

Daptomycin-nonsusceptible (DAP-NS) Staphylococcus aureus often exhibits gain-in-function mutations in the mprF gene (involved in positive surface charge maintenance). Standard β-lactams, although relatively inactive against methicillin-resistant S. aureus (MRSA), may prevent the emergence of mprF mutations and DAP-NS. We determined if β-lactams might also impact DAP-NS isolates already possessing an mprF mutation to revert them to DAP-susceptible (DAP-S) phenotypes and, if so, whether this is associated with specific penicillin-binding protein (PBP) targeting.
KEYWORDS: cationic peptide, methicillin-resistant Staphylococcus aureus, mprF, penicillin-binding protein
ABSTRACT
Daptomycin-nonsusceptible (DAP-NS) Staphylococcus aureus often exhibits gain-in-function mutations in the mprF gene (involved in positive surface charge maintenance). Standard β-lactams, although relatively inactive against methicillin-resistant S. aureus (MRSA), may prevent the emergence of mprF mutations and DAP-NS. We determined if β-lactams might also impact DAP-NS isolates already possessing an mprF mutation to revert them to DAP-susceptible (DAP-S) phenotypes and, if so, whether this is associated with specific penicillin-binding protein (PBP) targeting. This study included 25 DAP-S/DAP-NS isogenic, clinically derived MRSA bloodstream isolates. MICs were performed for DAP, nafcillin (NAF; PBP-promiscuous), cloxacillin (LOX; PBP-1), ceftriaxone (CRO; PBP-2), and cefoxitin (FOX; PBP-4). Three DAP-NS isolates were selected for a 28-day serial passage in subinhibitory β-lactams. DAP MICs and time-kill assays, host defense peptide (LL-37) susceptibilities, and whole-genome sequencing were performed to associate genetic changes with key phenotypic profiles. Pronounced decreases in baseline MICs were observed for NAF and LOX (but not for CRO or FOX) among DAP-NS versus DAP-S isolates (“seesaw” effect). Prolonged (28-d) β-lactam passage of three DAP-NS isolates significantly reduced DAP MICs. LOX was most impactful (∼16-fold decrease in DAP MIC; 2 to 0.125 mg/liter). In these DAP-NS isolates with preexisting mprF polymorphisms, accumulation of additional mprF mutations occurred with prolonged LOX exposures. This was associated with enhanced LL-37 killing activity and reduced surface charge (both mprF-dependent phenotypes). β-lactams that either promiscuously or specifically target PBP-1 have significant DAP “resensitizing” effects against DAP-NS S. aureus strains. This may relate to the acquisition of multiple mprF single nucleotide polymorphism (SNPs), which, in turn, affect cell envelope function and metabolism.
INTRODUCTION
Staphylococcus aureus has developed resistance to virtually every class of antimicrobials (1, 2). Although daptomycin (DAP) has been effective in treating methicillin-resistant S. aureus (MRSA) infections, including those refractory to vancomycin (VAN), a number of published clinical reports document the in vivo development of DAP nonsusceptibility (DAP-NS) during treatment (3, 4). Currently, the Infectious Diseases Society of America (IDSA) recommends DAP plus β-lactam therapy as a principal option for the treatment of such persistent MRSA infections (1). This recommendation reflects previous findings suggesting oxacillin (or nafcillin) can enhance the antimicrobial effects of DAP and prevent or delay the emergence of the DAP-NS (5, 6).
The characterization of a number of DAP-NS MRSA isolates demonstrates that mutations within cell wall-associated and global regulatory genes contribute to the DAP-NS phenotype (5). However, the most clinically relevant and frequent mutations cluster within recognized DAP-NS “hot spots” of the multipeptide resistance factor (mprF) gene (5, 6). The S. aureus mprF locus is responsible for increasing the synthesis of cationic lysyl-phosphatidylglycerol (L-PG) and its translocation into the outer cytoplasmic membrane (CM) leaflet; this presumably results in an increase in net positive surface charge and repulsion of the calcium-DAP complex, preventing insertion of the cationic oligomer (6, 7). Interestingly, DAP-NS isolates with these hot spot mprF mutations can become “resensitized” in vitro to DAP when additional mprF point mutations are gained. Isolates with such genotypes are associated with a loss of mprF functionality in terms of reduced lysinylation of PG and/or its outer CM translocation (8, 9).
A common finding among DAP-NS isolates is a paradoxical increase in β-lactam susceptibility, a phenomenon termed the DAP-β-lactam “seesaw effect” (10, 11). Although the seesaw mechanism has not been fully elucidated, it appears to involve at least two genes which impact either the cell wall stimulon (vraSR) and/or PBP2a chaperoning, folding, and CM localization (prsA) (11, 12). The synergy of DAP and β-lactams exploits this effect through a combination of enhanced DAP binding to the cell wall divisome, as well as blockade of specific penicillin-binding proteins (PBPs) (4, 13, 14). The latter effect has been shown to be most potent with β-lactams that inhibit PBP-1. This monofunctional transpeptidase is responsible for cell division and separation, and it locates at the divisome of S. aureus (15). The cell divisome is also where DAP binding focally occurs to exert its most potent CM depolarization, as well as essential cell division protein mislocalization (16). Given these elegantly linked mechanisms of DAP-β-lactam combined activity, we hypothesized that exposure to β-lactams might potentially resensitize DAP-NS S. aureus to DAP. In this study, DAP-NS isolates were passaged in β-lactams possessing specific versus promiscuous PBP-binding profiles and then evaluated for their resulting phenotypic and genotypic modifications.
This study was presented in part at the 18th International Symposium on Staphylococci and Staphylococcal Infection in Copenhagen, Denmark, August 2018.
RESULTS
Antibiotic susceptibilities.
The susceptibilities to the tested antibiotics in the 25 DAP-susceptible (DAP-S) and DAP-nonsusceptible (DAP-NS) isolate pairs and their previously identified mprF mutations are provided in Table 1 (14, 17–19). Overall, baseline DAP MICs ranged from 0.19 to 0.75 mg/liter in the DAP-S isolates and 2 to 4 mg/liter for DAP-NS isolates derived in vivo following DAP treatment. The isolate JKD6005 displayed a DAP MIC of 1 mg/liter by broth dilution and 2 mg/liter by Etest, which is similar to the reported value (20). However, this isolate was derived in vivo from a patient receiving VAN treatment only, and it lacks an mprF mutation, so this discordance is not unexpected. Overall, the seesaw effect between elevated DAP MICs and lower β-lactam MICs was apparent among many β-lactams tested (Fig. 1). A ≥4-fold decrease in the MICs for at least one β-lactam in the DAP-NS versus DAP-S isolates occurred in 48% of pairs, most notably with the PBP-1 specific cloxacillin (LOX) (44% of pairs). Analysis of the correlation between DAP-NS and β-lactam susceptibilities revealed a significant negative association between DAP and LOX MICs (P = 0.032), while nafcillin (NAF), meropenem (MEM), and ceftriaxone (CRO) susceptibilities demonstrated similar trends toward negative association but lacked statistical significance (P ≥ 0.118) (Fig. 1).
TABLE 1.
Isolate characteristicsa
| Isolate | mprF SNP | MIC (mg/liter) of: |
|||||
|---|---|---|---|---|---|---|---|
| DAP | NAF | LOX | MEM | CRO | FOX | ||
| J01 | 0.5 | 32 | 16 | 8 | 128 | 64 | |
| J03 | T345I | 2 | 16 | 2 | 4 | 128 | 64 |
| D592 | 0.5 | 256 | 512 | 512 | 256 | 256 | |
| D712 | L341S | 2 | 128 | 512 | 512 | 256 | 256 |
| JKD6004 | 0.5 | 256 | 512 | 512 | 512 | 512 | |
| JKD6005 | 2 | 128 | 512 | 512 | 512 | 512 | |
| C1 | 0.19b | 16 | 32 | 32 | 4 | 32 | |
| C2 | L826F | 2 | 16 | 2 | 16 | 16 | 32 |
| C3 | 0.5 | 32 | 64 | 32 | 128 | 32 | |
| C4 | P314L | 4 | 32 | 2 | 16 | 16 | 128 |
| C5 | 0.25 | 32 | 2 | 32 | 128 | 32 | |
| C6 | T345A | 3b | 32 | 0.125 | 16 | 32 | 32 |
| C7 | 0.5 | 64 | 16 | 32 | 64 | 64 | |
| C8 | 3b | 64 | 2 | 32 | 8 | 32 | |
| C9 | 0.5 | 32 | 1 | 16 | 32 | 32 | |
| C10 | L826F | 3b | 16 | 0.063 | 8 | 4 | 32 |
| C13 | 0.75b | 4 | 4 | 16 | 16 | 64 | |
| C14 | T472K | 4 | 0.25 | 0.125 | 2 | 4 | 32 |
| C15 | 0.75b | 64 | 256 | 32 | 32 | 64 | |
| C16 | M347R | 4 | 32 | 32 | 16 | 16 | 128 |
| C17 | 0.5 | 64 | 128 | 32 | 64 | 32 | |
| C18 | L341S | 4 | 64 | 64 | 32 | 64 | 64 |
| C19 | 0.38b | 128 | 64 | 32 | 64 | 64 | |
| C21 | L826F | 4 | 32 | 0.5 | 8 | 16 | 32 |
| C22 | 0.5 | 8 | 0.125 | 4 | 8 | 64 | |
| C23 | 4 | 64 | 64 | 32 | 128 | 128 | |
| C24 | 0.5 | 4 | 4 | 8 | 64 | 32 | |
| C25 | S295L | 3 | 0.25 | 0.25 | 4 | 16 | 32 |
| C26 | 0.38b | 128 | 256 | 128 | 512 | 64 | |
| C27 | T345K | 2 | 128 | 512 | 128 | 512 | 128 |
| C30 | 0.25 | 32 | 8 | 8 | 16 | 32 | |
| C31 | L826F | 2 | 32 | 2 | 4 | 4 | 32 |
| C32 | 0.5 | 4 | 2 | 4 | 32 | 32 | |
| C33 | S337L | 2 | 4 | 4 | 4 | 16 | 16 |
| C34 | 0.38b | 64 | 32 | 8 | 32 | 64 | |
| C35 | 4 | 64 | 2 | 8 | 32 | 32 | |
| C36 | 0.5 | 128 | 512 | 128 | 128 | 64 | |
| C37 | V351E | 3b | 1 | 0.25 | 8 | 16 | 32 |
| C38 | 0.75b | 32 | 16 | 8 | 16 | 64 | |
| C39 | L826F | 3b | 32 | 16 | 8 | 8 | 64 |
| C40 | 0.25 | 16 | 0.5 | 8 | 4 | 32 | |
| C41 | M347R | 3b | 2 | 0.25 | 16 | 16 | 32 |
| C42 | 0.75b | 16 | 4 | 8 | 64 | 64 | |
| C43 | S337L | 3b | 8 | 2 | 8 | 16 | 64 |
| C46 | 0.38b | 32 | 8 | 16 | 32 | 16 | |
| C47 | L826F | 3b | 128 | 8 | 32 | 32 | 64 |
| C48 | 0.5 | 16 | 0.25 | 8 | 4 | 32 | |
| C49 | T345I | 2 | 16 | 0.25 | 32 | 16 | 64 |
| C50 | 0.5 | 64 | 512 | 128 | 256 | 128 | |
| C51 | T345I | 2 | 64 | 128 | 128 | 128 | 64 |
These include previously identified mprF SNPs (149) and DAP and β-lactam susceptibilities in DAP-S and DAP-NS isolates. Results include both broth dilution and Etest method confirmation.
Etest result is displayed when discriminate differences are found between broth dilutions (e.g., 2 to 4 mg/liter).
FIG 1.

DAP-β-lactam seesaw effect demonstrated by correlation of DAP and β-lactam MICs in all 50 isolates. *, P = 0.032.
Serial passage.
DAP-NS isolates J03, D712, C25, and JKD6005 were passaged in triplicate daily for 28 d in exposure arms of no antibiotic (media alone), NAF, LOX, CRO, or cefoxitin (FOX). The selection of these isolates for passage was based on their containing common but distinct hot spot mutations within the bifunctional domain of MprF for J03 (T345I), D712 (L341S), and C25 (S295L) (6). The isolate JKD6005 was selected as a “negative” control to interrogate the importance of preexisting mprF mutations, as it contained a wild-type mprF sequence. Figure 2 displays the DAP MIC fold change over the 28-d exposures with or without different β-lactams. Of note, in isolates with preexisting mprF mutations, passage in β-lactams was often able to resensitize the isolate to DAP. This occurred as early as day 7 of passage and continued throughout the 28-d exposure. Overall, LOX was most effective at resensitizing isolates to DAP, followed by NAF. In isolate C25, CRO was also highly effective in DAP resensitization, but a limited resensitizing effect of this agent was noted in other isolates. Enhanced DAP susceptibility with β-lactam passage did not occur in JKD6005 lacking a preexisting mprF polymorphism. This strain does contain a mutation in WalR (YycF/VicR), an essential response regulator implicated in both DAP-NS and the seesaw effect, so this may play a role in preventing resensitization (20). The DAP-β-lactam seesaw effect was present in two of these three DAP-NS strains used for long-term β-lactam passage (J03 and C25). In these two latter strains, DAP resensitization postpassage was accompanied by at least a 2-fold MIC increase in the respective β-lactam used for passage (Table 2).
FIG 2.

Serial passage in isolates J03 (A), D712 (B), C25 (C), and JKD6005 (D) with no antibiotic or β-lactams. Data represent median DAP MIC changes over 28 days with different exposures.
TABLE 2.
Daptomycin susceptibility and mprF polymorphisms with β-lactam passage after 28 daysa
| Isolate | Passage | Replicate | DAP MIC (mg/liter) | β-lactam MIC (mg/liter)b | mprF SNP | MprF domain | div1b mutation | rpoB/C mutation |
|---|---|---|---|---|---|---|---|---|
| J01 | None | 0.5 | ||||||
| J03 | None | 2 | T345I | Bifunctional | rpoB S464P | |||
| Media | i | 2 | None | |||||
| Mediaa | ii | 1 | Y325H | Bifunctional | ||||
| Mediaa | iii | 1 | R437P | Synthase | ||||
| CROc | ii | 0.75 | 512 | V152G | Translocase | |||
| LOXc | ii | 0.125 | 32 | R788L | Synthase | Q425d | ||
| LOXc | iii | 0.125 | 32 | R788L | Synthase | Q415d | ||
| D592 | None | 0.5 | ||||||
| D712 | None | 2 | L341S | Bifunctional | ||||
| Media | i | 2 | None | |||||
| Media | ii | 1 | None | |||||
| Media | iii | 2 | None | |||||
| NAFe | iii | 1 | 256 | S337P | Bifunctional | |||
| CROe | i | 2 | 2,048 | M609T | Synthase | rpoC A567V | ||
| CROe | iii | 0.5 | 2,048 | G389A | Synthase | rpoB G767C | ||
| FOXe | ii | 0.5 | 512 | F657L | Synthase | |||
| LOXe | i | 0.5 | 1,024 | S136L | Translocase | |||
| LOXe | ii | 0.5 | 1,024 | S136L | Translocase | |||
| C24 | None | 0.5 | - | - | ||||
| C25 | None | 3–4 | S295L | Bifunctional | ||||
| Mediaf | i | 1 | S825d | Synthase | ||||
| Mediaf | ii | 1 | S825d | Synthase | ||||
| Media | iii | 2 | None | - | ||||
| FOXf | Iii | 1 | 64 | A315S | Bifunctional | |||
| LOXf | i | 0.125 | 8 | L84d | Translocase | A420E | ||
| LOXf | ii | 0.125 | 8 | L84d | Translocase | |||
| LOXf | iii | 0.125 | 8 | L84d | Translocase | E416d |
For the β-lactam-exposed strains, only the passages with additional mprF mutations (versus DAP-NS parent strain) are presented.
β-lactam MIC represents the MIC in the respective β-lactam after passage.
Passage isolates maintained MprF T345I and RpoB S464P.
Nonsense stop-gain mutation resulting in premature end of translation.
Passage isolates maintained MprF L341S.
Passage isolates maintained MprF S295L.
Whole-genome sequencing.
The isolates selected for serial passage were sequenced at day zero prior to β-lactam passage and then at the end of treatment (day 28). Passage isolates maintained the preexisting mprF mutations identified in the J03, D712, and C25 backgrounds and gained additional mutations in mprF, a cell division gene (div1b), the beta and beta′ subunits of the RNA polymerase (rpoBC), and several genes associated with metabolic function (Table 2; Table S1 in the supplemental material). Of particular interest was the accumulation of additional mprF mutations. This was observed in all three isolate backgrounds with LOX passage; the passage isolates with these genotypes also demonstrated increased sensitivity to DAP, with up to 32-fold difference in susceptibility (e.g., MIC changes from 3 to 4 mg/liter to 0.125 mg/liter). The largest shifts in DAP susceptibility were also associated with concomitant gains of div1b mutations. Mutations in mprF were not identified in the JKD6005 background with any β-lactam passage, indicating that a preexisting mprF mutation may be necessary for β-lactams to induce this latter effect.
To determine the temporal relationship between these identified mutations and phenotype, interim-passaged strains at days 7, 14, and 21 were also whole-genome sequenced. This identified, first, that no new mutations occurring in any passaged strains during this 7- to 21-day period compared to those identified at day 28 of passage (Table 2 and Table S1). Second, this also determined the approximate time of appearance and sustainability of these mutations throughout continued passage. With mprF, div1b, and rpoB/C mutations, this interim sequencing identified that mprF single nucleotide polymorphisms (SNPs) were present at day 7, while div1b was first detected at day 21 and rpoB/C only at day 28. Table 3 represents the mprF SNP frequencies in strains J03 and D712 at days 7, 21, and 28 passaged in LOX. In both strains, replicates with high mprF allele frequency correlated with a DAP resensitization, while replicates with no mprF alternative allele frequency maintained DAP MICs >1 mg/liter. In J03 passaged in LOX, mutations in div1b occurred along with an additional mprF mutation. In SNP frequency analysis, this mutation was detected initially at day 21 (0.71 to 0.72 alternative allele frequency) and then at day 28 (0.99 to 1.00 alternative allele frequency). This div1b appearance aligned with the greatest DAP resensitization (MIC = 0.125 mg/liter) occurring at days 21 to 28.
TABLE 3.
mprF SNP frequencies in strains J03 and D712 at days 7, 14, 21, and 28 of LOX passagea
| Isolate | Passage | Replicate | Additional mprF SNP frequency at day: |
|||
|---|---|---|---|---|---|---|
| 7 | 14 | 21 | 28 | |||
| J03b | LOX | i | 0.58 | 0.74 | 0.11 | 0.00c |
| J03b | LOX | ii | 0.11 | 0.08 | 0.78 | 0.98 |
| J03b | LOX | iii | 0.51 | 0.61 | 0.79 | 0.99 |
| D712d | LOX | i | 0.91 | 0.67 | 0.86 | 1.00 |
| D712d | LOX | ii | 1.00 | 1.00 | 1.00 | 0.99 |
| D712d | LOX | iii | 0.00 | 0.00 | 0.00 | 0.00 |
Frequency of mapped reads containing the alternative allele is represented.
Passage isolates maintained MprF T345I.
Value of 0.00 indicates that all mapped reads contained the reference allele.
Passage isolates maintained MprF L341S.
Time-kill assays.
To determine if the MIC reductions with β-lactam passage translated to enhanced DAP killing, isolates J01/J03 and J03 passaged in β-lactam for 28 days were evaluated in time-kill curves with DAP at 3.9 mg/liter. As displayed in Fig. 3, no significant growth defect occurred after the 28-day passage. Upon exposure to DAP, the DAP-S isolate J01 was rapidly killed, while the DAP-NS J03 isolate regrew in the presence of DAP. However, the β-lactam-passaged isolates were all significantly killed with DAP over the first 8 h of exposure, paralleling the substantive decline in DAP MICs on passage. The most rapid, extensive, and sustained DAP killing occurred with the J03 LOX passage isolate (P < 0.01 versus J03) and was similar in DAP killing to the susceptible wild-type isolate J01. Moreover, only the LOX-passaged isolate did not exhibit substantial regrowth between 24 and 48 h exposure to DAP. To assess the contribution of mprF along with other mutations developed during passage on DAP killing, J03-LOX (additional mprF plus div1b mutation), D712-LOX (additional mprF plus rpoB mutation), and JKD6005-LOX (no mprF mutation) were assessed in kill curves. As displayed in Fig. 4, DAP killing was enhanced with additional mprF mutations and most pronounced when with the div1b mutation. These results also indicate β-lactam passage alone does not substantially enhance DAP activity without additional mprF mutations.
FIG 3.

Time-kill curves. (A) Kill curves J01 and J03 versus DAP of 3.9 mg/liter. (B) Kill curves of J03-passaged isolates versus DAP of 3.9 mg/liter.
FIG 4.
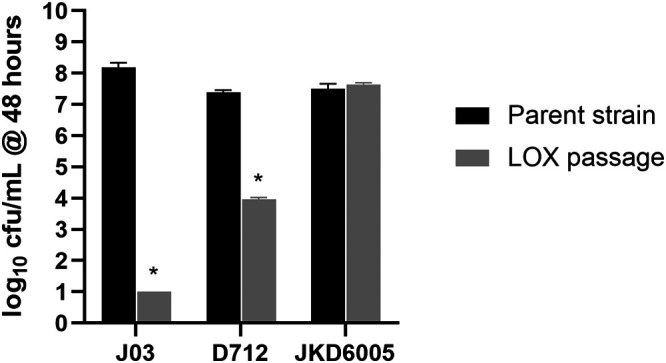
Bacteria quantification (CFU/ml) at 48 h in time time-kill curves with DAP 3.9 mg/liter against individual strains passaged in LOX. *, P < 0.001 versus parent strains.
LL-37 susceptibility.
The evolution of DAP-NS is often accompanied by increased resistance to killing by human host defense cationic peptides, such as LL-37 (18, 19). The J01/J03 pair and J03-passaged isolates were assessed for LL-37 susceptibility. Like previous studies, DAP-NS (J03) was associated with reduced LL-37 killing compared to DAP-S (J01) (18). Among the β-lactam-passaged isolates, the LOX-passaged strains exhibited the lowest survival profiles when exposed in vitro to LL-37 concentrations of either 2 mg/liter or 5 mg/liter (Table 4) (P < 0.01); the LL-37 susceptibility pattern was virtually identical to the parental DAP-S J01 isolate. The other β-lactam-passaged isolates exhibited similar LL-37 survival profiles to J03, except for CRO- and FOX-passaged isolates, which exhibited enhanced survival at the higher concentration of LL-37 (P < 0.05). Similar to time-kill curves with DAP, LL-37 killing was impacted by additional mprF plus other mutations. LL-37 killing was most pronounced in J03-LOX with additional mprF plus div1b mutations (Table 4). However, minimal killing with LL-37 of 2 to 5 mg/liter was observed against D712-LOX with additional mprF plus rpoB mutations (93% to 98% survival) and JKD6005-LOX with no additional mutations (89% to 98% survival).
TABLE 4.
In vitro susceptibility to DAP and host defense peptide (HDP) LL-37 in J01/J03 and J03 passage isolates
| Isolate | Passage | DAP MIC (mg/liter) | % survival (mean + SD) after 2 h of exposure to LL-37 concn of: |
|
|---|---|---|---|---|
| 2 mg/liter | 5 mg/liter | |||
| J01 | 0.5 | 82 ± 9 | 8 ± 1 | |
| J03 | 3 to 4 | 92 ± 21 | 46 ± 8 | |
| J03 | Media | 1 | 95 ± 2 | 61 ± 7a |
| NAF | 1 | 84 ± 8 | 51 ± 9 | |
| LOX | 0.125 | 75 ± 13a | 7 ± 10a | |
| CRO | 2 | 97 ± 1 | 63 ± 4a | |
| FOX | 2 | 98 ± 1 | 68 ± 6a | |
P < 0.05 versus DAP-NS J03.
DISCUSSION
Several studies have described the synergistic relationship between β-lactam antibiotics and both DAP and cationic host defense peptides (HDPs) (4, 21). This synergy with HDPs is thought to be advantageous in enhancing β-lactam treatment, and it may contribute to the unambiguously superior clinical outcomes with β-lactams over VAN in methicillin-susceptible S. aureus (MSSA) bacteremic syndromes (22, 23). Although prior studies have shown that β-lactams can suppress the evolution of DAP or VAN resistance in vitro (24–26), few studies have determined the impact of β-lactams to prevent emergence of resistance to these agents in vivo. In the case of DAP-NS, this impact has been linked to the ability of β-lactams to prevent emergence of mprF mutations (24, 25). However, to our knowledge, no report has conclusively documented the ability of β-lactams to resensitize clinically derived DAP-NS isolates to DAP. The results of our current investigation indicate that long-term β-lactam passage can, indeed, resensitize DAP-NS isolates to DAP; this event appears to be mediated, at least in part, through the accumulations of additional point mutations in mprF.
Based on our data and others, the PBP-binding profile of those β-lactams that seem to be associated with DAP synergy in MRSA are likely selective, not global (11, 12). We previously found that β-lactams that target PBP-1 either as part of promiscuous PBP binding (PBPs 1 to 4 and 2a) or via PBP-1 specifically result in highly synergistic interactions with DAP versus DAP-NS MRSA. This synergy does not appear to occur exclusively through enhanced DAP binding to the CM, but rather through a dual mechanistic effect at distinct β-lactam and DAP cell wall divisome targets (13).
In the current study, using a large collection of well-characterized DAP-S/DAP-NS isolate pairs derived from patients, we evaluated the ability of β-lactams with a range of PBP-binding specificity profiles to resensitize DAP-NS strains. The most profound synergistic effects with DAP occurred with LOX, a PBP-1-specific antibiotic, although similar trends were observed with NAF, MER, and CRO (Table 1; Fig. 1). We were somewhat surprised to find a substantial seesaw effect with CRO, a PBP-2-specific antibiotic (8/25 pairs [32%]). Of note, the seesaw effect among specific DAP-S/DAP-NS isolate-pairs was generally harmonious for LOX versus CRO. CRO MICs were the highest among all the β-lactams tested in the 25 DAP-S isolates (MIC50 = 64), perhaps allowing for a greater “window” for disclosing a seesaw relationship. In contrast, The DAP-S isolates were more susceptible to LOX and MEM (MIC50 = 16). This work emphasizes that the seesaw effect is variable among distinct β-lactams and strain specific and that inhibition of PBP-1 or PBP-2 particularly can foster a synergistic interaction with DAP versus DAP-NS isolates (27).
This work has potentially important clinical translational implications. As previously described, DAP-NS isolates derived from patients treated with DAP who failed DAP therapy quite frequently exhibited point mutations in mprF (20/25 isolates [80%] in this investigation) (5, 8, 28). Although DAP-NS can evolve without DAP treatment, this has been shown to occur primarily in VAN treatment in which cross-resistance to both VAN and DAP develops, most likely via a combination of a cell wall-thickening phenotype as well as distinct metabolic adaptations (29–32). In this study, we identified that prolonged β-lactam passage can reverse the elevated DAP MICs of DAP-NS isolates, resulting in some passaged isolates becoming up to 16-fold more DAP susceptible versus their respective DAP-NS isolates and 4-fold lower than the original DAP-S parental isolate. This effect was noted in all LOX-passaged isolates from backgrounds with preexisting mprF polymorphisms, while no significant resensitization occurred with prolonged β-lactam passage in JKD6005 possessing the wild-type mprF sequence. This analysis also delineated that these additional mprF mutations can occur as early as day 7 of passage and were directly related to this resensitization event as one of the first mutations identified. These data indicate that existing mprF perturbations may be essential for the β-lactam-induced DAP resensitization event to occur. Although the β-lactam passage was done without DAP in culture, there may be benefits to adding β-lactams to DAP treatment during prolonged courses of therapy to either prevent DAP-NS development or revert emerging DAP-NS subpopulations toward a more DAP-S phenotype (17). Of interest, we demonstrated the latter phenomenon occurring in a patient in which the DAP-NS phenotype reverted to DAP-S when ceftaroline was added to DAP treatment (33).
In a recent study by Yang and colleagues (8), introduction of dual-point mutations in mprF via genetic complementation substantially lowered DAP MICs compared to the DAP-NS host isolate, as well as the DAP-S parental isolate. These investigators incorporated combinations of two common hot spot mprF mutations in DAP-NS isolates, S295L plus L826F and T345A plus L826F, resulting in (i) enhanced DAP susceptibility, and (ii) evidence of reduced MprF functionality (i.e., significant reductions in outer CM L-PG flipping). In our current passage experiments, these same hot spot mutations were present in J03 (T345X) and C25 (S295L), while another common hot spot mprF mutation was present in isolate D712 (L341S). We were able to recapitulate the novel findings of the Yang group (8) “pharmacologically” by using distinct β-lactams to generate additional mprF mutations in these isolates. Notably, the PBP-1-specific LOX generated such additional mprF mutations in all three isolates. Isolates exposed to LOX with the same preexisting mprF mutations as studied by Yang et al. resulted in exquisite susceptibility to DAP (MIC = 0.125 mg/liter) in these dual-point mprF mutation, postpassage isolates. These additional point mutations occurred in either the synthase or translocase domain, while none were mapped to the bifunctional domain (Table 2). However, the present study presents an interesting association but does not indicate causality of these dual-point mprF SNPs. The changes associated with CM phospholipid composition and CM order with these dual-point mprF mutations and their causality for DAP-NS reversal are a focus of future studies.
One well-described mechanism of DAP action is via calcium-activated DAP oligomerization and subsequent CM depolarization. This mechanism recently was found to be linked to DAP binding at the divisome of the CM (16, 34). We previously identified that DAP-NS cells have increased divisome formation and increased PBP-1 transcription, indicating that this may be an adaptive response to DAP-induced CM damage (13).
In this present study, several mutations besides mprF occurred in β-lactam-passaged isolates (Table 2). Many of these mutations are associated with fundamental cellular metabolism, such as purine biosynthesis, sensor histidine kinases, asparaginyl-tRNA synthetase, and phosphoglycerate dehydrogenase (Table S1 in the supplemental material); in turn, these perturbations may well be expected during prolonged exposure to subinhibitory antibiotic levels (35, 36). Furthermore, mutations were identified postpassage in div1b, encoding a putative cell division protein. This occurred exclusively with isolates passaged in LOX and, when present, resulted in highly DAP-S variants (MIC of 0.125 mg/liter). We noted that β-lactam-passaged isolates were more susceptible to DAP killing; it should be emphasized that LOX-passaged isolates with a div1b mutation were killed to the detection limit within 8 h, faster than either the DAP-NS progenitor or DAP-S parental isolate (Fig. 3). We hypothesize that mutations within divisome proteins may alter the compensatory response mechanism to DAP, thus rendering isolates more DAP-S. Current work is in progress to identify the functional role of alterations in cell divisome proteins on DAP activity, including introduction of these novel div1b mutations via allelic exchange into a naive background strain(s).
There are certain limitations to our work to note. First, although a large and well-characterized MRSA strain collection was used for susceptibility screening, only four backgrounds were used in passage experiments. These four strains were phylogenetically diverse with USA300/ST8, USA100/ST5, and ST239 represented (17, 20, 37). Important trends emerged with these latter isolates, but further variability may be documented when additional isolates are evaluated. Our investigation did not evaluate a PBP-3-selective antibiotic such as cefaclor, although we have previously noted limited effects of this agent in combination with DAP (14). Moreover, the exact mechanistic basis for the CM changes observed in our resensitization studies require further clarification. Most interestingly, the mechanism(s) by which selected β-lactams induce DAP-NS isolates with preexisting mprF SNPs to accumulate additional mprF mutations remains to be elucidated. Finally, the mechanisms by which multiple mprF mutations render such strains significantly more DAP-S remain to be delineated. It does not appear that a general “growth” defect is in-play, as the LOX-postpassage DAP-S variant had growth kinetics similar to its DAP-NS progenitor. It should be noted that the growth and killing assays used quantitative CFU assays, which have some limitations to determine bacterial viability due to cell aggregation compared to turbidity measurements.
In summary, this study provides novel insights on the activity of β-lactam antibiotics in DAP-NS MRSA. It points toward an important role of PBP-1-targeting antibiotics to induce mutations that may potentially reverse or prevent the DAP-NS phenotype. These findings support the previous notion of β-lactam prevention of DAP-NS through inhibiting mprF mutation development (17). However, it also introduces the exciting notion that β-lactams can reverse DAP-NS by inducing additional mutations in signature genes related to DAP-NS, such as mprF.
MATERIALS AND METHODS
Bacterial isolates.
This study used a well-characterized collection of 50 clinical MRSA isolates that represent DAP-S and DAP-NS (MICs ≥ 2 mg/liter) pairs derived from bacteremic patients (Table 1). Previous publications with targeted or whole-genome sequence data have described these isolates in detail (5, 17, 20). This collection includes 22 DAP-S/DAP-NS pairs of clinical bloodstream isolates from the Cubist Pharmaceuticals Isolate Collection, two DAP-S/DAP-NS MRSA pairs from patients successfully treated with DAP plus nafcillin following DAP-NS emergence, and one DAP-S/DAP-NS MRSA pair isolated after vancomycin (VAN) treatment (these latter three isolates were kindly supplied by George Sakoulas and Benjamin Howden).
Antimicrobials and media.
The antibiotics used in this study and their PBP-binding profiles include nafcillin (NAF), PBP-nonselective; cloxacillin (LOX), PBP-1 selective; meropenem (MEM), PBP-1 selective; ceftriaxone (CRO), PBP-2 selective; and cefoxitin (FOX), PBP-4 selective. The β-lactams were purchased from Sigma-Aldrich (St. Louis, MO, USA). DAP was commercially purchased and its activity confirmed by quality control susceptibility testing against ATCC 29231 per Clinical and Laboratory Standards Institute (CLSI) guidelines, version M100 ED28:2018 (38). Mueller-Hinton broth II (MHB) (BD, Sparks, MD, USA) supplemented with 25 mg/liter calcium (as CaCl2), 12.5 mg/liter magnesium (as MgCl2), and 2% sodium chloride were used to grow S. aureus in liquid culture with β-lactams. All DAP assays used MHB with 50 mg/liter calcium as recommended (38).
Antibiotic susceptibility testing.
The MICs of all isolates (n = 25 pairs, 50 isolates) to DAP and β-lactams were determined in triplicate by broth microdilution according to the CLSI guidelines (38). DAP MICs were also confirmed by Etest. The passaged isolates were evaluated for DAP MIC in triplicate following 7, 14, 21, and 28 days of passage in each β-lactam. Visual inspection for MIC determination occurred following 18 to 24 h of incubation at 35°C. Isolate pairs with a positive seesaw effect were defined by a ≥ 4-fold decrease in β-lactam MIC in the DAP-NS isolate compared to its respective parental DAP-S strain.
Serial passage.
Four DAP-NS isolates were passaged in triplicate daily for 28 days in exposure arms of no antibiotic, NAF, LOX, CRO, or FOX. The free average concentration in human serum (fCavg) after standard dosing was used for β-lactam daily passage (2.6 mg/liter NAF, 1.4 mg/liter LOX, 24 mg/liter MEM, and 19 mg/liter CRO. If these concentrations were above the β-lactam MIC, then 0.5× MICs were used to allow for bacterial growth as previously described (14). On day zero, bacterial colonies from overnight growth on Mueller-Hinton agar were suspended in normal saline and turbidity adjusted to the equivalent of 0.5 McFarland standard. Each culture was diluted 1:100 into three replicates with fresh MHB25 plus 2% salt to a total volume of 1 ml and containing each β-lactam. Each sample was grown overnight at 37°C with shaking at 160 rpm. Following incubation, 10 μl of each culture was transferred into fresh media and placed back on the shaker to grow as previously described (17). This process continued for 28 consecutive days with subsequent DAP susceptibility testing on samples obtained on days 7, 14, 21, and 28 of passage.
Whole-genome sequencing of serial passage isolates.
Genomic changes in all isolates and replicates pre- and postpassage were analyzed by whole-genome sequencing. Genomic DNA was extracted with the Janus automated workstation, using the Chemagic DNA/RNA kit (Perkin Elmer). Unique dual-index libraries were prepared using the Nextera XT DNA preparation kit (Illumina), and libraries were sequenced on a NextSeq (Illumina) with 2 × 150-bp chemistry as per the manufacturer’s instructions. The short-read sequence data for all isolates were mapped to reference S. aureus J01 (RefSeq accession no. NZ_CP040619.1), D712 (RefSeq accession no. NZ_CP040665.1), or JKD6004 (RefSeq accession no. NZ_CP040622.1), using Snippy v4.4.3 (https://github.com/tseemann/snippy). Variants were called using a minimum mapped read depth of five and base call stringency of 90%. Allelic frequencies were calculated from the mapped alignment without application of the listed thresholds, displayed as the proportion of mapped reads containing the alternative allele compared to those containing the reference allele. Predicted protein consequences of variants were identified using snpEff v4.4 (39), with custom databases constructed from the above-listed RefSeq genomes and configured to use the bacterial and plastid codon table.
Time-kill assays.
Time-kill studies with DAP against the three DAP-NS isolates were conducted both prior to β-lactam passage and on the resulting isolates of each β-lactam passage. Time-kill studies were performed at 37°C in MHB50 containing DAP at the active unbound (free average concentration [ƒCavg]) of 3.9 mg/liter from a standard human dose of 6 mg/kg (40). This was determined with the formula ƒCavg = (ƒCmax + ƒCmin)/2 (14). The rationale for using this test concentration was to approximate a clinically relevant concentration that would be achieved regardless of variation in volume of distribution or patient-dependent clearance.
LL-37 susceptibility.
DAP-NS MRSA frequently exhibits cross-resistance to multiple host defense peptides (HDPs), especially those from either mammalian PMNs or platelets (18). We studied the relative HDP susceptibility profiles of one prototype isogenic DAP-S/DAP-NS isolate-set (J01-J03), including its β-lactam-postpassage variants using LL-37, a linear cathelicidin HDP found in mammalian PMNs and epithelium (41).
The LL-37 bactericidal assay was carried out in minimal liquid nutrient medium, phosphate-buffered saline (PBS; pH 7.4), and 1% brain heart infusion (BD, Sparks, MD, USA) by a 2-h time-kill method as previously described (18, 42). We used 2 to 5 μg/ml of the LL-37 peptide concentrations for the time-kill assay. These sublethal concentrations were employed on the basis of (i) their ability to decrease survival of the parental DAP-S isolates by <50% in preliminary studies, and (ii) peptide concentrations used in previous investigations of HDP-S. aureus interactions (18, 42). LL-37 was obtained from Peptide International, Louisville, KY. A final inoculum (103 CFU/ml) of S. aureus cells was used to assess LL-37, and S. aureus cells were incubated at 37°C. Post-2 h exposure, samples were collected and further processed for quantitative culture to evaluate the degree of killing. Final data were represented as mean percent surviving CFU/ml (± standard deviation [SD]). Since there is no bona fide resistance breakpoint for LL-37, we compared only the mean percent survivability ± SD of study isolates for statistical comparison. A minimum of three experiments were performed on distinct days.
Statistical analysis.
DAP MIC results were evaluated using the Wilcoxon rank sum test. A two-tailed Student's t test was used for statistical analysis of all other quantitative data. Spearman’s r was used to determine antibiotic susceptibility correlations. P values of ≤0.05 defined significance.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by a research grant from the National Institutes of Health (NIAID 1R01AI132627-02) to W.E.R. A.S.B. was supported in part by NIAID grant 1R01AI146078-01; N.N.M. was supported by an intramural grant (number 531604-01-00) from The Lundquist Institute-Harbor UCLA Medical Center, Torrance, CA.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak MJ, Talan DA, Chambers HF, Infectious Diseases Society of America. 2011. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 52:e18–e55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 2.Rodvold KA, McConeghy KW. 2014. Methicillin-resistant Staphylococcus aureus therapy: past, present, and future. Clin Infect Dis 58(Suppl 1):S20–S27. doi: 10.1093/cid/cit614. [DOI] [PubMed] [Google Scholar]
- 3.Capone A, Cafiso V, Campanile F, Parisi G, Mariani B, Petrosillo N, Stefani S. 2016. In vivo development of daptomycin resistance in vancomycin-susceptible methicillin-resistant Staphylococcus aureus severe infections previously treated with glycopeptides. Eur J Clin Microbiol Infect Dis 35:625–631. doi: 10.1007/s10096-016-2581-4. [DOI] [PubMed] [Google Scholar]
- 4.Dhand A, Bayer AS, Pogliano J, Yang SJ, Bolaris M, Nizet V, Wang G, Sakoulas G. 2011. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin Infect Dis 53:158–163. doi: 10.1093/cid/cir340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayer AS, Mishra NN, Cheung AL, Rubio A, Yang SJ. 2016. Dysregulation of mprF and dltABCD expression among daptomycin-non-susceptible MRSA clinical isolates. J Antimicrob Chemother 71:2100–2104. doi: 10.1093/jac/dkw142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayer AS, Mishra NN, Chen L, Kreiswirth BN, Rubio A, Yang SJ. 2015. Frequency and distribution of single-nucleotide polymorphisms within mprF in methicillin-resistant Staphylococcus aureus clinical isolates and their role in cross-resistance to daptomycin and host defense antimicrobial peptides. Antimicrob Agents Chemother 59:4930–4937. doi: 10.1128/AAC.00970-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 1277:139–158. doi: 10.1111/j.1749-6632.2012.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang SJ, Mishra NN, Kang KM, Lee GY, Park JH, Bayer AS. 2018. Impact of multiple single-nucleotide polymorphisms within mprF on daptomycin resistance in Staphylococcus aureus. Microb Drug Resist 24:1075–1081. doi: 10.1089/mdr.2017.0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oku Y, Kurokawa K, Ichihashi N, Sekimizu K. 2004. Characterization of the Staphylococcus aureus mprF gene, involved in lysinylation of phosphatidylglycerol. Microbiology 150:45–51. doi: 10.1099/mic.0.26706-0. [DOI] [PubMed] [Google Scholar]
- 10.Vignaroli C, Rinaldi C, Varaldo PE. 2011. Striking “seesaw effect” between daptomycin nonsusceptibility and beta-lactam susceptibility in Staphylococcus haemolyticus. Antimicrob Agents Chemother 55:2495–2496. doi: 10.1128/AAC.00224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renzoni A, Kelley WL, Rosato RR, Martinez MP, Roch M, Fatouraei M, Haeusser DP, Margolin W, Fenn S, Turner RD, Foster SJ, Rosato AE. 2017. Molecular bases determining daptomycin resistance-mediated resensitization to beta-lactams (seesaw effect) in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 61:e01634-16. doi: 10.1128/AAC.01634-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jousselin A, Manzano C, Biette A, Reed P, Pinho MG, Rosato AE, Kelley WL, Renzoni A. 2015. The Staphylococcus aureus chaperone prsA is a new auxiliary factor of oxacillin resistance affecting penicillin-binding protein 2A. Antimicrob Agents Chemother 60:1656–1666. doi: 10.1128/AAC.02333-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berti AD, Theisen E, Sauer JD, Nonejuie P, Olson J, Pogliano J, Sakoulas G, Nizet V, Proctor RA, Rose WE. 2016. Penicillin binding protein 1 is important in the compensatory response of Staphylococcus aureus to daptomycin-induced membrane damage and is a potential target for beta-lactam-daptomycin synergy. Antimicrob Agents Chemother 60:451–458. doi: 10.1128/AAC.02071-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berti AD, Sakoulas G, Nizet V, Tewhey R, Rose WE. 2013. Beta-lactam antibiotics targeting PBP1 selectively enhance daptomycin activity against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 57:5005–5012. doi: 10.1128/AAC.00594-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira SF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A. 2009. Evidence for a dual role of PBP1 in the cell division and cell separation of Staphylococcus aureus. Mol Microbiol 72:895–904. doi: 10.1111/j.1365-2958.2009.06687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pogliano J, Pogliano N, Silverman JA. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol 194:4494–4504. doi: 10.1128/JB.00011-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berti AD, Baines SL, Howden BP, Sakoulas G, Nizet V, Proctor RA, Rose WE. 2015. Heterogeneity of genetic pathways toward daptomycin nonsusceptibility in Staphylococcus aureus determined by adjunctive antibiotics. Antimicrob Agents Chemother 59:2799–2806. doi: 10.1128/AAC.04990-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra NN, McKinnell J, Yeaman MR, Rubio A, Nast CC, Chen L, Kreiswirth BN, Bayer AS. 2011. In vitro cross-resistance to daptomycin and host defense cationic antimicrobial peptides in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother 55:4012–4018. doi: 10.1128/AAC.00223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra NN, Bayer AS, Moise PA, Yeaman MR, Sakoulas G. 2012. Reduced susceptibility to host-defense cationic peptides and daptomycin coemerge in methicillin-resistant Staphylococcus aureus from daptomycin-naive bacteremic patients. J Infect Dis 206:1160–1167. doi: 10.1093/infdis/jis482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howden BP, McEvoy CR, Allen DL, Chua K, Gao W, Harrison PF, Bell J, Coombs G, Bennett-Wood V, Porter JL, Robins-Browne R, Davies JK, Seemann T, Stinear TP. 2011. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator walKR. PLoS Pathog 7:e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, Dhand A, Pogliano J, Yeaman MR, Hensler ME, Bayer AS, Nizet V. 2014. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med (Berl) 92:139–149. doi: 10.1007/s00109-013-1100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SH, Kim KH, Kim HB, Kim NJ, Kim EC, Oh MD, Choe KW. 2008. Outcome of vancomycin treatment in patients with methicillin-susceptible Staphylococcus aureus bacteremia. AAC 52:192–197. doi: 10.1128/AAC.00700-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fowler VG Jr, Boucher HW, Corey GR, Abrutyn E, Karchmer AW, Rupp ME, Levine DP, Chambers HF, Tally FP, Vigliani GA, Cabell CH, Link AS, DeMeyer I, Filler SG, Zervos M, Cook P, Parsonnet J, Bernstein JM, Price CS, Forrest GN, Fatkenheuer G, Gareca M, Rehm SJ, Brodt HR, Tice A, Cosgrove SE, S. aureus Endocarditis and Bacteremia Study Group. 2006. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 355:653–665. doi: 10.1056/NEJMoa053783. [DOI] [PubMed] [Google Scholar]
- 24.Zheng X, Berti AD, McCrone S, Roch M, Rosato AE, Rose WE, Chen B. 2017. Combination antibiotic exposure selectively alters the development of vancomycin intermediate resistance in Staphylococcus aureus. Antimicrob Agents Chemother 62:e02100-17. doi: 10.1128/AAC.02100-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berti AD, Wergin JE, Girdaukas GG, Hetzel SJ, Sakoulas G, Rose WE. 2012. Altering the proclivity towards daptomycin resistance in methicillin-resistant Staphylococcus aureus using combinations with other antibiotics. Antimicrob Agents Chemother 56:5046–5053. doi: 10.1128/AAC.00502-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta S, Singh C, Plata KB, Chanda PK, Paul A, Riosa S, Rosato RR, Rosato AE. 2012. Beta-lactams increase the antibacterial activity of daptomycin against clinical methicillin-resistant Staphylococcus aureus strains and prevent selection of daptomycin-resistant derivatives. Antimicrob Agents Chemother 56:6192–6200. doi: 10.1128/AAC.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kosowska-Shick K, McGhee PL, Appelbaum PC. 2010. Affinity of ceftaroline and other beta-lactams for penicillin-binding proteins from Staphylococcus aureus and Streptococcus pneumoniae. Antimicrob Agents Chemother 54:1670–1677. doi: 10.1128/AAC.00019-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayer AS, Mishra NN, Sakoulas G, Nonejuie P, Nast CC, Pogliano J, Chen KT, Ellison SN, Yeaman MR, Yang SJ. 2014. Heterogeneity of mprf sequences in methicillin-resistant Staphylococcus aureus clinical isolates: role in cross-resistance between daptomycin and host defense antimicrobial peptides. Antimicrob Agents Chemother 58:7462–7467. doi: 10.1128/AAC.03422-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howden BP, Johnson PD, Ward PB, Stinear TP, Davies JK. 2006. Isolates with low-level vancomycin resistance associated with persistent methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob Agents Chemother 50:3039–3047. doi: 10.1128/AAC.00422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui L, Ma X, Sato K, Okuma K, Tenover FC, Mamizuka EM, Gemmell CG, Kim MN, Ploy MC, El-Solh N, Ferraz V, Hiramatsu K. 2003. Cell wall thickening is a common feature of vancomycin resistance in Staphylococcus aureus. J Clin Microbiol 41:5–14. doi: 10.1128/JCM.41.1.5-14.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui L, Neoh HM, Shoji M, Hiramatsu K. 2009. Contribution of vraSR and graSR point mutations to vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother 53:1231–1234. doi: 10.1128/AAC.01173-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui L, Tominaga E, Neoh HM, Hiramatsu K. 2006. Correlation between reduced daptomycin susceptibility and vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother 50:1079–1082. doi: 10.1128/AAC.50.3.1079-1082.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose WE, Schulz LT, Andes D, Striker R, Berti AD, Hutson PR, Shukla SK. 2012. Addition of ceftaroline to daptomycin after emergence of daptomycin-nonsusceptible Staphylococcus aureus during therapy improves antibacterial activity. Antimicrob Agents Chemother 56:5296–5302. doi: 10.1128/AAC.00797-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller A, Wenzel M, Strahl H, Grein F, Saaki TNV, Kohl B, Siersma T, Bandow JE, Sahl HG, Schneider T, Hamoen LW. 2016. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc Natl Acad Sci U S A 113:E7077–E7086. doi: 10.1073/pnas.1611173113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berti AD, Shukla N, Rottier AD, McCrone JS, Turner HM, Monk IR, Baines SL, Howden BP, Proctor RA, Rose WE. 2018. Daptomycin selects for genetic and phenotypic adaptations leading to antibiotic tolerance in MRSA. J Antimicrob Chemother 73:2030–2033. doi: 10.1093/jac/dky148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dey S, Gudipati S, Giuliano C, Zervos MJ, Monk JM, Szubin R, Jorgensen SCJ, Sakoulas G, Berti AD. 2019. Reduced production of bacterial membrane vesicles predicts mortality in ST45/USA600 methicillin-resistant staphylococcus aureus bacteremia. Antibiotics (Basel) 9:2. doi: 10.3390/antibiotics9010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clinical and Laboratory Standards Institute. 2019. Performance standards for antimicrobial susceptibility testing. CLSI M100-ED29. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 38.Cingolani P, Platts A, Wang Le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dvorchik B, Arbeit RD, Chung J, Liu S, Knebel W, Kastrissios H. 2004. Population pharmacokinetics of daptomycin. Antimicrob Agents Chemother 48:2799–2807. doi: 10.1128/AAC.48.8.2799-2807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kahlenberg JM, Kaplan MJ. 2013. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol 191:4895–4901. doi: 10.4049/jimmunol.1302005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishra NN, Liu GY, Yeaman MR, Nast CC, Proctor RA, McKinnell J, Bayer AS. 2011. Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob Agents Chemother 55:526–531. doi: 10.1128/AAC.00680-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang KM, Mishra NN, Park KT, Lee GY, Park YH, Bayer AS, Yang SJ. 2017. Phenotypic and genotypic correlates of daptomycin-resistant methicillin-susceptible Staphylococcus aureus clinical isolates. J Microbiol 55:153–159. doi: 10.1007/s12275-017-6509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.