

Abstract
The insulinotropic actions of glucagon-like peptide 1 receptor (GLP-1R) in β-cells have made it a useful target to manage type 2 diabetes. Metabolic stress reduces β-cell sensitivity to GLP-1, yet the underlying mechanisms are unknown. We hypothesized that Glp1r expression is heterogeneous among β-cells and that metabolic stress decreases the number of GLP-1R–positive β-cells. Here, analyses of publicly available single-cell RNA-Seq sequencing (scRNASeq) data from mouse and human β-cells indicated that significant populations of β-cells do not express the Glp1r gene, supporting heterogeneous GLP-1R expression. To check these results, we used complementary approaches employing FACS coupled with quantitative RT-PCR, a validated GLP-1R antibody, and flow cytometry to quantify GLP-1R promoter activity, gene expression, and protein expression in mouse α-, β-, and δ-cells. Experiments with Glp1r reporter mice and a validated GLP-1R antibody indicated that >90% of the β-cells are GLP-1R positive, contradicting the findings with the scRNASeq data. α-cells did not express Glp1r mRNA and δ-cells expressed Glp1r mRNA but not protein. We also examined the expression patterns of GLP-1R in mouse models of metabolic stress. Multiparous female mice had significantly decreased β-cell Glp1r expression, but no reduction in GLP-1R protein levels or GLP-1R–mediated insulin secretion. These findings suggest caution in interpreting the results of scRNASeq for low-abundance transcripts such as the incretin receptors and indicate that GLP-1R is widely expressed in β-cells, absent in α-cells, and expressed at the mRNA, but not protein, level in δ-cells.
Keywords: cell sorting, flow cytometry, G protein–coupled receptor (GPCR), glucagon-like peptide 1 receptor (GLP-1R), GLP-1R antibody, glucose-dependent insulinotropic polypeptide receptor (GIPR), heterogeneity, incretin, islet, metabolism
The incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) are secreted from the gastrointestinal tract following a meal and augment insulin secretion, an effect that is blunted in individuals with type 2 diabetes (T2D) (1). Agonists of the GLP-1 receptor (GLP-1R) have been useful therapeutics for people with T2D (2), and recent iterations of drug development have coupled GLP-1R agonism in single peptides that also stimulate the GIP receptor (GIPR) (3). Although the GLP-1R and GIPR have distinct expression across various tissues (4), both receptors were originally identified in β-cells, where they mediate the principle action of incretins, insulin secretion. Studies in humans suggest additivity of GIP and GLP-1 on glucose-stimulated insulin secretion (5) and GIPR/GLP-1R dual agonists have greater glucose-lowering potency than GLP-1R agonists in mice and humans (6, 7). However, it is not clear whether these effects are because of joint stimulation of β-cells expressing both incretin receptors, or if populations of β-cells have specific expression of one or the other incretin receptors. Heterogeneity of incretin receptor expression among β-cells has not yet been rigorously tested, but this possibility has significant implications for physiology and therapeutics.
Estimates of incretin receptor expression across islet populations have been approached mainly with histological (8–10) and single-cell gene expression techniques. Immunostaining approaches have been limited by the lack of specific GLP-1R antibodies to label functional receptors (11) and the inherent lack of precision in distinguishing among individual cells on histological specimens. Several studies have compared the transcriptional profiles of islet α-, β-, and δ-cells in healthy and T2D subjects to gain insight into normal and pathological physiology. Application of single-cell RNA-Seq (scRNAseq) has demonstrated greater variability among islet cell types than previously appreciated, in addition to supporting distinct gene expression in the development of diabetes (12–14). This approach has the potential for understanding specific physiology, such as the incretin effect, and important clinical applications, such as the mechanism of action of multireceptor agonists.
Previous work has demonstrated that diabetes or metabolic stress decreases Glp1r expression in lysates from rodent islets (8) or cell lines (15). These preclinical findings are compatible with the reduced incretin effect among persons with T2D (16), and the blunted response of diabetic subjects to exogenous infusion of GLP-1 (17). However, to date the contribution of GLP-1R activity to abnormal β-cell function has been limited to the analysis of RNA expression, and it remains unclear if the levels of GLP-1R protein are reduced in dysglycemic states and, if so, whether this occurs in all β-cells. Although a validated GLP-1R antibody has recently been developed (9), detailed comparisons of Glp1r expression and GLP-1R protein content in individual islet cells have not been made.
In this paper, we describe a series of experiments to test the hypothesis that GLP-1R expression is heterogeneous in β-cells. The work starts with analysis of human and mouse single-cell RNA-Seq data to document the expression of the incretin receptor genes in islets cells. These data suggested significant heterogeneity in incretin receptor expression, which we then expanded upon with studies measuring Glp1r RNA expression and GLP-1R protein presence on individual islet α-, β-, and δ-cells. Finally, the role of reduced β-cell Glp1r transcription in metabolic stress was tested in the context of GLP-1R protein and activity measurements.
Results
Single-cell RNA-Seq data suggest incretin receptor expression is heterogeneous in β-cells
To test whether incretin receptors were heterogeneously expressed in β-cells, transcriptomes from publicly available human (12, 13, 18–21) and mouse (22) scRNAseq datasets were analyzed to determine the expression patterns of GLP1R/Glp1r and GIPR/Gipr. The expression of incretin receptors in single β-cells was calculated (Table 1) and plotted to visualize the extent of co-expression (Fig. 1). In human β-cells, GLP1R/GIPR co-expression was variable across datasets, ranging from no co-expression (19) to up to ∼27% of β-cells expressing both receptors (13). All human datasets demonstrated a significant number of β-cells that did not express either incretin receptor (range: 7–92%) (Table 1). Fewer datasets were available for mouse β-cells; however, available data (22) suggest that although ∼56% of β-cells had both incretin receptors, 25% expressed Glp1r only, 9% expressed Gipr only, and 9% of β-cells did not express either. Across species and platforms, scRNAseq suggests considerable heterogeneity in the expression of incretin receptors in β-cells, with a surprising number of β-cells expressing either only a single receptor or none at all.
Table 1.
GLP1R/Glp1r and GIPR/Gipr expression in β-cells from published scRNAseq datasets
Number of β-cells (% of total) shown for β-cell expressing either, both, or neither incretin receptor.
| Study | Data Source | Platform | Species | β-cell number | GLP1R+ GIPR− | GLP1R− GIPR+ | GLP1R+ GIPR+ | GLP1R− GIP R− |
|---|---|---|---|---|---|---|---|---|
| Baron et al. (18) | GSE84133* | inDrop | Human | 2507 | 187 (7.5%) | 216 (8.6%) | 30 (1.2%) | 2074 (82.7%) |
| Lawlor et al. (12) | GSE86469* | Fluidigm C1 | Human | 258 | 8 (3.1%) | 184 (71.3%) | 48 (18.6%) | 18 (7.0%) |
| Grün et al. (19) | GSE81076* | CEL-Seq | Human | 161 | 24 (14.9%) | 1 (0.6%) | 0 (0%) | 136 (84.5%) |
| Muraro et al. (20) | GSE85241* | CEl-Seq2 | Human | 445 | 132 (29.7%) | 9 (2.0%) | 6 (1.3%) | 298 (67.0%) |
| Segerstolpe et al. (13) | E-MTAB-5061* | Smart-Seq2 | Human | 308 | 100 (32.5%) | 22 (7.1%) | 83 (26.9%) | 103 (33.4%) |
| Xin et al. (21) | GSE114297 | 10 × Genomics | Human | 7361 | 510 (6.9%) | 98 (1.3%) | 10 (0.1%) | 6743 (91.6%) |
| Tabula Muris (22) | Tabula muris github | Smart-Seq2 | Mouse | 449 | 115 (25.6%) | 42 (9.4%) | 254 (56.6%) | 38 (8.5%) |
* The dataset was provided by SeuratData.
Figure 1.

A and B, heterogeneous expression of GLP1R/Glp1r and GIPR/Gipr in β-cells from published scRNAseq datasets in human (A) and mouse (B). Each circle represents an individual β-cell.
The Glp1r promoter is active in both β- and δ-cells, but not α-cells
To test the hypothesis that β-cell Glp1r expression is heterogeneous, Glp1r reporter mice were generated by crossing Glp1r-Cre mice (23) with mTmG reporter (24) mice (Glp1r:mTmG). This reporter model is a constitutive reporter and GFP+ (Cre+) cells reflect those that express Glp1r promoter activity at any stage of development. Islet cells from Glp1r:mTmG mice were isolated, dispersed, and separated by FACS into GFP+ (Cre+) and tdTomato (Tom+, Cre−) populations (Fig. 2A). Quantification of the distinct cell populations demonstrated that 78% of islets cells were GFP+ and 20% were Tom+ (Fig. 2B). A small percentage (2%) of cells were GFP+/Tom+, but given their low abundance these were not analyzed further. Only GFP+ cells expressed Glp1r mRNA, validating the model (Fig. 2C). To identify the types of cells constituting GFP+ and Tom+ populations, expression of genes specific to α-, β-, and δ-cells were measured by qPCR and expressed relative to whole islet levels. Glucagon (Gcg), defining α-cells, was highly enriched in Tom+ cells and nearly absent in the GFP+ cells (Fig. 2D). Conversely, insulin II (Ins2) and somatostatin (Sst), markers of β- and δ-cells, respectively, were enriched in GFP+ cells and low/absent in Tom+ cells (Fig. 2, E and F). These findings suggest that Glp1r expression coincides with markers for β- and δ-cells, but not α-cells in mouse islets, which aligns with other reports (25). Moreover, Ins2 expression was nearly undetectable in Tom+ cells, suggesting the number of potential Glp1r-negative β-cells in mouse islets is very low. Although it is possible that an Ins2 signal is diluted by the abundance of α-cells in the Tom+ population, the data from mouse scRNAseq dataset (22) (Table 1) would suggest that ∼18% of β-cells are Glp1r negative. This percentage of cells should compose enough of the Tom+ population to produce an Ins2 signal, which was not the case here. This indicates that the number of Glp1r-negative β-cells is substantially lower than 18%, a result incompatible with the hypothesis of heterogeneous Glp1r expression in β-cells generated by the scRNAseq data.
Figure 2.

Glp1r promoter activity is enriched in β-and δ-cells. A, gating strategy to separate tdTomato+ (Tom+) and GFP+ islet cells from Glp1r:mTmG mice by FACS. B, quantification of Tom+ (red circles), GFP+ (green circles), and GFP+/Tom+ (yellow circles) cells from individual mice. C–F, qPCR in Tom+ and GFP+ cells for expression of (C) Glp1r, (D) Gcg, (E) Ins2, and (F) Sst. All qPCR data are normalized to gene expression in whole islet lysates. Comparisons between Tom+ and GFP+ cells were compared by paired t test, ****, p < 0.0001. Each circle represents an individual mouse and data are presented as mean ± S.E.
Glp1r is highly expressed in β-cells enriched from WT mouse islets
To support the results of the Glp1r:mTmG model, a complementary approach was used to assess Glp1r expression in WT islet cells. First, enriched populations of islet cells were separated by FACS (Fig. 3A) based on endogenous FADPH-based fluorescence and side scatter (26, 27). Using this approach, the distribution of cells collected from islets of 17 mice was 22% α-cells, 65% β-cells, and 13% δ-cells (Fig. 3B). These percentages align with those obtained by the Glp1r:mTmG model (Fig. 2B) as well as with previous studies estimating islet cell composition in fixed tissue (28). The enriched populations of cells from 8 of the mice were validated with qPCR to determine expression of Gcg, Ins2, and Sst (Fig. 3, C–E). Gcg and Sst expression were virtually exclusive to the α- and δ-cell populations, respectively. Ins2 was most robustly expressed in β-cells. Because islet cells express relatively high levels of the genes for hormones that are not secreted (e.g. Ins2 in α- and δ-cells (25)), lower-expressing genes that are more specific for α-cells (somatostatin receptor 2, Sstr2) and β-cells (solute carrier family 2, member 2, Slc2a2; galanin receptor 1, Galr) (25) were also measured to confirm fidelity of the enriched populations. Expression of Sstr2 identified the α-cell population as highly enriched, as did the measures of Slc2a2 and Gal1r for β-cells (Fig. S1, A–C). Glp1r expression was highest in the enriched β-cell population, with lower, but detectable, measures in α- and δ-cells (Fig. 3F). All three endocrine populations have been reported to express the Glp1r at variable levels using a bulk RNA-Seq approach (28). These findings align with those reports and with the robust expression of Glp1r in β- and δ-cells in the Glp1r:mTmG mice. However, the detectable expression of Glp1r in enriched α-cells of WT mice contrasts with the lack of promoter activity in α-cells from Glp1r:mTmG mice.
Figure 3.

Glp1r is highly expressed in enriched populations of β-cells from WT mouse islets. A, gating strategy to separate enriched populations of α-cells (purple), β-cells (orange), and δ-cells (green) from WT islets by autofluorescence (FITC-Area) and side scatter (SSC-Area). B, quantification of cells separated by this method. C–F, enriched populations were verified for enrichment of (C) Gcg in α-cells, (D) Ins2 in β-cells, (E) Sst in δ-cells, and (F) Glp1r expression in enriched populations. All qPCR data are normalized to gene expression in whole islet lysates. Populations were compared by one-way ANOVA with Tukey's multiple comparisons test for pairwise comparisons between α-, β-, and δ-cells. Significant differences are denoted by *, p ≤ 0.05; **, p < 0.01; ****, p < 0.0001. Each circle represents an individual mouse and data are presented as mean ± S.E.
GLP-1R protein is detectable in nearly all β-cells
The estimates of Glp1r expression patterns in β-cells assessed by scRNAseq datasets did not align with those produced by with qPCR in FACS-separated islet cells. This discrepancy may reflect methodological shortcomings of scRNAseq when targeting genes expressed at low abundance (29). To rectify the discordant findings and provide an important linkage between Glp1r transcription and translation, an assay to measure protein expression of GLP-1R on individual, live islet cells was developed using antibody Glp1R0017 (9) conjugated to an allophycocyanin (APC) fluorophore. All experiments used the IgG-APC control to set gating parameters (GLP-1R− and GLP-1R+) and serve as an index of nonspecific binding. Dispersed islet cells were treated with GLP1R-APC or IgG-APC and staining determined in enriched β-cells from control and Glp1rβcell−/− mice. β-cells from WT mice had >90% GLP-1R+ staining, whereas staining in the cells from Glp1rβcell−/− mice was comparable to the IgG control (Fig. 4A). As a complementary approach, GLP1R-APC binding was also tested in Glp1r:mTmG mice to compare Glp1r promoter activity with protein expression. Over 90% of GFP+ cells stained GLP-1R+ and virtually all (>97%) of Tom+ cells did not stain with the antibody (GLP-1R−) (Fig. 4B). These staining characteristics were consistent with qPCR measures of Glp1r expression in these discrete cell populations (Fig. 4C). Collectively, these data demonstrate specificity of GLP1R-APC for GLP-1R protein and its suitability for flow cytometry applications. Moreover, there appears to be a strong overall concordance of Glp1r promoter activity and protein expression. However, a small population of cells (<10%) were GLP-1R− despite being GFP+.
Figure 4.
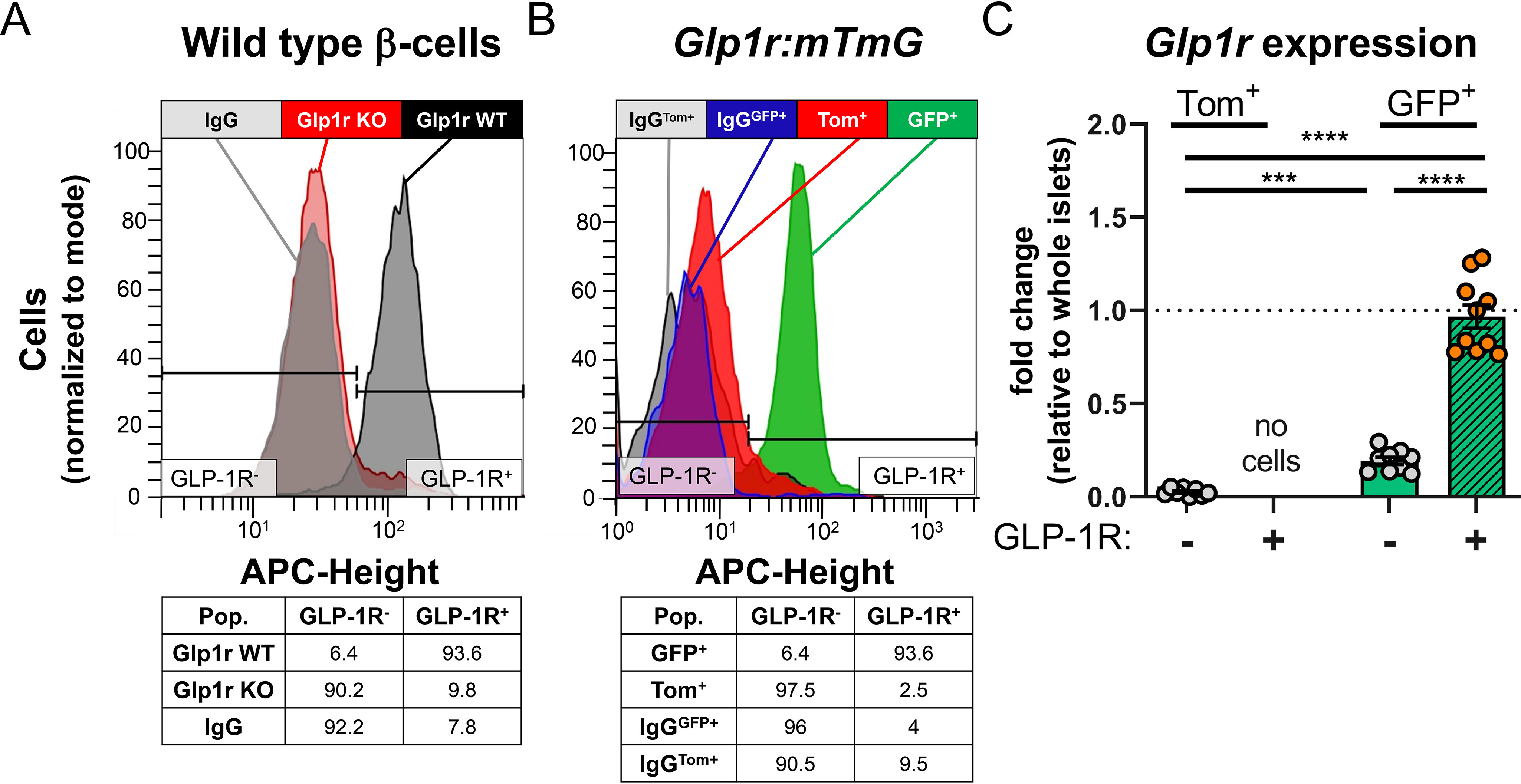
GLP1R-APC is specific for GLP-1R protein detection in islet cells. A, representative histogram of β-cells from Glp1r WT (black, Glp1rβcell+/+, n = 1) and Glp1r KO (red, Glp1rβcell−/−, n = 3) mice stained with GLP1R-APC; IgG-APC staining control is shown in gray and population statistics are below. B, representative histogram of islet cells from Glp1r:mTmG mice (n = 10) stained with GLP1R-APC in Tom+ (red) and GFP+ (green) populations. IgG-APCTom+ and IgG-APCGFP+ staining controls are shown in gray and blue, respectively with population statistics below. C, Glp1r expression in Tom+/GLP1R-APC−, GFP+/GLP1R-APC−, GFP+/GLP1R-APC+ populations from Glp1r:mTmG mice incubated with GLP1R-APC. A and B, histograms are normalized to mode. C, Glp1r expression is normalized to whole islet lysates. Populations were compared by mixed effects analysis and Tukey's multiple comparisons test was used for pairwise comparisons. Significant differences are denoted by ***, p < 0.001; ****, p < 0.0001. Each circle represents an individual mouse, and data are presented as mean ± S.E.
To characterize the population of cells from Glp1r:mTmG islets that were GFP+ but had no GLP-1R immunostaining, gene expression was measured by qPCR in each cell population. Consistent with the findings shown in Fig. 2D, Gcg expression was enriched in Tom+ cells (Fig. 5A), indicating this population of cells are α-cells (no Glp1r promoter activity; no immunodetection of GLP-1R). Ins2 was highest in the GFP+ cells that stained GLP-1R+ (Fig. 5B), consistent with this population being β-cells (high Glp1r promoter activity; high immunodetection of GLP-1R). Interestingly, Sst was highly enriched in GFP+ cells that were GLP-1R− (Fig. 5C), indicating that these are δ-cells. Thus, FACS separation was able to generate β-cells with robust staining of GLP-1R, but did not yield GLP-1R+ δ-cells.
Figure 5.
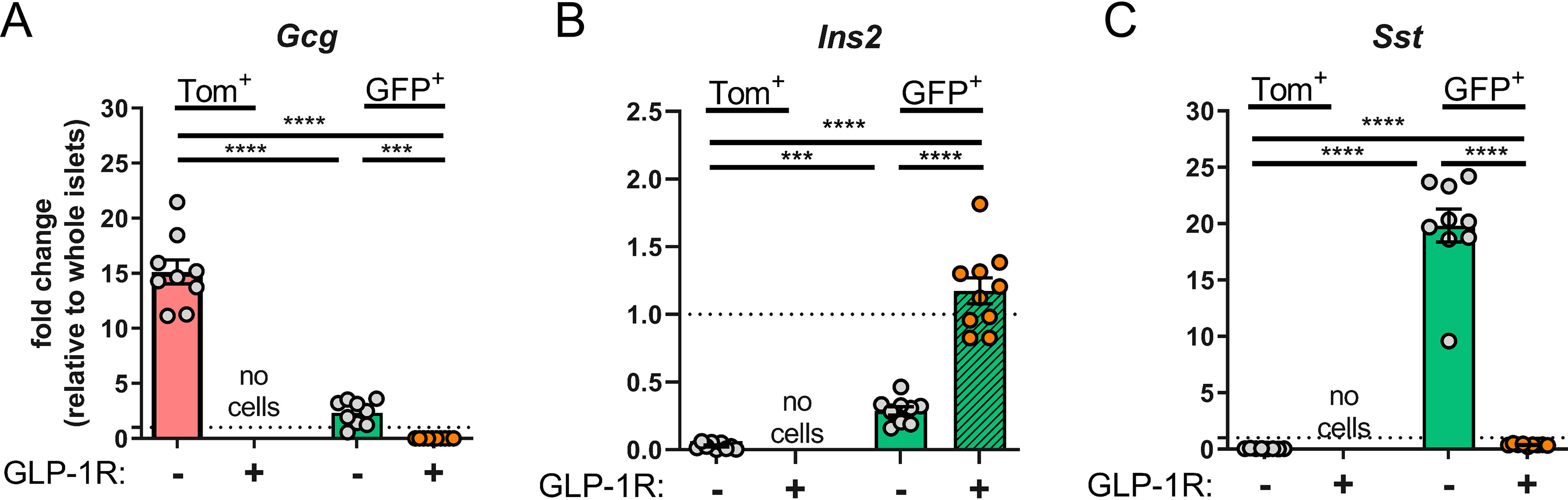
qPCR of FACS-sorted populations from Glp1r:mTmG mice incubated with GLP1R-APC. A–C, sorted populations from Tom+/GLP1R-APC− (red bar/gray circles), GFP+/GLP1R-APC− (green bar/gray circles), and GFP+/GLP1R-APC+ (green bar/orange circles) were analyzed for expression of (A) Gcg, (B) Ins2, and (C) Sst. Tom+/GLP1R-APC+ cells were not acquired. All qPCR data are normalized to gene expression in whole islet lysates. Populations were compared by mixed effects analysis and Tukey's multiple comparisons test was used for pairwise comparisons. Significant differences are denoted by ***, p < 0.001; ****, p < 0.0001. Each circle represents an individual mouse and data are presented as mean ± S.E.
In parallel experiments, islets from WT mice were evaluated for GLP1R-APC staining in enriched α-, β-, and δ-cell populations (Fig. 6). In α-cells, the IgG-APC control produced a positive signal in 7.1% of the cells (an index of background staining), whereas the GLP1R-APC produced a positive signal in 16.8% of the cells, a difference of only borderline statistical significance (Fig. 6A) (p = 0.07), compatible with, but not definitive for, a small population of GLP-1R+ α-cells. In contrast, the vast majority of β-cells (89.7%) stained positive for GLP1R-APC (Fig. 6B). Finally, in the δ-cell population, 7.4% of the cells stained positive for GLP1R-APC versus 2.9% with IgG (Fig. 6C) (p < 0.05). Thus, analysis of GLP-1R–positive cells in enriched populations from WT islets align with the findings from the Glp1r:mTmG islets; GLP-1R-APC robustly stains the majority of β-cells but only produces a signal in small minorities of α- or δ-cells.
Figure 6.

GLP-1R staining is robust in enriched β-cells by flow cytometry. A, GLP1R-APC staining in α-cells (purple) did not reach statistical significance (p = 0.07). B, β-cells had robust GLP1R-APC staining (orange). C, δ-cells had a slightly greater GLP1R-APC staining (green) than IgG-APC control. Representative histograms of staining in α-, β-, and δ-cells are shown with IgG-APC (gray) displayed for each cell type. Population statistics are reported below histograms as a percentage of enriched cells (S.E.). n = 7 experiments. ****, p < 0.0001; *, p < 0.05, paired t test.
Finally, the different populations of islets cells were characterized by gene expression based on FACS separation (Fig. 3A) and GLP1R-APC staining (Fig. 6). In addition to α-cells (GLP-1R−), β-cells (GLP-1R+), and δ-cells (GLP-1R−), a group of GLP-1R− cells characterized as β-cells by autofluorescence were collected and analyzed. Similar to the flow cytometry analysis (Fig. 6), FACS sorting of islets cells produced a GLP-1R-APC signal in the majority of β-cells (Fig. S2, B and E), but only a small proportion of α- and δ-cells (Fig. S2, A–D and F). Given the low abundance of GLP-1R+ α- and δ-cells, there was not enough cellular material to get consistent, sufficient amounts of RNA for qPCR in these populations. Gcg, Ins2, and Sst were highest in the α-, β- and δ-cell populations, respectively (Fig. S2, G–J). Both GLP-1R+ and GLP-1R− β-cell populations expressed similar levels of Ins2 (Fig. S2H). Interestingly, Glp1r expression was robust in both β-cell populations, but not in α- or δ-cells (Fig. S2J). Small but detectable increases in Gcg and Sst were observed in GLP-1R– β-cells, suggesting either small numbers of α- or δ-cells contaminating this gating or that a subset of GLP-1R– β-cells are multihormonal expressing cells. Taken together, the FACS and flow cytometry assays of WT islet cells demonstrate minimal GLP-1R staining in enriched α- and δ-cell populations, but nearly uniform staining in enriched β-cells.
Metabolic stress reduces Glp1r expression but not GLP-1R levels or activity
Islets from young mice fed a standard rodent diet produced β-cells with robust and nearly ubiquitous staining for GLP-1R (Fig. 6B and Fig. S2, B and E). Previous work has shown that hyperglycemia in pancreatectomized rats (8) and cultured MIN6 cells (15) decreases Glp1r expression. Moreover, the insulin secretion response to physiological levels of exogenous GLP-1 is decreased in people with T2D (17). These measurements have not been extended to the resolution of individual β-cells. Thus, male mice were fed a 60% high-fat diet for 4 weeks to determine whether this metabolic stress reduces Glp1r expression in individual β-cells, and whether this translates to differences in the proportion of GLP-1R+ β-cells. Both WT and Glp1r:mTmG mice were included to provide independent measures of Glp1r activity. High-fat feeding increased body weight and ambient glycemia in both mouse lines (Fig. S3, A and B), consistent with induction of metabolic stress. However, the expression of Glp1r or Gipr, which has also been shown to be reduced by hyperglycemia (8), in enriched β-cell populations did not decrease in either mouse model (Fig. S3, C, D, F, G). Moreover, the number of GLP-1R–positive β-cells remained unchanged in both the WT (Fig. S3E) and Glp1r:mTmG mice (Fig. S3, E and H). These results do not conform to previous studies, possibly because a period of longer than 4 weeks of high-fat feeding, or more extreme hyperglycemia, is required to reduce Glp1r expression.
To induce metabolic stress through an alternative approach, the expression of GLP-1R was compared in multiparous (MP) and nulliparous (NP) female mice. Multiparity is associated with increased adiposity and impaired glucose tolerance (30). Moreover, multiparity has been shown to be a physiological stress that reduces β-cell mass in mice through dedifferentiation of β-cells into an α-cell–like phenotype (31). This model was used to test the hypothesis that a dedifferentiating β-cell could present an Ins+:Glp1r− profile and produce a heterogeneous population of β-cells with respect to GLP-1R expression. MP mice were significantly heavier than NP mice (Fig. 7A) but had similar ambient glycemia (Fig. 7B). Glp1r and Gipr expression in enriched β-cells was significantly reduced in MP mice (Fig. 7, C and D). However, the number of GLP-1R–positive β-cells in islets from MP mice was similar to NP mice, and the GLP-1R was present in nearly all β-cells (Fig. 7E). To test whether reduced Glp1r expression translated into reduced GLP-1R activity, insulin secretion from perifused NP and MP islets was measured in response to increasing concentrations of GLP-1. These concentrations were based on our prior studies testing GLP-1–mediated insulin secretion, where the EC50 of GLP-1 was 0.03 nm and maximal response was achieved at 1 nm (32). MP mice had elevated glucose-stimulated insulin secretion (Fig. 7F), likely reflecting insulin resistance and metabolic stress. However, the response to GLP-1, measured as the relative increase over GSIS, was comparable between NP and MP mice (Fig. 7, F and G). In contrast, the reduced expression of β-cell Gipr in islets from MP mice (Fig. 7D) was associated with a reduced insulin secretory response to GIP in perifused islets compared with NP islets (Fig. 7, H and I). These data demonstrate that decreased Glp1r message did not reflect reduction of GLP-1R protein or activity in MP animals, demonstrating robust maintenance of functional receptors despite a physiological challenge.
Figure 7.

GLP-1R and GIPR expression and function in metabolically stressed, multiparous female mice. A and B, weight (A) and blood glucose (B) at sacrifice in nulliparous (NP, white) or multiparous (MP, yellow) mice. C–E, Glp1r (C), Gipr expression (D), and GLP1R-APC (E) staining in enriched β-cells from NP and MP mice. F, insulin secretion from NP and MP islets perifused with increasing concentrations of GLP-1. G, GLP-1 response normalized to glucose-stimulated insulin secretion (GSIS, n = 9 per group). H, insulin secretion from NP and MP islets perifused with increasing concentrations of GIP. I, GIP response normalized to GSIS (n = 6 per group). *, p < 0.05, t test.
Discussion
The incretin axis, long recognized as an important regulator of glucose tolerance, has taken on increased significance as a target of drug development in recent years. Yet, the factors that regulate incretin receptor expression in β-cells remain incompletely understood. As one example, the question of whether all β-cells have similar expression and activity of the GLP-1R has been widely assumed but rarely tested. Moreover, the mechanisms that cause decreased GLP-1 sensitivity in T2D remain unexplained. Here, we observed considerable heterogeneity in the expression patterns of GLP1R/Glp1r in human and mouse β-cells from scRNAseq datasets. However, further investigation using Glp1r promoter activity, gene expression, protein expression, and ultimately GLP-1R function, demonstrates discordance between measures of gene expression and actual receptor content in islet cells. Based on our results it appears that the majority of adult mouse β-cells express GLP-1R on their plasma membrane. Moreover, GLP-1R function is not necessarily reflected by the level of Glp1r gene expression. These findings are consistent with a model whereby GLP-1R signaling is a uniform and resilient feature of healthy β-cells.
Single cell RNA-Seq provides tremendous breadth in the analysis of β-cell gene expression, albeit at a cost of potential false-negative reads, usually because of limits in the cDNA library preparation or the sequencing depth of that library (29, 33). The findings presented here exemplify one of these limitations. In published datasets of islet scRNAseq (12, 13, 18–22) the majority of human β-cells did not express at least one of the incretin receptors, whereas the majority of mouse β-cells express considerable heterogeneity in incretin receptor expression and both had a surprising paucity of cells positive for both receptors (Fig. 1 and Table 1). However, this dichotomous pattern of Glp1r expression was not confirmed with more directed experiments that, in fact, support the presence of the receptor on most β-cells. Thus, the results in this paper are consistent with previous cautions that scRNAseq has limited precision for detecting low-expressing transcripts such as GLP1R/Glp1r (29, 33).
A key reagent in this line of investigation was an effective and well-validated antibody for the GLP-1R (9). After conjugation to a fluorophore, Glp1R0017 binds specifically to GLP-1R in live, dispersed β-cells as demonstrated by our studies using Glp1rβcell−/− mice as negative controls. Moreover, GLP1R-APC labeled 97% of islet cells that had Glp1r promoter activity (i.e. GFP+ cells), and virtually none that did not (Tom+), providing a second, independent measure of its specificity in this application. In the present study, GFP+/GLP-1R+ cells had β-cell markers and most of the enriched β-cells bound GLP1R-APC. We interpret these results as demonstrating that nearly all β-cells in an adult mouse express the GLP-1R, a conclusion previously advanced in reports of studies using immunostaining of fixed sections of pancreas from mice (9) and islets from humans (10, 34), as well as gene expression in dispersed islet cells from mice (35). Our findings add to this established literature with a more definitive approach that mitigates some of the methodological limits of previous work.
In addition to profiling the expression patterns of GLP-1R on β-cells, our approach also enabled the investigation of expression patterns in α- and δ-cells. Recent islet cell transcriptomics datasets also demonstrate very low Glp1r in mouse α-cells (25, 36), although others have reported α-cell GLP-1R (37, 38). Although a numerically greater number of α-cells bound GLP1R-APC compared with IgG-APC in our flow cytometry experiments (Fig. 6), we were unable to acquire enough cells to perform a reliable qPCR analysis of this population (Fig. S2). Contamination of the α-cell pool with β-cells is possible in this experiment, as others have noted (29). Because of technical difficulties, a live/dead stain was not used in these assays and because the cells were not sorted, it is not clear whether dying or dead β-cells contaminated α- and δ-cell populations. Regardless, our findings indicate little or no GLP-1R on α-cells, consistent with the data from both the reporter and the gene expression studies, and in line with a recent human study that reported <0.5% of α-cells stained with a GLP-1R antibody (10). Thus, the bulk of current evidence suggests that any actions of GLP-1 on α-cell function are likely to be indirect.
Given that most studies do not support significant GLP-1R in α-cells, the consensus to explain GLP-1 action to decrease glucagon release has rested on a paracrine model. The most common explanation involves GLP-1 stimulation of somatostatin release from δ-cells with secondary inhibition of α-cells. For example, treatment of perfused pancreata with pharmacological antagonists of somatostatin receptors mute the inhibitory effect of GLP-1 on glucagon secretion, implicating a role for δ-cells to mediate this response (39, 40). Likewise, exposure of cultured human islets to the GLP-1R agonist liraglutide reduces drug-induced glucagon secretion, an effect which is abolished by an SSTR2 inhibitor (34). Although there is little histological data available that demonstrates localization of GLP-1R on δ-cells, especially in mouse islets where these cells are relatively infrequent, recent studies in mouse (38) and human (34) islets suggest some colocalization. The observations reported here neither support nor challenge a role of somatostatin to mediate GLP-1 effects on glucagon release. However, they do bring into question whether this is mediated by a direct action of GLP-1 on δ-cells. Despite clear concurrence of Sst expression and Glp1r promoter activity in Glp1r:mTmG mice, GLP1R-APC staining in enriched δ-cells was only slightly greater than that of IgG-APC. Attempts to sort GLP-1R+ δ-cells did not provide sufficient material for qPCR analysis. Although GLP1R-APC is specific and stains β-cells convincingly, it remains possible that our staining parameters did not allow for the detection of the GLP-1R in δ-cells. Alternatively, it is possible that the Glp1r promoter is active in δ-cells during development, but not expressed or translated in the adult mouse. Reconciling the observation that robust Glp1r expression in δ-cells does not translate to more than minimal GLP-1R protein levels in isolated δ-cells warrants future investigation.
To test the plasticity of GLP-1R gene and protein expression, multiparity was used as a means to induce metabolic stress (30), based on the models others have reported to identify islet cell plasticity and β- to α-cell transitions (31, 41). In rodents and, to a lesser extent, humans (42, 43), pregnancy induces transcriptional changes often associated with increased β-cell mass (44, 45), which return to prepregnancy levels shortly after parturition (46, 47). Mice lacking GLP-1R fail to increase β-cell mass during pregnancy (48), suggesting an important role for GLP-1R signaling during gestation. The MP model had a second advantage as a model that may increase the population of islet cells that others have described as co-expressing Gcg, Ins, and Glp1r (35, 49). Importantly, the MP model induced a decrease in β-cell Glp1r that was similar in magnitude to other studies (8, 15). However, the decreased levels of Glp1r expression did not translate into altered expression of GLP-1R protein on the plasma membrane in live β-cells. Moreover, the action of GLP-1R to stimulate insulin secretion in MP mice remained intact, in line with studies finding similar GLP-1 sensitivity in lean and obese humans (50). These data indicate that RNA expression is not sufficient to determine changes in GLP-1R protein/activity.
Taken together with the other findings reported in this paper, the results from the MP mice suggest nearly universal expression of GLP-1R and continued GLP-1R function in this distinct setting of metabolic stress. However, this model does support potential dynamic regulation of Gipr. Most previous work indicates that the GIPR is expressed in α-, β-, and δ-cells, in contrast to what we show here for the GLP-1R. Although our general approach to studying GLP-1R on islet cells is applicable to the GIPR, we have not yet found a suitable antibody for labeling and cell sorting. Although we have shown here that gene expression does not necessarily reflect protein expression, it should be noted that MP mice had decreased sensitivity to exogenous GIP (Fig. 6), suggesting potential differences in incretin receptor regulation following metabolic stress. These divergent effects on incretin action have been reported in mice (48) and humans (51). Given universal expression of GLP-1R in β-cells and interest in co-agonists for both receptors to treat T2D (3, 52) and the different responses of these receptors to ligand (53), understanding the interplay between GLP-1R and GIPR is of interest.
There are some caveats to consider with the data presented in this paper. First, although our scRNAseq data were similar between mouse and human, our subsequent studies were limited to mouse islets. Thus, it remains to be determined whether the distribution of GLP-1R among islet cells described here would differ in human specimens. Although others have observed similar staining patterns in whole islet sections (10, 34), this question has not been addressed at the level of individual islet cells. Second, despite demonstrations of Glp1R0017 specificity provided here and in a prior paper (9), this validation has been mostly with β-cells, and we cannot exclude the possibility that our findings in δ-cells is because of different antibody binding in the two cell types or differences in the cellular localization of GLP-1R. Western blotting may allow for detecting intracellular GLP-1R, but we did not validate Glp1R0017 for this application. Third, our measures of GLP-1R heterogeneity among islet cells used a dichotomous definition, e.g. present or absent. We cannot determine from our results the variation of receptor expression across the population of β-cells in a mouse islet. Finally, we used MP mice because of their decreased Glp1r expression and general metabolic stress; however, we did not control for age compared with NP mice or for important reproductive factors such as estrus cycle or time since last pregnancy. Our studies do not address whether age contributes to the discrepancies observed between human and mouse β-cell findings. These factors could also have implications for incretin receptor modulation because of changes in β-cell proliferation and apoptosis (45).
In summary, the findings reported here demonstrate that the majority of β-cells express the GLP-1R, refuting the hypothesis suggested by scRNAseq analyses that a significant percentage of these cells are receptor negative. We also show that WT, adult α-cells neither express Glp1r nor contain GLP-1R in the plasma membrane to any significant extent. Mouse δ-cells express the Glp1r, at least at some time during development, but have limited membrane receptors in adulthood. The discrepancy between gene expression and amount of active receptors is also seen in MP mice, which down-regulate Glp1r but maintain normal membrane receptor content and intact responsiveness to GLP-1. Overall, our results support a model whereby direct signaling of GLP-1 is limited to β-cells in adult mouse islets and not modified during metabolic stress.
Experimental procedures
Single-cell RNA-Seq data analysis
Single-cell RNA-Seq data were obtained from seven published studies (12, 13, 18–22) (Table 1). The datasets were all analyzed in Seurat v3 (54), namely, expression was normalized by log normalization method. Pancreatic islet cell identities were taken from the results of each study, and β-cells were extracted for GPCR co-expression analysis.
GPCR expression in human and mouse β-cells
GPCR gene annotation was obtained from HUGO Gene Nomenclature Committee. GPCRs detected in more than 10 β-cells were included in the analysis. Pairwise co-expression pattern was examined for all the detected GPCRs in β-cells. The normalized expression of GLP1R (Glp1r in mouse) and GIPR (Gipr in mouse) was used to illustrate co-expression patterns in the human and mouse β-cells. Each dataset has specific expression units according to its single-cell sequencing platform.
Animals
C57Bl/6 mice (WT) were maintained through our internal breeding colony. Glp1r:mTmG mice were generated by crossing Glp1r-ires-cre (23) with Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J reporter mice (24) and maintained on a mixed background. These mice express endogenous, membrane-bound tdTomato fluorescence. Following cre-mediated recombination, tdTomato is excised and GFP is transcribed and expressed on the plasma membrane (24). To test GLP-1R antibody specificity, Glp1r:Gcgrβ-cell−/− were produced by breeding Glpr1fl/flGcgrfl/fl mice with MIPcreERT (MIP-Cre) mice to generate inducible, β-cell–specific knockouts. Control (Glpr1fl/flGcgrfl/fl:MIP-Cre+/+) and knockout mice (Glpr1fl/flGcgrfl/fl:MIP-CreCre/+) were treated with tamoxifen by oral gavage at 6 weeks of age and mice were used at least 4 weeks after treatment as reported previously (32). High-fat diet–fed mice received 60% high-fat diet (HFD, Research Diets D12492) for 4 weeks. Multiparous (≥4 pregnancies) female mice were maintained on standard breeder chow (Laboratory Diet 5058). All other mice received standard rodent chow (Laboratory Diet 5053). Animal experiments were conducted in accordance with Duke IACUC guidelines.
Islet isolation
Islets were isolated by inflating the pancreas with collagenase type V (0.8 mg/ml) in Hank's Balanced Salt Solution injected retrograde through the pancreatic duct. Digestion occurred at 37°C and was stopped with application of ice-cold RMPI (2 mm l-glutamine, 0.25% BSA). Islets were separated from pancreatic tissue using a histopaque gradient and allowed to recover in RMPI (11.1 mm glucose, 10% FBS, 1% penicillin/streptomycin) overnight before experiments were performed.
Islet dispersion
After overnight recovery, 70-100 islets were collected from each mouse and rinsed once in PBS before incubation with Accutase (Sigma, A6964) for 12-15 min at 37°C with intermittent vortexing. Digestion was stopped with addition of cold RPMI and dispersed islet cells were centrifuged for 3 min, 350 × g, at 4°C. RPMI was aspirated and islets were washed with sorting buffer (RPMI 1640 without phenol red (11835030), 11.1 mm glucose, 1% FBS, 1% penicillin/streptomycin, 2 μm HEPES, 10 units/milliliter DNase). Islets not receiving antibody staining were washed again in sorting buffer before FACS; islets receiving antibody staining were processed as described below.
GLP-1R antibody conjugation and staining
Glp1R0017 (9) and control hIgG1 (MedImmune) were conjugated to an APC fluorophore with a commercially available kit (Abcam, ab201807) per the manufacturer's instructions. Briefly, antibodies were mixed with APC modifier reagent and incubated overnight, in the dark, at room temperature with APC. Then, quenching reagent and sodium azide (0.05% final concentration) was added to yield a concentration of 1.25 mg/ml of Glp1R00017-APC (GLP1R-APC) and hIgG1-APC (IgG-APC). Conjugated antibodies were stored at 4°C for up to 2 months. For FACS experiments, dispersed islet cells were incubated with GLP1R-APC or IgG-APC control antibodies (10 μg/ml) and Hoechst 33342 (10 μm) for 90 min rocking at 4°C in the dark. For flow cytometry experiments, dispersed islet cells were incubated only with GLP1R-APC or IgG-APC control antibodies (10 μg/ml) for 30 min rocking at 4°C in the dark. Following antibody incubation, cells were spun down and washed three times with sorting buffer before FACS or flow cytometry analysis.
Flow cytometry
Dispersed islet cells were transported on ice and filtered through 30 μm mesh prior to FACS using a Beckman-Coulter MoFlo Astrios or analyzed using Attune NxT Analyzer (Thermo Fisher A24863). Forward and side scatter were used to separate single cells from debris and doublets. For FACS, live islet cells (Hoechst−) from Glp1r:mTmG mice were separated into GFP+ and tdTomato+, and by autofluorescence and side scatter, into α-, β-, and δ-cell populations for WT mice (26, 27) into TRIzol. For flow analyzer experiments, WT islets were gated similarly, but without Hoechst stain because of technical limitations of our instrument. Islets treated with antibody were sorted based on APC staining, with IgG-APC+ cells used to set the negative gating control (GLP-1R−) and fluorescence greater than this was classified as GLP-1R+. Where possible, cell percentages reported in this manuscript are calculated from FACS sorts (i.e. absolute number of cells sorted to TRIzol). Post sort analysis of FACS files in FlowJo (v.10.6.2) is used to present flow cytometry plots and in instances where cells numbers were not collected or cell numbers were not reported. Analysis methods are described in the legend of each figure.
RNA extraction, DNA synthesis, RT-PCR
Whole islets and sorted cells were collected into TRIzol for RNA extraction and cDNA was synthesized from 100 ng RNA (Thermo Fisher cat. no. 4368814). qPCR was run using Taqman reagents and primers (Table S1). Data were analyzed by calculating CT and each gene of interest was normalized to cyclophilin A. Data are shown as -fold change relative to whole islet lysates in control animals.
Islet perifusion
For islet perifusion, 75 islets were handpicked and loaded into 0.275-ml chambers containing KRPH (140 mm NaCl, 4.7 mm KCl, 1.5 mm CaCl2, 1 mm NaH2PO4, 1 mm MgSO4, 5 mm HEPES, 2 mm NaHCO3, 1% fatty acid–free BSA) in 2.7 mm glucose. Prior to all experiments, KRPH with 2.7 mm glucose was perifused at a rate of 200 μl/min for 48 min to equilibrate using the BioRep Perifusion system. Following equilibration, experimental conditions were applied and perifusate was collected each minute. GLP-1 (Bachem, cat. no. 4030663) and GIP (Phoenix Pharmaceuticals, cat. no. 027-27) were reconstituted according to manufacturer's instructions and diluted in KRPH prior to experiment. Perifusate insulin concentrations were measured with AlphaLISA (Perkin Elmer).
Statistics
Data in figures are presented as mean ± S.E., and data in the text are presented as mean ± S.D. Analysis was done using GraphPad Prism (v. 8.3). Pairwise comparisons are stated throughout the text, and mice were compared within genetic background. Differences between >2 groups were compared by one-way ANOVA (with mouse repeated) or mixed effect model where values were missing. Post hoc tests used Tukey's multiple comparisons test to determine significance. Differences between cell populations within the same mouse were compared by paired t test and differences between two groups were compared with unpaired t test. Incretin responses were normalized to glucose-stimulated insulin secretion and a two-way ANOVA was run to test for the effect of parity, incretin dose, and parity*dose interaction term. Sidak's multiple comparisons test was used to compare responses between groups within dose. Tests are described in the legend of each figure.
Data availability
The datasets generated during and analyzed during the current study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank Lynn Martinek and Nancy Martin of the Duke Cancer Institute Flow Cytometry Shared Resource for extensive assistance and support with FACS experiments; Jim White for assistance with flow cytometry experiments; Derek Nunez for thoughtful discussions regarding these data; and Matthew Coghlan, Julie Moyers, and Ruth Gimeno for supporting this work.
This article contains supporting information.
Author contributions—S. M. G., J. E. C., and D. A. D. conceptualization; S. M. G. and Y. X. data curation; S. M. G., Y. X., and J. G. formal analysis; S. M. G., J. E. C., and D. A. D. funding acquisition; S. M. G., J. E. C., and D. A. D. validation; S. M. G., E. C. R., B. M. C., M. E. C., K. E., and B. S. investigation; S. M. G. visualization; S. M. G., Y. X., K. W. S., and J. E. C. methodology; S. M. G., Y. X., J. E. C., and D. A. D. writing-original draft; S. M. G., J. E. C., and D. A. D. project administration; S. M. G., Y. X., E. C. R., B. M. C., M. E. C., K. E., B. S., P. R., K. W. S., J. T., J. G., J. E. C., and D. A. D. writing-review and editing; Y. X., M. E. C., P. R., and J. G. resources; J. T., J. G., J. E. C., and D. A. D. supervision.
Funding and additional information—This work was supported by NIDDK, National Institutes of Health Grants T32-DK007012 and F32-DK121420 (to S. M. G), F32-DK-116542 (to M. E. C.), T32-DK007012 (to K. E.), R01 DK123075 (to J. E. C.), and R01 DK101991 (to D. A. D.); Carlsberg Foundation Grant CF16-0996 and Lundbeck Foundation Grant 2016-2394 (to B. S.). This work was also supported by career development award from the American Diabetes Association Grant 1-18-JDF-017 (to J. E. C.). J. E. C. is a Borden Scholar. A portion of this work was supported by Eli Lilly and Company through the Lilly Research Award Program (LRAP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—Y. X. and J. G. are currently employed by Vertex Pharmaceuticals. P. R. is an employee of AstraZeneca. K. W. S. is an employee of Eli Lilly. A portion of this work was supported by Eli Lilly and Company through the Lilly Research Award Program (LRAP).
- T2D
- type 2 diabetes
- APC
- allophycocyanin
- NP
- nulliparous
- MP
- multiparous
- ANOVA
- analysis of variance
- qPCR
- quantitative PCR.
References
- 1. Nauck M., Stöckmann F., Ebert R., and Creutzfeldt W. (1986) Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29, 46–52 10.1007/BF02427280 [DOI] [PubMed] [Google Scholar]
- 2. Drucker D. J., and Nauck M. A. (2006) The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368, 1696–1705 10.1016/S0140-6736(06)69705-5 [DOI] [PubMed] [Google Scholar]
- 3. Capozzi M. E., DiMarchi R. D., Tschöp M. H., Finan B., and Campbell J. E. (2018) Targeting the incretin/glucagon system with triagonists to treat diabetes. Endocrine Rev. 39, 719–738 10.1210/er.2018-00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Campbell J. E., and Drucker D. J. (2013) Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17, 819–837 10.1016/j.cmet.2013.04.008 [DOI] [PubMed] [Google Scholar]
- 5. Nauck M. A., Bartels E., Orskov C., Ebert R., and Creutzfeldt W. (1993) Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7-36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J. Clin. Endocrinol. Metab. 76, 912–917 10.1210/jcem.76.4.8473405 [DOI] [PubMed] [Google Scholar]
- 6. Coskun T., Sloop K. W., Loghin C., Alsina-Fernandez J., Urva S., Bokvist K. B., Cui X., Briere D. A., Cabrera O., Roell W. C., Kuchibhotla U., Moyers J. S., Benson C. T., Gimeno R. E., D'Alessio D. A., et al. (2018) LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol. Metab. 18, 3–14 10.1016/j.molmet.2018.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frias J. P., Bastyr E. J., Vignati L., Tschöp M. H., Schmitt C., Owen K., Christensen R. H., and DiMarchi R. D. (2017) The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 26, 343–352.e2 10.1016/j.cmet.2017.07.011 [DOI] [PubMed] [Google Scholar]
- 8. Xu G., Kaneto H., Laybutt D. R., Duvivier-Kali V. F., Trivedi N., Suzuma K., King G. L., Weir G. C., and Bonner-Weir S. (2007) Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: Possible contribution to impaired incretin effects in diabetes. Diabetes 56, 1551–1558 10.2337/db06-1033 [DOI] [PubMed] [Google Scholar]
- 9. Biggs E. K., Liang L., Naylor J., Madalli S., Collier R., Coghlan M. P., Baker D. J., Hornigold D. C., Ravn P., Reimann F., and Gribble F. M. (2018) Development and characterisation of a novel glucagon like peptide-1 receptor antibody. Diabetologia 61, 711–721 10.1007/s00125-017-4491-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramracheya R., Chapman C., Chibalina M., Dou H., Miranda C., González A., Moritoh Y., Shigeto M., Zhang Q., Braun M., Clark A., Johnson P. R., Rorsman P., and Briant L. J. B. (2018) GLP-1 suppresses glucagon secretion in human pancreatic alpha-cells by inhibition of P/Q-type Ca2+ channels. Physiol. Rep. 6, e13852 10.14814/phy2.13852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Panjwani N., Mulvihill E. E., Longuet C., Yusta B., Campbell J. E., Brown T. J., Streutker C., Holland D., Cao X., Baggio L. L., and Drucker D. J. (2013) GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE−/− mice. Endocrinology 154, 127–139 10.1210/en.2012-1937 [DOI] [PubMed] [Google Scholar]
- 12. Lawlor N., George J., Bolisetty M., Kursawe R., Sun L., Sivakamasundari V., Kycia I., Robson P., and Stitzel M. L. (2017) Single-cell transcriptomes identify human islet cell signatures and reveal cell-type–specific expression changes in type 2 diabetes. Genome Res. 27, 208–222 10.1101/gr.212720.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Segerstolpe Å., Palasantza A., Eliasson P., Andersson E.-M., Andréasson A.-C., Sun X., Picelli S., Sabirsh A., Clausen M., Bjursell M. K., Smith D. M., Kasper M., Ämmälä C., and Sandberg R. (2016) Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 10.1016/j.cmet.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xin Y., Kim J., Okamoto H., Ni M., Wei Y., Adler C., Murphy A. J., Yancopoulos G. D., Lin C., and Gromada J. (2016) RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 24, 608–615 10.1016/j.cmet.2016.08.018 [DOI] [PubMed] [Google Scholar]
- 15. Rajan S., Dickson L. M., Mathew E., Orr C. M., Ellenbroek J. H., Philipson L. H., and Wicksteed B. (2015) Chronic hyperglycemia downregulates GLP-1 receptor signaling in pancreatic β-cells via protein kinase A. Mol. Metab. 4, 265–276 10.1016/j.molmet.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nauck M. A., Heimesaat M. M., Orskov C., Holst J. J., Ebert R., and Creutzfeldt W. (1993) Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Invest. 91, 301–307 10.1172/JCI116186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kjems L. L., Holst J. J., Vølund A., and Madsbad S. (2003) The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 52, 380–386 10.2337/diabetes.52.2.380 [DOI] [PubMed] [Google Scholar]
- 18. Baron M., Veres A., Wolock S. L., Faust A. L., Gaujoux R., Vetere A., Ryu J. H., Wagner B. K., Shen-Orr S. S., Klein A. M., Melton D. A., and Yanai I. (2016) A single-cell transcriptomic map of the human and mouse pancreas reveals inter-and intra-cell population structure. Cell Syst. 3, 346–360.e4 10.1016/j.cels.2016.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grün D., Muraro M. J., Boisset J. C., Wiebrands K., Lyubimova A., Dharmadhikari G., van den Born M., van Es J., Jansen E., Clevers H., de Koning E. J. P., and van Oudenaarden A. (2016) De novo prediction of stem cell identity using single-cell transcriptome data. Cell Stem Cell 19, 266–277 10.1016/j.stem.2016.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muraro M. J., Dharmadhikari G., Grün D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M. A., Carlotti F., de Koning E. J. P., and van Oudenaarden A. (2016) A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394.e3 10.1016/j.cels.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xin Y., Dominguez Gutierrez G., Okamoto H., Kim J., Lee A. H., Adler C., Ni M., Yancopoulos G. D., Murphy A. J., and Gromada J. (2018) Pseudotime ordering of single human β-cells reveals states of insulin production and unfolded protein response. Diabetes 67, 1783–1794 10.2337/db18-0365 [DOI] [PubMed] [Google Scholar]
- 22. Tabula Muris Consortium. (2018) Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 10.1038/s41586-018-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Williams E. K., Chang R. B., Strochlic D. E., Umans B. D., Lowell B. B., and Liberles S. D. (2016) Sensory neurons that detect stretch and nutrients in the digestive system. Cell 166, 209–221 10.1016/j.cell.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muzumdar M. D., Tasic B., Miyamichi K., Li L., and Luo L. (2007) A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- 25. DiGruccio M. R., Mawla A. M., Donaldson C. J., Noguchi G. M., Vaughan J., Cowing-Zitron C., van der Meulen T., and Huising M. O. (2016) Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 5, 449–458 10.1016/j.molmet.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pipeleers D. G., and Pipeleers-Marichal M. A. (1981) A method for the purification of single A, B and D cells and for the isolation of coupled cells from isolated rat islets. Diabetologia 20, 654–663 10.1007/BF00257436 [DOI] [PubMed] [Google Scholar]
- 27. Van de Winkle M., Maes E., and Pipeleers D. (1982) Islet cell analysis and purification by light scatter and autofluorescence. Biochem. Biophys. Res. Commun. 107, 525–532 10.1016/0006-291X(82)91523-6 [DOI] [PubMed] [Google Scholar]
- 28. Cabrera O., Berman D. M., Kenyon N. S., Ricordi C., Berggren P.-O., and Caicedo A. (2006) The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. U.S.A. 103, 2334–2339 10.1073/pnas.0510790103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mawla A. M., and Huising M. O. (2019) Navigating the depths and avoiding the shallows of pancreatic islet cell transcriptomes. Diabetes 68, 1380–1393 10.2337/dbi18-0019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rebholz S. L., Jones T., Burke K. T., Jaeschke A., Tso P., D'Alessio D. A., and Woollett L. A. (2012) Multiparity leads to obesity and inflammation in mothers and obesity in male offspring. Am. J. Physiol. Endocrinol Metab. 302, E449–E457 10.1152/ajpendo.00487.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Talchai C., Xuan S., Lin H. V., Sussel L., and Accili D. (2012) Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234 10.1016/j.cell.2012.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Capozzi M. E., Svendsen B., Encisco S. E., Lewandowski S. L., Martin M. D., Lin H., Jaffe J. L., Coch R. W., Haldeman J. M., MacDonald P. E., Merrins M. J., D'Alessio D. A., and Campbell J. E. (2019) β Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 4, e126742 10.1172/jci.insight.126742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y. J., and Kaestner K. H. (2019) Single-cell RNA-Seq of the pancreatic islets—a promise not yet fulfilled? Cell Metab. 29, 539–544 10.1016/j.cmet.2018.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saponaro C., Gmyr V., Thevenet J., Moerman E., Delalleau N., Pasquetti G., Coddeville A., Quenon A., Daoudi M., Hubert T., Vantyghem M. C., Bousquet C., Martineau Y., Kerr-Conte J., Staels B., et al. (2019) The GLP1R agonist liraglutide reduces hyperglucagonemia induced by the SGLT2 inhibitor dapagliflozin via somatostatin release. Cell Rep. 28, 1447–1454.e4 10.1016/j.celrep.2019.07.009 [DOI] [PubMed] [Google Scholar]
- 35. Richards P., Parker H. E., Adriaenssens A. E., Hodgson J. M., Cork S. C., Trapp S., Gribble F. M., and Reimann F. (2014) Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 63, 1224–1233 10.2337/db13-1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adriaenssens A. E., Svendsen B., Lam B. Y., Yeo G. S., Holst J. J., Reimann F., and Gribble F. M. (2016) Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 59, 2156–2165 10.1007/s00125-016-4033-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., Parajuli K. R., Fava G. E., Gupta R., Xu W., Nguyen L. U., Zakaria A. F., Fonseca V. A., Wang H., Mauvais-Jarvis F., Sloop K. W., and Wu H. (2019) GLP-1 receptor in pancreatic α-cells regulates glucagon secretion in a glucose-dependent bidirectional manner. Diabetes 68, 34–44 10.2337/db18-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ast J., Arvaniti A., Fine N. H. F., Nasteska D., Ashford F. B., Stamataki Z., Koszegi Z., Bacon A., Jones B. J., Lucey M. A., Sasaki S., Brierley D. I., Hastoy B., Tomas A., D'Agostino G., et al. (2020) Super-resolution microscopy compatible fluorescent probes reveal endogenous glucagon-like peptide-1 receptor distribution and dynamics. Nat. Commun. 11, 467 10.1038/s41467-020-14309-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Heer J., Rasmussen C., Coy D. H., and Holst J. J. (2008) Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 51, 2263–2270 10.1007/s00125-008-1149-y [DOI] [PubMed] [Google Scholar]
- 40. Ørgaard A., and Holst J. J. (2017) The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia 60, 1731–1739 10.1007/s00125-017-4315-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuo T., Damle M., González B. J., Egli D., Lazar M. A., and Accili D. (2019) Induction of α cell–restricted Gc in dedifferentiating β cells contributes to stress-induced β-cell dysfunction. JCI Insight 5, e128351 10.1172/jci.insight.128351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Butler A. E., Cao-Minh L., Galasso R., Rizza R. A., Corradin A., Cobelli C., and Butler P. C. (2010) Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 53, 2167–2176 10.1007/s00125-010-1809-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baeyens L., Hindi S., Sorenson R. L., and German M. S. (2016) β-cell adaptation in pregnancy. Diabetes Obes. Metab. 18, Suppl 1, 63–70 10.1111/dom.12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xue Y., Liu C., Xu Y., Yuan Q., Xu K., Mao X., Chen G., Wu X., Brendel M. D., and Liu C. (2010) Study on pancreatic islet adaptation and gene expression during pregnancy in rats. Endocrine 37, 83–97 10.1007/s12020-009-9273-0 [DOI] [PubMed] [Google Scholar]
- 45. Rieck S., White P., Schug J., Fox A. J., Smirnova O., Gao N., Gupta R. K., Wang Z. V., Scherer P. E., Keller M. P., Attie A. D., and Kaestner K. H. (2009) The transcriptional response of the islet to pregnancy in mice. Mol. Endocrinol. 23, 1702–1712 10.1210/me.2009-0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scaglia L., Smith F. E., and Bonner-Weir S. (1995) Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology 136, 5461–5468 10.1210/endo.136.12.7588296 [DOI] [PubMed] [Google Scholar]
- 47. Rieck S., and Kaestner K. H. (2010) Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol. Metab. 21, 151–158 10.1016/j.tem.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moffett R. C., Vasu S., Thorens B., Drucker D. J., and Flatt P. R. (2014) Incretin receptor null mice reveal key role of GLP-1 but not GIP in pancreatic beta cell adaptation to pregnancy. PLoS One 9, e96863 10.1371/journal.pone.0096863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tornehave D., Kristensen P., Rømer J., Knudsen L. B., and Heller R. S. (2008) Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J. Histochem. Cytochem. 56, 841–851 10.1369/jhc.2008.951319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aulinger B. A., Vahl T. P., Wilson-Pérez H. E., Prigeon R. L., and D'Alessio D. A. (2015) β-cell sensitivity to GLP-1 in healthy humans is variable and proportional to insulin sensitivity. J. Clin. Endocrinol. Metab. 100, 2489–2496 10.1210/jc.2014-4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bonde L., Vilsbøll T., Nielsen T., Bagger J. I., Svare J. A., Holst J. J., Larsen S., and Knop F. K. (2013) Reduced postprandial GLP-1 responses in women with gestational diabetes mellitus. Diabetes Obes. Metab. 15, 713–720 10.1111/dom.12082 [DOI] [PubMed] [Google Scholar]
- 52. Finan B., Yang B., Ottaway N., Smiley D. L., Ma T., Clemmensen C., Chabenne J., Zhang L., Habegger K. M., Fischer K., Campbell J. E., Sandoval D., Seeley R. J., Bleicher K., Uhles S., et al. (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 21, 27–36 10.1038/nm.3761 [DOI] [PubMed] [Google Scholar]
- 53. Buenaventura T., Bitsi S., Laughlin W. E., Burgoyne T., Lyu Z., Oqua A. I., Norman H., McGlone E. R., Klymchenko A. S., Corrêa I. R. Jr., Walker A., Inoue A., Hanyaloglu A., Grimes J., Koszegi Z., et al. (2019) Agonist-induced membrane nanodomain clustering drives GLP-1 receptor responses in pancreatic beta cells. PLoS Biol. 17, e3000097 10.1371/journal.pbio.3000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M., Hao Y., Stoeckius M., Smibert P., and Satija R. (2019) Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and analyzed during the current study are available from the corresponding author upon reasonable request.