

Cefepime is commonly used in the intensive care unit (ICU) to treat bacterial infections. The time during which the free cefepime concentration is above the MIC (fT>MIC) should be optimized to increase the efficacy of the regimen. We aim to optimize the exposure of cefepime in ICU patients by using population pharmacokinetic (PK) modeling and simulations. Two data sets were included in this study. The first was a prospective study of pediatric patients who received cefepime at 50 mg/kg of body weight and had extensive PK sampling.
KEYWORDS: Monte Carlo simulation, cefepime, clinical therapeutics, population pharmacokinetics, precision dosing
ABSTRACT
Cefepime is commonly used in the intensive care unit (ICU) to treat bacterial infections. The time during which the free cefepime concentration is above the MIC (fT>MIC) should be optimized to increase the efficacy of the regimen. We aim to optimize the exposure of cefepime in ICU patients by using population pharmacokinetic (PK) modeling and simulations. Two data sets were included in this study. The first was a prospective study of pediatric patients who received cefepime at 50 mg/kg of body weight and had extensive PK sampling. The second study comprised retrospective data for adult ICU patients admitted to UF Health Shands Hospital who received cefepime and had their cefepime concentrations measured. The population PK model was developed, and simulations were performed, using Pmetrics. The target exposures were 100% fT>MIC and 100% fT>4×MIC. The studies included a total of 266 patients, and the mean ages were 3.9 years in the pediatric group and 55 years in adult group. More than half of the patients were males. The mean (standard deviation [SD]) creatinine clearance (CrCl) was 125 (93) ml/min. The mean (SD) daily dose for adults was 4.9 (1.6) g. Cefepime was well described by a two-compartment model with weight as a covariate on the volume of distribution and elimination rate constant (kel), and CrCl and age group as covariates on kel. At a MIC of 8 mg/liter, a cefepime loading dose of 4 g as an extended infusion followed by a 6-g continuous infusion was needed for good target attainment. In conclusion, prolonged or continuous infusions will be needed to achieve optimal cefepime exposure for ICU patients. Given the observed variability, therapeutic drug monitoring can help individualize therapy.
INTRODUCTION
Antimicrobial resistance is a worldwide problem that is putting millions of patients’ lives at risk (1). In the United States, resistant bacteria and fungi cause almost 3 million infections and more than 35,000 deaths annually. The Centers for Disease Control and Prevention have specified certain pathogens as associated with great threat to humans given their resistance patterns and mortality rates, including carbapenem-resistant Acinetobacter spp. and Enterobacteriaceae, extended-spectrum beta-lactamase-producing Enterobacteriaceae, and multidrug-resistant Pseudomonas aeruginosa (2). The existence of these resistant pathogens has created a problem for clinicians and has limited their options in treating such resistant bacterial infections, especially with the slow development of new antibiotics.
In the critical care setting, sepsis is considered a major problem. The number of sepsis cases is increasing, and the associated mortality rate is 25% globally (3–5). Part of the early management of sepsis is adequate antimicrobial therapy (6). Resistance can develop in such populations if there is suboptimal antimicrobial exposure that is insufficient to eradicate the pathogen (7, 8). In addition, there is ample evidence in the literature that intensive care unit (ICU) patients have great variability in antimicrobial exposure, given the dynamic changes happening continuously (9–11).
Beta-lactam antibiotics, such as cefepime, are prescribed frequently in the ICU for various Gram-negative bacterial infections. Since their bacterial kill is time dependent, beta-lactam therapy should be optimized by achieving a high percentage of the dosing interval during which the concentration of the free, unbound fraction of the drug is above the MIC (%fT>MIC). The doses currently suggested in the package insert of cefepime are insufficient to achieve the pharmacokinetic/pharmacodynamic (PK/PD) targets for therapeutic efficacy against common bacteria in the ICU (12–14). In this study, we aim to investigate the cefepime regimens associated with optimal exposure in critically ill patients by using population PK modeling and Monte Carlo simulations (MCS) to provide a guide for the best initial dosing regimens.
RESULTS
Population characteristics.
A total of 266 patients and 813 plasma samples were included in the study. Table 1 shows the baseline characteristics for both data sets. The mean (standard deviation [SD]) ages were 3.9 (4.7) years in the pediatric group and 55 (18.8) years in the adult group. More than half of the patients were males in both groups. In the adult group, the mean (SD) creatinine clearance (CrCl) was 125 (93) ml/min, and the mean (SD) daily dose was 4.9 (1.6) g. Cefepime was administered as an intermittent infusion (II) to 174 adult patients (and to all 36 pediatric patients), as an extended infusion (EI) to 47 patients, and as a continuous infusion (CI) to 9 patients. Figure 1 shows the local MIC data for P. aeruginosa.
TABLE 1.
Baseline characteristicsa
| Characteristic | Value for group |
|
|---|---|---|
| Adult (n = 230) | Pediatric (n = 36) | |
| Age (yr) | 55.0 (18.8) | 3.9 (4.7) |
| Male sex (no. [%]) | 131 (57.0) | 21 (58.3) |
| Wt (kg) | 86.6 (33.3) | 16.0 (16.1) |
| Serum creatinine concn (mg/dl) | 1.1 (1.0) | 0.4 (0.2) |
| Creatinine clearance (ml/min) | 124.9 (93.1) | 67.6 (38.9) |
| No. (%) with the following creatinine clearance (ml/min): | ||
| ≤30 | 13 (5) | 2 (6) |
| >30–60 | 41 (18) | 19 (53) |
| >60–90 | 43 (19) | 5 (14) |
| >90–120 | 41 (18) | 7 (19) |
| >120 | 92 (40) | 3 (8) |
| Plasma samples (no.) | 302 | 511 |
Data are presented as means (SD) unless otherwise specified.
FIG 1.
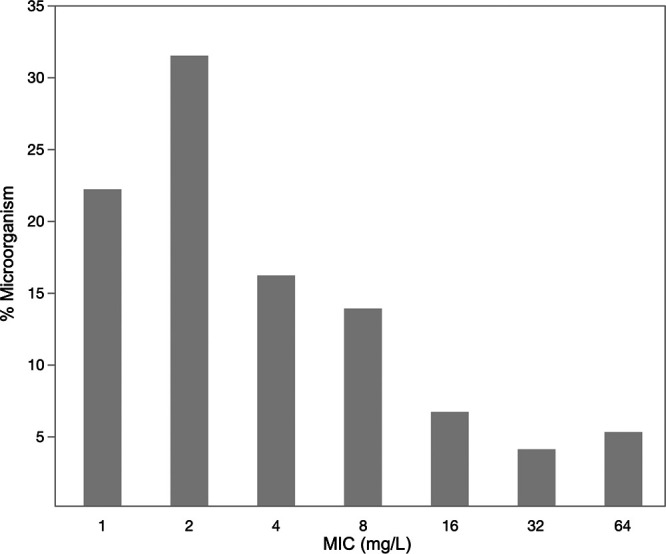
Local cefepime MIC distribution for Pseudomonas aeruginosa.
Population pharmacokinetic model.
The best population PK model describing the cefepime data was a two-compartment model with weight normalized to the mean (37 kg) on the volume of distribution (V) and the total elimination rate constant (kel), and CrCl normalized to the mean (80 ml/min) and age group (pediatric versus adult) as covariates on kel:
where Peds stands for pediatric group, with a value of 1 for the pediatric study and a value of 0 for the adult ICU group.
The results for pediatric simulations and the probability of target attainment (PTA) have been published previously (15–18). Table 2 shows the parameter estimates for the final model. The means (SDs) for the estimated cefepime kel were 0.28 (0.21) h−1 for adults and 1.10 (0.74) h−1 for pediatric patients; the mean (SD) estimated V was 7.75 (3.69) liters; and the mean (SD) estimated intercompartmental transfer rates (the rate constants for transfer from the central to the peripheral compartment [Kcp] and from the peripheral to the central compartment [Kpc]) were 1.53 (1.39) h−1 and 1.26 (1.23) h−1, respectively. Figure 2 shows the observed versus predicted population and individual concentrations of cefepime.
TABLE 2.
Population parameter estimates for final cefepime model
| Parameter | Median | 95% confidence interval | Shrinkage (%) |
|---|---|---|---|
| kel (h–1) | |||
| Adult | 0.32 | 0.14–0.36 | 44 |
| Pediatric | 1.03 | 0.79–1.18 | 69 |
| V (liters) | 6.71 | 6.04–7.22 | 43 |
| Kcp (h–1) | 1.16 | 0.62–1.74 | 65 |
| Kpc (h–1) | 1.33 | 0.04–1.65 | 63 |
FIG 2.

Observed versus predicted population (A) and individual (B) plots. (A) Observed versus predicted population cefepime concentrations. R2, 0.71; intercept, 9.31 mg/liter (95% confidence interval, 6.37 to 11.90 mg/liter); slope, 0.85 (95% confidence interval, 0.81 to 0.89); bias, 0.83; imprecision, 25.50. (B) Observed versus predicted individual cefepime concentrations. R2, 0.92; intercept, 0.16 mg/liter (95% confidence interval, 1.29 to 1.61 mg/liter); slope, 0.97 (95% confidence interval, 0.95 to 0.99); bias, 0.41; imprecision, 4.51.
Monte Carlo simulations.
Figures 3 and 4 show the PTA at targets of 100% fT>MIC and 100% fT>4×MIC, respectively, at different time points after simulations of different adult dosing regimens. For the target 100% fT>MIC, at a MIC of 1 mg/liter, all regimens had ≥90% target attainment after the first dose, 24 h, and 72 h of therapy, except for 2 g every 12 h (q12h). At a MIC of 2 mg/liter, 2 g every 12 h and an II every 8 h fell short of 90% target attainment at all times. An EI of cefepime at 2 g every 8 h gave an 89% PTA after the first dose, but the PTA exceeded 90% after 24 and 72 h of therapy. All other regimens had ≥90% target attainment at all times at a MIC of 2 mg/liter. After the first dose, with a MIC of 4 mg/liter, only an 8-g CI had a >90% PTA, while a 6-g CI, a 7-g CI, and an EI of 2 g every 6 h had 85%, 89%, and 89% PTA, respectively. Similar results were observed after 24 h at the same MIC. All these regimens had a >90% PTA after 72 h. With a MIC of 8 mg/liter, all the regimens had low target attainment percentages after the first dose and after 24 h. After 72 h, only CI regimens were successful in attaining ≥90% of the target at a MIC of 8 mg/liter. At a MIC of 16 mg/liter, the highest PTA reached was 81% after 72 h with an 8-g CI. A loading dose (LD) of 4 g infused over 4 h, followed by continuous infusion, improved the PTA at a MIC of 8 mg/liter in the first 24 h (Fig. 5A). At a MIC of 16 mg/liter, only an 8-g CI led to >80% target attainment after 72 h. After the simulation of a 4-g loading dose before the CI, 7-g and 8-g CIs led to >80% target attainment in the first 24 h of therapy at that MIC.
FIG 3.

Probability of target attainment (100% fT>MIC) for cefepime regimens in critically ill adult patients after the first dose (A), after the first 24 h (B), and after 72 h of regular dosing (C). Horizontal dashed lines indicate a 90% probability of target attainment. The mean (SD) creatinine clearance and weight used for the simulations were 116 (92) ml/min and 86 (34) kg, respectively. CI, continuous infusion; EI, extended infusion; II, intermittent infusion.
FIG 4.

Probability of target attainment (100% fT>4×MIC) for cefepime regimens in critically ill adult patients after the first dose (A), after the first 24 h (B), and after 72 h of regular dosing (C). Horizontal dashed lines indicate a 90% probability of target attainment. The mean (SD) creatinine clearance and weight used for the simulations were 116 (92) ml/min and 86 (34) kg, respectively. CI, continuous infusion; EI, extended infusion; II, intermittent infusion.
FIG 5.

Probability of target attainment for the first 24 h for 4 g cefepime infused over 4 h, followed by continuous infusion. The target was 100% fT>MIC (A) or 100% fT>4×MIC (B). Horizontal dashed lines indicate a 90% probability of target attainment. The mean (SD) creatinine clearance and weight used for the simulations were 116 (92) ml/min and 86 (34) kg, respectively. CI, continuous infusion.
For the target 100% fT>4×MIC (Fig. 4), only an 8-g CI led to >90% target attainment at a MIC of 1 mg/liter in the first 24 h, followed by the PTA for 6-g and 7-g CIs and for 2-g EIs every 6 h, which were between 80% and 90%. After 72 h, only those regimens led to >90% PTA at that MIC. At a MIC of ≥2 mg/liter, all regimens failed to lead to 90% target attainment in the first 24 h. After 72 h, only CI regimens led to a ≥90% PTA at a MIC of 2 mg/liter. The highest PTA reached at a MIC of 4 mg/liter after 72 h was 82% with an 8-g CI. Adding an LD before the CI improved target attainment at a MIC of 1 to 2 mg/liter, and the PTA exceeded 90% in the first 24 h but was around 80% at a MIC of 4 mg/liter (Fig. 5B).
Table 3 shows the initial dosing guidance for cefepime based on the simulations generated by our model using the target 100% fT>MIC. This table also reflects the breakpoints for each regimen and the CrCl. As the MIC and/or the CrCl increased, there was a need to introduce EI or CI regimens to achieve higher targets. Cefepime should be avoided, and another agent used, for patients with bacterial MICs of 16 mg/liter and a CrCl of >60 ml/min.
TABLE 3.
Guidance on initial cefepime dosing regimens to achieve 100% fT>MIC in critically ill adult patients based on different renal functions and MICs
| MIC (mg/liter) | Cefepime dosing regimena at the following creatinine clearance (ml/min): |
||||
|---|---|---|---|---|---|
| 10–30 | >30–60 | >60–90 | >90–120 | >120–150 | |
| 1 | 2 g q12h (II) | 2 g q12h (II) | 2 g q8h (II) | 2 g q8h (II) | 2 g q8h (EI) |
| 2 | 2 g q12h (II) | 2 g q8h (II) | 2 g q8h (II) | 2 g q8h (EI) | 6-g CI |
| 4 | 2 g q12h (II) | 2 g q8h (II) | 2 g q8h (EI) | 7- to 8-g CI | 8-g CI |
| 8 | 2 g q8h (II) | 2 g q8h (EI) | 2 g q6h (EI) | 4-g LD, 6-g CI | 4-g LD, 7-g CI |
| 16 | 2 g q6h (II) | 4-g LD, 6-g CI | 4-g LD, 6-g CIb | 4-g LD, 8-g CIb | 4-g LD, 8-g CIb |
CI, continuous infusion; EI, extended infusion over half the dosing interval; II, intermittent infusion over 30 min; LD, loading dose over 4 h.
Target attainment between 80% and 90%.
DISCUSSION
We described a cefepime population PK model in critically ill patients using a nonparametric approach, followed by simulations of different dosing regimens and effects at different time points during therapy. Although some regimens led to acceptable target attainment after 72 h, a high PTA (>90%) for 100% fT>MIC was not possible with most of the regimens at MICs of 4 to 8 mg/liter early in therapy, which is important for critically ill patients. Cefepime at 2 g every 8 h as an II led to <80% target attainment at MICs of ≥2 mg/liter. At the higher MIC, i.e., 4 mg/liter, at least 2 g every 6 h as an EI or one of the CI regimens was needed to achieve >80% of the target at any time during therapy. For an 8-mg/liter MIC, which is the current breakpoint for P. aeruginosa, a loading dose administered as an EI, followed by a CI regimen, is needed for >90% PTA. The PTA dropped to 80% with a 4-g loading dose followed by an 8-g CI at a MIC of 16 mg/liter, indicating that this is not a valid MIC target. and alternative agents should be chosen when this level of resistance is observed clinically. Also, this finding may support a lowering of the breakpoint for P. aeruginosa. The target of 100% fT>4×MIC was more difficult to achieve, and an LD followed by a CI was needed at MICs of 1 and 2 mg/liter.
Optimizing antimicrobial therapy early during sepsis in critically ill patients is important (6). This includes optimal target attainment with the first dose administered to the patients. Previous work on beta-lactams (in general) and cefepime (in particular) showed poor early target attainment with contemporary dosing regimens in ICUs. Huttner et al. reported a prospective beta-lactam PK study of 100 ICU patients with CrCl of >60 ml/min where drug concentrations were measured on days 1 to 3 and 5 (19). Cefepime, piperacillin, meropenem, and imipenem concentrations below or equal to 4, 4, 2, and 2 mg/liter, respectively, were considered subtherapeutic. The usual regimens were 2 g cefepime twice daily (2%), 4.5 g piperacillin-tazobactam three times daily (33%), 2 g meropenem three times daily (11%), and 500 mg imipenem four times daily (54%). Eighty-six percent of patients who received any beta-lactam had subtherapeutic trough values, and 40% of cefepime trough samples were subtherapeutic. Undetectable beta-lactam trough concentrations were reported in 27% of patients (19). In another prospective study of ICU patients, the authors evaluated the achievement of fT>4×MIC after the first dose of beta-lactams in patients with severe sepsis and septic shock, using the European Committee on Antimicrobial Susceptibility Testing breakpoint for P. aeruginosa. Eighty patients were included; they received 2 g cefepime (n = 19), 4.5 g piperacillin-tazobactam (n = 27), 2 g ceftazidime (n = 18), or 1 g meropenem (n = 16). The median %T>4×MIC for cefepime was 34%, and only 16% (n = 3) of patients who received cefepime achieved 70% T>4×MIC (20). Similarly, Lipman and colleagues found that 2 g cefepime achieved low trough values in most of the patients with normal kidney function after the first dose (13). In our study, we investigated cefepime regimens associated with optimal exposure from the first dose, but the clinical outcomes associated with early target attainment compared to steady state still need to be investigated in larger studies.
Different cefepime population PK models for ICU patients have been published, using different targets for simulations. Nicasio et al. published a cefepime PK model for 32 adult ICU patients who had ventilator-associated pneumonia (21). Theirs was a two-compartment model with CrCl on kel and weight on V as covariates. Using 50% fT>MIC as the target at steady state, the simulated regimen of 2 g every 8 h (3-h infusion) had PTA of 91.8%, 78.1%, and 50.3% at MICs of 8, 16, and 32 mg/liter, respectively, for patients with a CrCl of 50 to 120 ml/min. At a CrCl of 30 to 49 ml/min, 1 g every 8 h and 2 g every 12 h (II and EI) had around 90% PTA at a MIC of 8 mg/liter. For a CrCl of <30 ml/min, 1 g every 12 h and 2 g every 24 h (II and EI) had ≤90% PTA at a MIC of 8 mg/liter (21). Another cefepime model, published by Liu and colleagues, included pediatric patients, adult ICU patients, and febrile neutropenic adult cancer patients (18). Both weight and CrCl were included as covariates on V and kel, respectively. The final model was used to simulate 2 g cefepime every 8 h infused over 30 min, and a target of 68% fT>MIC was evaluated for the first 24 h of therapy. From a MIC of 2 mg/liter to a MIC of 4 mg/liter, the PTA dropped from >90% to <80% (18). A third model, developed by Roos et al., included 13 adult ICU patients and used 65% fT>MIC as a target (22). The final model was a three-compartment model, and creatinine clearance described cefepime clearance. The authors concluded that at least a 4-g CI was needed for >90% target attainment with P. aeruginosa (MIC, 8 mg/liter) (22). Although our model had shrinkage values of >30%, which might be due to sparse sampling as part of a clinical therapeutic drug monitoring (TDM) service, it combined pediatric and adult ICU patients, and, as in the previously published rich models, CrCl and weight were significant covariates on kel and V, respectively. We simulated different cefepime dosing regimens, including II, EI, and CI, and used 100% fT>MIC and 100% fT>4×MIC as targets. Only CI regimens and an EI of 2 g every 6 h reached around 90% PTA at a MIC of 4 mg/liter using the target 100% fT>MIC. In contrast to the previous studies, 2 g cefepime every 8 h (II and EI) did not lead to high target attainment at a MIC of ≥4 mg/liter. This is most likely due to the use of a 100% fT>MIC target, in addition to the variability in population between our study and others. All the previous studies, as well as ours, highlight the variability in cefepime PK parameters for ICU patients, which will affect target attainment for each patient (9, 10). The results generated and the dosing regimens suggested by the simulations can serve only for initial dosing in critically ill patients. Dose individualization via TDM should follow so as to optimize therapy for each patient.
In addition to the PK variability, there was also variability in the target specified in previous clinical work on cefepime, with a common range of 50% to 74% fT>MIC; very few studies evaluated 100% fT>MIC. Preclinical studies on cephalosporins suggested that these antimicrobials have a static effect at >30% to 40% fT>MIC and maximal killing effect at 60% to 70% (23, 24), while a trough-to-MIC ratio of >3.8 was needed for resistance suppression (25). On the other hand, Rhodes et al. used population PK models to generate cefepime exposures for 180 patients and identified >68% and >74% fT>MIC as breakpoints associated with improved survival in patients with Gram-negative bacterial bloodstream infections (26). Abdul-Aziz et al. evaluated the achievement of 100% fT>MIC and clinical outcomes for 140 ICU patients randomized to receive piperacillin, meropenem, or cefepime as a CI or II. In the CI arm, more patients achieved the PK/PD target, the clinical cure rate was higher, and the median number of ventilator-free days was higher than in the II arm (27). McKinnon and colleagues evaluated PK/PD data for 76 patients who received cefepime or ceftazidime in two clinical trials (28). Patients who achieved 100% T>MIC had a significantly higher clinical cure rate and more microbiological eradication than those who did not (28). A higher target, fT>4×MIC, was assessed in a smaller study for 20 patients with Gram-negative bacterial infections and was found to be associated with higher microbiological success (29). Even if targets are achieved at the plasma level, clinicians should be aware whether the suggested targets, especially in vivo and in vitro, are achievable at the site of infection. As an example, pneumonia is one of the most common infections in the ICU (30), and beta-lactams and cefepime have been found to have variable and poor penetration to the epithelial lining fluid (ELF) and bronchial secretions in critically ill patients (10, 31). This indicates that higher plasma drug concentrations might be needed in order to achieve the desired exposure at the site of infection. Benítez-Cano et al. conducted a randomized PK trial on 31 patients with nosocomial pneumonia receiving 3 g or 6 g meropenem as a CI and modeled both measured plasma and intrapulmonary meropenem concentrations. Their simulations showed that a 6-g CI is required to achieve the desired targets at the ELF (32). In addition to a higher efficacy profile, the achievement of higher plasma drug concentrations can be associated with toxicity. Neurotoxicity is one of the adverse events that researchers have been trying to correlate with plasma cefepime exposure; however, current data are limited due to retrospective study design, trough-only concentrations, total or calculated unbound concentrations, difficulty in defining the event, and lack of control for other covariates affecting this event, which is common in ICUs (33–36). In our simulations, the probability of achieving a total trough concentration in plasma of ≥20 mg/liter, which has been suggested as the threshold for cefepime-induced neurotoxicity in previous literature, was highest with CI regimens (70 to 80%) compared with EI (45 to 50%) and II (20 to 40%). More work is needed in this area to weigh the risks and benefits and to optimize cefepime exposure, and TDM can play an important role in maximizing efficacy while limiting toxicity.
Only the unbound drug molecule can pass through the blood capillary wall and reach the interstitial fluid and then the site of infection (37). Studies reporting the degree of cefepime protein binding are limited. One study reported the large variability in the percentage of unbound cefepime compared to the total concentration in patients’ samples, with a median of 61% (range, 52% to 99%) (38). Consequently, assuming the unbound fraction to be a fixed value for all patients might be misleading, and measuring unbound concentrations clinically will be important for optimizing therapy and assessing target attainment.
Our study has a number of limitations. It was a retrospective study with variable cefepime sampling times and numbers of samples as part of the TDM service, which sometimes included measuring only one sample per patient. We measured the total cefepime concentration and assumed the unbound fraction, which does not consider the variability in protein binding among patients, especially in the ICU setting. Finally, we quantified the cefepime concentration in the plasma, which might differ from the concentrations at different sites of infection. Prospective PK studies evaluating the achievement of higher targets in plasma and quantifying the free cefepime concentration at the site of infection may address these problems.
Conclusions.
Cefepime was well described by a two-compartment model with CrCl and weight as covariates on kel and V, respectively. Using 100% fT>MIC as the target, simulations showed that 2 g cefepime given as an II q8h, as an EI q8h, or as an EI q6h and an LD followed by CI had >90% PTA at MICs of 1, 2, 4, and 8 mg/liter, respectively. At a MIC of 16 mg/liter, we recommend an alternative antibiotic. At the target of 100% fT>4×MIC, CI regimens were needed to achieve >90% PTA at MICs of 1 to 2 mg/liter. Initial dosing guidance was suggested, and TDM should follow to further optimize and individualize therapy.
MATERIALS AND METHODS
This was a PK study combining two different existing ICU data sets. The first included pediatric patients admitted to Rainbow Babies and Children’s Hospital. Patients between the ages of 2 months and 18 years were enrolled prospectively and received 50 mg of cefepime/kg of body weight intravenously (i.v.) over 30 min every 8 h. Patients were excluded if they were allergic to beta-lactam antibiotics or if they had central nervous system involvement, human immunodeficiency virus infection, cystic fibrosis, endocarditis, lung abscess, osteomyelitis, severe burns (≥20% full thickness), an infected prosthesis, an absolute granulocyte count of <500/mm3, or a serum creatinine level of >2 mg/dl. Blood was sampled at 0, 0.5, 0.75, 1, 2, 4, 6, and 8 h after the beginning of the i.v. infusion. High-performance liquid chromatography was used for plasma cefepime quantification, with a lower limit of quantification of 0.1 mg/liter. The coefficient of variation (CV) for within-day precision and accuracy was <5%, and the between-day CV was <10% (39, 40).
The second data set was from a retrospective study of adult ICU patients at UF Health Shands Hospital. Patients who were 18 years of age or older, received cefepime, had cefepime concentrations reported as part of routine clinical care, and were admitted to the medical, surgical, cardiac, or neurological ICU between 2016 and 2018 were included. Patients receiving renal replacement therapy were excluded. Cefepime at 2 g q8h was administered as an II (over 30 min) or EI (over 3 to 4 h), or 6 g was given as a CI. The dose was adjusted for patients with renal impairment according to their CrCl. Typically, blood sampling for TDM was requested for peak and trough concentrations. The cefepime concentration was quantified at the Infectious Disease Pharmacokinetics Laboratory using validated liquid chromatography with tandem mass spectrometry assays. The range of quantification was 2 to 100 mg/liter. Intra- and interbatch precision were 3.72 to 10.98% and 3.30 to 9.79%, respectively. Intra- and interbatch accuracy were 94.10 to 109.5% and 90.84 to 106.09%, respectively.
Both data sets included age, sex, weight, serum creatinine levels, cefepime regimens, and serum cefepime concentrations. CrCl was calculated using the Cockcroft-Gault equation (41) to find a reasonable renally based descriptor of cefepime PK.
The studies included were approved by the Institutional Review Boards at the participating sites, and written consent for study participation was obtained from a parent or legal guardian for each patient in the prospective pediatric study.
PK analysis and MCS.
The nonparametric adaptive grid (NPAG) platform in Pmetrics, v1.5.2, was used to build the cefepime population PK model and perform the MCS (42). One- and two-compartment models were tested, and the best-fit model was chosen. The covariates tested were weight, sex, age, and CrCl; they were added on PK parameters in a forward stepwise fashion, starting with the lowest P value. Models were examined on every step, and the final models were compared based on the Akaike information criterion (AIC), the coefficient of determination (R2) of observed versus predicted plots for both population and Bayesian models, imprecision, and bias. We accounted for assay error (standard deviation) and environmental noise using error polynomials as function of observed concentration [standard deviation = C0 + (C1 × observed concentration)] using C0 (intercept) and C1 (slope) values of 1 and 0.1, respectively. A gamma multiplicative error model was used to estimate residual error, and a value of 5 was entered initially; this was dropped to 2 later on.
For adults, 1,000 subjects were simulated for each of the following regimens: 2 g every 6 or 8 h, either as an II or as an EI; 2 g every 12 h as an II; and 6 g, 7 g, and 8 g as a CI, with or without a 4-g loading dose over 4 h. For simulation purposes, the EI time was defined as half of the dosing interval. The MICs chosen for simulation were 1, 2, 4, 8, and 16 mg/liter. The PK/PD targets specified were 100% fT>MIC and 100% fT>4×MIC, and the unbound fraction of cefepime was assumed to be 80% (43). The PTA was calculated for the first dose (from time zero to the end of the dosing interval), the first 24 h of therapy, and after 72 h of therapy (from 72 h to the end of the dosing interval). The breakpoint was considered the highest MIC at which the simulated regimen was able to attain at least 90% of the target (44). In order to suggest an initial cefepime regimen, different regimens were simulated for CrCl values of 10 to 30, >30 to 60, >60 to 90, >90 to 120, and >120 to 150 ml/min at the MICs specified above.
ACKNOWLEDGMENTS
We thank Eric Rubido, Yang Zhao, Jack Guerci, Lauren Wong, Daniel Lee, and Rachel Shaddock for helping with data collection. We also acknowledge all the clinical pharmacists, nurses, and physicians who assisted in obtaining and assessing beta-lactam concentrations.
Marc Scheetz received a research grant from Allecra. No funding was provided from Allecra for this study. Kenneth P. Klinker is a current employee of Merck & Co., Inc., Kenilworth, NJ. At the time of this research, he was employed by the University of Florida. No funding or support was provided from Merck for this study.
REFERENCES
- 1.World Health Organization. 2015. Global action plan on antimicrobial resistance. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2019. Antibiotic resistance threats in the United States. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 3.Fleischmann C, Scherag A, Adhikari NKJ, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K, International Forum of Acute Care Trialists. 2016. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 193:259–272. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 4.Kaukonen K-M, Bailey M, Suzuki S, Pilcher D, Bellomo R. 2014. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA 311:1308–1316. doi: 10.1001/jama.2014.2637. [DOI] [PubMed] [Google Scholar]
- 5.McPherson D, Griffiths C, Williams M, Baker A, Klodawski E, Jacobson B, Donaldson L. 2013. Sepsis-associated mortality in England: an analysis of multiple cause of death data from 2001 to 2010. BMJ Open 3:e002586. doi: 10.1136/bmjopen-2013-002586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche JD, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, MacHado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, Simpson SQ, Singer M, Thompson BT, Townsend SR, Van Der Poll T, Vincent JL, Wiersinga WJ, Zimmerman JL, Dellinger RP. 2017. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med 43:304–377. doi: 10.1007/s00134-017-4683-6. [DOI] [PubMed] [Google Scholar]
- 7.Tam VH, Schilling AN, Neshat S, Poole K, Melnick DA, Coyle EA. 2005. Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa. Antimicrob Agents Chemother 49:4920–4927. doi: 10.1128/AAC.49.12.4920-4927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts JA, Kruger P, Paterson DL, Lipman J. 2008. Antibiotic resistance—what’s dosing got to do with it? Crit Care Med 36:2433–2440. doi: 10.1097/CCM.0b013e318180fe62. [DOI] [PubMed] [Google Scholar]
- 9.Chapuis TM, Giannoni E, Majcherczyk PA, Chioléro R, Schaller MD, Berger MM, Bolay S, Décosterd LA, Bugnon D, Moreillon P. 2010. Prospective monitoring of cefepime in intensive care unit adult patients. Crit Care 14:R51. doi: 10.1186/cc8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonçalves-Pereira J, Póvoa P. 2011. Antibiotics in critically ill patients: a systematic review of the pharmacokinetics of β-lactams. Crit Care 15:R206. doi: 10.1186/cc10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts JA, Abdul-Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, Hope WW, Farkas A, Neely MN, Schentag JJ, Drusano G, Frey OR, Theuretzbacher U, Kuti JL, International Society of Anti-Infective Pharmacology and the Pharmacokinetics and Pharmacodynamics Study Group of the European Society of Clinical Microbiology and Infectious Diseases. 2014. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis 14:498–509. doi: 10.1016/S1473-3099(14)70036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koomanachai P, Bulik CC, Kuti JL, Nicolau DP. 2010. Pharmacodynamic modeling of intravenous antibiotics against gram-negative bacteria collected in the United States. Clin Ther 32:766–779. doi: 10.1016/j.clinthera.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Lipman J, Wallis SC, Rickard C. 1999. Low plasma cefepime levels in critically ill septic patients: pharmacokinetic modeling indicates improved troughs with revised dosing. Antimicrob Agents Chemother 43:2559–2561. doi: 10.1128/AAC.43.10.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crandon JL, Bulik CC, Kuti JL, Nicolau DP. 2010. Clinical pharmacodynamics of cefepime in patients infected with Pseudomonas aeruginosa. Antimicrob Agents Chemother 54:1111–1116. doi: 10.1128/AAC.01183-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Courter JD, Kuti JL, Girotto JE, Nicolau DP. 2009. Optimizing bactericidal exposure for β-lactams using prolonged and continuous infusions in the pediatric population. Pediatr Blood Cancer 53:379–385. doi: 10.1002/pbc.22051. [DOI] [PubMed] [Google Scholar]
- 16.Ellis JM, Kuti JL, Nicolau DP. 2005. Use of Monte Carlo simulation to assess the pharmacodynamics of β-lactams against Pseudomonas aeruginosa infections in children: a report from the OPTAMA program. Clin Ther 27:1820–1830. doi: 10.1016/j.clinthera.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 17.Shoji K, Bradley JS, Reed MD, Van Den Anker JN, Domonoske C, Capparelli EV. 2016. Population pharmacokinetic assessment and pharmacodynamic implications of pediatric cefepime dosing for susceptible-dose-dependent organisms. Antimicrob Agents Chemother 60:2150–2156. doi: 10.1128/AAC.02592-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J, Neely M, Lipman J, Sime F, Roberts JA, Kiel PJ, Avedissian SN, Rhodes NJ, Scheetz MH. 5 March 2020. Development of population and Bayesian models for applied use in patients receiving cefepime. Clin Pharmacokinet doi: 10.1007/s40262-020-00873-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huttner A, Von Dach E, Renzoni A, Huttner BD, Affaticati M, Pagani L, Daali Y, Pugin J, Karmime A, Fathi M, Lew D, Harbarth S. 2015. Augmented renal clearance, low β-lactam concentrations and clinical outcomes in the critically ill: an observational prospective cohort study. Int J Antimicrob Agents 45:385–392. doi: 10.1016/j.ijantimicag.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 20.Taccone FS, Laterre P-F, Dugernier T, Spapen H, Delattre I, Witebolle X, De Backer D, Layeux B, Wallemacq P, Vincent J-L, Jacobs F. 2010. Insufficient β-lactam concentrations in the early phase of severe sepsis and septic shock. Crit Care 14:R126. doi: 10.1186/cc9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicasio AM, Ariano RE, Zelenitsky SA, Kim A, Crandon JL, Kuti JL, Nicolau DP. 2009. Population pharmacokinetics of high-dose, prolonged-infusion cefepime in adult critically ill patients with ventilator-associated pneumonia. Antimicrob Agents Chemother 53:1476–1481. doi: 10.1128/AAC.01141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roos JF, Bulitta J, Lipman J, Kirkpatrick CMJ. 2006. Pharmacokinetic-pharmacodynamic rationale for cefepime dosing regimens in intensive care units. J Antimicrob Chemother 58:987–993. doi: 10.1093/jac/dkl349. [DOI] [PubMed] [Google Scholar]
- 23.Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–12. doi: 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- 24.Craig WA. 2003. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect Dis Clin North Am 17:479–501. doi: 10.1016/s0891-5520(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 25.Tam VH, Chang K-T, Zhou J, Ledesma KR, Phe K, Gao S, Van Bambeke F, Sánchez-Díaz AM, Zamorano L, Oliver A, Cantón R. 2017. Determining β-lactam exposure threshold to suppress resistance development in Gram-negative bacteria. J Antimicrob Chemother 72:1421–1428. doi: 10.1093/jac/dkx001. [DOI] [PubMed] [Google Scholar]
- 26.Rhodes NJ, Kuti JL, Nicolau DP, Van Wart S, Nicasio AM, Liu J, Lee BJ, Neely MN, Scheetz MH. 2016. Defining clinical exposures of cefepime for Gram-negative bloodstream infections that are associated with improved survival. Antimicrob Agents Chemother 60:1401–1410. doi: 10.1128/AAC.01956-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdul-Aziz MH, Sulaiman H, Mat-Nor MB, Rai V, Wong KK, Hasan MS, Abd Rahman AN, Jamal JA, Wallis SC, Lipman J, Staatz CE, Roberts JA. 2016. Beta-lactam infusion in severe sepsis (BLISS): a prospective, two-centre, open-labelled randomised controlled trial of continuous versus intermittent beta-lactam infusion in critically ill patients with severe sepsis. Intensive Care Med 42:1535–1545. doi: 10.1007/s00134-015-4188-0. [DOI] [PubMed] [Google Scholar]
- 28.McKinnon PS, Paladino JA, Schentag JJ. 2008. Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. Int J Antimicrob Agents 31:345–351. doi: 10.1016/j.ijantimicag.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Tam VH, McKinnon PS, Akins RL, Rybak MJ, Drusano GL. 2002. Pharmacodynamics of cefepime in patients with Gram-negative infections. J Antimicrob Chemother 50:425–428. doi: 10.1093/jac/dkf130. [DOI] [PubMed] [Google Scholar]
- 30.Papazian L, Klompas M, Luyt CE. 2020. Ventilator-associated pneumonia in adults: a narrative review. Intensive Care Med 46:888–906. doi: 10.1007/s00134-020-05980-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heffernan AJ, Sime FB, Lipman J, Dhanani J, Andrews K, Ellwood D, Grimwood K, Roberts JA. 2019. Intrapulmonary pharmacokinetics of antibiotics used to treat nosocomial pneumonia caused by Gram-negative bacilli: a systematic review. Int J Antimicrob Agents 53:234–245. doi: 10.1016/j.ijantimicag.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 32.Benítez-Cano A, Luque S, Sorlí L, Carazo J, Ramos I, Campillo N, Curull V, Sánchez-Font A, Vilaplana C, Horcajada JP, Adalia R, Bermejo S, Samsó E, Hope W, Grau S. 2020. Intrapulmonary concentrations of meropenem administered by continuous infusion in critically ill patients with nosocomial pneumonia: a randomized pharmacokinetic trial. Crit Care 24:55. doi: 10.1186/s13054-020-2763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boschung-Pasquier L, Atkinson A, Kastner LK, Banholzer S, Haschke M, Buetti N, Furrer DI, Hauser C, Jent P, Que YA, Furrer H, Babouee Flury B. 2020. Cefepime neurotoxicity: thresholds and risk factors. A retrospective cohort study. Clin Microbiol Infect 26:333–339. doi: 10.1016/j.cmi.2019.06.028. [DOI] [PubMed] [Google Scholar]
- 34.Beumier M, Casu GS, Hites M, Wolff F, Cotton F, Vincent JL, Jacobs F, Taccone FS. 2015. Elevated β-lactam concentrations associated with neurological deterioration in ICU septic patients. Minerva Anestesiol 81:497–506. [PubMed] [Google Scholar]
- 35.Rhodes NJ, Kuti JL, Nicolau DP, Neely MN, Nicasio AM, Scheetz MH. 2016. An exploratory analysis of the ability of a cefepime trough concentration greater than 22 mg/L to predict neurotoxicity. J Infect Chemother 22:78–83. doi: 10.1016/j.jiac.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Lamoth F, Buclin T, Pascual A, Vora S, Bolay S, Decosterd LA, Calandra T, Marchetti O. 2010. High cefepime plasma concentrations and neurological toxicity in febrile neutropenic patients with mild impairment of renal function. Antimicrob Agents Chemother 54:4360–4367. doi: 10.1128/AAC.01595-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jager NGL, van Hest RM, Lipman J, Roberts JA, Cotta MO. 2019. Antibiotic exposure at the site of infection: principles and assessment of tissue penetration. Expert Rev Clin Pharmacol 12:623–634. doi: 10.1080/17512433.2019.1621161. [DOI] [PubMed] [Google Scholar]
- 38.Al-Shaer MH, Alghamdi WA, Graham E, Peloquin CA. 2019. Meropenem, cefepime, and piperacillin protein binding in patient samples. Ther Drug Monit 42:129–132. doi: 10.1097/FTD.0000000000000675. [DOI] [PubMed] [Google Scholar]
- 39.Reed MD, Yamashita TS, Knupp CK, Veazey JM, Blumer JL. 1997. Pharmacokinetics of intravenously and intramuscularly administered cefepime in infants and children. Antimicrob Agents Chemother 41:1783–1787. doi: 10.1128/AAC.41.8.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbhaiya RH, Forgue ST, Shyu WC, Papp EA, Pittman KA. 1987. High-pressure liquid chromatographic analysis of BMY-28142 in plasma and urine. Antimicrob Agents Chemother 31:55–59. doi: 10.1128/aac.31.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cockcroft DW, Gault MH. 1976. Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 42.Neely MN, Van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. doi: 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bristol-Myers Squibb. 2007. Maxipime (cefepime) package insert. Bristol-Myers Squibb, New York, NY.
- 44.Mouton JW, Brown DFJ, Apfalter P, Cantón R, Giske CG, Ivanova M, MacGowan AP, Rodloff A, Soussy CJ, Steinbakk M, Kahlmeter G. 2012. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: the EUCAST approach. Clin Microbiol Infect 18:E37–E45. doi: 10.1111/j.1469-0691.2011.03752.x. [DOI] [PubMed] [Google Scholar]