

The factors driving fine-scale composition and dynamics of gut microbial communities are poorly understood. In this study, we used metagenomic amplicon deep sequencing to decipher the strain dynamics of two key members of the honey bee gut microbiome. Using this high-throughput and cost-effective approach, we were able to confirm results from previous large-scale whole-genome shotgun (WGS) metagenomic sequencing studies while also gaining additional insights into the community dynamics of two core members of the honey bee gut microbiome. Moreover, we were able to show that cryptic strains are not responsible for the observed variations in microbiome composition across bees.
KEYWORDS: community dynamics, honey bee, microbiome, strain diversity
ABSTRACT
Host-associated microbiomes can be critical for the health and proper development of animals and plants. The answers to many fundamental questions regarding the modes of acquisition and microevolution of microbiome communities remain to be established. Deciphering strain-level dynamics is essential to fully understand how microbial communities evolve, but the forces shaping the strain-level dynamics of microbial communities remain largely unexplored, mostly because of methodological issues and cost. Here, we used targeted strain-level deep sequencing to uncover the strain dynamics within a host-associated microbial community using the honey bee gut microbiome as a model system. Our results revealed that amplicon sequencing of conserved protein-coding gene regions using species-specific primers is a cost-effective and accurate method for exploring strain-level diversity. In fact, using this method we were able to confirm strain-level results that have been obtained from whole-genome shotgun sequencing of the honey bee gut microbiome but with a much higher resolution. Importantly, our deep sequencing approach allowed us to explore the impact of low-frequency strains (i.e., cryptic strains) on microbiome dynamics. Results show that cryptic strain diversity is not responsible for the observed variations in microbiome composition across bees. Altogether, the findings revealed new fundamental insights regarding strain dynamics of host-associated microbiomes.
IMPORTANCE The factors driving fine-scale composition and dynamics of gut microbial communities are poorly understood. In this study, we used metagenomic amplicon deep sequencing to decipher the strain dynamics of two key members of the honey bee gut microbiome. Using this high-throughput and cost-effective approach, we were able to confirm results from previous large-scale whole-genome shotgun (WGS) metagenomic sequencing studies while also gaining additional insights into the community dynamics of two core members of the honey bee gut microbiome. Moreover, we were able to show that cryptic strains are not responsible for the observed variations in microbiome composition across bees.
INTRODUCTION
There is increasing evidence that deciphering strain-level diversity is crucial for understanding the impact of microbiomes on host health. However, the fine-scale community dynamics within host microbiomes is still poorly characterized and understood. Our lack of understanding of host-associated microbial community dynamics can be attributed to two main factors. (i) Most methodologies lack the accuracy to fully evaluate strain-level diversity. (ii) Host-associated microbial communities are generally very complex, making it challenging to disentangle strain-level dynamics (1, 2). A common method used in microbiome studies is amplicon sequencing of the 16S rRNA gene, which fails to provide an accurate picture of species, and especially strain-level, diversity within a community. Whole-genome shotgun (WGS) metagenomic sequencing can provide insights about strain diversity. However, WGS metagenomics requires a high sequencing depth to recover strain diversity, making it expensive to apply to large numbers of samples and virtually incapable of capturing low-frequency species and strains, especially in complex communities. An alternative but rarely used method for assessing strain-level diversity employs amplicon sequencing of species-specific protein-coding gene markers (3–6), similar to multilocus sequence typing (MLST) but applied to natural communities. We will refer to this method as metagenomic amplicon strain typing (MAST). By assessing nucleotide variation in core protein-coding genes, MAST can identify strain diversity for each individual species in a community. Therefore, this approach can be used to determine the population structure and dynamics of different microbial strains across samples, locations, times, and conditions. However, one limitation of this approach is that it requires specific primers for each species of the community. Therefore, it cannot easily be applied to entire complex communities or to species for which reference genomes are not available.
We used the honey bee (Apis mellifera) as a model system to study the population dynamics of host-associated microbiomes using the MAST method. The honey bee is an ideal model system for tackling fundamental questions about microbial community dynamics because its microbiome is simple (eight species make up ∼95% of the community), conserved (five core species are found in all honey bees globally), and vertically transmitted (from bee to bee) (7–10). Despite the simplicity and conservation of the honey bee gut microbial community at the species level, a large amount of strain-level variation has been shown to exist within each of the core species (4, 11–13). The strain-level diversity within honey bees and bumble bees has been investigated mostly through WGS metagenomics and isolate or single-cell sequencing (11, 12, 14–20), though one study used the MAST method to investigate strain-level diversity of Snodgrassella alvi in honey bees and bumble bees (4). From these studies, it has been shown that individual bees from the same colony harbor different strains, resulting in very high strain-level diversity within a population, i.e., colony (4, 11–13). There is evidence that strains can be dominant in an individual bee and absent from another within the same colony (4, 12) and that within an individual, closely related strains generally coexist together (12). The high strain-level diversity does not seem to correspond to variation in age or sampling time, and strains are not unique to geographic location, e.g., the same strains have been found in bees from different countries (4, 12). Moreover, strains within the same species often possess different metabolic capabilities, making individual communities functionally distinct (14–16, 20–22). Although WGS metagenomic studies have revealed a lot about strain-level dynamics in the honey bee gut, they were unable to rule out the possibility that low-frequency variants within the population could explain the strain-level population dynamics. Likewise, the previous study which used the MAST method to investigate strain diversity designed their marker (MinD) to capture Snodgrassella diversity across both bumble bees and honey bees (4), limiting their ability to capture fine-scale strain variation because of the high level of divergence of Snodgrassella, which is likely composed of multiple species across these different bee hosts.
Here, we used MAST on protein-coding gene markers for two of the most dominant core members of the honey bee gut microbiome, Snodgrassella alvi and Gilliamella spp., to evaluate strain composition in four honey bee colonies from four different locations in the United States. We were able to confirm the results of previous strain-level studies while also gaining additional insights. Importantly, we were able to explore the impact of low-frequency variants on microbiome dynamics and show that cryptic strain diversity is not responsible for the observed variations in microbiome composition across bees. We identified that strain composition is far from random in honey bees, where several strains are frequently associated together, while others almost never co-occur in the same host. We found that many bees from the same hive harbor highly divergent strain compositions, while bees sampled across different geographic locations (different states in the United States) frequently harbor similar strain communities. Strains present within an individual honey bee typically possess very high sequence similarity, and our results indicate that they did not originate from within-host diversification.
RESULTS
Analysis of the MAST markers.
We analyzed the strain composition of two core members of the honey bee gut microbiome: S. alvi and Gilliamella spp. Honey bees were sampled from a single colony from four different locations in the United States (numbers of sampled bees per location, 103 for Texas, 21 for Tennessee, 9 for Utah, and 11 for Florida). DNA was extracted from the gut, and amplicon sequencing was performed for four markers (guaA and gluS for S. alvi and pflA and rimM for Gilliamella spp.) on each individual bee (6). These markers were designed to target only Snodgrassella and Gilliamella in honey bees (A. mellifera), since creating markers that also capture bumble bee (Bombus spp.) strains resulted in a lack of resolution (6).
Sequenced markers were quality filtered to avoid overprediction due to sequencing errors (see Materials and Methods). Sequences were aligned, and strains were considered different if they possessed ≥1 nucleotide difference along the marker. The coverage depth of each marker (average of 46,000 reads per marker and per sample) allowed us to identify strains with high confidence (see Materials and Methods and Table S1 and Fig. S1 in the supplemental material). Using this approach, we identified more than 200 different strains for each marker across all bees analyzed.
Average read coverage for each marker by sampling location. Download FIG S1, PDF file, 0.2 MB (172.3KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Total number of reads obtained for each sample for each marker. NA represents samples that reads were not obtained due to issues with DNA extraction or sequencing. Download Table S1, XLSX file, 0.01 MB (15.1KB, xlsx) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We also built the core genome phylogenies and marker sequence phylogenies (amplicon region of each gene used for MAST) using all publicly available genomes of S. alvi and Gilliamella spp. This allowed us to determine whether the marker sequence phylogenies were congruent with the species phylogenies. The resolution of the marker sequence phylogenies is low due to the limited number of base pairs (less than 500 bp for each marker), but overall, all four marker phylogenies were consistent with the core genome phylogenies (Fig. S2).
Phylogenetic trees of A. mellfiera-associated S. alvi and Gilliamella strains available on NCBI. (A) Core genome reference phylogeny of S. alvi, (B) MAST gluS marker phylogeny, and (C) MAST guaA marker phylogeny. (D) Core genome reference phylogeny, (E) MAST rimM marker phylogeny, and (F) MAST pflA marker phylogeny. Blue shading is used to delineate the different Gilliamella species or phylotypes. Phylogenetic trees were constructed with RAxML v8 with a GAMMA + GTR model. Download FIG S2, PDF file, 0.3 MB (436.5KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strain diversity of S. alvi and Gilliamella spp.
Since designing our MAST markers (6), the number of sequenced genomes for Gilliamella has more than doubled and Gilliamella has been split into at least two, possibly three, distinct species (12, 23). Thus, we first checked to see whether our four markers captured the entire strain diversity of S. alvi and Gilliamella spp. To this aim, we built the phylogeny for each marker using our MAST-derived sequences and the corresponding sequences of the publicly available genomes. Our results indicate that both markers gluS and guaA captured all the diversity of sequenced S. alvi from A. mellifera (Fig. S3), which is thought to be the sole Snodgrassella species present in A. mellifera (12). In contrast, our phylogenetic analysis indicates that the two Gilliamella markers offer different resolutions: the rimM marker captures strain diversity across all sequenced Gilliamella species, whereas pflA captures the strain diversity of G. apicola only (Fig. S4). For clarity, the results of the two Gilliamella markers will be presented and discussed in parallel, since one marker provides a genus-level resolution of strain diversity and the other depicts the strain diversity within the G. apicola species.
Phylogenetic tree of A. mellifera-associated S. alvi strains based on gluS and guaA MAST markers. The phylogeny was made using the amplified regions of each gene marker from the strains sequenced in this study (green font) as well as the sequenced genomes available on NCBI as of July 2020 (black font and highlighted in green). The trees were constructed with RAxML v8 with a GAMMA + GTR model. Download FIG S3, PDF file, 0.4 MB (338.3KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic tree of A. mellifera-associated Gilliamella strains based on pflA and rimM MAST markers. The phylogeny was made using the amplified regions of each gene marker from the strains sequenced in this study (blue font) as well as the sequenced genomes available on NCBI as of July 2020 (black font and highlighted in blue). The trees were constructed with RAxML v8 with a GAMMA + GTR model. Download FIG S4, PDF file, 0.4 MB (450.9KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We identified an average of five to six strains for both S. alvi and Gilliamella spp. per bee based on three markers (guaA, gluS, and rimM), while the G. apicola-specific marker (pflA) yielded an average of 16 strains per bee (Fig. 1). For S. alvi and G. apicola, any two strains were found to differ by seven to nine single nucleotide polymorphisms (SNPs) on average (Fig. S5A). As expected, the Gilliamella species marker (rimM) displayed higher sequence diversity across strains (∼20 SNPs), due to the fact that it captures multiple species (Fig. S5A).
FIG 1.
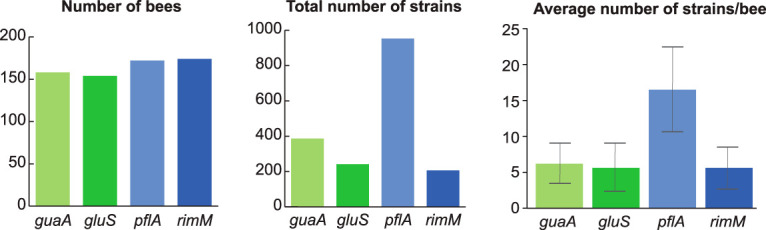
Number of strains identified with the four markers. (Left) Total number of bees analyzed for each marker. (Middle) Total number of strains identified with each marker. (Right) Average number of strains identified per bee. The standard deviations are indicated by error bars. The markers of S. alvi and Gilliamella spp. are represented in green and blue, respectively.
(A) Average number of SNPs across strains estimated with the four gene markers. The markers of S. alvi and G. apicola are represented in green and blue, respectively. (B) Resampling analysis of strain composition. For each marker, the average number of strains was estimated for different numbers of randomly sampled bees (nonredundant). The y axis shows the average number of strains found in 100 combinations of randomly sampled bees. The x axis indicates the number of nonredundant resampled bees included in each combination. Download FIG S5, PDF file, 0.2 MB (253.5KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We estimated to what extent our procedure was capable of capturing the entire strain diversity of S. alvi, G. apicola, and Gilliamella spp. To do so, we conducted a resampling analysis of the bees used to infer strain diversity and built saturation curves based on each marker (Fig. S5B). Results indicate that a substantial fraction of the strain diversity of these bacteria has been captured by our analysis for the four hives sampled from the four states. However, the saturation curves do not plateau, indicating that we did not capture the entire strain diversity of Gilliamella spp. or S. alvi. Results likely reflect heterogeneous strain compositions across bees and suggest that a larger strain diversity exists for these two symbionts of the honey bee gut.
Geography does not drive strain composition of the bee gut microbiome.
We tested whether strain distribution was biased across the hives from the four geographic locations (Texas, Utah, Florida, or Tennessee). We compared the number of distinct strains observed for each location and compared that to the number of expected strains under randomization (i.e., each bee was randomly allocated a state). The number of expected strains under the random distribution was not significantly different from the observed distribution for S. alvi and G. apicola markers (P > 0.1, chi-square test, Fig. 2) but was significant for the Gilliamella marker (P = 0.005, chi-square test, Bonferroni correction). Overall, these results suggest that strains of S. alvi and G. apicola are not associated with specific locations and that most strains are likely to be found across the United States.
FIG 2.
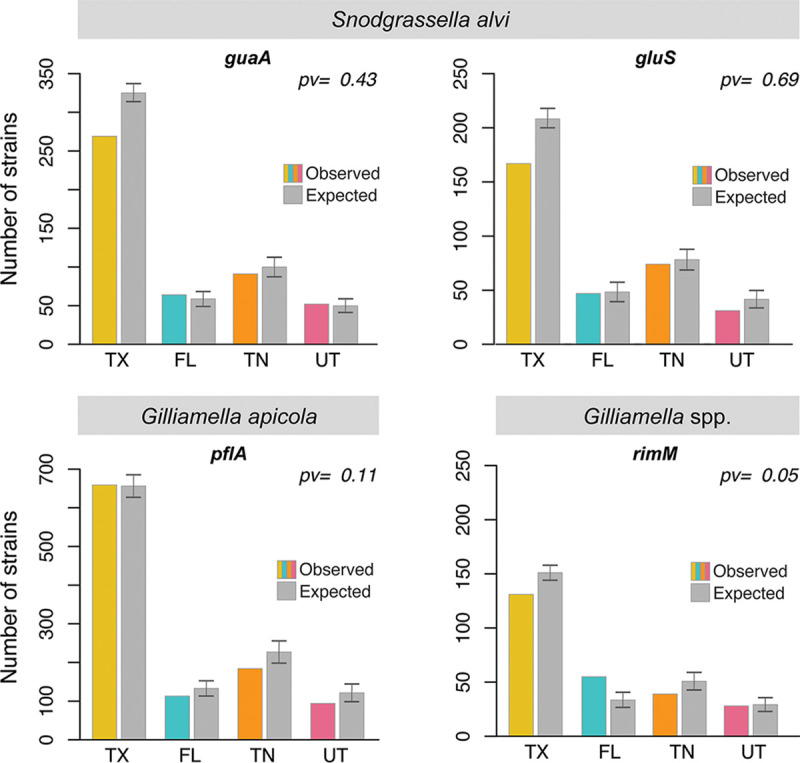
Number of observed strains versus number of randomly expected strains for each location. For each marker, the number of strains is indicated for each location (color bars). The number of strains for each location was compared to the number of strains expected at each location under random expectation (gray bars). The random expectation was obtained by randomly reallocating each bee to a different location. The error bars represent standard deviations. pv, P value.
In order to further analyze the strain composition (presence/absence) within bees from different hives and locations, we performed principal coordinate analyses using unweighted UniFrac (24) on the four amplicons. The principal coordinate analyses did not show clustering based on hive location for any of the markers or species, indicating that bees from the same hive are not more similar in strain composition than bees sampled from other hives in different states. In contrast, this analysis revealed that some bees sampled from hives at various locations present similar strain compositions (Fig. 3).
FIG 3.

Principal coordinate analyses (PCoA) of strain composition for each bee analyzed based on the four markers and geographic location. Each symbol represents the value for an individual bee. The colors and shapes of the symbols represent the location of each bee (Texas, Utah, Tennessee, or Florida). The analysis was conducted by comparing the composition of strains for all four markers (see Materials and Methods).
Bacterial strains are not randomly associated within bees.
To further decipher the distribution of strains across bees and geographic locations, we analyzed the patterns of strain co-occurrence within bees. Consistent with previous findings (12), we observed that several bees tended to present either very similar strain compositions or very different strain compositions (Fig. 4). Similar results were observed when representing the co-occurrence of strains (Fig. S6). For all markers, we found that some strains almost exclusively co-occur together within the same bees while others never or rarely co-occur (Fig. S6). To further test this, we randomly reassorted the strains across bees and counted the number of times the randomly shuffled strains co-occurred. We found that the distribution of strain associations was significantly different from the randomized association for S. alvi and G. apicola markers (for guaA, gluS, and pflA, P < 10−7, P < 0.003, and P < 10−12, respectively, chi-square tests with Bonferroni corrections). The test was not found significant for the Gilliamella rimM marker (P = 0.43, chi-square test).
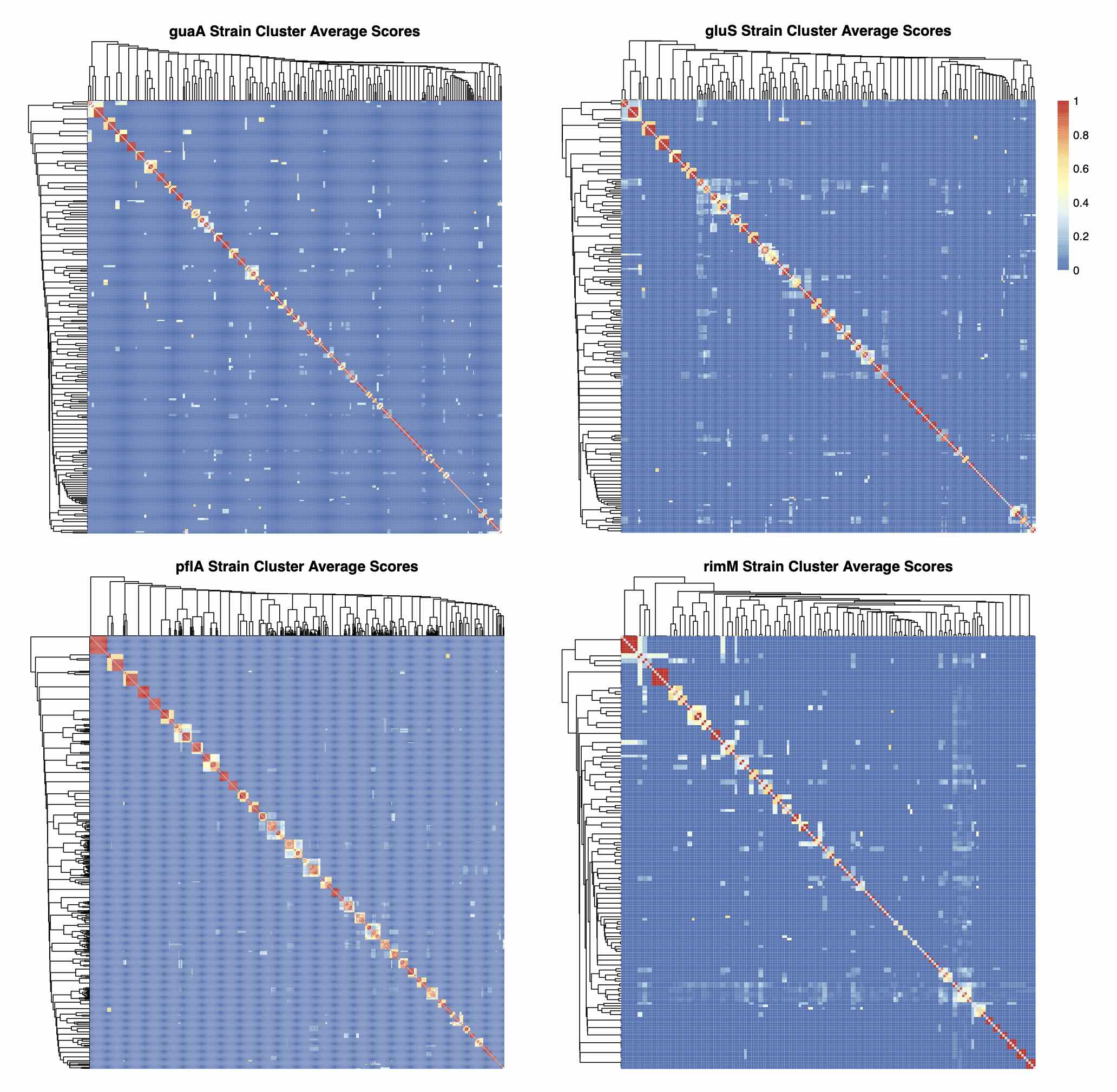
FIG 4.

Heatmaps of strain composition across bees for each marker. Each line and column represent a bee. A score of 1.0 represents 100% co-occurrence (the bees are composed of the exact same strains), and 0.0 represents no co-occurrence (the bees do not share any common strains).
Heatmaps of strain co-occurrence for each marker. Each line and column represent a strain. A score of 1.0 represents 100% co-occurrence (the strains are always found together), and 0.0 represents no co-occurrence (the strains are never found together). Download FIG S6, JPG file, 0.3 MB (355KB, jpg) .
{kind=link}
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Overall, we identified around 40 strain clusters (strains that frequently co-occur in the same bees), with each cluster containing approximately three to four strains, with the exception of the G. apicola marker pflA where a higher strain diversity was captured (Fig. S7A). We found that the vast majority of bees were composed of a single cluster of strains (Fig. S7B). When comparing the strains within each cluster, we found that most strains from the same cluster typically differ by a single SNP or very few SNPs (Fig. S7C). For each of the four markers, strains from the same cluster were more similar to each other than strains from different clusters (P < 10−15, Wilcoxon test, Fig. 5). Consistent with previous evidence (12), most individual bees were found to possess only a single cluster of closely related strains.
FIG 5.
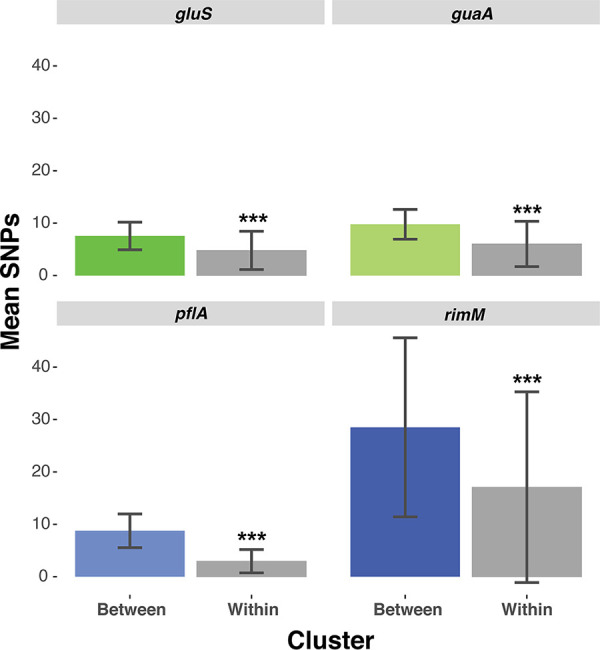
Barplots comparing sequence similarity (mean number of SNPs) between clusters (colored boxes) versus across clusters (gray boxes). All four Wilcoxon tests were significant (***, P < 10−15).
(A) Number of strains per cluster. (B) Number of strain clusters per bee. (C) Average number of SNPs within strain clusters. (D) Number of strain clusters obtained for each marker with MCL using different inflation parameters (I = 1.2, 2.0, 4.0, and 6.0). The scores used for the clustering measure the co-occurrence of strains across bees (see Materials and Methods). Download FIG S7, PDF file, 0.5 MB (499KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The fact that bees usually contain a single cluster of strains and that these clusters are composed of closely related strains could indicate that each bee was originally colonized by a single strain which subsequently diversified in its host. However, the distribution of strains across bees rejects this model. Under a model of colonization from a single strain followed by within-host diversification, we would expect that strains evolving within each bee would acquire independent mutations at different sites. Instead, we observed that the exact same strains are often found across hives and locations (Fig. 6), indicating that they did not evolve from a single colonizing strain. This pattern indicates that the multiple closely related strains within a strain cluster are frequently acquired together. Not only do some bees from different locations harbor the same strain clusters, within those clusters, bees from different locations often share the same strains in similar relative abundance (Fig. 6). These results indicate that strain composition and relative abundance are not random because the observed patterns are conserved across bees sampled from remote geographic locations.
FIG 6.

Examples of strain compositions in bees across locations and markers. Each color represents a different strain. Several strains were not shown by colors when too many strains were present in the same graph (n > 12).
We tested whether the strains of S. alvi and Gilliamella spp. presented specific patterns of interspecies associations. We reasoned that strains from different species living in the same niche might be engaged in specific relationships (e.g., some strains might engage in mutualistic relationships, while others might be antagonists), and this would result in nonrandom associations between the strains of the two species. To test this, we analyzed the patterns of strain co-occurrence based on the four marker pairs: guaA-pflA, guaA-rimM, gluS-pflA, and gluS-rimM. As in the previous analysis, we randomly shuffled the strains across bees and estimated whether the association of strains deviated from the random expectation. We did not find any evidence of interspecies association between strains for the four pairs of markers (not significant, chi-square tests), suggesting that interspecies interactions do not result in specific strain associations in our samples.
Sequencing errors cannot account for the patterns of strain distribution.
We investigated whether PCR amplification artifacts and/or sequencing errors can account for the observed patterns of strain distribution. Indeed, PCR amplification errors can occur during the first amplification cycles and potentially lead to the inference of SNPs at high frequency in the samples (25). Although this process likely leads to an overestimation of inferred strains (when reaching ≥1% frequency), it is unlikely to affect the patterns of strain distribution, i.e., it is unlikely to lead to the exact same sequence variant in different bees. To test this, we simulated the extreme scenario, where the total diversity of strain sequences observed in our data set was introduced exclusively by PCR artifacts. We simulated sequences in silico for each of the four markers based on the number of strains detected in each bee. Each strain sequence was generated from the reference sequence by introducing an average number of SNPs based on the average number of SNPs observed between the strains found in one bee following a Poisson process (∼5,000 distinct sequences across markers and samples). Following this procedure, we did not observe a single case of sequence convergence across all samples. These results indicate that, although we might slightly overestimate the number of strains due to PCR errors, this is highly unlikely to account for the patterns of strain distribution across our samples.
Convergent evolution cannot account for the patterns of strain distribution.
We tested whether selective constraints could account for the patterns of strain distribution observed in this study. Indeed, purifying selection imposes strong constraints on sequence evolution, and one could argue that this could lead to similar sequences by convergent substitutions when sequences are short and many variants are analyzed. We used the relative rates of sequence evolution across codon positions of the four markers to simulate the evolution of sequences, starting from the reference sequence of each marker, while introducing independent mutations until we obtained the total strain diversity captured in our study. The relative rates of codon evolution were determined by the relative allelic diversity across codon positions observed in each of our four markers. As expected for protein-coding genes, we observed that the third codon position was the least constrained by selection; 71 to 82% of the SNPs were observed at this position across markers. The second codon position was the most constrained; 4 to 10% of the SNPs were observed at this position across markers. Finally, the first codon position was found to present an intermediate level of diversity; 12 to 20% of the SNPs were observed at this position across markers. By simulating sequences with these imposed rates of substitution across codon positions, we did not observe a single case of sequence convergence across the simulated sequences, indicating that purifying selection is unlikely to lead to convergent sequence evolution in our markers.
Cryptic strains do not explain the patterns of strain composition.
The fact that bees from different hives and states present similar strain profiles, whereas many bees from the same hive have completely different strain compositions, suggests that there are complex strain dynamics in the honey bee microbiota. Several hypotheses could explain these patterns (see Discussion). The current data set does not allow us to test all hypotheses; however, we can test the hypothesis that cryptic strain diversity remains present in all bees. One possible explanation of the observed patterns is that each bee could potentially contain a much higher and undetected strain diversity. Thus, our results could be simply due to variations in strain frequencies across bees instead of independent strain acquisitions. All results presented above were generated under stringent filters to avoid overestimation of strain diversity due to sequencing errors, but it is possible that many low-frequency or very low-frequency strains (i.e., cryptic strains) remained unnoticed. Due to the high sequencing depth for each marker, we were able to test this hypothesis. We specifically searched for the presence of our set of identified strains in the set of filtered-out reads in bees where these strains were not detected under our threshold of 1% frequency. For this analysis, a strain was identified even if supported by a single read. We then compared the diversity of strains that would be observed under these loose criteria compared to our original prediction (Fig. 7). As anticipated, we identified the presence of additional strains across a higher proportion of bees. On average, we observed a fivefold increase in the prevalence of each strain across bees, but as mentioned, many likely correspond to sequencing errors. Nevertheless, this procedure did not allow us to recover each strain in every sample (Fig. 7), indicating that most strains are limited to a subset of bee hosts (typically <30%). Of course, we cannot exclude the possibility that additional cryptic strains would be recovered by increasing sequencing depth, but our sequencing depth should allow us, in theory, to detect strains present at 0.00002% frequency on average. Thus, our results support the view that bees present distinct strain compositions and that the distribution of strain clusters is due to mechanisms of transmission and selection, not simply variations in strain frequencies.
FIG 7.

Prevalence of rare strains across bees with relaxed filters. The x axis represents the prevalence of each strain in our sample of bees (percentage of bees where the strain has been identified). The y axis shows the prevalence of each strain when redefining the strains with relaxed filters: i.e., the strain is considered present even when found <1% frequency in the bee (see Materials and Methods).
DISCUSSION
We applied a targeted approach to assess the strain-level diversity of the honey bee microbiome. By deep sequencing variable regions of protein-coding genes, we were able to decipher the strain dynamics of two core members of the honey bee microbiome. Although the present study did not attempt to evaluate the diversity of the entire community, we were able to gain additional insights on the strain dynamics of two key members of the honey bee gut microbiome. MAST sequencing allowed us to explore certain hypotheses more thoroughly than would have been possible using shotgun metagenomic sequencing or by sequencing individual isolates. For instance, we were able to search for strains potentially present at frequency as low as 0.00002%. While it is virtually impossible to prove that strains are completely missing from a sample, our results provide higher confidence that cryptic strains are truly absent from our samples and do not account for the observed patterns of strain dynamics.
Overall, our results on strain diversity of S. alvi and Gilliamella spp. are consistent with previous studies (4, 12). We observed that strain composition within the bee microbiome is not random. Our strain cluster analysis revealed that the same groups of strains frequently co-occur together and that, most of the time, a bee contains only closely related strains. As reported in reference 12, different Gilliamella spp. (based on the rimM marker) do co-occur in a single bee, but generally only closely related strains within each “species” co-occur. These patterns might be attributed to strain-strain incompatibilities due to resource competition or strain warfare. Alternatively, specific sets of strains could be filtered or selected by the host environment (i.e., the immune system) as previously proposed (26). Interestingly, co-occurring strains typically differed by only a single nucleotide in the marker region. At first, these patterns appeared compatible with a scenario of colonization by a single strain, followed by within-host diversification by mutations. Such a scenario has been observed for some dominant species of the human microbiome (27, 28). However, the consistent presence of the exact same strains across different bees (including bees from different hives and states) indicates that these patterns are not due to within-host diversification. Rather, within-host diversification of a single strain would generate various strains through random mutations affecting different sequence sites (i.e., it is extremely unlikely that random mutations would produce the exact same alleles in multiple bees).
The co-occurrence of strains predominantly composed of closely related strains goes against the intuitive explanation that related strains must compete against one another for the same resources. One possible explanation of these patterns could be due to strain warfare. Bacteria frequently secrete compounds to kill other bacteria but must be immune to their own secreted chemicals such as toxin-antitoxin systems (29, 30), and some strains of S. alvi have been shown to encode type VI secretion systems (20). These mechanisms would lead to the elimination of competitors by a single strain; however, recently diverged strains would also encode the same genetic arsenal, and as a result, closely related strains would be immune to the same compounds. As strains diverge from each other over time, their warfare systems would eventually evolve and would not confer cross-immunity. A similar mechanism could be proposed through bacteriophage predation since closely related strains would present similar sensitivities and immunities to bacteriophages (31, 32). Note that in the case of temperate bacteriophages, lysogenic bacteria could encode a prophage themselves, and the occasional reactivation of this prophage could eliminate the nonlysogenic strains, while protecting the prophage-encoding strains (33) (prophages must confer superinfection immunity to be viable [34, 35]). Alternatively, the consistent association of closely related strains could result from the establishment of codependence between strains (i.e., processes similar to the Black Queen hypothesis but limited to closely related strains) (36, 37). For instance, bacteria require the secretion of multiple proteins in the environment to establish biofilms or other structures (38–40). Assuming a simple system with two secreted proteins A and B, the loss of gene A in one clone and the loss of gene B in another clone would establish a situation of codependence between these two newly formed strains. Over time, this situation could potentially lead to the loss by drift of the original strain carrying both gene copies A and B. This scenario of balancing selection could potentially explain why closely related strains are maintained together. This could also explain why the relative frequencies of these strains also appear conserved across sampled bees, since secreted proteins would need to be secreted at similar concentrations to interact with one another efficiently.
Finally, our results showed that bees from the same hive often present dissimilar strain compositions, whereas many bees from different hives and states present similar strain compositions. This observation further suggests that strain composition of the bee gut microbiome is not shaped by neutral processes. We showed that these patterns do not seem compatible with the presence of a higher undetected strain diversity within each bee microbiome (the presence of cryptic strains). However, we cannot completely exclude the possibility that this result could be due to bee migration between hives located in different states or founding queens coming from the same breeders. Commercial hives are often moved across the United States for seasonal field fertilization, and this process could lead to frequent migration of bees across states. Moreover, many bee colonies are founded by sister queens reared by the same operation and shipped with workers. Although we know that the Florida bees were from a commercial colony that was frequently treated with antibiotics and other chemicals, the Tennessee bees were from an organic colony that had not been treated with any chemicals for more than 20 years, the Texas bees were taken from a research colony that had not been treated with any chemicals for more than 2 years, and the Utah bees were sampled from a feral colony that had not been managed for at least 10 years, we lack precise data regarding how these hives were initially founded. Nevertheless, colony migration and the initial founding queens are not likely to account for the observed strain dynamics, since previous studies have demonstrated that the same strains are found in bees from different countries (4, 12). Additionally, age and lifestyle (nurse versus forager) does not seem to be a factor in determining strain composition (12). Thus, previous studies as well as our results support the hypothesis that different strain profiles might be selected by different bee genotypes. A honey bee queen can mate with up to 59 different drones, and on average mates with about 14 drones (41), creating a colony that consists of a wide range of different genotypes. If strain composition is associated with particular genotypes, this could explain the variation within a single hive (26). Finally, these patterns of strain composition could also be driven by colonization order and strain warfare. This scenario implies that the first colonizing strain—which might be random—would prevent the future colonization of certain strains but allow other strains to colonize (42). Because closely related strains are often found associated together, this process would not be driven by niche competition but by direct strain antagonism mediated by warfare systems such as toxin-antitoxin systems, type VI secretion system effectors, and prophages.
One major limitation of the MAST method is the need for sequenced genomes representative of the diversity of each species to develop accurate markers. As previously noted, the markers used in this study were designed using a limited set of genomes (6), which resulted in one of our markers (rimM) capturing all honey bee-associated Gilliamella species while the other (pflA) captured the diversity of only G. apicola. Both markers provided useful information, but it is obvious from our results that the marker that captures all Gilliamella species comes at the cost of reduced strain resolution, e.g., the pflA marker recovered more than 900 strains, while rimM recovered only ∼200 strains. Therefore, MAST is a powerful method for analyzing strain diversity, but genome availability and marker design are critical factors in the accuracy of strain detection. Another caveat of using the MAST method is that identical protein-coding amplicon sequences do not necessarily imply that the entire genomes are 100% identical. Therefore, MAST cannot technically determine whether strains are identical across samples. However, the MAST method is a good proxy for strain similarity and composition and as more strains are sequenced, we will be able to determine whether MAST-inferred strains are truly identical and if not, how much they differ in gene content and average nucleotide identity (ANI).
Conclusions.
Overall, our results revealed that strain composition within the honey bee gut is complex despite the overall simplicity of the microbiome. Using the MAST approach, we were able to characterize the population dynamics of two host-associated microbes at a level not feasible with other methods. Future studies using controlled conditions (e.g., genotype, age, life history), will help reveal the factors that shape strain-level dynamics in the honey bee gut microbiome and provide answers to fundamental questions about the population genetics of host-associated microbiomes.
MATERIALS AND METHODS
Bee sampling and processing.
Bees were sampled from a single colony from each of the following locations: Texas, Tennessee, Florida, and Utah (see Table S1 in the supplemental material) and stored in 100% ethanol at 4°C. Bees from Texas were used in our previous study (6) and were sampled directly from the University of Texas (UT) Austin campus. Bees from Utah, Tennessee, and Florida were obtained from collaborators. Therefore, the sample size of bees from Texas was much larger than that of the other locations. The guts of all bees were dissected using sterile forceps and then homogenized. DNA was extracted using a cetyltrimethylammonium bromide (CTAB) bead-beating protocol described in reference 43. PCR was performed on the extracted DNA using the guaA, gluS, pflA, and rimM primers designed in reference 6 attached to Illumina adaptors. Triplicate 25-μl reactions were carried out with 0.25 μl Phusion high-fidelity DNA polymerase (New England Biolabs), 5 μl of Phusion HF buffer (New England Biolabs), 1.25 μl of dimethyl sulfoxide (DMSO), 1 μl (each) 10 μM primer, 14.5 μl H2O, and 1 μl of template DNA in buffer. The cycling conditions consisted of 98°C for 30 s, 25 cycles with 1 cycle consisting of 98°C for 10 s, 59°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 7 min. The amplicons were pooled and cleaned using AMPure XP beads (Beckman Coulter). The Genome Sequencing and Analysis Center at the University of Texas at Austin performed the barcoding and sequencing using Illumina MiSeq 2X300.
Strain inference and identification of strain clusters.
Paired-end reads were merged together using FLASH v1.2.11 (44) with default parameters. Reads with low quality were discarded (average Phred score of <30), and the remaining reads were aligned against the reference sequence (G. apicola wkB1 or S. alvi wkB2) of each marker with BOWTIE2 v2.2.8 (45). Reads that aligned with gaps were excluded. Single nucleotide polymorphisms (SNPs) were identified, and each sequence was considered a potential strain. For each marker, a strain was inferred if present at >1% frequency in at least one sample (this threshold of 1% corresponds to an average of 460 reads per strain per sample). The total number of strains identified for each marker are provided in Table S2A. The number of strains identified in each sample are shown in Table S2B and the detailed description of all the strains is provided in Data Set S1 in the supplemental material.
(A) Number of strains detected for each marker. (B) Number of strains per sample (bee) for each marker. NA represents samples that reads were not obtained due to issues with DNA extraction or sequencing. Download Table S2, XLSX file, 0.01 MB (14.7KB, xlsx) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Individual strain frequencies for all samples and makers. Download Data Set S1, XLSX file, 0.4 MB (424.3KB, xlsx) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strain clusters were defined based on the frequency of strain co-occurrence within the same bee. For each pair of strains defined across the four markers, we defined a score S of co-occurrence as the number of bees containing both strains divided by the average number of bees containing either strain: , with bAB the number of bees containing both strains A and B, bA the number of bees containing strain A, and bB the number of bees containing strain B. These scores were used to build the heatmaps and the clusters with MCL v14-137 (46) with different inflation parameters: I = 1.2 (minimum), 2.0, 4.0, and 6.0. With I = 1.2, most strains were assembled into very few clusters (<20) composed of >30 strains per cluster. The three other inflation parameters yielded rather consistent clustering patterns—the number of clusters varied from 40 to 106 clusters across markers guaA, gluS, pflA, and rimM and inflation parameters (Fig. S7D). The different clustering parameters yielded similar results. We conducted the analysis with inflation parameter I = 2.0, since it yielded consistent numbers of clusters across gene markers (42 to 64 clusters for guaA, gluS, and pflA).
Sequence analysis.
Unweighted UniFrac analysis was conducted with QIIME 1.9.1 (47) using the four gene markers gluS, guaA, pflA, and rimM. One sequence representative of each strain was selected to construct the phylogenetic tree of each marker. Each phylogenetic tree was then constructed using BIONJ (48) with the Jukes-Cantor model implemented in Seaview v4 (49). Analysis of reference genomes was conducted by downloading all reference genomes of Gilliamella available in GenBank (June 2019). The core genome was identified as follows. Briefly, orthologous genes were identified by pairwise genome comparison with Usearch Global (50). Two genes were considered orthologs when they showed ≥70% protein identity and ≥80% length conservation. Genes were then grouped into gene families by transitivity (i.e., two orthologous genes necessarily belong to the same family). Gene families with paralogs were excluded from the core genome. Additionally, gene families containing “double outliers” were excluded from the core genome. Double outliers were defined as sequences that present an abnormally low or high sequence identity score relative (i) to the other sequences of the gene family and (ii) to the average identity score of the other core genes of the corresponding genome. In both cases, outliers were defined as gene families containing at least one sequence with an identity score lower or higher than 1.5 × IQR (interquartile range) compared to (i) the other sequences of the gene family and (ii) to the average sequence identity of the other core genes of the corresponding genome. The scripts used to build the core genomes have been assembled into a computer package freely available on GitHub: https://github.com/lbobay/CoreCruncher. The sequences of each gene family were then aligned with MAFFT v7 (51) and back translated in silico into the nucleotide sequence of each corresponding genome. The different gene alignments were then merged into a single core genome concatenate.
Phylogenies.
We used the entire set of genomes of Snodgrassella alvi and Gilliamella spp. from Apis mellifera to extract the four gene sequences guaA, gluS, pflA, and rimM from each genome, The sequences of each gene were aligned with MAFFT v7 (51), and the region of each marker was extracted. A representative sequence of each strain identified above was added to each marker alignment. Alignments were manually inspected, and no misaligned sequences were observed. For each marker alignment, phylogenetic trees were built with RAxML v8 (52) with a GAMMA + GTR model and 100 fast bootstrap replicates. The core genome phylogeny was run on the concatenate of the core genome using the same program and the same parameters.
Simulations.
Simulations were initiated with the reference sequence of each marker. We simulated 144 sets of sequences to mimic the number of bees in our data set. For each bee, we simulated a number of sequences that closely matches the average number of distinct strains observed per bee (6 strains per bee for guaA, gluS, and rimM; 16 strains per bee for pflA). Each strain was generated following a Poisson process by introducing a number of SNPs that matches the average number of SNPs observed across strains found in the same bee (Fig. 1). Simulations were conducted with CoreSimul (53) either by introducing SNPs randomly along each sequence to test the impact of PCR artifacts or by introducing SNPs with different probabilities across codon positions to test the effect of purifying sequence on strain inference. The relative probability of substitutions across codon positions was defined empirically based on the distribution of observed SNPs across the distinct strains inferred by each marker: 0.12, 0.10, and 0.78 for guaA, 0.17, 0.04, and 0.79 for gluS, 0.14, 0.04, and 0.82 for pflA, and 0.20, 0.09, and 0.71 for rimM at the first, second, and third codon positions, respectively. The scripts used for the simulations are available at https://github.com/lbobay/CoreSimul.
Data availability.
All raw sequencing reads are deposited in NCBI Sequence Read Bioproject under accession numbers PRJNA562505 and PRJNA415093. All other data generated are included in the supplemental material files.
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation under grant DEB-1930776 awarded to L.-M.B., by the National Institute of General Medical Sciences of the National Institutes of Health under award R01GM132137 awarded to L.-M.B., and by the National Science Foundation under grant DEB-1930776 awarded to K.R.
We thank Nancy Moran and all members of the Moran Lab at UT Austin for constructive suggestions and other various forms of support for this research. We also thank Kim Hammond for assistance with beekeeping, Carrie Stott for performing some of the phylogenetic analyses, and the UT Genome Sequencing and Analysis Facility for sequencing services.
REFERENCES
- 1.Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. 2017. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res 27:626–638. doi: 10.1101/gr.216242.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, Kota K, Sunyaev SR, Weinstock GM, Bork P. 2013. Genomic variation landscape of the human gut microbiome. Nature 493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caro-Quintero A, Ochman H. 2015. Assessing the unseen bacterial diversity in microbial communities. Genome Biol Evol 7:3416–3425. doi: 10.1093/gbe/evv234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powell JE, Ratnayeke N, Moran NA. 2016. Strain diversity and host specificity in a specialized gut symbiont of honey bees and bumble bees. Mol Ecol 25:4461–4471. doi: 10.1111/mec.13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, Pusey AE, Peeters M, Hahn BH, Ochman H. 2016. Cospeciation of gut microbiota with hominids. Science 353:380–382. doi: 10.1126/science.aaf3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raymann K, Bobay L-M, Moran NA. 2018. Antibiotics reduce genetic diversity of core species in the honeybee gut microbiome. Mol Ecol 27:2057–2066. doi: 10.1111/mec.14434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bobay L-M, Raymann K. 2019. Population genetics of host-associated microbiomes. Curr Mol Biol Rep 5:128–139. doi: 10.1007/s40610-019-00122-y. [DOI] [Google Scholar]
- 8.Kwong WK, Moran NA. 2015. Evolution of host specialization in gut microbes: the bee gut as a model. Gut Microbes 6:214–220. doi: 10.1080/19490976.2015.1047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng H, Steele MI, Leonard SP, Motta EVS, Moran NA. 2018. Honey bees as models for gut microbiota research. Lab Anim (NY) 47:317–325. doi: 10.1038/s41684-018-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwong WK, Moran NA. 2016. Gut microbial communities of social bees. Nat Rev Microbiol 14:374–384. doi: 10.1038/nrmicro.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel P, Stepanauskas R, Moran NA. 2014. Hidden diversity in honey bee gut symbionts detected by single-cell genomics. PLoS Genet 10:e1004596. doi: 10.1371/journal.pgen.1004596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellegaard KM, Engel P. 2019. Genomic diversity landscape of the honey bee gut microbiota. Nat Commun 10:446. doi: 10.1038/s41467-019-08303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellegaard KM, Engel P. 2016. Beyond 16S rRNA community profiling: intra-species diversity in the gut microbiota. Front Microbiol 7:1475. doi: 10.3389/fmicb.2016.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwong WK, Engel P, Koch H, Moran NA. 2014. Genomics and host specialization of honey bee and bumble bee gut symbionts. Proc Natl Acad Sci U S A 111:11509–11514. doi: 10.1073/pnas.1405838111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engel P, Martinson VG, Moran NA. 2012. Functional diversity within the simple gut microbiota of the honey bee. Proc Natl Acad Sci U S A 109:11002–11007. doi: 10.1073/pnas.1202970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng H, Nishida A, Kwong WK, Koch H, Engel P, Steele MI, Moran NA. 2016. Metabolism of toxic sugars by strains of the bee gut symbiont Gilliamella apicola. mBio 7:e01326-16. doi: 10.1128/mBio.01326-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwong WK, Moran NA. 2013. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order 'Enterobacteriales' of the Gammaproteobacteria. Int J Syst Evol Microbiol 63:2008–2018. doi: 10.1099/ijs.0.044875-0. [DOI] [PubMed] [Google Scholar]
- 18.Kwong WK, Mancenido AL, Moran NA. 2014. Genome sequences of Lactobacillus sp. strains wkB8 and wkB10, members of the Firm-5 clade, from honey bee guts. Genome Announc 2:e01176-14. doi: 10.1128/genomeA.01176-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engel P, Kwong WK, Moran NA. 2013. Frischella perrara gen. nov., sp. nov., a gammaproteobacterium isolated from the gut of the honeybee, Apis mellifera. Int J Syst Evol Microbiol 63:3646–3651. doi: 10.1099/ijs.0.049569-0. [DOI] [PubMed] [Google Scholar]
- 20.Steele MI, Kwong WK, Whiteley M, Moran NA. 2017. Diversification of type VI secretion system toxins reveals ancient antagonism among bee gut microbes. mBio 8:e01630-17. doi: 10.1128/mBio.01630-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonilla-Rosso G, Engel P. 2018. Functional roles and metabolic niches in the honey bee gut microbiota. Curr Opin Microbiol 43:69–76. doi: 10.1016/j.mib.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Kešnerová L, Mars RAT, Ellegaard KM, Troilo M, Sauer U, Engel P. 2017. Disentangling metabolic functions of bacteria in the honey bee gut. PLoS Biol 15:e2003467. doi: 10.1371/journal.pbio.2003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludvigsen J, Porcellato D, Amdam GV, Rudi K. 2018. Addressing the diversity of the honeybee gut symbiont Gilliamella: description of Gilliamella apis sp. nov., isolated from the gut of honeybees (Apis mellifera). Int J Syst Evol Microbiol 68:1762–1770. doi: 10.1099/ijsem.0.002749. [DOI] [PubMed] [Google Scholar]
- 24.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kebschull JM, Zador AM. 2015. Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res 43:e143. doi: 10.1093/nar/gkv717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. 2014. Human genetics shape the gut microbiome. Cell 159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garud NR, Good BH, Hallatschek O, Pollard KS. 2019. Evolutionary dynamics of bacteria in the gut microbiome within and across hosts. PLoS Biol 17:e3000102. doi: 10.1371/journal.pbio.3000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao S, Lieberman TD, Poyet M, Kauffman KM, Gibbons SM, Groussin M, Xavier RJ, Alm EJ. 2019. Adaptive evolution within gut microbiomes of healthy people. Cell Host Microbe 25:656–667.e8. doi: 10.1016/j.chom.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goeders N, Van Melderen L. 2014. Toxin-antitoxin systems as multilevel interaction systems. Toxins (Basel) 6:304–324. doi: 10.3390/toxins6010304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Melderen L, Saavedra De Bast M. 2009. Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet 5:e1000437. doi: 10.1371/journal.pgen.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez-Valera F, Martin-Cuadrado A-B, Rodriguez-Brito B, Pasić L, Thingstad TF, Rohwer F, Mira A. 2009. Explaining microbial population genomics through phage predation. Nat Rev Microbiol 7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 32.Thingstad T, Lignell R. 1997. Theoretical models for the control of bacterial growth rate, abundance, diversity and carbon demand. Aquat Microb Ecol 13:19–27. doi: 10.3354/ame013019. [DOI] [Google Scholar]
- 33.Brown SP, Le Chat L, De Paepe M, Taddei F. 2006. Ecology of microbial invasions: amplification allows virus carriers to invade more rapidly when rare. Curr Biol 16:2048–2052. doi: 10.1016/j.cub.2006.08.089. [DOI] [PubMed] [Google Scholar]
- 34.Susskind MM, Botstein D. 1978. Repression and immunity in Salmonella phages P22 and L: phage L lacks a functional secondary immunity system. Virology 89:618–622. doi: 10.1016/0042-6822(78)90204-0. [DOI] [PubMed] [Google Scholar]
- 35.Susskind MM, Botstein D. 1980. Superinfection exclusion by lambda prophage in lysogens of Salmonella typhimurium. Virology 100:212–216. doi: 10.1016/0042-6822(80)90571-1. [DOI] [PubMed] [Google Scholar]
- 36.Morris JJ, Lenski RE, Zinser ER. 2012. The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. mBio 3:e00036-12. doi: 10.1128/mBio.00036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris JJ, Papoulis SE, Lenski RE. 2014. Coexistence of evolving bacteria stabilized by a shared Black Queen function. Evolution 68:2960–2971. doi: 10.1111/evo.12485. [DOI] [PubMed] [Google Scholar]
- 38.Crespi BJ. 2001. The evolution of social behavior in microorganisms. Trends Ecol Evol 16:178–183. doi: 10.1016/s0169-5347(01)02115-2. [DOI] [PubMed] [Google Scholar]
- 39.Nogueira T, Rankin DJ, Touchon M, Taddei F, Brown SP, Rocha EPC. 2009. Horizontal gene transfer of the secretome drives the evolution of bacterial cooperation and virulence. Curr Biol 19:1683–1691. doi: 10.1016/j.cub.2009.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nogueira T, Touchon M, Rocha EPC. 2012. Rapid evolution of the sequences and gene repertoires of secreted proteins in bacteria. PLoS One 7:e49403. doi: 10.1371/journal.pone.0049403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarpy DR, Delaney DA, Seeley TD. 2015. Mating frequencies of honey bee queens (Apis mellifera L.) in a population of feral colonies in the Northeastern United States. PLoS One 10:e0118734. doi: 10.1371/journal.pone.0118734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wexler AG, Goodman AL. 2017. An insider’s perspective: Bacteroides as a window into the microbiome. Nat Microbiol 2:17026. doi: 10.1038/nmicrobiol.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raymann K, Shaffer Z, Moran NA. 2017. Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biol 15:e2001861. doi: 10.1371/journal.pbio.2001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gascuel O. 1997. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol 14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 49.Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 50.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 51.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bobay L-M. 2020. CoreSimul: a forward-in-time simulator of genome evolution for prokaryotes modeling homologous recombination. BMC Bioinformatics 21:264. doi: 10.1186/s12859-020-03619-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Average read coverage for each marker by sampling location. Download FIG S1, PDF file, 0.2 MB (172.3KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Total number of reads obtained for each sample for each marker. NA represents samples that reads were not obtained due to issues with DNA extraction or sequencing. Download Table S1, XLSX file, 0.01 MB (15.1KB, xlsx) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic trees of A. mellfiera-associated S. alvi and Gilliamella strains available on NCBI. (A) Core genome reference phylogeny of S. alvi, (B) MAST gluS marker phylogeny, and (C) MAST guaA marker phylogeny. (D) Core genome reference phylogeny, (E) MAST rimM marker phylogeny, and (F) MAST pflA marker phylogeny. Blue shading is used to delineate the different Gilliamella species or phylotypes. Phylogenetic trees were constructed with RAxML v8 with a GAMMA + GTR model. Download FIG S2, PDF file, 0.3 MB (436.5KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic tree of A. mellifera-associated S. alvi strains based on gluS and guaA MAST markers. The phylogeny was made using the amplified regions of each gene marker from the strains sequenced in this study (green font) as well as the sequenced genomes available on NCBI as of July 2020 (black font and highlighted in green). The trees were constructed with RAxML v8 with a GAMMA + GTR model. Download FIG S3, PDF file, 0.4 MB (338.3KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic tree of A. mellifera-associated Gilliamella strains based on pflA and rimM MAST markers. The phylogeny was made using the amplified regions of each gene marker from the strains sequenced in this study (blue font) as well as the sequenced genomes available on NCBI as of July 2020 (black font and highlighted in blue). The trees were constructed with RAxML v8 with a GAMMA + GTR model. Download FIG S4, PDF file, 0.4 MB (450.9KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Average number of SNPs across strains estimated with the four gene markers. The markers of S. alvi and G. apicola are represented in green and blue, respectively. (B) Resampling analysis of strain composition. For each marker, the average number of strains was estimated for different numbers of randomly sampled bees (nonredundant). The y axis shows the average number of strains found in 100 combinations of randomly sampled bees. The x axis indicates the number of nonredundant resampled bees included in each combination. Download FIG S5, PDF file, 0.2 MB (253.5KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Heatmaps of strain co-occurrence for each marker. Each line and column represent a strain. A score of 1.0 represents 100% co-occurrence (the strains are always found together), and 0.0 represents no co-occurrence (the strains are never found together). Download FIG S6, JPG file, 0.3 MB (355KB, jpg) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Number of strains per cluster. (B) Number of strain clusters per bee. (C) Average number of SNPs within strain clusters. (D) Number of strain clusters obtained for each marker with MCL using different inflation parameters (I = 1.2, 2.0, 4.0, and 6.0). The scores used for the clustering measure the co-occurrence of strains across bees (see Materials and Methods). Download FIG S7, PDF file, 0.5 MB (499KB, pdf) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Number of strains detected for each marker. (B) Number of strains per sample (bee) for each marker. NA represents samples that reads were not obtained due to issues with DNA extraction or sequencing. Download Table S2, XLSX file, 0.01 MB (14.7KB, xlsx) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Individual strain frequencies for all samples and makers. Download Data Set S1, XLSX file, 0.4 MB (424.3KB, xlsx) .
Copyright © 2020 Bobay et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
All raw sequencing reads are deposited in NCBI Sequence Read Bioproject under accession numbers PRJNA562505 and PRJNA415093. All other data generated are included in the supplemental material files.