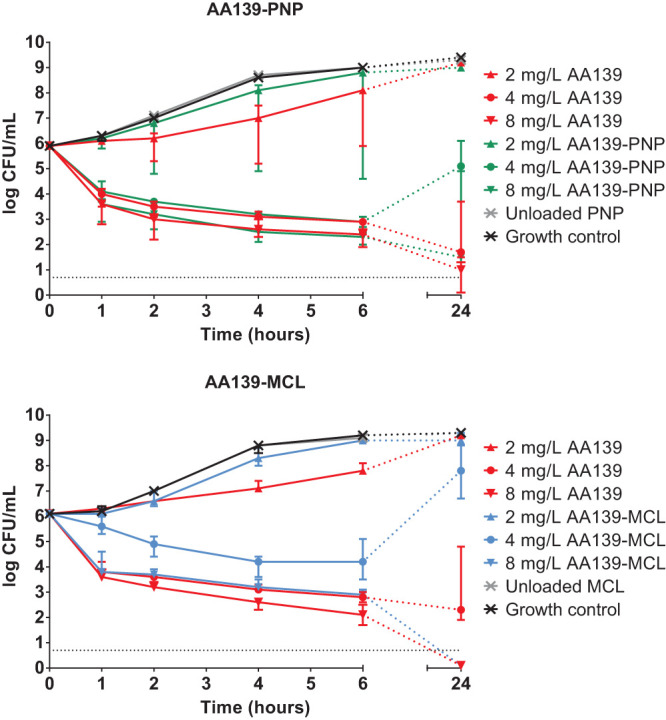
Concentration- and time-dependent antimicrobial activity of AA139, AA139-PNP, or AA139-MCL against K. pneumoniae ESBL. Bacterial cultures of K. pneumoniae ESBL were exposed to 2-fold increasing concentrations of free AA139, AA139-PNP, or AA139-MCL for 24 h at 37°C under shaking conditions. Shown are the median ± range of triplicate experiments. The dashed gray line indicates the lower limit of quantification being log 0.7.
Biodistribution of AA139, AA139-PNP, and AA139-MCL in uninfected rats.
Positron emission tomography-computed tomography (PET-CT) image sequences were obtained for [124I]NaI (control), [124I]AA139, [124I]AA139-PNP, and [124I]AA139-MCL (Fig. 2) in groups of 4 rats. The images reveal that the biodistribution patterns of [124I]AA139, [124I]AA139-PNP, and [124I]AA139-MCL significantly differ from that of the control. When [124I]NaI was administered, it was rapidly cleared from the lungs, and almost no signal was observed in this organ after 3 hours. On the contrary, a significant fraction of [124I]AA139 and both [124I]AA139 nanomedicines were still visible within the lungs at the same time point. Of note, the images reveal that the residence time of [124I]AA139-PNP and [124I]AA139-MCL is longer than the residence time of free [124I]AA139 since, for the first two, a clear signal within the lungs is still visible at 9, 15, and 24 hours after administration. The elimination pathways of the different species suggest excretion via urine and the gastrointestinal tract.
FIG 2.

PET-CT coronal images showing the biodistribution of radiolabeled compounds after endotracheal aerosolization in uninfected rats. Shown are PET-CT images obtained at different time points of [124I]NaI (control) (A), [124I]AA139 (B), [124I]AA139-PNP (C), and [124I]AA139-MCL (D). PET images have been overlaid with CT images of the same animals for accurate location of the radioactive signal.
The deposition of free AA139 among the five lung lobes and its distribution to blood plasma in uninfected rats immediately after endotracheal aerosolization were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) bioanalysis (see Fig. S2 in the supplemental material). Total deposition in the lungs (ca. ∼88% of delivered dose) was high but showed considerable variation between the lung lobes, with most AA139 found in the relatively large left lobe and least AA139 found in the smallest right middle lobe. Total distribution to blood plasma (ca. ∼2% of delivered dose) was low. These findings matched our in vivo imaging results, as well as the findings from an earlier study in which the deposition of 2-deoxy-2-[18F]fluoro-d-glucose (a clinically used radiotracer) after the same administration method was studied (33).
Residence time of AA139, AA139-PNP, and AA139-MCL in lungs of uninfected rats.
Quantification of the radioactivity in the lungs confirmed the trends observed in these in vivo sequences. To determine the biological half-life (t1/2) of the radiolabeled compounds in the rat lungs, the concentrations of radioactivity in the lungs at different time points were fitted to a monoexponential decay equation (Fig. 3). The residence time in lungs of free [124I]AA139 (t1/2 = 1.71 h) was longer by 21.6% when administered as [124I]AA139-PNP (t1/2 = 2.08 h) and longer by 82.4% when administered as [124I]AA139-MCL (t1/2 = 3.12 h). The residence time of the control [124I]NaI in lungs was very short (t1/2 = 0.09 h).
FIG 3.
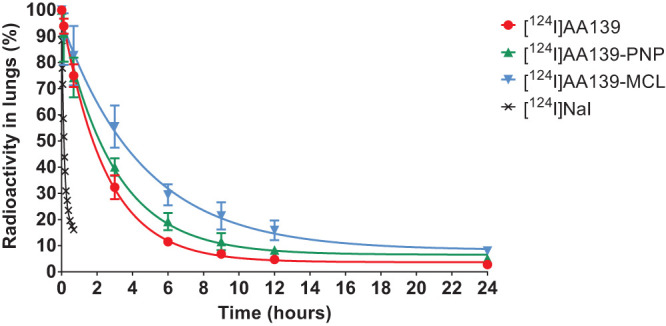
Time-dependent concentration of radioactivity in rat lungs after endotracheal aerosolization of radiolabeled compounds. Shown are the mean ± standard deviation of four experiments as well as monoexponential curves fitted to the experimental data.
Limiting dose of AA139, AA139-PNP, and AA139-MCL in uninfected rats.
Rats were administered 2-fold increasing doses of free AA139, AA139-PNP, AA139-MCL, or normal saline solution (NSS) by endotracheal aerosolization in groups of 11 rats. As shown in Table 1, administration of NSS did not lead to any signs of acute toxicity or discomfort. Free AA139 was not tolerated when administered at doses above the MTD of 0.25 mg/rat (∼1 mg/kg of body weight). AA139-PNP could be safely administered at the maximum feasible dose (MFD) of 0.5 mg/rat (∼2 mg/kg), which was the highest possible dose that could be given due to technical limitations. AA139-MCL could be safely administered at the MTD of 0.5 mg/rat (∼2 mg/kg) but induced acute toxicity in terms of abnormal breathing at higher doses. Analysis of blood biomarkers showed no indications of acute toxicity at limiting doses (see Fig. S3 in the supplemental material).
TABLE 1.
Limiting dose of AA139, AA139-PNP, or AA139-MCL
| Treatmenta | Limiting dose (mg/rat) [mg/kg] | Dose-limiting toxicity |
|---|---|---|
| AA139 | 0.25 (∼1)b | Acute toxicity (abnormal breathing) observed at 0.5 mg/rat |
| AA139-PNP | 0.5 (∼2)c | Maximum feasible dose due to technical limitations |
| AA139-MCL | 0.5 (∼2)b | Acute toxicity (abnormal breathing) observed at 1 mg/rat |
| NSS | Well tolerated at this vol |
Treatment was administered to groups of 11 rats via endotracheal aerosolization of a 100-μl suspension. Rats were regularly checked for signs of acute toxicity over 24 h. When no acute toxicity was observed, the dose was increased 2-fold, until acute toxicity was observed or until the dose could not be further increased due to technical limitations. MTD, maximum tolerated dose. MFD, maximum feasible dose.
MTD.
MFD.
Early bacterial killing activity of AA139, AA139-PNP, and AA139-MCL in infected rats.
At 24 hours after initiation of infection, rats in groups of 6 were treated with a single dose of free AA139, AA139-PNP, or AA139-MCL at the MTD of free AA139 (0.25 mg/rat; ∼1 mg/kg) by endotracheal aerosolization. Rats were sacrificed at 2, 6, and 24 hours after administration to determine bacterial counts in the rat lungs and blood biomarkers (Fig. 4). In rats treated with NSS as a placebo, bacterial numbers increased almost 100-fold within 24 hours. Following treatment with free AA139, bacterial counts exhibited a robust decrease by 6 hours after administration but had rebounded at 24 hours to bacterial counts similar to the placebo group. Administration of AA139-PNP resulted in a rapid decrease of bacterial counts within 2 hours after administration but began to rebound at 6 hours postdose, generating similar bacterial counts to the placebo group by 24 hours postdose. In contrast, treatment with AA139-MCL resulted in a milder, but prolonged, bacterial decrease which persisted for at least 24 hours. Rats treated with unloaded PNP or unloaded MCL had bacterial counts similar to the placebo treatment, showing no bacterial decrease during treatment. Analysis of blood biomarkers showed no indications of acute toxicity of any treatment (see Fig. S4 in the supplemental material). Early in vivo bacterial killing activity experiments were performed to screen those potential nanomedicine candidates that showed promising in vitro bactericidal activity. However, only a limited number of rats were used in these screening experiments (34), and consequently, statistical evaluation of these results could not be performed.
FIG 4.
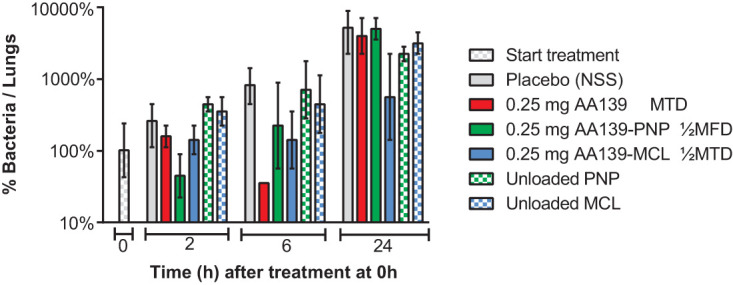
Early bacterial killing activity of AA139, AA139-PNP, or AA139-MCL in rats with ESBL pneumonia-septicemia. At 24 hours after initiation of infection, rats were treated with a single dose of free AA139, AA139-PNP, or AA139-MCL at 0.25 mg (ca. ∼1 mg/kg). Rats were sacrificed at 2, 6, and 24 hours after administration and were dissected. Shown are the mean ± range of 2 rats per time point per treatment group. MTD, maximum tolerated dose; MFD, maximum feasible dose.
Therapeutic efficacy of AA139, AA139-PNP, and AA139-MCL in infected rats.
At 24 hours after initiation of infection, rats in groups of 12 were treated with free AA139, AA139-PNP, or AA139-MCL once-daily (q24h) for 10 days. Placebo-treated rats reached humane endpoints within 7 days after start of treatment (Fig. 5). The treatment outcomes were statistically evaluated and compared; all P values can be found in Table 2. Infected rats were monitored every 12 hours for the duration of the experiment. No signs of acute toxicity were observed following administration of compounds at the limiting dose q24h for 10 days. Rats treated with free AA139 at MTD (0.25 mg/rat; ∼1 mg/kg), unloaded PNP, or unloaded MCL did not show an improvement in survival compared with placebo-treated rats. Rats treated with AA139-PNP at MFD (0.5 mg/rat; ∼2 mg/kg) showed significantly improved survival compared with rats treated with free AA139 at MTD, unloaded PNP, or placebo. Rats treated with AA139-PNP at one-half of MFD did not show significantly improved survival. Rats treated with AA139-MCL at MTD (0.5 mg/rat; ∼2 mg/kg) or at one-half of MTD showed significantly improved survival compared with rats treated with free AA139 at MTD, unloaded MCL, or placebo. No significant differences in therapeutic efficacy were found between AA139-PNP and AA139-MCL.
FIG 5.
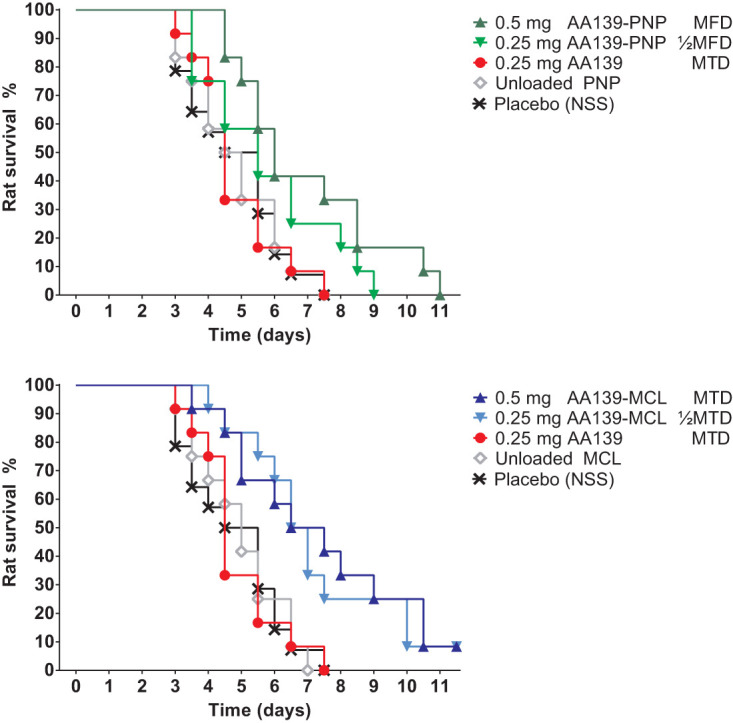
Therapeutic efficacy of AA139, AA139-PNP, or AA139-MCL in rats with ESBL pneumonia-septicemia. At 24 hours after initiation of infection, groups of 12 rats were treated once-daily (q24h) for 10 days with free AA139, AA139-PNP, or AA139-MCL. Shown are the survival curves (Kaplan-Meier) representing rats reaching humane endpoints. MTD, maximum tolerated dose; MFD, maximum feasible dose.
TABLE 2.
Statistical evaluation of rat survival after treatment with AA139, AA139-PNP, or AA139-MCL
| Treatment | Treatment dose (mg) |
P value by treatmenta |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo (NSS) | Unloaded |
AA139 |
|||||||
| PNP | MCL | Free | PNP |
MCL |
|||||
| 0.25 mg | 0.25 mg | 0.5 mg | 0.25 mg | 0.5 mg | |||||
| Placebo (NSS) | |||||||||
| Unloaded-PNP | 0.9329 | ||||||||
| Unloaded-MCL | 0.9689 | 0.9778 | |||||||
| AA139 | 0.25 | 0.9772 | 0.9879 | 0.9747 | |||||
| AA139-PNP | 0.25 | 0.0911 | 0.1218 | 0.1680 | 0.1208 | ||||
| 0.5 | 0.0131 | 0.0156 | 0.0161 | 0.0079 | 0.5966 | ||||
| AA139-MCL | 0.25 | 0.0043 | 0.0074 | 0.0061 | 0.0069 | 0.1140 | 0.7208 | ||
| 0.5 | 0.0055 | 0.0068 | 0.0085 | 0.0056 | 0.0997 | 0.5966 | 0.7635 | ||
Statistical evaluation using the log-rank (Mantel-Cox) test of rat survival after once-daily (q24h) administration of free AA139, AA139-PNP, or AA139-MCL over 10 days. Significant P values (P < 0.05) are indicated in bold.
DISCUSSION
The clinical utility of many AMPs is currently limited by several implementation barriers, such as toxic side effects (8) and short biological half-lives due to susceptibility to proteases (8, 9). However, these implementation barriers could potentially be offset by the development of nanomedicine formulations based on AMPs (13, 19). Additionally, the direct delivery of antibiotic-nanomedicines to the lungs may provide therapeutic advantages compared with conventional administration methods (19, 20). To investigate this hypothesis, the novel AMP AA139 was successfully entrapped in two nanocarriers (PNP or MCL) to generate two novel nanomedicines (AA139-PNP and AA139-MCL). The in vitro antimicrobial activity of both nanomedicines was found to be comparable to free AA139 at multiple concentrations, suggesting that AA139 retained its bactericidal activity despite formulation in PNP or MCL nanocarriers, a promising characteristic for further in vivo studies. Further studies into the mechanisms of entrapment and subsequent release of AA139 from the nanomedicines were barred, as we did not succeed in the successful labeling of AA139 for nuclear magnetic resonance (NMR) studies.
In uninfected rats, biodistribution studies showed that the residence time in the lungs of both AA139-PNP and AA139-MCL was longer than that observed with free AA139. Specifically, AA139-PNP exhibited ca. ∼20% longer residence time in the lung than free AA139. The residence time of AA139-MCL in the rat lung was observed to be even longer (at ca. ∼80%) than that in free AA139. As discussed by Kumari et al., many types of PNP-based drug delivery systems exhibit controlled release mechanisms, although this depends on the particular type of PNP utilized (35). Furthermore, Gill et al. previously reported that entrapping drugs in MCLs facilitates sustained drug release in the lung (32). Regarding the limiting dose, both nanomedicines could be safely administered at twice the dose of free AA139. Although the dose is not a particularly high increase, this finding does demonstrate that nanomedicine formulations can result in reduced toxic side effects, consequently allowing the administration of higher doses of AMPs during treatment. Other investigators have previously discussed the reduction of toxic side effects as a benefit of drug nanoformulations, which has been reported for both PNP (36) and MCL (30).
The early bacterial killing activity of the nanomedicines by single-dose administration was investigated in rats with fatal pneumonia-septicemia caused by multidrug-resistant K. pneumoniae. AA139-PNP showed a rapid but short-lasting bacterial killing effect, whereas AA139-MCL showed a slow but sustained bacterial killing effect, reflecting the difference in biological half-lives between AA139-PNP and AA139-MCL, as observed in our biodistribution studies. Taken together, our findings showed that similar doses (0.25 mg/rat; ∼1 mg/kg) of free AA139, AA139-PNP, and AA139-MCL had comparable bacterial killing activity over the first 6 hours postdose; although bactericidal activity persisted through 24 hours only for AA139-MCL, bacterial counts following the other two treatments rebounded to placebo levels by 24 hours postdose. As these studies were used for an early assessment of in vivo bacterial killing activity in the development pipeline, only a limited number of rats were used to reduce and refine the use of experimental animals (34). As a consequence, evaluation of the early bacterial killing results with statistical significance calculations could not be performed. However, these experiments have been included (i) to provide a preliminary look at the effect of AA139-nanomedicines on the bacterial load in the lungs over time, (ii) to further support conclusions that can be drawn from the TKK assays and biodistribution studies, (iii) to showcase how the early development pipeline of novel nanomedicines is structured, and (iv) because the dissemination of animal experimental results should be a professional standard for ethical animal research (37).
Further studies assessed the therapeutic efficacy of the nanomedicines by once-daily (q24h) administration for 10 days at the limiting dose of both nanomedicines (0.5 mg/rat; ∼2 mg/kg) or at the MTD of free AA139 (0.25 mg/rat; ∼1 mg/kg). Both nanomedicines exhibited a statistically significant improvement in therapeutic efficacy when dosed at their limiting dose compared with free AA139 dosed at its MTD. Although not particularly high, the 2-fold increase in limiting dose was sufficient to demonstrate a potential advantage of nanomedicine formulations in minimizing potential toxic side effects which may be associated with particular AMPs when treating patients. In addition, AA139-MCL showed significantly improved therapeutic efficacy at one-half of MTD as well, suggesting that the superior residence time of AA139-MCL in the lung resulted in further improved therapeutic efficacy in a daily dosing schedule. The lung residence time as well as the early bacterial killing activity of AA139-nanomedicines in the lung indicate that more frequent administration could have resulted in improved therapeutic efficacy. In our pilot therapeutic efficacy experiment, twice-daily (q12h) administration of AA139-nanomedicines was used; however, rats subjected to this schedule showed acute toxicity (abnormal breathing of rats) after several days which lead to experiment termination. The q24h dosing schedule was well tolerated and was therefore chosen to perform the therapeutic efficacy studies.
Future pharmacokinetic studies of the two AA139-nanomedicines may reveal more appropriate dosing schedules which could lead to improvement of their therapeutic efficacy. The present study shows that although both nanomedicines resulted in significantly improved therapeutic efficacy in terms of increased rat survival time compared with free AA139, survival of all infected rats was not achieved. In relation to this, it should be noted that the pneumonia-septicemia rat model used in this study is representative of multidrug-resistant bacterial infections that are associated with very high mortality rates in patients and are particularly challenging to treat successfully (38). Furthermore, there is published evidence that rats may be more sensitive to the innate immune activation known to be caused by AMPs, including histamine release (39–41). This makes the rat a good model for evaluating potential AMP toxicity decreases using nanomedicine formulation, as the tolerability of AMPs in rats is generally low. Of note, single and repeated AA139 aerosol administrations of up to 20 mg/kg/day have been well-tolerated in uninfected mice (Adenium Biotech ApS, personal communication).
A practical limitation in the experimental set-up was the maximum volume of compounds (100 μl) that could be administered to the rats by endotracheal aerosolization in order to avoid reflux of compound after administration. This meant that only restricted doses could be administered to the rats, in this way preventing the demonstration of a more optimal therapeutic effect. In addition, the animals must be under anesthesia for the administration which results in a low respiratory rate and therefore a low efficiency of drug delivery, restricting the dose that can be administered. In patients, inhalation of antibiotics would be the preferred route of administration, as inhalation allows higher doses of antibiotic to be delivered to the lungs over a longer period of time (42). In an attempt to improve aerogenic treatment in rats, we previously tested two aerosol inhalation systems for their capacity to administer the described nanomedicines to uninfected rats (33). Although an improved uniform distribution of compound in the lungs was achieved, the results also indicated that only <0.1% of compound was deposited in the lungs, compared with ∼85% deposition using endotracheal aerosolization (33). Based on these results, a decision was made to utilize the more efficient endotracheal aerosolization in our efficacy studies in rats.
Free AA139 has been shown to be potent in in vivo efficacy in studies using different infection models, including a murine pneumonia model (26; Adenium Biotech ApS, personal communication). In the present study, both AA139-nanomedicines showed significantly improved therapeutic efficacy in terms of an extended rat survival time. While these results are promising, survival of all infected rats was not obtained. Various factors could explain the lack of survival of infected rats receiving free AA139 or nanomedicine, including (i) a suboptimal administration frequency, (ii) the severity of the present infection model in rats, (iii) the general sensitivity of rats to AMP toxicity, or (iv) the limitations of endotracheal aerosolization as an administration method. Due to these factors, a maximum therapeutic effect (in terms of survival of all infected rats) could unfortunately not be achieved. Importantly, it is clear that the choice of animal model and study protocol are of significant importance for the success of preclinical studies involving AMPs and should be considered during the drug development process. In addition, future studies on AA139-nanomedicines should further refine these antimicrobial candidates for the treatment of bacterial infections to show more clinically significant improvements over free AA139 in different experimental set-ups.
In conclusion, the present study demonstrates that AMP nanomedicines may reduce some of the main barriers to the clinical use of underutilized AMP antibiotics, primarily by ameliorating potential toxic side effects and prolonging the biological half-life of AMP at the site of infection, while maintaining the AMP antimicrobial activity. Our results support the further evaluation of AA139-based nanomedicines as potential novel therapeutic agents for the treatment of multidrug-resistant Gram-negative pulmonary infections.
MATERIALS AND METHODS
Peptides, nanocarriers, chemical agents, and solvents.
AA139 in Ringer’s acetate solution was obtained from Adenium Biotech ApS (Copenhagen, Denmark). PNPs were produced at CIDETEC (Donostia-San Sebastián, Spain). MCLs were produced at Utrecht University (Utrecht, The Netherlands). Polyethylene glycosylated distearyl phosphatidyl ethanolamine (DSPE-PEG2000) Na-salt was purchased from Lipoid GmbH, Ludwigshafen, Germany. Ultrapure water (18MΩcm) was generated using a Milli-Q system (MilliporeSigma, Bedford, MA, USA). Dextran, glycidyl methacrylate (GMA), dimethyl sulfoxide (DMSO), 3-mercaptopropionic acid (3-MPA), and 2,2′-(ethylenedioxy)diethanethiol were of analytical grade and purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA). Phosphate-buffered saline (PBS) was purchased from Scharlab SL (Barcelona, Spain).
Preparation of AA139 nanomedicines.
AA139 was entrapped in the two different nanoparticles PNP or MCL to prepare two distinct AA139 nanomedicines; PNPs were used to prepare polymeric nanoparticulate AA139 nanomedicines (AA139-PNP), and MCLs were used to prepare lipid-core micellar AA139 nanomedicines (AA139-MCL).
For preparation of AA139-PNP, dextran-based PNPs functionalized with 3-MPA were obtained as previously described in the literature (27). To prepare AA139-PNP, loading of AA139 to PNPs was carried out by noncovalent interaction with an AA139-to-PNP mass ratio of 10%. In brief, 100 mg AA139 was incubated with 1 g PNPs in 25 ml of saline solution at a pH of 7.2 for 15 h at room temperature. This was the maximum amount of AA139 that could be used, as proportions of AA139 greater than 10% resulted in aggregation, as larger particles sizes were observed by dynamic light scattering (DLS). The crude complex was then purified by membrane ultrafiltration using Vivaspin 500 10-kDa molecular weight cutoff (MWCO) polyethersulphone (PES) centrifugal concentrators (GE Healthcare, Chicago, USA) and an aqueous saline solution at pH of 7.2 to eliminate any unbound AA139. The maximum loading, established as the maximum concentration of peptide at which no peptide was collected in the filtrate, was 0.1 mg AA139/1 mg PNP. The AA139-PNP particle dispersions (corresponding to 5 mg/ml AA139 and 50 mg/ml PNP) in normal saline solution (NSS) of 0.9% (wt) NaCl had a mean size of 20 nm, as determined by DLS, and were negatively charged. High-performance liquid chromatography (HPLC) studies demonstrated that the nanomedicines were stable for at least 1 year when frozen at –20°C. Size measurement of AA139-PNP after freeze-thaw conditions demonstrated that the diameter of the nanomedicines was not affected by these storage conditions. Further dilution of this pristine dispersion of AA139-PNP in NSS for use in in vitro and in vivo experiments did not show any sign of instability.
For the preparation of AA139-MCL, entrapment of AA139 in MCL was carried out during the formation of MCLs, which were prepared by using a thin lipid film hydration method. In brief, a thin lipid film of DSPE-PEG2000 was prepared by dissolving the lipid in MeOH in a glass, round-bottom flask and evaporating the organic solvent under reduced pressure. Residual solvent was removed under a nitrogen flow. The lipid film was hydrated at 30°C in PBS containing the AA139 peptide at a 1:3 molar ratio of the peptide to DSPE-PEG2000. This was the maximum amount of AA139 that could be used, as higher proportions of AA139 resulted in aggregation, as determined by DLS. The eluted product was collected in 100-μl fractions. AA139-MCL has a size of 15.4 (±1.5) nm according to DLS measurement and a near neutral zeta-potential of −2.1 (±2.2) mV, as was measured from MCL prepared in 10 mM HEPES-buffered saline (HBS) buffer. The loading was between 10 and 13 mg/ml AA139 in 11 to 14 mM DSPE-PEG2000, with a loading efficiency of 100% (±4%). Further dilution of this pristine dispersion of AA139-MCL in NSS for use in in vitro and in vivo experiments did not show any sign of instability.
The preparation of unloaded PNPs and MCLs for control experiments was identical to the preparation process of their respective AA139-nanomedicines. The concentration of unloaded PNPs and MCLs was adjusted with NSS after the preparation process to ensure that the administered concentration of unloaded PNPs and MCLs was identical to the administered concentration of nanoparticles in the nanomedicine formulations.
All nanomedicine preparations were transported to partner locations in insulated thermal boxes with cooling elements frozen at –20°C to ensure that sample temperatures did not exceed –5°C. A selection of samples were sent back to the original partner faculties (CIDETEC or Utrecht University) for reanalysis to confirm stability under these transportation conditions.
Bacterial strains.
The ESBL-positive strain K. pneumoniae ESBL EMC2003 (referred to in this publication as K. pneumoniae ESBL) used in this study has previously been characterized in terms of its genotype and antimicrobial susceptibility profile and was used to establish pneumonia-septicemia in rats (38). The stability of the plasmid-containing K. pneumoniae ESBL strain was confirmed through five consecutive passages in Mueller-Hinton II (MH-II) broth (Becton, Dickinson BV, Vianen, The Netherlands). The virulence of the bacterial strain was maintained by rat lung passage every 12 months.
Concentration- and time-dependent antimicrobial activity of AA139, AA139-PNP, and AA139-MCL in vitro.
The antimicrobial activity of AA139, AA139-PNP, and AA139-MCL was compared using the time-kill kinetics (TKK) assay as previously described (25). Three 2-fold increasing antibiotic concentrations were used, representing (i) the lowest concentration at which AA139 showed bacterial growth inhibition; (ii) the MIC of AA139; and (iii) the lowest concentration at which AA139 led to complete bacterial killing after 24 hours of antibiotic exposure. These AA139 concentrations were 2 mg/liter, 4 mg/liter, and 8 mg/liter, respectively. AA139-PNP (corresponding to 20 mg/liter, 40 mg/liter, and 80 mg/liter of PNP) and AA139-MCL (corresponding to 2.2 μM, 4.5 μM, and 9 μM MCL) were tested at the same AA139 concentrations based on the AA139 content of the nanomedicines. Samples were 10-fold serially diluted and subcultured onto MH-II agar plates (Becton, Dickinson BV) for CFU counts after 24 hours at 37°C.
Animals.
Female Sprague-Dawley rats bred at Janvier Labs (Le Genest-Saint-Isle, France) were used for the in vivo imaging studies (age, 6 to 8 weeks). Specific-pathogen-free (SPF) male strain RP/AEur/RijHsd albino rats bred at the Laboratory Animals Center of Erasmus University Medical Center Rotterdam (Erasmus MC) were used for the in vivo tolerability and infection studies. Rats (age, 10 to 18 weeks; body weight, 250 to 350 g) were housed individually in ventilated cages with food and water ad libitum. Rats were randomly allocated to experimental groups once they reached the appropriate age and body weight. Group sizes were based on estimates of the hazard ratio. Euthanasia was applied by CO2 exposure when humane endpoints were reached or at termination of experiments. Infected rats were monitored every 12 hours for 11 days to assess the disease progression and signs of acute toxicity, as well as predefined humane endpoints by changes in body temperature and body weight and by external symptoms of disease, including ungroomed appearance, pallor, nose bleeding, lack of reactivity, inactivity, instability, or abnormal breathing. Rat survival was based on rats reaching humane endpoints, at which point euthanasia was applied by CO2 exposure. The rat survival parameter thus reflects rats reaching humane endpoints. After dissection, bacteria isolated from lung and blood were identified by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry (Bruker Daltonics, Bremen, Germany) to rule out coinfection as a preestablished exclusion criterium.
Ethics.
Animals were maintained and handled in accordance with the Guidelines for Accommodation and Care of Animals (European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes). All animal procedures were performed in accordance with either the Dutch Animal Experimentation Act (BWBR0003081) or the Spanish policy for animal protection (RD53/2013), both of which meet the requirements of the European Union Animal Directive (2010/63/EU). Experimental procedures were approved by either the Ethical Committee of the Asociación Centro de Investigación Cooperativa en Biomateriales (CIC biomaGUNE) or by the Institutional Animal Care and Use Committee of the Erasmus MC. The current study was designed to abide by the three Rs principles of animal research (replace, reduce, and refine) whenever possible (34) and was written to conform to the animal research: reporting of in vivo experiments (ARRIVE) guidelines for reporting animal research (43).
Endotracheal aerosolization of AA139, AA139-PNP, and AA139-MCL in rats.
Rats were anesthetized with 3% to 5% isoflurane (IsoFlo; Abbott Laboratories, Lake Bluff, IL, USA) in pure O2 for 5 minutes to ensure deep sedation. Animals were placed on a rodent workstand (Hallowell EMC, Pittsfield, MA, USA) in supine position inclined at a 45° angle. Endotracheal aerosolization was performed using a MicroSprayer-syringe assembly for rat (Penn-Century, Inc., Wyndmoor, PA, USA). A small animal laryngoscope (Penn-Century, Inc.) was used for visualization of the epiglottis, ensuring correct positioning of the tip just above the carina. A predefined volume of nanomedicine suspension was administered by endotracheal aerosolization, and rats remained in place for 10 seconds after administration to allow for inhalation.
Radiolabeling of AA139, AA139-PNP, and AA139-MCL for imaging studies.
The radio-iodination of AA139 was carried out by electrophilic aromatic substitution on the tyrosine residues. For that purpose, 1-mg/ml AA139 solution was incubated with [124I]NaI (Perkin Elmer, Inc., Waltham, MA, USA) in 0.2 M sodium acetate buffer solution (50 μl; pH 5.5) for 2 hours at 25°C in the presence of Iodo-beads (Thermo Fisher Scientific, Waltham, MA, USA). The crude reaction mixture was purified by retention in a Sep-Pak C18 plus light cartridge (Waters Corporation, Milford, MA, USA) and subsequent elution using 1 ml of 0.1% aqueous trifluoroacetic acid (TFA) solution/ethanol (20/80). The solvent was evaporated and the residue reconstituted with 0.2 M sodium acetate buffer solution (300 μl; pH 5.5). Chemical and radiochemical purity were determined by HPLC with radioactive detection (radio-HPLC), using a Mediterranea Sea 18 (5 μm; 15 by 0.46 cm) column (Teknokroma Analítica SA, Barcelona, Spain) as the stationary phase and 0.1% solution of TFA in water (A) and 0.1% solution of TFA in acetonitrile (B) as mobile phase, using the following gradient: initial, 90% (A) and 10% (B); 4 min, 85% (A) and 15% (B); 6 min, 75% (A) and 25% (B); 10 min, 75% (A) and 25% (B); 16 min, 5% (A) and 95% (B); 18 min, 5% (A) and 95% (B); 20 min, 90% (A) and 10% (B); and 22 min, 90% (A) and 10% (B). Overall decay-corrected radiochemical yield was 62% ± 2%, and the molar activity was within the range of 0.5 to 1.5 GBq/μmol at the end of the synthesis.
Radiochemical stability of the 124I-labeled AA139 was assessed by incubation in different media at 37°C, including moderately acidic conditions (sodium acetate buffer, pH of 5.5). At different time points, samples were withdrawn and analyzed by radio-HPLC using the experimental conditions described above. Radiochemical stability was directly calculated from chromatographic profiles. [124I]AA139 was stable in moderately acidic conditions similar to the infected lung environment for 48 h and did not lead to detachment of 124I from AA139 (see Fig. S1 in the supplemental material).
Labeled [124I]AA139-PNP and [124I]AA139-MCL nanomedicines were prepared as described above using [124I]AA139. For the preparation of [124I]AA139-MCL nanomedicines, Illustra Nap-5 columns prepackaged with the Sephadex G-25 DNA grade solution (GE Healthcare, Chicago, IL, USA) were preconditioned with a sodium acetate buffer solution (10 ml; 0.02 M; pH of 5.5) and then used for final purification by size exclusion chromatography, using PBS (10 mM; 0.8% NaCl; pH of 7.4) as the mobile phase.
The radiochemical yields of labeled [124I]AA139-PNP and [124I]AA139-MCL were calculated as the ratio between the amount of radioactivity present in [124I]AA139 nanomedicines and the starting amount of [124I]AA139. When the [124I]AA139-to-PNP mass ratio was set to 1:10, quantitative attachment of AA139 to PNP was achieved and radiochemical yields close to 60% were achieved after purification of [124I]AA139-PNP. [124I]AA139 was efficiently attached to PNP as well as efficiently incorporated within MCL. Incorporation of [124I]AA139 into MCL resulted in >95% radiochemical yields in the initial fractions of [124I]AA139-MCL.
Imaging studies of AA139, AA139-PNP, and AA139-MCL in uninfected rats.
Rats were administered 50 μl (ca. 1.85 MBq) of [124I]AA139, [124I]AA139-PNP, or [124I]AA139-MCL by endotracheal aerosolization in groups of 4 rats. One group of 2 rats was administered with an equivalent amount of [124I]NaI in saline solution as the control. Immediately after endotracheal aerosolization, animals were positioned in an eXploreVista computerized tomography (CT) and positron emission tomography (PET) small animal preclinical imaging system (GE Healthcare). A PET scan was acquired over 40 minutes, while anesthesia was maintained using 1% to 2% isoflurane in pure O2 and body temperature was maintained with a homeothermic blanket control unit (Bruker BioSpin GmbH, Karlsruhe, Germany) to prevent hypothermia. Animals were then allowed to recover from anesthesia and were returned to their cages. Imaging sessions were repeated at 3, 6, 9, 15, and 24 hours after administration.
After each PET acquisition, a CT scan (X-ray energy, 40 kV; intensity, 140 μA) was performed for a later attenuation correction application in the image reconstruction, as well as for unambiguous localization of the radioactive signal. Random and scatter corrections were also applied to the reconstructed image (filtered back projection reconstruction algorithm), generating a 175- by 175- by 220-voxel image, with a 2-mm axial full width at half maximum (FWHM) spatial resolution in the center of the field of view (FOV). After reconstruction, images were analyzed using πMOD analysis software (version 3.4; PMOD Technologies, Ltd., Zürich, Switzerland). Volumes of interest (VOIs) were manually delineated in the whole lungs, and the concentration of radioactivity in the lungs was determined for each compound and time point.
Deposition of AA139 in lungs and blood of uninfected rats.
Deposition of AA139 in lungs and its distribution in blood plasma were determined in 6 rats which were sacrificed immediately after endotracheal aerosolization of 1 mg AA139 in 100 μl. The five lung lobes were collected separately, weighed, and homogenized in 2 ml NSS using the gentleMACS Octo dissociator (Milteny Biotec BV, Leiden, The Netherlands). Blood plasma was obtained from whole blood collected via cardiac puncture in lithium-heparin tubes (Sarstedt BV, Etten-Leur, The Netherlands), and total rat blood plasma volume was estimated based on body weight (44). AA139 concentrations were determined at Covance Laboratories (Harrogate, UK) using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) detection bioanalytical assay.
Limiting dose of AA139, AA139-PNP, and AA139-MCL in uninfected rats.
For determining the maximum tolerated dose (MTD) or maximum feasible dose (MFD), rats were administered 100 μl of AA139, AA139-PNP, or AA139-MCL by endotracheal aerosolization in groups of 11 rats, starting at a low dose of 0.125 mg AA139 based on the AA139 content of the nanomedicines (corresponding to 1.25 mg/ml AA139, 12.5 mg/ml PNP, and 0.14 mM MCL). A total of 100 μl of normal saline solution (NSS) was used as a control treatment. After administration, animals were placed on an electric heating pad to emerge from anesthesia before being placed back in cages. Rats were then regularly checked for signs of acute toxicity over 24 hours. After 24 hours, rats were sacrificed, and blood plasma was obtained from whole blood collected via cardiac puncture in lithium-heparin tubes to assess blood biomarkers for acute toxicity, namely, alanine aminotransferase (ALAT), aspartate aminotransferase (ASAT), creatinine, and blood urea nitrogen (BUN), at the Department of Clinical Chemistry, Erasmus MC. When no acute toxicity was observed, the dose was increased 2-fold, either until acute toxicity was observed (in which case the lower dose was determined as the MTD) or until the dose could not be further increased due to technical limitations, thereby reaching the MFD.
Rat model of pneumonia-septicemia.
Bilateral K. pneumoniae ESBL pneumonia-septicemia was induced as described previously (38). In short, suspensions of washed bacteria in the logarithmic phase of growth were used to prepare inocula. After intubation and cannulation of the trachea under anesthesia, rats were held in a vertical position. Rat lungs were inoculated with 60 μl PBS containing 2 × 106 K. pneumoniae ESBL, followed by inhalation.
Early bacterial killing activity of AA139, AA139-PNP, and AA139-MCL in infected rats.
K. pneumoniae ESBL pneumonia-septicemia was induced in groups of 6 rats. At 24 hours after initiation of infection, rats were administered 100 μl of AA139, AA139-PNP, or AA139-MCL by endotracheal aerosolization administered at the same dose which was the MTD of free A139. Rats were administered 100 μl of unloaded PNP, MCL, or NSS as a control treatment. Rats were sacrificed at 0 hours (before start of treatment) and at 2, 6, and 24 hours after start of treatment, with 2 rats per time point per treatment group. Lungs were homogenized in 5 ml NSS using the gentleMACS Octo Dissociator (Milteny Biotec BV). Lung homogenates were diluted and subcultured onto MH-II agar plates for CFU count after 20 hours of incubation of plates at 37°C. Blood plasma was obtained for determination of blood biomarkers of acute toxicity as described above.
Therapeutic efficacy of AA139, AA139-PNP, and AA139-MCL in infected rats.
K. pneumoniae ESBL pneumonia-septicemia was induced in groups of 12 rats. At 24 hours after the initiation of infection, rats were administered 100 μl of AA139, AA139-PNP, or AA139-MCL once-daily (q24h) by endotracheal aerosolization. The treatments were all administered at their respective limiting dose, as well as at the MTD of free AA139. Rats were administered 100 μl of unloaded PNP, unloaded MCL, or NSS as control treatments. The disease progression was monitored twice-daily (q12h) for 10 days. Rats having reached humane endpoints were euthanized and dissected to check for the presence of K. pneumoniae ESBL in lungs and blood.
Statistics.
Statistical significance of bacterial growth inhibition was determined by interpolating four-parameter sigmoidal growth curves and performing extra-sum-of-squares F-tests for the slope in Prism 5.01 (GraphPad, Inc., San Diego, CA, USA). Kaplan-Meier rat survival curves were generated, and statistical differences in rat survival rates were calculated using the log rank test in Prism 5.01.
Supplementary Material
ACKNOWLEDGMENTS
The analysis of blood plasma was performed by the Clinical Pharmacology and Toxicology Laboratory at the Erasmus University Medical Center, Rotterdam, The Netherlands. The LC-MS/MS bioanalytical procedure was performed by Covance Laboratories, Harrogate, United Kingdom.
This work was financially supported by the European Union’s Seventh Programme for Research, Technological Development and Demonstration under grant agreement no. 604434 PneumoNP.
This study was performed in collaboration with CIDETEC, who are the patent holders of AA139-PNP. It was approved for publication by all consortium members of the PneumoNP project, including Adenium Biotech ApS, who are the patent holders of AA139.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Marshall DC, Goodson RJ, Xu Y, Komorowski M, Shalhoub J, Maruthappu M, Salciccioli JD. 2018. Trends in mortality from pneumonia in the Europe union: a temporal analysis of the European detailed mortality database between 2001 and 2014. Respir Res 19:81. doi: 10.1186/s12931-018-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Welte T, Torres A, Nathwani D. 2012. Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax 67:71–79. doi: 10.1136/thx.2009.129502. [DOI] [PubMed] [Google Scholar]
- 3.Sader HS, Farrell DJ, Flamm RK, Jones RN. 2014. Antimicrobial susceptibility of Gram-negative organisms isolated from patients hospitalised with pneumonia in US and European hospitals: results from the SENTRY Antimicrobial Surveillance Program, 2009–2012. Int J Antimicrob Agents 43:328–334. doi: 10.1016/j.ijantimicag.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Bassetti M, Welte T, Wunderink RG. 2016. Treatment of Gram-negative pneumonia in the critical care setting: is the beta-lactam antibiotic backbone broken beyond repair? Crit Care 20:19. doi: 10.1186/s13054-016-1197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler MS, Blaskovich MA, Cooper MA. 2017. Antibiotics in the clinical pipeline at the end of 2015. J Antibiot (Tokyo) 70:3–24. doi: 10.1038/ja.2016.72. [DOI] [PubMed] [Google Scholar]
- 6.Lewis K. 2013. Platforms for antibiotic discovery. Nat Rev Drug Discov 12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 7.Hancock RE, Sahl H-G. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 8.Marr AK, Gooderham WJ, Hancock RE. 2006. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol 6:468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Pini A, Falciani C, Bracci L. 2008. Branched peptides as therapeutics. Curr Protein Pept Sci 9:468–477. doi: 10.2174/138920308785915227. [DOI] [PubMed] [Google Scholar]
- 10.Fox JL. 2013. Antimicrobial peptides stage a comeback. Nature Biotechnol 31:379–382. doi: 10.1038/nbt.2572. [DOI] [PubMed] [Google Scholar]
- 11.Zazo H, Colino CI, Lanao JM. 2016. Current applications of nanoparticles in infectious diseases. J Control Release 224:86–102. doi: 10.1016/j.jconrel.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Biswaro LS, da Costa Sousa MG, Rezende T, Dias SC, Franco OL. 2018. Antimicrobial peptides and nanotechnology, recent advances and challenges. Front Microbiol 9:855. doi: 10.3389/fmicb.2018.00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huh AJ, Kwon YJ. 2011. “Nanoantibiotics”: a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J Control Release 156:128–145. doi: 10.1016/j.jconrel.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Wenzler E, Fraidenburg DR, Scardina T, Danziger LH. 2016. Inhaled antibiotics for Gram-negative respiratory infections. Clin Microbiol Rev 29:581–632. doi: 10.1128/CMR.00101-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang W, Peters JI, Williams RO III. 2008. Inhaled nanoparticles—a current review. Int J Pharm 356:239–247. doi: 10.1016/j.ijpharm.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Patton JS, Byron PR. 2007. Inhaling medicines: delivering drugs to the body through the lungs. Nat Rev Drug Discov 6:67–74. doi: 10.1038/nrd2153. [DOI] [PubMed] [Google Scholar]
- 17.Sung JC, Pulliam BL, Edwards DA. 2007. Nanoparticles for drug delivery to the lungs. Trends Biotechnol 25:563–570. doi: 10.1016/j.tibtech.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Bailey MM, Berkland CJ. 2009. Nanoparticle formulations in pulmonary drug delivery. Med Res Rev 29:196–212. doi: 10.1002/med.20140. [DOI] [PubMed] [Google Scholar]
- 19.Ritsema JA, van der Weide H, Te Welscher YM, Goessens WH, Van Nostrum CF, Storm G, Bakker-Woudenberg IA, Hays JP. 2018. Antibiotic-nanomedicines: facing the challenge of effective treatment of antibiotic-resistant respiratory tract infections. Future Microbiol 13:1683–1692. doi: 10.2217/fmb-2018-0194. [DOI] [PubMed] [Google Scholar]
- 20.Andrade F, Rafael D, Videira M, Ferreira D, Sosnik A, Sarmento B. 2013. Nanotechnology and pulmonary delivery to overcome resistance in infectious diseases. Adv Drug Deliv Rev 65:1816–1827. doi: 10.1016/j.addr.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frima HJ, Gabellieri C, Nilsson M-I. 2012. Drug delivery research in the European Union’s Seventh Framework Programme for Research. J Control Release 161:409–415. doi: 10.1016/j.jconrel.2012.01.044. [DOI] [PubMed] [Google Scholar]
- 22.Huang JX, Neve S, Elliot AG, Cain AK, Boinett CJ, Zuegg J, Steen J, Ramu S, Kavanaugh AM, Pelingon R, Butler MS, Blaskovich MA, Cooper MA. 2014. Novel arenicin-3 peptide antibiotics with broad-spectrum activity against MDR Gram-negative bacterial act via dual mode of actions, poster F-256. 54th International Conference of Antimicrobial Agents and Chemotherapy (ICAAC) and Infectious Diseases Society of America (IDSA) Joint Meeting, Washington, DC. [Google Scholar]
- 23.Neve S, Lociuro S, Morrissey I, Hawser S, Nordkild P. 2014. The arenicin-3 derived clinical candidate AA139 shows potent activity against Gram-negative pathogens, poster F-261. Abstr 54th ICAAC Meeting, Washington, DC, 5 to 9 September 2014. [Google Scholar]
- 24.Cain AK, Boinett CJ, Barquist L, Neve S, Huang JX, Elliot AG, Butler MS, Blaskovich MA, Parkhill J, Cooper MA. 2014. Transposon directed insertion-site sequencing (TraDIS) to elucidate the mode of action of the antimicrobial arenicin-3 (Arn-3), poster F-256. 54th International Conference of Antimicrobial Agents and Chemotherapy (ICAAC) and Infectious Diseases Society of America (IDSA) Joint Meeting, Washington, DC. [Google Scholar]
- 25.van der Weide H, Vermeulen-de Jongh DMC, van der Meijden A, Boers SA, Kreft D, Ten Kate MT, Falciani C, Pini A, Strandh M, Bakker-Woudenberg IAJM, Hays JP, Goessens WHF. 2019. Antimicrobial activity of two novel antimicrobial peptides AA139 and SET-M33 against clinically and genotypically diverse Klebsiella pneumoniae isolates with differing antibiotic resistance profiles. Int J Antimicrob Agents 54:159–166. doi: 10.1016/j.ijantimicag.2019.05.019. [DOI] [PubMed] [Google Scholar]
- 26.Lociuro S. 2014. Oral presentation: arenicin antimicrobial peptides (novel AMPs with a novel mode of action). 54th International Conference of Antimicrobial Agents and Chemotherapy (ICAAC) and Infectious Diseases Society of America (IDSA) Joint Meeting, Washington, DC. [Google Scholar]
- 27.Gracia R, Marradi M, Cossío U, Benito A, Pérez-San Vicente A, Gómez-Vallejo V, Grande H-J, Llop J, Loinaz I. 2017. Synthesis and functionalization of dextran-based single-chain nanoparticles in aqueous media. J Mater Chem B 5:1143–1147. doi: 10.1039/c6tb02773c. [DOI] [PubMed] [Google Scholar]
- 28.Torchilin VP. 2001. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release 73:137–172. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 29.Kröger APP, Paulusse JM. 2018. Single-chain polymer nanoparticles in controlled drug delivery and targeted imaging. J Control Release 286:326–347. doi: 10.1016/j.jconrel.2018.07.041. [DOI] [PubMed] [Google Scholar]
- 30.Sawant RR, Torchilin VP. 2010. Multifunctionality of lipid-core micelles for drug delivery and tumour targeting. Mol Membr Biol 27:232–246. doi: 10.3109/09687688.2010.516276. [DOI] [PubMed] [Google Scholar]
- 31.Murgia X, Pawelzyk P, Schaefer UF, Wagner C, Willenbacher N, Lehr C-M. 2016. Size-limited penetration of nanoparticles into porcine respiratory mucus after aerosol deposition. Biomacromolecules 17:1536–1542. doi: 10.1021/acs.biomac.6b00164. [DOI] [PubMed] [Google Scholar]
- 32.Gill KK, Nazzal S, Kaddoumi A. 2011. Paclitaxel loaded PEG5000–DSPE micelles as pulmonary delivery platform: formulation characterization, tissue distribution, plasma pharmacokinetics, and toxicological evaluation. Eur J Pharm Biopharm 79:276–284. doi: 10.1016/j.ejpb.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 33.Cossío U, Gómez-Vallejo V, Flores M, Gañán-Calvo B, Jurado G, Llop J. 2018. Preclinical evaluation of aerosol administration systems using positron emission tomography. Eur J Pharm Biopharm 130:59–65. doi: 10.1016/j.ejpb.2018.05.037. [DOI] [PubMed] [Google Scholar]
- 34.Balls M, Goldberg AM, Fentem JH, Broadhead CL, Burch RL, Festing MF, Frazier JM, Hendriksen CF, Jennings M, van der Kamp M. 1995. The three Rs: the way forward: the report and recommendations of ECVAM Workshop 11. Altern Lab Anim 23:838–866. [PubMed] [Google Scholar]
- 35.Kumari A, Yadav SK, Yadav SC. 2010. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces 75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Jain AK, Swarnakar NK, Godugu C, Singh RP, Jain S. 2011. The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials 32:503–515. doi: 10.1016/j.biomaterials.2010.09.037. [DOI] [PubMed] [Google Scholar]
- 37.Wieschowski S, Biernot S, Deutsch S, Glage S, Bleich A, Tolba R, Strech D. 2019. Publication rates in animal research. Extent and characteristics of published and non-published animal studies followed up at two German university medical centres. PLoS One 14:e0223758. doi: 10.1371/journal.pone.0223758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Weide H, ten Kate MT, Vermeulen-de Jongh DMC, van der Meijden A, Wijma RA, Boers SA, van Westreenen M, Hays JP, Goessens WHF, Bakker-Woudenberg I. 2020. Successful high-dosage monotherapy of tigecycline in a multidrug-resistant Klebsiella pneumoniae pneumonia–septicemia model in rats. Antibiotics 9:109. doi: 10.3390/antibiotics9030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brzezińska-Błaszczyk E, Czuwaj M, Kuna P. 1987. Histamine release from mast cells of various species induced by histamine releasing factor from human lymphocytes. Agents Actions 21:26–31. doi: 10.1007/BF01974916. [DOI] [PubMed] [Google Scholar]
- 40.Decorti G, Klugmann FB, Candussio L, Furlani A, Scarcia V, Baldini L. 1989. Uptake of adriamycin by rat and mouse mast cells and correlation with histamine release. Cancer Res 49:1921–1926. [PubMed] [Google Scholar]
- 41.Brown KL, Hancock RE. 2006. Cationic host defense (antimicrobial) peptides. Curr Opin Immunol 18:24–30. doi: 10.1016/j.coi.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 42.Brain JD, Knudson DE, Sorokin SP, Davis MA. 1976. Pulmonary distribution of particles given by intratracheal instillation or by aerosol inhalation. Environ Res 11:13–33. doi: 10.1016/0013-9351(76)90107-9. [DOI] [PubMed] [Google Scholar]
- 43.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. 2010. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee H, Blaufox M. 1985. Blood volume in the rat. J Nucl Med 26:72–76. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.