

DNA Pol α transfers parental histones to lagging strands.
Abstract
How parental histones, the carriers of epigenetic modifications, are deposited onto replicating DNA remains poorly understood. Here, we describe the eSPAN method (enrichment and sequencing of protein-associated nascent DNA) in mouse embryonic stem (ES) cells and use it to detect histone deposition onto replicating DNA strands with a relatively small number of cells. We show that DNA polymerase α (Pol α), which synthesizes short primers for DNA synthesis, binds histone H3-H4 preferentially. A Pol α mutant defective in histone binding in vitro impairs the transfer of parental H3-H4 to lagging strands in both yeast and mouse ES cells. Last, dysregulation of both coding genes and noncoding endogenous retroviruses is detected in mutant ES cells defective in parental histone transfer. Together, we report an efficient eSPAN method for analysis of DNA replication–linked processes in mouse ES cells and reveal the mechanism of Pol α in parental histone transfer.
INTRODUCTION
Faithful duplication of both genetic and epigenetic information is fundamental to the reproduction and evolution of all living organisms. Nucleosome, the basic unit of eukaryotic chromatin, is composed of an octamer of histones wrapped around with ~147–base pair (bp) DNA. Posttranslational modifications (PTMs) of histones play important roles in many cellular processes including gene transcription and DNA replication (1). Several histone PTMs, including trimethylation of H3 lysine 9 (H3K9me3) and lysine 27 (H3K27me3), have been shown to be transmitted to daughter cells during mitosis, even through meiosis, to maintain gene expression states (2–5). Moreover, nucleosome positions of parental histones are preserved following DNA replication in both yeast and likely in mouse embryonic stem (ES) cells (6, 7). However, how epigenetic information encoded by histone modifications is inherited during mitotic cell divisions is largely unknown.
Eukaryotic DNA replication initiates at specific genomic sites during S phase in a highly orchestrated process (8, 9). In budding yeast, DNA replication initiates at well-defined adenine- and thymine-rich (AT-rich) autonomously replicating sequences. In contrast, replication origins in mammalian cells lack consensus sequence and site-specific information (10). Instead, most of DNA replication origins in higher eukaryotic cells initiate as broad initiation zones (11–13). During G1 phase, these origins are recognized by the origin recognition complex, which loads two heterohexamers of minichromosome maintenance (MCM) proteins (MCM2-7) to form the pre-replication complex. The MCM2-7 hexamers are subsequently activated in early S phase and form two active replicative helicases consisting of Cdc45-MCM-GINS (CMG) at two replication forks, which travel bidirectionally on leading strand templates and unwind the double-stranded template DNA. DNA polymerase α (Pol α) then generates a primer to initiate DNA synthesis at both strands, with DNA Pol ε synthesizing most of the leading strands and Pol δ replicating the lagging strands or Okazaki fragments (14).
During DNA replication, nucleosomes ahead each fork are rapidly disassembled to allow for efficient DNA synthesis. Immediately following replication, replicated DNA is assembled into nucleosomes by two pathways, de novo deposition of newly synthesized H3-H4 and the transfer of parental histone H3-H4 tetramers. In general, nucleosome assembly is a step-wise process with the deposition of H3-H4 tetramers first and followed by rapid deposition of H2A-H2B. It is known that nascent H3-H4 is deposited with the help of various histone chaperones (15, 16). For example, in yeast, nascent histone H3-H4 dimer is escorted by Asf1 for acetylation of lysine 56 (H3K56ac) by Rtt109 (17, 18). Asf1 then hands over H3-H4 dimers to downstream chaperones including the CAF-1 (chromatin assembly factor 1) complex, which deposit nascent (H3-H4)2 tetramers for nucleosome formation (15). The transfer of parental histones, which guides the re-establishment of epigenetic landscape (19), however, is much less understood. Recently, several replisome components are shown to harbor histone chaperone activities and facilitate the transfer of parental H3-H4 onto replicating DNA strands. For instance, using the eSPAN (enrichment and sequencing of protein-associated nascent DNA) method in budding yeast, Dpb3 and Dpb4, two subunits of Pol ε, have been shown to interact with H3-H4 and promote parental histone transfer to leading strand (20). Moreover, human POLE3 and POLE4, the functional homologs of Dpb3 and Dpb4, display histone chaperone activity in vitro (21). MCM2, a subunit of the MCM helicase, contains a conserved histone-binding motif (HBM) at its N terminus (22–24). Mcm2 mutations defective in histone binding show defects in the transfer of parental histones to lagging strands in budding yeast (25). Using SCAR-seq (sister chromatids after replication by DNA sequencing), a method that uses the same principle as eSPAN, it has been shown that MCM2 mutant defective in histone binding in mouse ES cells also show defects in the transfer of modified parental histones (26), indicating a conserved role of MCM2 in parental histone transfer.
The active CMG complex travels along the leading strand template with the N termini of MCM subunits facing the parental nucleosomes (27–29). In addition, the CMG helicase is connected to lagging strands through adaptor protein Ctf4, which forms a homotrimeric complex and interacts with GINS and Pol1, the catalytic subunit of DNA primase Pol α (30, 31). We show that Ctf4 mutants that cannot bind Pol1 or Pol1 mutant that cannot bind Ctf4 compromise the transfer of parental H3-H4 in a manner similar to the Mcm2 mutant defective in binding to H3-H4 (25). However, it is still not known how Pol1 modulates parental histone transfer. Recent studies indicate that both yeast Pol1 and mammalian POLA1 contain a conserved HBM that binds to H2A-H2B (32), raising a possibility that this potential histone-binding activity of Pol α facilitates the transfer of parental H3-H4.
Here, we reported an updated version of the eSPAN method by using modified CUT&Tag/ACT-seq, a recently developed technology to digest and tag genomic DNA bound by proteins of interest using protein A–fused transposase Tn5 (pA-Tn5) (33, 34), followed by enrichment of nascent DNA using bromodeoxyuridine (BrdU) immunoprecipitation (IP). Using this new eSPAN method, we can profile histone distributions at leading or lagging strands with much fewer starting cells. Moreover, we show that POLE3 and POLE4 facilitate the transfer of parental H3-H4 to leading strands in mouse ES cells. Furthermore, POLA1 interacts with H3-H4 preferentially over H2A-H2B, and the Pol α–H3-H4 interaction is important for the transfer of parental H3-H4 onto replicating lagging strands. Last, MCM2 and Pol α mutants defective in parental histone transfer show defects in the silencing of endogenous retroviral elements (ERVs) in mouse ES cells, which may contribute to increased expression of some nearby genes. Together, these studies describe an easy and efficient eSPAN method, which is powerful to study DNA replication–coupled processes that have the intrinsic difference between leading and lagging strands. Moreover, we have revealed the mechanism whereby Pol α mediates the parental histone transfer.
RESULTS
Identification of efficient origins in mouse ES cells using OK-seq
Replication origins/initiation zones are needed for analysis of the eSPAN datasets. Therefore, we profiled replication origins in mouse ES cells using Okazaki fragment sequencing (OK-seq) (13). We identified 2591 and 2288 initiation zones in two independent replicates, respectively, with 1835 of them overlapped (fig. S1A). In addition, the OK-seq bias profiles had a good correlation with published datasets both genome-wide and around origins (fig. S1B) (26), indicating the reproducibility and reliability of our identified origins. For the downstream analysis, we selected 1548 origins whose genomic localization is less than 3 kb apart in the two repeats. These origins showed a strong lagging strand bias in both repeats and were ranked on the basis of replication efficiency (fig. S1, C and D).
A modified eSPAN to monitor histone distributions at DNA strands in mouse ES cells
We have previously reported the eSPAN technique for measuring the relative amount of a protein at leading and lagging strands of DNA replication forks in budding yeast (35). The first step of eSPAN involves chromatin immunoprecipitation (ChIP), which requires a large number of cells that may be challenging in mammalian systems. Recently, other laboratories have shown that the CUT&Tag/ACT-seq can generate the same information as ChIP-seq but with a much smaller number of cells (33, 34). Therefore, we replaced the ChIP step of the eSPAN procedure with a modified CUT&Tag/ACT-seq to preserve strand-specific information. Briefly, pA-Tn5 loaded with one oligonucleotide adaptor was targeted to specific chromatin regions by antibodies against a protein of interest. After addition of Mg2+, pA-Tn5 digested and tagmented chromatin locally (Fig. 1A). A second adaptor is then ligated to the 3′ end of the purified DNA fragments so that the strand-specific information is preserved. A small fraction of the DNA was processed directly for library preparation (CUT&Tag/ACT-seq) (top, Fig. 1A). Most of the tagmented DNA was denatured and immunoprecipitated using antibodies against BrdU, a nucleotide analog incorporated into newly synthesized DNA, and subsequent library preparation and sequencing. In this way, we could generate both CUT&Tag/ACT-seq and eSPAN libraries with 1 million mouse ES cells for most histone marks and as low as 50 thousand cells for H4K20me2 (see below).
Fig. 1. Development of the eSPAN method to analyze proteins at replication forks in mammalian cells.
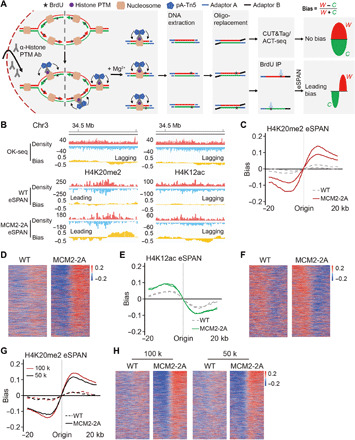
(A) A graphic diagram of the eSPAN workflow. Ab, antibody. (B) Snapshots of OK-seq reads density and bias and H4K20me2 and H4K12ac eSPAN reads density and bias in WT and MCM2-2A mouse ES cells at selected initiation zone regions on chromosome 3. Red and blue tracks indicate sequencing reads of Watson (W) and Crick (C) strands, respectively. Bias is calculated using the formula (W − C)/(W + C). (C and E) Average bias of H4K20me2 (C) and H4K12ac (E) eSPAN (n = 1548) in WT and MCM2-2A mouse ES cells with two repeats is shown for each genotype. (D and F) Representative heatmap of H4K20me2 (D) and H4K12ac (F) eSPAN biases in WT and MCM2-2A mouse ES cells at each of the 1548 initiation zones ranking from the most efficient (top) to the least efficient (bottom) ones based on OK-seq bias. (G) Average bias of H4K20me2 eSPAN (n = 1548) in 100 thousand (red) and 50 thousand (black) of WT and MCM-2A mouse ES cells. (H) Representative heatmap of H4K20me2 eSPAN biases in different amounts of WT and MCM2-2A mouse ES cells at each of the 1548 initiation zones ranking from the most efficient (top) to least efficient (bottom) ones.
H4K20me2 and histone H4 lysine 12 acetylation (H4K12ac) are found on parental and newly synthesized histone H4, respectively (36, 37). Therefore, we analyzed a genome-wide distribution of H4K20me2 and H4K12ac using CUT&Tag/ACT-seq and their association with replicating strands using eSPAN. Because of lack of H4K20me2 ChIP-seq profiles in mouse ES cells, we first compared the global H4K20me2 enrichment among wild-type (WT) and various mutant cells. At a single chromosome locus, H4K20me2 CUT&Tag/ACT-seq peaks exhibited similar patterns among WT and all the mutant cells (fig. S2A). In addition, genome-wide enrichment of H4K20me2 among two replicates of WT and the mutants had strong correlations (fig. S2C). Similar results were obtained for H4K12ac (fig. S2, B and D). Thus, these results indicate that the modified CUT&Tag/ACT-seq procedures generate reproducible chromatin localization profiles of these two histone modifications, a prerequisite for the success of the eSPAN method.
Next, we analyzed the bias patterns of both H4K20me2 and H4K12ac CUT&Tag/ACT-seq and eSPAN around 1548 replication origins. We observed that H4K20me2 and H4K12ac eSPAN signals in WT cells showed a slight bias toward the leading and lagging strand, respectively, with a markedly smaller amplitude than OK-seq bias (Fig. 1B), consistent with the previous observations on the association of H4K20me2 with replicating DNA strands (26). Moreover, an MCM2 mutant defective in histone binding (MCM2-2A) exacerbated the leading strand bias of H4K20me2 and lagging strand bias of H4K12ac eSPAN (Fig. 1, B to F). Furthermore, the eSPAN bias of H4K20me2 in both WT and MCM2-2A mutant cells closely resembled that detected by SCAR-seq (fig. S3A). The biases for both parental and new histone eSPAN were more pronounced around efficient origins than those less efficient ones based on the ranked OK-seq bias profile (Fig. 1, D and F). We also calculated the average bias of CUT&Tag/ACT-seq signals around origins and observed that both H4K20me2 and H4K12ac CUT&Tag/ACT-seq displayed no bias in either WT or MCM2-2A mutant cells (fig. S3B), suggesting that the H4K20me2 and H4K12ac eSPAN biases are not from the method itself. Moreover, the average signal intensity of H4K20me2 and H4K12ac eSPAN around origins and total levels of these two histone modifications were comparable between WT and MCM2-2A cells (fig. S3, C and D), suggesting that MCM2-2A has no apparent effects on the overall histone modification levels. Together, these results indicate that the eSPAN method reported here can efficiently monitor histone segregation in mouse ES cells using a relatively small number of cells.
eSPAN in low number of mouse ES cells
To further test the sensitivity of the eSPAN procedure, we tested smaller numbers of starting mouse ES cells (100 thousand and 50 thousand) for H4K20me2 eSPAN in both WT and MCM2-2A mutant cells. H4K20me2 eSPAN peaks showed a consistent slight leading bias with different amounts of WT cells, which was greatly exacerbated in MCM2-2A mutant cells (Fig. 1, G and H). The biases generated in small number of cells were highly correlated with that of 1 million cells (fig. S3, E to G), indicating that the updated eSPAN method was reproducible and highly sensitive even with very low number of cells. Together, we have a robust eSPAN method in mouse ES cells.
POLE3 and POLE4 knockout impairs parental histone transfer
We have previously shown that Dpb3 and Dpb4, two subunits of leading strand DNA polymerase, interact with H3-H4 and promote the transfer of parental histones to leading strands in budding yeast (20). However, whether mammalian POLE3 and POLE4, the functional homologs of Dpb3 and Dpb4, respectively, also function in the parental histone transfer was not known. We therefore analyzed the impact of POLE3 and POLE4 deletion on histone segregation in mouse ES cells. Deletion of POLE3 and POLE4 in mouse ES cells by CRISPR-mediated knockout (KO) was confirmed by sequencing and immunoblotting (fig. S4, A and B). We observed that deletion of POLE3 or POLE4 had no apparent effects on cell cycle progression (fig. S4C), as well as the levels of H4K20me2 and H4K12ac (figs. S3D and S4D).
In contrast to WT or MCM2-2A mutant cells, the H4K20me2 eSPAN peaks in both POLE3 and POLE4 KO cells showed a strong lagging strand bias (Fig. 2, A to C), whereas H4K12ac eSPAN peaks showed a reduced lagging strand bias based on the inspection of H4K12ac eSPAN at an individual origin (Fig. 2A) and analysis of average bias ratio of the selected origins (Fig. 2, D and E) compared to WT cells. In addition, H4K20me2 and H4K12ac CUT&Tag/ACT-seq signals in POLE3 and POLE4 KO cells had no bias (fig. S4E). These results are consistent with the idea that the transfer of parental H3-H4 to leading strands is compromised in POLE3 and POLE4 deletion cells. Thus, mouse POLE3 and POLE4, similar to their functional homologs in yeast cells, facilitate the transfer of parental H3-H4 onto leading strands during replication.
Fig. 2. POLE3 and POLE4 promote the transfer of parental H3-H4 to leading strands in mouse ES cells.
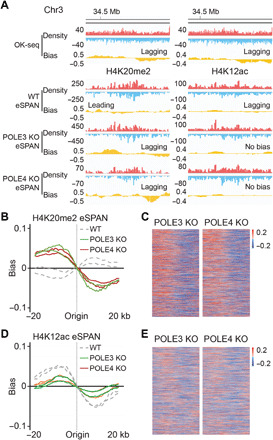
(A) Snapshots of OK-seq reads density and bias and H4K20me2 and H4K12ac eSPAN reads density and bias in WT, POLE3 KO, and POLE4 KO mouse ES cells at selected initiation zones on chromosome 3. (B and D) Average bias of H4K20me2 (B) and H4K12ac (D) eSPAN (n = 1548) in WT, POLE3 KO, and POLE4 KO mouse ES, each with two repeats. (C and E) Representative heatmap of H4K20me2 (C) and H4K12ac (E) eSPAN biases at each of 1548 initiation zones in POLE3 KO and POLE4 KO mouse ES cells. The initiation zones were ranked from the most efficient (top) to the least efficient (bottom).
Mutation of Pol α HBM impairs parental histone transfer to lagging strand
We have shown that Pol1 mutant that cannot bind Ctf4, which connects Pol1 to the CMG helicase on leading strand, is defective in the transfer of parental H3-H4 to lagging strands in budding yeast (25). However, it was not known how Pol1 is involved in this process. Paradoxically, it was also reported that both Pol1 and POLA1, the catalytic subunit of mammalian Pol α primase, interact with histones H2A-H2B (32). Therefore, we tested whether the potential histone-binding activity of POLA1 is involved in histone deposition in mouse ES cells. Briefly, we mutated two conserved amino acids to alanine (POLA1-2A) in mouse ES cells using the CRISPR-Cas9 method (fig. S5A). The POLA1-2A mutant ES cells had no morphological changes and had the same cell cycle profile as that of WT ES cells (fig. S5B). H4K20me2 and H4K12ac eSPAN peaks in POLA1-2A mutant cells showed a strong leading and lagging strand bias, respectively (Fig. 3, A to C, E, and F). The H4K20me2 and H4K12ac bias patterns in POLA1-2A cells are similar to those in MCM2-2A cells, which can also be reproduced in a low number of cells (fig. S5C). These results indicate that POLA1 may function in the transfer of H3-H4 to lagging strands through its interactions with histones.
Fig. 3. Mutating the Pol α HBM impairs parental histone transfer to lagging strand in mouse ES cells.
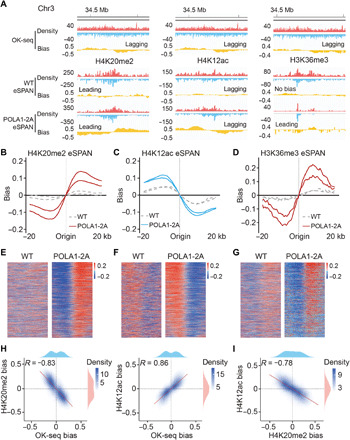
(A) Snapshots of OK-seq reads density and bias and H4K20me2, H4K12ac, and H3K36me3 eSPAN reads density and bias in WT and POLA1-2A mouse ES cells at selected origin regions on chromosome 3. (B to D) Average bias of H4K20me2 (B), H4K12ac (C), and H3K36me3 (D) eSPAN results at selected initiation zones (n = 1548) in WT and POLA1-2A mouse ES cells, each with two repeats shown. (E to G) Representative heatmap of H4K20me2 (E), H4K12ac (F), and H3K36me3 (G) eSPAN in WT and POLA1-2A mouse ES cells. (H) H4K20me2 and H4K12ac eSPAN biases in POLA1-2A mouse ES cells showed a strong anticorrelation and correlation with OK-seq bias, respectively. Spearman’s rank correlation coefficient and the density distribution were shown. (I) H4K20me2 and H4K12ac eSPAN biases in POLA1-2A mouse ES cells showed reverse correlation.
Nucleosome assembly, in general, is a step-wise process with the deposition of histone H3-H4 tetramers first followed by rapid deposition of H2A-H2B (38, 39). How does mutating a H2A-H2B binding motif of POLA1 affect the transfer of H3-H4? To answer this question, we first analyzed the distribution of H3K36me3, which also marks parental histones but occupies distinct chromatin regions than H4K20me2, using eSPAN. Analysis of H3K36me3 CUT&Tag/ACT-seq datasets in both WT and POLA1-2A mutant cells indicates that H3K36me3 CUT&Tag/ACT-seq peaks correlated strongly with published H3K36me3 ChIP-seq signals genome wide (fig. S5, D and E) (40), suggesting that our method and the H3K36me3 antibody used are reliable to detect its chromatin locations. H3K36me3 eSPAN peaks in WT cells displayed a weak bias toward the leading strand, and the bias was markedly increased in POLA1-2A mutant cells (Fig. 3, A, D, and G). Moreover, the H3K36me3 CUT&Tag/ACT-seq peaks had no bias in either WT or POLA1-2A mutant cells (fig. S5F). Last, the levels of H4K20me2, H4K12ac, and H3K36me3 around origins were not altered markedly in POLA1-2A mutant cells compared to WT (figs. S3D and S5G). Together, these results provide additional evidence that POLA1-2A mutant cells affect the transfer of parental H3-H4 to lagging strands.
To explore whether the asymmetrical histone segregation is associated with DNA replication, we calculated the correlation between histone segregation bias and OK-seq bias. As shown in Fig. 3H, H4K12ac eSPAN bias correlated positively with bias, whereas H4K20me2 and H3K36me3 eSPAN bias showed strong anticorrelation with OK-seq bias (Fig. 3H and fig. S5H). Origins with higher replication efficiencies had bigger histone biases toward both strands, suggesting that the asymmetric histone deposition is associated with DNA replication. Last, the biases of H4K12ac and H4K20me2 were strongly anticorrelated in POLA1-2A mutant cells (Fig. 3I), consistent with the idea that new H3-H4 can replace parental H3-H4 at lagging strands in the POLA1-2A cells defective in parental H3-H4 transfer to lagging strands. Together, these results show that similar to the MCM2-2A mutant defective in histone binding, mutating the HBM of POLA1 also impairs parental histone transfer to lagging strands.
Mutations of yeast Pol1 HBM also impair the transfer of parental H3-H4 to lagging strand
To provide additional evidence that mutations at the Pol1 HBM affect the transfer of H3-H4 to lagging strands, we determined whether mutating the HBM of yeast Pol1 (Pol1-2A2) also affect parental histone transfer in budding yeast (fig. S6A) (20, 25). In budding yeast, H3 lysine 4 tri-methylation (H3K4me3) and H3 lysine 56 acetylation (H3K56ac) mark parental and new histone H3, respectively. Unlike in mammalian cells, H3K4me3 and H3K56ac eSPAN peaks showed a slight bias toward lagging and leading strands in WT budding yeast, respectively (Fig. 4, A to E), consistent with a previous report (20). In contrast, in pol1-2A2 cells, H3K4me3 and H3K56ac eSPAN peaks at individual origins exhibited a strong bias toward the leading and lagging strands, respectively, except for the +1 and −1 nucleosomes around the origins (Fig. 4, A to E). These bias patterns were opposite from what we observed in WT yeast cells, and the bias ratio was also much more pronounced (Fig. 4, C and E), a phenomenon similar to what we detected in mcm2-3A and Pol1 mutant defective in binding to Ctf4 (20). Together, these results indicate that the pol1-2A2 mutant with mutations at the HBM affects transfer of parental histone H3-H4 onto replicating DNA strands in yeast.
Fig. 4. The Pol1 mutant defective in histone binding shows defects in parental histone transfer to lagging strand in budding yeast.
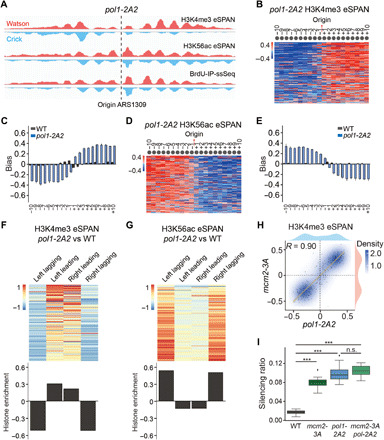
(A) Snapshot of BrdU-IP-ssSeq, H3K56ac, and H3K4me3 eSPAN peaks surrounding the ARS1309 origin in pol1-2A2 mutant cells. Red and blue tracks represent sequencing reads of Watson and Crick strands, respectively. (B and D) Heatmap representing the bias ratio and pattern of H3K4me3 (B) and H3K56ac (D) eSPAN peaks in pol1-2A2 mutant cells at each of the 20 individual nucleosomes surrounding each of the 134 early DNA replication origins ranked from top to bottom based on replication efficiency. Individual nucleosomes are represented by a circle, and their positions are indicated (−10 to +10), with each row representing one origin. (C and E) The average bias ratio of H3K4me3 (C) and H3K56ac (E) eSPAN peaks in WT and pol1-2A2 mutant cells at each of the 20 individual nucleosomes of the 134 early replication origins. Data were shown as means ± SEM from two independent replicates. (F and G) The relative amount of H3K4me3 (F) and H3K56ac (G) in pol1-2A2 compared to WT strains at each of the 134 origins (heatmap, top) and the average (bottom) of these origins. (H) H3K4me3 eSPAN bias patterns in pol1-2A2 showed a strong correlation with that of mcm2-3A. Spearman’s rank correlation coefficient and the density distribution were shown. (I) Effects of the pol1-2A2 and mcm2-3A mutation alone and in combination on the silencing loss at the HML locus. Data were plotted as means ± SEM from three independent repeats. ***P < 0.001; n.s., not significant; P > 0.05.
Next, we calculated the relative amounts of H3K4me3 in pol1-2A2 cells on both strands compared to WT cells. Notably, we observed a dramatic decrease of H3K4me3 on the lagging strand and an increase of H3K4me3 on the leading strand in pol1-2A2 cells compared to WT yeast cells, with the reduction on the lagging strand being more pronounced than the increase on the leading strand (Fig. 4F). A similar calculation was also performed using H3K56ac eSPAN, and the opposite effects were observed for the relative amount of new H3K56ac (Fig. 4G), suggesting that pol1-2A2 mutation indeed impairs parental histone transfer onto lagging strands. Last, the impact of pol1-2A2 on parental histone transfer was similar to that of mcm2-3A (Fig. 4H). These results support the idea that MCM2 functions through Pol α, which deposits parental histones to lagging strands via direct interaction with histones. Consistent with this idea, the impact of MCM2-2A and POLA1-2A mutations on the distribution of H4K20me2 and H4K12ac on replicating DNA strands also correlated with each other in mouse ES cells (fig. S6, B and C). Together, these results show that mutations at the HBM of Pol α in both yeast and mouse ES cells affect the transfer of parental H3-H4 to lagging strands.
POLA1 preferentially binds to H3 and H4
To understand how mutations at the HBM of Pol1, which is reported to bind H2A-H2B, affected parental histone transfer of H3-H4, we constructed three POLA1 N-terminal fragments of different lengths (N0, 1 to 55 amino acids; N1, 1 to 110 amino acids; N2, 1 to 200 amino acids) and purified the recombinant proteins for in vitro glutathione S-transferase (GST) pull-down assays. We observed, however, that POLA1 N1 and N2 fragments interacted with H3-H4 preferentially over H2A-H2B, whereas the N0 fragment did not bind to histones (Fig. 5A). The binding of POLA1 to H3-H4 was detectable even in high salt concentrations (350 mM NaCl), whereas its interaction with H2A-H2B was hardly detected (Fig. 5, A and B, and fig. S6D). Consistent with our observations, isothermal titration calorimetry (ITC) measurements, which were performed under much higher protein concentrations (110 μM in ITC versus 10 nM in GST pull-down assays), showed that mouse POLA1 N1 fragment bound H3-H4 tetramers and H2A-H2B with a dissociation constant (Kd) of 7.9 and 18 μM at 500 mM NaCl, respectively (Fig. 5, C and D). Moreover, the POLA1-2A mutation significantly reduced the association of POLA1 with H3-H4 (Fig. 5E). Similarly, yeast Pol1 N-terminal fragments (N1, 1 to 110 amino acids; N2, 1 to 200 amino acids) also interacted with H3-H4 preferentially over H2A-H2B, and the pol1-2A2 mutation markedly reduced the interaction of Pol1 with H3-H4 (Fig. 5F and fig. S6E). Together, these results demonstrate that the N terminus of Pol1 in yeast and mouse ES cells binds to core histones, with a preference for histone H3-H4 tetramers. These results were opposite from the report that the Pol1 HBM binds H2A-H2B (32). One possible explanation for the contradicting results is that we used purified histone octamers in the in vitro binding assays, whereas HeLa nuclear extracts were used in the previous study, which may contain other proteins including histone chaperones, in the binding assays.
Fig. 5. Pol α preferentially interacts with histone H3-H4 tetramers.
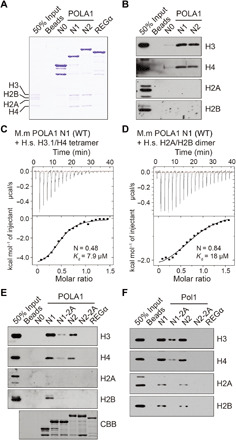
(A and B) In vitro interactions between GST-POLA1 N-terminal fragments and histones. Proteins associated with GST-POLA1 N-terminal fragments were visualized by Coomassie Brilliant Blue (CBB) staining (A) and immunoblotting (B). GST beads and GST Regα were used as negative controls for the assays. (C and D) ITC results using POLA1 N1 fragment and H3.1/H4 tetramer (C) or H2A/H2B dimer (D). Top, raw data; bottom, fitted curves. The N value in (C) (N = 0.48) indicated that one POLA1 N1 molecule binds two copies of H3-H4 dimer (i.e., one tetramer). M.m, Mus musculus; H.s., Home sapiens. (E) Mutations at the HBM of POLA1 reduce its interaction with histone H3-H4. Proteins associated with GST-POLA1 N-terminal fragments were visualized by immunoblotting. GST beads and GST REGα were used as negative controls for the assays. (F) In vitro interactions between yeast GST-Pol1 N-terminal fragments and histones based on GST pull-down assays and Western blot analysis of indicated proteins.
Mutants defective in parental histone transfer affect transcription
In budding yeast, the pol1-2A2 mutant strain shows partial defects in transcription silencing at the silent mating-type locus (32). Using the CRASH (Cre-reported altered states of heterochromatin) assay (41), we analyzed the impact of this mutant on the stability of silent chromatin. We observed a significant increase in the rates of silencing loss at the HML locus in both pol1-2A2 and mcm2-3A mutant strains compared to WT cells (Fig. 4I). The pol1-2A2/mcm2-3A double mutant cells exhibited a similar silencing loss compared to the single mutants (Fig. 4I), consistent with the idea that Pol1 and Mcm2 work in the same pathway in parental histone transfer.
The impact of defects in parental histone transfer on mammalian chromatin integrity was not known. Therefore, we analyzed how POLA1-2A and MCM2-2A mutants affected the expression of coding genes and noncoding repetitive elements in mouse ES cells using RNA sequencing (RNA-seq). Compared to WT cells, 2015 genes showed abnormal expression in POLA1-2A mutant cells, with 542 genes up-regulated and 1473 genes down-regulated (Fig. 6A and fig. S7A). In MCM2-2A cells, about 229 and 341 genes were up- and down-regulated, respectively (Fig. 6B and fig. S7B). In addition, genes altered in MCM2-2A mutant cells overlapped significantly with those altered in POLA1-2A mutant cells for both the up-regulated (101, P = 4.58 × 10−105) and down-regulated (234, P = 2.84 × 10−217) ones (Fig. 6C). Gene ontology analysis on the differentially expressed genes revealed that similar pathways including stress response, cell movement, and development were affected in both MCM2-2A and POLA1-2A mutant cells (fig. S7, C and D).
Fig. 6. Cells defective in parental histone transfer show defects in gene transcription and ERV silencing.
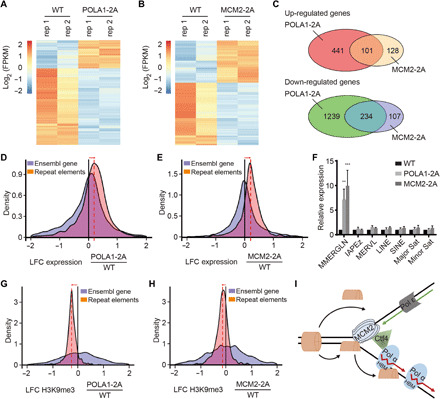
(A and B) Cluster analysis of differentially expressed genes between WT and POLA1-2A (A) and MCM2-2A (B) mutant mouse ES cells by RNA-seq. Results from two independent repeats (rep 1 and rep 2) were shown. FPKM, fragments per kilobase of transcript per million mapped reads. (C) Venn diagram of up-regulated (top) and down-regulated (bottom) genes between POLA1-2A and MCM2-2A mutant mouse ES cells. (D and E) Changes in the transcription of transposable elements (TEs) in POLA1-2A (D) and MCM2-2A (E) relative to WT mouse ES cells. Histograms show the distributions of the log2 fold change (LFC) of repeat element expression in each mutant relative to WT cells. Orange, transcription of repetitive elements (Repbase with RNA-seq; POLA1-2A n = 2; MCM2-2A n = 2). Blue, transcription of Ensembl genes. (F) Expression analysis of repetitive elements in WT, POLA1-2A, and MCM2-2A mouse ES cells by quantitative PCR. MMERGLN-int (ERV1), IAPEz-int (ERVK), and MERVL-int (ERVL) representing different families of ERVs were tested. Data were plotted as means ± SEM from at least three independent repeats. **P < 0.01 and ***P < 0.001. (G and H) Changes in the enrichment of H3K9me3 in POLA1-2A (G) and MCM2-2A (H) relative to WT mouse ES cells. Histograms show the distributions of the LFC of H3K9me3 density in POLA1-2A and MCM2-2A mutants relative to WT cells. (I) A model for the role of POLA1 in parental histone transfer.
We also examined the expression of transposable elements (TEs), including ERVs, which are normally silenced in mammalian cells (42). Using a previously described method to analyze the expression of both TEs and coding genes (43), we observed a global increase of TE in both POLA1-2A and MCM2-2A cells compared to coding genes (Fig. 6, D and E). Most of these TEs were ERVs, including ERVK and ERV1 (table S1). However, when individual ERVs were analyzed, we observed that only a handful of specific ERVs showed a statistical increase in expression in either POLA1-2A or MCM2-2A mutant cells (fig. S7, E and F). Moreover, the up-regulated ERVs in the two mutant cells also significantly overlapped with each other (fig. S7G). On average, the up-regulated TEs had higher copy numbers in the genome than those unchanged TEs in the mutant cells (fig. S7, H and I). Last, the increased expression of ERV1, but not other repetitive elements or non–long terminal repeat retrotransposons (short and long interspersed nuclear elements), in POLA1-2A and MCM2-2A mutant cells were confirmed by reverse transcription polymerase chain reaction (RT-PCR) (Fig. 6F). These results suggest that silencing of most ERVs may be lost in some but not all POLA1-2A and MCM2-2A mutant cells.
ERVs, which represent about 10% of the mouse genome, are normally silenced (42). Multiple mechanisms including DNA methylation, H3K27me3, and H3K9me3 are involved in ERV silencing, with H3K9me3 playing a dominant role in ES cells (44). Therefore, we analyzed the impact of POLA1-2A and MCM-2A mutations on H3K9me3 at ERV loci using CUT&Tag/ACT-seq. Compared with WT mouse ES cells, these mutations led to a decrease of H3K9me3 in CUT&Tag/ACT-seq reads mapping to the ERVs (Fig. 6, G and H) while having little impact on the global H3K9me3 levels (fig. S7J). However, not all up-regulated ERVs showed a significant reduction of H3K9me3 (table S1), which may be due to the fact that different classes of ERVs are regulated by different mechanisms in mouse ES cells (45). In addition, the expression of 55 genes that are involved in regulating H3K9me3, including all the writers, erasers, and classic readers, was not significantly down-regulated in the POLA1-2A and MCM2-2A mutant cells (fig. S8, A and B). These results suggest that the abnormal transcription of some ERVs is directly linked to changes in H3K9me3 at their chromatin, but not the expression of H3K9me3 regulators.
It has been previously shown that reduction of silencing at TEs leads to increased expression of nearby genes (46). Therefore, we asked whether the observed increase in gene expression in POLA1-2A and MCM2-2A cells was linked to increased ERV expression. We observed that the genomic distance between up-regulated genes and up-regulated TEs in these two mutant cells was shorter than that with unchanged TEs (fig. S8, C and D). Moreover, genes flanking up-regulated TEs were more likely to be up-regulated in both mutant cells compared to genes far away from these TEs (fig. S8, E and F). Together, these results suggest that the increased gene expression detected in POLA1-2A and MCM2-2A mutant cells is likely due, at least in part, to the loss of silencing of nearby ERVs.
DISCUSSION
Development of eSPAN method in mammalian cells
Previously, we reported the eSPAN method (35), which makes it possible to analyze the distribution of proteins including histones on leading and lagging strands at DNA replication forks in budding yeast. Using this method, we have gained valuable mechanistic insights into replication-linked processes where strand-specific information is needed. For instance, we observed that proliferating cell nuclear antigen is unloaded from lagging strands during replication stress (35). Moreover, DNA replication checkpoint kinase Rad53 couples leading and lagging strand DNA synthesis (47). Last, we found two pathways that facilitate the transfer of parental H3-H4 onto leading and lagging strands, respectively, in budding yeast (20, 25). However, the eSPAN method, similar to traditional ChIP-seq procedures, suffers from low yield and cumbersome procedures for single-stranded DNA (ssDNA) library preparation. Here, we described an updated version of the eSPAN method in mammalian cells, which is much simpler, more reliable, and cost-effective than our previous eSPAN procedures or SCAR-seq (26, 35). The new eSPAN procedure introduces a modified version of the CUT&Tag/ACT-seq method in which pA-Tn5 loaded with one sequencing adaptor instead of two in the original CUT&Tag/ACT-seq method is used for tagmentation. To preserve strand-specific information, the second adaptor is ligated through oligo-replacement.
Using this improved method, we can monitor both the chromatin association of histone modifications (CUT&Tag/ACT-seq) and the distribution of these modifications on replicating DNA strands (eSPAN) in mammalian cells starting from 50 thousand cells in a strand-specific way (Fig. 1, G and H), whereas SCAR-seq starts with 20 million cells and requires many more reads for calculation (26). In theory, the new method will allow us to study DNA replication–linked processes in rare cell populations, such as germ cells, or even at a single-cell level if optimized further, as tagmentation-based sequencing methods have been extensively tested in single-cell studies (33, 34, 48, 49). Another major advantage over the traditional eSPAN procedures and SCAR-seq is that the current eSPAN protocol greatly reduces the amount of time and work, as the experiment from cell collection to sequencing can be completed in only 2 days instead of 1 week. Furthermore, the current method does not involve library preparation kits. Therefore, this improved eSPAN approach can be easily adopted in studies of processes linked to DNA replication in addition to nucleosome assembly. For example, in Drosophila germ cells, most parental histones are inherited by daughter cells that retain “stemness,” whereas the differentiated cells contain mostly new histones (50, 51). Furthermore, asymmetric cell division of cancer stem cells is associated with cancer progression and treatment (52). We expect that the eSPAN method reported here will be useful in addressing questions related to histone distributions on replicating DNA strands in these processes.
We observed previously that on newly replicating chromatin, nucleosomes formed with parental H3-H4 tetramers are more stable and resistant to micrococcal nuclease digestion than nucleosomes formed with newly synthesized H3-H4 (25). We speculate that this could be due to parental H3-H4 being transferred to their original positions. It has been shown that parental nucleosomes can remember their positions following DNA replication in budding yeast (6). Moreover, mutating Dpb3 and Mcm2, two chaperones involved in the transfer of parental H3-H4 to leading and lagging strands, respectively, results in a loss of nucleosome position memory in yeast (6). In mouse ES cells, parental histones at repressed chromatin domains are redistributed locally to their starting genomic locations during DNA replication, whereas active chromatin regions show more dynamic patterns (7). It would be interesting to determine whether POLE3/POLE4, MCM2, and POLA1 are also important for the memory of nucleosome positions in mouse ES cells.
Pol α transfers parental histones to lagging strand through direct binding to histone H3-H4 tetramers
We have shown previously that Pol1, the catalytic subunit of Pol α primase on lagging strands, functions in the same pathway as MCM2, a subunit of the CMG helicase that travels along leading strand template, in the transfer of parental H3-H4 to lagging strands in yeast cells (25). However, how Pol α is involved in this process remains unclear. Recently, it has been shown that Pol1 contains a conserved HBM and interacts with H2A-H2B (32). In this study, we demonstrated that mutations at this region of Pol α impair the transfer of parental H3-H4 onto replicating lagging strands in both yeast and mouse ES cells. These unexpected results prompted us to analyze how Pol1 interacts with histones. We show that Pol1 in yeast and POLA1 in mouse ES cells interact with histone H3-H4, preferentially over H2A-H2B, and mutations at the conserved HBM of both Pol1 and POLA1 lead to reduced binding to H3-H4 (Fig. 5). The apparent discrepancy of both studies suggests the possibilities that Pol α interacts with both H3-H4 and H2A-H2B, but its affinity toward H3-H4 and H2A-H2B may be dynamically regulated in cells and/or may be dependent on PTM status and binding partners. Supporting this idea, we noticed that nuclear extracts were used in the previously reported in vitro binding assays (32), whereas purified histone octamers were used in our study. In addition, the Kd value for POLA1-H2A-H2B interaction is markedly different from that reported in (32). This difference arises largely from the different salt concentrations used in these two studies (500 versus 150 mM NaCl), as we obtained much similar Kd value to that reported in (32) when we measured Kd at 150 mM salt (fig. S6F). We also noticed that the amino acid sequence of the N terminus of mouse POLA1 used in our study differs from that of the human POLA1 used in (32) by 15 amino acids, which may also contribute to this difference. Nonetheless, our study indicates that Pol1 also functions as a histone chaperone for H3-H4 to facilitate their transfer onto lagging strands (Fig. 6I).
MCM2 and POLA1 mutants defective in parental histone transfer affect gene expression and ERV silencing in mouse ES cells
In budding yeast, mutants defective in parental histone transfer show a significant increase in the rate of loss of transcriptional silencing at heterochromatin regions (20, 25, 53). However, heterochromatic regions in budding yeast do not contain H3K9 methylation found at heterochromatin in Schizosaccharomyces pombe and higher eukaryotic cells. It was not known whether mutations in genes involved in parental histone deposition also affect gene expression and heterochromatin silencing in mammalian cells. Here, we provide the first line of evidence that MCM2-2A and POLA1-2A mutants defective in parental histone transfer lead to abnormal expression of a small number of the coding genes. The genes altered in MCM2-2A and POLA1-2A mutant cells overlap significantly with each other. These results provide additional support to the idea that MCM2 and POLA1 function in the same pathway in parental histone transfer.
In addition to coding genes, we also observed that in both MCM2-2A and POLA1-2A mutant ES cells, the expression of selected ERVs increased with a concomitant reduction of H3K9me3 at the ERVs (Fig. 6, D to H). Furthermore, we found that genes up-regulated in MCM2-2A and POLA1-2A cells are associated with the loss of ERV silencing. ERVs represent a significant fraction of mammalian genomes, reflecting their effective integration into germ cells during multiple viral epidemics occurring over evolutionary time scales (54). These elements are transcriptionally silenced through conserved mechanisms including H3K9me3 (44). Silenced chromatin domains are maintained by local redistribution of parental histones (7), suggesting that altered parental histone deposition observed in MCM2-2A and POLA1-2A cells contributes to the altered ERV silencing. Previously, it has been shown that depletion of CAF-1, the histone chaperone involved in deposition of new H3-H4 onto replicating DNA, results in increased expression of ERV transgenes (55, 56). Therefore, it would be interesting to determine whether CAF-1, MCM2, and POLA1 function in parallel in silencing of ERVs in mouse ES cells.
In general, reactivation of ERVs is associated with genome instability (42). In cancer cells, reactivation of these proviruses can sensitize tumors to immunotherapy, likely due to an increase in expression of neoantigens (57). Moreover, the reactivated ERVs can also serve as virus mimic to induce antiviral immune responses against the “infected” tumor cells (58, 59). Therefore, elucidating the mechanisms of parental histone transfer will not only increase our understanding of epigenetic inheritance but also open new windows for cancer immunotherapy in the future.
MATERIALS AND METHODS
Cell culture, antibodies, and reagents
Mouse E14 ES cells (a gift from T. Fazzio, University of Massachusetts Medical School, Worcester, MA) were cultured in Dulbecco’s modified Eagle’s medium (Corning) supplemented with 15% (v/v) fetal bovine serum, 1% penicillin/streptomycin (Invitrogen), 1 mM sodium pyruvate (Cellgro), 2 mM l-glutamine (Cellgro), 1% MEM nonessential amino acids (Invitrogen), 55 μM β-mercaptoethanol (Sigma-Aldrich), and mouse leukemia inhibitory factor (10 ng/ml) on gelatin-coated dishes in a 37°C incubator with a humidified, 5% CO2 atmosphere. The antibodies used in this study include anti-H4K20me2 (Diagenode, C15200205), anti-H4K12ac (Abcam, ab46983), anti-H3K36me3 (Active Motif, 61021), anti-H3K4me3 (Abcam, ab8580), and anti-BrdU (BD Biosciences, 555627). Other antibodies including anti-H3K56ac, anti-H3, anti-H4, anti-H2A, and anti-H2B were generated by our lab using a synthetic peptide or full-length recombinant proteins.
Yeast culture and strains
Yeast cells were synchronized and cultured following the standard protocol (60). Briefly, yeast cells were synchronized by adding α factor (Sigma-Aldrich) at 5 mg/ml to an exponentially growing culture in yeast extract, peptone, and dextrose (YPD) medium at OD600 (optical density at 600 nm) around 0.4 to 0.5. Cells were arrested for 3 hours at 25°C and then released into fresh YPD medium containing BrdU (400 mg/ml) (Sigma-Aldrich, 5002) for 30 min. All strains are available upon request.
Gene editing
CRISPR-Cas9–guided gene editing was performed following the standard protocol (61). Briefly, oligos were synthesized (IDT, Coralville, IA, USA) and inserted into pX459 vector. Oligos targeting MCM2-2A were used the same as previously reported (26), and the others were detailed in table S1. Plasmids were transfected into mouse ES cells by transfection using Lipofectamine 3000 (Invitrogen). After puromycin (2 μg/ml) selection for 2 days, single-cell clones were picked and expanded for further characterization. Genetic mutations were confirmed by Sanger sequencing.
In vitro binding assay
GST fusion proteins were expressed in BL-21 RIL competent Escherichia coli cells by 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) induction at 25°C for 4 to 6 hours. E.coli cells were lysed in lysis buffer [5 mM tris-HCl (pH 8.0), 500 mM NaCl, 5% glycerol, 2 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine, and benzonase (5 U/ml)] and bound to Glutathione Sepharose 4B beads (GE Healthcare). Beads were extensively washed with lysis buffer, and proteins were eluted for the binding assays. Native histone octamers were purified from HeLa cells, as previously described (62). Briefly, nuclei were extracted in high-salt extraction buffer [10 mM tris-HCl (pH 7.4), 750 mM NaCl, and 0.2 mM PMSF]. Nonhistone fractions and DNA were further removed by ultracentrifugation. For the binding assays, histone octamers were incubated at a final concentration of 10 nM with GST fusion proteins in binding buffer [20 mM tris-HCl (pH 8.0), 150 mM NaCl, 5% glycerol, and 0.1% Triton X-100] at 4°C for 4 hours. Beads were washed three times with wash buffer [20 mM tris-HCl (pH 8.0), 400 mM NaCl, 5% glycerol, and 0.1% Triton X-100], boiled for 5 min, and analyzed by Coomassie Brilliant Blue (CBB) staining or immunoblotting.
pA-Tn5 protein purification
pA-Tn5 plasmid was transformed into T7 Express lysY/Iq competent E. coli cells (New England Biolabs, C3013) and induced with 0.25 mM IPTG (Sigma-Aldrich) at 18°C for 6 hours or overnight. Cells were lysed in lysis buffer [50 mM tris-HCl (pH 8.0), 1 M NaCl, 20 mM imidazole, 0.1% Triton X-100, 1 mM DTT, 1 mM PMSF, and 1 mM benzamidine] and bound to Ni-NTA beads at 4°C for 4 hours. pA-Tn5 proteins were eluted with elution buffer [50 mM tris-HCl (pH 8.0), 1 M NaCl, 125 mM imidazole, and 0.1% Triton X-100]. Protein concentration and purity were quantified after diluted with equal volume of 100% glycerol.
Enrichment and sequencing of protein-associated nascent DNA
eSPAN in yeast was performed exactly as previously described (35). For the modified eSPAN in mammalian cells, exponentially growing mouse ES cells were first treated with BrdU at 50 μM for 20 to 40 min. Cells were harvested, washed, and bound to concanavalin A–coated magnetic beads. Primary antibodies were incubated with cells in antibody buffer [20 mM Hepes-NaOH (pH 7.5), 150 mM NaCl, 0.5 mM spermidine, 2 mM EDTA, 0.1% bovine serum albumin, 0.08% digitonin, and 1× proteinase inhibitor cocktail] overnight at 4°C. Secondary antibodies and preassembled pA-Tn5 complex were sequentially bound. Tagmentation was performed at 37°C for 1 hour with gentle shaking. Reactions were stopped by mixing in 20 mM EDTA, 0.1% SDS, and proteinase K (0.1 mg/ml) and incubating at 50°C for 1 hour or 37°C overnight with gentle shaking. The supernatants were purified using ChIP DNA concentrator columns (Zymo Research, D5205). The eluents were proceeded to an oligo-replacement reaction as detailed in (63). Briefly, the DNA samples were mixed with 0.5 mM deoxynucleotide triphosphate mix, 0.5 μM mosaic end adaptor B, and 1× Ampligase buffer and incubated in an annealing program (50°C, 1 min; 45°C, 10 min; ramp to 37°C at 0.1°C/s and hold). T4 DNA polymerase and Ampligase were added to the reaction and incubated at 37°C for 1 hour. The reaction products (5 to 10%) were saved as CUT&Tag/ACT-seq samples, and the others were boiled for 5 min and immediately chilled on ice. The samples were further diluted with ice-cold BrdU IP buffer [1× phosphate-buffered saline (PBS) and 0.0625% Triton X-100] and mixed with E. coli transfer RNA and BrdU antibodies at 4°C for 2 hours. Twenty-five microliters of prewashed protein G beads (GE Healthcare, 17-0618-02) was added to each sample and rotated at 4°C for 1 hour. After extensive wash, beads were incubated with elution buffer [50 mM tris-HCl (pH 8.0), 10 mM EDTA, and 1% SDS] at 65°C for 15 min. The supernatants were purified with ChIP DNA concentrator columns, and library PCR was performed using standard Illumina Nextera Dual Indexing primers. Samples were pooled and sequenced using paired-end sequencing on Illumina NextSeq 500 platforms at Columbia University Genome Center.
Okazaki fragment sequencing
OK-seq was performed as previously described with some modifications (13). Briefly, 100 million mouse E14 ES cells were labeled with 20 μM 5-ethynyl-2′-deoxyuridine (EdU) (Sigma-Aldrich, 900584) for 2 min. After EdU pulse, cells were washed with ice-cold PBS, collected by scraping, and centrifuged at 400g for 10 min. Genomic DNA was extracted by a routine proteinase K/phenol extraction/ethanol precipitation procedure and dissolved in 6 ml of TE buffer [10 mM tris-HCl (pH 8.0) and 1 mM EDTA]. DNA was further denatured at 98°C for 10 min, chilled on ice for 10 min, and separated equally into six Ultra-clear tubes (Beckman, 344059) containing 10 ml of 5 to 30% (w/v) glucose gradient [10 mM tris-HCl (pH 8.0), 1 mM EDTA, and 100 mM NaCl]. After centrifuging at 26,000g for 17 hours at 20°C, fractions with <200-bp DNA fragments were collected and concentrated using Amicon Ultra-15 Centrifugal Filter (Millipore). Buffers were exchanged with 5 ml of deoxyribonuclease (DNase)–free H2O and concentrated to 360 μl. Click reaction was performed by sequentially mixing 100 mM potassium phosphate buffer (pH 7), 10% dimethyl sulfoxide, 0.5 mM biotin-TEG (triethyleneglycol) azide, 0.5 mM tris(3-hydroxypropyltriazolylmethyl)amine, 0.1 mM CuSO4, and 10 mM sodium ascorbate and incubating for 1 hour at room temperature. Reaction products were precipitated with ethanol and dissolved in 90 μl of DNase-free H2O. RNA was removed by adding 10 μl of 2.5 M NaOH and incubating at 37°C for 30 min, followed by adding 10 μl of 2.5 M acetic acid for neutralization. DNA samples were then purified on Micro Bio-Spin Columns with Bio-Gel P-30 (Bio-Rad) and incubated with 300 μg of MyOne Streptavidin T1 beads (Invitrogen) in binding buffer [5 mM tris-HCl (pH 7.5), 0.5 mM EDTA, and 1 M NaCl] for 1 hour at room temperature to capture biotinylated DNA. Uncaptured DNA were then removed by washing three times with wash buffer [5 mM tris-HCl (pH 7.5), 0.5 mM EDTA, 1 M NaCl, and 0.05% Tween]. Parental DNA strands were removed by incubating with 150 mM NaOH for 5 min, followed by sequential wash for three times with 150 mM NaOH, twice with TET buffer [10 mM tris-HCl (pH 8.0), 1 mM EDTA, and 0.05% Tween], once with TE buffer [10 mM tris-HCl (pH 8.0) and 1 mM EDTA]. The final DNA products were eluted by boiling at 95°C for 5 min in elution buffer [95% formamide and 10 mM EDTA (pH 8.2)] and purified with ethanol precipitation into 18 μl of EB buffer [10 mM tris-HCl (pH 8.5)]. The ssDNA libraries were prepared using the Accel-NGS 1S Plus DNA library kit (Swift Bioscience, 10096) and sequenced using paired-end sequencing on Illumina NextSeq 500 platforms at the Columbia University Genome Center.
RT-PCR analysis
Total RNA was isolated using an RNeasy Plus Mini kit (Qiagen). One microgram of total RNA was used for RT with random hexamers (Invitrogen). Real-time quantitative PCR was performed in triplicates for each sample with SYBR Green PCR Master Mix on the CFX96 platform (Bio-Rad Laboratories). Primers used were listed in table S2.
RNA sequencing
Total RNA samples were purified the same as for RT-PCR. RNA-seq library preparation and deep sequencing were performed by the Columbia University Genome Center. Two or three replicates for each sample were sequenced. Raw data were aligned to the mouse genome and to gene annotations from RefSeq using TopHat v2.05.
Cell cycle analysis
Exponentially growing mouse ES cells were collected and fixed in 70% ethanol at 4°C with rotation overnight. Cell pellets were resuspended in PBS containing RNase A (50 μg/ml) and incubated at 37°C after washing twice with PBS. Cells were then stained with propidium iodide and analyzed by an LSR II flow cytometer (BD Biosciences).
Histone extraction
Histone was obtained by standard acid extraction protocol to detect histone modifications. Briefly, about 5 million cells were collected, and cell pellets were incubated with hypotonic buffer [10 mM tris-HCl (pH 8.0), 1 mM KCl, 1.5 mM MgCl2, 1 mM DTT, and protease inhibitor] rotating at 4°C for 30 min after washing once with hypotonic buffer. The pellets were washed three times with hypotonic buffer and resuspended in 400 μl of 0.2 N of HCl and rotate at 4°C for 30 min. After centrifugation, 100 μl of trichloroacetic acid was added to the supernatant and incubated on ice for 30 min. The pellet was then washed by cold acetone twice. Histone was dissolved in ddH2O for immunoblotting.
ITC assay
Purified proteins were dialyzed overnight against ITC buffer [20 mM tris-HCl (pH 8.0), 500 mM NaCl, and 5% glycerol] and diluted with the same buffer to achieve the desired concentrations. All measurements were performed using an ITC200 titration calorimeter (MicroCal) at 25°C. A pre-drop of 0.4 μl and subsequent 19 2.0-μl drops of mPOLA1-N1 (800 μM) were stepwise injected into the calorimetric cell containing hH2A-H2B (106 μM) or hH3.1-H4 (115 μM) at intervals of 120 s. The stirring speed remained constant at 700 rpm to thoroughly mix the samples in the cell. The raw data were analyzed and fitted into a one-site binding model using Origin 7.0 software (MicroCal). The relationships between change in enthalpy (ΔH), entropy (ΔS), and binding energy (ΔG) were calculated as: ΔG = ΔH − TΔS.
CRASH assay
Silencing-loss rates in WT and mutant yeast strains were measured using the CRASH assay, as previously reported (25, 41). Briefly, 10 colonies of each strain were grown separately in YPD medium to saturation, diluted to OD600 = 0.01 in YPD, and continued growing for 5 hours at 30°C. For each colony, 50,000 cells were collected using a BD Fortessa cytometer. The silencing-loss rates at the HML loci were calculated by dividing the number of red fluorescent protein–positive (RFP+)/green fluorescent protein–positive (GFP+) cells (cells that have recently undergone the Cre-mediated recombination leading to GFP expression but still contain RFP) by the total number of cells with the potential to lose silencing (RFP+ cells).
Genome alignment
All the CUT&Tag/ACT-seq, eSPAN, OK-seq, and RNA-seq libraries were constructed in a strand-specific way and sequenced using a paired-end method on Illumina platforms. First, adaptor sequences of all raw reads were removed by Cutadapt, and reads of <10 nt were also removed. CUT&Tag/ACT-seq, eSPAN, and OK-seq data were mapped to yeast (sacCer3) and mouse (mm10) reference genome according to the species using Bowtie2 software (64). RNA-seq data of mouse ES cells were mapped to the mouse (mm10) reference genome using STAR software (65). Only paired-end reads with both ends mapped correctly were kept for further analysis. Except for repeat element analysis, all multimapped reads were removed using SAMtools (MAPQ < 40) for further analysis (66). For the CUT&TAG/ACT-seq, eSPAN, and OK-seq libraries, duplicate reads were removed using Sambamba software (67).
Identification of replication initiation zones by OK-seq
Identification of initiation zones follows the method described previously (13). Briefly, the genome-wide OK-seq bias value was calculated in each bin of 1 kb. Bias = (W − C)/(W + C), where W and C are the number of reads mapped on Watson and Crick strand in each bin, respectively. A four-state HMM (Hidden Markov model) was used to detect the initiation zone (“Up” state), termination zone (“Down” state), and constant bias (“Flat1” and “Flat2” states) from the bias profile. A sliding window of 60 kb was used when detecting the HMM states, and ΔBiasn = (Biasn + 1 − Biasn)/2 was calculated between adjacent windows to predict the state of each sliding window. Two repeats of our OK-seq samples are calculated independently, and then 1548 initiation zones that were overlapped in two repeats and closer than 3 kb were selected for further analysis.
eSPAN and CUT&Tag/ACT-seq bias calculation
The bias of each histone modification eSPAN and CUT&Tag/ACT-seq at selected replication origins was computed from trimmed, mapped, unique reads in each bin using the formula Bias = (W − C)/(W + C), where W and C are the number of reads mapped on Watson and Crick strand in each bin, respectively. The bias value ranges from [−1,1] to quantify the histone segregation equality. Read coverage in each bin was calculated using the R package of Rsamtools. In mouse ES cells, the bias was calculated in each bin of 5 kb, and the bias in yeast cells was calculated on the basis of nucleosome positions annotated previously (68). In mouse ES cells, the bias was further smoothed by flanking five bins for visualization.
To calculate the relative amounts of parental or new histones at each strand between mutant and WT strains for yeast cells, the following calculation was applied. First, the corresponding BrdU-IP-ssSeq peaks were called to identify replication regions using SICER software (69). Then, the eSPAN signals were split into left and right based on the identified replication origins. Each replication region can be split into four quadrants: Watson strand at the left (WL) and the Crick strand at the right (CR), which are lagging strands; Crick strand at the left (CL) and the Watson strand at the right (WR), which are leading strands. On each replication quadrant, the eSPAN raw reads were normalized to the total number of mapped reads and the corresponding BrdU signal to quantify the histone enrichment per unit of newly synthesized DNA. Last, the normalized eSPAN signal was used to calculate the relative amount of histone enrichment between mutant and WT cells as follows: relative amount = (norm:eSPANmut − norm:eSPANwt)/norm:eSPANwt × 100%.
Analysis of gene expression and histone modifications at repeat elements
To quantify the reads at repetitive regions of the genome, it is challenging due to the mapping uncertainty of those multimapped reads from highly similar sequences in the genome. To allow for the multimapped reads mapping up to 100 locations, RNA-seq data of mouse ES cells were mapped to the mouse (mm10) reference genome by STAR software with the parameter “-- outFilterMultimapNmax 100 --winAnchorMultimapNmax 100” (65). The coding gene annotation was downloaded from GENCODE, and the repetitive elements annotation was downloaded from University of California, Santa Cruz Table Browser track “RepeatMasker” (70, 71). To achieve robust results for ERV gene expression, we repeated the analysis using two approaches when counting reads. First, we used TEtranscripts, a specialized tool to assign ambiguously multimapped reads in an expectation-maximization algorithm (72). Second, we used featureCounts to assign the multimapped reads in a fractional way with parameters “allowMultiOverlap = T, countMultiMappingReads = T, fraction = T” (73). For both of two approaches, read counts at the coding gene and ERV genes were combined together for analysis of differential gene expression using DESeq2 (74). The differential genes resulted from two independent approaches were quite consistent, and the cutoffs for differential genes were false discovery rate < 0.05 and |fold change| > 1.5.
To analyze the enrichment of a histone modification using the CUT&Tag/ACT-seq datasets, we performed differential enrichment analysis similarly to differential expression analysis. First, sequencing reads at coding genes were counted at each gene promoter [upstream of 1000 bp to downstream of 200 bp from TSS (transcription start site)]. In addition, for ERV genes, read counts were summed from all repetitive elements for each ERV. Then, we obtained a count table of histone enrichment on coding gene promoters (~20,000 genes) and ERV gene bodies (~1000 ERVs) using featureCounts with parameters “allowMultiOverlap = T, countMultiMappingReads = T, fraction = T” (73). Last, the fold change of histone enrichment between mutant and WT cells was calculated by DESeq2 (74).
Statistical analysis
The statistical test was performed using R software. The coefficient of correlation was calculated using Spearman’s rank correlation method.
Supplementary Material
Acknowledgments
We thank K. Zhao, T. Fazzio, and S. Henikoff for help on CUT&Tag/ACT-seq. We thank S. Goff for advices on ERV and critical reading of this manuscript. Funding: This work is supported by NIH grants GM R35118015 (to Z.Z.), K99GM134180 (to A.S.-C.), and in part through the NIH/NCI Cancer Center Support Grant P30CA013696 to the Herbert Irving Comprehensive Cancer Center. This work is also funded by grants from the Ministry of Science and Technology of China (2019YFA0508900 and 2018YFE0203301) and Natural Science Foundation of China (31521002) to R.-M.X. Author contributions: Z.L., X.H., and Z.Z. conceived the project. Z.L., A.S.-C., X.X., and H.Z. performed experiments. X.H. performed the data analysis. C.-l.C. helped with the OK-seq analysis. S.G., W.-S.Y., and R.-M.X. performed the ITC experiments. Z.Z. supervised the study. Z.L. and Z.Z. wrote the manuscript with comments from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The accession number for the sequencing data reported in this paper is GEO: GSE142996. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eabb5820/DC1
REFERENCES AND NOTES
- 1.Allis C. D., Jenuwein T., The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Ragunathan K., Jih G., Moazed D., Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348, 1258699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang B., Zheng H., Huang B., Li W., Xiang Y., Peng X., Ming J., Wu X., Zhang Y., Xu Q., Liu W., Kou X., Zhao Y., He W., Li C., Chen B., Li Y., Wang Q., Ma J., Yin Q., Kee K., Meng A., Gao S., Xu F., Na J., Xie W., Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537, 553–557 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Wang X., Moazed D., DNA sequence-dependent epigenetic inheritance of gene silencing and histone H3K9 methylation. Science 356, 88–91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia W., Xu J., Yu G., Yao G., Xu K., Ma X., Zhang N., Liu B., Li T., Lin Z., Chen X., Li L., Wang Q., Shi D., Shi S., Zhang Y., Song W., Jin H., Hu L., Bu Z., Wang Y., Na J., Xie W., Sun Y.-P., Resetting histone modifications during human parental-to-zygotic transition. Science 365, 353–360 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Schlissel G., Rine J., The nucleosome core particle remembers its position through DNA replication and RNA transcription. Proc. Natl. Acad. Sci. U.S.A. 116, 20605–20611 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escobar T. M., Oksuz O., Saldaña-Meyer R., Descostes N., Bonasio R., Reinberg D., Active and repressed chromatin domains exhibit distinct nucleosome segregation during DNA replication. Cell 179, 953–963.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell S. P., Dutta A., DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71, 333–374 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Burgers P. M. J., Kunkel T. A., Eukaryotic DNA replication fork. Annu. Rev. Biochem. 86, 417–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fragkos M., Ganier O., Coulombe P., Méchali M., DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 16, 360–374 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Ganier O., Prorok P., Akerman I., Méchali M., Metazoan DNA replication origins. Curr. Opin. Cell Biol. 58, 134–141 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Machida Y. J., Hamlin J. L., Dutta A., Right place, right time, and only once: Replication initiation in metazoans. Cell 123, 13–24 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Petryk N., Kahli M., d'Aubenton-Carafa Y., Jaszczyszyn Y., Shen Y., Silvain M., Thermes C., Chen C.-L., Hyrien O., Replication landscape of the human genome. Nat. Commun. 7, 10208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeeles J. T. P., Janska A., Early A., Diffley J. F. X., How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol. Cell 65, 105–116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serra-Cardona A., Zhang Z., Replication-coupled nucleosome assembly in the passage of epigenetic information and cell identity. Trends Biochem. Sci. 43, 136–148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ransom M., Dennehey B. K., Tyler J. K., Chaperoning histones during DNA replication and repair. Cell 140, 183–195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han J., Zhou H., Horazdovsky B., Zhang K., Xu R.-M., Zhang Z., Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315, 653–655 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Driscoll R., Hudson A., Jackson S. P., Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315, 649–652 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reverón-Gómez N., González-Aguilera C., Stewart-Morgan K. R., Petryk N., Flury V., Graziano S., Johansen J. V., Jakobsen J. S., Alabert C., Groth A., Accurate recycling of parental histones reproduces the histone modification landscape during DNA replication. Mol. Cell 72, 239–249.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu C., Gan H., Serra-Cardona A., Zhang L., Gan S., Sharma S., Johansson E., Chabes A., Xu R.-M., Zhang Z., A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 361, 1386–1389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bellelli R., Belan O., Pye V. E., Clement C., Maslen S. L., Skehel J. M., Cherepanov P., Almouzni G., Boulton S. J., POLE3-POLE4 is a histone H3-H4 chaperone that maintains chromatin integrity during DNA replication. Mol. Cell 72, 112–126.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishimi Y., Ichinose S., Omori A., Sato K., Kimura H., Binding of human minichromosome maintenance proteins with histone H3. J. Biol. Chem. 271, 24115–24122 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Huang H., Strømme C. B., Saredi G., Hödl M., Strandsby A., González-Aguilera C., Chen S., Groth A., Patel D. J., A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat. Struct. Mol. Biol. 22, 618–626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foltman M., Evrin C., De Piccoli G., Jones R. C., Edmondson R. D., Katou Y., Nakato R., Shirahige K., Labib K., Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep. 3, 892–904 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Gan H., Serra-Cardona A., Hua X., Zhou H., Labib K., Yu C., Zhang Z., The Mcm2-Ctf4-Polα axis facilitates parental histone H3-H4 transfer to lagging strands. Mol. Cell 72, 140–151.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petryk N., Dalby M., Wenger A., Stromme C. B., Strandsby A., Andersson R., Groth A., MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Georgescu R., Yuan Z., Bai L., de Luna Almeida Santos R., Sun J., Zhang D., Yurieva O., Li H., O'Donnell M. E., Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc. Natl. Acad. Sci. U.S.A. 114, E697–E706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noguchi Y., Yuan Z., Bai L., Schneider S., Zhao G., Stillman B., Speck C., Li H., Cryo-EM structure of Mcm2-7 double hexamer on DNA suggests a lagging-strand DNA extrusion model. Proc. Natl. Acad. Sci. U.S.A. 114, E9529–E9538 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douglas M. E., Ali F. A., Costa A., Diffley J. F. X., The mechanism of eukaryotic CMG helicase activation. Nature 555, 265–268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon A. C., Zhou J. C., Perera R. L., van Deursen F., Evrin C., Ivanova M. E., Kilkenny M. L., Renault L., Kjaer S., Matak-Vinković D., Labib K., Costa A., Pellegrini L., A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature 510, 293–297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gambus A., van Deursen F., Polychronopoulos D., Foltman M., Jones R. C., Edmondson R. D., Calzada A., Labib K., A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase α within the eukaryotic replisome. EMBO J. 28, 2992–3004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evrin C., Maman J. D., Diamante A., Pellegrini L., Labib K., Histone H2A-H2B binding by Pol α in the eukaryotic replisome contributes to the maintenance of repressive chromatin. EMBO J. 37, e99021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter B., Ku W. L., Kang J. Y., Hu G., Perrie J., Tang Q., Zhao K., Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun. 10, 3747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaya-Okur H. S., Wu S. J., Codomo C. A., Pledger E. S., Bryson T. D., Henikoff J. G., Ahmad K., Henikoff S., CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu C., Gan H., Han J., Zhou Z.-X., Jia S., Chabes A., Farrugia G., Ordog T., Zhang Z., Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol. Cell 56, 551–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jasencakova Z., Scharf A. N. D., Ask K., Corpet A., Imhof A., Almouzni G., Groth A., Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell 37, 736–743 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Saredi G., Huang H., Hammond C. M., Alabert C., Bekker-Jensen S., Forne I., Reverón-Gómez N., Foster B. M., Mlejnkova L., Bartke T., Cejka P., Mailand N., Imhof A., Patel D. J., Groth A., H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 534, 714–718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alabert C., Groth A., Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 13, 153–167 (2012). [DOI] [PubMed] [Google Scholar]
- 39.MacAlpine D. M., Almouzni G., Chromatin and DNA replication. Cold Spring Harb. Perspect. Biol. 5, a010207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinberg D. N., Papillon-Cavanagh S., Chen H., Yue Y., Chen X., Rajagopalan K. N., Horth C., McGuire J. T., Xu X., Nikbakht H., Lemiesz A. E., Marchione D. M., Marunde M. R., Meiners M. J., Cheek M. A., Keogh M.-C., Bareke E., Djedid A., Harutyunyan A. S., Jabado N., Garcia B. A., Li H., Allis C. D., Majewski J., Lu C., The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dodson A. E., Rine J., Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae. eLife 4, e05007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Padeken J., Zeller P., Gasser S. M., Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 31, 12–19 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Solovyov A., Vabret N., Arora K. S., Snyder A., Funt S. A., Bajorin D. F., Rosenberg J. E., Bhardwaj N., Ting D. T., Greenbaum B. D., Global cancer transcriptome quantifies repeat element polarization between immunotherapy responsive and T cell suppressive classes. Cell Rep. 23, 512–521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung D. C., Lorincz M. C., Silencing of endogenous retroviruses: When and why do histone marks predominate? Trends Biochem. Sci. 37, 127–133 (2012). [DOI] [PubMed] [Google Scholar]
- 45.He J., Fu X., Zhang M., He F., Li W., Abdul M. M., Zhou J., Sun L., Chang C., Li Y., Liu H., Wu K., Babarinde I. A., Zhuang Q., Loh Y.-H., Chen J., Esteban M. A., Hutchins A. P., Transposable elements are regulated by context-specific patterns of chromatin marks in mouse embryonic stem cells. Nat. Commun. 10, 34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karimi M. M., Goyal P., Maksakova I. A., Bilenky M., Leung D., Tang J. X., Shinkai Y., Mager D. L., Jones S., Hirst M., Lorincz M. C., DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gan H., Yu C., Devbhandari S., Sharma S., Han J., Chabes A., Remus D., Zhang Z., Checkpoint kinase Rad53 couples leading- and lagging-strand DNA synthesis under replication stress. Mol. Cell 68, 446–455.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q. H., Xiong H. Q., Ai S. S., Yu X. H., Liu Y. X., Zhang J. J., He A. B., CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell 76, 206–216.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Ai S., Xiong H., Li C. C., Luo Y., Shi Q., Liu Y., Yu X., Li C., He A., Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol. 21, 1164–1172 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Tran V., Lim C., Xie J., Chen X., Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338, 679–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wooten M., Snedeker J., Nizami Z. F., Yang X., Ranjanl R., Urban E., Kim J. M., Gall J., Xiao J., Chen X., Asymmetric histone inheritance via strand-specific incorporation and biased replication fork movement. Nat. Struct. Mol. Biol. 26, 732–743 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lytle N. K., Barber A. G., Reya T., Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 18, 669–680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saxton D. S., Rine J., Epigenetic memory independent of symmetric histone inheritance. eLife 8, e51421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoye J. P., Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 10, 395–406 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Yang B. X., El Farran C. A., Guo H. C., Yu T., Fang H. T., Wang H. F., Schlesinger S., Seah Y. F. S., Goh G. Y. L., Neo S. P., Li Y., Lorincz M. C., Tergaonkar V., Lim T.-M., Chen L., Gunaratne J., Collins J. J., Goff S. P., Daley G. Q., Li H., Bard F. A., Loh Y.-H., Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 163, 230–245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng L., Zhang X., Wang Y., Gan H., Xu X., Lv X., Hua X., Que J., Ordog T., Zhang Z., Chromatin Assembly Factor 1 (CAF-1) facilitates the establishment of facultative heterochromatin during pluripotency exit. Nucleic Acids Res. 47, 11114–11131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rooney M. S., Shukla S. A., Wu C. J., Getz G., Hacohen N., Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiappinelli K. B., Strissel P. L., Desrichard A., Li H., Henke C., Akman B., Hein A., Rote N. S., Cope L. M., Snyder A., Makarov V., Budhu S., Slamon D. J., Wolchok J. D., Pardoll D. M., Beckmann M. W., Zahnow C. A., Merghoub T., Chan T. A., Baylin S. B., Strick R., Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roulois D., Loo Yau H., Singhania R., Wang Y., Danesh A., Shen S. Y., Han H., Liang G., Jones P. A., Pugh T. J., O'Brien C., De Carvalho D. D., DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.M. J. Dunham, M. J. Dunham, M. R. Gartenberg, G. W. Brown, Methods in Yeast Genetics and Genomics : A Cold Spring Harbor Laboratory Course Manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, ed. 2015, 2015), pp. xvii, 233 p. [Google Scholar]
- 61.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Holt C., Brandt W. F., Greyling H. J., Lindsey G. G., Retief J. D., Rodrigues J. d. A., Schwager S., Sewell B. T., Isolation and characterization of histones. Methods Enzymol. 170, 431–523 (1989). [DOI] [PubMed] [Google Scholar]
- 63.Wang Q., Gu L., Adey A., Radlwimmer B., Wang W., Hovestadt V., Bähr M., Wolf S., Shendure J., Eils R., Plass C., Weichenhan D., Tagmentation-based whole-genome bisulfite sequencing. Nat. Protoc. 8, 2022–2032 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tarasov A., Vilella A. J., Cuppen E., Nijman I. J., Prins P., Sambamba: Fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brogaard K., Xi L., Wang J.-P., Widom J., A map of nucleosome positions in yeast at base-pair resolution. Nature 486, 496–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zang C., Schones D. E., Zeng C., Cui K., Zhao K., Peng W., A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jurka J., Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 16, 418–420 (2000). [DOI] [PubMed] [Google Scholar]
- 71.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J. M., Sisu C., Wright J., Armstrong J., Barnes I., Berry A., Bignell A., Sala S. C., Chrast J., Cunningham F., Domenico T. D., Donaldson S., Fiddes I. T., Girón C. G., Gonzalez J. M., Grego T., Hardy M., Hourlier T., Hunt T., Izuogu O. G., Lagarde J., Martin F. J., Martínez L., Mohanan S., Muir P., Navarro F. C. P., Parker A., Pei B., Pozo F., Ruffier M., Schmitt B. M., Stapleton E., Suner M.-M., Sycheva I., Uszczynska-Ratajczak B., Xu J., Yates A., Zerbino D., Zhang Y., Aken B., Choudhary J. S., Gerstein M., Guigó R., Hubbard T. J. P., Kellis M., Paten B., Reymond A., Tress M. L., Flicek P., GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jin Y., Tam O. H., Paniagua E., Hammell M., TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31, 3593–3599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eabb5820/DC1