

Current antileishmanial treatment is hampered by limitations, such as drug toxicity and the risk of treatment failure, which may be related to parasitic drug resistance. Given the urgent need for novel drugs, the Drugs for Neglected Diseases initiative (DNDi) has undertaken a drug discovery program, which has resulted in the identification of aminopyrazoles, a highly promising antileishmanial chemical series. Multiple experiments have been performed to anticipate the propensity for resistance development.
KEYWORDS: Leishmania, ABC transporters, aminopyrazoles, resistance
ABSTRACT
Current antileishmanial treatment is hampered by limitations, such as drug toxicity and the risk of treatment failure, which may be related to parasitic drug resistance. Given the urgent need for novel drugs, the Drugs for Neglected Diseases initiative (DNDi) has undertaken a drug discovery program, which has resulted in the identification of aminopyrazoles, a highly promising antileishmanial chemical series. Multiple experiments have been performed to anticipate the propensity for resistance development. Resistance selection was performed by successive exposure of Leishmania infantum promastigotes (in vitro) and intracellular amastigotes (both in vitro and in golden Syrian hamsters). The stability of the resistant phenotypes was assessed after passage in mice and Lutzomyia longipalpis sandflies. Whole-genome sequencing (WGS) was performed to identify mutated genes, copy number variations (CNVs), and somy changes. The potential role of efflux pumps (the MDR and MRP efflux pumps) in the development of resistance was assessed by coincubation of aminopyrazoles with specific efflux pump inhibitors (verapamil, cyclosporine, and probenecid). Repeated drug exposure of amastigotes did not result in the emergence of drug resistance either in vitro or in vivo. Selection at the promastigote stage, however, was able to select for parasites with reduced susceptibility (resistance index, 5.8 to 24.5). This phenotype proved to be unstable after in vivo passage in mice and sandflies, suggesting that nonfixed alterations are responsible for the elevated resistance. In line with this, single nucleotide polymorphisms and indels identified by whole-genome sequencing could not be directly linked to the decreased drug susceptibility. Copy number variations were absent, whereas somy changes were detected, which may have accounted for the transient acquisition of resistance. Finally, aminopyrazole activity was not influenced by the MDR and MRP efflux pump inhibitors tested. The selection performed does not suggest the rapid development of resistance against aminopyrazoles in the field. Karyotype changes may confer elevated levels of resistance, but these do not seem to be stable in the vertebrate and invertebrate hosts. MDR/MRP efflux pumps are not likely to significantly impact the activity of the aminopyrazole leads.
INTRODUCTION
Visceral leishmaniasis (VL) is a disease generally caused by the protist parasites Leishmania infantum and Leishmania donovani, transmitted by the bite of infected sandflies (1, 2). The Leishmania parasite has a dimorphic life cycle, with a motile promastigote stage in the vector and a nonmotile amastigote stage in host myeloid cells. Infection can disseminate to the liver, spleen, and bone marrow, causing severe symptoms, such as prolonged fever, hepatosplenomegaly, pancytopenia, progressive anemia, and weight loss. VL can, therefore, be fatal without adequate treatment (3). Currently available therapies are limited, as they either lack sufficient effectiveness, show important levels of toxicity, or have to be administered parenterally. Drug resistance might also be a concern and could affect the efficacy of current and future new drugs in the field. Antimonials (Sb) have been the therapeutic option of choice for several decades but are no longer effective in the Indian subcontinent due to widespread resistance (4). Miltefosine (MIL), currently the only available oral drug, is confronted with increasing treatment failure rates (5–9). A few clinical isolates with reduced susceptibility to amphotericin B (AmB) have also been found (10). The emergence of drug resistance can often be ascribed to the ability of the parasite to overcome the cytotoxic effects of drugs either by causing mutations that alter the drug target or the uptake system or by overexpressing efflux pumps (11). A well-known efflux pump family is the ATP-dependent binding cassette (ABC) transporter superfamily (ABC transporters), which is located in extra- or intracellular membranes and which can extrude a wide variety of substrates. The upregulation of these transporters has already been linked to drug resistance in Leishmania (12, 13). More particularly, the upregulation of ABCC3 (MRPA or PGPA) and ABCC7 (PRP1) (14–16) and ABCB1 (MDR or PGP) (17) contributes significantly to various degrees of Sb resistance. Also, the reported AmB resistance has been linked to the overexpression of ABCB1 (10). Other efflux pumps of the same superfamily, such as ABCG4 and ABCG6, have also been identified as playing a role in drug resistance (18, 19). Additionally, host cell ABC transporters can codetermine the susceptibility of intracellular Leishmania stages (20), as these may regulate intracellular drug availability. For example, reduced drug exposure in Sb-resistant parasites was linked to the upregulation of the ABCC1 pump in macrophages (21). One approach to studying the involvement of ABC transporters in Leishmania drug resistance is the use of specific efflux pump inhibitors to reverse resistance (22–24).
To summarize, there is a crucial need for the development of novel drugs that are affordable, easy to use, and safe and that preferably have an alternative mode of action to the currently available antileishmanials. The latter is important for the possible implementation of combination therapies, which could result in improved efficacy, reduced side effects, a lower risk of resistance emergence, and shorter treatment durations (2, 25–27). Several new drug classes that have good efficacy against Leishmania have been discovered, including nitroimidazoles, oxaboroles, proteasome inhibitors, cdc2-related kinase 12 (CRK-12) inhibitors, and aminopyrazoles (APs) (25). Simultaneously, efforts to determine their properties and mode of action have been or are being undertaken (25, 28–30). However, aside from the pharmacodynamics in vitro and in vivo (28, 31), not much is known about the antileishmanial action of the AP class. One study has identified mitogen-activated protein kinases (MAPK) and cdc2-related kinases (CRK) to be potential target proteins of a diaminopyrimidine in Trypanosoma brucei and Leishmania major (32). Other distant analogues have been used in oncology and shown to inhibit human cyclin-dependent kinases (CDK) (33–35) and posttranslational modification processes (36). In Plasmodium falciparum, mutations in a P-type ATPase were shown to confer resistance to GNF-Pf4492, a pyrazole urea (37). All these studies have been performed with distant analogues and may provide only some indication for the antileishmanial effects. Additionally, it is essential to explore the possibility of the rapid development of resistance against novel compounds, as this may severely impact a drug’s clinical life span once it is widely used in the field. The number of alternatives in the drug discovery pipeline is rather scarce, and this kind of information is important to safeguard novel drugs from the emergence of resistance, to guide dosing regimens, and to predict the importance of incorporation in a combination therapy.
The aim of this laboratory study was therefore (i) to evaluate the propensity of VL species to develop resistance to the new AP lead series, (ii) to characterize the resistant strains obtained, and (iii) to check the involvement of the major efflux pumps MDR and MRP in both the parasite and the macrophage host cell.
RESULTS
In vitro and in vivo resistance selection on the amastigote stage.
Selection for resistance to the AP series was performed to evaluate the possible propensity toward resistance to these compounds in the field and to allow mode-of-action/resistance studies. Both in vitro and in vivo intracellular resistance selection procedures have been shown to be successful in previous studies (38, 39). In vitro resistance selection by five successive cycles of exposure of intracellular L. infantum amastigotes (using laboratory strain MHOM/MA/67/ITMAP263 [ITMAP263] and field strain MHOM/FR/09/LEM4038 [LEM4038]) to the AP lead compound DNDI-1044 did not result in significant differences in 50% inhibitory concentration (IC50) values between the wild-type (WT) parent and the selected parasites (Table 1). Similarly, resistance selection of the lab strain (ITMAP263) in golden Syrian hamsters failed to induce a shift in the IC50 for DNDI-1044 when the IC50s for the wild-type parent (0.46 ± 0.09 μM) and the ex vivo amastigotes collected after five successive selection cycles (0.42 ± 0.07 μM) were compared.
TABLE 1.
In vitro susceptibility of intracellular amastigotes to DNDI-1044 after each in vitro selection cyclea
| Cycle | Mean IC50 (μM) ± SEM |
|
|---|---|---|
| ITMAP263 | LEM4038 | |
| Wild type | 0.19 ± 0.08 | 0.35 ± 0.07 |
| Cycle 1 | 0.21 ± 0.02 | 0.43 ± 0.08 |
| Cycle 2 | 0.60 ± 0.07 | 0.27 ± 0.04 |
| Cycle 3 | 0.45 ± 0.09 | 0.36 ± 0.11 |
| Cycle 4 | 0.53 ± 0.15 | 0.56 ± 0.17 |
| Cycle 5 | 0.24 ± 0.08 | 0.86 ± 0.30 |
The results are based on two independent replicates run in duplicate.
In vitro resistance selection on the promastigote stage.
As resistance selection in the relevant host amastigote stage failed, an attempt was made in the extracellular insect promastigote stage using the L. infantum MHOM/FR/96/LEM3323 (LEM3323) clinical isolate. Selection for resistance to the two selected APs was successful within approximately 60 days. Drug exposure could be increased in a stepwise fashion to a maximum of 20 μM and 45 μM for DNDI-1044 and DNDI-8012, respectively (Fig. 1). Higher drug concentrations led to parasite death. The stepwise selection showed a gradual increase in the IC50 upon each successive passage, with a significant increase being seen after 4 passages in the extracellular assay (see Fig. S1 in the supplemental material). Selection for resistance to both DNDI-1044 and DNDI-8012 was therefore successful, with the resistance indexes (RI) being 24.5 and 5.8, respectively. Cross-resistance was observed for both resistant lines (Table 2). An intracellular susceptibility assay was performed to assess resistance at the amastigote stage. Only the DNDI-1044-selected resistant line (LEM3323Cl4/1044) retained its resistant and cross-resistant phenotype at the amastigote stage (Table 2; Fig. S2). These resistant strains selected in the promastigote stage were used for further characterization.
FIG 1.
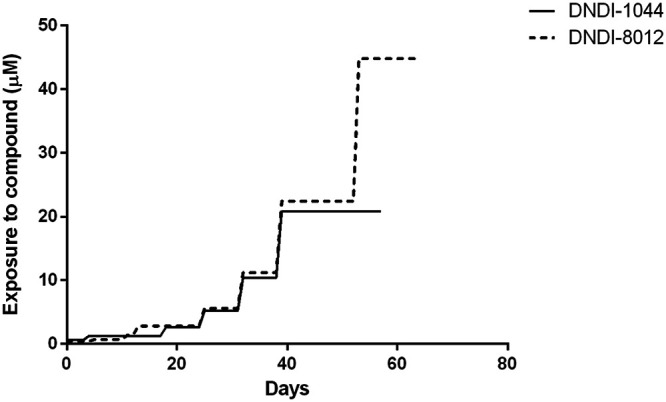
Overview of the stepwise generation of extracellular promastigote lines resistant to two APs.
TABLE 2.
Resistance index for the AP-selected lines after the highest exposurea
| Compound | Extracellular promastigote assay |
Intracellular amastigote assay |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild type vs LEM3323Cl4/1044 |
Wild type vs LEM3323Cl4/8012 |
Wild type vs LEM3323Cl4/1044 |
Wild type vs LEM3323Cl4/8012 |
|||||||||
| Mean IC50 (μM) ± SEM |
RI | Mean IC50 (μM) ± SEM |
RI | Mean IC50 (μM) ± SEM |
RI | Mean IC50 (μM) ± SEM |
RI | |||||
| Wild type | LEM3323Cl4/1044 | Wild type | LEM3323Cl4/8012 | Wild type | LEM3323Cl4/1044 | Wild type | LEM3323Cl4/8012 | |||||
| DNDI-1044 | 0.20 ± 0.04 | 4.90 ± 0.55 | 24.5 | 0.34 ± 0.11 | 2.78 ± 0.55 | 8.2 | 0.44 ± 0.12 | 2.01 ± 0.46 | 4.6 | 0.29 ± 0.01 | 0.37 ± 0.01 | 1.3 |
| DNDI-8012 | 0.52 ± 0.02 | 7.55 ± 1.30 | 14.5 | 0.62 ± 0.12 | 3.62 ± 0.31 | 5.8 | 0.54 ± 0.10 | 2.20 ± 0.35 | 4.1 | 0.22 ± 0.00 | 0.19 ± 0.01 | 0.9 |
The results are based on two independent replicates run in duplicate. The intracellular assay with LEM3323Cl4/8012 was performed only once in duplicate, as no differences were observed. RI, resistance index.
Generation of clones.
The microdrop technique was used to obtain individual clones of the two resistant strains, to reduce variability in further experiments. A total of nine clones (LEM3323Cl4/8012Cl1 [8012Cl1] to LEM3323Cl4/8012Cl9 [8012Cl9]) were generated from the DNDI-8012-selected line (LEM3323Cl4/8012), and seven more clones (LEM3323Cl4/1044Cl1 [1044Cl1] to LEM3323Cl4/1044Cl7 [1044Cl7]) were obtained from the DNDI-1044-selected resistant L. infantum line (LEM3323Cl4/1044). Despite the slight decrease in RI that could be observed after the cloning procedure, the resistance profile in the promastigote parasite stage was maintained for both selected lines (Fig. 2; Table 3). One line, namely, LEM3323Cl4/1044Cl7, did not show a resistant phenotype. Cross-resistance was observed for all tested APs, which also included the new lead DNDI-5561. No resistant phenotype was observed for the intracellular amastigotes of the LEM3323Cl4/8012 clones, which was expected, given the susceptible characteristic of the polyclonal parent line. The polyclonal LEM3323Cl4/1044-selected line, on the contrary, did show a resistant phenotype with an RI of ±4 for intracellular amastigotes. However, the full resistant phenotype could not be maintained with some of the clones displaying an intermediate resistant phenotype (clones LEM3323Cl4/1044Cl1, LEM3323Cl4/1044Cl2, LEM3323Cl4/1044Cl3, and LEM3323Cl4/1044Cl5). The LEM3323Cl4/1044Cl1 line was the first clone to be generated and was used in further experiments. In the resistant clones, cross-resistance was observed for all tested APs (Fig. 2; Table 4).
FIG 2.

Comparison of in vitro intracellular amastigote (A and C) and extracellular promastigote (B and D) susceptibility to the selected AP series between the wild-type parent (LEM3323Cl4) and the successfully cultured adapted clones of the DNDI-1044-selected (A and B) and DNDI-8012-selected (C and D) resistant lines. Results are expressed as the mean IC50 ± SEM and are based on two independent replicates run in duplicate (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). For DNDI-8012-generated clones, only a few clones were selected for amastigote susceptibility assessment.
TABLE 3.
Resistance index for the AP-selected lines in the extracellular promastigote assaya
| Compound | Wild type vs LEM3323Cl4/1044Cl1 |
Wild type vs LEM3323Cl4/8012Cl2 |
||||
|---|---|---|---|---|---|---|
| Mean IC50 (μM) ± SEM |
RI | Mean IC50 (μM) ± SEM |
RI | |||
| Wild type | LEM3323Cl4/1044Cl1 | Wild type | LEM3323Cl4/8012Cl2 | |||
| DNDI-1044 | 0.50 ± 0.06 | 2.79 ± 0.61 | 5.6 | 0.50 ± 0.06 | 2.52 ± 0.81 | 5.0 |
| DNDI-8012 | 0.67 ± 0.12 | 3.48 ± 0.75 | 5.2 | 0.67 ± 0.12 | 3.70 ± 1.17 | 5.5 |
| DNDI-5561 | 0.82 ± 0.16 | 4.45 ± 0.93 | 5.4 | 0.82 ± 0.16 | 6.57 ± 0.76 | 8.0 |
Results are based on two independent replicates run in duplicate. RI, resistance index.
TABLE 4.
Resistance index for the AP-selected lines in the intracellular amastigote assaya
| Compound | Wild type vs LEM3323Cl4/1044Cl1 |
Wild type vs LEM3323Cl4/8012Cl2 |
||||
|---|---|---|---|---|---|---|
| Mean IC50 (μM) ± SEM |
RI | Mean IC50 (μM) ± SEM |
RI | |||
| Wild type | LEM3323Cl4/1044Cl1 | Wild type | LEM3323Cl4/8012Cl2 | |||
| DNDI-1044 | 0.18 ± 0.05 | 0.61 ± 0.08 | 3.4 | 0.21 ± 0.03 | 0.20 ± 0.01 | 1.0 |
| DNDI-8012 | 0.30 ± 0.09 | 0.90 ± 0.10 | 3.0 | 0.28 ± 0.02 | 0.42 ± 0.02 | 1.5 |
| DNDI-5561 | 0.10 ± 0.003 | 0.37 ± 0.09 | 3.6 | 0.16 ± 0.01 | 0.28 ± 0.10 | 1.8 |
Results are based on two independent replicates run in duplicate. RI, resistance index.
Intracellular multiplication of the resistant lines.
Given that drug-resistant traits could affect parasite infectivity, the amastigote multiplication ratio was calculated over a period of 7 days to evaluate the intracellular expansion of the resistant lines compared to that of the wild type (Fig. 3). The number of amastigotes in at least 50 macrophages was counted, and the ratio was used to determine the infection ratio (IR) for each time point. The amastigote multiplication ratio was determined by normalization using the initial infection index at 24 h postinfection (hpi). The IR and the percentage of infected macrophages at 24 hpi can be found in Table S2. A slight decrease in amastigote multiplication could be noted for the DNDI-1044-selected resistant clone, whereas the DND-8012-selected strain did not show significant differences from the WT.
FIG 3.
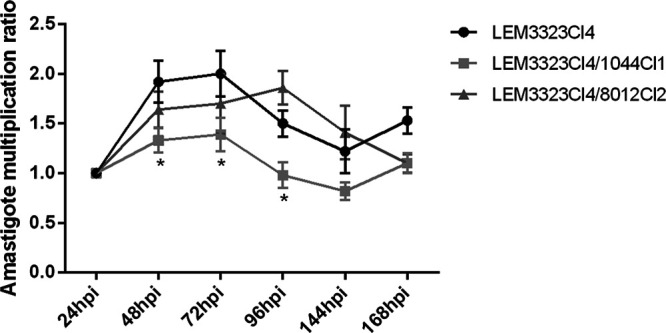
Comparison of the in vitro amastigote multiplication ratio between the wild-type parent (LEM3323Cl4) and the selected resistant lines. Results are expressed as the mean amastigote multiplication ratio ± SEM and are based on one experiment run in sextuplet (*, P < 0.05).
Stability of the resistant phenotype.
To assess the stability of the resistant phenotype, the generated resistant clones LEM3323Cl4/1044Cl1 and LEM3323Cl4/8012Cl2 were used for infection in both mouse and sandfly. The susceptibility was assessed before and after in vivo passage and compared to that of the wild-type line passaged under the same conditions. This experiment could give some insights into the possibility that the resistant phenotype would be sustained in the field. The fully resistant phenotype of promastigotes, which was clearly present prior to the in vivo passage, was lost after infection in mouse and sandfly. This loss was complete for LEM3323Cl4/1044Cl1, but an intermediate resistant phenotype could still be observed for LEM3323Cl4/8012Cl2 (Table 5).
TABLE 5.
Comparison of in vitro extracellular promastigote susceptibility to the selected AP series before and after in vivo passage (mouse and sandfly) between the wild type and the generated clones
| Time relative to passage | Compound | Mean IC50 (μM) ± SEMa |
||
|---|---|---|---|---|
| Wild type | LEM3323Cl4/1044Cl1 | LEM3323Cl4/8012Cl2 | ||
| Original | DNDI-1044 | 0.46 ± 0.05 | 3.37 ± 0.54 (7.3)**** | 2.63 ± 0.25 (5.7)*** |
| DNDI-8012 | 0.66 ± 0.09 | 4.12 ± 0.89 (6.2)**** | 3.81 ± 0.43 (5.7)**** | |
| DNDI-5561 | 0.80 ± 0.08 | 9.28 ± 1.01 (11) | 7.68 ± 1.09 (9.6)* | |
| After mouse passage | DNDI-1044 | 0.61 ± 0.09 | 0.64 ± 0.10 (1.0) | 1.86 ± 0.15 (3.0) |
| DNDI-8012 | 0.73 ± 0.15 | 1.01 ± 0.06 (1.4) | 2.68 ± 0.29 (3.7) | |
| DNDI-5561 | 0.68 ± 0.05 | 0.95 ± 0.16 (1.4) | 2.18 ± 0.54 (3.2) | |
| After sandfly passage | DNDI-1044 | 0.51 ± 0.07 | 0.55 ± 0.10 (1.1) | 1.36 ± 0.27 (2.6) |
| DNDI-8012 | 0.78 ± 0.12 | 0.85 ± 0.13 (1.1) | 1.76 ± 0.17 (2.2)* | |
| DNDI-5561 | 0.74 ± 0.08 | 0.76 ± 0.07 (1.0) | 1.20 ± 0.39 (1.6) | |
Results are based on three independent replicates run in duplicate. The resistance index (RI) is given in parentheses and was always calculated with respect to the corresponding wild type. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
Comparative WGS.
Comparative whole-genome sequencing (WGS) was performed to compare both resistant strains to the WT line to unravel possible mechanisms which caused the occurrence of the resistant phenotype. A coverage of at least 121 times was obtained for all the sequenced reads. A first analysis was performed to determine the presence of single nucleotide polymorphisms (SNPs) and indels. A total of 176 and 104 nonsynonymous SNPs in coding genes was identified in LEM3323Cl4/1044Cl1 and LEM3323Cl4/8012Cl2, respectively, compared to the sequence of the reference genome, and these were absent in the wild-type line. Ten and 12 genes, respectively, were found to be mutated in a homozygous manner. Twenty genes were commonly mutated in both resistant lines, and five of these were homogeneously mutated (Table 6). Surprisingly, the same mutations were detected in all listed genes for both resistant clones, suggesting that these mutations are most likely to occur as natural polymorphisms in the parental strain. As the resistant phenotype was found to be unstable after mouse and sandfly passage, additional analysis was conducted to evaluate the possible presence of copy number variations (CNVs) in both resistant lines (see the supplemental material). No significant deletions or amplifications were noted; however, the resistant lines demonstrated aneuploidy compared to the ploidy of the parent line (Table 7).
TABLE 6.
Overview of common mutated genes in both LEM332Cl4/1044Cl1 and LEM3323Cl4/8012Cl2a
| LEM3323Cl4 strain | Gene identifier | Position of mutation | Ref/mut nucleotide | Allele frequency | Ref/mut AA | Gene function |
|---|---|---|---|---|---|---|
| 1044Cl1 | LinJ.03.0410 | 156 | GA/G | Hom. | NA | 60S acidic ribosomal protein P2, putative |
| 8012Cl2 | 156 | GA/G | Hom. | NA | ||
| 1044Cl1 | LinJ.10.0390 | 172 | G/A | Hom. | E/K | Folate biopterin transporter, putative |
| 8012Cl2 | 172 | G/A | Hom. | E/K | ||
| 1044Cl1 | LinJ.12.0070 | 1277 | G/C | Het. | R/P | Hypothetical protein, unknown function |
| 8012Cl2 | 1277 | G/C | Het. | R/P | ||
| 1044Cl1 | LinJ.14.1180 | 2511 | G/C | Het. | E/D | Kinesin K39, putative |
| 8012Cl2 | 1885 | T/G | Het. | S/A | ||
| 8012Cl2 | 2511 | G/C | Het. | E/D | ||
| 1044Cl1 | LinJ.14.1190 | 3770 | A/C | Het. | E/A | Kinesin K39, putative |
| 8012Cl2 | 3770 | A/C | Het. | E/A | ||
| 8012Cl2 | 5699 | G/A | Hom. | S/N | ||
| 1044Cl1 | LinJ.15.0290 | 2429 | T/G | Het. | L/R | Hypothetical protein, conserved |
| 8012Cl2 | 561 | C/CGTCTCGGAAGCGGAGTCGCTCTCAGCCGCG | Het. | NA | ||
| 1044Cl1 | LinJ.18.1290 | 283 | T/G | Het. | S/A | Hypothetical protein, conserved |
| 8012Cl2 | 283 | T/G | Het. | S/A | ||
| 1044Cl1 | LinJ.19.1690 | 957 | AGCGCCCCAGCCGAGCGAGGCGGCGCCGGTGTCTGCAGTGGAGGCTCTGCCTCCGACGCCTGCCGAGTGCGCATCTGAGGCG/A | Het. | NA | Hypothetical protein |
| 8012Cl2 | 957 | AGCGCCCCAGCCGAGCGAGGCGGCGCCGGTGTCTGCAGTGGAGGCTCTGCCTCCGACGCCTGCCGAGTGCGCATCTGAGGCG/A | Het. | NA | ||
| 1044Cl1 | LinJ.27.2060 | 294 | T/TG | Het. | NA | Hypothetical protein, unknown function |
| 8012Cl2 | 294 | T/TG | Het. | NA | ||
| 1044Cl1 | LinJ.29.0270 | 546 | T/G | Het. | E/D | Hypothetical protein, conserved |
| 8012Cl2 | 546 | T/G | Het. | E/D | ||
| 8012Cl2 | 544 | C/G | Het. | E/Q | ||
| 1044Cl1 | LinJ.29.2240 | 1267 | G/A | Hom. | L/F | Hypothetical protein, conserved |
| 8012Cl2 | 1267 | G/A | Hom. | L/F | ||
| 1044Cl1 | LinJ.31.1290 | 3139 | G/C | Het. | R/G | P-glycoprotein e ABCC4 |
| 8012Cl2 | 3350 | A/C | Het. | L/R | ||
| 8012Cl2 | 3139 | G/C | Het. | R/G | ||
| 1044Cl1 | LinJ.32.1310 | 827 | G/C | Het. | S/T | Ubiquitin hydrolase, putative; cysteine peptidase Clan CA family C19, putative |
| 8012Cl2 | 827 | G/C | Het. | S/T | ||
| 1044Cl1 | LinJ.33.3230 | 5873 | A/G | Het. | Y/C | Hypothetical protein, conserved |
| 8012Cl2 | 5873 | A/G | Het. | Y/C | ||
| 1044Cl1 | LinJ.34.0710 | 2417 | T/C | Het. | K/R | Flagellar attachment zone protein, putative |
| 8012Cl2 | 2417 | T/C | Het. | K/R | ||
| 1044Cl1 | LinJ.34.2650 | 544 | T/G | Het. | S/A | Amastin-like surface protein, putative |
| 8012Cl2 | 544 | T/G | Het. | S/A | ||
| 1044Cl1 | LinJ.35.0490 | 11527 | G/A | Het. | A/T | Proteophosphoglycan ppg4 |
| 8012Cl2 | 11527 | G/A | Het. | A/T | ||
| 1044Cl1 | LinJ.35.0510 | 721 | G/A | Hom. | A/T | Proteophosphoglycan ppg4 |
| 8012Cl2 | 721 | G/A | Het. | A/T | ||
| 8012Cl2 | 733 | C/G | Het. | L/V | ||
| 1044Cl1 | LinJ.35.0520 | 6859 | G/A | Het. | G/S | Proteophosphoglycan ppg4 |
| 8012Cl2 | 6859 | G/A | Het. | G/S | ||
| 1044Cl1 | LinJ.35.0540 | 685 | T/C | Het. | C/R | Proteophosphoglycan 5 |
| 8012Cl2 | 685 | T/C | Het. | C/R |
Ref/mut, reference/mutated; Hom., homozygous; Het., heterozygous; NA, not applicable due to frameshift; AA, amino acid.
TABLE 7.
Overview of the ploidy of the wild-type line and both resistant clonesa

Chromosomal copy numbers are indicated with numbers and shades of blue ranging from light blue (mono- and disomic) to dark blue (tri- and tetrasomic). WT, wild type.
Involvement of efflux pump inhibitors.
MDR and MRP are well-known efflux pumps, which are often implicated in resistance. Their involvement was therefore determined using efflux pump inhibitors. The ABC transport inhibitors were first evaluated for their cytotoxicity on MRC5 cells and their intrinsic inhibitory effect on intracellular amastigotes and extracellular promastigotes (Table S3). Test concentrations used for the coadministration of verapamil (8 μM), cyclosporine (1.5 μM for L. infantum and 2 μM for L. donovani), and probenecid (700 μM) were selected for the susceptibility assays with the lead compounds. Under these conditions, inhibition by the ABC transporter inhibitors remained under 15%, 10%, and 20% respectively. As part of the assay validation, verapamil and probenecid were confirmed to increase the susceptibility to the trivalent form of Sb (SbIII), but no impact was observed with cyclosporine (Fig. 4). When the aminopyrazoles were coincubated with the ABC inhibitors, no alterations in the IC50 for intracellular and extracellular parasites were noted (Fig. 4). The involvement of MRP-based efflux at the host cell level was also evaluated using the fluorescent substrate 5(6)-carboxy-2′-7′-dichlorofluorescein (CDCF). CDCF efflux was inhibited by the MRP inhibitor probenecid and the MRP substrate SbIII, particularly in noninfected macrophages (Fig. S3). In contrast, the aminopyrazoles did not impact the efflux of CDCF by uninfected and L. infantum-infected macrophages, suggesting the absence of substrate-level competition.
FIG 4.

Effect of coincubation of verapamil, cyclosporine, and probenecid with the aminopyrazoles for L. infantum (A and C) and L. donovani (B and D) in the intracellular amastigote (A and B) and extracellular promastigote (C and D) susceptibility assays. Results are based on two independent repeats run in duplicate and are expressed as the mean IC50 ± standard mean of error (SEM) (*, P < 0.05).
DISCUSSION
In particular areas where VL is endemic and where the use of Sb and MIL has been implemented, the effectiveness of these drugs has been gradually decreasing, as reflected by posttreatment relapses (40). It is therefore a prerequisite for any new drug under development to have a novel mode of action and a low propensity for rapid resistance development. The aminopyrazoles constitute a series, newly developed by DNDi, which has already been shown not to have cross-resistance with the current antileishmanial drugs (28). The present study attempted to experimentally select and characterize AP-resistant strains and to evaluate the putative involvement of specific efflux pumps.
To generate experimental resistance in Leishmania, two approaches may be adopted: either selection of resistance on the extracellular insect stage, the promastigote, or selection on the intracellular host stage, the amastigote. The first is more regularly performed, as it is easier, cheaper, and quicker (30, 41, 42), but a major disadvantage is that it does not mimic the real interaction between the parasite and the drug in field situations. However, obtaining resistant promastigotes may still be very useful to deconvolute drug mechanisms that are shared between parasite life cycle stages. An alternative approach relies on resistance selection on the more relevant intracellular amastigote stage (38, 39). Using such a strategy, amastigote-selected drug-resistant parasite lines have already been successfully obtained for MIL and paromomycin (38, 39, 43). Selection for resistance to MIL proved to be very tedious and was successful in vitro for only one particular clinical isolate (43), which may reflect the situation in the field, where MIL-resistant parasite phenotypes are very scarce (44). In contrast, paromomycin resistance selection was alarmingly fast both in vitro and in vivo in multiple Leishmania strains, which advocates for its vigilant use, especially in monotherapy (38, 39). Exposure of amastigotes to APs in vitro and in vivo for 5 successive selection cycles failed to demonstrate changes in drug susceptibility, suggesting a low likelihood of rapid induction of resistance in the field. On the contrary, selection on promastigotes was successful after several rounds of exposure to increasing drug concentrations, allowing further phenotypic and genotypic characterization. Previous studies have demonstrated that the infectivity of drug-resistant parasites can be affected (45–49), and this was found to be decreased in one of the AP-resistant strains.
Leishmania parasites have developed several adaptation mechanisms to overcome elimination by drug exposure. Genetic changes, such as the introduction of SNPs and indels in drug targets or transporters, can be at the basis of a stable resistance (50–54). The AP-selected strains were found not to have a stable resistant phenotype after in vivo passage, suggesting that nonfixed alterations are responsible for the elevated resistance observed. Gene modulation through DNA CNVs is another way for the parasite to overcome the cytotoxic effects of a compound by specifically amplifying or deleting certain loci coding for genes involved in the drug’s mode of action or mode of resistance (55–60). In this study, the repeated exposure to APs did not give rise to CNVs; however, changes in the ploidy could be noted. Aneuploidy has been shown to be very common in Leishmania and has been linked to the development of resistance in a number of cases (58, 60–65). Ploidy has also been demonstrated to be rather plastic and adaptive to as little as just one in vivo passage (66). This could explain the resistant phenotype observed in the AP-selected parasites and the subsequent loss of resistance after a single mouse and sandfly passage. Additional generation of a larger panel of resistant parasite lines in combination with WGS could help establish the relevance of ploidy and further pinpoint the chromosomes that are most likely involved in the transient AP resistance.
Because the MDR and MRP pumps are known to confer elevated SbIII and AmB resistance in Leishmania parasites (10, 16, 17), this study assessed their involvement in the activity of APs. For this purpose, the AP susceptibility of nonresistant strains in combination with reference ABC transport inhibitors was evaluated; probenecid is known to reverse the effects of MRP pumps (67, 68), while verapamil and cyclosporine inhibit efflux mediated by both the MRP and MDR pumps (69–71). Treatment with SbIII was included as a positive control (21, 72–75) and confirmed an increased susceptibility of extracellular and intracellular parasites in combination with verapamil and probenecid, although verapamil is considered not to efficiently inhibit the MDR pumps in Leishmania (76). Due to host cell toxicity, cyclosporine was included at a low concentration, which may explain the observed lack of an effect. The different response of L. donovani toward probenecid in combination with SbIII could be explained by differences in pump expression between L. infantum and L. donovani (17, 77, 78). No significant effects could be noted after coincubation of the APs with the ABC transporter inhibitors in both extracellular and intracellular assays. The APs were also unable to inhibit the efflux of the fluorescent ABC efflux substrate 5(6)-carboxy-2′-7′-dichlorofluorescein.
To conclude, the DNDi aminopyrazole lead series is promising when it comes to the likelihood of drug resistance development in the field, since repeated exposure of amastigotes to the compounds did not result in decreased susceptibility. Resistance selection of the promastigotes revealed that karyotypic changes may confer elevated levels of resistance, but these did not seem to be stable in the vertebrate and invertebrate hosts. Additionally, the AP lead compounds tested do not appear to be substrates of the MDR or MRP pumps of the parasite and macrophage host cell tested, reducing the risk of resistance development through this common pathway.
MATERIALS AND METHODS
Ethics statement.
The use of laboratory rodents was carried out in accordance with all mandatory guidelines (EU directives, including Revised Directive 2010/63/EU on the protection of animals used for scientific purposes, which came into force on 1 January 2013, and the Declaration of Helsinki [in its latest version]) and was approved by the Ethical Committee of the University of Antwerp, Antwerp, Belgium (UA-ECD 2011-77, revised 2015).
Parasite cultures.
Promastigotes of two laboratory strains, MHOM/MA/67/ITMAP263 (Leishmania infantum) and MHOM/ET/67/L82 (L. donovani), were cultured at 25°C in HOMEM (Gibco, Life Technologies) supplemented with 10% inactivated bovine serum (iFBS). Both strains were also available as ex vivo amastigotes obtained from the spleens of heavily infected donor hamsters (38). Additionally, two L. infantum field strains, MHOM/FR/09/LEM4038 and MHOM/FR/96/LEM3323, available as promastigotes, were obtained from HIV-infected patients (79).
Animals.
Female Swiss mice, BALB/c mice, and female golden hamsters were purchased from Janvier (Le Genest Saint Isle, France). Food for laboratory rodents (Carfil, Arendonk, Belgium) and drinking water were available ad libitum. Before the start of the in vivo experiments, the hamsters were randomly allocated to experimental units of 3 animals each.
Test substances.
For the in vitro experiments, the aminopyrazoles (see Table S1 in the supplemental material) were formulated in 100% dimethyl sulfoxide (DMSO) at 20 mM (stock solution). The efflux pump inhibitors verapamil (an MDR and MRP inhibitor), cyclosporine (a broad-specificity efflux inhibitor), and probenecid (an MRP inhibitor) (Sigma-Aldrich, Diegem, Belgium) were formulated in 100% DMSO at 20 mM, except for probenecid, which was diluted in phosphate-buffered saline (PBS) after the addition of ethanol (2%) and NaOH at 50 mM. Stock solutions were further diluted in demineralized water. In all in vitro assays, the final in-test concentration of DMSO did not exceed 1%. For the in vivo experiments, DNDI-1044 was formulated in 1% (wt/vol) methylcellulose (4,000 centipoise) and 5% (vol/vol) Tween 80 in water; miltefosine (MIL) was formulated in water.
Intracellular susceptibility assay.
To obtain primary peritoneal macrophages, Swiss mice were stimulated by intraperitoneal injection of 1 ml of a 2% starch suspension in PBS 48 h prior to cell collection. Animals were euthanized with a CO2 overdose, and upon removal of the skin, 10 ml of RPMI 1640 (Life Technologies) was injected into the peritoneal cavity to collect the macrophages, which were then seeded into 96-well plates at a final concentration of 30,000 cells/well in 100 μl of RPMI 1640 medium supplemented with 5% iFBS, 2% penicillin-streptomycin, and 1% l-glutamine (Life Technologies). After 24 h, the cells were infected with metacyclic promastigotes (infection ratio, 15:1), and 2-fold drug dilutions were added 24 h later. The test plates were incubated for 96 h at 37°C in 5% CO2, after which the cells were fixed with methanol and Giemsa stained for microscopic determination of the IC50 values, based on the reduction in the amastigote burden in the treated cells compared to that in the untreated control cells.
Extracellular susceptibility assay.
Log-phase promastigotes (3 days old) were counted in a Kova counting chamber and diluted to a concentration of 106 promastigotes/well in a 96-well plate. Dilutions of the test compounds were then added to the promastigote plates. The highest in-test concentrations and dilutions of all the compounds were as described above. Drug exposure covered a 72-h period without renewal of the culture medium. Parasite proliferation was assessed with the resazurin assay. Drug activity was measured as the percent reduction in the mean fluorescence compared to that for the nontreated control wells.
In vitro intracellular resistance selection.
Intracellular amastigotes were repeatedly exposed to DNDI-1044, as previously described (39). In brief, primary peritoneal mouse macrophages were infected with metacyclic L. infantum promastigotes (MHOM/FR/09/LEM4038 and MHOM/MA/67/ITMAP263) in two duplicate 96-well plates and exposed to increasing drug concentrations. One plate was Giemsa stained to determine the drug susceptibility by microscopic evaluation, while the other plate was used for promastigote backtransformation (PBT). PBT was evaluated by releasing residual viable amastigotes and allowing their transformation into promastigotes in HOMEM at 25°C. Promastigotes were collected from the highest concentration and further expanded in routine culture. Infection and PBT cycles were repeated until resistance could be observed or for a maximum of five successive passages. Statistically significant differences between the different passages were analyzed using two-way analysis of variance (ANOVA), and the results were considered statistically significantly different if P was <0.05.
In vitro extracellular resistance selection.
Log-phase promastigotes of the Leishmania infantum clinical isolate MHOM/FR/96/LEM3323 (3 days old) were counted and diluted to a concentration of 1 × 106 promastigotes/ml containing 0.5× the IC50 of DNDI-1044 or DNDI-8012. Promastigotes were left to recover from drug exposure without renewal of the medium. Upon complete recovery, a next selection round was initiated by subculturing at twice the drug concentration of the previous selection round. Thus, promastigotes were gradually exposed to increasing drug concentrations until their growth was irreversibly inhibited. A sample from each selection cycle was cryopreserved in liquid nitrogen upon supplementation with 10% glycerol. Finally, intracellular and extracellular susceptibility assays were performed to assess the phenotypic acquisition of resistance as a result of the selection procedure. Statistically significant differences between the different passages were analyzed using two-way analysis of variance (ANOVA), and the results were considered statistically significantly different if P was <0.05.
In vivo resistance selection.
The in vivo selection of drug resistance was performed as previously described (38). Briefly, L. infantum spleen-derived amastigotes (MHOM/MA/67/ITMAP263) were diluted to prepare an infection inoculum containing 2 × 107 amastigotes in 100 μl PBS. All animals were treated orally for 5 days starting from 21 days postinfection (dpi), after which the animals were closely monitored for signs of relapse. Upon relapse, amastigotes were collected and used for infection of new naive hamsters, which received the same treatment regimen at 21 dpi. This procedure was repeated until evaluation of the in vitro IC50 values revealed a reduced drug susceptibility or for a maximum of 5 treatment/relapse cycles. A treatment regimen of 50 mg/kg of body weight twice a day (b.i.d.) was chosen, based on previously published results, to ascertain a parasite clearance of more than 95% in the main target organs (28). DNDI-1044 at 50 mg/kg b.i.d. was previously shown to result in an amastigote burden reduction of 97.1% and 98.6% 10 days after the end of treatment in liver and spleen, respectively. Statistically significant differences between the different passages were analyzed using two-way analysis of variance (ANOVA). Results were considered statistically significantly different if P was <0.05.
Generation of clones.
The microdrop technique was used to generate clones of the resistant lines to reduce genetic variability in the population (80). A dilution of the culture was prepared in HOMEM in a 96-well plate, from which microdrops were taken. A total of 8 μl HOMEM was placed on the sidewall of a new well plate before placing the drop in the middle of the well. The drop was then microscopically evaluated for the presence of a single promastigote(s), and 100 μl of HOMEM and 100 μl of spent medium were added to the well. The plates were incubated at 25°C and observed weekly for promastigote growth under a microscope. Once growth was established, the promastigotes were transferred into culture flasks and subcultured twice weekly according to standard procedures.
Intracellular multiplication.
Amastigote multiplication was compared between the resistant clones and the wild-type parent line for assessment of a possible loss or gain of infectivity accompanied by the resistant phenotype. Peritoneal macrophages were collected and infected with metacyclic promastigotes at an infection ratio of 2:1 as described previously (43). Parasites were preconditioned in acidic HOMEM (pH 5.4) 24 h prior to infection to ascertain that all promastigotes were at the same metacyclic stage (81). The infected macrophages were washed twice with RPMI 1640 after 24 h of infection to exclude the possibility of reinfection by noninternalized promastigotes. The medium on the cells was replaced every 4 days to ensure macrophage viability. Amastigote multiplication was evaluated every day for a period of 7 days postinfection by microscopy evaluation after Giemsa staining. The infection index was calculated by dividing the total number of counted amastigotes by the total number of counted macrophages. The amastigote multiplication ratio was then calculated by dividing the infection index of every time point by the infection ratio at 24 hpi, thereby correcting for baseline infectivity differences.
Stability of the resistant phenotype after sandfly passage.
The stability of the resistant phenotype was assessed for the most resistant clones compared to that for the wild-type parent line after passage in the vector. Two- to 5-day-old female Lutzomyia longipalpis sandflies were fed through a chick skin membrane on a mixture containing heparin-treated mouse blood and 5 × 106 logarithmic-phase promastigotes per ml. Blood-fed sandflies were separated 24 h later and maintained at 26°C with 30% (vol/vol) sugar solution. Seven days after feeding, the flies were anesthetized with CO2, and their midguts were dissected and crushed after addition of 50 μl of PBS. The released parasites were then incubated in acidic HOMEM (pH 5.4) supplemented with 20% iFBS, 1% gentamicin, 2% penicillin-streptomycin, and 2% spent medium. After 24 to 48 h of incubation, parasites were subcultured and susceptibility testing was performed in both promastigotes and intracellular amastigotes.
Stability of the resistant phenotype after mouse passage.
The stability of the resistant phenotype was also assessed after mouse passage. Infection was performed by injecting 1 × 108 metacyclic promastigotes into the tail vein of 8-week-old BALB/c mice. After 3 weeks of infection, the mice were euthanized and PBT was performed from the liver, spleen, and bone marrow. This entailed incubating a small piece of organ (spleen, liver, bone marrow from the femur) in acidic HOMEM (pH 5.4) supplemented with 20% iFBS, 1% gentamicin, 2% penicillin-streptomycin, and 2% spent medium. After 5 days of incubation, promastigotes were subcultured and susceptibility testing was subsequently performed.
Comparative whole-genome sequencing.
Genomic DNA was prepared from the L. infantum resistant clones (1044Cl1 and 8012Cl2) and the wild-type (WT) parent line. DNA was quantified fluorometrically, and 2 μg of material was used for library preparation and sequencing on an Illumina NovaSeq 6000 sequencer with a NovaSeq 6000 S2 reagent kit (200 cycles). The reads obtained were aligned against the L. infantum JPCM5 reference genome (Tritryp_v9) using BWA-MEM software. Single nucleotide polymorphisms (SNPs) and indels were identified using GATK software. SNPs and indels that were present in the WT line but that were not located in coding sequences or that did not generate an amino acid alteration in the resistant lines (synonymous mutations) were not considered. To determine the influence of the mutation, the allele frequency (homozygous/heterozygous), the impact of the mutation (as a property of the altered amino acid and indel), and the presence of the mutated gene in both resistant lines were taken into account. Genes with identical mutations in both resistant lines and genes harboring several mutations (≥3) were considered less important, as they likely reflect natural polymorphisms. Additionally, the copy number variations (CNVs) in each chromosome were determined by calculating the ratio of the number of normalized reads from the resistant line compared to the number from the susceptible parent line.
Intracellular efflux susceptibility assay.
Macrophages were infected with spleen-derived L. infantum (MHOM/MA/67/ITMAP263) or L. donovani (MHOM/ET/67/L82) amastigotes at an infection ratio of 5:1. After 2 h, the medium was replaced with 220 μl of fresh RPMI 1640 medium containing the aminopyrazole and the ABC transporter inhibitor. The latter was added at a concentration below the IC50, as determined in the standard intracellular amastigote susceptibility assay (Table S2). The DNDi compounds were diluted 4-fold, with the highest in-test concentration being 10 μM. Drug activity was evaluated as described above in the intracellular susceptibility assay, and IC50 values were compared for conditions with and without an ABC transporter inhibitor.
Extracellular efflux susceptibility assay.
Log-phase promastigotes (3 days old) of L. infantum and L. donovani were diluted to a concentration of 106 promastigotes/well in a 96-well plate. Dilutions of the reference compounds and aminopyrazoles were then added either with or without the ABC transporter inhibitor. The IC50 of the inhibitor was first determined by a standard promastigote susceptibility assay (Table S2). Drug activity was determined as described above for the extracellular susceptibility assay, and IC50 values were compared for conditions with and without an ABC transporter inhibitor.
Flow cytometric analysis of drug efflux.
The accumulation of 5(6)-carboxy-2′-7′-dichlorofluorescein (CDCF; Sigma-Aldrich) was evaluated upon incubation of macrophages (2 × 105 cells/measurement) with 160 μM CDCF in RPMI 1640 without phenol red for 1 h at 4°C. When experiments were performed with Leishmania-infected macrophages, the cells were first seeded in a T75 culture flask in RPMI 1640 medium and infected with ex vivo amastigotes for 24 h. After incubation, the cells were washed twice with cold medium and an aliquot was taken to quantify CDCF uptake by flow cytometry in the fluorescein isothiocyanate channel (FACSCalibur flow cytometer; Becton, Dickinson). Dye retention in macrophages was evaluated after 2 h of incubation at 37°C in the absence (negative control) or presence (positive control) of probenecid at 4 mM alongside the DNDi compounds and SbIII at 10 μM. To assess cell viability, 7-amino actinomycin D (7-AAD; Biosciences) was added. Data analysis and determination of the mean fluorescence intensity were carried out with Flow Jo X software.
Supplementary Material
ACKNOWLEDGMENTS
We thank An Matheeussen, Margot Desmet, Pim-Bart Feijens, and Mandy Vermont for their excellent technical assistance in running the in vitro and in vivo experiments. Special thanks go to L. Lachaud (Laboratoire de Parasitologie-Mycologie et Centre National de Référence des Leishmanioses, Montpellier, France) for providing the L. infantum field strain MHOM/FR/09/LEM4038.
This work was funded by the Fonds Wetenschappelijk Onderzoek (grants G051812N and 12I0317N), a research fund of the University of Antwerp (grant TT-ZAPBOF 33049), and a CIHR Foundation grant to M.O. M.O. is the holder of a Canada Research Chair. For the work described in this paper, DNDi received financial support from the following donors: the Dutch Ministry of Foreign Affairs (DGIS), the Netherlands; UK Aid, United Kingdom; the Federal Ministry of Education, and Research (BMBF) through KfW, Germany; The Bill & Melinda Gates Foundation (BMGF), USA; the Global Health Innovative Technology Fund (GHIT Fund), Japan; and the World Health Organization Special Program for Research and Training in Tropical Diseases (WHO-TDR). DNDi also thanks Médecins sans Frontières International (MSF) and the Swiss Agency for Development and Cooperation, Switzerland, for contributing to its overall mission.
The donors had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript. LMPH is part of COST Action CM1307 (Targeted chemotherapy toward diseases caused by endoparasites) and is a partner of the Antwerp Drug Discovery Network (ADDN; www.addn.be) and the Excellence Centre Infla-Med (www.uantwerpen.be/infla-med).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Hussain H, Al-Harrasi A, Al-Rawahi A, Green IR, Gibbons S. 2014. Fruitful decade for antileishmanial compounds from 2002 to late 2011. Chem Rev 114:10369–10428. doi: 10.1021/cr400552x. [DOI] [PubMed] [Google Scholar]
- 2.Rajasekaran R, Chen Y. 2015. Potential therapeutic targets and the role of technology in developing novel antileishmanial drugs. Drug Discov Today 20:958–968. doi: 10.1016/j.drudis.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Sundar S, Singh A. 2016. Recent developments and future prospects in the treatment of visceral leishmaniasis. Ther Adv Infect Dis 3:98–109. doi: 10.1177/2049936116646063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry MR, Prajapati VK, Menten J, Raab A, Feldmann J, Chakraborti D, Sundar S, Fairlamb AH, Boelaert M, Picado A. 2015. Arsenic exposure and outcomes of antimonial treatment in visceral leishmaniasis patients in Bihar, India: a retrospective cohort study. PLoS Negl Trop Dis 9:e0003518. doi: 10.1371/journal.pntd.0003518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sundar S, Singh A. 2018. Chemotherapeutics of visceral leishmaniasis: present and future developments. Parasitology 145:481–489. doi: 10.1017/S0031182017002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rijal S, Ostyn B, Uranw S, Rai K, Bhattarai NR, Dorlo TP, Beijnen JH, Vanaerschot M, Decuypere S, Dhakal SS, Das ML, Karki P, Singh R, Boelaert M, Dujardin JC. 2013. Increasing failure of miltefosine in the treatment of kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin Infect Dis 56:1530–1538. doi: 10.1093/cid/cit102. [DOI] [PubMed] [Google Scholar]
- 7.Ostyn B, Hasker E, Dorlo TP, Rijal S, Sundar S, Dujardin JC, Boelaert M. 2014. Failure of miltefosine treatment for visceral leishmaniasis in children and men in South-East Asia. PLoS One 9:e100220. doi: 10.1371/journal.pone.0100220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sundar S, Singh A, Rai M, Prajapati VK, Singh AK, Ostyn B, Boelaert M, Dujardin JC, Chakravarty J. 2012. Efficacy of miltefosine in the treatment of visceral leishmaniasis in India after a decade of use. Clin Infect Dis 55:543–550. doi: 10.1093/cid/cis474. [DOI] [PubMed] [Google Scholar]
- 9.Deep DK, Singh R, Bhandari V, Verma A, Sharma V, Wajid S, Sundar S, Ramesh V, Dujardin JC, Salotra P. 2017. Increased miltefosine tolerance in clinical isolates of Leishmania donovani is associated with reduced drug accumulation, increased infectivity and resistance to oxidative stress. PLoS Negl Trop Dis 11:e0005641. doi: 10.1371/journal.pntd.0005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purkait B, Kumar A, Nandi N, Sardar AH, Das S, Kumar S, Pandey K, Ravidas V, Kumar M, De T, Singh D, Das P. 2012. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother 56:1031–1041. doi: 10.1128/AAC.00030-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Salcedo JA, Unciti-Broceta JD, Valverde-Pozo J, Soriano M. 2016. New approaches to overcome transport related drug resistance in trypanosomatid parasites. Front Pharmacol 7:351. doi: 10.3389/fphar.2016.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouellette M, Legare D, Papadopoulou B. 2001. Multidrug resistance and ABC transporters in parasitic protozoa. J Mol Microbiol Biotechnol 3:201–206. [PubMed] [Google Scholar]
- 13.Perez-Victoria JM, Parodi-Talice A, Torres C, Gamarro F, Castanys S. 2001. ABC transporters in the protozoan parasite Leishmania. Int Microbiol 4:159–166. doi: 10.1007/s10123-001-0031-2. [DOI] [PubMed] [Google Scholar]
- 14.El Fadili K, Messier N, Leprohon P, Roy G, Guimond C, Trudel N, Saravia NG, Papadopoulou B, Légaré D, Ouellette M. 2005. Role of the ABC transporter MRPA (PGPA) in antimony resistance in Leishmania infantum axenic and intracellular amastigotes. Antimicrob Agents Chemother 49:1988–1993. doi: 10.1128/AAC.49.5.1988-1993.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legare D, Richard D, Mukhopadhyay R, Stierhof YD, Rosen BP, Haimeur A, Papadopoulou B, Ouellette M. 2001. The Leishmania ATP-binding cassette protein PGPA is an intracellular metal-thiol transporter ATPase. J Biol Chem 276:26301–26307. doi: 10.1074/jbc.M102351200. [DOI] [PubMed] [Google Scholar]
- 16.Leprohon P, Legare D, Ouellette M. 2009. Intracellular localization of the ABCC proteins of Leishmania and their role in resistance to antimonials. Antimicrob Agents Chemother 53:2646–2649. doi: 10.1128/AAC.01474-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreira DS, Neto RLM, Andrade JM, Santi AMM, Reis PG, Frezard F, Murta S. 2013. Molecular characterization of the MRPA transporter and antimony uptake in four New World Leishmania spp. susceptible and resistant to antimony. Int J Parasitol Drugs Drug Resist 3:143–153. doi: 10.1016/j.ijpddr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castanys-Munoz E, Alder-Baerens N, Pomorski T, Gamarro F, Castanys S. 2007. A novel ATP-binding cassette transporter from Leishmania is involved in transport of phosphatidylcholine analogues and resistance to alkyl-phospholipids. Mol Microbiol 64:1141–1153. doi: 10.1111/j.1365-2958.2007.05653.x. [DOI] [PubMed] [Google Scholar]
- 19.Castanys-Munoz E, Perez-Victoria JM, Gamarro F, Castanys S. 2008. Characterization of an ABCG-like transporter from the protozoan parasite Leishmania with a role in drug resistance and transbilayer lipid movement. Antimicrob Agents Chemother 52:3573–3579. doi: 10.1128/AAC.00587-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seral C, Carryn S, Tulkens PM, Van Bambeke F. 2003. Influence of P-glycoprotein and MRP efflux pump inhibitors on the intracellular activity of azithromycin and ciprofloxacin in macrophages infected by Listeria monocytogenes or Staphylococcus aureus. J Antimicrob Chemother 51:1167–1173. doi: 10.1093/jac/dkg223. [DOI] [PubMed] [Google Scholar]
- 21.Chowdhury S, Mukhopadhyay R, Saha S, Mishra A, Sengupta S, Roy S, Majumder HK. 2014. Flavone-resistant Leishmania donovani overexpresses LdMRP2 transporter in the parasite and activates host MRP2 on macrophages to circumvent the flavone-mediated cell death. J Biol Chem 289:16129–16147. doi: 10.1074/jbc.M113.539742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caballero E, Manzano JI, Puebla P, Castanys S, Gamarro F, San Feliciano A. 2012. Oxazolo[3,2-a]pyridine. A new structural scaffold for the reversal of multi-drug resistance in Leishmania. Bioorg Med Chem Lett 22:6272–6275. doi: 10.1016/j.bmcl.2012.07.100. [DOI] [PubMed] [Google Scholar]
- 23.Pérez-Victoria JM, Bavchvarov BI, Torrecillas IR, Martínez-García M, López-Martín C, Campillo M, Castanys S, Gamarro F. 2011. Sitamaquine overcomes ABC-mediated resistance to miltefosine and antimony in Leishmania. Antimicrob Agents Chemother 55:3838–3844. doi: 10.1128/AAC.00065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pérez-Victoria JM, Cortés-Selva F, Parodi-Talice A, Bavchvarov BI, Pérez-Victoria FJ, Muñoz-Martínez F, Maitrejean M, Costi MP, Barron D, Di Pietro A, Castanys S, Gamarro F. 2006. Combination of suboptimal doses of inhibitors targeting different domains of LtrMDR1 efficiently overcomes resistance of Leishmania spp. to miltefosine by inhibiting drug efflux. Antimicrob Agents Chemother 50:3102–3110. doi: 10.1128/AAC.00423-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alves F, Bilbe G, Blesson S, Goyal V, Monnerat S, Mowbray C, Ouattara GM, Pecoul B, Rijal S, Rode J, Solomos A, Strub-Wourgaft N, Wasunna M, Wells S, Zijlstra EE, Arana B, Alvar J. 2018. Recent development of visceral leishmaniasis treatments: successes, pitfalls, and perspectives. Clin Microbiol Rev 31:e00048-18. doi: 10.1128/CMR.00048-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatelain E, Ioset JR. 2011. Drug discovery and development for neglected diseases: the DNDi model. Drug Des Devel Therapy 5:175–181. doi: 10.2147/DDDT.S16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sundar S, Chakravarty J. 2015. An update on pharmacotherapy for leishmaniasis. Expert Opin Pharmacother 16:237–252. doi: 10.1517/14656566.2015.973850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van den Kerkhof M, Mabille D, Chatelain E, Mowbray CE, Braillard S, Hendrickx S, Maes L, Caljon G. 2018. In vitro and in vivo pharmacodynamics of three novel antileishmanial lead series. Int J Parasitol Drugs Drug Resist 8:81–86. doi: 10.1016/j.ijpddr.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wall RJ, Rico E, Lukac I, Zuccotto F, Elg S, Gilbert IH, Freund Y, Alley MRK, Field MC, Wyllie S, Horn D. 2018. Clinical and veterinary trypanocidal benzoxaboroles target CPSF3. Proc Natl Acad Sci U S A 115:9616–9621. doi: 10.1073/pnas.1807915115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyllie S, Roberts AJ, Norval S, Patterson S, Foth BJ, Berriman M, Read KD, Fairlamb AH. 2016. Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in Leishmania. PLoS Pathog 12:e1005971. doi: 10.1371/journal.ppat.1005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Bocxlaer K, Caridha D, Black C, Vesely B, Leed S, Sciotti RJ, Wijnant GJ, Yardley V, Braillard S, Mowbray CE, Ioset JR, Croft SL. 2019. Novel benzoxaborole, nitroimidazole and aminopyrazoles with activity against experimental cutaneous leishmaniasis. Int J Parasitol Drugs Drug Resist 11:129–138. doi: 10.1016/j.ijpddr.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mercer L, Bowling T, Perales J, Freeman J, Nguyen T, Bacchi C, Yarlett N, Don R, Jacobs R, Nare B. 2011. 2,4-Diaminopyrimidines as potent inhibitors of Trypanosoma brucei and identification of molecular targets by a chemical proteomics approach. PLoS Negl Trop Dis 5:e956. doi: 10.1371/journal.pntd.0000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu XJ, DePinto W, Bartkovitz D, So SS, Vu BT, Packman K, Lukacs C, Ding Q, Jiang N, Wang K, Goelzer P, Yin X, Smith MA, Higgins BX, Chen Y, Xiang Q, Moliterni J, Kaplan G, Graves B, Lovey A, Fotouhi N. 2006. Discovery of [4-amino-2-(1-methanesulfonylpiperidin-4-ylamino)pyrimidin-5-yl](2,3-difluoro-6- methoxyphenyl)methanone (R547), a potent and selective cyclin-dependent kinase inhibitor with significant in vivo antitumor activity. J Med Chem 49:6549–6560. doi: 10.1021/jm0606138. [DOI] [PubMed] [Google Scholar]
- 34.DePinto W, Chu XJ, Yin X, Smith M, Packman K, Goelzer P, Lovey A, Chen Y, Qian H, Hamid R, Xiang Q, Tovar C, Blain R, Nevins T, Higgins B, Luistro L, Kolinsky K, Felix B, Hussain S, Heimbrook D. 2006. In vitro and in vivo activity of R547: a potent and selective cyclin-dependent kinase inhibitor currently in phase I clinical trials. Mol Cancer Ther 5:2644–2658. doi: 10.1158/1535-7163.MCT-06-0355. [DOI] [PubMed] [Google Scholar]
- 35.Rana S, Sonawane YA, Taylor MA, Kizhake S, Zahid M, Natarajan A. 2018. Synthesis of aminopyrazole analogs and their evaluation as CDK inhibitors for cancer therapy. Bioorg Med Chem Lett 28:3736–3740. doi: 10.1016/j.bmcl.2018.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY. 2016. Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg Med Chem Lett 26:1218–1223. doi: 10.1016/j.bmcl.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 37.Flannery EL, McNamara CW, Kim SW, Kato TS, Li F, Teng CH, Gagaring K, Manary MJ, Barboa R, Meister S, Kuhen K, Vinetz JM, Chatterjee AK, Winzeler EA. 2015. Mutations in the P-type cation-transporter ATPase 4, PfATP4, mediate resistance to both aminopyrazole and spiroindolone antimalarials. ACS Chem Biol 10:413–420. doi: 10.1021/cb500616x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hendrickx S, Mondelaers A, Eberhardt E, Delputte P, Cos P, Maes L. 2015. In vivo selection of paromomycin and miltefosine resistance in Leishmania donovani and L. infantum in a Syrian hamster model. Antimicrob Agents Chemother 59:4714–4718. doi: 10.1128/AAC.00707-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hendrickx S, Boulet G, Mondelaers A, Dujardin JC, Rijal S, Lachaud L, Cos P, Delputte P, Maes L. 2014. Experimental selection of paromomycin and miltefosine resistance in intracellular amastigotes of Leishmania donovani and L. infantum. Parasitol Res 113:1875–1881. doi: 10.1007/s00436-014-3835-7. [DOI] [PubMed] [Google Scholar]
- 40.Zulfiqar B, Shelper TB, Avery VM. 2017. Leishmaniasis drug discovery: recent progress and challenges in assay development. Drug Discov Today 22:1516–1531. doi: 10.1016/j.drudis.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Victoria FJ, Castanys S, Gamarro F. 2003. Leishmania donovani resistance to miltefosine involves a defective inward translocation of the drug. Antimicrob Agents Chemother 47:2397–2403. doi: 10.1128/aac.47.8.2397-2403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreira W, Leprohon P, Ouellette M. 2011. Tolerance to drug-induced cell death favours the acquisition of multidrug resistance in Leishmania. Cell Death Dis 2:e201. doi: 10.1038/cddis.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hendrickx S, Mondelaers A, Eberhardt E, Lachaud L, Delputte P, Cos P, Maes L. 2015. Intracellular amastigote replication may not be required for successful in vitro selection of miltefosine resistance in Leishmania infantum. Parasitol Res 114:2561–2565. doi: 10.1007/s00436-015-4460-9. [DOI] [PubMed] [Google Scholar]
- 44.Vanaerschot M, Dumetz F, Roy S, Ponte-Sucre A, Arevalo J, Dujardin JC. 2014. Treatment failure in leishmaniasis: drug-resistance or another (epi-) phenotype? Expert Rev Anti Infect Ther 12:937–946. doi: 10.1586/14787210.2014.916614. [DOI] [PubMed] [Google Scholar]
- 45.Eberhardt E, Bulte D, Van Bockstal L, Van den Kerkhof M, Cos P, Delputte P, Hendrickx S, Maes L, Caljon G. 2019. Miltefosine enhances the fitness of a non-virulent drug-resistant Leishmania infantum strain. J Antimicrob Chemother 74:395–406. doi: 10.1093/jac/dky450. [DOI] [PubMed] [Google Scholar]
- 46.Hendrickx S, Beyers J, Mondelaers A, Eberhardt E, Lachaud L, Delputte P, Cos P, Maes L. 2016. Evidence of a drug-specific impact of experimentally selected paromomycin and miltefosine resistance on parasite fitness in Leishmania infantum. J Antimicrob Chemother 71:1914–1921. doi: 10.1093/jac/dkw096. [DOI] [PubMed] [Google Scholar]
- 47.Natera S, Machuca C, Padrón-Nieves M, Romero A, Díaz E, Ponte-Sucre A. 2007. Leishmania spp.: proficiency of drug-resistant parasites. Int J Antimicrob Agents 29:637–642. doi: 10.1016/j.ijantimicag.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 48.Turner KG, Vacchina P, Robles-Murguia M, Wadsworth M, McDowell MA, Morales MA. 2015. Fitness and phenotypic characterization of miltefosine-resistant Leishmania major. PLoS Negl Trop Dis 9:e0003948. doi: 10.1371/journal.pntd.0003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanaerschot M, Maes I, Ouakad M, Adaui V, Maes L, De Doncker S, Rijal S, Chappuis F, Dujardin JC, Decuypere S. 2010. Linking in vitro and in vivo survival of clinical Leishmania donovani strains. PLoS One 5:e12211. doi: 10.1371/journal.pone.0012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laffitte MCN, Leprohon P, Hainse M, Legare D, Masson JY, Ouellette M. 2016. Chromosomal translocations in the parasite Leishmania by a MRE11/RAD50-independent microhomology-mediated end joining mechanism. PLoS Genet 12:e1006117. doi: 10.1371/journal.pgen.1006117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laffitte MCN, Leprohon P, Legare D, Ouellette M. 2016. Deep-sequencing revealing mutation dynamics in the miltefosine transporter gene in Leishmania infantum selected for miltefosine resistance. Parasitol Res 115:3699–3703. doi: 10.1007/s00436-016-5195-y. [DOI] [PubMed] [Google Scholar]
- 52.Mondelaers A, Sanchez-Canete MP, Hendrickx S, Eberhardt E, Garcia-Hernandez R, Lachaud L, Cotton J, Sanders M, Cuypers B, Imamura H, Dujardin JC, Delputte P, Cos P, Caljon G, Gamarro F, Castanys S, Maes L. 2016. Genomic and molecular characterization of miltefosine resistance in Leishmania infantum strains with either natural or acquired resistance through experimental selection of intracellular amastigotes. PLoS One 11:e0154101. doi: 10.1371/journal.pone.0154101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez-Victoria FJ, Gamarro F, Ouellette M, Castanys S. 2003. Functional cloning of the miltefosine transporter—a novel P-type phospholipid translocase from Leishmania involved in drug resistance. J Biol Chem 278:49965–49971. doi: 10.1074/jbc.M308352200. [DOI] [PubMed] [Google Scholar]
- 54.Pérez-Victoria FJ, Sánchez-Cañete MP, Castanys S, Gamarro F. 2006. Phospholipid translocation and miltefosine potency require both L. donovani miltefosine transporter and the new protein LdRos3 in Leishmania parasites. J Biol Chem 281:23766–23775. doi: 10.1074/jbc.M605214200. [DOI] [PubMed] [Google Scholar]
- 55.Beverley SM. 1991. Gene amplification in Leishmania. Annu Rev Microbiol 45:417–444. doi: 10.1146/annurev.mi.45.100191.002221. [DOI] [PubMed] [Google Scholar]
- 56.Brotherton MC, Bourassa S, Leprohon P, Legare D, Poirier GG, Droit A, Ouellette M. 2013. Proteomic and genomic analyses of antimony resistant Leishmania infantum mutant. PLoS One 8:e81899. doi: 10.1371/journal.pone.0081899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, de Doncker S, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Schonian G, Stark O, Sundar S, Vanaerschot M, Hertz-Fowler C, Dujardin JC, Berriman M. 2011. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res 21:2143–2156. doi: 10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leprohon P, Legare D, Raymond F, Madore E, Hardiman G, Corbeil J, Ouellette M. 2009. Gene expression modulation is associated with gene amplification, supernumerary chromosomes and chromosome loss in antimony-resistant Leishmania infantum. Nucleic Acids Res 37:1387–1399. doi: 10.1093/nar/gkn1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ouellette M, Papadopoulou B. 1993. Mechanisms of drug-resistance in Leishmania. Parasitol Today 9:150–153. doi: 10.1016/0169-4758(93)90135-3. [DOI] [PubMed] [Google Scholar]
- 60.Ubeda JM, Legare D, Raymond F, Ouameur AA, Boisvert S, Rigault P, Corbeil J, Tremblay MJ, Olivier M, Papadopoulou B, Ouellette M. 2008. Modulation of gene expression in drug resistant Leishmania is associated with gene amplification, gene deletion and chromosome aneuploidy. Genome Biol 9:R115. doi: 10.1186/gb-2008-9-7-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shaw CD, Lonchamp J, Downing T, Imamura H, Freeman TM, Cotton JA, Sanders M, Blackburn G, Dujardin JC, Rijal S, Khanal B, Illingworth CJR, Coombs GH, Carter KC. 2016. In vitro selection of miltefosine resistance in promastigotes of Leishmania donovani from Nepal: genomic and metabolomic characterization. Mol Microbiol 99:1134–1148. doi: 10.1111/mmi.13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ubeda JM, Raymond F, Mukherjee A, Plourde M, Gingras H, Roy G, Lapointe A, Leprohon P, Papadopoulou B, Corbeil J, Ouellette M. 2014. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol 12:e1001868. doi: 10.1371/journal.pbio.1001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar P, Lodge R, Raymond F, Ritt JF, Jalaguier P, Corbeil J, Ouellette M, Tremblay MJ. 2013. Gene expression modulation and the molecular mechanisms involved in nelfinavir resistance in Leishmania donovani axenic amastigotes. Mol Microbiol 89:565–582. doi: 10.1111/mmi.12298. [DOI] [PubMed] [Google Scholar]
- 64.Mukherjee A, Boisvert S, do Monte-Neto RL, Coelho AC, Raymond F, Mukhopadhyay R, Corbeil J, Ouellette M. 2013. Telomeric gene deletion and intrachromosomal amplification in antimony-resistant Leishmania. Mol Microbiol 88:189–202. doi: 10.1111/mmi.12178. [DOI] [PubMed] [Google Scholar]
- 65.Yasur-Landau D, Jaffe CL, David L, Doron-Faigenboim A, Baneth G. 2018. Resistance of Leishmania infantum to allopurinol is associated with chromosome and gene copy number variations including decrease in the S-adenosylmethionine synthetase (METK) gene copy number. Int J Parasitol Drugs Drug Resist 8:403–410. doi: 10.1016/j.ijpddr.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dumetz F, Imamura H, Sanders M, Seblova V, Myskova J, Pescher P, Vanaerschot M, Meehan CJ, Cuypers B, De Muylder G, Spath GF, Bussotti G, Vermeesch JR, Berriman M, Cotton JA, Volf P, Dujardin JC, Domagalska MA. 2017. Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. mBio 8:e00599-17. doi: 10.1128/mBio.00599-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barrand MA, Bagrij T, Neo SY. 1997. Multidrug resistance-associated protein: a protein distinct from P-glycoprotein involved in cytotoxic drug expulsion. Gen Pharmacol 28:639–645. doi: 10.1016/s0306-3623(96)00284-4. [DOI] [PubMed] [Google Scholar]
- 68.Chauhan IS, Rao GS, Singh N. 2019. Enhancing the copy number of Ldrab6 gene in Leishmania donovani parasites mediates drug resistance through drug-thiol conjugate dependent multidrug resistance protein A (MRPA). Acta Trop 199:105158. doi: 10.1016/j.actatropica.2019.105158. [DOI] [PubMed] [Google Scholar]
- 69.Mahringer A, Fricker G. 2016. ABC transporters at the blood-brain barrier. Expert Opin Drug Metab Toxicol 12:499–508. doi: 10.1517/17425255.2016.1168804. [DOI] [PubMed] [Google Scholar]
- 70.Bellamy WT. 1996. P-glycoproteins and multidrug resistance. Annu Rev Pharmacol Toxicol 36:161–183. doi: 10.1146/annurev.pa.36.040196.001113. [DOI] [PubMed] [Google Scholar]
- 71.Kaur J, Dey CS. 2000. Putative P-glycoprotein expression in arsenite-resistant Leishmania donovani down-regulated by verapamil. Biochem Biophys Res Commun 271:615–619. doi: 10.1006/bbrc.2000.2680. [DOI] [PubMed] [Google Scholar]
- 72.Austrup J, Ntais P, Christodoulou V, Dedet JP, Pratlong F, Antoniou M. 2014. Frequency of MDR1-related p-gp overexpression in Greek Leishmania isolates. Parasitol Res 113:1225–1232. doi: 10.1007/s00436-014-3761-8. [DOI] [PubMed] [Google Scholar]
- 73.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. 2006. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 74.Messaritakis I, Christodoulou V, Mazeris A, Koutala E, Vlahou A, Papadogiorgaki S, Antoniou M. 2013. Drug resistance in natural isolates of Leishmania donovani s.l. promastigotes is dependent of Pgp170 expression. PLoS One 8:e65467. doi: 10.1371/journal.pone.0065467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Valiathan R, Dubey ML, Mahajan RC, Malla N. 2006. Leishmania donovani: effect of verapamil on in vitro susceptibility of promastigote and amastigote stages of Indian clinical isolates to sodium stibogluconate. Exp Parasitol 114:103–108. doi: 10.1016/j.exppara.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 76.Perez-Victoria JM, Di Pietro A, Barron D, Ravelo AG, Castanys S, Gamarro F. 2002. Multidrug resistance phenotype mediated by the P-glycoprotein-like transporter in Leishmania: a search for reversal agents. Curr Drug Targets 3:311–333. doi: 10.2174/1389450023347588. [DOI] [PubMed] [Google Scholar]
- 77.Rai S, Bhaskar Goel SK, Nath Dwivedi U, Sundar S, Goyal N. 2013. Role of efflux pumps and intracellular thiols in natural antimony resistant isolates of Leishmania donovani. PLoS One 8:e74862. doi: 10.1371/journal.pone.0074862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dos Reis PG, do Monte-Neto RL, Melo MN, Frézard F. 2017. Biophysical and pharmacological characterization of energy-dependent efflux of Sb in laboratory-selected resistant strains of Leishmania (Viannia) subgenus. Front Cell Dev Biol 5:24. doi: 10.3389/fcell.2017.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lachaud L, Bourgeois N, Plourde M, Leprohon P, Bastien P, Ouellette M. 2009. Parasite susceptibility to amphotericin B in failures of treatment for visceral leishmaniasis in patients coinfected with HIV type 1 and Leishmania infantum. Clin Infect Dis 48:e16–e22. doi: 10.1086/595710. [DOI] [PubMed] [Google Scholar]
- 80.Hendrickx S, da Luz RAI, Bhandari V, Kuypers K, Shaw CD, Lonchamp J, Salotra P, Carter K, Sundar S, Rijal S, Dujardin JC, Cos P, Maes L. 2012. Experimental induction of paromomycin resistance in antimony-resistant strains of L. donovani: outcome dependent on in vitro selection protocol. PLoS Negl Trop Dis 6:e1664. doi: 10.1371/journal.pntd.0001664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.da Luz RI, Vermeersch M, Dujardin JC, Cos P, Maes L. 2009. In vitro sensitivity testing of Leishmania clinical field isolates: preconditioning of promastigotes enhances infectivity for macrophage host cells. Antimicrob Agents Chemother 53:5197–5203. doi: 10.1128/AAC.00866-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.