

Abstract
Many microbial secondary metabolites are produced by multienzyme complexes comprising nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs). The ketosynthase (KS) domains of polyketide synthase normally catalyze the decarboxylative Claisen condensation of acyl and malonyl blocks to extend the polyketide chain. However, the terminal KS domain in tenuazonic acid synthetase 1 (TAS1) from the fungus Pyricularia oryzae conducts substrate cyclization. Here, we report on the unique features of the KS domain in TAS1. We observed that this domain is monomeric, not dimeric as is typical for KSs. Analysis of a 1.68-Å resolution crystal structure suggests that the substrate cyclization is triggered via proton abstraction from the active methylene moiety in the substrate by a catalytic His-322 residue. Additionally, we show that TAS1 KS promiscuously accepts aminoacyl substrates and that this promiscuity can be increased by a single amino acid substitution in the substrate-binding pocket of the enzyme. These findings provide insight into a KS domain that accepts the amino acid–containing substrate in an NRPS–PKS hybrid enzyme and provide hints to the substrate cyclization mechanism performed by the KS domain in the biosynthesis of the mycotoxin tenuazonic acid.
Keywords: biosynthesis, polyketide, mycotoxin, fungi, natural product
We previously reported that tetramic acid (2,4-pyrrolidinedione) derivative mycotoxin tenuazonic acid (TeA) is synthesized by tenuazonic acid synthetase 1 (TAS1) in Pyricularia oryzae. TAS1 is the first reported fungal nonribosomal peptide synthetase–polyketide synthase (NRPS–PKS) hybrid enzyme that consists of an NRPS module of domains C-A-PCP (C, condensation; A, adenylation; PCP, peptidyl carrier protein) and a terminal PKS ketosynthase (KS) domain. Tetramic acid ring formation by Dieckmann cyclization in the course of TeA biosynthesis is predicted to be catalyzed by the KS domain (1) (Fig. 1A).
Figure 1.
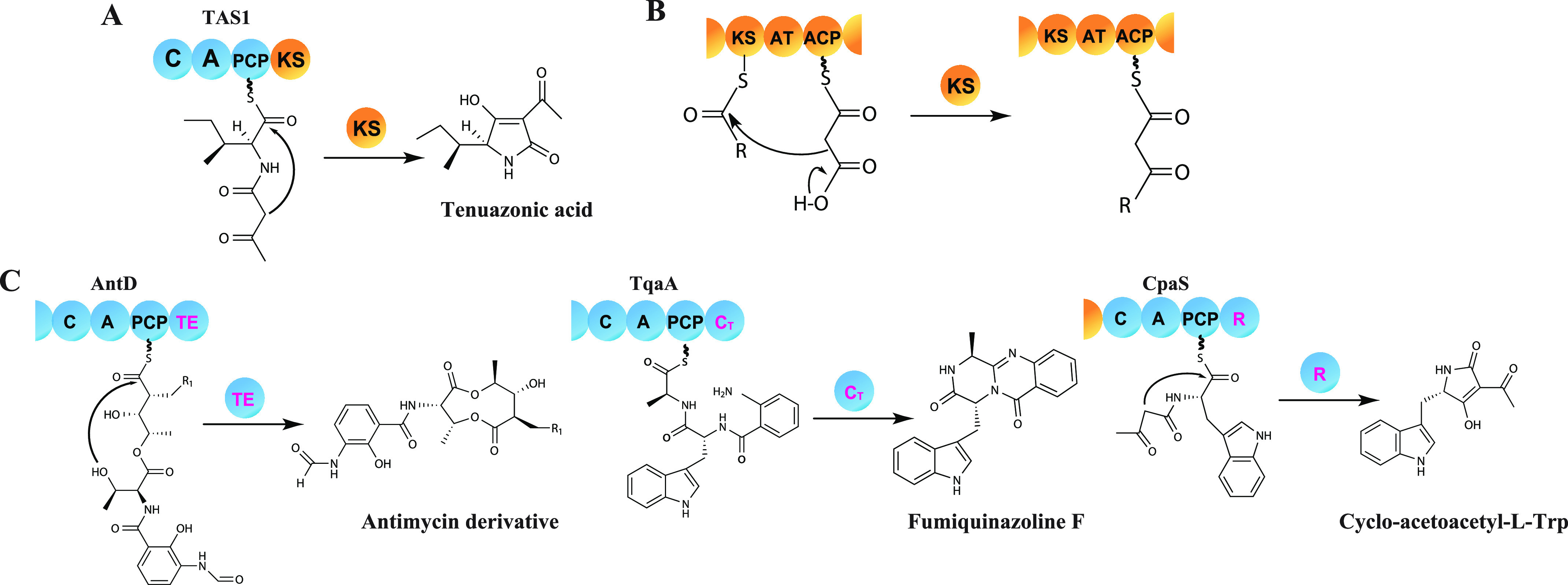
Substrate cyclization by terminal cyclization domains and Claisen condensation by the KS domain. A, proposed cyclization model by the KS domain in TAS1. B, classic function of KS domains in PKS that leads to C–C bond formation by Claisen condensation for ketide extension. C, release through cyclization by the TE domain in bacterial NRPS (AntD), cyclization by the CT domain in fungal NRPS (TqaA), and reduction by the R domain in fungal PKS–NRPS hybrid (CpaS), which is followed by spontaneous cyclization. NRPS domains are colored blue and PKS domains are yellow.
The KS domain is an essential domain of modular type I PKS systems that normally carries out decarboxylative Claisen condensation of acyl and malonyl blocks to yield linear polyketide backbones (2) (Fig. 1B). However, exceptional KS domains have also been reported to have a noncanonical role in type I PKS systems. PKS RhiE-KS3 plays a key role in vinylogous chain branching in the course of rhizoxin biosynthesis without ketide chain elongation (3). A KS domain which has one His residue of the catalytic triad mutated has been reported to only carry out substrate transfer to the next domain in the course of FR901464 biosynthesis (4). Additionally, in type III PKSs homodimers of KS domains catalyze ketide extension and substrate cyclization in a single catalytic pocket (5). Nevertheless, a KS domain with a cyclization and not ketide extension role has only been reported in TAS1.
NRPSs have a monomeric structure (6), but the EM reconstruction of full-length type I PKS module shows an overall homodimeric architecture within which the KS domains associate (7) and KS domain dimerization is a common feature in all reported PKS structure (8). Additionally, there is a report showing PKSs to be dimers that rely on biochemistry, not structure (9). However, there is no report on the association state of the amino acid–containing substrate-accepting KS domain in the NRPS–PKS hybrid enzyme like TAS1.
Bioactive natural products, biosynthesized by the multidomain enzymes NRPS and PKS, often possess a cyclic portion (10, 11). Because of the beneficial properties such as cell permeability and biological activity, cyclic natural products have attracted considerable attention in the field of therapeutic agent development (12–14). Generally, bacterial NRPSs utilize the terminal thioesterase (TE) domain to conduct substrate cyclization, whereas fungal NRPSs use the terminal condensation-like domain for product cyclization. Additionally, the terminal reductase-like cyclization domain in fungal PKS–NRPS hybrid enzymes known that related to substrate cyclization (15–17) (Fig. 1C). However, the precise cyclization mechanism at the molecular level of all the reported cyclization domains remains unknown.
Herein, we propose the mechanism of cyclization for tetramic acid ring formation by the KS domain of TAS1 in the course of TeA biosynthesis using crystallographic and mutational analyses and demonstrate broad substrate specificity and characteristic association state of TAS1 KS.
Results
TAS1 KS is a monomer in aqueous solution
We first wished to determine the oligomerization state of the TAS1 KS domain in solution. To obtain the excised KS domain, a gene construct encoding only the KS domain from TAS1 was heterologously expressed in Escherichia coli. This KS domain was purified, had its affinity tags removed, and was confirmed to be active (Fig. S1). Intriguingly, size-exclusion chromatography showed TAS1 KS domain to be in monomeric state in aqueous solution (Fig. 2). To determine whether this state is distinctive to TAS1 KS or common to amino acid–containing substrate-accepting KS, we also performed size-exclusion chromatography analysis of OzmQ-KS1 protein, which accepts aminoacyl substrate but preforms traditional elongation in the hybrid oxazolomycin biosynthetic gene cluster (18). Interestingly, OzmQ-KS1 showed a dimeric state in aqueous solution, unlike TAS1 KS (Fig. 2), suggesting that the monomeric state is characteristic of TAS1 KS.
Figure 2.

Size-exclusion chromatography of TAS1 KS and OzmQ-KS1. BSA and carbonic anhydrase were used as size markers.
Overall crystal structure of the TAS1 KS domain
To investigate the unique TAS1 KS domain that has unprecedented cyclization function in the course of TeA biosynthesis, we determined its crystal structure and compared it to that of other previously reported canonical KS proteins. TAS1 KS domain protein was crystallized and its structure solved at 1.68 Å resolution (Fig. 3A). The TAS1 KS domain shows a thiolase fold typical of canonical KS domain structures (19). Interestingly, the overall structure of the TAS1 KS domain was more similar to that of the type I PKS KS domain (20), with only ketide extension function (root mean square deviation of 1.30 Å), than to that of the type III PKS domain (21), with ketide extension and cyclization functions (root mean square deviation of 3.01 Å) (Fig. S2). Our previous study indicated that the TAS1 KS domain is closely classified to type I PKS KS domain as a new independent clade in phylogenetic analysis (1), and the obtained crystal structure model supports this classification. Superposed structures of TAS1 KS and canonical type I PKS KS from 6-deoxyerythronolide B synthase (PDB ID: 2HG4) (20) revealed considerable similarity except for the α-helix structure covering the substrate-binding pocket, which is only present in the canonical type I PKS KS structure (Fig. 3, B and C). Type I PKS KS crystal structures without this α-helix have been described; and these KS domains are all located after the NRPS module, suggesting that these KS domains accept amino acid–containing substrates (22, 23) (Fig. S3). Absence of this helical structure may be a hallmark of KS domains that accept more bulky substrates which contain amino acid.
Figure 3.

Crystal structure of TAS1 KS. A, the overall structure of TAS1 KS (PDB ID: 6KOG). B, superposed structure of TAS1 KS (gold) and canonical type I PKS KS domain (purple, 6DEBS module 5 KS, PDB ID: 2HG4). C, the enlarged substrate-binding pocket of the superposed structure of TAS1 KS and 6DEBS module 5 KS domain. The side chains of catalytic triad and Ser-324 of TAS1 KS are shown as stick models.
Mutational analysis of TAS1 KS
Substrate-binding pocket structure of the TAS1 KS is similar to that of type I PKS KS. The catalytic triad of type I PKS KS domain is Cys-His-His (24), and TAS1 KS has a similar Cys-His-Asn (Fig. S4). To evaluate the catalytic role of the catalytic triad in cyclization, we analyzed the TeA-producing activity of KS mutants. The C179A mutant did not show TeA-producing activity (Fig. 4A). This is not unexpected because intermediate translocation from the upstream acyl carrier protein to the catalytic Cys residue of KS is the initial step of KS function in PKS system (25). Therefore the result indicates that in WT TAS1, the PCP-bound intermediate is analogously transferred to Cys-179, and that the C179A mutant could not load the intermediate for further reaction. Mutations H322A and N376A decreased TeA-producing activity to 6 and 9% of WT KS, respectively. Next, we tested mutation of Ser-324 because it is situated in the highly conserved region adjacent to the catalytic triad and not fully conserved between TAS1 KS and type I PKS KS, where it is a Thr (Fig. S4). We, therefore, tested the S324A mutant, which also decreased TeA-producing activity to 4% of WT KS (Fig. 4A). These results indicate that the tested residues are important for the enzymatic reaction of TAS1 KS.
Figure 4.

Substrate cyclization mechanism analysis of TAS1 KS. A, mutant activity analysis. Cell-free synthesized TAS1 C-A-PCP domain and purified KS domain protein were used for analysis. In vitro reaction conditions are described in “Experimental procedures.” All samples were assayed in triplicate and averaged with S.D. ND, not detected. B, in silico docking simulation using TAS1 KS as a receptor and N-acetoacetyl-l-Ile as a ligand covalently bound onto residue Cys-179. C, proposed model for substrate cyclization.
As mentioned, TAS1 KS shows two substitutions of conserved residues compared with type I PKS KS, His to Asn-376 and Thr to Ser-324. To determine whether these replacements affect cyclization, we tested N376H and S324T single mutants and N376H/S324T double mutant. The N376H and S324T mutants and the N376H/S324T double mutant showed 31%, 67%, and 31% activities compared with WT, respectively (Fig. 4A). The results indicate that substitutions H376N and T324S are not essential for the cyclization function of TAS1 KS. The secondary structure of all the mutants used in this study was confirmed using CD analysis (Fig. S5).
Docking simulation of TAS1 KS with the substrate
To gain a deeper insight into the substrate and the key amino acid–residue interaction, we attempted co-crystallization of TAS1 KS and an artificial substrate, N-acetoacetyl-l-Ile-SNAC (1); however, despite various efforts, we could not obtain a co-crystal. Therefore, we conducted in silico docking simulation using the crystallographic structure of TAS1 KS as a receptor and N-acetoacetyl-l-Ile covalently bound to Cys-179 as substrate ligand. Through the docking simulation, we obtained the most stable docking conformation of the ligand with the lowest binding energy. In this conformation, the covalently bound substrate was positioned near the two catalytically important residues (His-322 and Ser-324), and a hydrogen bond occurred between the carbonyl oxygen of the acetoacetyl moiety of the bound ligand and the hydroxyl group of residue Ser-324 (Fig. 4, B and C). Because the Nε atom of His-322 is close to the methylene moiety of the bound ligand, His-322 likely abstracts the methylene proton to produce a carbanion intermediate that is stabilized by two adjacent carbonyl groups. (Fig. 4, B and C). Therefore, based on the crystal structure and docking simulation of TAS1 KS, we hypothesize that covalently bound N-acetoacetyl-l-Ile onto Cys-179 is properly positioned by hydrogen bonding to Ser-324, and then the methylene proton abstraction by His-322 triggers a nucleophilic attack on the thioester carbonyl to produce the tetramic acid ring and TeA release (Fig. 4C).
Verification of the function of His-322
To test our hypothesis from the docking simulation, we constructed the H322F mutant because Phe does not have proton donor or acceptor atoms, and thus, if our hypothesis is valid, H322F should not produce TeA. The results show that H322F was indeed unable to produce TeA (Fig. 4A). However, we cannot exclude the possibility that the bulky side chain of Phe-322 hampered substrate loading onto Cys-179, thus preventing TeA production. To verify these possibilities, we conducted a MALDI-TOF MS analysis to detect covalently bound protein-substrate complexes. The artificial substrate N-acetoacetyl-l-Ile-SNAC was used for activity analysis of TAS1 KS domain in our previous report (1). However, we cannot discriminate between substrate-loaded and -unloaded protein using this substrate because the loaded substrate onto WT KS domain cyclized then released as a TeA. Thus, we synthesized N-2,2-dimethylacetoacetyl-l-Ile-SNAC, an analog of the original substrate containing dimethyl moiety instead of active methylene moiety that prevents hydrogen abstraction for cyclization and product release (Figs. S6 and S13–S19). Covalently bound protein–substrate complexes were detected in WT and H322F, whereas no complex was observed in C179A. These results show the possibilities that the H322F mutant can load but not cyclize the substrate (Fig. 5), and that proton abstraction from the active methylene moiety is a crucial step for cyclization.
Figure 5.

Substrate loading analysis using MALDI-TOF MS. MALDI mass spectrum of KS and mutant proteins with or without N-2,2-dimethylacetoacetyl-l-Ile-SNAC reacted for 12 h in 2 mm Tris-HCl, pH 7.5. The m/z of the detected protein–substrate complex is shown in red characters. Spectrum peaks with and without substrate are shown in green and yellow, respectively. Theoretical molecular weight (Da): KS WT, 47,959; C179A, 47,927; and H322F, 47969. The molecular weight of N-2,2-dimethylacetoacetyl-l-Ile, 227.
Enhancement of TeA production after a single amino acid substitution
We also tested another residue, Glu-378, which is located near the center of the catalytic pocket in TAS1 KS (Fig. S4). Interestingly, the E378A mutant showed more than 2-fold higher TeA production than WT. We assumed that the increase in activity might be because of a resolution of an electrostatic imbalance caused between a nonpolar Ile of the substrate and a polar Glu residue or elimination of steric hindrance coming from the Glu residue. To further investigate, we constructed mutants with nonpolar and polar amino acid substitutions at position 378 (E378L and E378Q). These mutants showed similar TeA producing capability to WT, indicating that resolution of the electrostatic imbalance is not the reason for increased activity of E378A (Fig. 4A). We also constructed mutants with larger and smaller amino acid substitutions, E378F and E378G. E378F was approximately as active as WT, but E378G produced 3-fold more TeA (Fig. 4A), suggesting that elimination of the steric hindrance resulting from Glu-378 increased KS activity. To check substrate loading on the E378A mutant, MALDI-TOF MS analysis was conducted. The detected peak corresponding to the protein–substrate complex of E378A was higher than that of WT (Fig. S7). It is possible that the increased activity of E378A is related to substrate-loading efficiency. Using N-acetoacetyl-l-Ile-SNAC, the Km value was 0.41 ± 0.04 mm and 0.34 ± 0.05 mm for E378A and WT, respectively, and the kcat value for E378A was (141 ± 7.2) × 10−3 min−1, an ∼44-fold increase compared with WT, which was (3.67 ± 0.3) × 10−3 min−1 (Fig. S8). However, the exact mechanism underlying the increased activity of E378A remains unclear. Further studies using di-domain crystal structure analysis of PCP-KS of TAS1 with the tethered substrate will be helpful to clarify the substrate-loading mechanism from PCP to the KS domain. The other amino acid–accepting ketosynthase domain of bacillaene synthase, BaeJ KS1, shows high substrate specificity through significant selectivity against bulkier substituents at the α position of the amino acid–containing substrate (26). Thus, we predicted that elimination of the steric hindrance in the E378A mutant might result in the acceptance of substrates with amino acids larger than Ile. To confirm the prediction, N-acetoacetyl-l-Phe-SNAC and N-acetoacetyl-l-Trp-SNAC were synthesized (Figs. S6, S13, and S20–S27). The results show that these substrates were successfully cyclized by E378A (Fig. 6 and Figs. S9 and S10). Interestingly, although weaker than E378A, WT KS also cyclized the N-acetoacetyl-l-Phe-SNAC (Fig. 6). TeA analogs containing Leu and Val instead of Ile were already reported in the culture broth of TeA producer Alternaria tenuis (27). We also tested 20 amino acids as substrates with the purified full-length TAS1 enzyme (1) and the product was only detected in three amino acids (Ile, Leu, Val) (Fig. S11). This observation suggests that TAS1 KS domain can accept at least the three above amino acids, as well as the Phe and may indicate that TAS1 KS has intrinsic tolerance for a broad range of substrates.
Figure 6.
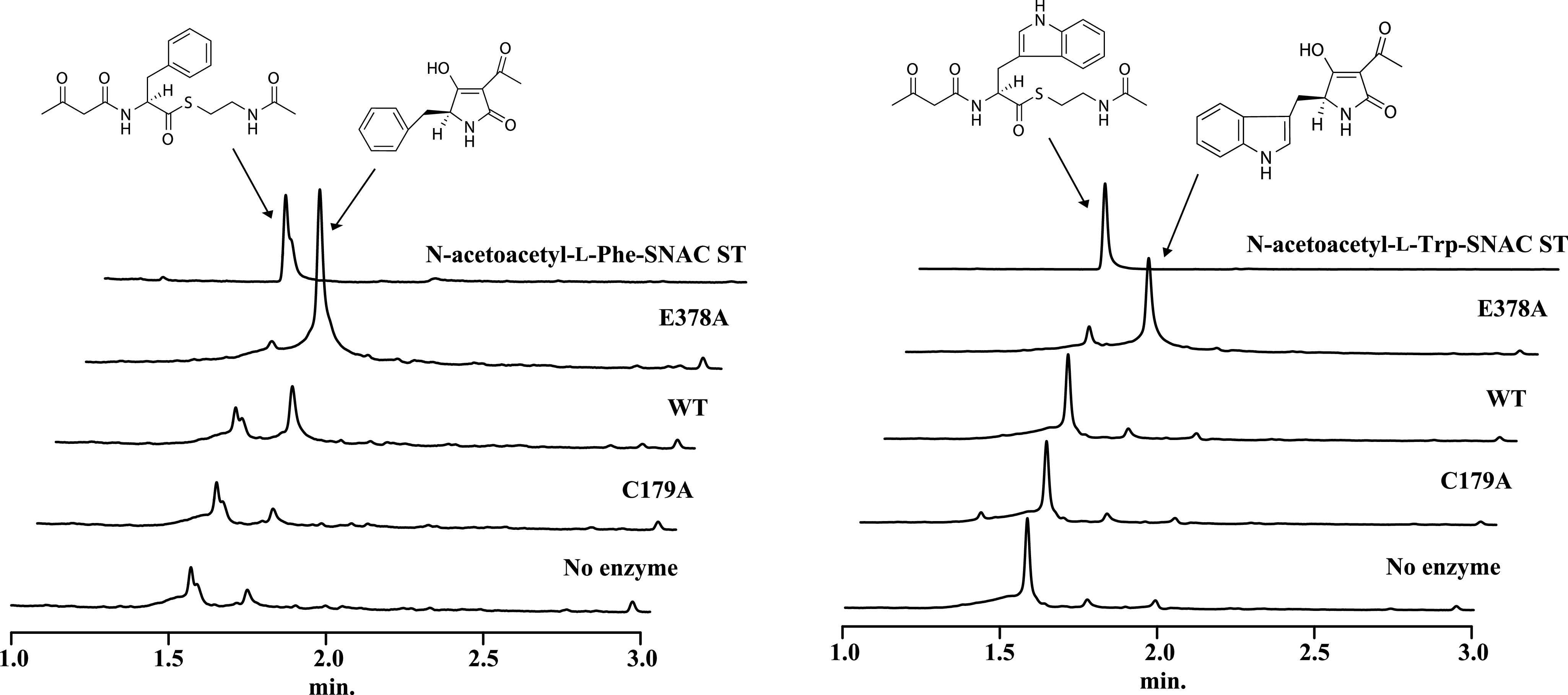
Cyclization capability analysis using SNAC substrates. N-acetoacetyl-l-Phe-SNAC and N-acetoacetyl-l-Trp-SNAC were used as a substrate for cyclization capability analysis with KS WT and mutants proteins. ST, standard.
Discussion
A huge number of structurally diverse tetramic acid ring-containing natural products have been discovered to date in bacteria, fungi, and marine organisms and have shown a wide range of bioactivities such as antimicrobial, antiviral, and antitumor activities (28). To date, four different enzymes or domains responsible for tetramic acid ring formation have been reported, in addition to TAS1 KS. In fungi, tetramic acid–containing natural products are generally biosynthesized by a PKS–NRPS hybrid enzyme, and tetramic acid ring structure formation is reportedly related to the terminal cyclization domain that has a sequence similarity to short-chain dehydrogenase/reductases (29, 30). In bacteria, tetramic acid moieties containing secondary metabolites are biosynthesized by PKS–NRPS hybrid enzymes or cooperation of continuous type I PKS and NRPS modular enzymes, and three different tetramic acid ring formation patterns were reported. First, the TE domain located at the end of the PKS–NRPS assembly lines mediates Dieckmann cyclization in HSAF, frontalamides, and ikarugamycins biosynthesis (31–33). Second, tetramic acid ring formation through the action of stand-alone TE-like Dieckmann cyclase was reported in triandamycin biosynthesis (34). Third, in the course of pyrroindomycin biosynthesis, two genes that PyrD3 homologous to various acyltransferase E2 subunits of 2-oxoacid dehydrogenase complexes and PyrD4 belonging to the α/β-hydrolase superfamily are demonstrated to be involved in tetramic acid ring formation (35). However, the detailed molecular mechanism of all the reported enzymes and domains, including TAS1 KS, that catalyze tetramic acid ring formation is unclear. In this study, we elucidated one part of the mechanism of tetramic acid ring formation in natural product biosynthesis.
The TAS1 KS domain showed a monomeric state in contrast to other canonical type KSs, which are present in a dimeric state. KS domain deletion experiments from the multidomain PKS show that the dimeric feature of the KS domain is crucial for dimerization of the full-length PKS (36). Although we still do not know the association state of the full length of TAS1, the KS domain in this enzyme is seems quite different from other classical KSs.
From docking simulations, the role of His-322 and Ser-324 residues in the cyclization process was predicted. However, the role of Asn-376 in the course of substrate cyclization remains unclear in this docking model. TeA has cis-type peptide bonds, and thus we assume that deprotonated trans-N-acetoacetyl-l-Ile by His-322 isomerize to cis-N-acetoacetyl-l-Ile, and then a nucleophilic attack of the carbanion on the thioester carbonyl carbon occurs to produce TeA (Fig. S12). Most stable docking conformations using the cis-N-acetoacetyl-l-Ile as a ligand show the di-ketide part of the substrate shifting the direction to the Asn-376 residue, and distance between Asn-376 and di-ketide is 2.9 Å, adequate for hydrogen bonding (Fig. S12). The Asn-376 residue might contribute to the stabilization of the cis-N-acetoacetyl-l-Ile form for the nucleophilic attack to produce TeA.
Nine amino acids (Ala, Cys, Gly, Ile, Leu, Met, Phe, Pro, Val) do not contain the hydrogen acceptor or donor atoms. To test the role of His-322, we made two nondeprotonatable His-322 mutants, H322A and H322F. We thought that Ala could be representative of amino acids smaller than His and that Phe could be representative of amino acids bigger than His. Thus, we predicted that if Phe does not hinder substrate loading, then other amino acids will not either. In fact, we also tried to make the H322L mutant but we could not get access to the soluble protein. The hydrophobic leucine substitute may change the hydrophobicity of protein, but at this time, we do not know the exact reason why H322L mutant protein was insoluble. The nondeprotonatable mutation H322F did not exhibit TeA production capacity despite its ability to load the substrate. However, H322A retained a minuscule TeA production ability despite Ala not having proton abstraction capacity. These observations suggest that proton abstraction from the substrate by His-322 is not the sole mechanism for substrate cyclization. It cannot be ruled out that cis-N-acetoacetyl-l-Ile is loaded onto Cys-179 and deprotonation occurs to produce TeA. H322A mutant has a more broadening substrate-binding pocket than WT by substitution of smaller amino acid, and we observed that the artificial substrate N-acetoacetyl-l-Ile-SNAC is slightly cyclized in aqueous solution. Slow cyclization may, therefore, occur even in the absence of deprotonation by amino acid in the substrate-binding pocket.
Because of their potent bioactivities, broadening the structural diversity of natural products using domain engineering in PKS and NRPS has been attempted (37–39). Amino acid–containing substrate-accepting KSs have strict substrate specificities (26), which occasionally hindered domain engineering (40). Our data suggest that PKS systems that incorporate various amino acids into products may be possible to engineer using TAS1 KS mutant E378A, a domain with broad substrate specificity.
In this study, we illustrate a possible mechanism of substrate cyclization by the KS domain in TAS1 with its unique features and suggest an approach to increase the structural diversity of natural products comprised of polyketide and peptide using TAS1 KS.
Experimental procedures
Chemicals
Carbenicillin and kanamycin were purchased from Nacalai (Kyoto, Japan) and acetoacetyl-CoA, l-isoleucine, l-phenylalanine, l-tryptophan, TeA, and ATP were purchased from Sigma-Aldrich. All other reagents were of analytical grade.
Strains and culture conditions
Strains and plasmids used in this study are listed in Table S1. E. coli was grown in lysogeny broth (LB) at 37°C, and transformation was performed according to standard methods (41). E. coli-transformant selection was carried out using kanamycin or carbenicillin (50 µg ml−1) added to LB medium and 2% agar added for plate culture if needed.
DNA manipulation
DNA isolation and manipulation were performed according to standard methods (41). PCR was performed with a DNAEngine Peltier thermal cycler (Bio-Rad). KOD-Plus-neo DNA polymerase (Toyobo, Osaka, Japan) was used for DNA amplification via PCR. DNA fragments were isolated from agarose gels using a gel extraction kit (Qiagen, Valencia, CA, USA). DNA fragment ligation for vector construction was carried out using an In-Fusion HD cloning system (Clontech, Palo Alto, CA, USA). Plasmids were extracted from E. coli and purified with a QIAprep Spin Miniprep kit (Qiagen). DNA sequences were analyzed using an automated 3730xl capillary DNA analyzer (Applied Biosystems, Foster City, CA, USA) with a BigDye terminator version 3.1 kit (Applied Biosystems).
Plasmid construction for KS domain and mutants
The KS domain region of TAS1 (3481–4809 nucleotides) was amplified from plasmid YEp352-TAS1 (1) via PCR using TAS1 KS_F and TAS1 KS_R primers, and the amplified fragment was purified and inserted into the pET42b vector fragment that amplified using primers pET42b_F and pET42b_R located between the thrombin recognition site and the termination codon via infusion reaction to construct the KS protein expression plasmid pET42b_KS. Mutant KS protein expression vectors were constructed using primers containing the appropriate mutation. For example, the C179A_R primer, which introduces a TGC to GCC mutation at the Cys-179 position, and TAS1 KS_F primer used to amplify the fragment 1 containing KS from the start codon to the Cys-179 residue from pET42b_KS. The C179_F and pET42b_R primers were used to amplify fragment 2, containing the vector region and the KS part from Cys-179 to the termination codon from pET42b_KS. Fragments 1 and 2 were gel-purified, cloned using the In-Fusion cloning system to yield pET42b_C179A, and then transformed into DH5α. The inserted sequence of all constructed plasmids was confirmed by sequencing. Primers OzmQ_KS_F and OzmQ_KS_R were used to amplify the OzmQ_KS1 gene from pAPC109406.106 (generously donated by Dr. Ben Shen, Scripps Research Institute) (23). The amplified fragment was purified and then inserted into the pET42b vector used for TAS1 KS expression plasmid construction. The primers used in this study are listed in Table S1.
Protein expression and purification
To express KS protein, the constructed plasmid pET42b_KS was transformed into E. coli BL21 (DE3) pLysS. After overnight culture at 37°C in LB with Km (50 µg ml−1), 1 ml cultured cells was inoculated into 50 ml Overnight Express Autoinduction System (Novagen, Madison, WI, USA) with Km (50 µg ml−1) and cultured at 25°C for 12 h. After growth, the cells were harvested via centrifugation (5000 × g) for 15 min. The collected cells were suspended in 5 ml protein extraction buffer (10 mm Tris-HCl pH 8.0, 1 mm DTT, 0.1% Triton X-100, and 15% glycerol) and sonicated five times on ice for 30 s each with 30-s intervals. Cellular debris was removed via centrifugation (8000 × g) for 30 min at 4 °C. The supernatant was purified on a nickel-nitrilotriacetic acid beads (Qiagen) column as previously described (1). The purified protein was digested with thrombin (1 unit/0.3 mg protein) at 4°C for 60 h to separate nontag protein. Digested protein was treated with benzamide Sepharose beads (GE Healthcare) to exclude thrombin, and then His-tagged GST protein was eliminated with nickel-nitrilotriacetic acid beads column from the tag-free KS protein. KS mutant proteins were expressed and purified using the same method. Purified proteins were stored at −80°C until use. The C-A-PCP domain protein was expressed using a wheat germ cell–free system as previously described (1).
Protein crystallization and structure determination
Purified TAS1 KS domain protein was concentrated to 12 mg ml−1 in crystallization buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1 mm DTT) using a 30-kDa cutoff concentrator (Millipore, Bedford, MA, USA). Crystallization was performed using the sitting drop vapor diffusion method. Crystallization droplet was prepared by mixing 1 μl of concentrated TAS1 KS domain protein solution and reservoir solution (20 mm NaCl, 1.65 m ammonium sulfate, 0.1 m 2-(N-morpholino)ethanesulfonic acid (MES), pH 6.5, 5% methanol) at 20°C. Single crystals were obtained within 2 days. Obtained crystals were flash-soaked in cryoprotectant buffer (120 mm NaCl, 0.1 m MES, pH 6.5, 1.7 m ammonium sulfate, 24% glycerol, 5% methanol) and immediately flash-cooled using a cryo-stream operated at 100 K. X-ray diffraction data were collected at BL41XU at SPring-8 (Sayo, Japan). Crystals were exposed to 1.00000 Å X-ray for 0.1 s and 0.1 degree oscillation per frame. Collected datasets were indexed and scaled using XDS package and AIMLESS from the CCP4 suite. Crystal structure was determined with molecular replacement using Phaser MR. Molecular replacement solution was obtained from CurL KS domain that is part of curacin A polyketide synthase (PDB ID code 4MZ0) (42) as a search model. Model building and refinement were performed using Phenix and Coot. The overall geometry in the final structure was good, with 98% of residues in favored regions of the Ramachandran plot and only one outlier (0.12%). The data collection and refinement statistics are summarized in Table S2. Molecular images shown in this study were prepared using PyMOL.
In vitro enzyme assay and product detection
TeA producing activity assay with TAS1 KS and C-A-PCP domain was conducted at 25°C for 2 h in 100 μl reaction mixture containing 1 μm KS protein, 50 μl crude soluble C-A-PCP domain protein synthesized in a cell-free system, 1 μm purified 4′-phosphopantetheinyl transferase from P. oryzae, 80 μm CoA, 2 mm MgCl2, 50 mm Tris-HCl, pH 7.5, 1 mm ATP, 5% glycerol, 10 mm TCEP, 1 mm isoleucine, and 1 mm acetoacetyl CoA. For analysis of artificial substrate cyclization capability, 5% glycerol, 5 mm TCEP, and 0.25 mm N-acetoacetyl-l-Phe or Trp-SNAC were reacted at 25°C for 2 h. To determine the kinetic parameters, the concentration of N-acetoacetyl-l-Ile-SNAC varied from 0.01 to 0.75 mm. The kinetic parameters Km and kcat were determined using nonlinear regression to fit the data to the Michaelis-Menten equation using SigmaPlot (Systat software). All the enzyme reactions were performed in triplicate. After the enzyme reaction, the reaction mixtures were extracted with 4 volumes of ethanol, evaporated with an N2 stream, and finally dissolved in 200 μl methanol for ultra-performance liquid chromatography/MS (UPLC/MS) analysis of TeA production. UPLC/MS analysis was carried out with an Acquity UPLC H-Class system (Waters Alliance, Milford, MA, USA) equipped with a mass spectrometer (API 3200, Applied Biosystems). The UPLC conditions were as follows: column, XTerra MSC18 (Waters), 5 μm (2.1 × 150 mm); flow rate, 0.6 ml/min; solvent A, water containing 0.05% formic acid; solvent B, acetonitrile. After the sample injection into a column equilibrated with 5% solvent B, then developed with a linear gradient from 5 to 100% solvent B over the course of 3.5 min and kept at 100% solvent B for another 1.5 min. Mass spectra were collected in the electrospray ionization-positive and electrospray ionization-negative modes.
SNAC-artificial substrates synthesis
All solvents were of reagent grade. Reactions were monitored by TLC with 0.25 mm E. Merck (Darmstadt, Germany) precoated silica gel plates. E. Merck 0.5 mm precoated silica gel was used for preparative TLC (PTLC). Kanto chemical silica gel 60 N (spherical, neutral) (40-50 μm) was used for flash chromatography. 1H NMR spectra were recorded on a JEOL JNM-ECA-500 (500 MHz) (JEOL, Tokyo, Japan) spectrometer in CDCl3 with tetramethylsilane (0 ppm) as internal standard and in CD3OD (3.30 ppm). 13C NMR spectra were recorded on a JEOL JNM-ECA-500 (125 MHz) spectrometer in CDCl3 as solvent and internal standard (77.0 ppm) and in CD3OD (49.0 ppm). The following abbreviations were used to explain the multiplicities: s, single; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. High resolution mass spectra were recorded on a Waters IMS Vion QT of system coupled to a Waters UPLC H-class (column: Waters UPLC BEH C18, 50 mm × 2.1 mm, particle size 1.7 mm, ionization method: electron spray ionization).
(S)-S-(2-acetamidoethyl) 2-amino-3-phenylpropanethioate TFA salt (3a)
Compound 3a was prepared starting from N-(tert-butoxycarbonyl)-l-phenyalanine using a modified procedure (43). A solution of N-(tert-butoxycarbonyl)-l-phenyalanine 1a (106.1 mg, 0.4 mmol) in dehydrated THF (5 ml) was stirred at room temperature under a nitrogen atmosphere. A solution of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC•HCl, 76.7 mg, 0.4 mmol) and 1-hydroxybenzotriazole (HOBt, 54.0 mg, 0.4 mmol) in dehydrated THF (5 ml) and then N-acetylcysteamine (42.6 μl, 0.4 mmol) was added. After the mixture was stirred for 1 h, K2CO3 (27.6 mg, 0.2 mmol) was added and the resulting mixture was stirred for 30 min. The solvent was evaporated under vacuum and the residue was dissolved in ethyl acetate (20 ml). The organic layer was washed with saturated aqueous NaHCO3 solution and brine, dried over Na2SO4, and concentrated. The crude product was purified using silica gel column chromatography (n-hexane:EtOAc = 1:2) to give 2a (125.6 mg, 86%) as a colorless solid. The SNAC derivative 2a (115.0 mg, 0.31 mmol) was dissolved in TFA (1 ml) and CH2Cl2 (2 ml). The mixture was stirred for 1 h at room temperature and concentrated under reduced pressure. The residue was twice taken up in CH2Cl2 and concentrated to remove TFA to obtain 3a (111.4 mg, 94%).
2a: 1H NMR (500 MHz, CDCl3) δ 1.42 (s, 9H), 1.95 (s, 3H), 3.03 (m, 3H), 3.13 (dd, J = 14.0, 5.5 Hz, 1H), 3.40 (m, 2H), 4.59 (ddd, J = 8.0, 7.0, 4.5 Hz, 1H), 4.92 (d, J = 8.0 Hz, 1H), 5.81 (brs, 1H), 7.17 (dd, J = 6.5, 1.0 Hz, 2H), 7.27 (m, 1H), 7.32 (dd, J = 6.5, 6.5 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 23.1, 28.3, 28.6, 38.1, 39.1, 61.2, 80.6, 127.2, 128.7, 129.3, 136.6, 155.1, 170.4, 201.5. 3a: 1H NMR (500 MHz, CD3OD) δ 1.93 (s, 3H), 3.11 (m, 2H), 3.14 (ddd, J = 13.0, 7.0, 7.0 Hz, 1H), 3.29 (dd, J = 13.0, 7.0 Hz, 1H), 3.35 (m, 2H), 4.48 (dd, J = 7.0. 7.0 Hz, 1H), 7.29 (d, J = 7.5 Hz, 2H), 7.32 (dd, J = 7.5, 7.5 Hz, 1H), 7.37 (dd, J = 7.5, 7.5 Hz, 2H). 13C NMR (125 MHz, CD3OD) δ 22.5, 29.7, 38.7, 39.5, 61.4, 117.7 (q, J = 289.6 Hz), 129.0, 130.2, 130.6, 135.0, 162.1 (q, J = 35.8 Hz), 173.0, 197.2.
(S)-S-(2-acetamidoethyl) 2-(3-oxobutanamido)-3-phenylpropanethioate (4a)
A stirred solution of 3a (13.3 mg, 0.03 mmol) in dehydrated ethanol (1 ml) was cooled at 0–5°C under a nitrogen atmosphere. Sodium ethoxide (4.8 mg, 0.07 mmol) and diketene (5.3 μl, 0.07 mmol) were added. The resulting mixture was stirred for 2 h at room temperature, neutralized with saturated aqueous NH4Cl solution, and evaporated under reduced pressure. The residue was dissolved in ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The crude product was purified with PTLC to provide 4a (9.2 mg, 76%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 1.95 (s, 3H), 2.22 (s, 3H), 3.01 (dd, J = 7.5, 7.5 Hz, 2H), 3.04 (dd, J = 14.0, 8.0 Hz, 1H), 3.19 (dd, J = 14.0, 5.5 Hz, 1H), 3.39 (m, 2H), 3.40 (d, J = 17.0 Hz, 1H), 3.42 (d, J = 17.0 Hz, 1H), 4.90 (ddd, J = 8.0. 8.0, 5.5 Hz, 1H), 7.20 (d, J = 7.5 Hz, 2H), 7.27 (ddd, J = 7.5, 7.5, 1.5 Hz, 1H), 7.32 (dd, J = 7.5, 7.5 Hz, 2H), 7.60 (brd, 8.0 Hz). 13C NMR (125 MHz, CDCl3) δ 23.2, 28.7, 31.0, 38.0, 39.0, 48.9, 60.3, 127.3, 128.7, 129.3, 135.4, 165.7, 170.4, 200.0, 204.1 HRMS (ESI): calcd for C17H22NaN2O4S [M+Na]+ 373.1193; found, 373.1196.
(S)-S-(2-acetamidoethyl) 2-amino-3-(1H-indol-3-yl)propanethioate TFA salt (3b)
Compound 3b was prepared starting from N-(tert-butoxycarbonyl)-l-tryptophan via 2b using the procedure described for 3a. 2b: a colorless solid. 93% yield. 1H NMR (500 MHz, CDCl3) δ 1.43 (s, 9H), 1.86 (s, 3H), 2.94 (ddd, J = 13.5, 5.5, 5.5 Hz, 1H), 2.98 (ddd, J = 13.5, 5.5, 5.5 Hz, 1H), 3.23 (dd, J = 14.5, 5.5 Hz, 1H), 3.31 (m, 2H), 3.34 (dd, J = 14.5, 5.5 Hz, 1H), 4.64 (ddd, J = 8.0, 5.5, 5.5 Hz, 1H), 5.12 (d, J = 8.0 Hz, 1H), 5.62 (brs, 1H), 7.07 (d, J = 2.5 Hz, 2H), 7.13 (dd, J = 8.0, 8.0 Hz, 1H), 7.21 (dd, J = 8.0, 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 8.46 (brs, 1H). 13C NMR (125 MHz, CDCl3) δ 23.0, 28.0, 28.3, 28.5, 39.0, 60.4, 60.7, 80.5, 109.5, 111.3, 118.7, 119.7, 122.3, 123.3, 127.5, 136.1, 155.2, 170.6, 202.3. 3b: a colorless oil. 86% yield. 1H NMR (500 MHz, CD3OD) δ 1.91 (s, 3H), 3.06 (ddd, J = 13.5, 7.0, 7.0 Hz, 1H) 3.12 (ddd, J = 13.5, 7.0, 7.0 Hz, 1H), 3.31 (m, 3H), 3.49 (dd, J = 15.0, 6.0 Hz, 1H), 4.47 (dd, J = 7.5. 6.0 Hz, 1H), 7.07 (dd, J = 7.5, 7.5 Hz, 1H), 7.14 (dd, J = 8.0, 7.5 Hz, 2H), 7.21 (s, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 7.5 Hz, 1H). 13C NMR (125 MHz, CD3OD) δ 22.5, 29.2, 29.8, 39.5, 60.9, 107.3, 112.7, 117.7 (q, J = 286.1 Hz), 118.9, 120.4, 123.0, 125.7, 128.2, 138.3, 162.6 (q, J = 34.6 Hz), 173.5, 197.6.
(S)-S-(2-acetamidoethyl) 3-(1H-indol-3-yl)-2-(3-oxobutanamido)propanethioate (4b)
Compound 4b was prepared from 3b using the procedure described for 4a. 4b: a colorless solid. 93% yield. 1H NMR (500 MHz, CDCl3) δ 1.84 (s, 3H), 2.20 (s, 3H), 2.93 (ddd, J = 14.0. 6.0, 6.0 Hz, 1H), 2.96 (ddd, J = 14.0. 6.0, 6.0 Hz, 1H), 3.27 (m, 3H), 3.36 (dd, J = 14.5, 6.0 Hz, 1H), 3.39 (s, 2H), 4.94 (ddd, J = 14.0. 6.0, 6.0 Hz, 1H), 5.55 (brs. 1H), 7.12 (dd, J = 8.0, 8.0 Hz, 2H), 7.15 (d, J = 1.5 Hz, 1H), 7.20 (dd, J = 8.0, 8.0 Hz, 1H), 7.37 (d, 8.0 Hz, 1H), 7.56 (d, 8.0 Hz, 1H), 7.62 (d, 8.0 Hz). 13C NMR (125 MHz, CDCl3) δ 23.0, 27.9, 28.7, 31.0, 38.8, 49.2, 59.7, 109.3, 111.4, 118.6, 119.7, 122.3, 123.5, 127.4, 136.1, 165.8, 170.4, 200. HRMS (ESI): calcd for C19H23NaN3O4S [M+Na]+ 412.1302; found, 412.1306.
(2S,3S)-S-(2-acetamidoethyl) 2-(2,2-dimethyl-3-oxobutanamido)-3-methylpentanethioate (6)
A solution of (2S,3S)-S-(2-acetamidoethyl) 2-amino-3-methylpentanethioate 2,2,2-trifluoroacetate 4c (1) (34.6 mg, 0.1 mmol) and 2,2-dimethyl-3-oxobutanoic acid 5 (44, 45) (13.0 mg, 0.1 mmol) in dehydrated THF (1 ml) was stirred at room temperature under a nitrogen atmosphere, and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (38.4 mg, 0.2 mmol) and triethylamine (27.7 mg, 0.2 mmol) were added. After the mixture was stirred for 1 h, the mixture was concentrated under reduced pressure. The residue was purified by SiO2 column chromatography (CHCl3: MeOH = 20: 1) to give 6 (3.6 mg, 17%) as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 0.92 (d, J = 7.5 Hz, 1H), 0.99 (d, J = 7.5 Hz, 1H), 1.11 (m, 1H), 1.41 (m, 1H), 1.46 (s, 3H), 1.47 (s, 3H), 1.97 (s, 3H), 2.05 (m, 1H), 2.25 (s, 3H), 3.04 (dd, J = 6.5, 6.5 Hz, 2H), 3.42 (m, 2H), 4.58 (dd, J = 8.5, 4.5 Hz, 1H), 5.84 (brs, 1H), 6.45 (brd, J = 8.0 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 11.5, 15.9, 22.6, 22.7, 23.2, 24.4, 26.6, 28.4, 37.3, 39.3, 55.9, 64.0, 170.3, 172.1, 200.4, 209.6. HRMS (ESI): calcd for C16H28NaN2O4S [M+Na]+ 367.1662; found, 367.1669. The purity of the final compounds, 4a, 4b, and 6, were estimated over 95% from the NMR and UPLC data.
CD spectra analysis of KS and mutants
The secondary structure of all the purified WT and mutants proteins used in this study was determined at 25°C. CD spectra from 195 to 250 nm were collected as the average of four scans using JASCO J-1500 spectrophotometer (JASCO, Tokyo, Japan) in a 1-mm path length cuvette at a speed of 20 nm min−1 and a bandwidth of 1 nm. For all measurements, 0.1 mg ml−1 protein was used.
Size-exclusion chromatography
The buffer containing TAS1 KS protein was exchanged with 50 mm MES, pH 6.5, 5% glycerol, and 5 mm TCEP and concentrated to 1.2 mg ml−1 in an Amicon Ultra-4 centrifugal filter with a cut-off of 30,000 Da (Millipore). The buffer for purified OzmQ was exchanged with the same buffer and concentrated to 0.6 mg ml−1. These samples were analyzed using gel-filtration chromatography at 0.5 ml/min flow rate with a HiLoad 16/600 Superdex 75 pg column (GE Healthcare) and chromatograms were recorded measuring absorbance at 280 nm. As molecular weight standard proteins, BSA (66 kDa) and carbonic anhydrase (29 kDa) in 50 mm potassium phosphate buffer, pH 7.4, 200 mm NaCl, and 10% glycerol were also similarly analyzed.
MALDI-TOF MS analysis of substrate-loaded KS
Substrate-loading reactions were conducted in 20 mm Tris-HCl, pH 7.5, 50 mm tris(2-carboxyethyl)phosphine (TCEP), 5% glycerol with 50 μm N-2,2-dimethylacetoacetyl-l-Ile-SNAC and 0.5 μm KS protein at 25°C for 12 h. After the reaction, samples were analyzed using MALDI-TOF MS with a Bruker rapifleX MALDI-TOF system (Brucker Daltonics, Billeria, MA, USA). A sinapinic acid (trans-3,5-dimethoxy-4-hydroxycinnamic acid, Bruker Daltonics)–saturated solution in 33% acetonitrile/67% water containing 0.1% TFA was prepared. A 3-hydroxypicolinic acid (Bruker Daltonics)–saturated solution was also prepared as described above. A matrix solution was prepared with 3:1 volumes of each solution. A 0.5 μl volume of each sample was mixed with 0.5 μl of the matrix solution and air-dried at room temperature on the MALDI plate for analysis.
Docking simulation of N-acetoacetyl-l-Ile
Like other canonical PKS KS domains, the substrate of the TAS1 KS domain should be loaded onto Cys-179, and Dieckmann cyclization should occur to produce tenuazonic acid. To search for an optimized conformation of the substrate bound to Cys-179, N-acetoacetyl-l-Ile was covalently attached to Cys-179 to make a thioester bond in silico and docking simulation was conducted using AutoDock 4.2 as follows. The initial structures of trans-N-acetoacetyl-l-Ile and cis-N-acetoacetyl-l-Ile were generated and optimized with Chem3D Pro. The carboxyl group of N-acetoacetyl-l-Ile was substituted with methylthiol group, and the methylthiol moiety of the substrate was overlapped with the side chain of Cys-179 to make a covalent thioester bond using MGLTools1.5.6. Hydrogen atoms were added to the ligand and receptor molecules and PDBQT files were created using Autodock 4.2. The grid box size was set to 60 × 60 × 60 Å3. One hundred Lamarckian genetic algorithm runs were performed. Cluster analysis was performed at the end of the simulation. Solutions that were within a 1-Å root mean square deviation of each other were grouped in the same group, and clusters were ranked according to their lowest energy member.
Data availability
The atomic coordinates and structure factors of the reported crystal structures have been deposited in the Protein Data Bank under code 6KOG. The authors declare that all other data supporting the findings of this study are available within the paper and its supporting information.
Supplementary Material
Acknowledgments
The pAPC109406.106 plasmid was kindly provided by Dr. B. Shen, Scripps Research Institute. We thank Dr. T. Nogawa for HR-ESI-TOF-MS analysis. We thank Dr. T.M. Schmeing and Mr. I. Shron for helpful discussion.
This article contains supporting information.
Author contributions—C.-S. Y., K. N., and T. H. formal analysis; C.-S. Y. investigation; C.-S. Y., T. S., and N. D. methodology; C.-S. Y. and S. N. writing-original draft; K. N., T. H., and S. N. data curation; T. M., S. N., and H. O. conceptualization; T. M. supervision; T. M. and H. O. writing-review and editing; S. N. and H. O. project administration; H. O. funding acquisition.
Funding and additional information—This work was supported by JSPS KAKENHI Grants JP17H06412, JP17K07784, and JP18H03945 and by the NARO Bio-oriented Technology Research Advancement Institution (Research program on development of innovative technology) (to H. O.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- TeA
- tenuazonic acid
- NRPS
- nonribosomal peptide synthetase
- PKS
- polyketide synthase
- KS
- ketosynthase
- TE
- thioesterase
- PCP
- peptidyl carrier protein
- UPLC
- ultra-performance liquid chromatography
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Yun C.-S., Motoyama T., and Osada H. (2015) Biosynthesis of the mycotoxin tenuazonic acid by a fungal NRPS-PKS hybrid enzyme. Nat. Commun. 6, 8758 10.1038/ncomms9758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hertweck C. (2009) The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 48, 4688–4716 10.1002/anie.200806121 [DOI] [PubMed] [Google Scholar]
- 3. Bretschneider T., Heim J. B., Heine D., Winkler R., Busch B., Kusebauch B., Stehle T., Zocher G., and Hertweck C. (2013) Vinylogous chain branching catalysed by a dedicated polyketide synthase module. Nature 502, 124–128 10.1038/nature12588 [DOI] [PubMed] [Google Scholar]
- 4. He H.-Y., Tang M.-C., Zhang F., and Tang G.-L. (2014) Cis-double bond formation by thioesterase and transfer by ketosynthase in FR901464 biosynthesis. J. Am. Chem. Soc. 136, 4488–4491 10.1021/ja500942y [DOI] [PubMed] [Google Scholar]
- 5. Katsuyama Y., and Ohnishi Y. (2012) Type III polyketide synthases in microorganisms. Methods Enzymol. 515, 359–377 10.1016/B978-0-12-394290-6.00017-3 [DOI] [PubMed] [Google Scholar]
- 6. Tanovic A., Samel S. A., Essen L.-O., and Marahiel M. A. (2008) Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321, 659–663 10.1126/science.1159850 [DOI] [PubMed] [Google Scholar]
- 7. Dutta S., Whicher J. R., Hansen D. A., Hale W. A., Chemler J. A., Congdon G. R., Narayan A. R. H., Håkansson K., Sherman D. H., Smith J. L., and Skiniotis G. (2014) Structure of a modular polyketide synthase. Nature 510, 512–517 10.1038/nature13423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weissman K. J. (2015) Uncovering the structures of modular polyketide synthase. Nat. Prod. Rep. 32, 436–453 10.1039/c4np00098f [DOI] [PubMed] [Google Scholar]
- 9. Kao C., Pieper M. R., Cane D. E., and Khosla C. (1996) Evidence for two catalytically independent clusters of active sites in a functional modular polyketide synthase. Biochemistry 35, 12363–12368 10.1021/bi9616312 [DOI] [PubMed] [Google Scholar]
- 10. Fischbach M. A., and Walsh C. T. (2006) Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 106, 3468–3496 10.1021/cr0503097 [DOI] [PubMed] [Google Scholar]
- 11. Walsh C. T. (2004) Polyketide and nonribosomal peptide antibiotics: Modularity and versatility. Science 303, 1805–1810 10.1126/science.1094318 [DOI] [PubMed] [Google Scholar]
- 12. Abdalla M. A., and McGaw L. J. (2018) Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 23, 2080 10.3390/molecules23082080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zorzi A., Deyle K., and Heinis C. (2017) Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 38, 24–29 10.1016/j.cbpa.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 14. Driggers E. M., Hale S. P., Lee J., and Terrett N. K. (2008) The exploration of macrocycles for drug discovery—an underexploited structural class. Nat. Rev. Drug. Discov. 7, 608–624 10.1038/nrd2590 [DOI] [PubMed] [Google Scholar]
- 15. Trauger J. W., Kohli R. M., Mootz H. D., Marahiel M. A., and Walsh C. T. (2000) Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 407, 215–218 10.1038/35025116 [DOI] [PubMed] [Google Scholar]
- 16. Gao X., Haynes S. W., Ames B. D., Wang P., Vien L. P., Walsh C. T., and Tang Y. (2012) Cyclization of fungal nonribosomal peptides by a terminal condensation-like domain. Nat. Chem. Biol. 8, 823–830 10.1038/nchembio.1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boettger D., and Hertweck C. (2013) Molecular diversity sculpted by fungal PKS-NRPS hybrids. Chembiochem 14, 28–42 10.1002/cbic.201200624 [DOI] [PubMed] [Google Scholar]
- 18. Zhao C., Coughlin J. M., Ju J., Zhu D., Wendt-Pienkowski E., Zhou X., Wang Z., Shen B., and Deng Z. (2010) Oxazolomycin biosynthesis in Streptomyces albus JA3453 featuring an “acyltransferase-less” type I polyketide synthase that incorporates two distinct extender units. J. Biol. Chem. 285, 20097–20108 10.1074/jbc.M109.090092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai S.-C., and Ames B. D. (2009) Structural enzymology of polyketide synthases. Methods Enzymol. 459, 17–47 10.1016/S0076-6879(09)04602-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang Y., Kim C.-Y., Mathews I. I., Cane D. E., and Khosla C. (2006) The 2.7-Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U.S.A. 103, 11124–11129 10.1073/pnas.0601924103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mori T., Yang D., Matsui T., Hashimoto M., Morita H., Fujii I., and Abe I. (2015) Structural basis for the formation of acylalkylpyrones from two β–ketoacyl units by the fungal type III polyketide CsyB. J. Biol. Chem. 290, 5214–5225 10.1074/jbc.M114.626416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gay D. C., Wagner D. T., Meinke J. L., Zogzas C. E., Gay G. R., and Keatinge-Clay A. T. (2016) The LINKS motif zippers trans-acyltransferase polyketide synthase assembly lines into a biosynthetic megacomplex. J. Struct. Biol. 193, 196–205 10.1016/j.jsb.2015.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lohman J. R., Ma M., Osipiuk J., Nocek B., Kim Y., Chang C., Cuff M., Mack J., Bigelow L., Li H., Endres M., Babnigg G., Joachimiak A., Phillips G. N. Jr., and Shen B. (2015) Structural and evolutionary relationships of “AT-less” type I polyketide synthase ketosynthases. Proc. Natl. Acad. Sci. U.S.A. 112, 12693–12698 10.1073/pnas.1515460112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keatinge-Clay A. T. (2012) The structure of type I polyketide synthases. Nat. Prod. Rep. 29, 1050–1073 10.1039/c2np20019h [DOI] [PubMed] [Google Scholar]
- 25. Robbins T., Kapilivsky J., Cane D. E., and Khosla C. (2016) Roles of conserved active site residues in the ketosynthase domain of an assembly line polyketide synthase. Biochemistry 55, 4476–4484 10.1021/acs.biochem.6b00639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kohlhaas C., Jenner M., Kampa A., Briggs G. S., Afonso J. P., Piel J., and Oldham N. J. (2013) Amino acid-accepting ketosynthase domain from a trans-AT polyketide synthase exhibits high selectivity for predicted intermediate. Chem. Sci. 4, 3212–3217 10.1039/c3sc50540e [DOI] [Google Scholar]
- 27. Gatenbeck S., and Sierankiewicz J. (1973) Microbial production of tenuazonic acid analogues. Antimicrob. Agents Chemother. 3, 308–309 10.1128/aac.3.2.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mo X., Li Q., and Ju J. (2014) Naturally occurring tetramic acid products: Isolation, structure elucidation and biological activity. RSC Adv. 4, 50566–50593 10.1039/C4RA09047K [DOI] [Google Scholar]
- 29. Liu X., and Walsh C. T. (2009) Cyclopiazonic acid biosynthesis in Aspergillus sp.: Characterization of a reductase-like R* domain in cyclopiazonate synthetase that forms and releases cyclo-acetoacetyl-l-tryptophan. Biochemistry 48, 8746–8757 10.1021/bi901123r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sims J. W., and Schmidt E. W. (2008) Thioesterase-like role for fungal PKS-NRPS hybrid reductive domain. J. Am. Chem. Soc. 130, 11149–11155 10.1021/ja803078z [DOI] [PubMed] [Google Scholar]
- 31. Lou L., Qian G., Xie Y., Hang J., Chen H., Zaleta-Rivera K., Li Y., Shen Y., Dussault P. H., Liu F., and Du L. (2011) Biosynthesis of HSAF, a tetramic acid-containing macrolactam from Lysobacter enzymogenes. J. Am. Chem. Soc. 133, 643–645 10.1021/ja105732c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blodgett J. A. V., Oh D.-C., Cao S., Currie C. R., Kolter R., and Clardy J. (2010) Common biosynthetic origins for polycyclic tetramate macrolactams from phylogenetically diverse bacteria. Proc. Natl. Acad. Sci. U.S.A. 107, 11692–11697 10.1073/pnas.1001513107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang G., Zhang W., Zhang Q., Shi T., Ma L., Zhu Y., Li S., Zhang H., Zhao Y.-L., Shi R., and Zhang C. (2014) Mechanistic insights into polycycle formation by reductive cyclization in ikarugamycin biosynthesis. Angew. Chem. Int. Ed. 53, 4840–4844 10.1002/anie.201402078 [DOI] [PubMed] [Google Scholar]
- 34. Gui C., Li Q., Mo X., Qin X., Ma J., and Ju J. (2015) Discovery of a new family of Dieckmann cyclases essential to tetramic acid and pyridine-based natural products biosynthesis. Org. Lett. 17, 628–631 10.1021/ol5036497 [DOI] [PubMed] [Google Scholar]
- 35. Wu Q., Wu Z., Qu X., and Liu W. (2012) Insights into pyrroindomycin biosynthesis reveal a uniform paradigm for tetramate/tetronate formation. J. Am. Chem. Soc. 134, 17342–17345 10.1021/ja304829g [DOI] [PubMed] [Google Scholar]
- 36. Witkowski A., Joshi A., and Smith K. S. (2004) Characterization of the β-carbon processing reactions of the mammalian cytosolic fatty acid synthase: Role of the central core. Biochemistry 43, 10458–10466 10.1021/bi048988n [DOI] [PubMed] [Google Scholar]
- 37. Klaus M., and Grininger M. (2018) Engineering strategies for rational polyketide synthase design. Nat. Prod. Rep. 35, 1070–1081 10.1039/c8np00030a [DOI] [PubMed] [Google Scholar]
- 38. Bozhüyük K. A. J., Fleischhacker F., Linck A., Wesche F., Tietze A., Niesert C.-P., and Bode H. B. (2018) De novo design and engineering of non-ribosomal peptide synthetases. Nat. Chem. 10, 275–281 10.1038/nchem.2890 [DOI] [PubMed] [Google Scholar]
- 39. Awakawa T., Fujioka T., Zhang L., Hoshino S., Hu Z., Hashimoto J., Kozone I., Ikeda H., Shin-Ya K., Liu W., and Ikuro A. (2018) Reprogramming of the antimycin NRPS-PKS assembly lines inspired by gene evolution. Nat. Commun. 9, 3534 10.1038/s41467-018-05877-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams G. J. (2013) Engineering polyketide synthase and nonribosomal peptide synthetases. Curr. Opin. Struct. Biol. 23, 603–612 10.1016/j.sbi.2013.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sambrook J., Fritsch E. F., and Maniatis T. (2001) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 42. Whicher J. R., Smaga S., Hansen D. A., Brown W. C., Gerwick W. H., Sherman D. H., and Smith J. L. S. (2013) Cyanobacterial polyketide synthase docking domains: A tool for engineering natural product biosynthesis. Chem. Biol. 20, 1340–1351 10.1016/j.chembiol.2013.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ehmann D. E., Trauger J. W., Stachelhaus T., and Walsh C. T. (2000) Aminoacyl-SNACS as small-molecule substrates for the condensation domains of nonribosomal peptide synthetases. Chem. Biol. 7, 765–772 10.1016/S1074-5521(00)00022-3 [DOI] [PubMed] [Google Scholar]
- 44. Marshall F. J., and Cannon W. N. (1956) Alkylation of α-substituted acetoacetic esters. J. Org. Chem. 21, 245–247 10.1021/jo01108a605 [DOI] [Google Scholar]
- 45. Muthusamy S., and Sivaguru M. (2013) Synthesis of conformationally restricted C2 symmetric macrodiolides via head to tail dimerization of carbonyl ylides. Tetrahedron Lett. 54, 6810–6813 10.1016/j.tetlet.2013.10.007 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors of the reported crystal structures have been deposited in the Protein Data Bank under code 6KOG. The authors declare that all other data supporting the findings of this study are available within the paper and its supporting information.