

A sulfide oxidase mediates therapeutic actions of H2S by targeting mitochondrial electron flow to induce mitochondrial uncoupling.
Abstract
Hydrogen sulfide (H2S) is a gasotransmitter and a potential therapeutic agent. However, molecular targets relevant to its therapeutic actions remain enigmatic. Sulfide-quinone oxidoreductase (SQR) irreversibly oxidizes H2S. Therefore, SQR is assumed to inhibit H2S signaling. We now report that SQR-mediated oxidation of H2S drives reverse electron transport (RET) at mitochondrial complex I, which, in turn, repurposes mitochondrial function to superoxide production. Unexpectedly, complex I RET, a process dependent on high mitochondrial membrane potential, induces superoxide-dependent mitochondrial uncoupling and downstream activation of adenosine monophosphate–activated protein kinase (AMPK). SQR-induced mitochondrial uncoupling is separated from the inhibition of mitochondrial complex IV by H2S. Moreover, deletion of SQR, complex I, or AMPK abolishes therapeutic effects of H2S following intracerebral hemorrhage. To conclude, SQR mediates H2S signaling and therapeutic effects by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Moreover, SQR is a previously unrecognized target for developing non-protonophore uncouplers with broad clinical implications.
INTRODUCTION
Although classically viewed as an environmental toxicant, hydrogen sulfide (H2S) is now considered an endogenous gasotransmitter that modulates numerous physiological and pathological processes. Moreover, H2S is a potentially versatile therapeutic agent, and several H2S donors have been in clinical trials for treating inflammatory and cardiovascular diseases (1–3). However, molecular targets relevant to therapeutic actions of H2S remain enigmatic. The effects of signaling molecules are supposed to be mediated specifically by a limited number of cellular targets (4). However, the dominant candidate mechanism of H2S signaling, i.e., sulfhydration of target proteins by H2S-derived polysulfides, is nonspecific, since 10 to 25% cellular proteins are sulfhydrated (5). It has been suggested that H2S is not a “true” signaling molecule but acts as an antioxidant to modulate various activities (6). Moreover, H2S is well established as a potent inhibitor of mitochondrial complex IV comparable to cyanide. It is unclear whether therapeutic mechanisms of H2S are separated from the toxic effect of H2S.
Sulfide-quinone oxidoreductase (SQR) is located in mitochondria and initiates irreversible oxidation of H2S. Thus, SQR is long assumed to inhibit H2S signaling. Since SQR has an extremely high activity of H2S oxidation, it has been questioned whether H2S can be maintained at the steady concentrations required for signaling (7). By oxidation of H2S, SQR transfers electrons from H2S to oxidized coenzyme Q (CoQ). Then, electrons are either forwardly transferred to oxygen via mitochondrial complexes III and IV or transferred to oxidized nicotinamide adenine dinucleotide (NAD+) via reverse electron transport (RET) at mitochondrial complex I (8). While the former pathway is bioenergetic (9), the latter pathway is assumed to be essential for detoxification of H2S when H2S at high concentrations inhibits complex IV (8). The normal function of complex I is to create the mitochondrial membrane potential (ΔΨm) that drives adenosine triphosphate (ATP) synthesis. In contrast, complex I produces mitochondrial superoxide [reactive oxygen species (ROS)] via RET (10–13). ROS is an important mediator of cellular signaling (14). Notably, mitochondrial ROS dissipates ΔΨm by activating uncoupling proteins (UCPs), leading to mitochondrial uncoupling (15, 16). Thus, we hypothesize that SQR mediates H2S signaling by driving complex I RET to induce ROS-dependent mitochondrial uncoupling. However, complex I RET is currently thought to be incompatible with mitochondrial uncoupling since high ΔΨm is required to drive complex I RET (10–13). Here, we reported that SQR oxidation of H2S preferentially drove complex I RET, which, in turn, induced ROS-dependent mitochondrial uncoupling and downstream activation of adenosine monophosphate–activated protein kinase (AMPK). Moreover, SQR mediated therapeutic effects of H2S via AMPK activation following intracerebral hemorrhage (ICH). Thus, SQR is a previously unrecognized molecular target relevant to therapeutic actions of H2S. Moreover, targeting SQR opens up the opportunity of developing non-protonophore uncouplers for clinical translation, given that currently identified mitochondrial uncouplers exclusively act as protonophores to induce uncoupling and display intolerable toxicity (17, 18).
RESULTS
Exogenous and endogenous H2S induces mitochondrial uncoupling
Mitochondrial uncoupling reduces intracellular ATP levels and, thereby, activates AMPK (17). On the basis of our finding that H2S activates AMPK to inhibit neuroinflammation in microglia (19), we hypothesize that H2S induces mitochondrial uncoupling in microglia. ADT-OH [5-(4-hydroxyphenyl)-3H-1, 2-dithiole-3-thione] is a widely used slow-releasing H2S donor that requires mitochondrial enzymes to release H2S (20). Like the protonophore uncoupler carbonyl cyanide p-trifluromethoxyphenylhydrazone (FCCP), ADT-OH (50 to100 μM) enhanced cellular oxygen consumption rate (OCR) in the presence of oligomycin (Fig. 1, A and B, and fig. S1A) and dose-dependently reduced intracellular ATP levels and increased adenosine diphosphate (ADP)/ATP ratios in primary microglia (Fig. 1, C and D). Moreover, ADT-OH treatment for 30 min reduced ΔΨm, as indicated by JC-1 staining (Fig. 1, E and F). These are the hallmark properties of mitochondrial uncouplers. Tetramethylrhodamine, methyl ester (TMRM) staining further showed that ADT-OH treatment reduced ΔΨm within 30 min, which was reversible even after 24 hours of treatment (fig. S1, B and C). The structurally unrelated donor 5a [N-(benzoylthio)benzamide] releases H2S in the presence of cellular cysteine (21). Like ADT-OH, 5a at 50 μM induced mitochondrial uncoupling in primary microglia (fig. S1, D to G). ADT-OH also induced mitochondrial uncoupling in BV2 microglia, as evidenced by enhanced OCR in the presence of oligomycin (fig. S1, H and I). Next, we examined the H2S-releasing kinetics of ADT-OH and 5a. In contrast to the instant release pattern of the inorganic donor NaHS, the release of H2S from ADT-OH and 5a was continuous after they were added into BV2 microglia (fig. S1J). ADT-OH and 5a at the concentrations used to induce mitochondrial uncoupling released similar concentrations of H2S in similar time frames in BV2 microglia (fig. S1J). We further incubated BV2 microglia in medium containing ZnCl2, which can block the effects of free H2S (22). ZnCl2 not only abolished ADT-OH–induced reduction in ΔΨm and intracellular ATP levels (fig. S2, A and B) but also blocked ADT-OH–induced enhancement of OCR in the presence of oligomycin in BV2 microglia (fig. S2, C and D). ZnCl2 also abolished 5a-induced reduction in ΔΨm and intracellular ATP levels (fig. S2, E and F). These results suggested that these donors induced mitochondrial uncoupling via H2S.
Fig. 1. Exogenous and endogenous H2S induces mitochondrial uncoupling.
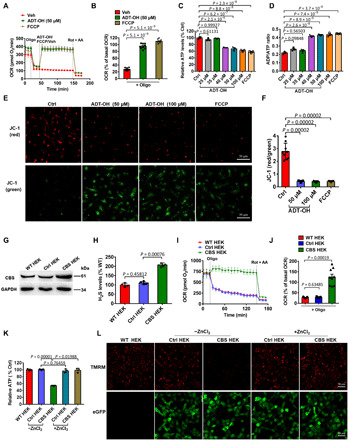
(A to F) Real-time change and statistical analysis of OCR (A and B; n = 9), ATP levels (C; n = 3), ADP/ATP ratios (D; n = 3), and ΔΨm (E, representative images of JC-1 staining; F, statistical analysis; n = 9) in primary microglia treated with the H2S donor ADT-OH or the protonophore uncoupler FCCP. For OCR assay, oligomycin (Oligo), rotenone (Rot), and antimycin A (AA) were added at the indicated time points. ATP/ADP and ΔΨm were assessed at 30 min after treatment with ADT-OH or FCCP. Control (Ctrl), vehicle (Veh)–treated cells. (G) Protein levels of the H2S synthase cystathionine β-synthase (CBS) in wild-type human embryonic kidney (HEK) 293 cells (WT HEK), control HEK293 cells stably expressing enhanced green fluorescent protein (eGFP) (Ctrl HEK), and recombinant HEK293 cells stably overexpressing CBS and eGFP (CBS HEK); three independent replicates. (H) Relative H2S levels in medium collected from WT HEK, Ctrl HEK, or CBS HEK cultures (n = 4). (I to K) Real-time change and statistical analysis of OCR (I and J; n = 9), and ATP levels (K; n = 3) in WT HEK, Ctrl HEK, and CBS HEK cells. ZnCl2 was added to block the effects of free H2S. (L) ΔΨm in WT HEK, Ctrl HEK, and CBS HEK cells with/without ZnCl2 (three independent replicates). Red, tetramethylrhodamine, methyl ester (TMRM) fluorescence; green, eGFP fluorescence. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
By constructing recombinant human embryonic kidney (HEK) 293 cells overexpressing the H2S synthase cystathionine β-synthase (CBS) (1), we further confirmed that endogenously produced H2S induced mitochondrial uncoupling. CBS overexpression enhanced endogenous H2S levels (Fig. 1, G and H). Oligomycin did not reduce OCR in CBS-overexpressing HEK293 versus wild-type or control HEK293 cells expressing enhanced green fluorescent protein (eGFP) (Fig. 1, I and J). Moreover, overexpression of CBS reduced intracellular ATP levels and ΔΨm, and these effects were abolished by ZnCl2 (Fig. 1, K and L).
H2S-induced mitochondrial uncoupling requires SQR and is separable from the inhibition of complex IV by H2S
SQR protein and mRNA were abundantly expressed in mouse primary microglia and astrocytes but not in primary neurons (Fig. 2, A and B). SQR was also expressed in human-derived HEK293 cells (fig. S3A). Unlike FCCP, ADT-OH (50 to 100 μM) did not induce mitochondrial uncoupling in primary neurons (Fig. 2, C to G). Conversely, in the primary neurons ectopically expressing SQR (fig. S3B), ADT-OH (50 to 100 μM) induced mitochondrial uncoupling, as indicated by enhanced OCR in the presence of oligomycin (Fig. 2, H and I), reduced intracellular ATP levels (Fig. 2J), and ΔΨm (fig. S3D). Consistently, lentiviral knockdown of SQR in primary microglia (fig. S3C) abolished ADT-OH–induced mitochondrial uncoupling (Fig. 2, K to M, and fig. S3E). Knockdown of SQR also abolished 5a (50 μM)–induced reduction in ΔΨm and intracellular ATP levels in primary microglia (fig. S3, F and G). To conclude, H2S induced mitochondrial uncoupling via SQR.
Fig. 2. H2S-induced mitochondrial uncoupling requires SQR and is separated from the inhibition of complex IV by H2S.
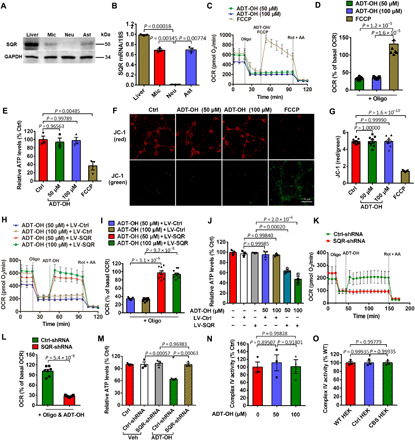
(A and B) Expression levels of SQR protein (A) and mRNA (B) in mouse primary microglia (Mic), neurons (Neu), and astrocytes (Ast). Liver: positive control. (C to G) ADT-OH failed to induce mitochondrial uncoupling in primary neurons, as indicated by real-time change and statistical analysis of OCR (C and D; n = 6), ATP levels (E; n = 3), and ΔΨm (F, representative images of JC-1 staining; G, statistical analysis; n = 9). Ctrl, Veh-treated neurons. (H to J) Real-time change and statistical analysis of OCR (H and I; n = 9), and ATP levels (J; n = 3) in primary neurons infected with lentivirus expressing SQR (LV-SQR) or control lentivirus (LV-Ctrl) and then treated with ADT-OH. (K to M) Real-time change and statistical analysis of OCR (K and L; n = 7), and ATP levels (M; n = 3) in primary microglia infected with lentivirus expressing SQR-shRNA (short hairpin RNA) or nonsense shRNA (Ctrl-shRNA) and then treated with ADT-OH. Ctrl, cells without lentiviral infection. (N) Complex IV activity in BV2 microglia treated with ADT-OH. (O) Complex IV activity in WT HEK, Ctrl HEK, and CBS HEK cells; n = 3.
As a toxicant, H2S potently inhibits complex IV. However, at the concentrations used to induce mitochondrial uncoupling, ADT-OH (50 to 100 μM) and 5a (50 μM) did not inhibit complex IV in BV2 microglia (Fig. 2N and fig. S3H). Moreover, ADT-OH (50 to 100 μM) and 5a (50 μM) enhanced the basal OCR and the extracellular acidification rate (ECAR) in BV2 microglia (fig. S3, I and J), further supporting that ADT-OH and 5a at these concentrations induced mitochondrial uncoupling and did not inhibit complex IV. Similarly, CBS overexpression did not inhibit complex IV in HEK293 cells (Fig. 2O). Moreover, there was a mild trend of an elevation of basal OCR and ECAR in HEK cells overexpressing CBS versus control or wild-type HEK cells (fig. S3, K and L). ADT-OH is insoluble in aqueous medium beyond 100 μM (19). Notably, 5a at 100 to 150 μM inhibited complex IV in microglia (fig. S3H). Consistently, 5a at 100 μM considerably reduced basal OCR (fig. S3I) and did not enhance OCR in the presence of oligomycin in BV2 microglia (fig. S3M). Collectively, these results suggested that H2S did not inhibit complex IV at the concentrations used to induce mitochondrial uncoupling and vice versa.
It is thought that cells lacking SQR like neurons are exquisitely sensitive to the inhibition of OCR by H2S. However, at the concentrations used to induce mitochondrial uncoupling, ADT-OH (50 to 100 μM) and 5a (50 μM) did not inhibit basal OCR of primary neurons (fig. S4A). Consistently, like ADT-OH at 50 to 100 μM, 5a at 50 μM did not reduce ATP levels (fig. S4B) and ΔΨm (fig. S4C) in primary neurons. These data further supported that ADT-OH and 5a at these concentrations required SQR to induce mitochondrial uncoupling and did not inhibit complex IV. Consistent with the inhibition of complex IV by 5a at 100 μM, 5a at 100 μM suppressed basal OCR of primary neurons (fig. S4A). Moreover, 5a (100 to 150 μM) reduced intracellular ATP levels (fig. S4B) and ΔΨm (fig. S4C) in primary neurons lacking SQR. These data suggested that 5a at high concentrations reduced ATP levels by inhibiting complex IV. Notably, incubating neurons with ADT-OH or 5a at 50 to 100 μM for 2 hours did not reduce neuronal viability (fig. S4D). It is possible that H2S is metabolized into nontoxic products via alternative mechanisms in neurons lacking SQR.
SQR-mediated oxidation of H2S drives complex I RET to repurpose mitochondrial function to ROS production
Complex I RET is driven by a highly reduced CoQ pool and high ΔΨm (23). Theoretically, oxidation of H2S by SQR transfers electrons from H2S to CoQ, resulting in an enhanced CoQ reduction. Consistent with the theoretic prediction, treating microglia with ADT-OH or 5a for 10 min at the concentration used to induce mitochondrial uncoupling enhanced CoQH2/CoQ ratios (Fig. 3, A and B). Moreover, treating microglia with ADT-OH for 10 min elevated ΔΨm, which preceded the drop in ΔΨm occurring at 25 min after treatment with ADT-OH (Fig. 3, C and D). Thus, H2S simultaneously elevated ΔΨm, and the reduced pool of CoQH2 before ΔΨm was decreased by H2S. Mitochondrial superoxide (ROS) burst is a hallmark of complex I RET (10). ADT-OH treatment persistently elevated mitochondrial ROS levels in BV2 microglia for at least 24 hours, and the elevation of ROS was abolished by rotenone (Fig. 3E), the inhibitor of complex I RET (10, 12, 24). Moreover, knockdown of reduced form of NAD+ (NADH) dehydrogenase (ubiquinone) Fe-S protein 3 (NDUFS3), a subunit of complex I, also abolished ADT-OH–induced mitochondrial ROS burst in HEK293 cells (Fig. 3, F and G). Collectively, these results suggested that H2S induced complex I RET and consequently repurposed mitochondrial function to ROS production.
Fig. 3. SQR-mediated oxidation of H2S drives complex I RET to repurpose mitochondrial function to ROS production.
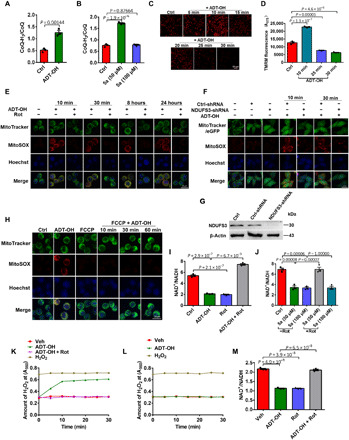
(A and B) CoQH2/CoQ ratios in BV2 microglia treated with ADT-OH (50 μM; A) or 5a (B); n = 3. (C and D) Representative images of the change in ΔΨm of BV2 microglia after treatment with ADT-OH (C) and quantification data (D; n = 3). (E) Mitochondrial ROS levels in BV2 microglia simultaneously treated with ADT-OH (50 μM) and/or Rot for the indicated periods of time. (F) Mitochondrial ROS levels in HEK293 cells infected with lentivirus expressing NDUFS3-shRNA or Ctrl-shRNA for 2 days and then treated with ADT-OH for the indicated periods of time. (G) NDUFS3 knockdown efficiency in HEK293 cells. (H) Mitochondrial ROS levels in BV2 microglia pretreated with 20 nM FCCP and then treated with ADT-OH for the indicated periods of time. (I and J) Mitochondrial NAD+/reduced form of NAD+ (NADH) ratios in BV2 microglia treated with ADT-OH (50 μM) or 5a with or without Rot; n = 3. (K and L) H2O2 levels in microglia-derived mitochondria (K) or neuron-derived mitochondria (L) following treatment with ADT-OH with or without Rot. H2O2, positive control. (M) NAD+/NADH ratios in microglia-derived mitochondria treated with ADT-OH and/or Rot.
The production of mitochondrial ROS via complex I RET tightly depends on high ΔΨm, and even a slight decrease in ΔΨm induced by FCCP at nanomolar concentrations dramatically reduces ROS generation via complex I RET (13). Consistently, pretreatment of BV2 microglia with 20 nM FCCP abolished ADT-OH–induced mitochondrial ROS burst (Fig. 3H), further supporting that ADT-OH induced ROS generation via complex I RET. As we showed above, treatment with ADT-OH reduced ΔΨm within 25 min, but ADT-OH–induced mitochondrial ROS burst lasted for 24 hours. Theoretically, ADT-OH cannot induce ROS generation via complex I RET after the drop in ΔΨm. As expected, ADT-OH–induced ROS generation after the drop in ΔΨm was not inhibited by FCCP added at 1 hour of ADT-OH treatment (fig. S5A). Notably, pretreating microglia with FCCP to prevent ADT-OH–induced complex I RET also inhibited ROS generation at 1 hour after ADT-OH treatment (Fig. 3H). This indicated that complex I RET induced by ADT-OH before the drop in ΔΨm was also required for ADT-OH–induced sustained ROS production after the drop in ΔΨm.
Next, we investigated whether endogenous H2S drove complex I RET. CBS-overexpressing HEK293 cells were cultured overnight. Notably, ΔΨm of CBS-overexpressing HEK293 cells recovered within 35 min and then dropped again at 50 min after we replaced the overnight culture medium with fresh medium (fig. S5, B and C). Endogenous H2S production enhanced by CBS overexpression was likely responsible for the change in ΔΨm of CBS-overexpressing cells after medium replacement. ΔΨm of CBS-overexpressing HEK293 cells was elevated to a level higher than that of wild-type or control HEK293 cells before it dropped again at 50 min of medium replacement (fig. S5, B and C). Thus, like exogenous H2S, endogenous H2S also elevated ΔΨm before it reduced ΔΨm. Moreover, the reduced CoQ pool was enhanced in CBS-overexpressing HEK293 cells versus wild-type or control HEK293 cells (fig. S5D). Overexpression of CBS also induced mitochondrial ROS burst that was inhibited by rotenone (fig. S5E). Collectively, the results suggested that both exogenous and endogenous H2S drove complex I RET and consequently repurposed mitochondrial function to ROS production.
Theoretically, H2S cannot induce ROS generation via complex I RET after H2S reduces ΔΨm below what is required to drive complex I RET. Notably, sustained ROS generation induced by ADT-OH at the later time points (8 or 24 hours) was abolished by the complex I inhibitor rotenone and the complex II inhibitors malonate and atpenin A5, which were added at 8 or 24 hours following addition of ADT-OH (fig. S5F). It is reported that ROS generation via deactive complex I is inhibited by rotenone but insensitive to FCCP (25, 26). Whether H2S induced sustained ROS generation via deactive complex I and other mitochondrial complexes after the drop in ΔΨm needs further investigation.
The decrease in mitochondrial NAD+/NADH ratios is another surrogate marker for complex I RET (24). As expected, ADT-OH or 5a at the concentration used to induce mitochondrial uncoupling decreased mitochondrial NAD+/NADH ratios in BV2 microglia (Fig. 3, I and J). Notably, 5a at 100 μM inhibited complex IV and also reduced mitochondrial NAD+/NADH ratios (Fig. 3J). Unlike inhibition of complex IV, complex I RET requires complex I activity to reduce mitochondrial NAD+/NADH ratios (24). The complex I inhibitor rotenone reversed the reduction in mitochondrial NAD+/NADH ratios in the microglia treated with ADT-OH or 5a at 50 μM (Fig. 3, I and J) but not in the microglia treated with 100 μM 5a (Fig. 3J). These results suggested that H2S drove complex I RET at the concentration that induced mitochondrial uncoupling.
Last, we confirmed that H2S induced complex I RET via SQR in isolated mitochondria. To indicate complex I RET, H2O2 generation was assessed. At 10 min of ADT-OH treatment, H2O2 generation was induced in mitochondria isolated from BV2 microglia, and the induction was inhibited by rotenone (Fig. 3K). In contrast, ADT-OH failed to enhance H2O2 generation in mitochondria isolated from primary neurons lacking endogenous SQR (Fig. 3L). Moreover, ADT-OH treatment for 10 min decreased NAD+/NADH ratios in mitochondria isolated from BV2 microglia, and the decrease was reversed by rotenone (Fig. 3M). To conclude, SQR-mediated oxidation of H2S drove complex I RET and consequently repurposed mitochondrial function to ROS generation.
H2S induces mitochondrial uncoupling via complex I RET and ROS-dependent activation of UCP2
To investigate whether H2S induced mitochondrial uncoupling via complex I RET, we used rotenone, the inhibitor of complex I RET (10, 12, 24). Rotenone abolished ADT-OH (50 μM)–induced elevation of OCR in oligomycin-treated microglia (Fig. 4, A and B). Rotenone itself decreased intracellular ATP levels and ΔΨm (Fig. 4, C to E). Notably, rotenone reversed the decrease in intracellular ATP levels and ΔΨm in primary microglia induced by ADT-OH (50 μM) but not in that induced by FCCP (Fig. 4, C to E). Similarly, rotenone abolished 5a (50 μM)–induced mitochondrial uncoupling in BV2 microglia (fig. S6, A to D). Next, we blocked complex I RET by knocking down the complex I subunit NDUFS3. Knockdown of NDUFS3 in HEK293 cells inhibited complex I activity, as evidenced by the reduction in basal OCR (Fig. 4, F and G). Knockdown of NDUFS3 abolished ADT-OH–induced mitochondrial uncoupling in HEK293 cells (Fig. 4, F to I). Rotenone also abolished the CBS overexpression–induced mitochondrial uncoupling in HEK293 cells (fig. S6, E to H). Thus, both pharmacological and genetic inhibition of complex I abolished H2S-induced mitochondrial uncoupling. Moreover, inhibiting complex IV with potassium cyanide (KCN) abolished ADT-OH–induced increase in OCR in the presence of oligomycin in BV2 microglia (fig. S6, I and J). This implicated that forward electron transfer was occurring when H2S induced mitochondrial uncoupling.
Fig. 4. H2S induces mitochondrial uncoupling via complex I RET and UCP2.
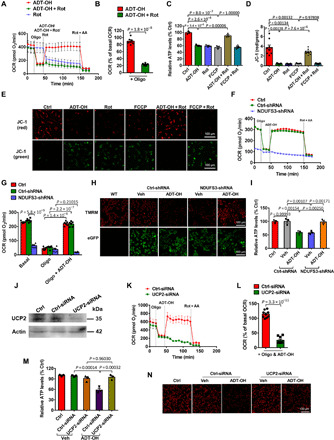
(A to E) The complex I RET inhibitor Rot abolished ADT-OH (50 μM)–induced mitochondrial uncoupling in primary microglia, as indicated by OCR (A, real-time change; B, statistical results; n = 6), ATP levels (C; n = 3), and ΔΨm (D, statistical results; n = 9; E, representative images of JC-1 staining). For OCR assay, the reagents were added, as indicated, except that Oligo was not added to the cells treated with Rot alone. Ctrl, Veh-treated cells. (F to I) Knockdown of the complex I subunit NDUFS3 abolished ADT-OH (50 μM)–induced mitochondrial uncoupling in HEK293 cells, as indicated by OCR (F, real-time change; G, statistical results; n = 6), ΔΨm (H, representative images of TMRM staining; green, eGFP fluorescence in NDUFS3-shRNA–infected or Ctrl-shRNA–infected HEK293 cells; three independent replicates), and ATP levels (I; n = 3). To indicate mitochondrial uncoupling, ATP levels and ΔΨm were assessed at 30 min after ADT-OH and/or Rot treatment. (J) UCP2 knockdown efficiency in BV2 microglia (three independent replicates). (K to N) Knockdown of UCP2-abolished ADT-OH (50 μM)–induced mitochondrial uncoupling in BV2 microglia, as indicated by OCR (K, real-time change; L, statistical results; n = 9), intracellular ATP levels (M; n = 3), and ΔΨm (N, representative images of TMRM staining; three independent replicates).
We showed that both H2S and the complex I inhibitor rotenone suppress ATP synthesis. Unexpectedly, H2S donors at the concentration used to induce mitochondrial uncoupling maintained cellular ATP levels in rotenone-treated cells. H2S stimulates glycolysis (27). We investigated whether ADT-OH promoted glycolysis to maintain ATP levels in rotenone-treated cells by assessing lactate accumulation. ADT-OH at 50 μM inhibited lactate production in rotenone-treated microglia (fig. S6K), suggesting that ADT-OH inhibited glycolysis in the presence of rotenone. H2S may serve as an energetic substrate via SQR-mediated oxidation of H2S, which bypasses complex I and is coupled to ATP synthesis by directly feeding electrons from H2S to mitochondrial complex III and IV (9). ADT-OH enhanced OCR in rotenone-treated microglia (fig. S6, L and M), implicating that SQR-mediated oxidation of H2S was coupled to ATP synthesis when complex I RET was inhibited by rotenone. The results further supported that SQR-mediated oxidation of H2S preferentially drove complex I RET to induce mitochondrial uncoupling unless complex I RET was inhibited.
To investigate whether H2S-induced complex I RET led to mitochondrial uncoupling via ROS-dependent activation of UC2, we used MitoTEMPO, a mitochondria-targeted antioxidant (11). Pretreatment of microglia with MitoTEMPO abolished ADT-OH–induced mitochondrial ROS burst (fig. S7A) and uncoupling (fig. S7, B to E). UCPs are the direct downstream mediators of ROS-dependent mitochondrial uncoupling (15, 16). UCP2 is expressed in microglia (28). Notably, knockdown of UCP2 blocked ADT-OH–induced mitochondrial uncoupling in BV2 microglia (Fig 4, J to N). Genipin specifically suppresses ROS-dependent activation of UCP2 (29). Genipin also abolished ADT-OH–induced mitochondrial uncoupling in microglia (fig. S7, F to I). Collectively, these results suggested that ROS-dependent activation of UCP2 mediated H2S-induced mitochondrial uncoupling in microglia.
The protonophoric ability of chemical uncouplers like FCCP can be tested by induction of swelling of nonrespiring liver mitochondria in the presence of rotenone (30). To exclude the possibility that ADT-OH directly functions as a protonophore to induce uncoupling and the possibility that protonophore uncouplers also require SQR to induce uncoupling, we performed mitochondrial swelling assay using mitochondria isolated from the livers of global SQR knockout mice. As a protonophore uncoupler, FCCP should induce mitochondrial swelling regardless of SQR expression. FCCP induced swelling of the mitochondria from SQR knockout mice (fig. S7J), supporting that SQR is not required for the protonophoric ability of chemical uncouplers. Since ADT-OH induced mitochondrial uncoupling via SQR-mediated RET, we expected that SQR deletion and rotenone would block the protonophoric ability of ADT-OH. As expected, ADT-OH did not induce swelling of the mitochondria deficient in SQR (fig. S7J). The results supported that ADT-OH was not a protonophore uncoupler. Instead, the protonophoric ability of ADT-OH was mediated by SQR and complex I.
H2S activates AMPK via SQR-induced mitochondrial uncoupling
Activation of AMPK is a potential mechanism underlying H2S signaling and therapeutic effects (31). Notably, mitochondrial uncoupling is an important mechanism underlying AMPK activation (17). Thus, we asked whether H2S-induced activation of AMPK was mediated by SQR-induced mitochondrial uncoupling. Unlike FCCP, ADT-OH (50 to 100 μM) failed to activate AMPK in primary neurons lacking endogenous SQR (fig. S8A). Conversely, ADT-OH (50 to 100 μM) activated AMPK in primary neurons ectopically expressing SQR (fig. S8B). Consistently, knockdown of SQR in primary microglia or BV2 microglia attenuated AMPK activation enhanced by ADT-OH (fig. S8, C and D) or by 50 μM 5a (fig. S8E). Moreover, inhibiting complex I RET with rotenone attenuated activation of AMPK enhanced by ADT-OH in microglia (fig. S8, F and G) and of that enhanced by CBS overexpression in HEK293 cells (fig. S8H). Since SQR and complex I RET were required for H2S-induced mitochondrial uncoupling, these results implicated that SQR-mediated oxidation of H2S enhanced activation of AMPK by driving complex I RET to induce mitochondrial uncoupling. Moreover, since mitochondrial ROS cannot activate AMPK directly (32), it is unlikely that SQR-mediated oxidation of H2S activated AMPK via mitochondrial ROS directly.
SQR mediates therapeutic effects of H2S via AMPK activation
H2S inhibits inflammation, and H2S donors have been in clinical trials for treating inflammatory diseases (1, 3). Notably, AMPK activation is an important mechanism underlying the inhibition of inflammation (11, 19, 33). Since SQR-mediated oxidation of H2S induced mitochondrial uncoupling and consequently activated AMPK, we hypothesized that SQR mediated therapeutic effects of H2S on inflammation via AMPK activation. To test the hypothesis, we used ICH-induced neuroinflammation as a model system. ICH is initiated by the rupture of cerebral vessels, followed by the entry of blood components into the cerebral parenchyma to elicit multiple pathogenic cascades, including microglia/macrophage-mediated neuroinflammation (34, 35). To mimic ICH-induced neuroinflammation, cultured microglia were treated with lysate of red blood cells (RBC) (35). ADT-OH at 50 μM inhibited RBC lysate–induced expression of proinflammatory mediators in BV2 microglia (Fig. 5, A to D) and primary microglia (fig. S9, A and B), and the inhibition was abolished by knockdown of SQR. ADT-OH enhanced the activation of AMPK in RBC lysate–treated BV2 microglia (Fig. 5, E and F) and primary microglia (fig. S9, C and D), which was also attenuated by SQR knockdown. Moreover, AMPK knockdown in BV2 microglia abolished the inhibitory effects of ADT-OH on RBC lysate–induced expression of proinflammatory mediators (Fig. 5, G to J). H2S-induced mitochondrial uncoupling was dependent on complex I RET. To block complex I RET, we knocked down the mitochondrial complex I subunit NDUFS3. Knockdown of NDUFS3 abolished the inhibitory effects of ADT-OH on the RBC lysate–enhanced expression of proinflammatory mediators in BV2 microglia (Fig. 5, K to M). NDUFS3 knockdown also blunted the ADT-OH–enhanced AMPK activation (Fig. 5, N to P). Moreover, the small interfering RNA (siRNA) against SQR, AMPK, or NDUFS3 did not affect basal inflammatory gene expression in microglia (fig. S9E). Collectively, the in vitro results suggested that SQR mediated the therapeutic effects of H2S by targeting mitochondrial electron flow at complex I to activate AMPK. AP39 is a derivative of ADT-OH with a mitochondrion-targeting moiety. If the beneficial effects of ADT-OH against neuroinflammation were mainly due to RET at mitochondrial complex I, then AP39 would recapitulate anti-inflammatory effects of ADT-OH at lower concentrations. As expected, AP39 at lower concentrations inhibited inflammation in RBC lysate–treated microglia compared to ADT-OH (fig. S9, F to H).
Fig. 5. SQR mediates therapeutic effects of H2S by targeting mitochondrial electron transfer at complex I to activate AMPK in vitro.
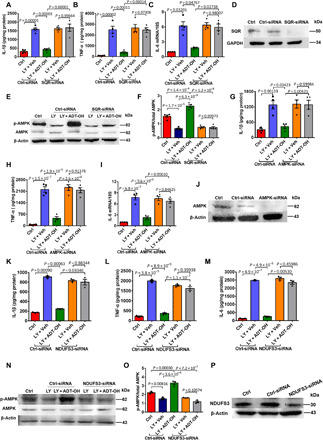
(A to C) Protein (n = 4) or mRNA (n = 3) levels of proinflammatory mediators in BV2 microglia transfected with SQR-siRNA (small interfering RNA) or nonsense siRNA (Ctrl-siRNA) for 2 days and then treated with the RBC lysate (LY) and ADT-OH/Veh. (D) SQR knockdown efficiency in BV2 microglia. (E and F) Immunoblot images of AMPK activation in BV2 microglia transfected with SQR-siRNA or Ctrl-siRNA and then treated with LY and ADT-OH/Veh (E) and quantification data (F). (G to I) Protein or mRNA levels of proinflammatory mediators in BV2 microglia transfected with AMPK-siRNA or Ctrl-siRNA and then treated with LY and ADT-OH/Veh; n = 4. (J) AMPK knockdown efficiency in BV2 microglia. (K to M) Protein levels of proinflammatory mediators in BV2 microglia transfected with NDUFS3-siRNA or Ctrl-siRNA and then treated with LY and ADT-OH/Veh; n = 4. (N and O) Immunoblot images of AMPK activation in BV2 microglia transfected with NDUFS3-siRNA or Ctrl-siRNA and then treated with LY and ADT-OH/Veh (N) and quantification data (O). (P) NDUFS3 knockdown efficiency in BV2 microglia.
To further address whether SQR mediated therapeutic actions of H2S in vivo, we used the CRISPR-Cas9 technique to generate SQR knockout mice. Since we found that global SQR knockout mice died before 2 months old, we generated mice with microglia/macrophage-specific deletion of SQR (Cx3cr1Cre:Sqrfl/fl; Fig. 6A). Cx3cr1Cre:Sqrfl/fl mice did not show any overt abnormalities. Immunoblot analysis of SQR expression in primary microglia derived from Cx3cr1Cre:Sqrfl/fl mice confirmed efficient ablation of SQR protein (Fig. 6B). In the mouse ICH model induced by injecting collagenase into the left striatum (35), ADT-OH ameliorated brain edema (Fig. 6C), reduced proinflammatory mediator expression in the ipsilateral striatum (Fig. 6, D to F), and improved neurological deficits (Fig. 6, G to I) in the control mice (Sqr1fl/fl) at 3 days after ICH. As expected, microglia/macrophage-specific deletion of SQR abolished the therapeutic effects of ADT-OH following ICH (Fig. 6, C to I). Moreover, ADT-OH enhanced AMPK activation in the striatum of the control mice following ICH, and ADT-OH–enhanced activation of AMPK following ICH was abolished in mice with microglia/macrophage-specific deletion of SQR (Fig. 6J).
Fig. 6. SQR mediates therapeutic effects of H2S via AMPK activation following ICH in vivo.
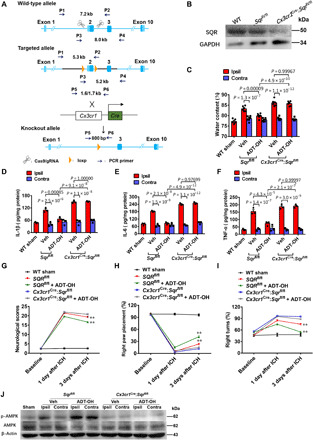
(A) Schema illustrating generation of mice with microglia/macrophage-specific deletion of SQR (Cx3cr1cre:Sqrfl/fl). (B) Immunoblot analysis of SQR expression on primary microglia from Cx3cr1cre:Sqrfl/fl mice, floxed control mice (Sqrfl/fl), and wild-type mice (WT); three independent replicates. (C) Brain edema assessed by measuring water content in the ipsilateral (hemorrhagic) striatum (Ipsil) and contralateral striatum (Contra) at 3 days following ICH (n = 6). WT sham, sham-operated wild-type mice. ADT-OH (50 mg/kg per day) was administered for 3 days, starting at 3 hours after ICH. (D to F) Protein levels of interleukin-1β (IL-1β), IL-6, and tumor necrosis factor–α (TNF-α) in the striatum 3 days following ICH (n = 4). (G to I) Neurological deficits assessed by the neurological score, forelimb placement, and corner test following ICH (n = 6). **P < 0.01, indicating a statistically significant difference between the control mice treated with Veh (Sqrfl/fl) and the control mice treated with ADT-OH (Sqrfl/fl + ADT-OH) at 3 days after ICH. No statistical difference was detected between Veh-treated Cx3cr1cre:Sqrfl/fl mice (Cx3cr1cre:Sqrfl/fl) and ADT-OH–treated Cx3cr1cre:Sqrfl/fl mice (Cx3cr1cre:Sqrfl/fl + ADT-OH). (J) AMPK activation in the striatum of the mice treated with ADT-OH or Veh at 3 days following ICH (three independent replicates).
To address potential off-target effects during microglial differentiation in germline Cx3cr1Cre:Sqrfl/fl mice, we used an alternative approach. To do this, endogenous SQR expression was knocked down in vivo by injecting lentivirus expressing SQR short hairpin RNA (shRNA) into the left striatum of adult mice (fig. S9I). Then, ICH was induced by injecting autologous blood into the left striatum at 14 days after lentiviral injection. Lentiviral knockdown of endogenous SQR in the left striatum abolished the therapeutic effects of ADT-OH following ICH (fig. S9, J to O). Moreover, SQR knockdown attenuated ADT-OH–enhanced activation of AMPK in the ipsilateral striatum (fig. S9P). Collectively, we provided evidence that SQR mediated the therapeutic effects of H2S via AMPK activation following ICH.
DISCUSSION
In this study, we provided evidence that SQR mediated the therapeutic effects of H2S by driving complex I RET to induce mitochondrial uncoupling. The discovery widens the function of SQR, from sulfide metabolism to cellular signaling. Current research shows that SQR-mediated oxidation of H2S is coupled to mitochondrial ATP synthesis by feeding electrons from H2S to mitochondrial complexes III and IV (9). In contrast, we showed herein that SQR-mediated oxidation of H2S repurposed mitochondrial function from ATP synthesis to ROS production by driving complex I RET. Consequently, oxidation of H2S by SQR induced ROS-dependent mitochondrial uncoupling. SQR-mediated oxidation of H2S induces complex I RET in colonocytes that are physiologically exposed to high concentrations of H2S (8). Functionally, H2S-induced complex I RET may be essential for detoxification of H2S when H2S at high concentrations inhibits complex IV (8). However, we found that H2S donors did not drive complex I RET at high concentrations that inhibited complex IV (Fig. 3, B and J, and fig. S3H). Moreover, SQR-mediated oxidation of H2S drove complex I RET in microglia that are not exposed to high levels of H2S physiologically. SQR-mediated oxidation of H2S not only mediated H2S signaling via mitochondrial uncoupling but also mediated therapeutic effects of H2S via uncoupling-induced activation of AMPK. The finding is in contrast to the long-held view that SQR extinguishes H2S signaling and effects (7, 8). Since mitochondrial uncoupling and downstream activation of AMPK regulate numerous pathophysiological processes, SQR-induced mitochondrial uncoupling likely represents a common mechanism underlying diverse effects of H2S, including beneficial effects of endogenous H2S (36) and therapeutic effects of H2S donors.
Our results suggested that H2S-induced mitochondrial uncoupling was dependent on complex I RET. Since high ΔΨm is required to drive complex I RET (10–12), complex I RET is currently thought to be incompatible with mitochondrial uncoupling. In contrast to the assumption, both genetic and pharmacological inhibition of complex I RET abolished H2S-induced mitochondrial uncoupling (Fig. 4, A to I), supporting that H2S-induced complex I RET led to mitochondrial uncoupling. Mechanistically, complex I RET initiated generation of mitochondrial ROS, and ROS-dependent activation of UCP2 in turn led to H2S-induced mitochondrial uncoupling in microglia. Numerous studies show that H2S reduces oxidative stress and ROS. Here, we showed that H2S induced mitochondrial uncoupling by enhancing mitochondrial ROS. ROS-dependent mitochondrial uncoupling may represent an important mechanism underlying the inhibitory effects of H2S on oxidative stress. In addition to UCPs, mitochondrial ADP/ATP carrier proteins, i.e., adenine nucleotide translocases (ANT), also mediate mitochondrial uncoupling (37). Whether ANT proteins contribute to H2S-induced mitochondrial uncoupling needs further investigation. To conclude, SQR-mediated oxidation of H2S initiated mitochondrial ROS generation by driving complex I RET, which, in turn, induced ROS-dependent mitochondrial uncoupling via UCPs (e.g., UCP2 and ANT) and consequently activated AMPK (fig. S10). This likely represents a new mechanism underlying therapeutic actions of H2S.
Complex I RET led to H2S-induced uncoupling by enhancing mitochondrial ROS. Notably, after H2S-induced uncoupling reduced ΔΨm below what is required to drive complex I RET, H2S continued to induce ROS generation via alternative mechanisms (fig. S5F). One limitation of the study was that we did not uncover the mechanisms underlying sustained ROS generation induced by H2S after the drop in ΔΨm. Notably, complex I RET, which was initiated before the drop in ΔΨm, was also required for H2S-induced sustained ROS generation after the drop in ΔΨm (Fig. 3H). This further supported the dependence of H2S-induced mitochondrial uncoupling on complex I RET. Nevertheless, in addition to complex I RET, other mechanisms also contributed to H2S-induced mitochondrial uncoupling and therapeutic actions of H2S, which need further investigation.
In vitro results suggested that SQR mediated the therapeutic effects of H2S via complex I RET and AMPK activation. Consistently, in vivo deletion of SQR in microglia/macrophages abolished the therapeutic effects of ADT-OH and ADT-OH–enhanced AMPK activation in a mouse ICH model (Fig. 6 and fig. S9). The in vivo results also suggested that SQR was important for therapeutic actions of H2S. However, further research is needed to investigate whether therapeutic actions of H2S involve complex I RET in vivo. Moreover, mitochondrial uncoupling exerts pleiotropic effects. Although AMPK activation is a major mechanism downstream of mitochondrial uncoupling, we cannot exclude the possibility that SQR-induced uncoupling mediated the therapeutic actions of H2S via other mechanisms.
Our discovery has translational significance beyond H2S biomedicine. Mitochondrial uncoupling extends life span and is attractive for treating various diseases, such as metabolic diseases and neurodegenerative diseases (17, 18). Despite great efforts that have been dedicated to seek safe chemical uncouplers for more than 80 years, protonophore uncouplers are still the only class of uncouplers identified so far (17, 18). Protonophore uncouplers display intolerable toxicity for clinical use. Current research focuses on developing new chemical structures or controlled-release formulations of protonophore uncouplers (17, 18). Unlike these approaches, our study opens up the opportunity of targeting SQR to develop non-protonophore uncouplers with broad implications for clinical translation.
Although there are inconsistent results about inhibitory effects of H2S on mitochondrial electron transfer (38), it is generally thought that H2S donors have too narrow therapeutic index due to the inhibition of complex IV by H2S (7). We showed herein that SQR-induced mitochondrial uncoupling mediated therapeutic actions of H2S, which was separable from the toxic effect of H2S on complex IV. Thus, it is theoretically possible to develop safe H2S donors. Moreover, due to the cell-specific expressing pattern of SQR, H2S donors represent a class of uncouplers with cellular specificity. In conclusion, SQR is a previously unrecognized target relevant to therapeutic actions of H2S. Moreover, SQR is a druggable target for developing non-protonophore mitochondrial uncouplers with broad implications for clinical translation.
MATERIALS AND METHODS
Construction of recombinant HEK293 cells overexpressing CBS
HEK293 cells (Cell Bank of Chinese Academy of Sciences, Shanghai, China) were seeded on 24-well plates and infected with lentivirus coexpressing CBS complementary DNA (NM_144855) and eGFP or control lentiviruses expressing eGFP only (GeneChem Inc., Shanghai, China). At 48 hours after infection, cells were incubated with medium containing puromycin (2 μg/ml) for selection of recombinant cells. After the GFP-positive cells were purified for at least four passages with puromycin-resistant selection, cells were almost 100% positive for eGFP under fluorescent microscopy. The recombinant cells were designated as CBS HEK cells or control HEK cells, respectively. Recombinant cells were kept in liquid nitrogen for further use.
Cellular OCR assay
OCR was assayed, as reported previously (11). Briefly, cells were plated on 24-well seahorse plates at a density of 105 cells per well. One well per row contained only supplemented medium without cells, serving as the background control. Before the assay, cells were washed and incubated with 500 μl of glucose-supplemented XF assay buffer per well. Then, plates were placed in a CO2-free incubator for 30 min. Cells were not pretreated with H2S donors or FCCP. All reagents were added at the indicated time points. Briefly, the H2S donors, FCCP (0.5 μM), oligomycin, the mitochondria-targeted ROS scavenger MitoTEMPO (1.5 mM), UCP2 inhibitor genipin (20 nM), ZnCl2 (800 μM), rotenone, and antimycin A were added to the appropriate port of injector plates and injected into cells, as indicated in each experiment. Plates were run on the Seahorse XF-24. OCR values (picomole of O2 per min) were measured in real time. Rotenone and antimycin A were added to inhibit mitochondrial complexes I and III. Thus, OCR in the presence of rotenone and antimycin A represents the nonmitochondrial OCR, which was subtracted from the raw values of basal OCR or OCR in the presence of oligomycin and/or the H2S donors or FCCP.
Determination of cellular CoQH2/CoQ ratios with HPLC-MS/MS
High-performance liquid chromatography (HPLC)–tandem mass spectrometry (MS/MS) was used to determine cellular CoQH2/CoQ ratios. CoQ10 and CoQ9 are the dominant forms of CoQ in human and rodents, respectively. Ratios of the reduced and oxidized forms of CoQ10 (CoQ10H2/CoQ10) in human-derived HEK293 cells were measured using CoQ9 (J&L Scientific) as the internal standard. Ratios of CoQ9H2/CoQ9 in mouse-derived BV2 cells were measured using CoQ4 as the internal standard. Briefly, BV2 microglia or HEK293 cells were seeded on 10-cm dishes (106 cells per dish) and cultured under normal conditions. Before assay, BV2 microglia were treated with H2S donors at the indicated concentrations for 10 min. To prevent oxidation of CoQH2, all solvents were prepurged with nitrogen before use. Cells were then washed with phosphate-buffered saline (PBS), harvested, and lysed on ice with n-propanol containing the internal standard CoQ9 (0.2 μg/ml) or CoQ4 (0.2 μg/ml). After vortex mixing for 2 min, the solution was centrifuged at 4500 rpm for 3 min at 4°C. The liquid phase was extracted three times with hexane. The organic phase containing hexane and n-propanol was pooled and evaporated to dryness under nitrogen. Extracts were reconstituted with a methanol-hexane mixture (95:5 v/v). HPLC-MS/MS analysis was performed using a Thermo TSQ vantage 3000 HPLC hyphenated with a TQD triple quadrupole mass spectrometer with an electrospray ionization ion source. Chromatographic separation was achieved using a Symmetry C18 analytical column (2.1 mm by 100 mm, 3.5-μm particle size; Waters, Japan). During the HPLC run, the system was maintained at 99% eluent A [methanol and 1-propanol (205 ml/45 ml) containing 5 mM ammonium acetate] and 1% eluent B [methanol and 1-propanol (125 ml/125 ml) containing 5 mM ammonium acetate], ramped to 100% eluent B at 0.5 to 1 min, maintained at 100% B until 5 min, and then ramped back to 99% A and 1% B at 5 to 7 min. MS was performed in a positive ion mode with the ion source spray voltage at +3500 V and temperature at 350°C. The sheath gas was 40 arbitrary units. Selective reaction monitoring was used for quantification of both reduced and oxidized CoQ. LCquan software was used for data acquisition and analysis. The mass/charge ratio (m/z) transitions were as follows: m/z 882.5 to 150.8 and 196.8 corresponding to the ammonium adduct of CoQ10H2; m/z 863.5 to 148.8 and 196.8 corresponding to CoQ10; m/z 814.400 to 153.8 and 196.9 corresponding to the ammonium adduct of CoQ9H2; m/z 795.500 to 150.8 and 196.8 corresponding to CoQ9; and m/z 455.314 to 95.1 and 181.0 corresponding to CoQ4.
Measurement of mitochondrial NAD+/NADH ratios in cultured cells
Mitochondrial NAD+/NADH was measured to indicate complex I RET (24). Crude mitochondrial fractions were isolated from cultured cells for assessment of NAD+/NADH ratios. Briefly, BV2 microglia were cultured on 10-cm dishes at a density of 106 cells per dish. Cells were treated with the H2S donors and/or rotenone before the assay. Then, cells were homogenized in 500 μl of lysis buffer [50 mM tris-HCl (pH 7.4), 225 mM mannitol, 75 mM saccharose, 1 mM phenylmethylsulfonyl fluoride, and 10 μl of protease inhibitor mixture], followed by centrifugation at 600g for 10 min to remove cellular debris. Supernatants were collected and centrifuged at 7000g for 10 min to obtain mitochondrial pellets. For the assay of NAD+/NADH, mitochondrial pellets were lysed with a base buffer (0.2 M sodium hydroxide supplemented with 1% dodecyltrimethylammonium bromide). The ratio of NAD+/NADH was assayed per the manufacturer’s instructions (Promega).
Assessment of mitochondrial ROS generation in cultured cells
BV2 microglia were seeded on 24-well plates at a density of 105 cells per well. Before the assay, BV2 microglia were treated with ADT-OH (50 μM), rotenone (10 nM), FCCP (20 nM), MitoTEMPO (1.5 mM), the UCP2 inhibitor genipin (20 nM), the mitochondrial complex II inhibitor malonate (1 mM), or atpenin A5 (10 μM), as indicated in each experiment. Then, microglia were incubated with 5 μM MitoSOX and 50 nM MitoTracker Green. For measurement of ROS in HEK293 cells, wild-type HEK293 cells were seeded on 24-well plates at a density of 105 cells per well and then infected with lentivirus expressing NDUFS3-shRNA or Ctrl-shRNA. At 2 days after infection, cells were treated with ADT-OH for the indicated periods of time and then incubated with 5 μM MitoSOX and 50 nM MitoTracker Green. To assess mitochondrial ROS in HEK293 cells stably overexpressing CBS or control HEK293 cells stably expressing eGFP, cells were seeded on 24-well plates at the density 105 cells per well. Then, cells were treated with rotenone or vehicle for the indicated periods of time, followed by incubation with 5 μM MitoSOX and 50 nM MitoTracker Green. After staining with MitoSOX and MitoTracker Green, cells were washed, fixed in 4% formaldehyde, and further incubated with Hoechst 33342 (10 μM). Fluorescence was visualized under confocal microscopy (Zeiss LSM 700, Germany) with constant parameters to acquire all images.
Assessment of ROS generation in isolated mitochondria
Production of the superoxide anion radical (O2•−) was assessed by measuring the release of H2O2 from mitochondria with the Amplex Red/horseradish peroxidase (HRP) system (13). In the system, the conversion of nonfluorescent Amplex Red into fluorescent resorufin by H2O2 is catalyzed by HRP. The reaction buffer contained 50 μM Amplex Red, HRP (6 U/ml), 3 mM Hepes-free acid, 1 mM EGTA, 120 mM KCl, 0.3% bovine serum albumin, and superoxide dismutase (30 U/ml) for conversion of O2•− into H2O2. A total of 100 μl of reaction buffer was added to microplate wells and prewarmed at 37°C for 10 min. Mitochondria (35 μg) isolated from BV2 microglia or primary neurons were resuspended in 100 μl of buffer containing ADT-OH and/or rotenone. The reaction was started by adding mitochondria into the reaction buffer. Resorufin fluorescence was continually monitored at 560 nm for excitation and at 590 nm for emission using a fluorescence microplate reader. Freshly prepared H2O2 was added to mitochondria and served as the positive control.
Determination of mitochondrial complex IV activity
The activity of mitochondrial complex IV was determined colorimetrically following oxidation of reduced cytochrome C as a decrease in the absorbance at 550 nm using a commercial kit (BioVision Inc., Milpitas, CA, USA). BV2 microglia or HEK293 cells were seeded on 10-cm dishes (106 cells per dish). BV2 microglial cells were treated with the H2S donors or vehicle at 30 min before the assay. Cells were washed with PBS and harvested. Complex IV activity was assessed per the manufacturer’s manual. Cellular protein concentrations were measured with a bicinchoninic acid kit for normalizing complex IV activity. Results were presented as the percentages of the complex IV activity of control cells treated with vehicle (for microglia) or that of wild-type cells (for HEK293 cells).
Generation and identification of global SQR knockout mice and mice with microglia/macrophage-specific deletion of SQR
CRISPR-Cas9–mediated genome engineering technique was used to generate conditional Sqr-knockout mice on C57BL/6 background, in which the exon 2 of Sqr (ENSMUST00000005953.10) was flanked by loxP sites. The presence of the 3′ arm (5.2 kb) and 5′ arm (5.3 kb) and the mutant allele [300 base pairs (bp)] and wild-type allele (250 bp) was confirmed by polymerase chain reaction (PCR) genotype analysis and DNA sequencing of PCR amplicons. To generate global SQR knockout mice, mice heterozygous to loxP-flanked (“floxed”’; fl) Sqr alleles (Sqrfl/+) were crossed with EIIacre mice (Shanghai Model Organisms, Shanghai, China). In F1 offspring that were heterozygously Sqr and Cre positive (EIIaCre:Sqr+/−), the adenovirus EIIa promoter drove expression of Cre recombinase in early mouse embryos and thereby resulted in germ line deletion of the loxP-flanked Sqr gene. EIIaCre:Sqr+/− mice were crossed with wild-type mice to generate mice heterozygous to Sqr and deficient in EIIaCre allele (Sqr+/−). Last, Sqr+/− mice were intercrossed to generate global knockout mice (Sqr−/−). To identify Sqr−/− mice, the presence of the mutant allele (800 bp) and absence of WT allele (1600 bp) in Sqr−/− mice were confirmed by PCR genotype analysis. Sqr−/− mice were much smaller than age-matched wild-type mice and died before 2 months old. Sqr−/− mice were used before 2 months old.
For generation of mice with microglia/macrophage-specific deletion of SQR, Cx3crCre mice, in which expression of Cre is driven by the Cx3cr1 promoter in microglia/macrophages, were purchased from the Jackson laboratory (025524). Experimental animals were generated by crossing Sqrfl/+mice with the heterozygous Cx3crCre mice. Then, F1 offspring that were heterozygously floxed and Cre positive (Cx3crCre:Sqrfl/+) were crossed with the mice homozygous to loxP-flanked Sqr alleles (Sqrfl/fl). The experimental genotypes were microglia/macrophage-specific SQR-knockout mice (Cx3cr1Cre:Sqrfl/fl) and floxed control (Sqrfl/fl). The littermates were used for experiments. Genotypes of mice were confirmed by PCR analysis.
In vitro neuroinflammation model associated with ICH
To mimic ICH-induced neuroinflammation, cultured microglia were treated with RBC lysate, as previously reported (35). Briefly, blood (0.5 ml) drawn from mice was centrifuged at 3000g for 5 min. Plasma and buffy coat were discarded. To lyse RBCs, packed RBCs were frozen in liquid nitrogen for 5 min followed by thawing at 37°C. Ten microliters of the RBC lysate was used to treat microglia seeded on 24-well plates.
Mouse ICH models
All animal protocols were conducted according to the regulations of the Animal Care and Use Committee of Soochow University. Adult male wild-type ICR mice, male SQR knockout mice on C57BL/6 background, or control floxed littermates were subjected to ICH at 2 months old. ICH was induced by injecting collagenase or autologous blood into the left striatum under anesthesia, as reported (35). For collagenase-induced ICH, a cranial burr hole was drilled. A needle (26s gauge) was stereotaxically inserted into the left striatum at the following coordination: 0.5 mm anterior to the bregma, 2 mm lateral to the midline, and 3.5 mm below the skull. Collagenase VII-S [saline (0.03 U/μl); Sigma-Aldrich, St. Louis, MO, USA] was infused at 0.1 μl/min, or 1 μl of saline was infused for sham surgery. To induce ICH with autologous blood, mice were positioned onto a stereotaxic frame (model 500; Kopf Instruments, Tujunga, CA, USA). A 30-gauge needle was inserted stereotaxically into the left striatum. Autologous whole blood (15 μl) drawn from the tail vein without anticoagulant treatment was infused at 2 μl/min. Then, the needle was remained in the position for 10 min to prevent reflux. For sham surgery, mice were infused with the same volume of saline. After ICH induction, mice were randomized to receive daily injection of vehicle (10% dimethyl sulfoxide in corn oil) or ADT-OH (50 mg/kg per day) via the intraperitoneal route for 3 days, starting at 3 hours after ICH. No animals died before assessment, and no animals were excluded from the analysis.
Assessment of neurological deficits
Neurological deficits were blindly assessed with the neurological score test, forelimb placement test, and corner turn test. For assessing neurological scores, each mouse was given points on the basis of the following tests: body symmetry, gait, climbing, circling behavior, symmetry in front limbs, compulsory circling, and whisker response. The points were summed to produce a total score for each mouse. The higher scores indicated, the more severe the deficits. For the forelimb placement test, the respective vibrissae of each mouse were stimulated by gently brushing the vibrissae on a corner edge of a countertop. Intact mice quickly placed the forelimb ipsilateral to the stimulated vibrissae on the countertop. Depending on the severity of injury following ICH, placement of the forelimb contralateral to the injured brain was impaired in response to vibrissa stimulation. Each mouse was tested 10 times. The number of the trials in which a mouse appropriately placed its forelimb was expressed as a percentage of the total trial number. The corner turn test was performed by forcing mice into a corner with a 30° angle. When mice turned, the choice of direction was recorded. Intact mice turned with no preference, while hemorrhagic mice preferentially turned toward the non-impaired (right) side. Each mouse was tested 10 times. The number of right turns was presented as a percentage of total trial number.
Statistical analysis
One-way analysis of variance (ANOVA) was applied for multiple comparisons and two-tailed Student’s t tests were used for pairwise comparisons using SPSS Statistics 17.0. For ANOVA, the homogeneity of variance was evaluated with the Levene’s test. If the results were similar, then post hoc Tukey analysis was performed. Otherwise, data were analyzed by the Games-Howell’s test. Data of behavioral tests were analyzed by two-way ANOVA. Data were expressed as means ± SEM. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Shanghai Model Organisms for generation of SQR knockout mice and Y. Q. Xiao for support in cell cultures. Funding: This work was supported by the National Natural Science Foundation of China (81671310, 81971119, 81571124, 81471336, and 81371278), Priority Academic Program Development of the Jiangsu Higher Education Institutions (PAPD), Suzhou Clinical Research Center of Neurological Disease (Szzx201503), and the Jiangsu Key Laboratory grant (BM2013003). Author contributions: J.C. and J.J. conceived the concept, designed the research, interpreted data, and wrote the manuscript. Z.-c.H. and Y.E.C. designed the research and interpreted data. Z.W., M.Z., C. Hu, F.X., Y.S., J.J., J.Z., J.L., M.H., Y.L., G.A., C. Ho, and Y.C. conducted the research and/or analyzed data. Competing interests: J.C., J.J., and G.A. are inventors on a patent application related to this work filed to the National Intellectual Property Administration, People’s Republic of China (no. 201810156633.X; filed 20 April 2018). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eaaz5752/DC1
REFERENCES AND NOTES
- 1.Wallace J. L., Wang R., Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 14, 329–345 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Wallace J. L., Vaughan D., Dicay M., MacNaughton W. K., de Nucci G., Hydrogen sulfide-releasing therapeutics: Translation to the clinic. Antioxid. Redox Signal. 28, 1533–1540 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Wallace J. L., Nagy P., Feener T. D., Allain T., Ditrói T., Vaughan D. J., Muscara M. N., de Nucci G., Buret A. G., A proof-of-concept, phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br. J. Pharmacol. 177, 769–777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang R., Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Paul B. D., Snyder S. H., H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 13, 499–507 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Hancock J. T., Whiteman M., Hydrogen sulfide and cell signaling: Team player or referee? Plant Physiol. Biochem. 78, 37–42 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Bouillaud F., Blachier F., Mitochondria and sulfide: A very old story of poisoning, feeding, and signaling? Antioxid. Redox Signal. 15, 379–391 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., Bouillaud F., Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 1797, 1500–1511 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Goubern M., Andriamihaja M., Nübel T., Blachier F., Bouillaud F., Sulfide, the first inorganic substrate for human cells. FASEB J. 21, 1699–1706 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N., Smith A. C., Eyassu F., Shirley R., Hu C.-H., Dare A. J., James A. M., Rogatti S., Hartley R. C., Eaton S., Costa A. S., Brookes P. S., Davidson S. M., Duchen M. R., Saeb-Parsy K., Shattock M. J., Robinson A. J., Work L. M., Frezza C., Krieg T., Murphy M. P., Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mills E. L., Kelly B., Logan A., Costa A. S., Varma M., Bryant C. E., Tourlomousis P., Däbritz J. H., Gottlieb E., Latorre I., Corr S. C., McManus G., Ryan D., Jacobs H. T., Szibor M., Xavier R. J., Braun T., Frezza C., Murphy M. P., O’Neill L. A., Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scialò F., Sriram A., Fernández-Ayala D., Gubina N., Lõhmus M., Nelson G., Logan A., Cooper H. M., Navas P., Enríquez J. A., Murphy M. P., Sanz A., Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robb E. L., Hall A. R., Prime T. A., Eaton S., Szibor M., Viscomi C., James A. M., Murphy M. P., Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 293, 9869–9879 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong H., Chandel N. S., Regulation of redox balance in cancer and T cells. J. Biol. Chem. 293, 7499–7507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Echtay K. S., Roussel D., St-Pierre J., Jekabsons M. B., Cadenas S., Stuart J. A., Harper J. A., Roebuck S. J., Morrison A., Pickering S., Clapham J. C., Brand M. D., Superoxide activates mitochondrial uncoupling proteins. Nature 415, 96–99 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Mills E. L., Pierce K. A., Jedrychowski M. P., Garrity R., Winther S., Vidoni S., Yoneshiro T., Spinelli J. B., Lu G. Z., Kazak L., Banks A. S., Haigis M. C., Kajimura S., Murphy M. P., Gygi S. P., Clish C. B., Chouchani E. T., Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560, 102–106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao H., Zhang Y., Zeng X., Shulman G. I., Jin S., Niclosamide ethanolamine–induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med. 20, 1263–1269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry R. J., Zhang D., Zhang X.-M., Boyer J. L., Shulman G. I., Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X., Cao Y., Ao G., Hu L., Liu H., Wu J., Wang X., Jin M., Zheng S., Zhen X., Alkayed N. J., Jia J., Cheng J., CaMKKβ-dependent activation of AMP-activated protein kinase is critical to suppressive effects of hydrogen sulfide on neuroinflammation. Antioxid. Redox Signal. 21, 1741–1758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee M., Tazzari V., Giustarini D., Rossi R., Sparatore A., Del Soldato P., McGeer E., McGeer P. L., Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: Potential for treating Parkinson disease. J. Biol. Chem. 285, 17318–17328 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y., Wang H., Xian M., Cysteine-activated hydrogen sulfide (H2S) donors. J. Am. Chem. Soc. 133, 15–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mimoun S., Andriamihaja M., Chaumontet C., Atanasiu C., Benamouzig R., Blouin J. M., Tomé D., Bouillaud F., Blachier F., Detoxification of H2S by differentiated colonic epithelial cells: Implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid. Redox Signal. 17, 1–10 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Chouchani E. T., Pell V. R., James A. M., Work L. M., Saeb-Parsy K., Frezza C., Krieg T., Murphy M. P., A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Lee S., Tak E., Lee J., Rashid M. A., Murphy M. P., Ha J., Kim S. S., Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 21, 817–834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernansanz-Agustín P., Ramos E., Navarro E., Parada E., Sánchez-López N., Peláez-Aguado L., Cabrera-García J. D., Tello D., Buendia I., Marina A., Egea J., López M. G., Bogdanova A., Martínez-Ruiz A., Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 12, 1040–1051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts P. G., Hirst J., The deactive form of respiratory complex I from mammalian mitochondria is a Na+/H+ antiporter. J. Biol. Chem. 287, 34743–34751 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Longchamp A., Mirabella T., Arduini A., MacArthur M. R., Das A., Treviño-Villarreal J. H., Hine C., Ben-Sahra I., Knudsen N. H., Brace L. E., Reynolds J., Mejia P., Tao M., Sharma G., Wang R., Corpataux J.-M., Haefliger J.-A., Ahn K. H., Lee C.-H., Manning B. D., Sinclair D. A., Chen C. S., Ozaki C. K., Mitchell J. R., Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell 173, 117–129.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Simone R., Ajmone-Cat M. A., Pandolfi M., Bernardo A., De Nuccio C., Minghetti L., Visentin S., The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J. Neurochem. 135, 147–156 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Zhang C.-Y., Parton L. E., Ye C. P., Krauss S., Shen R., Lin C.-T., Porco J. A. Jr., Lowell B. B., Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced β cell dysfunction in isolated pancreatic islets. Cell Metab. 3, 417–427 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Kanemoto N., Okamoto T., Tanabe K., Shimada T., Minoshima H., Hidoh Y., Aoyama M., Ban T., Kobayashi Y., Ando H., Inoue Y., Itotani M., Sato S., Antidiabetic and cardiovascular beneficial effects of a liver-localized mitochondrial uncoupler. Nat. Commun. 10, 2172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M., Tang W., Zhu Y. Z., An update on AMPK in hydrogen sulfide pharmacology. Front. Pharmacol. 8, 810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinchy E. C., Gruszczyk A. V., Willows R., Navaratnam N., Hall A. R., Bates G., Bright T. P., Krieg T., Carling D., Murphy M. P., Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 293, 17208–17217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y. P., Brown J. R., Sag D., Zhang L., Suttles J., Adenosine 5’-monophosphate–activated protein kinase regulates IL-10–mediated anti-inflammatory signaling pathways in macrophages. J. Immunol. 194, 584–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y., Wang Y., Wang J., Anne Stetler R., Yang Q.-W., Inflammation in intracerebral hemorrhage: From mechanisms to clinical translation. Prog. Neurobiol. 115, 25–44 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Wang Y.-C., Zhou Y., Fang H., Lin S., Wang P.-F., Xiong R.-P., Chen J., Xiong X.-Y., Lv F.-L., Liang Q.-L., Yang Q.-W., Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 75, 876–889 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Hine C., Harputlugil E., Zhang Y., Ruckenstuhl C., Lee B. C., Brace L., Longchamp A., Treviño-Villarreal J. H., Mejia P., Ozaki C. K., Wang R., Gladyshev V. N., Madeo F., Mair W. B., Mitchell J. R., Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160, 132–144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertholet A. M., Chouchani E. T., Kazak L., Angelin A., Fedorenko A., Long J. Z., Vidoni S., Garrity R., Cho J., Terada N., Wallace D. C., Spiegelman B. M., Kirichok Y., H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 571, 515–520 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umemura K., Kimura H., Hydrogen sulfide enhances reducing activity in neurons: Neurotrophic role of H2S in the brain? Antioxid. Redox Signal. 9, 2035–2041 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Módis K., Coletta C., Erdélyi K., Papapetropoulos A., Szabo C., Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 27, 601–611 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Huang G., Chen Y., Lu H., Cao X., Coupling mitochondrial respiratory chain to cell death: An essential role of mitochondrial complex I in the interferon-β and retinoic acid-induced cancer cell death. Cell Death Differ. 14, 327–337 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eaaz5752/DC1