

Abstract
The advent of endothermy is a defining feature of mammalian and avian evolution, achieved through continuous fine-tuned homeostatic regulation of body temperature and metabolism1,2. However, when challenged by food deprivation or harsh environmental conditions, many mammalian species initiate adaptive energy-conserving survival strategies, including torpor and hibernation, during which their body temperature decreases far below its homeostatic setpoint3–5. How homeothermic mammals initiate and regulate these extraordinary hypothermic states remains largely unknown. Here, we discover that entry into mouse torpor, a fasting-induced state with greatly decreased metabolic rate and body temperature as low as 20°C6, is regulated by neurons in the medial and lateral preoptic area of the hypothalamus. We show that re-stimulation of neurons activated during a previous bout of torpor is sufficient to initiate key features of torpor, even in animals that are not calorically restricted. Among these neurons we identify a population of glutamatergic Adcyap1+ cells whose activity accurately determines when animals naturally initiate and exit torpor, and whose inhibition disrupts the natural process of torpor entry, maintenance and arousal. Taken together, we discover a specific neuronal population in the mouse hypothalamus that serves as a core regulator of torpor. This work forms the basis for future explorations of mechanisms and circuitry regulating extreme hypothermic and hypometabolic states, enabling genetic access to monitor, initiate, manipulate and study these ancient adaptations of homeotherm biology.
Torpor and hibernation are some of the most fascinating adaptations of warm-blooded animals, endowing them with the ability to survive harsh environments otherwise incompatible with life7,8. While constituting complex multifaced behaviors9–11, perhaps the most striking feature of these states is the profound decrease in core body temperature far below its tightly controlled homeostatic setpoint3,4 . In this regard, several regions in the mammalian brain, including the preoptic area12, the dorsomedial hypothalamus13, and raphe nuclei,14 have been implicated in coordinating temperature regulation2. Specific electrophysiologically- and/or molecularly-defined cellular components of homeostatic thermoregulation have been identified, including neurons sensitive to changes in ambient temperatures15–18 and/or local brain temperature19–21. However, while a picture of the circuitry underpinning normal thermoregulation is beginning to emerge1,2, how animals disengage or circumvent these conserved homeostatic mechanisms in response to environmental challenges to enter profoundly hypothermic states such as torpor and hibernation remains a central mystery of homeotherm biology.
To study the mechanisms underlying the initiation of these adaptive hypothermic states, we employed a model of fasting-induced torpor in laboratory mice (Mus musculus). Mice placed in environments devoid of food and colder than their ~30°C thermoneutral point22,23 alternate between two survival strategies – higher-risk food-seeking behavior24 and energy-conserving torpor5,6,25. Mouse torpor is a complex natural behavior characterized by repeated bouts of profoundly reduced core body temperature (as low as 20°C), along with decreased movement, sensory perception, breathing, heart rate, and metabolic rate4–6,10,26–28. To study fasting-induced torpor, mice were housed at 22°C and implanted with telemetric temperature probes. While fed animals maintained a core body temperature (Tb) higher than 35.1 ± 0.2°C, all 24-hour food-restricted animals experienced one or more bouts of torpor, which we characterize by a precipitous drop in core body temperature (>1°C/20 min), a period of deep hypothermia (Tb of 24–35°C) lasting up to several hours, and finally arousal from torpor (Fig. 1a). Each torpor bout was preceded by a 45.1 ± 4.6% decrease in metabolic rate and accompanied by reduced movement (73.4 ± 7.0% reduction, Fig. 1a–c, Extended Data Fig. 1a). While circadian rhythms28, leptin signaling27,29, sympathetic nervous system activity, and adipose tissue thermogenesis26,30 have all been shown to modulate torpor, the mechanisms by which animals trigger and regulate this natural hypothermic state remain unknown.
Figure 1: Neuronal activity induces key features of torpor.

a) Core body temperature (Tb) and gross motor activity [arbitrary units] of a representative non-fasted or fasted mouse over 24 hours. Fasted mice enter torpor, while non-fasted mice do not. Gray/white indicate 12 hours of darkness/light. Dashed line indicates minimum Tb observed in non-fasted mice. b, c) Minimum Tb and metabolic rate (VO2 – volume of oxygen consumed [l/h]) in non-fasted (fed) and fasted mice (n = 7 animals, p = 6e-4, p = 1e-5, respectively). d) Schema for gaining genetic control over torpor-regulating neurons. Neurons active during torpor in FosTRAP; LSL-Gq-DREADD mice are TRAPed by 4-OHT administration and chemogenetically re-stimulated 7 days later in non-fasted mice by CNO treatment. e, f) CNO-induced reactivation of 4-OHT-TRAPed neurons active during torpor entry triggers a decrease in Tb characteristic of mouse torpor (fast-TRAP, n = 14 animals). The same mice injected with PBS (fast-PBS, n = 8 animals), or control mice in which neurons were not TRAPed (no-TRAP, n = 6 animals) or were TRAPed during a non-torpid state (fed-TRAP, n = 9 animals), did not decrease Tb upon CNO administration. e) Dashed line indicates the onset of CNO or PBS administration, shading indicates 95% confidence interval of Tb. f) Minimum Tb following CNO administration is lower in fast-TRAP (n = 14) animals compared to no-TRAP (n = 6, p = 6.2e-4), fed-TRAP (p = 2.4e-6) or fast-PBS (p = 2.5e-5) animals. All box plots indicate mean ± s.e.m. P-values calculated by two-tailed Mann-Whitney U test, *** p < 0.001.
Torpor-associated circuit activity
In principle, entry into torpor could be triggered by circulating factors capable of reducing metabolic rate and/or changes in thermoregulatory neural circuit activity. Consistent with the idea that altered circuit activity contributes to torpor entry26, staining for c-Fos, a marker of neuronal activity-induced transcription31, followed by whole-brain imaging and machine learning-enabled registration of c-Fos signal to the Allen Mouse Brain Atlas32 (Methods), revealed multiple brain regions that are active during fasting-induced torpor. As might be expected, we observed neuronal activity in brain regions regulating hunger, feeding, and energy balance33,34 as well as in thermoregulatory areas1,2,35 and a large number of other brain regions, indicating that brain circuits are engaged as fasted mice enter torpor and thus may potentially drive this process (Extended Data Fig. 1b–f).
To determine if neural circuit activity is sufficient to induce torpor phenotypes independent of caloric restriction, we employed genetic tools that allow the expression of a chemically-activated receptor Gq-DREADD (Gq-coupled Designer Receptor Exclusively Activated by Designer Drug) specifically in the neurons that are active as mice enter torpor. This approach allows the re-activation of the putative torpor-regulating neurons by the simple administration of the Gq-DREADD-activating synthetic ligand clozapine N-oxide (CNO) to the mice, in order to determine if the re-activation of these neurons alone, without caloric restriction, is sufficient to induce torpor-associated phenotypes. For this experiment, we used mice harboring a tamoxifen-dependent form of Cre recombinase driven from the Fos locus (Fos2A-iCreERT2, TRAP236) together with an allele of the Gq-coupled receptor that is expressed in a Cre-dependent manner (R26-LSL-Gq-DREADD37). When these “FosTRAP-Gq” mice are fasted to enter torpor, the neurons that are active and thus potentially mediate torpor entry induce Fos and CreERT2. When these animals are exposed to tamoxifen, the CreERT2 recombines the R26-LSL-Gq-DREADD allele, leading to the persistent expression of the Gq-DREADD and allowing these specific “TRAPed” neurons to be activated at a later time by administration of CNO (Fig. 1d).
FosTRAP-Gq mice (n = 14) were thus fasted and administered 4-hydroxy tamoxifen (4-OHT) as they entered natural torpor. After several days of recovery from fasting, these animals were administered the DREADD-activating ligand CNO to chemogenetically re-stimulate the neurons TRAPed during natural torpor (Fig. 1d, Methods). Remarkably, stimulation of neurons previously active during fasting/torpor was sufficient to induce the robust decrease in core body temperature and locomotor activity associated with natural torpor, despite the absence of caloric restriction. Importantly, this effect was dependent on CNO administration and on prior 4-OHT-mediated TRAPing in the fasted state (Fig. 1e, f, Extended Data Fig. 1g, h). Although we cannot exclude a contribution of either fasting-regulated neurons active prior to or following torpor or non-neuronal cells, this striking result suggests that systemic recapitulation of torpor-associated neuronal circuit activity is sufficient to acutely induce key behavioral and physiological features of torpor.
avMLPA neurons regulate features of torpor
To identify the brain areas labeled using the TRAP approach, we immunostained brain sections of these FosTRAP-Gq mice for the HA-tagged Gq-DREADD protein. Whole-brain imaging revealed widespread Gq-DREADD protein expression, with 190 differentially labeled regions identified between animals TRAPed in a fasted (fast-TRAPed) versus fed (fed-TRAPed) state (Extended Data Fig. 2i–k, Supplementary Table 1). A strong correlation across brain regions was observed between the number of c-Fos+ cells in torpid mice and the levels of Gq-DREADD expression in fast-TRAPed mice, suggesting that our TRAP approach labeled, as intended, neurons that are active and induce c-Fos during torpor (Extended Data Fig. 2l). Although in principle simultaneous activation of multiple neural populations across several distributed brain areas might be required to orchestrate torpor, we hypothesized that circuit activity within a single brain region might play a significant role in torpor regulation. To address this possibility, we designed a screen across brain regions identified by the Gq-DREADD staining in fast-TRAP mice. We stereotactically injected FosTRAP mice (that do not express an endogenous Gq-DREADD) with adeno-associated viruses expressing Cre-dependent Gq-DREADD fused to mCherry (AAV-DIO-Gq-mCherry), enabling injection area-restricted TRAPing, Gq-DREADD-mCherry expression, and subsequent chemogenetic re-stimulation of the fasting/torpor-active neurons within just the injected region (Fig. 2a). For these studies we focused on the torpor-associated drop in core body temperature, perhaps the most striking physiological aspect of natural torpor. FosTRAP mice (n = 54) were injected into different areas of the anterior hypothalamus, a region of the brain involved in thermoregulation and energy balance1,2 that showed substantial c-Fos expression during fasting-induced torpor (Extended Data Fig. 1). Following recovery, these “FosTRAPAAV-Gq” mice were fast/torpor-TRAPed, enabling persistent expression of the viral Cre-dependent Gq-DREADD-mCherry in fast/torpor-active neurons within the virally injected region. Several days later, we administered CNO to stimulate Gq-DREADD-expressing neurons selectively in the injected region of the hypothalamus to test whether their stimulation would decrease body temperature consistent with natural torpor. The reduction of core body temperature was correlated with the anatomical expression of the virally-derived Gq-DREADD-mCherry across 277 hypothalamic nuclei/areas (Fig. 2b, Methods). Leveraging the variability across injection sites in different animals (n = 54), this unbiased screen identified the anterior and ventral portion of the medial and lateral preoptic area (avMLPA) as key regions, with animals injected in the avMLPA exhibiting a large decrease in core body temperature in response to CNO as compared to animals in which these regions were not transduced (4.90 ± 0.68°C vs. 0.89 ± 0.25°C, p = 6.9e-9, Fig. 2c–e, Supplementary Table 2, Supplementary Table 3, Extended Data Fig. 2a–c). FosTRAPAAV-Gq animals injected selectively in the avMLPA, TRAPed during torpor, allowed sufficient time to recover, and then stimulated by CNO, showed a decrease in the metabolic rate by 30.4 ± 8.5% (p < 2.4e-3), and gross motor activity by 58.7 ± 5.2% (p < 1.5e-3), consistent with features observed in natural torpor (Fig. 2f, Extended Data Fig. 2d–g). Moreover, sectioning and staining brains for the virally-derived Cre-dependent Gq-DREADD-mCherry fusion protein revealed projections from torpor-TRAPed avMLPA (avMLPAtorpor) neurons to the dorsomedial hypothalamus (DMH), arcuate nucleus (ARC), periaqueductal gray (PAG), and raphe pallidus (RPa), regions known to modulate energy balance and adipose tissue thermogenesis, processes that are thought to be involved in torpor induction (Extended Data Fig. 2h–j)38,39. Together, these findings identify a brain area in which re-activation of torpor-associated neurons is sufficient to acutely induce torpor-like behavioral and physiological changes, and suggest that avMLPAtorpor neurons may represent a critical node in the circuit that regulates natural torpor entry.
Figure 2: Identification of brain regions regulating torpor.

a) Schema for identifying which hypothalamic regions contain torpor-regulating neurons. b) Quantification of AAV-DIO-Gq-DREADD-mCherry expression in mice TRAPed during fasting-induced torpor. 277 hypothalamic nuclei are plotted based on their AP coordinates relative to bregma (B). Fifty-four animals are ranked based on decrease in core body temperature (ΔTb) observed following chemogenetic stimulation of TRAPed neurons. ΔTb is correlated with viral expression in each region (Pearson correlation). c) Two regions in which viral expression did not, and three regions in which viral expression showed significant correlation to ΔTb (FDR-corrected q-value, Pearson correlation test, n = 54 animals). Minimum Tb was calculated across all animals grouped based on degree of viral expression into None, Partial or Complete. Box plot displays median, inter-quartile range (IQR), whiskers up to 1.5*IQR. d) Coronal section from an avMLPA-injected mouse (n = 15 animals). e, f) Chemogenetic re-stimulation of avMLPA TRAPed neurons (avMLPA-hit CNO, n = 15 animals), the same animals (n = 15) injected with PBS or control mice in which the avMLPA was missed (avMLPA-miss, n = 11 animals). e) Dashed line indicates CNO or PBS administration, gray shading indicates 95% confidence interval of Tb. f) Minimum Tb following CNO administration is lower in avMLPA-hit compared to avMLPA-miss (p = 1.8e-6) or avMLPA-hit mice injected with PBS (p = 5.8e-7). Two-tailed Mann-Whitney U test, *** p < 0.001. Box plot indicates mean ± s.e.m.
Molecular analysis of avMLPAtorpor neurons
The mammalian preoptic area houses an interconnected ensemble of cell types involved in temperature15–17,20, fluid36, and cardiovascular homeostasis40, as well as mating, parental behaviors41, and sleep42–44. To catalogue the diversity of neuronal cell types present in the avMLPA, and identify which among them are active and TRAPed during torpor, we adapted a high-throughput single-nucleus RNA-sequencing (snRNA-seq)-based strategy45 (Fig. 3a). Five FosTRAP mice were injected with AAV-DIO-Gq-DREADD-mCherry. Four of these animals were TRAPed during torpor, while one was kept as a non-TRAPed control. Their anteroventral preoptic area was dissected, dissociated, and 44,669 single nuclei were snRNA-sequenced at an average depth of 1286 genes and 2083 transcripts per nucleus (Extended Data Fig. 3a–e). Unsupervised graph-based clustering delineated major neuronal and non-neuronal cell classes, and further clustering of 28,103 neurons identified a remarkable diversity of 24 GABAergic, eight glutamatergic, three hybrid (GABAergic and glutamatergic), and one cholinergic neuronal cell type, consistent with large diversity of cell types present in the POA41 (Fig. 3b–e, Extended Data Fig. 4, Supplementary Table 4). The robustness of the obtained clusters was confirmed by subsampling analysis (Extended Data Fig. 3f). Importantly, 17,424 of the 28,103 sequenced neurons, representing all 36 cell types, expressed AAV-derived transcripts, consistent with broad tropism of AAV1 in the hypothalamus.
Figure 3: Molecular characterization of torpor-associated avMLPA neurons.

a) Schema for molecular characterization of avMLPATRAP cells. AAV-DIO-Gq-DREADD is injected into the avMLPA (n = 5 mice). Following TRAPing, the avMLPA is microdissected and snRNA-sequenced. b) UMAP plot of 39,562 nuclei from the avMLPA of five animals. Colors denote main cell types (names indicated). c) Marker gene expression across cell types (names abbreviated to first few letters). d) UMAP plot of 28,103 neuronal nuclei. Colors denote 36 neuronal subtypes. e) Marker gene expression across neuronal cell types. Cell types are organized on the basis of hierarchical clustering. Acronym comprises neuronal class (e - excitatory, i - inhibitory, h - hybrid, c - cholinergic) and cluster number, followed by select marker genes. f) UMAP plot of 17,424 neuronal nuclei that were transduced by the AAV (gray) and 342 neuronal nuclei that were TRAPed during torpor (red). g) Distribution of TRAPed neurons across all neuronal cell types. Box plots indicate mean ± s.e.m. (n = 4 animals). Yellow shading indicates cell types expressing Adcyap1 and Vglut2. Acronym comprises neuronal class and cluster number. c, e) Color denotes mean expression across all nuclei normalized to the highest mean across cell types. Size represents the fraction of nuclei in which the marker gene was detected.
We analyzed the expression of Gq-DREADD-mCherry transcripts as a means to identify which among the transduced neurons were TRAPed during torpor. This approach detected 342 torpor-TRAPed neurons among 15,056 transduced neurons in the four TRAPed mice and displayed a low false-positive and false-negative rate (Extended Data Fig. 5a–e, Methods). TRAPed neurons represented several avMLPA cell types, suggesting that multiple neuronal populations were active during fasting-induced torpor (Fig. 3f). However, the largest subset (42.6 ± 3.5%) of all torpor-TRAPed cells consisted of several populations of glutamatergic Adcyap1+ neurons (Fig. 3g, Extended Data Fig. 5f–i), a result that was subsequently confirmed using in situ hybridization methods (Extended Data Fig. 6a–g, Extended Data Fig. 7). Differential gene expression analysis between TRAPed and non-TRAPed Vglut2+ Adcyap1+ neurons identified markers of e5 neurons, consistent with preferential TRAPing of this molecularly-defined subtype of Adcyap1+ neurons (Supplementary Table 5).
Stimulation of torpor-associated neurons
Together with previous work describing distinct populations of GABAergic and glutamatergic warm-sensitive thermoregulatory neurons in the POA16–18,46, our results raise the possibility that Vglut2/Adcyap1-expressing avMLPA neurons (avMLPAVglut2/Adcyap1) regulate the drop in core body temperature associated with natural torpor. To directly test whether stimulation of these neurons is sufficient to phenocopy the drop in body temperature observed during natural torpor, we employed Vglut2-IRES-Cre and Adcyap1-2A-Cre mice and expressed the excitatory Gq-DREADD in avMLPAVglut2 or avMLPAAdcyap1 neurons (Fig. 4a). Chemogenetic activation of avMLPAAdcyap1 or avMLPAVglut2 neurons resulted in a rapid decrease in core body temperature (5.9 ± 0.4°C, 4.9 ± 1.0°C, respectively) and gross motor activity (58.7 ± 8.4%, 53.3 ± 11.8%, respectively, Fig. 4a–c, Extended Data Fig. 8a–f), that phenocopied the stimulation of torpor-TRAPed avMLPA neurons and effectively recapitulated the drop in core body temperature and activity observed during natural torpor. By contrast, chemogenetic activation of avMLPAVgat neurons via the use of Vgat-IRES-Cre mice led to no significant change in core body temperature (0.2 ± 0.3°C, p = 0.48, Fig. 4a–c, Extended Data Fig. 8b).
Figure 4: Sufficiency, necessity, and natural activity of avMLPA neuronal subpopulations during torpor.

a–c) Injection of AAV-DIO-Gq-DREADD and subsequent chemogenetic stimulation of avMLPAVgat, avMLPAVglut2, and avMLPAAdcyap1 neurons (n = 6, 5, 8 animals, respectively). a) Experimental schema. b) Mean core body temperature (Tb) following chemogenetic stimulation (dashed line). c) Mean Tb before and after chemogenetic stimulation of avMLPAVgat (p = 0.48), avMLPAVglut2 neurons (p = 7.9e-3), and avMLPAAdcyap1 (p = 1.6e-4). d) Schema for injection of AAV-DIO-TeLC to inhibit synaptic transmission. e, f) Tb of fed and fasted mice in which e) avMLPAVglut2 or f) avMLPAAdcyap1 neurons remained un-injected (Pre), were injected with a control AAV (Ctrl), or were injected with AAV-DIO-TeLC (TeLC). Colored lines indicate the mean across animals; gray shading indicates 95% confidence interval. Number of animals indicated in parenthesis. g) Schema for injection of AAV-DIO-GCaMP6s and fiber photometry recording from avMLPAAdcyap1 neurons. h–j) Example recording sessions in fasted mice showing Tb and normalized GCaMP6s signal. Example 20-minute trace spanning torpor entry (i) and torpor arousal (j). Colored bars indicate states (based on Tb); legend displayed in (h). Dashed line indicates transition between non-torpor and torpor states (Methods). k, l) Example photometry signal in non-torpor (k) and torpor (l). Baseline indicated in blue. m) Mean baseline (left) and peak frequency (right) of fiber photometry signal in one animal across states (legend displayed in (n), p-values indicated above plot). n) Difference in average baseline (left) and Log2 fold-change in average peak frequency (right) of fiber photometry signal between torpor states and non-torpor (n = 8 animals, p-values indicated above plots). All box plots indicate mean ± s.e.m. All p-values calculated by Mann-Whitney U test, two-tailed. n.s. represents non-statistically significant, *p < 0.05, ** p < 0.01, *** p < 0.001.
Silencing of torpor-associated neurons
While these findings are consistent with the idea that torpor-active avMLPAAdcyap1/Vglut2 cells are critical torpor-inducing neurons in the avMLPA, it remained possible that these cells simply constituted a part of the core circuitry controlling homeostatic body temperature as opposed to mediators of torpor entry per se. To directly address the role of avMLPAAdcyap1/Vglut2 neuronal activity in basal homeostatic thermoregulation as well as the natural process of fasting-induced torpor, we targeted the expression of a virally-encoded, Cre-dependent tetanus toxin light chain47 (AAV-DIO-TeLC), a derivative of a potent neurotoxin that eliminates synaptic transmission, to avMLPAAdcyap1 or avMLPAVglut2 neurons (Fig. 4d). Importantly, by expressing the Gq-DREADD and TeLC in the same neurons and stimulating them with CNO, we were able to first verify that TeLC expression effectively inhibited synaptic transmission in these cells insofar as it blocked the decrease in core body temperature associated with the chemogenetic stimulation (Extended Data Fig. 8g–i). Analysis of fed mice found that avMLPAAdcyap1 or avMLPAVglut2 silencing had no significant effect on normal homeostatic body temperature, including its circadian rhythm (Fig. 4e, f, Extended Data Fig. 8j–o). By contrast, mice in which avMLPAVglut2 neurons were selectively silenced showed profound disruption in fasting-induced torpor (Fig. 4e). Specifically, the core body temperature drop associated with fasting-induced torpor was significantly diminished upon AAV-DIO-TeLC injection (TeLC) as compared to that observed in the same mice prior to injection (Pre fast, p = 0.010) or in mice injected with a control AAV vector (Ctrl fast, p = 0.018, Extended Data Fig. 8j). The kinetics of the torpor-associated drop in body temperature were also significantly altered by avMLPAVglut2 silencing, with animals reaching their lowest body temperature after 22 ± 1 h instead of 14 ± 1h (Extended Data Fig. 8k, p = 9.2e-3). Silencing of avMLPAAdcyap1, a subset of avMLPAVglut2 neurons, also altered natural torpor (Fig. 4f, Extended Data Fig. 8l), replacing normal torpor bouts with a gradual decrease in core body temperature that took 6 h longer to reach a similar degree of hypothermia compared to normal torpor (Extended Data Fig. 8m, p < 7e-3). It is important to note that the injected site in some cases included areas extending beyond the avMLPA. In addition, by in situ hybridization we find that only a subset (43 ± 5%) of avMLPA Adcyap1+ neurons expressed TeLC, perhaps due to inefficient viral transduction or Cre-mediated recombination. This may have led to incomplete silencing of torpor-regulating avMLPAAdcyap1 neurons, and could be a potential reason for the incomplete elimination of fasting-induced torpor responses observed in this paradigm (Extended Data Fig. 8p, q). Nevertheless, these results suggest that the activity of avMLPAAdcyap1 neurons is necessary for the normal pattern of torpor thermoregulation characterized by rapid decrease in core body temperature, maintenance, and subsequent re-warming and that perhaps together with other glutamatergic neurons avMLPAAdcyap1 neurons are required for the depth of hypothermia observed during natural torpor.
avMLPAAdcyap1 neuronal dynamics in torpor
Active involvement of glutamatergic Adcyap1+ avMLPA neurons in torpor initiation should be reflected by acute torpor-associated changes in avMLPAAdcyap1 neuronal firing patterns. Moreover, a characterization of avMLPA neuronal firing patterns during natural torpor might provide further insight into the underlying functional role of this circuit in torpor regulation. For example, natural avMLPA firing patterns might encode a caloric deficit, circadian time, rate of change in body temperature, time of acute torpor onset, or maintenance of torpor. To distinguish among these possibilities, we expressed the calcium reporter GCaMP6s48 in avMLPAAdcyap1 neurons, installed an optical fiber, and continuously monitored neuronal calcium transients over 7–12 hours in freely moving fasted mice as they entered torpor (Fig. 4g, Extended Data Fig. 9a, b, Methods). This analysis failed to reveal a gradual change in neuronal activity correlated with caloric restriction or circadian time, or a transient pattern of activity that correlated with only torpor onset. Instead, we observed a striking change in neuronal activity which coincided with torpor entry and persisted until the animals began to exit the torpid state (Fig. 4h–l, Extended Data Fig. 9c, d, Methods). This torpor-associated activity pattern was characterized by an 8.0 ± 1.8% decrease in the baseline signal (p = 3e-3) and a 19.0 ± 7.0-fold increase in the frequency of highly prominent Ca2+-dependent peaks (p = 2e-4, Fig. 4m, n, Extended Data Fig. 9d–g). Strikingly, this distinct pattern of avMLPAAdcyap1 neuronal activity alone was sufficient to accurately model and determine when animals entered, maintained, and exited torpor (Extended Data Fig. 9h–k), suggesting that avMLPAAdcyap1 neurons encode information specific to torpor entry and maintenance.
To exclude the possibility that the avMLPAAdcyap1 neurons are merely responding to an acute change in body temperature, we recorded their activity in animals in which hypothermia was artificially induced by administration of the adenosine receptor agonist N6-cyclohexyladenosine (CHA)49. Chemically induced hypothermia resulted in a similar decrease in baseline signal (7.2 ± 1.7%), perhaps due to decreased neuronal discharge at lower body temperature. Importantly, chemically induced hypothermia failed to produce prominent Ca2+-dependent peaks associated with fasting-induced torpor, suggesting that the large Ca2+-transients observed in avMLPAAdcyap1 neurons may encode information relevant specifically to torpor and not simply hypothermia or cooling (Extended Data Fig. 10a–e). Whether this pattern of Ca2+-transients reflects the existence of distinct neuronal subpopulations or perhaps burst firing of a single population of responsive neurons remains unclear, as does its function in the process of torpor entry.
To investigate whether torpor-active avMLPAAdcyap1 neurons are distinct from previously identified populations of warm-sensitive neurons in the POA16–18,46, we next challenged the mice in which we had previously observed torpor-related avMLPAAdcyap1 neuron activity with either warm (37°C) or cold (10°C) environments. Unlike previously described warm-sensitive neurons, avMLPAAdcyap1 neurons showed no significant changes in activity in the warm environment but did display sensitivity to a cold environment, suggesting the existence of several functionally distinct subpopulations of POAAdcyap1 neurons (Extended Data Fig. 10f–i). In summary, we show that avMLPAAdcyap1 neurons both contribute to and are necessary for the natural decrease in body temperature observed during torpor, and encode a unique pattern of broad and highly prominent Ca2+-transients as animals enter and sustain torpor, suggesting that the activity of these neurons is critical to the natural process of torpor initiation and maintenance in mice.
Discussion
Our study dissects the mechanisms underlying a complex naturalistic behavior by employing several recent technological advances, including the FosTRAP approach, machine-learning enabled image registration, single-nucleus RNA-sequencing, and long-term recordings of neuronal activity. We implicate specific neuronal cell types within a defined brain region in the regulation of torpor, one of the most extreme and poorly understood physiological adaptations in homeothermic animals. We discover that neural activity alone is sufficient to induce several key features of torpor, including decreased locomotion and profoundly lowered metabolic rate and core body temperature in animals which are not calorically restricted. We identify the avMLPA of the hypothalamus as a torpor control center, and Vglut2/Adcyap1+ neurons as a central torpor-regulating neuronal population. We observe that the activity of these neurons changes dramatically when animals naturally enter torpor and that this both contributes to and is necessary for the precipitous drop in core body temperature observed during natural torpor. How these torpor-regulating neurons integrate information about internal states and environmental experience remains to be understood. Glutamatergic avMLPAAdcyap1 neurons receive information about decreasing ambient temperatures, leading us to speculate that these neurons might integrate information about environmental conditions with information about the animal’s internal energy reserves to control when the animal enters torpor. Transcriptome-wide analysis of Vglut2+ Adcyap1+ neurons identified expression of the leptin receptor in two of the Vglut2+ Adcyap1+ neuronal subtypes raising the possibility that circulating leptin levels might modulate the activity of torpor-regulating neurons, providing a potential mechanism for how information about decreased energy reserves in fasted mice could be conveyed to torpor-regulating avMLPAVglut2/Adcyap1 neurons27,30 (Extended Data Fig. 4j). In addition, given that entry into torpor requires hours of fasting, and involves many neuronal populations in which the c-Fos transcription factor is induced, an interesting possibility is that activity-dependent gene transcription plays a mechanistic role in establishing a state permissive for torpor entry.
Since torpor is a dynamic and complex behavior featuring profound changes in body temperature, metabolic rate, locomotion, perception, breathing, and heart rate7,10, engagement of other neuronal populations and brain regions, including additional torpor-associated neurons identified here by the FosTRAP approach and snRNA-seq, will likely be involved in orchestrating the full program of natural torpor entry, maintenance, and arousal. Importantly, the elucidation of torpor-regulating neuronal circuitry will allow for a dissection of the mechanisms by which this circuit inhibits, circumvents, or inverts normal cold defensive thermoregulatory processes1,2,50. This should provide insight into why only certain mammalian species have the ability to enter torpor, and whether such a hypometabolic state could be induced in species that don’t typically enter torpor. For example, while rats and humans do not naturally enter torpor, recent studies have recapitulated attributes of torpor in rats39, raising the possibility of inducing a similar state in humans. Future investigations promise exciting advances through the study and manipulation of these ancient adaptations of homeotherm biology.
Methods:
Animals
Animal experiments were approved by the National Institute of Health and
Harvard Medical School Institutional Animal Care and Use Committee, following ethical guidelines described in the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. For initial torpor experiments, we used adult (6–10-week-old) C57BL/6J (The Jackson Laboratory, Stock # 000664) mice. To generate FosTRAP-Gq mice we crossed Fos2A-iCreER (TRAP2) (The Jackson Laboratory Stock # 030323) with R26-LSL-Gq-DREADD (The Jackson Laboratory Stock # 026220) and used adult (6–18-week-old) male and female F1 progeny. For viral injections we used Fos2A-iCreER (TRAP2) (The Jackson Laboratory Stock # 030323), Adcyap1-2A-Cre (The Jackson Laboratory Stock # 030155), Vglut2-ires-cre (The Jackson Laboratory Stock # 028863), and Vgat-ires-cre (The Jackson Laboratory Stock # 028862) mice. All mice were housed at 22°C under a standard 12 hr light/dark cycle.
No statistical methods were used to predetermine the sample size. Mice were randomly assigned to experimental groups before surgery. Where possible, investigators were blinded during analysis.
Telemetric monitoring of core body temperature and gross motor activity
Mice with genotypes of interest were singly housed and implanted abdominally with telemetric temperature and activity probes (Starr Life Science VV-EMITT-G2). After at least four days of recovery, mice were recorded in standard cages placed onto a radio frequency receiver platform (Starr Life Science ER4000). Core body temperature and gross motor activity were logged every 60 seconds.
Measurements of oxygen consumption
Oxygen consumption was measured using the Columbus Instruments Comprehensive Lab Animal Monitoring System (CLAMS) at the Brigham and Women’s Hospital or the Beth Israel Deaconess Medical Center Metabolic Core. Individual animals were placed into metabolic cages allowing for the measurement of oxygen consumption (VO2, Volume of O2 consumed per unit time). The CLAMS system is compatible with simultaneous measurements of core body temperature and gross motor activity using the implanted telemetric probes.
Torpor induction
Adult (6–18-week-old) animals were singly housed prior to induction of torpor. Each animal was moved to a new individual cage containing water and nesting material but devoid of bedding and food at the beginning of the dark cycle. Initial bouts of torpor were observed after approximately 8 hrs of fasting. Animals were returned to their standard cages containing food 24 hrs after the start of the fast. The ambient temperature of the facility was maintained at ~22°C.
TRAPing
To label neurons active during torpor, we used mice harboring a tamoxifen-dependent form of Cre recombinase (CreERT2) driven from the Fos locus (Fos2A-iCreERT2, FosTRAP36). When these mice are fasted to enter torpor, the neurons that are active induce Fos and CreERT2. When these mice are exposed to 4-hydroxytamoxifen (4-OHT, Sigma # H6278-10MG), the CreERT2 will translocate to the nucleus. Once translocated, CreERT2 can recombine the genomically encoded Cre-dependent R26-LSL-Gq-DREADD allele or the virally-introduced Cre-dependent AAV-DIO-Gq-DREADD-mCherry construct selectively in the Fos-expressing cells. This recombination leads to the persistent expression of the Gq-DREADD and allows for the activation of these specific “TRAPed” neurons at a later time by administration of Clozapine N-Oxide (CNO)51.
4-hydroxytamoxifen (4-OHT, Sigma Aldrich # H6278) solution was prepared by initially dissolving 10 mg 4-OHT in 500 μL of 100% ethanol, then adding 450 μL of a 1:4 mixture of castor oil: sunflower oil and vortexing. The ethanol was removed via vacuum centrifugation, and the remaining 4-OHT solution was diluted with the same 1:4 mixture of castor oil: sunflower oil to a final concentration of ~6.25 mg/mL. For TRAPing, each mouse was injected intraperitoneally with 50 mg/kg of 4-OHT during torpor entry.
Clozapine N-Oxide (CNO) solution was prepared by initially dissolving CNO hydrochloride (Sigma Aldrich # SML2304) in H2O to a stock solution of 100 mM. The stock solution was diluted with PBS to a final concentration of 0.6 mM, and ~250 μL were injected intraperitoneally per mouse for a final injection concentration of 2 mg/kg.
Immunofluorescent staining
Mice were sacrificed by transcardial perfusion of 10 mL PBS followed by 10 mL of 4% paraformaldehyde (PFA). Brains were extracted, post-fixed overnight with 4% PFA at 4°C and then embedded in PBS with 3% agarose. Brains were sliced on a vibratome (Leica VT1000S) into 50 μm coronal sections. Coronal sections were washed three times with PBS containing 0.3% TritonX-100 (PBST) and blocked for 1 hr at room temperature with PBST containing 5% donkey serum (blocking buffer). Sections were incubated overnight at 4°C with primary antibodies diluted in blocking buffer, washed again three times with PBST, and incubated for 1 hr at room temperature with secondary antibodies diluted in blocking buffer. After washing twice in PBST and once in PBS, samples were mounted onto SuperFrost Plus glass slides (VWR) using DAPI Fluoromount-G.
Detection of Fos+ and Gq-DREADD-HA+ cells
Tissues were processed as indicated in “Immunofluorescent staining” using the following reagents: Primary antibodies - rabbit anti-Fos antibody 1:2000 (Cedarlane # 226003(SY)) and rabbit anti-HA antibody 1:1000 (Cell Signaling Technology # 3724S). Secondary antibody - 1:500 donkey anti-rabbit 647 secondary antibody (Life Technologies # A31573).
Sections were imaged on an Olympus BX61VS microscope using a UPlanSApo 10×0.4 objective (Harvard NeuroDiscovery Center).
Image registration to the Allen Brain Atlas and analysis
Imaged immunofluorescent brain slices were converted to TIF format and organized in sequential anterior to posterior order. To quantify the average FosHA signal and count Fos+ cells over different brain regions, the brain slices were registered with the Allen Brain Atlas as described previously52–55, with some custom modifications. There are three main parts to the pipeline: pre-processing, stack registration, and plane alignment. The pre-processing stage includes downsizing image files, using a previously trained machine learning algorithm to compute the brain slice borders, generating a mask and edge map, and detecting puncta if necessary. The stack-registration stage aligns the dataset with itself using the vertical line of symmetry as a guide. In the plane assignment stage, each internally-registered brain slice is paired to the Allen Brain Atlas using an estimate of the bregma value and a method similar to sequence alignment using dynamic programming. After pairwise registration to the Allen Brain Atlas, experimental brain slices are further adjusted using local non-linear deformations to maximize the fit between the experimental image and the image from the Allen Brain Atlas. Then, the signal intensity and the number of spots are quantified for each brain region defined by the Allen Brain Atlas. Finally, the volume of each brain region is calculated so that the signal density (signal intensity divided by volume) and density of spots (number of spots divided by volume) could be determined.
Several parameters in the pipeline were adjusted to optimally process our experimental brain slices. For both Fos and FosHA signal quantification, a boundary erosion was radius “1” (after downsizing) was implemented to exclude quantification at the edge of the brain slices, where inaccuracies during the experimental preparation may occur. Estimated bregma values for the most anterior and posterior brain slices were also adjusted based on each sample. To detect individual Fos+ nuclei via Laplacian of Gaussian filtering of the image, we found and distinguished the local maxima that are Fos puncta from local maxima in the background. The distribution of local maxima in the background was computed, and the threshold distance to background distribution was set to ‘7’ to indicate that only spots 7 standard deviations away from the mean of the background distribution were selected. For selected spots (putative Fos+ nuclei), their correlation with the ideal circular spot was measured, and any spot with a correlation below 0.5 was eliminated. These parameters were ignored when FosTRAP-Gq-HA brain signal was quantified as the HA-signal was not nuclear and was instead distributed across the cell body and neuronal processes and thus could not be modeled and quantified as a circular spot.
Viral constructs
AAV8-hSyn-DIO-Gq-mCherry (Addgene # 44361-AAV8) and AAV1-Syn-Flex-GCaMP6s-WPRE-SV40 (Addgene # 100845-AAV1) were obtained from Addgene. AAV2/1-hSyn-Flex-TeLC-EYFP56 was prepared through Boston Children’s Hospital Viral Core. All viruses were diluted with PBS to a final concentration of 5E12 – 1E13 genome copies per mL before stereotaxic delivery into the mice brain.
Stereotactic viral injection and photometry fiber implantation
For injections, animals were anesthetized with 3% isoflurane and placed in a stereotaxic head frame (Kopf Instrument, model 1900). Coordinates AP +0.4 mm, ML +/− 0.5 mm, DV −5.1 mm relative to bregma, were used for all avMLPA injection. Unless otherwise specified, all experiments were carried out with bilateral injections. An air-based injection system built with Digital Manometer (Grainger # 9LHH8) was used to infuse virus. The virus was infused at approximately 100 nL/minute, and the needle was kept at the injection site for 10 min before withdrawing. For chemogenetic stimulation, ~25–100 nL of AAV8-hSyn-DIO-Gq-mCherry was delivered into the region of interest. For silencing, ~100 nL of AAV2/1-hsyn-Flex-TeLC-EYFP was delivered into the region of interest. For fiber photometry recordings, 200 nL of a mixture of AAV8-hSyn-DIO-Gq-mCherry and AAV1-Syn-Flex-GCaMP6s-WPRE-SV40 was delivered into the region of interest, either unilaterally or bilaterally. For fiber photometry recording, Mono Fiber-optic Cannulas (Doric, MFC_200/230-0.37_###_MF1.25_FLT) were implanted 200 μm above the injection site. The fiber was fixed to the skull with LOCTITE 454 INSTANT ADHESIVE and further covered by dental cement to ensure the stability of the implant.
Mapping of virally injected hypothalamic nuclei and correlation with chemogenetically induced changes in core body temperature
Viral injection, sample preparation, and imaging
To minimize initial differences in body weight across animals which might influence the effects on core body temperature and confound our screen across brain regions, we employed a cohort of fifty-four 6–10-week-old female FosTRAP mice. Mice were bilaterally injected with AAV8-hSyn-DIO-Gq-mCherry, allowed to recover for 4–10 days and then fasted and injected with 4-OHT during torpor to TRAP active neurons. Subsequently (3–14 days later), mice were administered CNO to stimulate fast-TRAPed neurons within the virally injected regions and their core body temperature and activity were recorded. Given that different mice were injected in different hypothalamic areas and that no two injections will be identical due to the subtle variabilities in the injection site and viral spread, all mice were sacrificed to map post-hoc the exact brain regions that were transduced and contained TRAPed neurons. To this end, mice were sacrificed by transcardial perfusion of 10 mL PBS followed by 10 mL of 4% paraformaldehyde (PFA). Brains were extracted, post-fixed overnight with 4% PFA at 4°C and then embedded in PBS with 3% agarose. Brains were sliced on a vibratome (Leica VT1000S) into 50 μm coronal sections, mounted onto glass slides and imaged on an Olympus BX61VS microscope using a UPlanSApo 10× 0.4 objective (Harvard NeuroDiscovery Center). AAV-DIO-Gq-DREADD-mCherry expression was detected by endogenous fluorescence.
Image analysis
Viral mCherry expression was quantified across 277 hypothalamic regions, noting semi-quantitatively for each region and each hemisphere whether the viral expression was 0 (none), 1 (minimal), 2 (partial), or 3 (total, Supplementary Table 3, Fig. 2b). The analysis was performed blinded to the effect on core body temperature that was previously observed in each animal.
Correlation with changes in core body temperature
To assign a single numeric value for each hypothalamic region in each animal, we added the semi-quantitative transduction values from the two hemispheres. For correlation analysis, we analyzed 226 of the 277 brain regions that were transduced in at least three animals. For each region independently, we determined the Pearson correlation across all animals between the viral expression in that region and the decrease in core body temperature that was observed. FDR corrected q-values are shown in Extended Data Fig. 2a–c. For each region independently, we also calculated the average (across animals) minimum temperature that was observed following the chemogenetic stimulation of all animals in which the region was hit (viral expression > 0) and all animals in which the regions were missed (viral expression = 0). Values are plotted in Extended Data Fig. 2a–c.
Nuclear isolation for snRNA-seq57–59
Mice were anesthetized with 3% isoflurane and transcardially perfused with cold choline dissection media. The brains were extracted and sectioned into 300 μm coronal sections. Sections containing the MPA were microdissected to isolate the avMLPA.
Single-nuclei suspensions were generated as described previously60, with minor modifications. The avMLPA was dissected and placed into a Dounce with homogenization buffer (0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 20 mM Tricine-KOH, pH 7.8, 1 mM DTT, 0.15 mM spermine, 0.5 mM spermidine, protease inhibitors). The sample was homogenized using a tight pestle with 10 strokes. IGEPAL solution (5%, Sigma) was added to a final concentration of 0.32%, and 5 additional strokes were performed. The homogenate was filtered through a 40-μm filter, and OptiPrep (Sigma) was added to a final concentration of 25% iodixanol. The sample was layered onto an iodixanol gradient and centrifuged at 10,000g for 18 minutes as previously described59,60. Nuclei were collected between the 30% and 40% iodixanol layers and diluted to 80,000–100,000 nuclei/mL for encapsulation. All buffers contained 0.15% RNasin Plus RNase Inhibitor (Promega) and 0.04% BSA.
avMLPA single-nucleus RNA-seq
snRNA-Seq library preparation and sequencing
Single nuclei were captured and barcoded for whole-transcriptome libraries using the 10X Genomics Chromium v2 platform according to the manufacturer’s recommendations, collecting one library of approximately 10,000 nuclei from each of the 5 animals. Briefly, single nuclei along with single primer-carrying hydrogels were captured into droplets using a microfluidic device. Each hydrogel carried oligodT primers with a unique cell-barcode. Nuclei were lysed and the cell barcode-containing primers released from the hydrogel, initiating reverse transcription and barcoding of all cDNA in each droplet. Next, the emulsions were broken and cDNA across ~10,000 nuclei pooled into the same library. The cDNA was amplified, fragmented and adapters were added for sequencing on a Nextseq 500 benchtop DNA sequencer (Illumina).
For enrichment of virally-derived transcripts, a fraction (1 μL) of the non-fragmented cDNA was PCR amplified. The forward primer (5’-GCATGGACGAGCTGTACA) was designed to anneal to the sequence uniquely present at the 3’ terminus of mCherry. The reverse primer (5’-CTACACGACGCTCTTCCG) was designed to anneal to the R1 sequence, uniquely present at the 5’ terminus, to the 10X barcode and UMI sequence which were introduced during the reverse transcription. The result of the PCR is preferential amplification of the viral-derived transcripts, while simultaneously retaining the cell-barcode sequence necessary to assign each viral transcript to a particular cell/nucleus. Following PCR amplification (18 cycles, Hot Start High-Fidelity Q5 polymerase, NEB # M0494S), the product was purified using 0.6X SPRI select reagent (Thermo Fisher Scientific # NC0406406) and 1 μL of the 50 μL eluent was used in a second PCR reaction. The forward primer for the second PCR (5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTgtaaggcgcgccataac) was designed to anneal to the sequence uniquely present between the mCherry and loxP sites of the AAV-DIO-Gq-DREADD-mCherry vector. In addition, this primer introduced the R2 sequence necessary for later library amplification. The reverse primer (5’-CTACACGACGCTCTTCCG) was the same as in the first PCR reaction. The result of the PCR is again preferential amplification of the viral-derived transcripts, while simultaneously retaining the cell-barcode sequence necessary to assign each viral transcript to a particular cell/nucleus. Following PCR amplification (18 cycles, Hot Start High-Fidelity Q5 polymerase, NEB # M0494S), all the libraries were indexed according to the 10X protocol, pooled, and sequenced on a Nextseq 500 benchtop DNA sequencer (Illumina).
snRNA-seq sample mapping and viral barcode deconvolution by cell
The 10X Genomics package cellranger 3.1.0 was used to map transcripts to the mm10 reference mouse genome. Since single-nucleus RNA-seq captures unspliced pre-mRNA as well as mature mRNA, we create a custom “pre-mRNA” gene annotation file, in which all features are re-coded as exons, allowing us to map unspliced reads to these artificial exons and to assign them to the corresponding genes. Feature barcoding with custom features was used to assign viral mRNA reads to cells and distinguish between the transcript sequence from the initial AAV-DIO-Gq-DREADD-mCherry vector and the transcript sequence produced by the Cre-recombined vector in TRAPed neurons.
Doublet removal, embedding, and identification of main cell classes
Microfluidic encapsulation of nuclei results in some droplets containing more than one nucleus. During reverse transcription, transcripts from co-encapsulated nuclei would be labeled with the same cell barcode, effectively creating a hybrid cell or a “doublet”. To bioinformatically detect and remove predicted doublets, each of the five libraries were independently processed using the scrublet package61 with the following commands:
scrub = scr.Scrublet(counts_matrix, expected_doublet_rate=0.10)
doublet_scores, predicted_doublets = scrub.scrub_doublets(min_counts=2,
min_cells=3,
min_gene_variability_pctl=85,
n_prin_comps=30)
Doublet-removal eliminated 2,037 cells, leaving 44,669 nuclei across five libraries for further processing. We recovered an average of 2,083 unique non-viral transcripts per nucleus, representing 1,286 unique genes (Extended Data Fig. 3). Data from all nuclei were analyzed simultaneously and virally-derived sequences were removed for the purposes of embedding, clustering and cell-type identification. The R software package Seurat 3.158,62 was used to assign cells to clusters. Any genes whose expression was present in fewer than three cells were removed from the analysis. Data was normalized with the number of transcripts per cell and the percent transcripts derived from the mitochondrial genes regressed using the SCTransform function with default parameters. Initially, the 3,000 most variable genes were identified.
Tissue dissociation has been shown to increase the expression of mitochondrial RNAs, ribosomal-protein RNAs, and immediate-early genes63–65. To minimize the influence of these tissue processing-induced genes on the identification and molecular characterization of avMLPA cells, we removed from the initial list of 3,000 most variable genes any genes that were identified as mitochondrial, ribosomal protein-encoding or were the immediate-early genes Fos, Fosb, Fosl1, Fosl2, Egr1, and Npas4. To further diminish any potential effects of tissue-dissociation-induced expression changes on clustering, we identified and removed from the list of variable genes any genes whose expression across all cells correlated with any mitochondrial, ribosomal protein-encoding or immediate-early genes listed above41,58,66. (Pearson correlation coefficient greater than 0.2 or smaller than −0.2). The final list contained 2,827 variable genes. Next, principal component analysis (PCA) was carried out using the RunPCA() function. The FindNeighbors() function, using the top 30 principal components (PCs) and the FindClusters(), was used to identify the initial 34 clusters. Clusters that were disproportionately derived from a single sample were removed leaving 39,562 cells. Based on the expression of known marker genes, we merged clusters that represented the same cell type. Our final list of cell types was: Glutamatergic neurons (Glut), GABAergic neurons (GABA), Cholinergic neurons (Chol), Astrocytes (Astro), Endothelial cells (Endo), Microglia (Micro), Oligodendrocytes (Oligo) and Oligodendrocyte precursor cells (OPCs) (Fig. 3b, c).
Identification of neuronal subtypes:
Cells classified as neurons (n = 28,103 cells) were additionally processed to identify neuronal subtypes in the avMLPA. The same Seurat 3.1.0 pipeline was used as described above. Clustering identified 24 GABAergic, 8 glutamatergic, 3 hybrid and 1 cholinergic neuronal cell types (Fig. 3d). The function FindAllMarkers(seurat_mat, only.pos = F, min.pct = 0.1, thresh.use = 0.25) uses a Bonferroni-corrected (across 23,967 genes in the dataset) two-tailed Mann–Whitney U test to perform differential gene expression analysis and identify markers of each cell type. The top 5 markers based on fold enrichment are plotted in Extended Data Fig. 4. The top 20 markers are displayed in Supplementary Table 4. Cross-referencing these markers with previously described markers for cell types in the MPA41 led to the annotation of the 36 neuronal cell types indicated in Fig. 3e.
Hierarchical tree construction
Neuronal cell types were clustered on the basis of average gene expression across all 2,954 variable genes. A distance matrix was then calculated in Euclidean space and hierarchical clustering was carried out using the function hclust and the ward.D method.
Identification of TRAPed neurons in snRNA-seq data
Feature barcoding with custom features identified for each nucleus the number of viral mRNA reads that were derived from the non-recombined AAV-DIO-Gq-DREADD-mCherry vector and the number of viral mRNA reads from the Cre-recombined AAV-DIO-Gq-DREADD-mCherry vector (Extended Data Fig. 5a). Neuronal nuclei/cells containing three or more virally-derived transcripts (17,424) were considered transduced by the AAV. Among them, 343 neuronal nuclei/cells contained three or more virally-derived transcripts whose mRNA sequence indicated that the vector had been recombined by Cre, suggesting that these cells were previously TRAPed and now express the Gq-DREADD-mCherry protein.
Differential gene expression between TRAPed and non-TRAPed Adcyap1+ neurons
Differential gene expression analysis was carried out between TRAPed (n = 139 cells) and non-TRAPed (n = 5,848 cells) Adcyap1+ neurons in the snRNA-seq dataset. The FindMarkers(PACAP.cells, ident.1 = “TRAP”, ident.2 = “Non-TRAP”, verbose = FALSE) function in the R software package Seurat 3.1 was used to perform a Bonferroni-corrected (across 23,967 genes in the dataset) two-tailed Mann–Whitney U test and identify differentially expressed genes.
Combined fluorescence in situ hybridization and immunofluorescence
Fluorescence in situ hybridization
Mice were sacrificed by transcardial perfusion of 10 mL PBS followed by 10 mL of 4% paraformaldehyde (PFA). Brains were extracted, post-fixed overnight with 4% PFA at 4ºC and then incubated in PBS with 30% sucrose for 48 hrs at 4ºC for cryoprotection. Brains were embedded in tissue freezing medium and frozen in liquid nitrogen-cooled 2-methylbutane. Brains were sliced on a cryostat (Leica CM 1950) into 20 μm sections, adhered to SuperFrost Plus slides (VWR), and immediately stored at −80ºC until use. Samples were processed according to the ACD RNAscope Fluorescent Multiplex Assay manual with the following modifications: 500 mL of 1x Antigen retrieval solution was heated to 99–100ºC and maintained at a uniform boil. The slides of fixed frozen brain slices, stored at −80ºC, were immediately placed into a slide rack and slowly submerged into the boiling 1x Antigen retrieval solution for 5 min. Immediately following, the slides were washed 3–5 times by moving the slide rack up and down in milli-q H2O and washed again in 100% EtOH at room temperature. A hydrophobic barrier was drawn around each slice and allowed to dry for 1 min at room temperature. From this point on, the procedure followed the standard in situ hybridization protocol for RNAscope Fluorescent Multiplex Assay.
Immunofluorescence
Before mounting, slides were washed 2x for 2 min in PBST (PBS + 0.01% Tween) and blocked by 30 min room temperature incubation with 1% BSA and 10% Donkey Serum to PBST. They were then incubated with primary antibody diluted in PBS + 1% BSA overnight at 4ºC and washed 3x for 5 min with PBST. Slides were subsequently incubated with secondary antibody diluted in PBS + 1% BSA for 1 hr at room temperature, washed with PBST 3x for 2 min at room temperature, stained with DAPI (RNAscope), and finally mounted with ProLong™ Gold antifade reagent.
Determining markers of torpor-TRAPed avMLPA neurons
Brains were processed as indicated in “Combined fluorescence in situ hybridization and immunofluorescence” using the following reagents: For the detection of mCherry-positive cells 1:300 rabbit anti-mCherry (Abcam # ab167453) was used as the primary antibody and 1:500 donkey anti-rabbit 568 (Life technologies Cat# AB_2534017) as the secondary antibody.
Sample imaging
Sections containing avMLPA were imaged on a Leica SPE confocal microscope using an ACS APO 20x/0.60 IMM CORR objective (Harvard NeuroDiscovery Center). Tiled MPA areas were imaged with a single optical section to avoid counting the same cell across multiple optical sections. Channels were imaged sequentially to avoid any optical crosstalk.
Image analysis
To determine the fraction of TRAPed mCherry+ cells that express each marker gene, in each image mCherry+ cells were manually marked while staying blinded to the in situ hybridization signals (Adcyap1, Vglut2, Vgat). Since mCherry was fused to the Gq-DREADD and thus largely membrane-bound, we reasoned that manually marking mCherry+ cells would provide a more accurate measurement compared to semi-automated algorithms that are optimized for a more focal (nuclear or cytoplasmic) signal. Following the identification of mCherry+ cells, for each cell we evaluated whether it appeared positive for markers detected by in situ hybridization, staying blinded to the identity of the marker that was being evaluated. To determine the fraction of marker+ cells that are mCherry+, we additionally counted the total number of marker+ cells.
Mapping anterograde projections of torpor-TRAPed avMLPA neurons
FosTRAP2 mice that were injected with AAV8-hSyn-DIO-Gq-mCherry and TRAPed during natural torpor were sacrificed 2 months later. Brains were processed as indicated in “Immunofluorescent staining” using the following reagents: Primary antibody - 1:300 rabbit anti-mCherry antibody (Abcam # ab167453). Secondary antibody - donkey anti-rabbit 647 secondary antibody (Life Technologies # A31573). Sections were imaged on an Olympus BX61VS microscope using a UPlanSApo 10×0.4 objective (Harvard NeuroDiscovery Center).
Silencing avMLPA neurons
The avMLPA of male and female Adcyap1-2A-Cre and Vglut2-Cre mice was injected with AAV-DIO-Gq-DREADD-mCherry and AAV-DIO-TeLC-EYFP (1:1 ratio). Control animals were injected with AAV-DIO-Gq-DREADD-mCherry and/or AAV-DIO-GCaMP6s. Following 5–14 days of recovery from surgery, animals were fasted to induce torpor and their core body temperature was monitored. The minimum core body temperature is shown in Fig. 4d–f, Extended Data Fig. 8j–m. To analyze the kinetics of torpor, we calculated the time it took each animal to reach minimum body temperature, requiring that the minimum body temperature is at least as low as the 10th percentile of the body temperature observed in control fed animals (33.2°C). In order to avoid counting smaller oscillations in body temperature in this analysis, we classified the animals as not having entered torpor if the body temperature of a fasted animal did not decrease below this threshold. In this case, we assigned the time it took to reach the minimum body temperature (“Time to Min. Tb”) to be 24 h, indicating that the animal did not decrease its body temperature below this required threshold during the 24 h recording.
Identification of Adcyap1+ cells that express TeLC-EYFP
Sample preparation
TeLC-silenced Adcyap1-2A-Cre mice were sacrificed and the brains were processed as indicated in “Combined fluorescence in situ hybridization and immunofluorescence” using the following reagents: For the detection of TeLC-EYFP-positive cells 1:1000 chicken anti-GFP antibody (Abcam, ab13970) was used as the primary antibody and 1:500 donkey anti-chicken 488 antibody (Jackson ImmunoResearch Laboratories, 703-545-155) as the secondary antibody.
Sample imaging
Sections containing avMLPA were imaged on a Leica SPE confocal microscope using an ACS APO 20x/0.60 IMM CORR objective (Harvard NeuroDiscovery Center). Tiled MPA areas were imaged with a single optical section to avoid counting the same cell across multiple optical sections. Channels were imaged sequentially to avoid any optical crosstalk.
Image analysis
To determine the fraction of ISH Adcyap1+ cells that are co-positive for EYFP, Adcyap1+ cells in each image were manually marked while staying blinded to the EYFP+ cells. Following the identification of Adcyap1+ cells, each cell was evaluated for whether it appeared positive for EYFP.
Fiber Photometry
Setup
A three-channel multi-fiber photometry system (Neurophotometrics Ltd.) was used for these experiments. Briefly, light from three different wavelength LEDs (470 nm and 560 nm in phase, and 415 nm out of phase) were bandpass filtered and directed down a fiber optic patch cord via a 20x objective. This was coupled to a fiber optic cannula implanted in the animal. Emitted GCaMP6s and mCherry fluorescence were collected through the same cannula and patch cord, split by a 532 longpass dichroic, bandpass filtered, and focused onto opposite sides of CMOS camera sensor.
Data were acquired and quantified using the open-source software Bonsai by drawing a region of interest around the two images (green and red) of the patch cord and calculating the mean pixel value. To perform longitudinal fiber photometry recordings, the duty cycle of the excitation light was decreased to 10% (interleaved 470 nm + 560 nm / 415 nm with 25 ms period at 4 Hz). LED light was delivered at the minimum power and resulted in about 15 μW of 470 nm light and 25 μW of total light at the tip of the patch cord. A pigtailed fiber-optic rotary joint (Doric FRJ_1×1_PT_200/220/LWMJ-0.37_1.0_FCM_0.15_FCM) was connected to the patch cord (Doric MFP_200/220/900-0.37_#.#_FC_MF1.25) to eliminate bending and coiling of the patch cord.
Fasting and CHA-administration photometry session
Animals were placed in custom-built cages to allow free movement during the entire recording session in the dark. The cages were placed onto radio frequency receiver platforms (Starr Life Science ER4000). Core body temperature and gross motor activity were logged every 10 seconds. For fasting-induced torpor sessions, only water and nesting material were provided. The fasting was initiated at the beginning of the dark cycle, while the recordings were started up to several hours following the onset of fasting. For CHA-administration sessions, animals were given access to excess food so as to be maintained in a fed state. CHA (0.2 mg/kg) was administered via intraperitoneal injection without pausing the recordings.
Analysis of torpor entry, maintenance, and arousal for fiber photometry
Within each recording session, a temperature threshold was determined under which the animal was considered to be in torpor. This threshold was set as 2°C below the top 95% percentile of the temperatures recorded during the entire session (35.6 ± 0.2°C, mean ± s.e.m.). Anytime the animal was in torpor (as determined by low core body temperature) and the core body temperature was decreasing by more than 0.05°C/minute the animal was considered to be entering torpor (Fig. 4i). Anytime the animal was in torpor (as determined by low core body temperature) and its core body temperature was increasing by more than 0.1°C/minute the animal was considered to be arousing from torpor (Fig. 4j).
Analysis of fiber photometry data
Custom-written MATLAB software was used to analyze photometry data. Background signal measurement (mean signal with excitation lights off) was first subtracted from all signals. To correct for photobleaching and heat-mediated LED decay, the isosbestic signal was fit with a biexponential that was then linearly scaled to the calcium-dependent signal. To calculate dF/F, we divided the signal by this scaled fit. A three-minute sliding window was applied to calculate the local baseline (10th percentile value) and standard deviation of the dF/F values (Extended Data Fig. 9d). Peaks were chosen based on prominence (top 1%) of all peaks identified with MATLAB findpeaks function. The body temperature dataset was linearly interpolated, such that the number of samples was equal to the number of photometry data points.
To plot the distribution of the baseline, peak frequency and standard deviation across non-torpor and different stages of torpor, the entire recording was separated into tiled 3-minute periods. For each period the average was calculated, and all the 3-minute time periods that overlap with each stage of torpor (torpor entry, torpor, torpor arousal) or non-torpor plotted as a box plot (mean ± 2 * s.e.m. Fig. 4m, n, Extended Data Fig. 9e–g).
Temperature challenge
Temperature challenge was set up and performed similarly to Tan et al.16 Food was provided in the chamber. Raw calcium-dependent GCaMP6s signal was smoothed over a 5-element moving average window, and the baseline F was defined as the average fluorescence of a 10-minute window at 25°C before the first ramp of temperature. dF/F was calculated by dividing the smoothed calcium-dependent GCaMP6s signal with the baseline signal.
Fiber photometry model
Since we observed striking, statistically significant changes in neural activity when animals were entering and maintaining torpor compared to non-torpid animals or animals that were arousing from torpor, we investigated whether fiber photometry data are sufficient to determine when animals were entering and maintaining torpor. We first extracted several features from our photometry data: baseline signal, frequency of large peaks, and the standard deviation, and calculated the average value for each of these features across a 3, 10, and 30-minute sliding window, resulting in a total of 9 distinct features. Using these 9 data features, we performed unsupervised k-means clustering across each of the recording sessions (n = 8). Silhouette scores were used to determine the optimum number of clusters (n = 2), suggesting that fiber photometry data during each recording session could be robustly grouped into two clusters. Next, we asked which of the features that were included in the k-means clustering contributed most to these clusters and observed that the standard deviation and the baseline calculated from the 3-minute sliding window were the main contributors. We, therefore, used these two features to cluster our fiber photometry data and investigate whether the clusters/states accurately correlate with the behavioral data for when animals are entering or maintaining torpor. To evaluate the specificity and sensitivity of our model, we cross-referenced the states/clusters generated by features of the neural data to torpid versus non-torpid periods defined by movement and body temperature. Model sensitivity was calculated by dividing the amount of time that the photometry data-based model accurately determined torpor entry/maintenance by the total amount of time that an animal spent entering or maintaining torpor (Extended Data Fig. 9j, k). Model specificity was calculated by dividing the amount of time that the photometry data-based model accurately determined torpor entry/maintenance by the total amount of time that the model calculated the animal entering or maintaining torpor (whether it was accurate or not, Extended Data Fig. 9j, k). To investigate what the accuracy of the model would have been by chance, we randomly shuffled the model output for each of the recording sessions and evaluated the sensitivity and specificity of this shuffled model (Extended Data Fig. 9k)
Extended Data
Extended Data Fig. 1: Torpor metabolic rate, brain-wide search for torpor-regulating cells and chemogenetic reactivation of FosTRAP-Gq mice.

a) Mean metabolic rate (VO2), body temperature (Tb), and gross motor activity (Act) of mice in torpor compared to mice that are fed or fasted yet not in torpor (n = 7, p-values indicated on graph). b) Schema for whole-brain reconstruction of c-Fos staining. c) Example brain slice of c-Fos staining in a fasted torpid mouse (n = 3 animals). d) 3D-reconstructed c-Fos-stained brain slices from a fasted torpid mouse. e) Average density of c-Fos-positive cells (number of cells divided by the volume of the region, n = 3 animals, Methods) across 179 brain regions that had on average at least 100 c-Fos+ cells. Paraventricular hypothalamus (PVH), a subregion of the preoptic area (POA), arcuate nucleus (ARC), dorsomedial hypothalamus (DMH), and paraventricular thalamus (PVT) are indicated. f) c-Fos staining of the PVH, xiphoid nucleus (Xi), POA, ARC, DMH, and PVT of fasted torpid mice (n = 3 animals). g) Mean core body temperature (Tb) over 4 hours following CNO administration is significantly lower in torpor-TRAP (n = 14 animals) compared to non-TRAP (n = 6 animals, p = 5.2e-5) and fed-TRAP (n = 9 animals, p = 2.9e-5) mice and compared to torpor-TRAP mice injected with PBS (n = 8 animals, p = 2.5e-5). h) Mean activity over 4 hours following CNO administration is significantly lower in torpor-TRAP (n = 14 animals) compared to non-TRAP (n = 6 animals, p = 5.2e-5) and fed-TRAP (n = 9 animals, p = 9.8e-3) mice and compared to torpor-TRAP mice injected with PBS (n = 8 animals, p = 2.5e-5). i) Coronal brain sections from FosTRAP, LSL-Gq-DREADD-HA mice TRAPed during fasting-induced torpor (fast-TRAP, n = 2 animals) or fed state (fed-TRAP, n = 4 animals) and immunostained for HA. Staining in selected brain areas (PVH, POA, ARC, DMH, and PVT) is shown. j) Volume-normalized signal intensity of HA staining across different hypothalamic nuclei in four fed-TRAP and two fast-TRAP mice. k) Brain-wide quantification of HA staining from four fed-TRAP and two fast-TRAP mice. Numerous (190/316) brain regions, including 32 hypothalamic areas, show increased Gq-DREADD-HA expression (> 2-fold) in fast-TRAP animals compared to fed-TRAP mice. Solid line indicates line of unity, dashed lines indicate two-fold differences. l) Correlation across brain regions between the number of c-Fos+ cells in torpid mice and the levels of Gq-DREADD expression in fast-TRAP mice (R = 0.83, p = 2.2e-16, Pearson correlation test, n = 316 regions). All box plots indicate mean ± s.e.m. All p-values represent two-tailed Mann-Whitney U tests, ** p < 0.01, *** p < 0.001.
Extended Data Fig. 2: Chemogenetic reactivation of torpor-TRAPed neurons in different hypothalamic regions and anterograde projections of torpor-TRAPed avMLPA neurons.

a-c) AAV-DIO-Gq-mCherry was injected into different hypothalamic regions of FosTRAP mice (n = 54 animals). Following TRAPing during torpor, we administered CNO and measured the effect of the re-activation of torpor-TRAPed neurons within the virally injected region on core body temperature. All animals were sacrificed, and the expression of the virally derived Gq-DREADD-mCherry was evaluated in each animal across 277 hypothalamic nuclei. a, b) Each circle represents one of the 277 hypothalamic nuclei, and the y-axis represents the -Log10 FDR-corrected q-value of the Pearson correlation (across 54 animals, q-values displayed in Table S3) between the viral expression in that nucleus and the decrease in Tb that was observed following CNO stimulation. Next, for each nucleus, 54 animals were grouped into those in which the nucleus was hit vs. missed. For each of the two groups of animals, the minimum body temperature following CNO administration was averaged and plotted. The minimum body temperature averaged across all animals in which the nucleus was hit is shown in (a) while the minimum body temperature averaged across all animals in which the nucleus was missed is shown in (b). c) For each nucleus and the corresponding two groups of animals, the minimum body temperature following CNO administration was plotted (“hit” group – y-axis, “missed” group – x-axis). Arrows indicate anterior MPA and LPO regions. When these regions were hit with the virus and the TRAPed neurons chemogenetically re-activated, the body temperature of the animal decreased, whereas when these regions were missed with the virus the body temperature did not decrease. d) Mean activity over 4 hours following CNO administration is significantly lower in avMLPA-hit (n = 15 animals) vs. avMLPA-missed mice (n = 11 animals, p = 1.4e-3), and compared to avMLPA-hit mice injected with PBS (n = 15 animals, p = 4.8e-6). e) Mean metabolic rate (VO2) over 4 hours following CNO administration is significantly lower in avMLPA-hit (n = 7 animals) compared to non-injected mice (n = 6 animals, p = 2.3e-3) or avMLPA-hit mice injected with PBS (n = 6 animals, p = 1.2e-3). f) Mean core body temperature (Tb) over 4 hours following CNO administration is significantly lower in avMLPA-hit (n = 15 animals) compared to avMLPA-missed mice (n = 11 animals, p = 2.6e-7) or avMLPA-hit mice injected with PBS (n = 15 animals, p = 9.0e-8). g) Minimum metabolic rate (VO2) over 4 hours following CNO administration is significantly lower in avMLPA-hit (n = 7 animals) compared to non-injected mice (n = 6 animals, p = 2.3e-3) or avMLPA-hit mice injected with PBS (n = 6 animals, p = 1.2e-3). h) Schematic showing projections of TRAPed avMLPAtorpor neurons. i, j) Gq-DREADD-mCherry fusion protein expression was used to visualize the projection of TRAPed avMLPAtorpor neurons across the brain (n = 4 animals). i) mCherry expression near the injection site (avMLPA). j) Representative images of projections to the MHb, medial habenula; PVT, paraventricular thalamus; DMH, dorsomedial hypothalamus; PAG, periaqueductal gray; ARC, arcuate nucleus; and RPa, raphe pallidus. Scale bars, 50 μm. All box plots indicate mean ± s.e.m. All p-values represent two-tailed Mann-Whitney U tests, ** p < 0.01, *** p < 0.001.
Extended Data Fig. 3: snRNA-seq metrics.

a, b) UMAP plot of 39,562 nuclei from the avMLPA of five animals. a) Colors denote cells derived from each animal. b) Colors denote number of unique transcripts (UMI) per nucleus. c) Relative contribution of each sample (n = 5 animals) towards the total cell population making up each main cell class. d) Violin plot of the distribution of UMIs/cell for each main cell class (Glutamatergic neurons n = 11,275 cells, GABAergic neurons n = 16,307 cells, Cholinergic neurons n = 521 cells, Astrocytes n = 3,479 cells, Endothelial cells n = 421 cells, Microglia n = 1,247 cells, Oligodendrocytes n = 4,718 cells and OPCs n = 1,594). e) Violin plot of the distribution of genes/cell for each main cell class. f) Number of neuronal clusters formed when different fractions (25%, 50%, 75%, 90%) of total neurons (n = 7,025, 14,051, 21,077, 25,292, respectively) are used for clustering. For each fraction a random subset of neurons was used and the analysis repeated ten times. Box plot indicates mean ± s.e.m. Violin plot indicates distribution from the lowest to the largest value.
Extended Data Fig. 4: Marker gene expression across neuronal cell types.

Color denotes mean expression across all nuclei normalized to the highest mean across cell types. Size represents the fraction of nuclei in which the marker gene was detected. Cell types are organized on the basis of hierarchical clustering across all variable genes. The five most unique makers are identified and plotted for each cell type unless a marker was identified across multiple cell types, in which case it was plotted only once.
Extended Data Fig. 5: Strategy for identifying TRAPed torpor-regulating neurons via snRNA-seq and gene expression of marker genes in the avMLPA.

a) Schema for identifying Cre-dependent AAV-DIO-Gq-DREADD-mCherry mRNA with or without recombination. Top: AAV-DIO-Gq-DREADD-mCherry vector map before (Cre-) and after Cre-mediated recombination (Cre+). Blue and white triangles surrounding the Gq-GREADD-mCherry indicate loxP sites. Black arrows indicate the binding site of the sequencing primer. ITR, inverted terminal repeats; WPRE, Woodchuck Hepatitis Virus post-transcriptional regulatory element; Poly-A, polyadenylation signal. Bottom: Due to the Cre-mediated inversion in the AAV-DIO-Gq-DREADD-mCherry vector, the mRNA transcript sequence 3’ of the sequencing primer is different after Cre-mediated recombination, allowing us to identify TRAPed cells during snRNA-sequencing as those cells in which the viral mRNA contains the recombined (Cre+) sequence. b) Quantification of the number of virally transduced cells in TRAPed (n = 4 animals) and non-TRAPed (n = 1 animal) samples. c) Quantification of the number of TRAPed cells in TRAPed (86 ± 27 cells) and non-TRAPed samples (1 cell). d) Quantification of percent of transduced cells that are TRAPed in TRAPed (2.3 ± 0.7%, n = 4 animals) and non-TRAPed samples (0.04%, n = 1 animal) based on snRNA-seq. e) Quantification of percent of TRAPed neurons in TRAPed samples (1.8 ± 0.3%, n = 4 animals) based on fluorescence in situ hybridization. f-h) Mean transcripts per cell across all neuronal cell types identified in snRNA-seq for f) Vgat (Slc32a1, marker of GABAergic neurons), g) Vglut2 (Slc17a6, marker of glutamatergic neurons), and h) Adcyap1 (adenylate cyclase-activating peptide 1). i) snRNA-seq indicates that e2, e5, e10, e11, 16 and e30 represent Vglut2+ Adcyap1+ cell types, while e22, e27, h33, h12 and h24 are Vglut2+ and Adcyap1−. Based on this categorization, 72.4 ± 2.2% of Vglut2+ neurons are Adcyap1+ (n = 5 animals). Box plot indicates mean ± s.e.m. j) Mean transcripts per cell across all neuronal cell types identified in snRNA-seq for Lepr. Adcyap1+ clusters e5 and e10 express Lepr. Bar graphs indicate mean, error-bars 2* s.e.m.
Extended Data Fig. 6: In situ hybridization analysis of torpor-regulating avMLPA neurons.
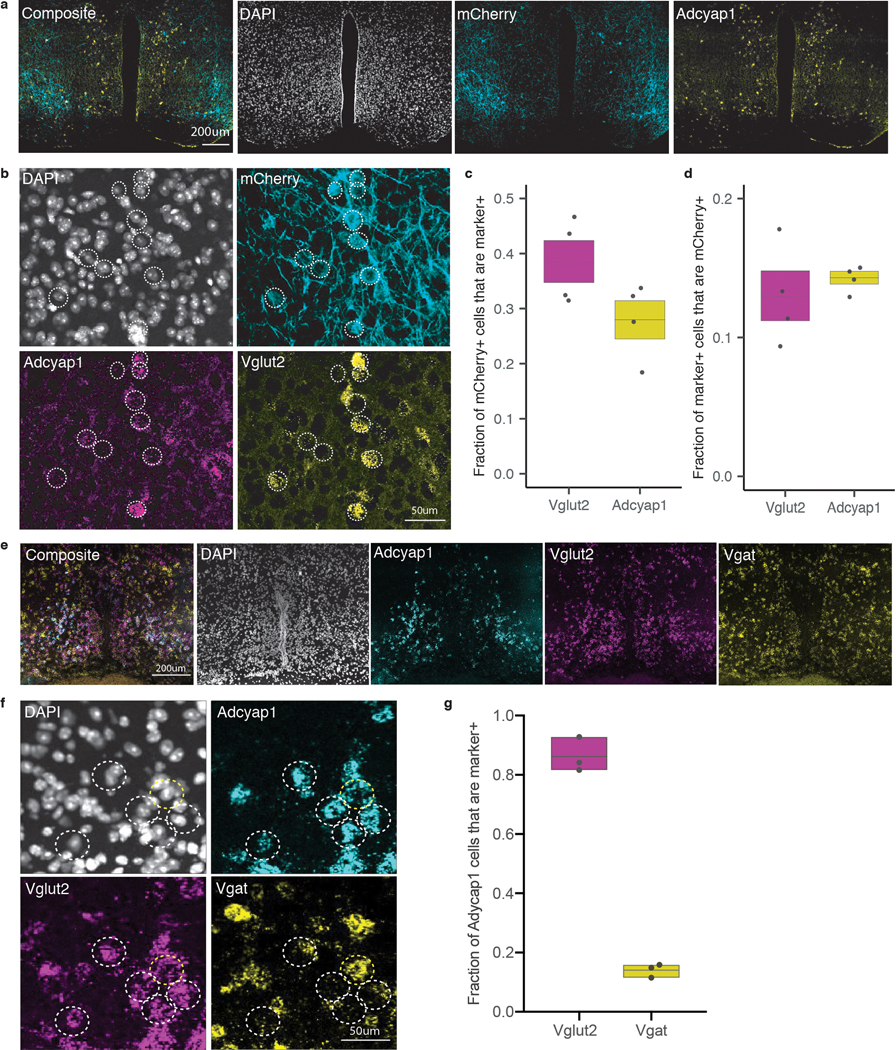
a) Coronal sections showing the avMLPA of FosTRAP mice (n = 4 animals) injected with AAV-DIO-Gq-DREADD-mCherry and torpor-TRAPed. Immunofluorescent staining against mCherry indicates the location of avMLPATRAP neurons (cyan), whereas in situ hybridization indicates the expression of the marker gene Adcyap1. b) High magnification images of staining shown in a) indicate the location of mCherry+ avMLPATRAP neurons (cyan), whereas in situ hybridization indicates the expression of marker genes Adcyap1 and Vglut2. Example avMLPATRAP mCherry+ cells are circled. Several mCherry+ cells express Adcyap1 and/or Vglut2. c) Quantification of the fraction of avMLPATRAP neurons that express Adcyap1 (28.8 ± 3.5%, n = 4 animals) and Vglut2 (38.5 ± 3.8%, n = 4 animals). d) Quantification of the fraction of avMLPAAdcyap1+ (14.3 ± 0.5%, n = 4 animals) and avMLPAVglut2+ (13.0 ± 1.8%, n = 4 animals) neurons that are torpor-TRAPed. e) Coronal section showing the avMLPA of FosTRAP mice. In situ hybridization shows cells that are positive for Adcyap1 (cyan), Vgat (yellow), and Vglut2 (purple). Composite image indicates co-expression of multiple markers. f) High magnification image with example Adcyap1+ cells circled. White circles indicate Adcyap1+ cells that are positive for Vglut2 and negative for Vgat, whereas yellow circles indicate all Adcyap1+ cells that are positive for Vgat (even if co-positive with Vglut2). g) The fraction of Adcyap1+ cells that are positive for Vglut2 or Vgat (82 ± 3% and 14 ± 1%, respectively, n = 3 animals). Scale bars are displayed. All box plots indicate mean ± s.e.m.
Extended Data Fig. 7: Expression pattern of Vgat, Vglut2, and Adcyap1 in the anterior POA.

Coronal sections adapted from Allen Mouse Brain Atlas (available from https://mouse.brain-map.org/). Anterior-posterior coordinates relative to Bregma are indicated for each set of images. Scale bars are displayed.
Extended Data Fig. 8: Chemogenetic stimulation and silencing of avMLPAVgat, avMLPAVglut2, or avMLPAAdcyap1 neurons.

a–c) Stereotaxic viral injection of AAV-DIO-Gq-DREADD and subsequent chemogenetic stimulation of avMLPAVgat (n = 6 animals), avMLPAVglut2 (n = 5 animals), or avMLPAAdcyap1 (n = 8 animals) neurons. a) Experimental schema. b) Minimum core body temperature of avMLPAVgat mice (orange p = 0.48), avMLPAVglut2 mice (light-blue, p = 8e-3), and avMLPAAdcyap1 mice (dark blue, p = 1.6e-4) before and after chemogenetic stimulation with CNO. c) Mean activity of the same avMLPAVgat mice (p = 0.24), avMLPAVglut2 mice (p = 0.032), and avMLPAAdcyap1 mice (p = 1.6e-4), before and after chemogenetic stimulation with CNO. d) Schema for unilateral stereotaxic viral injection of AAV-DIO-Gq-DREADD and subsequent chemogenetic stimulation of avMLPAAdcyap1 neurons. e, f) Changes in mean core body temperature following bilateral (n = 8 animals) and unilateral (n = 4 animals) chemogenetic stimulation of avMLPAAdcyap1 neurons. e) Dashed line indicates CNO administration. Colored lines indicate the mean core body temperature across animals; gray shading indicates the 95% confidence interval. f) Mean core body temperature of mice before and after bilateral (p = 1.6e-4) or unilateral (p = 0.03) chemogenetic stimulation of avMLPAAdcyap1 neurons. g) Schema for stereotaxic viral co-injection of AAV-DIO-TeLC and AAV-DIO-Gq-DREADD and subsequent chemogenetic stimulation of avMLPAVglut2 and avMLPAAdcyap1 neurons. h, i) Changes in mean core body temperature following chemogenetic stimulation of avMLPAVglut2 and avMLPAAdcyap1 neurons that either express the excitatory Gq-DREADD receptor (n = 6, n = 8 animals, respectively) or co-express the Gq-DREADD receptor and the tetanus toxin light chain construct that inhibits synaptic transmission (TeLC, n = 2, n = 4 animals, respectively). h) Dashed line indicates CNO administration. Colored lines indicate the mean core body temperature across animals; gray shading indicates the 95% confidence interval. i) Quantification of mean core body temperature over 4 hours following chemogenetic stimulation in avMLPAVglut2 and avMLPAAdcyap1 (p = 1e-6) neurons that either solely express the excitatory Gq-DREADD receptor or co-express the Gq-DREADD and the TeLC. j-o) Stereotactic injection of AAV-DIO-TeLC to inhibit synaptic transmission in avMLPAVglut2 and avMLPAAdcyap1 neurons. j, k) Core body temperature of fed and fasted Vglut2-IRES-Cre mice from Fig. 3e (animal numbers indicated on graph). j) Minimum Tb is not significantly different between Ctrl-fed and TeLC-fed (p = 0.72) mice. Minimum Tb is significantly lower in Ctrl-fast (p = 0.018), and Pre-fast (p = 0.01) compared to TeLC-fast mice, suggesting that avMLPAVglut2 activity is necessary for torpor. k) Time needed to reach the minimum body temperature in Fig. 3e is significantly longer in TeLC-fast compared to either Pre-fast or Ctrl-fast mice (p = 9.2e-3 for both sets). l, m) Body temperature of fed and fasted Adcyap1-2A-Cre mice from Fig. 3f (animal numbers indicated on graph). l) Minimum Tb is not significantly different between Ctrl-fed and TeLC-fed (p = 0.41) mice. Minimum Tb was not significantly different in TeLC-fast compared to Ctrl-fast (p = 0.71), and Pre-fast (p=0.19) mice. m) Time needed to reach the minimum body temperature in Fig 3f is significantly longer in TeLC-fast compared to Pre-fast (p = 2e-3) and Ctrl-fast mice (p = 5e-4). n, o) Core body temperature (measured in 1-minute intervals) of fed mice during the 12 h light and 12 h dark cycle in which avMLPAAdcyap1 (n) or avMLPAVglut2 (o) neurons were injected with either AAV-DIO-TeLC (TeLC), a control AAV (Ctrl), or remained un-injected (Pre). Core body temperature is significantly different between the dark and light cycle across n) Pre fed (n = 3 animals, n = 3,960 temperature data points, p = 2e-16), Ctrl fed (n = 2 animals, n = 2,640 temperature data points, p = 2e-16) and TeLC fed (n = 5 animal, n = 6,600 temperature data points, p = 2e-16) Vglut2-IRES-Cre mice as well as o), Pre fed (n = 4 animals, n = 5,280 temperature data points, p = 2e-16), Ctrl fed (n = 7 animals, n = 9,240 temperature data points, p = 2e-16) and TeLC fed (n = 8 animals, n = 10,560 temperature data points, p = 2e-16) Adcyap-2A-Cre mice. Box plot displays median, inter-quartile range (IQR), whiskers up to 1.5*IQR and any datapoints outside this range. p, q) Coronal section showing the avMLPA of Adcyap1-2A-Cre mice (n = 2 animals) injected with AAV-DIO-TeLC-EYFP. Immunofluorescent staining against EYFP indicates the location of silenced TeLC+ neurons (green), whereas in situ hybridization indicates the expression of the Adcyap1 mRNA. q) High magnification image with example Adcyap1+ cells circled. White circles indicate Adcyap1+ cells that co-express TeLC-EYFP (43 ± 5%, n = 2 animals), yellow circles indicate Adcyap1+ that do not co-express TeLC-EYFP. Scale bars are displayed. All p-values represent two-tailed Mann-Whitney U test. n.s. represents non-statistically significant, *p < 0.05, ** p < 0.01, *** p < 0.001. b–m) Box plots indicate mean ± s.e.m.
Extended Data Fig. 9: Fiber photometry setup, recordings, and torpor model.

a) Schema for fiber photometry setup. Three LED lights (415 nm, 470 nm, and 560 nm) were used as excitation light sources. For all recordings, 470 nm and 560 nm light sources were driven in phase, with 415 nm driven out of phase (Methods). The emitted signals were detected by a digital camera at the end of a patch cord. b) Example coronal brain slice from an Adcyap1-2A-Cre mouse co-injected with AAV-DIO-Gg-DREADD-mCherry and AAV-DIO-GCaMP6s and used for fiber photometry (n = 8 animals). The white dashed lines indicate the location of the optical fiber. Cells co-expressing GCaMP6s (green) and mCherry (red) appear yellow. c) Example fiber photometry recording (from mouse shown in b) displays the core body temperature (top) followed by three different signals (470 nm, 415 nm, and 560 nm). Here, the 470 nm signal represents calcium-dependent GCaMP6s signal, the 415nm represents the Ca2+-independent isosbestic GCaMP6s signal, and the 560 nm channel represents mCherry signal. The red line indicates the scaled fit of the Ca2+-independent 415 nm signal used to normalize the Ca2+-dependent 470 nm signal for Ca2+-independent changes in signal intensity. Both the 415 nm and 560 nm channels serve as controls for heat-mediated LED decay, bleaching of GCaMP6s, and movement artifacts. d) Recordings of a representative fasting session. (Top) core body temperature of mice during each recording session (dashed line indicates the threshold body temperature below which the animal is considered torpid); (second from top) raw Ca2+-dependent 470 nm GCaMP6s signal (red line indicates the scaled fit of the Ca2+-independent 415 nm signal used to normalize for bleaching or other Ca2+-independent changes in signal intensity); (third from the top) dF/F value relative to the Ca2+-independent scaled fit (blue line indicates the local baseline, which is determined as the 10th percentile of the dF/F value within a sliding three-minute interval); (fourth from the top) the standard deviation of the dF/F value calculated within a sliding three-minute interval; and (bottom) the dF/F values of the most prominent peaks identified (top 1% of all peaks in the session). e-g) Quantification of baseline dF/F [%] (e), standard deviation (f), and peak frequency [/min] (g) for non-torpid (yellow), torpor entry (light-blue), torpor (blue), and torpor arousal (teal) in eight individual animals across all three-minute intervals (left to right: n = 251, 62, 97, 17, 321, 46, 57, 15, 203, 52, 39, 19, 269, 44, 66, 18, 141, 30, 59, 2, 250, 57, 42, 5, 43, 31, 23, 7, 80, 44, 51, 8). All box plots indicate mean ± s.e.m. P-values greater than 0.05 are indicated. h) Example fiber photometry signal (top) clustered into two states and colored by state. State 0 corresponds to the animal being out of torpor or exiting torpor while state 1 corresponds to the animal entering or maintaining torpor. i) Core body temperature and motor activity are significantly lower during state 1 versus state 0 of the photometry-based model (n = 8 animals, p = 1.6e-4). j) The time that an animal spent in torpor (entry or maintenance) was accurately calculated by the photometry data-based model 82.3 ± 3.2% of the time (model sensitivity). Conversely, whenever the model determined that the animal was entering or maintaining torpor, its estimation was 88.4 ± 2.8% accurate (specificity). k) Model sensitivity and specificity were significantly lower (p = 1.6e-4, n = 8 animals) when the temporal relationship between the temperature and fiber photometry data was removed. All box plots indicate mean ± s.e.m. All p-values represent two-tailed Mann-Whitney U test. *p < 0.05, ** p < 0.01, *** p < 0.001.
Extended Data Fig. 10: Fiber photometry recordings of avMLPAAdcyap1 neurons in fed freely moving mice with CHA-induced hypothermia and changes in ambient temperature.

a) Fiber photometry recording data displayed as in Extended Data Fig. 8d. Dashed line indicates the time of CHA administration. b–d) Baseline (b), peak frequency (c), and standard deviation (d) of each animal before and after CHA administration across all recorded three-minute intervals (left to right: n = 69, 17, 63, 23, 140, 24, 161, and 37 time intervals). P-values indicated. e) Mean baseline is decreased following CHA treatment (p = 0.03, n = 4 animals). f) Schema for fiber photometry recording of avMLPAAdcyap1 neurons when mice are exposed to different environmental temperatures with food provided in the chamber. g) Mean GCaMP6s signal (n = 6 animals) of avMLPAAdcyap1 neurons as environmental temperature changes along a programmed sequence: 25°C → 37°C → 25°C → 10°C → 25°C. Gray shading indicates the 95% confidence interval. h) Example fiber photometry recording displays the ambient (chamber) temperature (top) followed by three different signals (470 nm, 415 nm, and 560 nm). Signals from 415 nm and 560 nm are used as controls for potential temperature effects on the photometry signal. i) Mean neuronal responses at different ambient temperatures. avMLPAAdcyap1 neurons are not sensitive to increases in the ambient temperature to 37°C (p = 0.59), and instead appear to be sensitive to a decrease in environmental temperature (n = 6 animals, p = 0.0021). All box plots indicate mean ± s.e.m. All p-values represent two-tailed Mann-Whitney U test. n.s. represents non-statistically significant, *p < 0.05, ** p < 0.01, *** p < 0.001.
Supplementary Material
Acknowledgements:
We thank Drs. B. Sabatini, M. Andermann, B. Lowell, C. Saper, and S. R. Datta for feedback on this work, members of the Sabatini and Regehr laboratories for reagents, members of the Greenberg, Sabatini, Andermann, Lowell, and Datta laboratories for discussions, and Amir Mina for technical assistance. This project relied on the Beth Israel Deaconess Medical Center Metabolic Core for experiments in metabolic cages, Boston Children’s Hospital Viral Core for AAV packaging, the Harvard NeuroDiscovery Center Enhanced Neuroimaging Core and the Neurobiology Imaging Facility (NINDS P30 Core Center Grant #NS072030) for imaging, the Data Analysis Core at Harvard Medical School for help with image analysis, and on the Research Instrumentation Core Facility for engineering support. S.S. wants to thank the support from Herchel Smith Fellowship. This work was supported by National Institute of Health Grants R01 NS028829 and BRAIN Initiative grant RF1 MH114081 to M.E.G., R01 DK107717 to A.S.B, and the Warren Alpert Distinguished Scholar Award to S.H.
Footnotes
Data availability:
RNA-Sequencing data have been deposited in the Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo), accession number GSE149344. Additional data supporting the findings of this study are available from the corresponding authors upon request.
Competing Interests:
Sage Aronson is founder and CEO of Neurophotometrics Ltd. which manufactures fiber photometry systems. All other authors declare no competing financial interests.
References:
- 1.Tan CL & Knight ZA Regulation of Body Temperature by the Nervous System. Neuron 98, 31–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison SF & Nakamura K. Central Mechanisms for Thermoregulation. Annu. Rev. Physiol 81, 285–308 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Heller HC & Hammel HT CNS control of body temperature during hibernation. Comp. Biochem. Physiol. A Physiol. 41, 349–359 (1972). [DOI] [PubMed] [Google Scholar]
- 4.Sunagawa GA & Takahashi M. Hypometabolism during Daily Torpor in Mice is Dominated by Reduction in the Sensitivity of the Thermoregulatory System. Sci. Rep 6, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geiser F. Metabolic Rate and Body Temperature Reduction During Hibernation and Daily Torpor. Annu. Rev. Physiol 66, 239–274 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Morton SR Torpor and Nest-Sharing in Free-Living Sminthopsis crassicaudata (Marsupialia) and Mus musculus (Rodentia). J. Mammal 59, 569–575 (1978). [Google Scholar]
- 7.Heldmaier G, Ortmann S. & Elvert R. Natural hypometabolism during hibernation and daily torpor in mammals. Respir. Physiol. Neurobiol 141, 317–329 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Hoffstaetter LJ, Bagriantsev SN & Gracheva EO TRPs et al.: a molecular toolkit for thermosensory adaptations. Pflugers Arch. 470, 745–759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes BM Freeze avoidance in a mammal: body temperatures below 0 degree C in an Arctic hibernator. Science 244, 1593–1595 (1989). [DOI] [PubMed] [Google Scholar]
- 10.Swoap SJ & Gutilla MJ Cardiovascular changes during daily torpor in the laboratory mouse. Am. J. Physiol.-Regul. Integr. Comp. Physiol 297, R769–R774 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Revel FG et al. The circadian clock stops ticking during deep hibernation in the European hamster. Proc. Natl. Acad. Sci 104, 13816–13820 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teague RS & Ranson SW The role of the anterior hypothalamus in temperature regulation. Am. J. Physiol.-Leg. Content 117, 562–570 (1936). [Google Scholar]
- 13.Zaretskaia MV, Zaretsky DV, Shekhar A. & DiMicco JA Chemical stimulation of the dorsomedial hypothalamus evokes non-shivering thermogenesis in anesthetized rats. Brain Res. 928, 113–125 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Schneeberger M. et al. Regulation of Energy Expenditure by Brainstem GABA Neurons. Cell 178, 672–685.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boulant JA & Hardy JD The effect of spinal and skin temperatures on the firing rate and thermosensitivity of preoptic neurones. J. Physiol 240, 639–660 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan CL et al. Warm-Sensitive Neurons that Control Body Temperature. Cell 167, 47–59.e15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu S. et al. Glutamatergic Preoptic Area Neurons That Express Leptin Receptors Drive Temperature-Dependent Body Weight Homeostasis. J. Neurosci. Off. J. Soc. Neurosci 36, 5034–5046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harding EC et al. A Neuronal Hub Binding Sleep Initiation and Body Cooling in Response to a Warm External Stimulus. Curr. Biol 28, 2263–2273.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama T, Eisenman JS & Hardy JD Single Unit Activity of Anterior Hypothalamus during Local Heating. Science 134, 560–561 (1961). [DOI] [PubMed] [Google Scholar]
- 20.Wang TA et al. Thermoregulation via Temperature-Dependent PGD2 Production in Mouse Preoptic Area. Neuron 103, 309–322.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feketa VV, Nikolaev YA, Merriman DK, Bagriantsev SN & Gracheva EO CNGA3 acts as a cold sensor in hypothalamic neurons. eLife 9, e55370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrington LP The heat regulation of small laboratory animals at various environmental temperatures. Am. J. Physiol.-Leg. Content 129, 123–139 (1940). [Google Scholar]
- 23.Reitman ML Of mice and men – environmental temperature, body temperature, and treatment of obesity. FEBS Lett. 592, 2098–2107 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Padilla SL et al. Agouti-related peptide neural circuits mediate adaptive behaviors in the starved state. Nat. Neurosci 19, 734–741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen TL, Kiersgaard MK, Sørensen DB & Mikkelsen LF Fasting of mice: a review. Lab. Anim 47, 225–240 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Hitrec T. et al. Neural control of fasting-induced torpor in mice. Sci. Rep 9, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavrilova O. et al. Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U. S. A 96, 14623–14628 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinne V. van der, Bingaman MJ, Weaver DR & Swoap SJ Clocks and meals keep mice from being cool. J. Exp. Biol 221, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Himms-Hagen J. Food restriction increases torpor and improves brown adipose tissue thermogenesis in ob/ob mice. Am. J. Physiol.-Endocrinol. Metab 248, E531–E539 (1985). [DOI] [PubMed] [Google Scholar]
- 30.Swoap SJ, Gutilla MJ, Liles LC, Smith RO & Weinshenker D. The full expression of fasting-induced torpor requires beta 3-adrenergic receptor signaling. J. Neurosci. Off. J. Soc. Neurosci 26, 241–245 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberg ME, Ziff EB & Greene LA Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science 234, 80–83 (1986). [DOI] [PubMed] [Google Scholar]
- 32.Copyright 2019. Allen Institute for Brain Science. Allen Mouse Brain Atlas. Available from: http://mouse.brain-map.org/.
- 33.Wu Q. et al. The Temporal Pattern of cfos Activation in Hypothalamic, Cortical, and Brainstem Nuclei in Response to Fasting and Refeeding in Male Mice. Endocrinology 155, 840–853 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Andermann ML & Lowell BB Toward a Wiring Diagram Understanding of Appetite Control. Neuron 95, 757–778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nectow AR et al. Identification of a Brainstem Circuit Controlling Feeding. Cell 170, 429–442.e11 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Allen WE et al. Thirst-associated preoptic neurons encode an aversive motivational drive. Science 357, 1149–1155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu H. et al. Cre-dependent DREADD (Designer Receptors Exclusively Activated by Designer Drugs) mice. Genes. N. Y. N 2000 54, 439–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geiser F, Currie SE, O’Shea KA & Hiebert SM Torpor and hypothermia: reversed hysteresis of metabolic rate and body temperature. Am. J. Physiol. Regul. Integr. Comp. Physiol 307, R1324–1329 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Tupone D, Madden CJ & Morrison SF Central Activation of the A1 Adenosine Receptor (A1AR) Induces a Hypothermic, Torpor-Like State in the Rat. J. Neurosci 33, 14512–14525 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinley MJ et al. The median preoptic nucleus: front and centre for the regulation of body fluid, sodium, temperature, sleep and cardiovascular homeostasis. Acta Physiol. 214, 8–32 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sherin JE, Shiromani PJ, McCarley RW & Saper CB Activation of ventrolateral preoptic neurons during sleep. Science 271, 216–219 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Chung S. et al. Identification of preoptic sleep neurons using retrograde labelling and gene profiling. Nature 545, 477–481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S-B & de Lecea L. The hypocretin (orexin) system: from a neural circuitry perspective. Neuropharmacology 167, 107993 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Habib N. et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Z-D et al. A hypothalamic circuit that controls body temperature. Proc. Natl. Acad. Sci. U. S. A 114, 2042–2047 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto M. et al. Reversible Suppression of Glutamatergic Neurotransmission of Cerebellar Granule Cells In Vivo by Genetically Manipulated Expression of Tetanus Neurotoxin Light Chain. J. Neurosci 23, 6759–6767 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen T-W et al. Ultra-sensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carlin JL et al. Hypothermia in mouse is caused by adenosine A1 and A3 receptor agonists and AMP via three distinct mechanisms. Neuropharmacology 114, 101–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tupone D, Cano G. & Morrison SF Thermoregulatory inversion: a novel thermoregulatory paradigm. Am. J. Physiol. - Regul. Integr. Comp. Physiol 312, R779–R786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexander GM et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cicconet M. & Hochbaum DR A Supervised, Symmetry-Driven, GUI Toolkit for Mouse Brain Stack Registration and Plane Assignment. bioRxiv 781880 (2019) doi: 10.1101/781880. [DOI] [Google Scholar]
- 53.Lein ES et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Oh SW et al. A mesoscale connectome of the mouse brain. Nature 508, 207–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renier N. et al. Mapping of Brain Activity by Automated Volume Analysis of Immediate Early Genes. Cell 165, 1789–1802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y. et al. Identifying local and descending inputs for primary sensory neurons. J. Clin. Invest 125, 3782–3794 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stroud H. et al. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hrvatin S. et al. A scalable platform for the development of cell-type-specific viral drivers. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wolock SL, Lopez R. & Klein AM Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 8, 281–291.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Butler A, Hoffman P, Smibert P, Papalexi E. & Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ilicic T. et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van den Brink SC et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Hrvatin S. et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat. Neurosci 21, 120–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buettner F. et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol 33, 155–160 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.