

抗凝血酶(AT)是一种丝氨酸蛋白酶抑制剂,通过结合活化凝血因子Ⅱ、Ⅸ、Ⅹ、Ⅺ、Ⅻ等丝氨酸蛋白酶活性中心的丝氨酸残基达到抗凝目的[1]。合成抗凝血酶蛋白的基因SERPINC1位于人类染色体1q25.1,长约13 480 bp。成熟的抗凝血酶是由432个氨基酸构成的糖蛋白[2]–[3]。遗传性抗凝血酶缺陷症是常染色体显性遗传,根据血浆中抗凝血酶活性(AT∶A)和抗凝血酶抗原(AT∶Ag)水平可分为Ⅰ型(AT∶A与AT∶Ag同步减少)、Ⅱ型(AT∶Ag正常而AT∶A降低)[4]。抗凝血酶缺陷或协同其他血栓形成危险因素的患者容易发生血栓性疾病[5]。本文报告一个遗传性抗凝血酶缺陷症家系的表型和基因分析结果并初步探讨其分子发病机制。
病例与方法
一、家系资料
先证者,男,21岁,2018年10月因“反复头痛2 d”入院。2年前有“深静脉血栓形成”病史。头颅CT显示“左侧横窦、乙状窦和直窦静脉窦血栓形成”;B超检查显示“右下肢深静脉血栓形成”。实验室检查:AT∶A 43%(参考值98%~119%),AT∶Ag 113 mg/L(参考值250~360 mg/L),D-二聚体12.73 mg/L(参考值0~0.50 mg/L),血浆凝血酶原时间(PT)、活化部分凝血活酶时间(APTT)、凝血酶时间(TT)、狼疮抗凝物、抗磷脂抗体、同型半胱氨酸等其他指标均无明显异常。患者最初使用肝素抗凝治疗,效果不显著,改用利伐沙班后头颅CT显示血栓范围较前缩小。先证者及家系其他成员3代8人肝、肾功能均无异常。先证者父亲和两位祖父有“深静脉血栓形成”病史,其余家庭成员无“深静脉血栓形成”病史。家系图见图1。
图1. 遗传性抗凝血酶缺陷症家系图.

二、健康对照组
为排除基因多态性,设立由150名健康体检者组成的健康对照组,男65名,女85名,中位年龄32(20~50)岁,肝肾功能正常、无其他基础性疾病。本研究通过本院伦理委员会审查,家系及健康对照组成员采样前均签署知情同意书。
三、实验方法
1.血浆凝血指标检测:标本采集后的上层血小板血浆在Stago STA-R全自动血凝仪上检测AT∶A、血浆蛋白C活性(PC∶A)、纤溶酶原活性(PLG∶A)和D-二聚体。应用免疫比浊法检测AT∶Ag含量(美国Beckman Coulter公司LX20PRO全自动生化分析仪)。所有操作步骤均严格按照试剂说明书进行。
2.DNA提取及PCR扩增:采用酚-氯仿法抽提受检者的外周血基因组DNA,参照文献[6]设计合成PCR引物。PCR体系为25 µl。反应条件:95 °C预变性5 min,95 °C变性30 s,57 °C退火30 s,72 °C延伸30 s(共30个循环),72 °C延伸10 min。
3.DNA序列分析:PCR扩增产物纯化后送上海桑尼生物工程有限公司直接测序,所用测序仪为ABI PRISM 3730。测序结果用Chromas软件与美国NCBI基因库所公布的抗凝血酶基因序列(Genbank X68793.1)进行比对,寻找基因突变位点。发现基因突变的序列则反向测序予以证实,再扩增其他家系成员相应外显子片段。采集健康对照组标本,同法抽提外周血基因组DNA,进行相应区域PCR扩增、测序,用于排除基因多态性。
4.PCR产物克隆测序:针对插入突变的DNA片段,在PCR扩增后进行电泳切胶回收纯化PCR产物,然后将PCR产物按克隆试剂盒要求装入PMD18-T克隆载体;再按说明书将已连接上目的DNA片段的PMD18-T Vector(即重组质粒)转化进入DH-5α感受态细胞;在LB固体培养基上涂布培养,经蓝白斑筛选挑取单个白色菌落接种于5 ml含Amp(1∶1000)的LB液体培养基中,在37 °C 300 r/min振荡培养16~20 h。然后提取重组质粒,用PMD18-T载体通用引物进行PCR扩增,电泳鉴定后进行测序分析。
四、生物信息学技术分析
采用Mutation Taster、Poly-Phen-2、PROVEAN和SIFT在线生物信息学软件分析突变氨基酸对蛋白质功能的影响。以蛋白数据库(PDB, http://www.rcsb.org/pdb/home/home/do.PDB ID:4XDE)中的3D结构为基础,用Swiss-Pdb Viewerversion 4.0.1软件和蛋白质相互作用计算器程序(http://pic.mbu.iisc.ernet.in)生成蛋白质模型,分析突变前后由氨基酸变化引起蛋白质空间结构的改变。
结果
1.凝血指标检测结果:先证者和其祖父、叔公、父亲、姑姑及妹妹AT∶A均降低为正常值的50%左右,其母亲AT∶A水平略下降至88%,上述家系成员AT∶Ag与AT∶A均平行减少(表1)。先证者的D-二聚体为12.73 mg/L。所有家系成员的PLG∶A和PC∶A均正常,并且PT、APTT、TT等其他凝血指标均在参考范围内。
表1. 遗传性抗凝血酶缺陷症家系成员实验室表型和基因型.
| 家系成员 | 年龄(岁) | PLG∶A(%) | PC∶A(%) | D-二聚体(mg/L) | AT∶A(%) | AT∶Ag(mg/L) | c.318_319insT | c.922G>T |
| 祖母(Ⅱ1) | 67 | 98 | 127 | 0.34 | 110 | 293 | 野生型 | 野生型 |
| 祖父(Ⅱ2) | 70 | 102 | 112 | 0.37 | 51 | 134 | 杂合子 | 野生型 |
| 叔公(Ⅱ3) | 68 | 87 | 95 | 0.26 | 47 | 125 | 杂合子 | 野生型 |
| 姑姑(Ⅲ1) | 41 | 75 | 88 | 0.30 | 45 | 119 | 杂合子 | 野生型 |
| 父亲(Ⅲ2) | 47 | 103 | 116 | 0.19 | 50 | 132 | 杂合子 | 野生型 |
| 母亲(Ⅲ3) | 46 | 93 | 97 | 0.16 | 88 | 232 | 野生型 | 杂合子 |
| 先证者(Ⅳ1) | 21 | 110 | 112 | 12.73 | 43 | 113 | 杂合子 | 杂合子 |
| 妹妹(Ⅳ2) | 23 | 89 | 92 | 0.19 | 51 | 135 | 杂合子 | 野生型 |
| 参考值 | 70~140 | 70~130 | 0~0.50 | 98~119 | 250~360 | |||
注:PLG∶A:纤溶酶原活性;PC∶A:血浆蛋白C活性;AT∶A:抗凝血酶活性;AT∶Ag:抗凝血酶抗原
2.抗凝血酶基因突变检测结果:先证者抗凝血酶基因检出2个突变:①2号外显子的c.318_319insT杂合插入突变;②5号外显子的c.922G>T杂合错义突变。先证者祖父、叔公、父亲、姑姑及妹妹检出c.318_319insT杂合突变,先证者母亲检出c.922G>T杂合突变;其祖母为野生型(表1)。上述两个抗凝血酶基因突变位点在健康对照组中均未检出,从而排除基因多态性。测序图见图2。
图2. 抗凝血酶基因c.318_319insT和c.922G>T突变测序图.
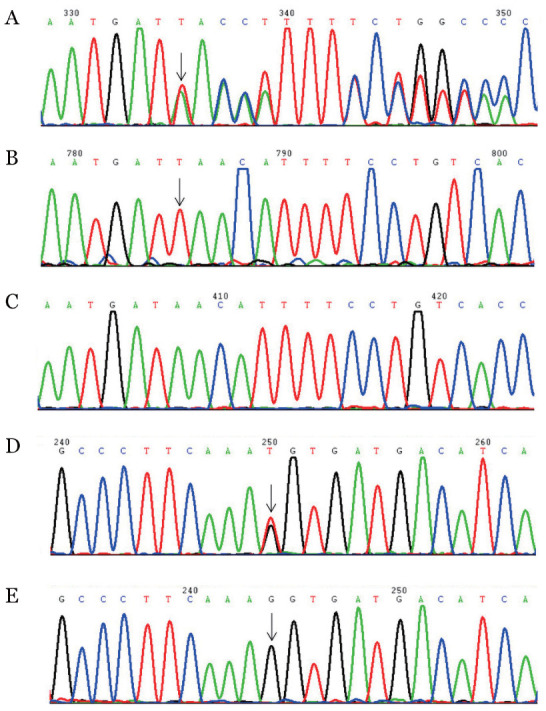
A:c.318_319insT正向测序结果;B:c.318_319insT的克隆序列;C:c.318_319insT野生型;D:c.922G>T突变型;E:c.922G>T野生型
3.生物信息学分析:四个生物信息学软件Mutation Taster、Poly-Phen-2、PROVEAN和SIFT对c.922G>T突变的预测结果分别为“致病的”、“良性的”、“有害的”和“影响蛋白功能”。c.318_319insT突变蛋白模型3D分析的结果显示插入该碱基会破坏74 Asp下游的正常阅读框,随后产生终止密码子(UAA),产生了截短蛋白(Asn75stop);c.922G>T突变显示Gly276被Cys276取代,导致侧链延伸,从而使Asp278与Cys276形成另一个氢键,同时引起Ala134和Ser138的空间位阻(图3)。
图3. 抗凝血酶蛋白模型分析图(绿色虚线为氢键,粉红虚线表示空间位阻).
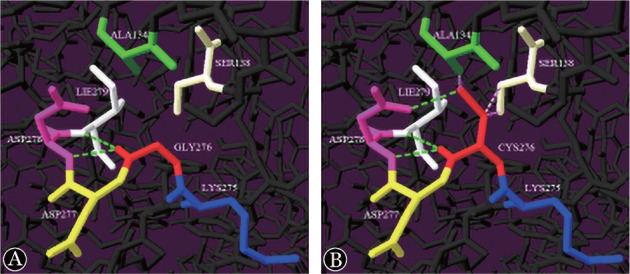
A:野生型;B:突变型
讨论
抗凝血酶在抗凝系统中起着非常重要的作用,当合成抗凝血酶的抗凝血酶基因发生基因突变并导致抗凝血酶蛋白量或者质的异常时,则会引起遗传性抗凝血酶缺陷症。该病是常染色体显性遗传,临床主要表现为反复血栓形成,以下肢深静脉血栓、肺栓塞最为常见,其临床发病率为1∶20 000~1∶2000。而在静脉血栓患者中,抗凝血酶缺陷症的发病率却高达1/200~1/20。抗凝血酶缺陷能够使血栓形成的危险增加20倍[7],属于易栓症的主要遗传性高危因素。截至目前,人类基因突变数据库共收录抗凝血酶基因突变419种,其中错义突变占56.1%,片段缺失或插入导致的框架转移突变占37.1%,其他类型突变占6.8%。
本例先证者的AT∶A水平降至43%,其他结果无明显异常,初步诊断为抗凝血酶缺陷症。通过先证者和家系成员的基因分析发现,先证者携带复合杂合突变(c.318_319insT和c.922G>T)且分别遗传自存在杂合突变的父亲和母亲。其父亲、姑姑、妹妹和两个祖父辈的AT∶A水平降低到大约50%且AT∶Ag与AT∶A平行减少,他们携带c.318_319insT杂合突变;其母亲的AT∶A水平降至88%,携带c.922G>T杂合突变;其祖母为野生型,AT∶A水平正常。查阅家庭成员资料,均无肝脏和肾脏疾病,同时这些人蛋白质C系统和其他凝血指标均正常,AT∶Ag与AT∶A平行减少,而且均携带相同突变位点,可推断两种杂合突变导致了先证者抗凝血酶活性和抗原同步降低,且该突变导致的遗传性抗凝血酶缺陷症表现为Ⅰ型遗传性抗凝血酶缺陷症。查阅突变数据库和相关文献资料,两个突变为未曾报道过的新突变。
抗凝血酶蛋白基因共由7个外显子和6个内含子构成,其中2号外显子编码位于氨基端的肝素结合位点区,6号外显子编码位于羧基端的蛋白酶抑制活性区,这两个区域是抗凝血酶蛋白分子表面重要的活性区[8]。抗凝血酶蛋白内还存在4个糖基化位点(Asn96、Asn135、Asn155、Asn192)以及3个二硫键(Cys8-Cys128、Cys21-Cys95、Cys247-Cys430),这些关键残基的改变会对抗凝血酶蛋白结构及功能产生影响[9]。本家系先证者存在c.318_319insT杂合插入突变,破坏了Asp74下游的正常阅读框,产生终止密码子(UAA),导致截短蛋白的产生,同时使糖基化位点Asn96消失和二硫键位点Cys21-Cys95断裂。有研究报道,p.Cys95Arg突变引起了Cys95-Cys21之间二硫键的断裂,使抗凝血酶蛋白分泌速率显著减慢并以低聚糖的形式滞留于细胞内质网是导致抗凝血酶缺陷症的原因[10]。我们发现的c.318_319insT突变同样导致Cys21和Cys95之间的二硫键断裂,因此,我们推测先证者c.318_319insT突变与抗凝血酶蛋白合成分泌减少有关。另外,c.318_319insT可引起的抗凝血酶蛋白的截短使得N-末端肝素结合区域不完整,故推测c.318_319insT可能会导致抗凝血酶和肝素之间的亲和力降低,且与先证者肝素抗凝疗效差的临床表现一致。
本研究通过蛋白质模型和四个在线生物信息学来分析c.922G>T杂合突变的可能影响。蛋白质模型分析显示Gly276被Cys276取代,导致侧链延伸,从而使Asp278与Cys276形成一个新的氢键,同时引起Ala134和Ser138的空间位阻。三个生物信息学软件程序分析表明Gly276Cys是一种有害突变,而PolyPhen-2分析表明Gly276Cys是一种良性突变;且Gly276Cys位于5号外显子,不在抗凝血酶的主要功能区域或肝素结合区域。本研究先证者的母亲携带Gly276Cys杂合子,AT∶A为88%,但没有血栓形成或凝血异常的临床表现。因此,我们推测Gly276Cys对该家族的抗凝血酶功能和活性可能没有显著影响。研究表明,若存在其他导致血栓形成的危险因素(高龄、高血压、长期制动、手术、妊娠、口服避孕药、肝脏损害、肾病综合征、营养不良、发育障碍、消耗增多等),血栓性疾病发生率将较单纯抗凝血酶缺陷增加数倍[11]。
本家系中先证者和其祖父、叔公、父亲、姑姑及妹妹都为c.318_319insT杂合插入突变,且先证者、其父亲和祖父、叔公都有“深静脉血栓形成”病史,对于已出现血栓疾病的患者应积极治疗,推荐使用口服抗凝药并定期检测凝血指标。姑姑和妹妹虽未表现出血栓形成倾向,但依然需要加强疾病的Ⅰ期预防,尤其是女性在妊娠期更应定期检测各项凝血指标,防止血栓形成造成流产。此外,先证者肝素抗凝效果不显著,提示对于遗传性抗凝血酶缺陷症患者,有必要进行基因诊断,从而指导临床用药。
综上所述,我们对一个Ⅰ型遗传性抗凝血酶缺陷症家系进行表型和基因变异分析,报道了两个新突变(c.318_319insT和c.922G>T),我们推测这些突变可能降低了抗凝血酶和肝素之间的亲和力并破坏其蛋白质的空间结构,导致AT∶A和AT∶Ag减少,但其确切分子致病机制有待进一步研究。
Funding Statement
基金项目:温州市科技局项目(Y20190471)
References
- 1.Quinsey NS, Greedy AL, Bottomley SP, et al. Antithrombin: in control of coagulation[J] Int J Biochem Cell Biol. 2004;36(3):386–389. doi: 10.1016/s1357-2725(03)00244-9. [DOI] [PubMed] [Google Scholar]
- 2.Arnaldi LA, Pretti FA, Zampieri JP, et al. Antithrombin deficiency in Brazilian patients with venous thrombosis: molecular characterization of a single splice site mutation, an insertion and a de novo point mutation[J] Thromb Res. 2001;104(6):397–403. doi: 10.1016/s0049-3848(01)00390-5. [DOI] [PubMed] [Google Scholar]
- 3.张 付华, 吴 竞生. 遗传性抗凝血酶缺乏症研究进展[J] 血栓与止血学. 2005;11(6):284–286. doi: 10.3969/j.issn.1009-6213.2005.06.015. [DOI] [Google Scholar]
- 4.Luxembourg B, Delev D, Geisen C, et al. Molecular basis of antithrombin deficiency[J] Thromb Haemost. 2011;105(4):635–646. doi: 10.1160/TH10-08-0538. [DOI] [PubMed] [Google Scholar]
- 5.Kamijima S, Sekiya A, Takata M, et al. Gene analysis of inherited antithrombin deficiency and functional analysis of abnormal antithrombin protein (N87D)[J] Int J Hematol. 2018;107(4):490–494. doi: 10.1007/s12185-017-2352-8. [DOI] [PubMed] [Google Scholar]
- 6.陈 琼, 陆 晔玲, 许 冠群, et al. I型遗传性抗凝血酶缺陷症两种新基因突变[J] 中华血液学杂志. 2010;31(3):145–148. doi: 10.3760/cma.j.issn.0253-2727.2010.03.001. [DOI] [Google Scholar]
- 7.Kamimoto Y, Wada H, Ikejiri M, et al. High frequency of decreased antithrombin level in pregnant women with thrombosis[J] Int J Hematol. 2015;102(3):253–258. doi: 10.1007/s12185-015-1822-0. [DOI] [PubMed] [Google Scholar]
- 8.Picard V, Nowak-Göttl U, Biron-Andreani C, et al. Molecular bases of antithrombin deficiency: twenty-two novel mutations in the antithrombin gene[J] Hum Mutat. 2006;27(6):600. doi: 10.1002/humu.9425. [DOI] [PubMed] [Google Scholar]
- 9.Silverman GA, Bird PI, Carrell RW, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature[J] J Biol Chem. 2001;276(36):33293–332966. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y, Ueda K, Ozawa T, et al. Intracellular accumulation of antithrombin Morioka (C95R), a novel mutation causing type I antithrombin deficiency[J] J Biol Chem. 2002;277(52):51058–51067. doi: 10.1074/jbc.M210231200. [DOI] [PubMed] [Google Scholar]
- 11.Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency--a model for conformational diseases[J] N Engl J Med. 2002;346(1):45–53. doi: 10.1056/NEJMra010772. [DOI] [PubMed] [Google Scholar]