

Fate mapping study via a new transgenic mouse line reveals monocyte-to-microglia conversion in development and after brain injury.
Abstract
Whether monocytes contribute to the brain microglial pool in development or after brain injury remains contentious. To address this issue, we generated CCR2-CreER mice to track monocyte derivatives in a tamoxifen-inducible manner. This method labeled Ly6Chi and Ly6Clo monocytes after tamoxifen dosing and detected a surge of perivascular macrophages before blood-brain barrier breakdown in adult stroke. When dosed by tamoxifen at embryonic day 17 (E17), this method captured fetal hematopoietic cells at E18, subdural Ki67+ ameboid cells at postnatal day 2 (P2), and perivascular microglia, leptomeningeal macrophages, and Iba1+Tmem119+P2RY12+ parenchymal microglia in selective brain regions at P24. Furthermore, this fate mapping strategy revealed an acute influx of monocytes after neonatal stroke, which gradually transformed into a ramified morphology and expressed microglial marker genes (Sall1, Tmem119, and P2RY12) for at least 62 days after injury. These results suggest an underappreciated level of monocyte-to-microglia transition in development and after neonatal stroke.
INTRODUCTION
From the beginning of microglia research, Rio-Hortega correctly deduced a mesodermal origin of this “third element of the neural centers” (1). Research in the past decade has confirmed his insight and illuminated that microglia in mice originate from the yolk sac (YS)–derived Runx+ primitive erythroid myeloid progenitors (EMPs) around embryonic day 7.5 (E7.5) 7.5 to E8, which migrate into the cephalic mesenchyme at E10.5, and expand inside the neuroepithelium perinatally with little or no inputs by peripheral monocytes that descended from the fetal hematopoietic stem cells (HSCs) (2–5). Furthermore, it has been suggested that the brain perivascular and leptomeningeal macrophages also derive from YS EMP–derived progenitors instead of the fetal monocytes (6). Although “a minor population of adult microglia, perhaps in a defined region of the brain, might derive from non-YS progenitors recruited later during development”, a scenario that may relate to the microglial heterogeneity has been discussed (5); there is no evidence to support substantial monocyte-to-microglia transition in development to date (7–9).
Monocytes in mice consist of two principal subsets, a long-lived CX3CR1+Ly6Clo subset patrolling in the blood and a short-lived CX3CR1−Ly6Chi subset that is actively recruited into the inflamed tissue (10–12). It is generally agreed that monocytes transform into “microglia” in adult brains only under defined host conditions, such as head-unshielded gamma irradiation or genetic deletion of the CSF1 receptor (CSF1R), but the monocyte derivatives after CSF1R deletion fail to express the microglial marker genes, such as Tmem119, P2RY12, and Sall1 (13–16). In the mouse model of experimental autoimmune encephalitis (EAE), peripheral monocytes invade the brain to promote neuroinflammation but do not contribute to the microglial pool (17, 18). In adult stroke, invading CCR2+ monocytes aggravate brain damage and then differentiate to M2-like macrophages to foster the resolution of inflammation (19–22). Although some infiltrating hematopoietic cells may become microglia-like cells after adult stroke (23), the identity of these blood cells and whether their derivatives express microglial markers remain unknown. Contrary to the findings in adult stroke, it was reported that acute macrophages after neonatal stroke are derived from the resident microglia, but not infiltrating monocytes (24). Given these conflicting reports, additional studies are warranted to elucidate the responses and outcomes of monocytes after neonatal cerebral ischemia.
To this end, we have used bacterial artificial chromosome (BAC) recombineering to generate CCR2-CreER mice that harbor an inducible Cre recombinase (CreER/T2) in the first exon of the CCR2 transgene. This genetic locus of CCR2 has been used to label Ly6Chi and ly6Clo monocytes in CCR2RFP knock-in mice, because the chemokine receptor CCR2 is monocyte specific for directing their migration into inflamed tissue (10, 11, 25). By crossing with an R26R–enhanced green fluorescent protein (EGFP) reporter line (Ai6) and after tamoxifen induction, the bitransgenic CCR2-CreER mice can be used to detect monocyte derivatives even if they no longer express the monocytic CCR2 marker, which is an advantage over CCR2RFP/+ mice for fate mapping. In addition, the CCR2-CreER–based fate mapping can label fetal monocytes, which is beyond the capabilities of bone marrow (BM) chimera and parabiosis methods. Our CCR2-CreER BAC transgenic line is similar to the previously reported CCR2-CreERT2-mKate2 knock-in mice (26). Last, after breeding with R26R-EGFP/Rpl10a mice, translating ribosome affinity purification (TRAP) can be used to determine the transcriptome of monocyte derivatives in bitransgenic CCR2-CreER mice (27). Using these methods, herein we report the evidence for sizable monocyte-to-microglia transition during development and neonatal stroke.
RESULTS
Monocytes become macrophages and CX3CR1+ microglia-like cells after neonatal stroke
To test whether peripheral monocytes contribute to the brain macrophages after neonatal stroke, we first used flow cytometry to analyze CD45+CD11b+ myeloid cells in brain parenchyma after the middle cerebral artery (MCA)–targeted photothrombosis in postnatal day 16 (P16) mice. We found an increase in neutrophils (CD45hiCD11b+Ly6G+) and monocytes/macrophages (CD45hi CD11b+Ly6G−), but not microglia (CD45intCD11b+Ly6G−) in the ipsilateral hemisphere at 72 hours after stroke (Fig. 1, A to C, n = 3 for each condition). We also sectioned the brains of CCR2RFP/+; CX3XR1GFP/+ mice (hereafter referred to as the R/G mice) and detected the influx of CCR2RFP+ monocytes, often coexpressing CX3CR1GFP+ with an ascending percentage from 24 to 48 to 72 hours after stroke (arrows in Fig. 1, D and E, n = 6 to 15 for each time point) (28). Furthermore, ameboid CCR2RFP+CX3CR1GFP+ cells were concentrated at the ischemic border, similar to the pattern in adult ischemic stroke (19), and expressed the macrophage/dendritic cell (Mφ/DC) marker major histocompatibility complex class II (MHC-II) (Fig. 1F, n = 15). The flow cytometry analysis of sham and poststroke R/G mouse brains at 72 hours after injury revealed two clear cell groups: The first is CX3CR1GFP+CCR2RFP− microglia that were MHC-II− in contralateral hemisphere and scantly MHC-II+ in the ipsilateral hemisphere (0.26%); the other is CX3CR1GFP+CCR2RFP+ cells that were MHC-II+Ly6C− (Mφ/DC, 22.2%) or MHC-II−Ly6Chi (monocytes, 59.6%) (Fig. 1G, n = 3 for each group; the gating strategy of flow cytometry and poststroke R/G mouse brain sections are shown in fig. S1). These results suggest that invading monocytes may convert to MHC-II+Ly6C− Mφ/DCs or CX3CR1GFP+CCR2RFP+ “microglia-like cells” in poststroke neonatal brains, but the final outcomes of invading monocytes cannot be established in R/G mice, owing to potential down-regulation of the CCR2RFP marker gene in transformed monocyte derivatives.
Fig. 1. Tracking monocyte derivatives after neonatal stroke in CCR2RFP/+; CX3CR1GFP/+ (R/G) mice.
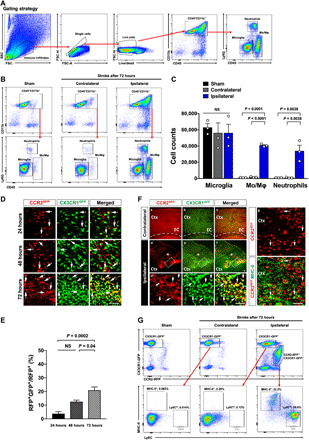
(A) The gating strategy for flow cytometry analysis used in (B). (B and C) Flow cytometry of the brain myeloid cells after photothrombosis in P16 mice. Shown in (B) are representative flow plots and in (C) are quantification results at 72 hours after stroke (n = 3 males). (D and E) Sections of the brains of R/G mice at 24, 48, and 72 hours after stroke (n = 6 to 15 of both genders). Arrows indicate CX3CR1GRP+CCR2RFP+ double-positive cells in the ipsilateral hemispheres in all time points. (F) At 72 hours after stroke, CCR2RFP+ monocytes (d and d′), CX3CR1GFP+ ameboid microglia (e and e′), and CX3CR1GRP+CCR2RFP+ cells (f and f′) were concentrated at the infarct border (asterisk) but absent in the contralateral hemisphere (a, b, and c). Many invading CCR2RFP+ monocytes expressed MHC-II (g and h) (n = 15 from both genders). (G) Flow cytometry of the R/G mouse brains at 72 hours after stroke. The CX3CR1GFP+ cells in the sham and contralateral hemisphere were negative for MHC-II and Ly6C. The CX3CR1GFP+CCR2RFP- cells in the ipsilateral hemisphere included few MHC-II+Ly6C− macrophages and MHC-II−Ly6C+ monocytes. In contrast, more CX3CR1GFP+CCR2RFP+ cells expressed the macrophage and monocyte markers. (n = 3 from 3 males for each). Data are presented as means ± SEM; analyses were performed using one-way ANOVA with Tukey’s post hoc test. Scale bars, 100 μm. Ctx, cerebral cortex; EC, entorhinal cortex; NS, not significant.
Fate mapping with CCR2-CreER(T2) mice labels both Ly6Chigh and Ly6Clow monocytes
Hence, we used BAC recombineering to generate CCR2-CreER mice that carry the CreER(T2) insertion in the first exon of the CCR2 transgene (Fig. 2A). We established three lines of CCR2-CreER mice (#21, #49, and #50) from >10 founders and crossed each line with the R26R-EGFP reporter mice to derive bitransgenic CCR2-CreERtg/+; R26R-EGFPtg/+ mice (hereafter referred to as bitransgenic CCR2-CreER mice). After tamoxifen induction (daily intraperitoneal injection of 5 mg/40 g body weight for 5 days), plentiful GFP+ cells were detected in the BM, spleen, and peripheral blood (PB) in all three lines of bitransgenic CCR2-CreER mice (Fig. 2B, n = 3 for each line). Because line #21 showed the highest percentage of CCR2+GFP+ cells among CCR2+ cells (i.e., efficiency) and GFP+ cells (i.e., specificity) in the BM and PB (Fig. 2, C and D, n = 3 for each line), it was used for further studies. In the BM of tamoxifen-induced bitransgenic CCR2-CreER mice, a high percentage of both CD115+CCR2+CD11b+Ly6Chi (88 ± 5%, means ± SEM) and CD115+CCR2+CD11b+Ly6Clo monocytes (78 ± 8%) expressed GFP, while no GFP+ cells were detected in mice without tamoxifen dosing (Fig. 2, E and F, n = 3). When bitransgenic CCR2-CreER mice were tamoxifen dosed at P14 and P15 and the brain was collected without challenge at P16, GFP+ cells were detected in the choroid plexus (CPx) and leptomeninges and absent in the cerebral cortex (Ctx) (Fig. 2, G to I, n = 4). These results suggest efficient labeling of monocytes with bitransgenic CCR2-CreER mice.
Fig. 2. Generation of CCR2-CreER(T2) mice for detecting monocytes and the monocyte derivatives.
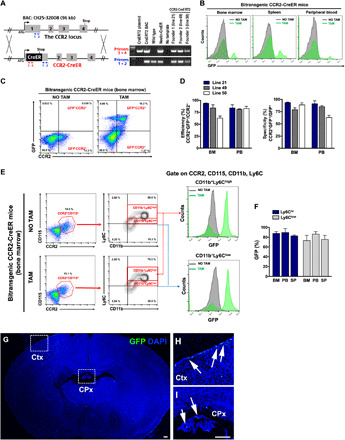
(A) Diagram of the BAC construct for transgenic CCR2-CreER(T2) mice, which includes the CCR2 promoter, CreERT2 cDNA, and the SV40 polyA signal. PCR genotyping confirmed the three founders used to established transgenic lines (#21, #49, and #50). (B) Bitransgenic CCR2-CreER mice (after being crossed with the R26R-EGFP, Ai6 mice) exhibited copious GFP+ cells in the BM, spleen, and PB after daily tamoxifen dosing for 5 days, but no GFP+ cells without tamoxifen. (C and D) Flow cytometry comparison of the efficiency (CCR2+GFP+/CCR2+) and specificity (CCR2+GFP+/GFP+) of monocyte-labeling in tamoxifen-dosed bitransgenic CCR2-CreER mice. Shown are means ± SEM (n = 3 for each line). (E and F) Flow cytometry showed high efficiency for labeling Ly6Chigh (88 ± 5%) and Ly6Clow monocytes (78 ± 8%) in bitransgenic CCR2-CreER mice (line #21). Data are presented as means ± SEM (n = 3). (G to I) Bitransgenic CCR2-CreER mice received tamoxifen induction at P14 and P15, and those analyzed at P16 showed no GFP+ cells in the brain parenchyma (n = 4). Magnified areas of squares (Ctx and CPx) are shown in (H) and (I). Scale bars, 100 μm.
Cerebral ischemia induces a surge of monocyte-derived perivascular macrophage before the BBB breakdown
The origin and turnover rate of perivascular macrophages (PVMs) remain controversial (6, 29). To investigate this issue, we injected tamoxifen to adult bitransgenic CCR2-CreER mice for 5 days, induced the MCA-targeted photothrombosis on the sixth day, and used intravital microscopy to search for GFP+ monocytes near the Rhodamine 6G dye–infused blood vessels at 24 or 72 hours after stroke (Fig. 3A). In the contralateral hemisphere, almost no GFP+ cells outside the blood vessels were detected, suggesting very slow or nil turnover of PVM in uninjured conditions (Fig. 3B and movie S1). Immunofluorescence labeling showed that the few GFP+ cells in the contralateral hemisphere were inside isolectin-B4 (IB4)+ cerebral vessels, indicating that they are circulating monocytes (Fig. 3C, n = 3). In contrast, abundant GFP+ cells were detected outside Rhodamine 6G dye–infused blood vessels in the ipsilateral peri-infarct area at 24-hour recovery (Fig. 3D, a to c, and movie S2; and Fig. 3D, d to f, and movie S3). These GFP+ monocytes and PVM showed a variety of morphology (round, ameboid, or spindle like) and motility, ranging from fast-moving motile cells (M; >12 μm/min), stationary-exploring cells (SE) that constantly extend and retract their processes, to inactive stationary cells (S) (Fig. 3D). Notably, although minute extravasation of Rhodamine 6G was evident at 72 hours (yellow signals in Fig. 3E), it was absent in the peri-infarct area at 24 hours (Fig. 3D, asterisk denotes the intravascular blood clots), suggesting that the surge of PVM precedes severe blood-brain barrier (BBB) damage in adult stroke. By 72 hours after stroke, two distinct behaviors of GFP+ monocyte derivatives were noticed. One is ameboid or spindle-like “resident” PVM outside the Rhodamine 6G–infused blood vessels (“R” in Fig. 3E, a to f, and movie S4). The other is a larger group of “exiting” PVMs that were increasingly labeled by Rhodamine 6G (presumably due to elevated BBB permeability) and then flushed into the blood stream (“E” in Fig. 3E, a to f; asterisk indicates intravascular blood clots).
Fig. 3. The surge of PVMs after cerebral ischemia.
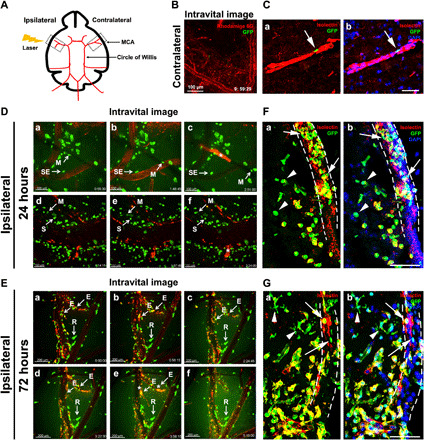
(A) Schematic diagram of the MCA-targeted photothrombotic stroke model. (B) Intravital imaging of the contralateral hemisphere in bitransgenic CCR2-CreER mice at 24 hours after stroke. See movie S1 (n = 3 from 3 males). Scale bar, 100 μm. (C) Immunostaining showed near absence of GFP+ monocyte derivatives outside the isolectin B4 (IB4)+ blood vessels in the contralateral hemisphere at 24 hours after stroke (n = 3 males). Scale bar, 100 μm. (D and E) Intravital imaging and videos of the peri-infarct area in bitransgenic CCR2-CreER mice at 24 hours after stroke [n = 3 males; D, a to c, movie S2; d to f, movie S3) or 72 hours (n = 3 males; E, a to f, movie S4). Note the minuscule perivascular leakage of Rhodamine 6G at 72 hours (E), but not 24 hours after stroke (D). Asterisks indicate intravascular blood clots. Scale bars, 100 μm (D) and 200 μm (E). (F and G) Immunostaining of GFP+ monocytes and IB4+ blood vessels at 24 hours (F) and 72 hours (G) in the peri-infarct area after stroke. Note the presence of intravascular (arrows) and extravascular GFP+ cells (arrowheads), and the more complex morphology of extravascular monocyte derivatives at 72 hours after stroke. n = 3 males for (F) and n = 4 males for (G). Scale bars, 100 μm. M, mobile cells; SE, stationary-exploring cells; S, stationary cells; R, resident PVMs; E, exiting cells.
Similar results were detected by immunofluorescence double labeling with IB4 in 24- or 72-hour poststroke bitransgenic CCR2-CreER mouse brains (Fig. 3, F and G, n = 3 for 24 hours, n = 4 for 72 hours). Although GFP+ intravascular monocytes (arrows) and PVM (arrowheads) were present at both time points, the majority of PVMs transformed from a round to ameboid morphology from 24 to 72 hours after stroke (Fig. 3, F and G). These results suggest an acute influx of PVM before severe BBB damage in cerebral ischemia.
CCR2+ progenitors produce border-associated macrophages and a subset of parenchymal microglia in development
It is a widely held view that parenchymal microglia and border-associated macrophages, with the exception of CPx macrophages, solely derive from the YS primitive EMPs (4, 6). To evaluate this notion, we injected tamoxifen to dam at E14 (5 mg/40 g body weight) and searched for GFP+ monocyte derivatives in the brains of bitransgenic CCR2-CreER pups at P2 (E14tam-P2). Without tamoxifen dosing, no GFP+ cells were present in the brain (Fig. 4A, n = 4). In contrast, copious GFP+ ameboid cells were detected in the subdural meninges, the CPx, and the ventricular aspect of the fornix, but absent in the corpus callosum (CC) (Fig. 4, B and C, n = 5). Many GFP+ monocyte derivatives in the meninges expressed the proliferative cell marker Ki67 (arrowheads in Fig. 4D). A smaller number of ramified GFP+ cells were also detected in brain parenchyma and labeled by the microglia marker anti-P2RY12 (arrows in Fig. 4E). The locations of GFP+ CCR2-monocyte derivatives in P2 brains are typical for those described as CX3CR1+ microglial progenitors (30) or “the fountains/nests of microglia” in the older literature (31).
Fig. 4. The progeny of fetal CCR2+ monocytes in bitransgenic CCR2-CreER mice.
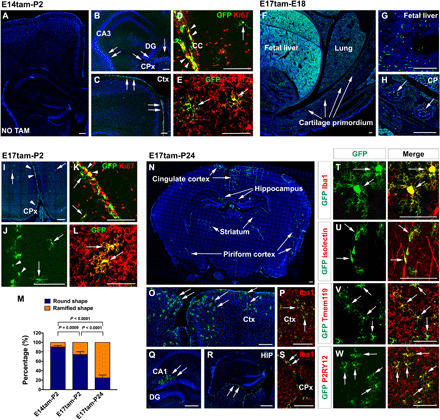
(A to E) Fate mapping CCR2+ monocytes in P2 mouse brains after tamoxifen dosing at E14 (n = 5). No GFP+ cells were labeled without tamoxifen dosing (A; n = 4). With tamoxifen, GFP+ cells were present in the CPx, the subdural meninges, and the fornix (arrows in B and C). GFP+ monocyte derivatives include Ki67+ cells in subdural meninges (arrowhead in D) and ramified microglial cells (arrows in D and E). The ramified monocyte derivatives were P2RY12+ (arrows in E). (F to H) With tamoxifen at E17, GFP+ cells were seen in the fetal liver and CP in E18 embryos (n = 6). (I to L) Fate mapping in P2 brains after tamoxifen induction at E17 (n = 5) showed GFP+ cells in the CPx and subdural meninges (arrowhead in I and J), and few ramified GFP+ cells (arrows in I to L). The progeny of CCR2+ monocytes included Ki67+ cells in subdural meninges (K) and P2RY12+ microglia (arrows in L). (M) Quantification of round- versus ramified-shaped progeny of fetal monocytes in E14tam-P2, E17tam-P2, and E17tam-P24 fate mapping (n = 5 for each). Data are shown as means ± SEM, one-way ANOVA with Tukey’s post hoc test. (N to W) Fate mapping monocyte derivatives in P24 brains after tamoxifen dosing at E17 showed Iba1+GFP+ microglia in the cingulate cortex, piriform cortex, striatum, and the hippocampus (N to Q and T; n = 5). Meningeal macrophages (R and S) and perivascular microglia (U) were visible. GFP+ ramified monocyte derivatives were Tmem119+ (V) and P2RY12+ (W). Scale bars, 200 μm (F to H), 100 μm (A to E, (I to L, and N to W).
Next, we administered tamoxifen to dam at E17 and examined the distribution of GFP+ derivatives in bitransgenic CCR2-CreER animals at E18 (E17tam-E18; Fig. 4, F to H), P2 (E17tam-P2; Fig. 4, I to L), or P24 (E17tam-P24; Fig. 4, N to W). We chose E17 for tamoxifen induction because the BM fetal hematopoiesis starts around E16 (32), and we concluded fate mapping at P24 as a previous study reported drastic reduction in brain-invading monocytes after P6 to near absence by P21 (33). In E17tam-E18 fate mapping, abundant GFP+ cells were found in the fetal liver, lung, and cartilage primordium (CP), but absent in the brain parenchyma (Fig. 4, F to H, and fig. S2, A to I; n = 6). In the E17tam-P2 tracing, many round GFP+ cells (arrowheads) were found in the CPx and subdural meninges, and a few ramified GFP+ cells (arrows) were also detected in brain parenchyma (Fig. 4, I and J, and fig. S2J; n = 5). The majority of round GFP+ cells were Ki67+, and the ramified GFP+ cells were labeled by the microglial marker P2RY12 (Fig. 4, K and L). Quantification showed a higher ratio of ramified-to-round GFP+ cells from E14tam-P2 to E17tam-P2 tracing, which was further increased in E17tam-P24 tracking (Fig. 4M, n = 5 for each group).
In E17tam-P24 fate mapping, a high percentage of GFP+ monocyte derivatives with a ramified morphology typical for parenchymal microglia were found in the cingulate cortex, the piriform cortex, the striatum, and the hippocampus (Fig. 4, N to S, n = 5). A smaller percentage of round, meninges-associated GFP+ cells were also detected (arrows in Fig. 4R), and both types of CCR2 monocyte derivatives expressed Iba1 (Fig. 4, P, S, and T). Furthermore, E17tam-P24 fate mapping labeled perivascular microglial cells attached to the IB4+ blood vessels (Fig. 4U). Last, the GFP+ parenchymal monocyte derivatives were labeled by anti-Tmem119 and anti-P2RY12, two microglial markers (Fig. 4, V and W) (34). These results suggest that, besides YS EMPs, fetal CCR2+ monocytes also contribute to parenchymal microglia and nonparenchymal macrophages in the mouse brain.
CCR2+ monocyte derivatives become proinflammatory macrophages and transform into ramified microglia-like cells after neonatal stroke
In the mouse EAE model, invading monocytes promote inflammation without conversion to the microglial pool, whereas monocyte-to-microglia transformation was reported after experimental meningitis, suggesting injury-dependent outcomes of monocyte derivatives (17, 35). To examine the fate of monocytes after neonatal stroke, we injected tamoxifen to bitransgenic CCR2-CreER mice at P14 and P15, induced the MCA photothrombosis at P16, and collected the brains at 3 days (Fig. 5, A to G), 30 days (Fig. 5, H to N), or 62 days (Fig. 5, O to U) after stroke to compare the morphology and properties of GFP+ monocyte derivatives (n = 6 for each time point).
Fig. 5. Morphological changes of monocyte derivatives from 3 to 62 days after neonatal stroke.
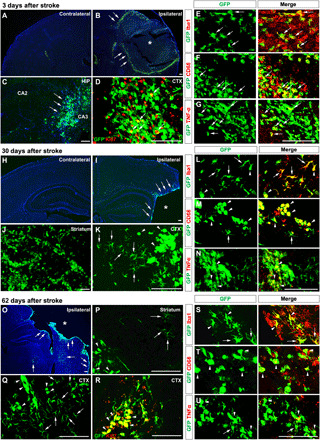
Bitransgenic CCR2-CreER mice received tamoxifen at P14 and P15 and photothrombosis at P16, followed by detection of the monocyte derivatives at 3 days (A to G; n = 6, 5 males and 1 female), 30 days (H to N; n = 6 males), or 62 days after stroke (O to U; n = 6 males). (A to G) At 3 days, GFP+ cells were absent in contralateral cortex (A) but populated in the ischemic border (arrows in B) and distant areas including hippocampus (arrows in C). Many GFP+ cells expressed the proliferative cell marker Ki67 (arrows in D), Iba1 (arrows in E), and the proinflammatory markers CD68 (F) and TNFα (G). (H to N) At 30 days, GFP+ monocyte derivatives were still restricted in the ipsilateral hemisphere (H to K). Many large round GFP+ monocyte derivatives coalesced and showed CD68 (arrowhead in M) and TNFα (arrowhead in N), but some showed a ramified morphology and anti-Iba1+ (arrows in L). (O to U) At 62 days, GFP+ monocyte derivatives were still restricted in the ipsilateral hemisphere (O to R). Many GFP+ monocyte derivatives expressed Ki67 (arrowhead in R). Fewer round-shaped monocyte derivatives coalesced or showed CD68 (arrowhead in T) and TNFα (arrowhead in U), but some showed a ramified microglial morphology and anti-Iba1 staining (arrows in S). Scale bars, 100 μm. Asterisk denotes the infarct core.
At 3 days after neonatal stroke, the contralateral hemisphere merely contained a few GFP+ cells in the CPx (Fig. 5A). In contrast, a large number of ameboid GFP+ cells were present in the ipsilateral hemisphere, predominantly around the ischemic border (arrows in Fig. 5B) but also forming clusters in the hippocampus (Fig. 5C). Many ameboid GFP+ derivatives expressed Ki67, Iba1, and the proinflammatory markers CD68 and tumor necrosis factor–α (TNFα) (arrows in Fig. 5, D to G). By 30 days after stroke, GFP+ cells disappeared in the contralateral hemisphere but remained abundant in the ipsilateral hemisphere (Fig. 5, H to K). Many GFP+ monocyte derivatives coalesced to form large clusters near the infarct border, but many remained individually distinguishable with a ramified morphology (arrows in Fig. 5K). The ramified GFP+ monocyte derivatives were Iba1+ (Fig. 5L), whereas the ameboid counterparts expressed the proinflammatory markers CD68 and TNFα (arrowhead in Fig. 5, M and N).
Similarly, by 62 days after stroke, GFP+ cells were still present in the ipsilateral hemisphere (Fig. 5, O to R). Many monocyte derivatives coalesced to form large GFP+ clusters near the infarct border in the striatum and the cortex, but many were individually distinguishable and showed a ramified morphology (arrows in Fig. 5, P and Q). Ameboid GFP+ cells expressed the proliferative cell marker Ki67 (arrows in Fig. 5R), whereas the ramified GFP+ monocyte derivatives were mostly Iba1+ (Fig. 5S). Some of the ameboid monocyte derivatives continued to express CD68 and TNFα (arrowhead in Fig. 5, T and U). These results suggest that a subset of infiltrating monocytes undergo “monocyte-to-microglia” transition in the poststroke developing brain.
Monocyte derivatives gradually express the M2-like markers and microglial marker genes after neonatal stroke
To confirm monocyte-to-microglia transition after neonatal stroke, we compared the expression of Tmem119 and P2RY12 by GFP+ monocyte derivatives in 3 or 30 days poststroke brain in bitransgenic CCR2-CreER mice. This experiment showed that, accompanied by a more ramified microglia-like morphology, GFP+ monocyte derivatives displayed stronger and more distinctive Tmem119 and P2RY12 immunosignals in 30 days poststroke brains (Fig. 6, A and B, n = 6 for each time point). Quantification of the mean fluorescence intensity (MFI) showed an increase from 83 ± 7 (3 days) to 133 ± 4 (30 days) for anti-Tmem119 signals, and from 75 ± 6 (3 days) to 143 ± 4 (30 days) for anti-P2RY12 signals in monocyte derivatives, whereas there was no difference in anti-Iba1 signals between 3 days (126 ± 4) and 30 days after stroke (131 ± 7) (Fig. 6C). We also confirm the expression of Sall1 mRNAs, another microglia-specific marker (36), in GFP+ monocyte derivatives at 30 days after stroke (Fig. 6D), and the expression of Tmem119 and P2RY12 in 62 days poststroke brains (fig. S3A). Of note, we used P16 bitransgenic CCR2-CreER pups of both genders in stroke experiments, because male and female mice of this age showed a similarly low level of estradiol in the blood (fig. S3B).
Fig. 6. Profiling the microglia and macrophage signature gene expression by monocyte derivatives in bitransgenic CCR2-CreER mouse brains.
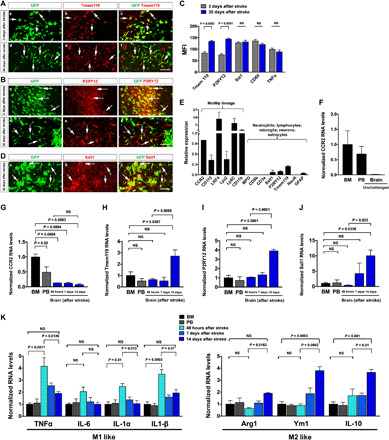
(A and B) GFP+ monocyte derivatives showed ascending Tmem119 and P2RY12 expression from 3 to 30 days after stroke (five males and one female for 3 days; six males for 30 days). (C) Mean fluorescence intensity (MFI) on CCR2+ derivatives from (A) and (B) (n = 6). Data are shown as means ± SEM, and the P value was determined by Student’s t test. (D) Sall1 mRNAs were present in GFP+ monocyte derivatives at 30 days after neonatal stroke (n = 3 males). (E) TRAP analysis showed amplification of monocyte-specific transcripts in the BM of tamoxifen-induced adult CCR2-CreERtg/+; R26R-EGFP/Rpl10Atg/+ mice (n = 3 males). (F) TRAP analysis of CCR2 mRNA levels in BM, PB, and brain at P16 (n = 3 males). (G to J) TRAP-based qPCR analysis showed gradual decline of CCR2 mRNAs and an inverse increase in Sall1, P2RY12, and Tmem119 mRNAs in monocyte derivatives in tamoxifen-dosed mouse brains from 48 hours to 7 days to 14 days after stroke compared with monocytes in BM and PB. Dare are shown as means ± SEM (n = 3 males), one-way ANOVA with Tukey’s post hoc test. (K) TRAP analysis showed gradual decline of M1-like cytokine and an inverse increase in M2-like mRNAs in brain monocyte derivatives from 48 hours to 7 days to 14 days after stroke, compared with monocytes in BM and PB. Data are shown as means ± SEM (n = 3 males), one-way ANOVA with Tukey’s post hoc test. Scale bars, 100 μm.
Next, we crossed CCR2-CreER mice with the R26R-EGFP/Rpl10A mice (TRAP mice) and used anti-EGFP/Rpl10A–based translating ribosome/mRNA affinity purification followed by reverse transcription quantitative polymerase chain reaction (RT-qPCR) to compare the expression of marker genes by monocytes and monocyte derivatives in the BM, PB, and 48 hours, 7 days, or 14 days poststroke brains (27). This analysis showed amplification of monocyte/macrophage transcripts in the BM and PB of tamoxifen-dosed P16 CCR2-CreERtg/+; R26R-EGFP/Rpl10Atg/+ mice (Fig. 6E and fig. S3, C and D; n = 3 for each). The TRAP analysis also showed a very low basal level of CCR2 mRNA in unchallenged P16 mouse brains, likely due to nil or minimal monocyte influx (Fig. 6F, n = 3). Similarly, compared with BM and PB, the normalized CCR2 expression by monocyte derivatives quickly declined in the poststroke neonatal brain, but the mRNAs for microglial markers CX3CR1, Tmem119, P2RY12, and Sall1 were all gradually increased in poststroke neonatal brains from 48-hour to 14-day recovery (Fig. 6, G to J, and fig. S3, E and F; n = 3 for each condition). The TRAP analysis also showed a trend of gradual reduction in M1-like markers [TNFα, IL1α, and interleukin-1β (IL-1β)] and an inverse increase in M2-like macrophage markers by monocyte derivatives from 48 hours to 14 days in poststroke brain (Fig. 6K, n = 3 for each time point) (37). These results support in situ reprogramming of monocyte derivatives after neonatal stroke (38).
DISCUSSION
CCR2-CreER mice as a genetic tool
Tamoxifen-inducible Cre-loxP recombination is a powerful research method, as evidenced by the popularity of CX3CR1CreER mice for fate mapping and genetic manipulation of microglia (12). In contrast, there is only one line of monocyte-specific CreER mice with few studies to date (26), despite the other monocyte fate mapping strategies all having limitations. For example, although CX3CR1CreER mice capture patrolling monocytes (CX3CR1+), they fail to label inflammatory monocytes (CX3CR1−Ly6Chi) (10, 11). The CCR2RFP/+ knock-in mice are a powerful tool for short-term tracking of monocytes, but if infiltrating monocytes stop expressing the CCR2 marker after in situ reprogramming as reported in a sterile liver injury model (38), they will “disappear” in the CCR2RFP/+ mouse brain tissue. Furthermore, although parabiosis and BM chimera are used in adult mice, they cannot label perinatal monocytes, due to a lengthy reconstitution period needed in both methods. In utero intraliver injection of lentiviral tracker has been used to label fetal HSCs, but this method is not monocyte specific, and the timing to initiate fate mapping is limited (33). Fate mapping by Ms4a3 expression traces monocyte-derived cells, but it also labels neutrophils, because Ms4a3 is expressed by the granulocyte-monocyte progenitors (39). In light of these limitations with current fate mapping methods, we decided to generate CCR2-CreER BAC transgenic mice to expand the repertoire of monocyte research.
Our results demonstrate that, after crossing with R26R-EGFP reporter mice, bitransgenic CCR2-CreER mice adeptly trace both inflammatory (CX3CR1−Ly6Chi) and patrolling monocytes (CX3CR1+Ly6Clo) (Fig. 2) and assist the visualization of PVMs that surge before severe BBB breakdown in cerebral ischemia (Fig. 3). The use of CCR2-CreER mice also enables us to fate map the progeny of fetal monocytes in the postnatal brains (Fig. 4), distinguish invading monocytes from the resident microglia (Fig. 5), and capture the changes in morphology and gene expression of monocyte derivatives in poststroke brains (Fig. 6). These results suggest that CCR2-CreER mice are a useful tool for monocyte-specific fate mapping and gene deletion.
Dual origin of microglia and border-associated macrophages
A widely held view posits that the parenchymal microglia and border-associated macrophages (except the CPx macrophages) are solely derived from YS EMPs without substantial inputs by the fetal HSC-derived monocytes (2–6). This scenario deviates from a layered ontogeny with successive waves of distinct progenitors during development of the other tissue-resident macrophages (4, 5). Yet, a recent study suggested that ~25% of the microglia population in the adult brain cortex are derived from Hoxb8+ progenitors in E12.5 fetal liver (7). This provocative finding contradicts the view of a sole YS origin for microglia and raises the possibility that fetal HSC-derived monocytes may contribute to microglia or border-associated macrophages.
Consistent with the layered ontogeny view, our results showed that following tamoxifen dosing in CCR2-CreER; Ai6 embryos at E14, ameboid monocytes populated the CPx, and subdural meninges, similar to the previously described fountains/nests of microglia, in the P2 mouse brains (30, 31). When tamoxifen dosed at E17 and the brains were examined at P24, many Iba1+Tmem119+P2RY12+ ramified microglia were identified in the cingulate cortex, the piriform cortex, the striatum, and the hippocampus. These locations overlap with, but not identical to, the distribution of Hoxb8+ progenitor–derived microglia (7). Furthermore, E17 tamoxifen dosing captured Iba1+ leptomeningeal macrophages and perivascular microglial cells in the P24 CCR2-CreER mouse brains. Together, our results and the previous finding suggest that the YS EMPs and fetal HSC-derived monocytes may form dual origins of parenchymal microglia and border-associated macrophages in the mouse brain (2–7).
Whether the dual origins contribute to the transcriptome heterogeneity and/or functional diversity of microglia warrants future research (8, 9). The mechanism that attracts monocytes to perinatal brains is unknown at present, but the MCP1-CCR2 axis is unlikely to play an essential role because neonatal brains express sizable MCP1 only after injury and because CCR2−/− mouse brain do not contain significantly fewer microglia and border-associated macrophages (6).
Monocyte-to-microglia reprogramming after brain injury
The combination of ontogeny and central nervous system environment appears to establish microglial identity for infiltrating monocytes in an insult-specific manner (15, 16). For example, peripheral monocytes engraft and differentiate into microglia in the brain after meningitis, but they do not contribute to the resident microglial pool after EAE (17, 35). In parabiosis or direct brain transplantation with CSF1R-deficinet mice, monocytes/macrophages acquire a microglia-like morphology but fail to express microglia signature genes, including Sall1, Tmem119, or P2RY12 (14, 15). Notably, the responses and fates of infiltrating monocytes after neonatal stroke remain unclear in the previous studies (24, 40).
Using CX3CR1GFP/+; CCR2RFP/+ (R/G) and bitransgenic CCR2-CreER mice for analysis, we showed an acute influx of monocytes within 24 hours onset of neonatal stroke. By 72 hours, CCR2+ monocytes were densely distributed at the ischemic border, similar to the findings in adult stroke (19), but also scattered in distant peri-infarct areas. Notably, these infiltrating monocytes had an ameboid cell shape at 3 days, but many transformed to a ramified microglia-like morphology at 30 days after stroke, correlated with up-regulation of microglial signature genes Tmem119, P2RY12, and Sall1 (14, 34, 36). Furthermore, the invading monocytes as a whole showed declining expression of M1-like proinflammatory cytokines and an inverse increase in M2-like markers, including IL-10, Arg1, and Ym1, from 48 hours to 14 days, following neonatal stroke (37). These results suggest biphasic functions of monocytes after neonatal cerebral ischemia (i.e., exacerbation of acute brain injury followed by promotion of the inflammation resolution) (19–22). Our results also suggest that a subset of invading monocytes may undergo in situ reprogramming to contribute to the microglial pool after neonatal stroke.
Conclusions and limitations of this study
In conclusion, the present study introduces CCR2-CreER BAC mice as a new tool for monocyte research and suggests a previously underappreciated level of monocyte-to-microglia transition in development and after neonatal stroke. This study, however, has several limitations. First, the full extent and spatial distribution of monocyte-derived microglia in developing mouse brains are yet to be determined. Second, whether there is a subacute wave of invading monocytes poststroke that differentiate directly into M2-like properties to promote the resolution of inflammation is uncertain. Last, single-cell transcriptome analysis of monocyte-derived microglia is warranted to assess their full differential plasticity and responses to immune stimuli (16, 41).
MATERIALS AND METHODS
Animals
C57BL/6J, CX3CR1GFP (JAX#005582), CCR2RFP (JAX#017586), and R26R-EGFP/RpL10A TRAP (JAX stock #022367) mice were purchased from the Jackson laboratory. CX3CR1GFP; CCR2RFP mice were obtained by mating CX3CR1GFP mice with CCR2RFP mice. To produce CCR2-CreER(T2) mice for fate mapping and inducible Cre/loxP gene deletion in monocyte derivatives. We have used a 96-kb mouse BAC (CH25-320O8) to insert the CreERT2 cassette, isolated from the pCAG-CreERT2 plasmid (AddGene #14797), in the first exon of the CCR2 locus. We have contracted the University of North Carolina Animal Models Core Facility to generate the BAC CCR2-CreERT2 transgene. The successfully engineered BAC will be digested and purified for microinjection into fertilized C57BL/6 eggs by the in-house transgenic animal facility at Emory University. The CCR2-CreER mice were then crossed with the ubiquitous R26R-EGFP Cre reporter line (Ai6, JAX stock #007906) or R26R-EGFP/RpL10A TRAP mice (JAX#022367). Most of the experiments were conducted in CCR2-CreER; R26R-EGFP mice (referred to as bitransgenic CCR2-CreER mice). All mice were housed in ventilated cages under standard laboratory conditions. All experiments were performed in accordance with animal protocols approved by the University of Virginia Animal Care and Use Committee in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines.
The following primer sequences were used for genotyping PCR:
Primer 1: ATG GAA GAC AAT AAT ATG TTA CCT CAG TTC
Primer 2: TTG CAG CAT AGT GAG CCC AGA ATG GTA ATG
Primer 3: GCTAA ACATG CTTCA TCGTC GG
Primer 4: GATCT CCGGT ATTGA AACTC CAGC
Flow cytometry analysis
To analyze the infiltrating immune cells in the brains after stroke, the mice were transcardially perfused with phosphate-buffered saline (PBS) extensively to remove blood cells in the circulation. The hemispheres from sham surgery animals and the contralateral and ipsilateral hemispheres from poststroke animals were harvested at 72 hours after stroke. The hemispheres were mechanically homogenized and the infiltrating immune cells were isolated with discontinued Percoll gradients (GE Healthcare). Moreover, to study the tamoxifen induction efficiency and specificity in adult bitransgenic CCR2-CreER mice, BM, PB, and spleen were harvested. Cell suspensions were stained with fluorochrome-conjugated antibodies including CD45 (30-F11, BioLegend), CD11b (M1/70, BioLegend), Ly-6G (1A8, BD Biosciences), Ly-6C (HK1.4, BioLegend), I-Ab (AF6-120.1, BioLegend), CCR2 (Clone #475301, R&D system), CD115 (T38-320, BD Biosciences), and LIVE/DEAD Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific). Subsequently, the stained suspensions were collected with flow cytometric analyses (Attune, Applied Biosystems) and analyzed with FlowJo v10.
Photothrombotic middle cerebral artery occlusion (MCAO)
Ten- to 12-week-old male mice were subjected to adult photothrombotic MCAO, as described (42). Mixed-gender mice were used for neonatal stroke, and the gender information is described in the figure legends. Briefly, bitransgenic CCR2-CreER mice or CCR2-CreER; R26R-TRAP mice were anesthetized with isoflurane and placed securely under a dissecting microscope. The proximal left MCA was exposed and directly irradiated with a green light laser (5 mW, 543.5 nm; Melles Griot, Carlsbad, CA) for 15 min, and Rose Bengal (5 mg/ml; Thermo Fisher Scientific) in saline was injected via the retro-orbital vein (25 mg/kg).
Immunohistochemistry
Brains from indicated littermates were fixed using 4% paraformaldehyde (PFA) at 4°C overnight with gentle agitation, cryopreserved in 30% sucrose, frozen, and finally stored at −20°C until use. For staining, 20-μm cryosections were made and incubated in blocking/permeabilization solution containing 3% normal goat serum (NGS) and 0.2% Triton-X in PBS. The sections were treated overnight with appropriate primary antibodies diluted in 1% NGS/0.2% Triton X-100/PBS followed by the corresponding secondary antibodies for 2 hours at room temperature. The following primary antibodies were used: rabbit anti-CD68 (Abcam); mouse anti-MHC-II (Abcam); rabbit anti-Ki67 (Abcam); isolectin GS-IB4, Alexa Fluor 594 conjugate (Invitrogen); rabbit anti-TNFα (Abcam); rabbit anti-Iba1 (Wako Chemicals USA Inc.); rabbit anti-Tmem119 (Abcam); and rat anti-P2RY12 (BioLegend).
Intravital confocal imaging
Real-time intravital imaging was recorded on a spinning disk confocal microscope (Visual Dynamix). Briefly, craniotomy of 3 to 5 mm in diameter was performed in the parietal bone of the skull while keeping the dura mater intact in adult bitransgenic CCR2-CreER mice. A cover glass was placed on the cranial window, and the cerebral vessels in the temporal cortex became visible under 20× water-immersion lens. Circulating leukocytes were labeled by 0.02% Rhodamine 6G (Thermo Fisher Scientific) in continuous infusion via the tail vein (2 μl/min) 5 min before intravital microscopy imaging. The image was postanalyzed using the Imaris software version 9 (Oxford instruments).
RNAscope assay
Mouse brain was freshly perfused and fixed in 4% PFA for 24 hours at 4°C. One day later, the brain was immersed in 30% sucrose, allowing the tissue to sink to the bottom of the container. The tissue was frozen in the optimal cutting temperature embedding media with dry ice. The blocks were sectioned by cutting 15-μm sections. RNA probe is commercially available from Advanced Cell Diagnostics (ACD). Here, we used probes against mouse Sall1 (ACD catalog no. 469661-C3), positive control probe (ACD catalog no. 310771) and negative probe (ACD catalog no. 310043), and then performed the assay by using the RNAscope Flurorescent Multiplex Reagent Kit (ACD catalog no. 320850) according to the manufacturer’s instructions.
Purification of cell type–specific mRNA and quantitative real-time PCR from CCR2-CreER; R26R-TRAP mice
BM, PB, and dissected mouse brains were immediately harvested in ice-cold dissection buffer following previously described procedures for the experiments [Heiman et al. (27)]. The rabbit anti-GFP antibodies, chromatin immunoprecipitation (ChIP) grade (Abcam, catalog no. ab290), were used for immunopurification. The RNA of the indicated samples was extracted using RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. Next, 1 μg of RNA was used for complementary DNA (cDNA) synthesis using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using a Bio-Rad CFX96 system (C1000 Thermal Cycler) and detected by SYBR Green master mix (Bio-Rad) as described previous (43). The following primer sequences were used for real-time PCR: glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-TGTAGACCATGTAGTTGAGGTCA-3′; LRF4, 5′-CCGACAGTGGTTGATCGACC-3′ and 5′-CCTCACGATTGTAGTCCTGCTT-3′; CD115, 5′-TGTCATCGAGCCTAGTGGC-3′ and 5′-GGTCCAAGGTCCAGTAGGG-3′; CCR2, 5′-GCCAGGACAGTTACCTTTGG-3′ and 5′- CGAAACAGGGTGTGGAGAAT-3′; Ly6C, 5′-GCAGTGCTACGAGTGCTATGG- 3′ and 5′- ACTGACGGGTCTTTAGTTTCCTT-3′; Otx2, 5′-TATCTAAAGCAACCGCCTTACG-3′ and 5′-GCCCTAGTAAATGTCGTCCTCTC-3′; LyZ2, 5′-GCTGACTGGGTGTGTTTAGC-3′ and 5′-TCCACGGTTGTAGTTTGTAGC-3′; CD11b, 5′-GGCTCCGGTAGCATCAACAA-3′ and 5′-ATCTTGGGCTAGGGTTTCTCT-3′; CX3CR1, 5′-TCTGGACTCACTACCTCATCAG-3′ and 5′-TCCGGTTGTTCATGGAGTTGG-3′; MPO, 5′-AGGGCCGCTGATTATCTACAT-3′ and 5′- CTCACGTCCTGATAGGCACA-3′; CD8b, 5′-CTCTGGCTGGTCTTCAGTATGA-3′ and 5′-TCTTTGCCGTATGGTTGGTTT-3′; CD3e, 5′-TCAGCCTCCTAGCTGTTGG-3′ and 5′-GTCAACTCTACACTGGTTCCTG-3′; Sall1, 5′-CTCAACATTTCCAATCCGACCC-3′ and 5′- GGCATCCTTGCTCTTAGTGGG-3′; P2RY12, 5′-TTTCAGATCCGCAGTAAATCCAA-3′ and 5′-GGCTCCCAGTTTAGCATCACTA-3′; Tmem119, 5′-CCTACTCTGTGTCACTCCCG-3′ and 5′-CACGTACTGCCGGAAGAAATC-3′; NeuN, 5′-CCCCTTGCCTAATACCCTTGA-3′ and 5′-GCCTCAGACATAGGTGGGATG-3′; GFAP, 5′-CGGAGACGCATCACCTCTG-3′ and 5′-TGGAGGAGTCATTCGAGACAA-3′; TNFα, 5′-CCACCACGCTCTTCTGTCTA-3′ and 5′-CTCCTCCACTTGGTGGTTTG-3′; IL-6, 5′-GGAGAGGAGACTTCACAGAGGAT-3′ and 5′-AGTGCATCATCGCTGTTCATAC-3′; IL-1α, 5′-CGAAGACTACAGTTCTGCCATT-3′ and 5′-GACGTTTCAGAGGTTCTCAGAG-3′;IL-1β, 5′-CTTTCGACAGTGAGGAGAATGAC-3′ and 5′-CAAGACATAGGTAGCTGCCACAG-3′; Arg1, 5′-CGCCTTTCTCAAAAGGACAG-3′ and 5′-CCAGCTCTTCATTGGCTTTC-3′; Ym1, 5′-ACCAGTTGGGCTAAGGACAG-3′ and 5′-TGGCCAGGAGAGTTTTTAGC-3′; IL-10, 5′-AAGGACCAGCTGGACAACAT-3′ and 5′-TCCTGAGGGTCTTCAGCTTC-3′. The samples were analyzed in triplicate and normalized versus the expression level of the GAPDH.
Statistical analysis
Data were summarized by mean and SEM. Statistical tests were done independently in Prism 7 and R 3.6.0 to ensure the consistency and are indicated in the figures. In short, Student’s t test for independent samples with unequal variances (i.e., Welch’s version of t test) was used to compare two groups, while one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons post hoc test were used to compare three or more groups. A P value of less than 0.05 was considered as significant. Power analysis and sample size determination were conducted in R under significance level of 0.05 and 80% statistical power. Figures were produced using Prism 7.
Supplementary Material
Acknowledgments
Funding: This work was supported by NIH grants (NS095064, NS100419, and NS108763 to C.-Y.K. and NS106592 to Y.-Y.S.) and an American Heart Association postdoctoral fellowship (#18POST34080334 to H.-R.C.). Author contributions: C.-Y.K. and H.-R.C. designed the study and wrote the manuscript. H.-R.C., Y.-Y.S., C.-W.C., Y.-M.K., I.S.K., J.C.S.-M., and M.R.S. conducted the experiments. Z.-R.T.L. performed the statistical analysis. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eabb2119/DC1
REFERENCES AND NOTES
- 1.Sierra A., Paolicelli R. C., Kettenmann H., Cien Años de Microglía: Milestones in a century of microglial research. Trends Neurosci. 42, 778–792 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., Mehler M. F., Conway S. J., Ng L. G., Stanley E. R., Samokhvalov I. M., Merad M., Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoeffel G., Chen J., Lavin Y., Low D., Almeida F. F., See P., Beaudin A. E., Lum J., Low I., Forsberg E. C., Poidinger M., Zolezzi F., Larbi A., Ng L. G., Chan J. K. Y., Greter M., Becher B., Samokhvalov I. M., Merad M., Ginhoux F., C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prinz M., Jung S., Priller J., Microglia biology: One century of evolving concepts. Cell 179, 292–311 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Ginhoux F., Guilliams M., Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Goldmann T., Wieghofer P., Jordão M. J. C., Prutek F., Hagemeyer N., Frenzel K., Amann L., Staszewski O., Kierdorf K., Krueger M., Locatelli G., Hochgerner H., Zeiser R., Epelman S., Geissmann F., Priller J., Rossi F. M. V., Bechmann I., Kerschensteiner M., Linnarsson S., Jung S., Prinz M., Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De S., Van Deren D., Peden E., Hockin M., Boulet A., Titen S., Capecchi M. R., Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development 145, dev152306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammond T. R., Dufort C., Dissing-Olesen L., Giera S., Young A., Wysoker A., Walker A. J., Gergits F., Segel M., Nemesh J., Marsh S. E., Saunders A., Macosko E., Ginhoux F., Chen J., Franklin R. J. M., Piao X., McCarroll S. A., Stevens B., Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginhoux F., Schultze J. L., Murray P. J., Ochando J., Biswas S. K., New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 17, 34–40 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Auffray C., Sieweke M. H., Geissmann F., Blood monocytes: Development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 27, 669–692 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Geissmann F., Jung S., Littman D. R., Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Yona S., Kim K.-W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D. A., Perlman H., Malissen B., Zelzer E., Jung S., Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mildner A., Schmidt H., Nitsche M., Merkler D., Hanisch U.-K., Mack M., Heikenwalder M., Brück W., Priller J., Prinz M., Microglia in the adult brain arise from Ly- 6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 10, 1544–1553 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Cronk J. C., Filiano A. J., Louveau A., Marin I., Marsh R., Ji E., Goldman D. H., Smirnov I., Geraci N., Acton S., Overall C. C., Kipnis J., Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 215, 1627–1647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett F. C., Bennett M. L., Yaqoob F., Mulinyawe S. B., Grant G. A., Gephart M. H., Plowey E. D., Barres B. A., A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98, 1170–1183.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Hove H., Martens L., Scheyltjens I., De Vlaminck K., Antunes A. R. P., De Prijck S., Vandamme N., De Schepper S., Van Isterdael G., Scott C. L., Aerts J., Berx G., Boeckxstaens G. E., Vandenbroucke R. E., Vereecke L., Moechars D., Guilliams M., Van Ginderachter J. A., Saeys Y., Movahedi K., A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 22, 1021–1035 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Ajami B., Bennett J. L., Krieger C., McNagny K. M., Rossi F. M. V., Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14, 1142–1149 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Yamasaki R., Lu H., Butovsky O., Ohno N., Rietsch A. M., Cialic R., Wu P. M., Doykan C. E., Lin J., Cotleur A. C., Kidd G., Zorlu M. M., Sun N., Hu W., Liu L. P., Lee J.-C., Taylor S. E., Uehlein L., Dixon D., Gu J., Floruta C. M., Zhu M., Charo I. F., Weiner H. L., Ransohoff R. M., Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533–1549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gliem M., Mausberg A. K., Lee J.-I., Simiantonakis I., van Rooijen N., Hartung H.-P., Jander S., Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 71, 743–752 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Miró-Mur F., Pérez-de-Puig I., Ferrer-Ferrer M., Urra X., Justicia C., Chamorro A., Planas A. M., Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav. Immun. 53, 18–33 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Fang W., Zhai X., Han D., Xiong X., Wang T., Zeng X., He S., Liu R., Miyata M., Xu B., Zhao H., CCR2-dependent monocytes/macrophages exacerbate acute brain injury but promote functional recovery after ischemic stroke in mice. Theranostics 8, 3530–3543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe S., Alexander M., Misharin A. V., Budinger G. R. S., The role of macrophages in the resolution of inflammation. J. Clin. Invest. 129, 2619–2628 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hess D. C., Abe T., Hill W. D., Studdard A. M., Carothers J., Masuya M., Fleming P. A., Drake C. J., Ogawa M., Hematopoietic origin of microglial and perivascular cells in brain. Exp. Neurol. 186, 134–144 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Denker S. P., Ji S., Dingman A., Lee S. Y., Derugin N., Wendland M. F., Vexler Z. S., Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J. Neurochem. 100, 893–904 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saederup N., Cardona A. E., Croft K., Mizutani M., Cotleur A. C., Tsou C.-L., Ransohoff R. M., Charo I. F., Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLOS ONE 5, e13693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croxford A. L., Lanzinger M., Hartmann F. J., Schreiner B., Mair F., Pelczar P., Clausen B. E., Jung S., Greter M., Becher B., The cytokine GM-CSF drives the inflammatory signature of CCR2+monocytes and licenses autoimmunity. Immunity 43, 502–514 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Heiman M., Schaefer A., Gong S., Peterson J. D., Day M., Ramsey K. E., Suárez-Fariñas M., Schwarz C., Stephan D. A., Surmeier D. J., Greengard P., Heintz N., A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung S., Aliberti J., Graemmel P., Sunshine M. J., Kreutzberg G. W., Sher A., Littman D. R., Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bechmann I., Kwidzinski E., Kovac A. D., Simbürger E., Horvath T., Gimsa U., Dirnagl U., Priller J., Nitsch R., Turnover of rat brain perivascular cells. Exp. Neurol. 168, 242–249 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Mizutani M., Pino P. A., Saederup N., Charo I. F., Ransohoff R. M., Cardona A. E., The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J. Immunol. 188, 29–36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kershman J., Genesis of microglia in the human brain. Arch. Neurol. Psych. 41, 24–50 (1939). [Google Scholar]
- 32.Mazo I. B., Massberg S., von Andrian U. H., Hematopoietic stem and progenitor cell trafficking. Trends Immunol. 32, 493–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Askew K., Li K., Olmos-Alonso A., Garcia-Moreno F., Liang Y., Richardson P., Tipton T., Chapman M. A., Riecken K., Beccari S., Sierra A., Molnár Z., Cragg M. S., Garaschuk O., Perry V. H., Gomez-Nicola D., Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 18, 391–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett M. L., Bennett F. C., Liddelow S. A., Ajami B., Zamanian J. L., Fernhoff N. B., Mulinyawe S. B., Bohlen C. J., Adil A., Tucker A., Weissman I. L., Chang E. F., Li G., Grant G. A., Gephart M. G. H., Barres B. A., New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U.S.A. 113, E1738–E1746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Djukic M., Mildner A., Schmidt H., Czesnik D., Brück W., Priller J., Nau R., Prinz M., Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain 129, 2394–2403 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Buttgereit A., Lelios I., Yu X., Vrohlings M., Krakoski N. R., Gautier E. L., Nishinakamura R., Becher B., Greter M., Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Ransohoff R. M., A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Dal-Secco D., Wang J., Zeng Z., Kolaczkowska E., Wong C. H. Y., Petri B., Ransohoff R. M., Charo I. F., Jenne C. N., Kubes P., A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 212, 447–456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z., Gu Y., Chakarov S., Bleriot C., Kwok I., Chen X., Shin A., Huang W., Dress R. J., Dutertre C.-A., Schlitzer A., Chen J., Ng L. G., Wang H., Liu Z., Su B., Ginhoux F., Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell 178, 1509–1525.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Faustino J., Chip S., Derugin N., Jullienne A., Hamer M., Haddad E., Butovsky O., Obenaus A., Vexler Z. S., CX3CR1-CCR2-dependent monocyte-microglial signaling modulates neurovascular leakage and acute injury in a mouse model of childhood stroke. J. Cereb. Blood Flow Metab. 39, 1919–1935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmid C. D., Melchior B., Masek K., Puntambekar S. S., Danielson P. E., Lo D. D., Sutcliffe J. G., Carson M. J., Differential gene expression in LPS/IFNγ activated microglia and macrophages: In vitro versus in vivo. J. Neurochem. 109 ( Suppl 1), 117–125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su E. J., Fredriksson L., Geyer M., Folestad E., Cale J., Andrae J., Gao Y., Pietras K., Mann K., Yepes M., Strickland D. K., Betsholtz C., Eriksson U., Lawrence D. A., Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat. Med. 14, 731–737 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang D., Sun Y.-Y., Nemkul N., Baumann J. M., Shereen A., Dunn R. S., Wills-Karp M., Lawrence D. A., Lindquist D. M., Kuan C.-Y., Plasminogen activator inhibitor-1 mitigates brain injury in a rat model of infection-sensitized neonatal hypoxia-ischemia. Cereb. Cortex 23, 1218–1229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eabb2119/DC1