

Abstract
Minimal residual disease (MRD) tracking by next generation sequencing of immunoglobulin sequences is moving towards clinical implementation in multiple myeloma. However, there is only sparse information available to address whether clonal sequences remain stable for tracking over time, and to what extent light chain sequences are sufficiently unique for tracking. Here, we analyzed immunoglobulin repertoires from 905 plasma cell myeloma and healthy control samples, focusing on the third complementarity determining region (CDR3). Clonal heavy and/or light chain expression was identified in all patients at baseline, with one or more subclones related to the main clone in 3.2 %. In 45 patients with 101 sequential samples, the dominant clonal CDR3 sequences remained identical over time despite differential clonal evolution by whole exome sequencing in 49 % of patients. The low frequency of subclonal CDR3 variants and absence of evolution over time in active multiple myeloma indicates that tumor cells at this stage are not under selective pressure to undergo antibody affinity maturation. Next, we establish somatic hypermutation and non-templated insertions as the most important determinants of light chain clonal uniqueness, identifying a potentially trackable sequence in the majority of patients. Taken together, we show that dominant clonal sequences identified at baseline are reliable biomarkers for long-term tracking of the malignant clone, including both IGH and the majority of light chain clones.
Keywords: Multiple myeloma, minimal residual disease, MRD, clonal tracking, immunoglobulin clonality, CDR3, next generation sequencing (NGS)
Introduction
The clonal B-cell origin of multiple myeloma has been successfully exploited for minimal residual disease (MRD) testing by next generation sequencing (NGS) of immunoglobulin heavy (IGH) and light chain V(D)J rearrangement sequences1–3.
Sequencing assays may target the full-length immunoglobulin variable region, while the commonly used clonoSEQ assay (Adaptive biotechnologies) focuses on the third complementarity determining region (CDR3)4–7. When applied to bone marrow aspirates, the sensitivity of NGS-based MRD assays is up to 1 tumor cell in 1,000,000 bone marrow cells (10−6)1,2. Non-invasive strategies are under development8–10. While the prognostic impact of molecular MRD testing has been thoroughly demonstrated, translation to wide clinical use will require further standardization based on understanding of the underlying biology2,3,11–13. There is limited information available to address whether clonal sequences remain stable for tracking over time, and to what extent light chain sequences are sufficiently unique for tracking.
Current molecular MRD testing strategies assume that clonal CDR3 sequences identified at baseline persist throughout the disease course without clonal evolution1,14. This has been supported by small studies of paired diagnosis and progression samples14,15. However, recent reports have identified CDR3-defined subclones at baseline or the time of MRD testing, raising the possibility of on-going evolution16,17. Indeed, the overall genomic landscape of multiple myeloma is characterized by subclonal heterogeneity and complex evolutionary patterns18–21. If subclones with evolved CDR3 sequences have the capacity to become dominant, this would raise concerns about the reliability of baseline sequences for long-term tracking.
Tracking of clonal IGK and IGL sequences is emerging as an important part of molecular MRD testing strategies, where previously the focus was almost entirely on IGH1,4. This is driven by two factors: 1) desire to identify a trackable clone in ~100 % of patients, including also those where no clonal IGH can be identified22,23; and 2) increasing use of all immunoglobulin alleles for simultaneous tracking in single-tube assays5. Using light chain sequences for tracking is not straightforward, because they are less diverse than IGH and therefore less specific for the tumor clone24. There is no published data on how “trackable” (i.e. sufficiently unique) sequences may be identified, which currently limits the applicability and interpretation of light chain tracking.
With molecular MRD testing moving into the clinic3, we were motivated to address the questions of CDR3 clonal evolution and the suitability of IGK and IGL clones for tracking. To accomplish this, we implemented a novel approach to tumor clonality analysis based on standard RNA sequencing data, compiling a large dataset of heavy and light chain CDR3 repertoires from 905 plasma cell myeloma and normal control samples from 841 individuals. Clonal diversity of CDR3 sequences was rare, and all dominant clonal sequences were stable over time in sequential samples despite substantial clonal shifts by whole exome sequencing. Light chain sequence complexity was the main determinant of uniqueness, allowing identification of a trackable IGK or IGL clone in the majority of patients. Taken together, we provide important insights supporting the clinical implementation of molecular MRD testing strategies.
Materials and Methods
Patients and public datasets
Thirty patients with plasma cell myeloma receiving care at MSKCC were included in the study. They were selected from a total of 177 patients included in a previous study of baseline clonality detection using the LymphoTrack assays (Invivoscribe, Inc, San Diego, CA)22. Patient selection was done to ensure representation across all immunoglobulin heavy and light chain classes, including patients without detectable complete immunoglobulin in serum. We selected 20 cases where LymphoTrack detected one or more clonal sequences, and 10 patients where LymphoTrack was negative. As a control cohort, we included 10 healthy individuals who donated bone marrow from bony material left over after hip replacement surgery. The use of patient samples and clinical data as well as healthy controls was approved by the IRB (09–141, 06–107 and 15–017) and subjects provided informed consent to participate.
Previously unpublished RNAseq data was analyzed from two additional cohorts. The first was from a clinical trial where patients with newly-diagnosed multiple myeloma received carfilzomib, lenalidomide and dexamethasone (NCT01402284), carried out at the National Institutes of Health (CRD-cohort)25. The second was from the Natural History Study of Monoclonal Gammopathy of Undetermined Significance and Smoldering Myeloma (NCT01109407), carried out at the National Cancer Institute (NCI-cohort).
Publicly available RNAseq data from 807 samples derived from 743 patients with multiple myeloma enrolled in the Multiple Myeloma Research Foundation (MMRF) CoMMpass trial was accessed through dbGaP (phs000748.v6.p4) and analyzed together with clinical data downloaded from the MMRF Research gateway (http://research.themmrf.org).
Bone marrow sample preparation
From our in-house cohort of 30 patients, we analyzed bone marrow mononuclear cells (MNC). These samples had variable tumor cell content, likely due to hemodilution in the late pulls of bone marrow aspirate reserved for research22. Control samples from hip replacement surgery were also MNCs, confirmed as normal by the MSK-IMPACT sequencing panel26. Samples sequenced in the CRD, NCI and CoMMpass cohorts were all CD138+ sorted tumor cells.
RNA sequencing
Four independent RNA sequencing datasets were analyzed as described in detail in the supplemental materials and methods. Briefly, poly-adenine based mRNA enrichment was used in all datasets except for the CDR-cohort, where ribo-depletion was performed. Sequencing of 83–100 bp paired end reads to a target depth of 80–120 million was performed on Illumina HiSeq instruments.
Immunoglobulin CDR3 extraction from RNAseq data
Adaptive immune receptor profiling from RNA sequencing data was initially developed to analyze immune infiltrates in solid tumors27–30. Here, we applied the same approach for the first time to plasma cell myeloma. Raw FASTQ files were analyzed using the freely available MiXCR software suite version 2.1.11, with settings optimized for RNAseq data as previously described (Supplemental Methods)27,28. In brief, MiXCR searches for reads aligning completely or partially to a catalogue of B- and T-cell receptor reference sequences. For downstream analysis we used only the third complementarity determining region (CDR3) sequences contained fully within a single read. Clonotype assembly based on CDR3 sequences was performed using standard settings. For diversity and evolution analysis we integrated data with and without clonotype clustering, in order to identify subclones differing by a single nucleotide. Somatic hypermutation analysis was based on mismatches reported between the CDR3 sequence and germline gene segments. As junctional insertion length we used the number of nucleotides of the CDR3 not aligned to any germline gene segment. V-J segment usage figures for visual data inspection were generated using vdjtools version 1.1.831.
DNA amplicon sequencing of IGH and IGK
V(D)J rearrangements of the IGH and IGK variable regions were assessed by the LymphoTrack assays and software (Invivoscribe Inc, San Diego, CA) in a three-step workflow as previously described: 1) PCR amplification, 2) NGS and 3) bioinformatics analysis and clonality calling22,23. In brief, for IGH we used PCR master mixes consisting of forward primers targeting variable (VH) region framework regions 1 to 3 (FR1–3) as well as several consensus reverse primers targeting the joining (JH) region. The IGK assay contains forward primers targeting conserved variable (VK) region and intron sequences, with reverse primers targeting joining (JK) and kappa deleted element (Kde) regions.
Clonal evolution patterns by exome sequencing
Single nucleotide variant data from whole exome sequencing, and segmented copy number data from whole exome and low-pass whole genome sequencing from the CoMMpass IA13 release were downloaded from the MMRF webpage.
Tumor purity was estimated for each sample as twice the 85th percentile of the allele frequencies of all called mutations with read coverage ≥100 in diploid regions. Cancer Cell Fraction was estimated using the formula CCF = AF (N/P + CN – N), where AF is the observed allele frequency, CN the cancer cell copy number (from exome data; if not available genome data was used), N is the germline copy number (1 for X or Y chromosomes in males, 2 otherwise), and P is purity. Estimated CCF above 100% were set to 100.
Patterns of clonal evolution were determined by manual review of somatic mutation and copy number data of sequential samples. Change in clonal dominance was defined as loss of one major subclone and acquisition of another.
Statistical analysis
Normality of variables was assessed by the Shapiro-Wilk test and quantile-quantile plots. Appropriate statistical summaries and tests are referenced throughout the manuscript.
In the uniqueness and prevalence analysis of clonal sequences, each clonal sequence was searched for in every available independent sample. To avoid cross-contamination, samples from the same dataset were only considered ‘independent’ if it was sequenced more than two batches before the sample in question. This procedure was developed based on cross-contamination analysis of the CoMMpass dataset (Figure S6). This resulted in a different number of samples considered independent for each clone, which we accounted for in downstream analysis. A clonal sequence was considered unique if it was not found in any of the samples searched. The prevalence was calculated by dividing the number of samples where the sequence was found, by the total number of samples searched. While there may still be traces of cross-contamination, this should not involve more than 1 % of samples, as indicated by the proportion of IGH clones found in independent samples.
To estimate the probability of a clonal sequence being unique given a number of samples searched, we calculated the mean proportion of unique clones in 1000 bootstrapping datasets for each sample sizes between 50 and 500. Bootstrapping mean with 95 % confidence intervals (CI) were reported for each sample size. Only clones with 500 or more samples available were included in this analysis.
Factors influencing the probability of uniqueness of light chain clones were estimated by multivariate logistic regression and reported as odds ratio (OR) with 95 % CI. Two-sided p-values < 0.05 were considered statistically significant. Analysis was done in R version 3.4.3.
Data sharing statement
Accession numbers for previously unpublished raw data as well as the full immune repertoires from all samples will be made available upon acceptance of the manuscript.
Results
Immunoglobulin CDR3 extraction from RNA sequencing data
The most diverse part of the immunoglobulin V(D)J region is the CDR3 sequence (Figure 1A). This is where variable (V), diversity (D) and joining (J) gene segments are joined together during V(D)J recombination of the heavy chain gene, while the light chain gene consists of only V and J segments1,4,32.
Figure 1.
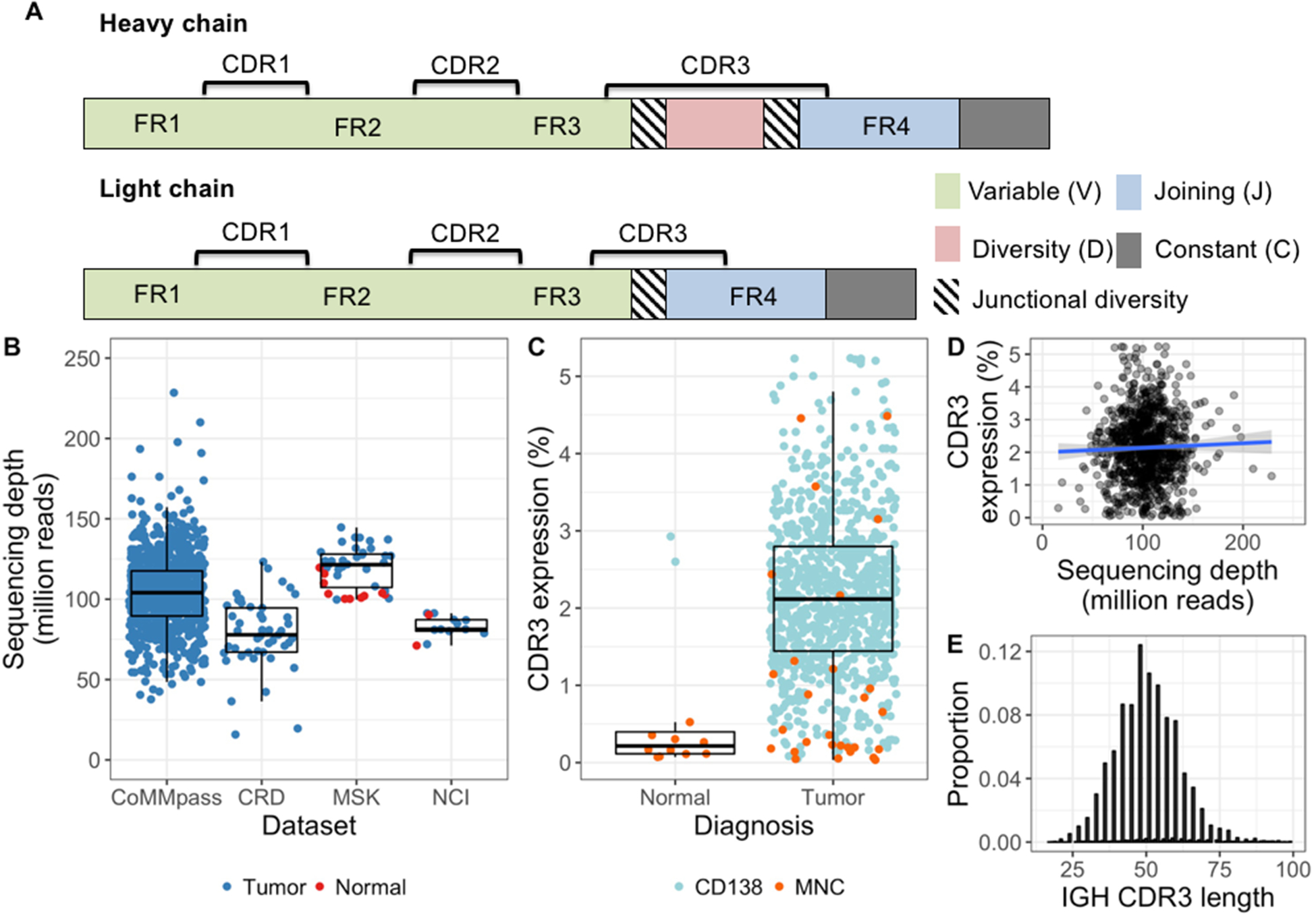
Immunoglobulin CDR3 extraction and expression. A, Schematic of the immunoglobulin heavy (above) and light (below) chain genes. The complementarity determining regions (CDR) are hotspots for somatic hypermutation, while framework (FR) regions are more stable and commonly used as PCR primer targets. Greatest diversity is found in the CDR3, which spans part of each V(D)J segment and contains random junctional diversity in the form of deletions and non-templated insertions. Light chains lack a Dsegment and their variable region is therefore less variable than the heavy chain gene. B, Sequencing depth of each bone marrow sample. Median and quartile range within each cohort are shown as box and whisker plots. C, The proportion of total reads mapping to immunoglobulin CDR3s (i.e. CDR3 expression) shown by sample type and the cell subset analyzed. Enrichment of CD138+ bone marrow cells resulted in considerably higher CDR3 expression in both tumors and normals, as compared with bone marrow mononuclear cells (MNCs). In tumor samples, the median difference between CD138+ cells and MNCs was 5-fold (2.1% and 0.39%, respectively; P < 0.001). D, CDR3 expression was not related to total coverage (spearman’s rho = 0.01, p = 0.73) and therefore suitable for comparison between samples. Linear regression line fitted to the data is shown in blue, with the surrounding shaded area representing the 95% confidence interval. E, There was no sign of length bias in extracted CDR3 sequences, as the top 10 IGH CDR3s from each sample showed the expected normal-approximate length distribution around a mean of 50.
To obtain a sufficiently large sample size for this study, we used a validated bioinformatic approach to extract repertoires of expressed immunoglobulin CDR3 sequences from four RNA sequencing (RNAseq) datasets27,28. Altogether we included 893 plasma cell myeloma samples and 12 normal bone marrow samples derived from 841 individuals (Table S1; Figure 1B). This included 807 samples from patients with multiple myeloma in the publicly available CoMMpass study. We extracted across samples a median of 2.15 million reads (1.37–2.9) containing a complete CDR3 sequence, corresponding to median 2.1 % of the total coverage (quartile range 1.4–2.8 %)(Figure 1C–E).
Distinct patterns of immunoglobulin CDR3 expression separate plasma cell tumors from normal bone marrow
Tumor samples were dominated by a single dominant CDR3 clonotype, constituting a median 81 % of the total immunoglobulin repertoire (quartile range 71–91 %), whereas normal bone marrow was distinctly polyclonal (top clonotype median 0.6 %, quartile range 0.5–1.3 %) (Figure 2A–C). Applying an 8 % cut-off for the dominant clonotype allowed perfect discrimination between tumors and normals.
Figure 2.
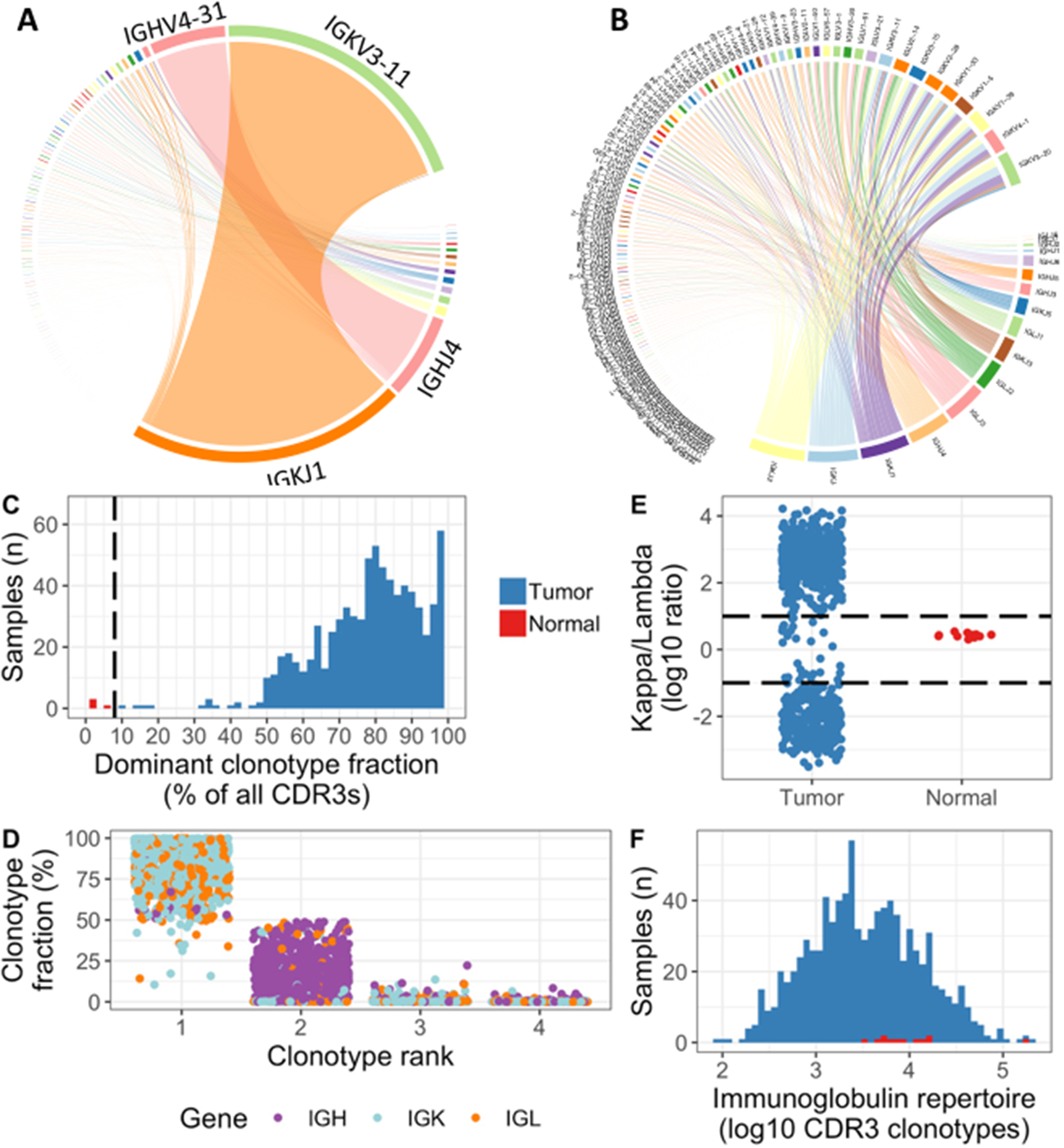
Clonotype dominance and skewed light chain repertoire in plasma cell tumors. A, Immunoglobulin repertoire in patient with multiple myeloma, highly dominated by clonal rearrangements of IGK (J1/V3–11) and IGH (J4/V4–31). Lines connect J gene segments in the lower right corner with V gene segments in the upper left, with thickness representing the proportion of immunoglobulin sequences with the respective V–J combination. B, Polyclonal immunoglobulin repertoire in CD138+ cells from normal bone marrow. C, Histogram showing the fraction of CDR3 reads made up by the dominant clonotype in each sample. The dotted line at 8% perfectly separates normals from tumors. D, The four most abundant clonotypes in each baseline sample, as percentage of total immunoglobulin CDR3 expression. The involved light chain was most commonly ranked 1st, with a heavy chain as 2nd. E, Ratio of total kappa to total lambda CDR3 expression plotted on a log10 scale, showing highly skewed repertoires in the vast majority of baseline tumor samples, whereas all the normal bone marrows are within a narrow window. F: Histogram showing the number of CDR3 clonotypes extracted from each tumor and normal sample on a log10 scale.
The predominant CDR3 expression pattern across IGH, IGK and IGL was dominated by the involved light chain, with lower expression of IGH and profound suppression of the uninvolved light chains (Figure 2D, S1). This was reflected in a skewed kappa/lambda gene expression ratio; while in normal bone marrow samples the ratio remained within a narrow range (mean 2.7, SD 0.39)(Figure 2E). Overall immunoglobulin diversity was also lower in tumor samples as compared with normal bone marrow (median 2973 vs 7682 unique CDR3 clonotypes, p = 0.003)(Figure 2F).
Clonal immunoglobulin CDR3 expression in all patients and minimal sequence diversity
To identify tumor-associated (i.e. “clonal”) heavy and light chain sequences in each sample, we developed criteria for clonality calling (supplemental methods, Figure S1, summarized in Table S2). We refer to the abundance of a clonal sequence as its’ clonal fraction, i.e. the number of reads containing the clonal CDR3 divided by the total number of CDR3 reads from that gene (i.e. IGH, IGK or IGL). This metric should not be understood as the proportion of cells harboring a given CDR3 sequence, as cellular levels of immunoglobulin mRNA vary greatly.
One or more clonal CDR3 sequence were identified at baseline in all 820 patients: Clonal light chain was detectable in 100 %, and IGH in 85 % (695/820) (Figure S2). Failure to identify a clonal IGH sequence was largely explained by low or undetectable serum M protein, with an area under the receiver operator curve of 0.934 (Figure S3). To predict successful IGH clonality detection by RNAseq, the highest sensitivity (91.6 %) and specificity (91.8 %) was achieved with an M protein threshold of 1.0 g/dL, yielding a positive predictive value of 98.6 % to successfully identify clonal IGH (Table S3).
Atypical clonal structure in the baseline sample was confirmed by manual review (supplemental methods) in 35 patients (4.2 %), following two main patterns: 1) subclonal heterogeneity with one or more additional CDR3 sequences related to the dominant clonal sequence; and 2) bi-clonal disease with expression of more than one light chain and/or heavy chain allele. Subclonality was the most common, present in 26 patients (3.2 %) when defined by an edit distance of 5 or lower between CDR3 sequences (i.e. one sequence can be made identical to the other within 5 substitutions, insertions or deletions)(Figure S4A, S5). Overall, 14 patients had an IGH subclone; 8 had a light chain subclone; and 4 had both. Bi-clonal disease was present with high confidence in 9 patients. The majority of these cases were consistent with a single clonal cell population expressing both alleles of either IGK, IGL, or one of each (Figure S4B). In one case there were two unrelated clonal IGH sequences as well as both IGK and IGL, consistent with two independent clonal populations (Figure S4C).
RNAseq shows good concordance with DNA amplicon sequencing and higher sensitivity for expressed clones
To validate our gene expression-based clonality data, we compared our results with standard DNA amplicon sequencing in 20 samples where clonality had been successfully characterized by the LymphoTrack assays (Invivosceibe, Inc; IGH FR1–3 and IGK)22. Identical clonal CDR3 sequences were identified in 16 cases (12 IGH; 1 IGK; and 3 with both IGH and IGK) (Table S4, Figure S6). Discrepancies could be explained by the unbiased capture of all expressed genes by RNAseq, while LymphoTrack is susceptible to false negatives due to PCR amplification bias, but has the added advantage of characterizing unproductively rearranged alleles (Table S4).
Next, we addressed the performance of RNAseq in 10 particularly challenging samples that were negative by LymphoTrack because of low tumor cell content and/or PCR amplification bias (Table S4)22. Clonal IGH was detectable in 7 of these samples by RNAseq. Two of the three remaining samples had undetectable serum M protein. Clonal light chain CDR3 was identified in all cases: Nine patients were kappa-restricted, and one was lambda-restricted. The light chain clones were clearly above the threshold for clonality calling (median 92 % of its respective light chain gene, range 27–99 %), with somewhat lower expression of IGH clones, as expected (median 85 %, range 10–94 %). This shows that the malignant plasma cells have sufficiently high immunoglobulin gene expression to be distinguished from the background even if they are rare in the sample.
No evidence for clonal evolution of multiple myeloma CDR3 sequences
To address the question of clonal evolution in immunoglobulin CDR3 sequences, we analyzed 101 sequential samples from 45 patients in the CoMMpass cohort (Table S5). All follow-up samples were obtained at a time of disease progression, except for 14 samples obtained during stable disease, partial response or very good partial response. Taking advantage of somatic copy number and single nucleotide variants data from whole exome sequencing, we also characterized the landscape of clonal evolution overall. The median time between the first sample and the last was 476 days (quartile range 290–638 days).
There was striking stability of CDR3 sequences over time: In every patient the dominant clonal heavy and light chain sequence remained identical. No related subclones emerged, and only one subclonal sequence became undetectable (Figure 3A). Conversely, exome-based clonality analysis revealed different patterns of clonal evolution as previously described, with 49 % of patients (22 out of 45) showing a major shift in clonal dominance from diagnosis to the follow-up sample (Figure 3B)18. This indicates that all genetically defined multiple myeloma subclones carry the same immunoglobulin CDR3 sequence.
Figure 3.
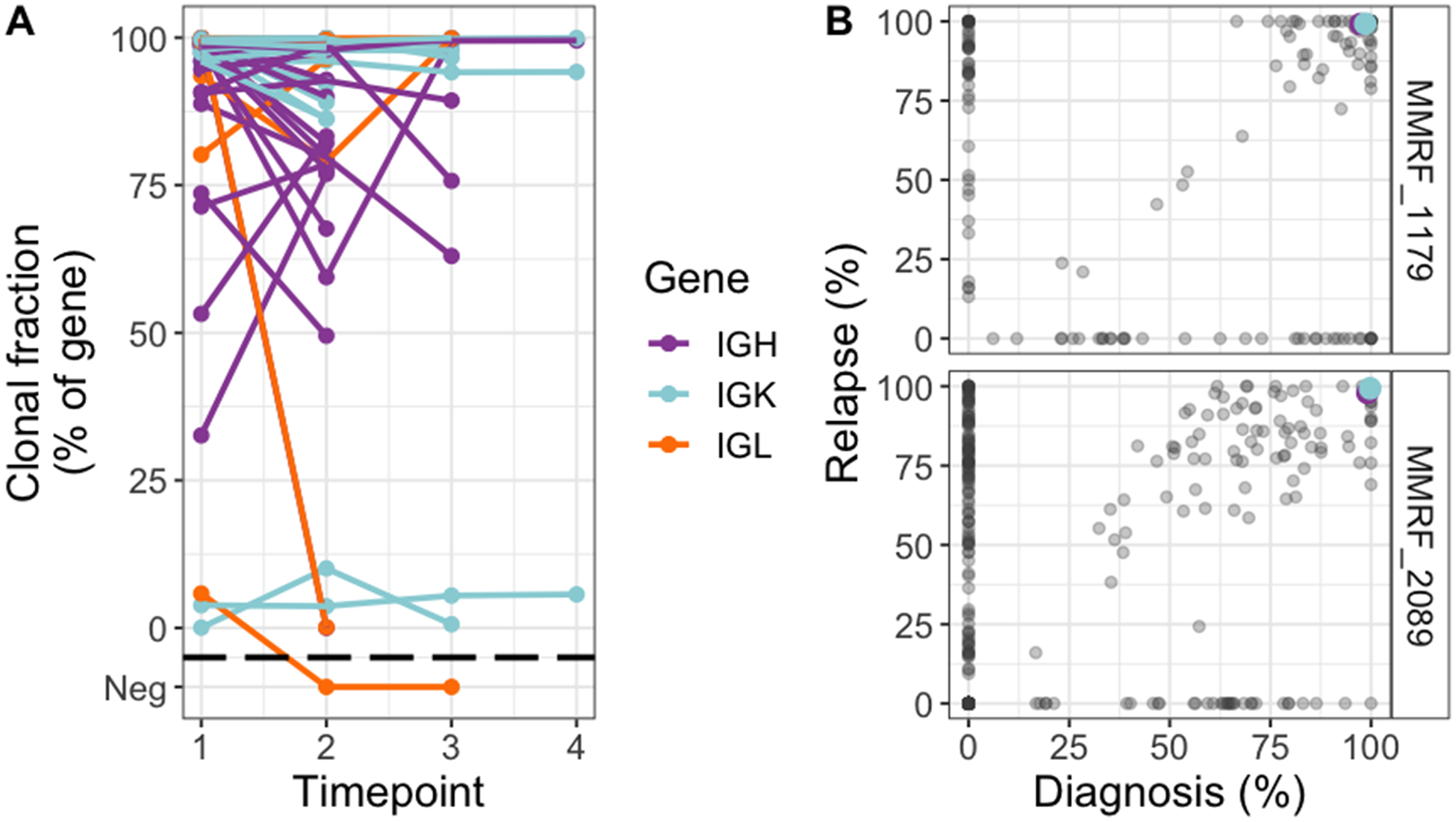
Stability of clonal immunoglobulin CDR3 sequences despite changing clonal dominance by exome sequencing. A, Tracking of clonal CDR3 sequences over time in sequential samples from the CoMMpass dataset. Included in this analysis were all CDR3 sequences that fulfilled clonality criteria on one or more time-point. Changes in clonal fraction of a single clone without corresponding subclonal changes likely reflects changes in the polyclonal background rather than clonal evolution. B, Two example patients where the dominant subclone changed from diagnosis to relapse, while the clonal IGH and IGK CDR3 sequences remained identical. Colored points are CDR3 clonal fractions at diagnosis (x-axis) and relapse (y-axis), plotted on the same scale as in A (i.e. clonal CDR3 expression). Somatic mutations are plotted on the same axes (gray points), represented by its cancer cell fraction (variant allele fraction corrected by tumor purity and local copy number). Gray dots along the diagonals have similar cancer cell fraction at both time-points; with the upper right corner cluster representing shared clonal mutations. Mutations clustered in the lower right corner were present in subclones that dominated at baseline and became undetectable at relapse. Conversely, mutations plotted in the upper left corner represent an undetectable subclone at diagnosis which became dominant at relapse. Taken together this shows a shift in clonal dominance in these patients as defined by exome sequencing, while the dominant CDR3 sequences remained identical.
Uniqueness of light chain CDR3 sequences is driven by somatic hypermutation and junctional diversity
The suitability of immunoglobulin CDR3 sequences for MRD tracking depends on the probability to encounter an identical sequence in the normal B-cell repertoire by chance. This could result in a (falsely) positive MRD result although no tumor cells are present. To mimic this setting and determine which factors influence the uniqueness of CDR3 sequences, we searched for each clonal sequence in a database of immunoglobulin CDR3 repertoires from unrelated samples (Methods, Figure S7).
First, we determined the probability that no identical CDR3 sequences are detected in a given number of independent samples (Figure 4A). As expected, >99 % of IGH clones were unique irrespective of how many samples were searched. Conversely, the lower diversity of light chain CDR3 sequences was reflected in lower probability of uniqueness for IGK and IGL sequences, which continued to decrease without reaching a plateau as we increased the sample size (Figure 4A). The prevalence distribution of light chain clones in independent samples was highly skewed towards 0, but with a long tail, and some sequences were present in >50 % of samples by chance (Figure 4B).
Figure 4.
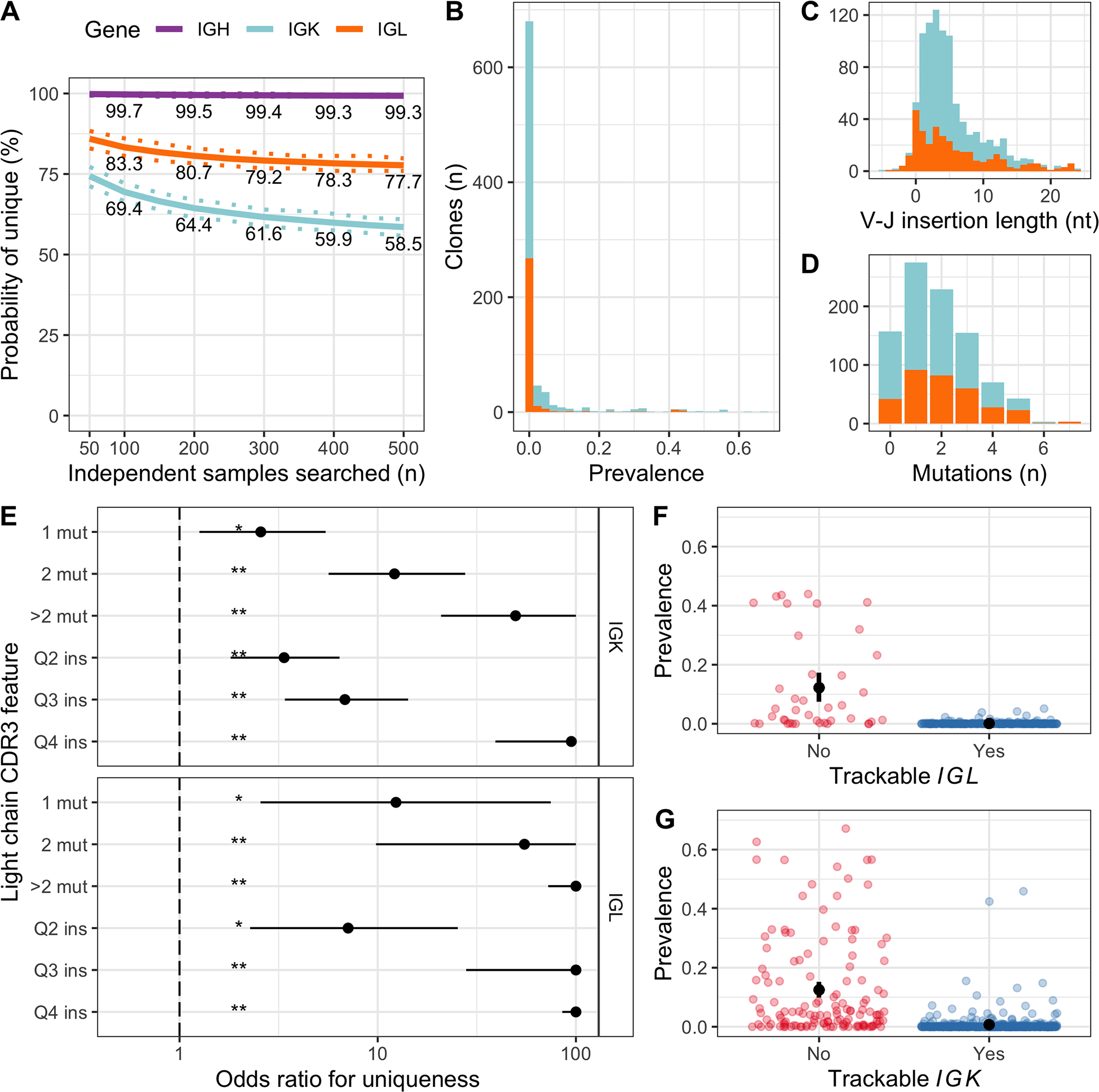
Uniqueness of IGK and IGL CDR3 sequences was determined by somatic mutations and V–J insertion length. A, Estimated probability of a clonal CDR3 being unique after searching a given number of independent samples. Continuous and dashed lines represent bootstrapping mean and 95% confidence intervals, respectively. Clonal sequences with 500 or more independent samples available to search were included in this analysis: 307 IGH sequences, 233 IGK and 129 IGL. B, Prevalence of each clonal IGK (light blue) and IGL (orange) CDR3 sequence in the population of independent samples, shown as a stacked histogram. Sequence prevalence (x-axis) was calculated as the number of samples where the sequence was identified, divided by the total number of samples searched. C, Stacked bar chart showing the V–J insertion length distribution for clonal IGK and IGL sequences. The median length was 4 nucleotides for both genes. D, Similar to C, showing the number of somatic mutations in the CDR3. The median was 1 for IGK and 2 for IGL. E, Results from two independent multivariate logistic regression models for IGK and IGL, where each clonal CDR3 sequence was a case and the outcome was whether the clone was unique (i.e. not found in any samples). Odds ratios (points) above 1 indicate increased probability of the clonal sequence being unique compared to the reference level, 95% confidence intervals are represented by horizontal lines and statistical significance is indicated by asterisks (**P < 0.001, *P < 0.05). Values were limited to 100. Only results from somatic mutation number (mut) and V–J insertion length (ins) are shown here, however the model also included V and J gene usage and CDR3 length, and was adjusted for the number of independent samples available to determine uniqueness. F, Displaying the prevalence of each clonal IGL CDR3 in independent samples (points), separated into trackable (blue) versus non-trackable (red) according to our simple criteria (>median mutation number or insertion length). Because the prevalence distribution was highly skewed, we used a bootstrapping approach to estimate the average prevalence in each group with 95% confidence intervals (black points with error bars). G, Similar to F, for IGK.
The most important predictors for uniqueness of IGK and IGL CDR3 sequences were the number of somatic mutations and the length of non-templated insertion in the V-J junctional region (Figures 4C–E and S8–9 and Tables S6–7). Any increase in mutations or insertions was associated with significantly increased odds of uniqueness for both IGK and IGL, and increasing CDR3 sequence complexity was associated with exponential increases in odds ratio (OR)(Figure 4E). By comparison, only four specific V and J gene segments showed statistically significant non-zero OR, all associated with reduced odds of being unique (Tables S6–7): IGKV2–29 (OR 0.035, 95 % CI 0.001–0.4, p = 0.01, n = 5); IGLJ3 (OR 0.33, 95 % CI 0.12–0.91, p = 0.03, n = 124); IGLV2–18 (OR 0.01, 95 % CI <0.0001–0.29, p = 0.008, n = 4); and IGLV3–1 (OR 0.05, 95 % CI <0.0001–0.46, p = 0.01, n = 34).
Identification of trackable light chain CDR3 sequences
Based on the above findings, we defined light chain clones as suitable for MRD tracking if they had >1 somatic mutation or above median V-J insertion length (Figure 4F–G). For IGL there were 263 clonal sequences (86.7 %) fulfilling the criteria, with a mean prevalence of 0.14 % (95 % CI 0.08–0.22 %) in independent samples. For IGK 413 sequences (76 %) fulfilled the criteria, with a mean prevalence of 0.7 % (95 % CI 0.41–1.09%). Taken together, these simple criteria identified clones with an average prevalence of less than 1 %, yielding a suitable light chain clone for tracking in 72 % of patients (591/820).
Discussion
Molecular MRD testing in multiple myeloma is increasingly being adopted in clinical trials as well as routine patient care3,11. However, there is only sparse information available to address the stability of clonal sequences, and to identify light chain sequences that are sufficiently tumor-specific for tracking. Here, we show that dominant CDR3 sequences remain stable over time despite clear evidence of differential clonal evolution by whole exome sequencing. This has important clinical implications for long-term disease tracking. Furthermore, we defined the main features underlying uniqueness of IGK and IGL CDR3 uniqueness, leading to identification of a trackable light chain clone in the majority of patients. Improved understanding of light chain clonal uniqueness will facilitate the use of these sequences for tracking in clinical assays.
Subclonal CDR3 sequences related to the dominant clone in multiple myeloma and precursor stages have been confirmed in several independent studies using different methodology, including 3.2 % in our large cohort of 820 patients16,17,33. Although rare, the presence of such subclones raises the possibility of changes in the dominant CDR3 from baseline to progression. This was not the case in any of the 45 patients with sequential samples in our study, including 22 patients who otherwise underwent major changes in the clonal structure of disease. Similarly, no change over time in the IGH CDR3 sequence was found in two previous studies totaling 36 patients analyzed by conventional PCR and Sanger sequencing. However, these previous studies did not evaluate the light chain sequences and lacked whole exome sequencing data for assessment of overall clonal evolution14,15. Taken together, the low frequency of subclonal CDR3 variants and absence of evolution over time in active multiple myeloma indicates that tumor cells at this stage are not under selective pressure to undergo antibody affinity maturation. Of immediate translational relevance, we confirm that dominant clonal CDR3 sequences identified at baseline are indeed reliable biomarkers for MRD tracking throughout the disease course.
Tracking of CDR3 sequences that are not sufficiently specific for the tumor cells may lead to false positive MRD results. This is more likely to happen with light chain clones than IGH because of their lower diversity (107 vs 1011), as confirmed by our data24,34. Diversity is generated particularly in the CDR3 region by: 1) V(D)J segment recombination; 2) deletions and non-templated insertions in the V(D)J junctions; and 3) somatic hypermutation32,35. Here, we establish the latter two processes as by far the most important determinants of sequence uniqueness. Based on these factors, we identified suitable clones for tracking in 72 % of patients that on average were present in less than 1 % of independent samples. Our findings support the use of light chain clonal tracking in addition to IGH for MRD testing in multiple myeloma and potentially other lymphoid malignancies. There is however one caveat, in that our RNA-based analysis of individuals with active disease will under-estimate the probability to encounter a given sequence by ultra-deep sequencing in the MRD setting. Validation studies will be required to standardize criteria for trackable sequences and quantify the expected false positive rate for specific MRD assays.
Strengths of this study include a large sample size of clonal heavy and light chain sequences, and follow-up samples with paired whole exome sequencing. This allowed statistical analysis of clonal uniqueness and demonstration of the disconnect between overall genomic evolution and CDR3 clonal stability. There are also limitations in the generalizability of results. To obtain a sufficiently large sample size, we used a gene expression-based approach to clonality analysis instead of the DNA amplicon-based assays that are currently the standard for MRD tracking. To ease the interpretation, we rigorously validated our RNA-based approach by comparing it to a standard DNA-based assay. Our findings support the use of RNAseq for clonality assessment in lymphoid tissues; however, this approach is not feasible for MRD testing.
Going forward, molecular MRD assays enable reliable long-term tracking of clonal heavy and light chain sequences with applicability approaching 100 %, which renders it an optimal strategy for routine clinical use.
Supplementary Material
Acknowledgments
The authors thank Maria Sirenko, Dominik Glodzik, Noushin Farnoud, Max Levine and Rachel Bashford-Rogers for discussions and feedback. Funding support for this study was provided by the Memorial Sloan Kettering Core Grant (P30 CA008748), the Perelman Family Foundation in collaboration with the Multiple Myeloma Research Foundation (MMRF), and the Intramural Research Program of the National Cancer Institute.
References
- 1.Ho C, Arcila ME. Minimal residual disease detection of myeloma using sequencing of immunoglobulin heavy chain gene VDJ regions. Semin Hematol. 2018;55(1):13–18. [DOI] [PubMed] [Google Scholar]
- 2.Perrot A, Lauwers-Cances V, Corre J, et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landgren O MRD Testing in Multiple Myeloma: From a Surrogate Marker of Clinical Outcomes to an Every-Day Clinical Tool. Semin Hematol. 2018;55(1):1–3. [DOI] [PubMed] [Google Scholar]
- 4.Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. The New England journal of medicine. 2018;378(6):518–528. [DOI] [PubMed] [Google Scholar]
- 6.Carlson CS, Emerson RO, Sherwood AM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nature communications. 2013;4:2680. [DOI] [PubMed] [Google Scholar]
- 7.van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98–3936. Leukemia. 2003;17(12):2257–2317. [DOI] [PubMed] [Google Scholar]
- 8.Rustad EH, Coward E, Skytoen ER, et al. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica. 2017;102(7):1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberle A, Brandt A, Voigtlaender M, et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica. 2017;102(6):1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazzotti C, Buisson L, Maheo S, et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood advances. 2018;2(21):2811–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landgren O, Rustad EH. Meeting report: Advances in minimal residual disease testing in multiple myeloma 2018. ADVANCES IN CELL AND GENE THERAPY. 2019;2(1):e26. [Google Scholar]
- 12.Munshi NC, Avet-Loiseau H, Rawstron AC, et al. Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-analysis. JAMA oncology. 2017;3(1):28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landgren O, Devlin S, Boulad M, Mailankody S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone marrow transplantation. 2016;51(12):1565–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puig N, Conde I, Jiménez C, et al. The predominant myeloma clone at diagnosis, CDR3 defined, is constantly detectable across all stages of disease evolution. Leukemia. 2015;29:1435. [DOI] [PubMed] [Google Scholar]
- 15.Ralph QM, Brisco MJ, Joshua DE, Brown R, Gibson J, Morley AA. Advancement of multiple myeloma from diagnosis through plateau phase to progression does not involve a new B-cell clone: evidence from the Ig heavy chain gene. Blood. 1993;82(1):202–206. [PubMed] [Google Scholar]
- 16.Munshi NC, Minvielle S, Tai Y-T, et al. Deep Igh Sequencing Identifies an Ongoing Somatic Hypermutation Process with Complex and Evolving Clonal Architecture in Myeloma. Blood. 2015;126(23):21–21. [Google Scholar]
- 17.Cowan G, Weston-Bell NJ, Bryant D, et al. Massive parallel IGHV gene sequencing reveals a germinal center pathway in origins of human multiple myeloma. Oncotarget. 2015;6(15):13229–13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nature communications. 2014;5:2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinhold N, Ashby C, Rasche L, et al. Clonal selection and double hit events involving tumor suppressor genes underlie relapse from chemotherapy: myeloma as a model. Blood. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egan JB, Shi CX, Tembe W, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120(5):1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rustad EH, Hultcrantz M, Yellapantula VD, et al. Baseline identification of clonal V(D)J sequences for DNA-based minimal residual disease detection in multiple myeloma. PloS one. 2019;14(3):e0211600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arcila ME, Yu W, Syed M, et al. Establishment of Immunoglobulin Heavy Chain Clonality Testing by Next-Generation Sequencing for Routine Characterization of B-Cell and Plasma Cell Neoplasms. J Mol Diagn. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briney B, Inderbitzin A, Joyce C, Burton DR. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korde N, Roschewski M, Zingone A, et al. Treatment With Carfilzomib-Lenalidomide-Dexamethasone With Lenalidomide Extension in Patients With Smoldering or Newly Diagnosed Multiple Myeloma. JAMA oncology. 2015;1(6):746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of Molecular Diagnostics. 2015;17(3):251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nature methods. 2015;12(5):380–381. [DOI] [PubMed] [Google Scholar]
- 28.Bolotin DA, Poslavsky S, Davydov AN, et al. Antigen receptor repertoire profiling from RNA-seq data. Nature biotechnology. 2017;35:908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorsson V, Gibbs DL, Brown SD, et al. The Immune Landscape of Cancer. Immunity. 2018;48(4):812–830.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mose LE, Selitsky SR, Bixby LM, et al. Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with V’DJer. Bioinformatics. 2016;32(24):3729–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shugay M, Bagaev DV, Turchaninova MA, et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLOS Computational Biology. 2015;11(11):e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunn-Walters D, Townsend C, Sinclair E, Stewart A. Immunoglobulin gene analysis as a tool for investigating human immune responses. Immunol Rev. 2018;284(1):132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zojer N, Ludwig H, Fiegl M, Stevenson FK, Sahota SS. Patterns of somatic mutations in VH genes reveal pathways of clonal transformation from MGUS to multiple myeloma. Blood. 2003;101(10):4137–4139. [DOI] [PubMed] [Google Scholar]
- 34.Soto C, Bombardi RG, Branchizio A, et al. High frequency of shared clonotypes in human B cell receptor repertoires. Nature. 2019;566(7744):398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Angelo S, Ferrara F, Naranjo L, Erasmus MF, Hraber P, Bradbury ARM. Many Routes to an Antibody Heavy-Chain CDR3: Necessary, Yet Insufficient, for Specific Binding. Frontiers in immunology. 2018;9:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.