

Abstract
Phosphatases of regenerating liver (PRLs) are markers of cancer and promote tumor growth. They have been implicated in a variety of biochemical pathways but the physiologically relevant target of phosphatase activity has eluded 20 years of investigation. Here, we show that PRL3 catalytic activity is not required in a mouse model of metastasis. PRL3 binds and inhibits CNNM4, a membrane protein associated with magnesium transport. Analysis of PRL3 mutants specifically defective in either CNNM-binding or phosphatase activity demonstrate that CNNM binding is necessary and sufficient to promote tumor metastasis. As PRLs do have phosphatase activity, they are in fact pseudo-pseudophosphatases. Phosphatase activity leads to formation of phosphocysteine, which blocks CNNM binding and may play a regulatory role. We show levels of PRL cysteine phosphorylation vary in response to culture conditions and in different tissues. Examination of related protein phosphatases shows the stability of phosphocysteine is a unique and evolutionarily conserved property of PRLs. The demonstration that PRL3 functions as a pseudophosphatase has important ramifications for the design of PRL inhibitors for cancer.
Keywords: autophosphorylation, magnesium, metastasis, cancer, phosphocysteine, cysteine phosphorylation, protein phosphatase, dual-specificity phosphoprotein phosphatase, pseudophosphatase, pseudoenzyme
Phosphatase of regenerating liver (PRLs) are small single-domain phosphatases within the family of protein-tyrosine phosphatases (PTP). The first member, PRL1, was identified based on its increased expression in regenerating liver and mitogen-treated cells (1). All three phosphatases are highly overexpressed in tumors and promote growth, motility, and cellular transformation both in culture and animal models (2–4). They are highly similar to each other (78 to 88% identity) and prenylated at their C terminus, which targets them to plasma and endosomal membranes (5–7). PRL1 crystallizes as a trimer and trimers of all three PRLs have been detected in solution by cross-linking (8, 9).
Catalysis by PTP phosphatases proceeds via a two-step mechanism involving transfer of the phosphate to the enzyme and subsequent hydrolysis to regenerate the active enzyme (10, 11). PRLs are unusual in that the second step is greatly slowed leading to the accumulation of the phosphocysteine intermediate both in in vitro and in vivo (12, 13). Unique among PTPs, the phosphocysteine form of PRLs has a lifetime of hours in vitro. The result is that PRLs exhibit burst kinetics with very low steady-state catalytic activity (8, 12). PRLs are also unusually sensitive to oxidation through formation of a disulfide bond between the catalytic cysteine and a neighboring cysteine residue, which further reduces their catalytic activity (8, 12, 14). Oxidation is a complex and relatively poorly understood regulatory mechanism of many protein phosphatases (15, 16).
The mechanism of PRL oncogenicity remains an enigma. A number of different signaling pathways have been implicated but without a clear consensus (17–19). As an alternative, PRLs were shown to directly bind and inhibit a class of Mg2+ transport-associated proteins, CBS domain divalent metal cation transport mediators (CNNMs) (4, 20). CNNMs are membrane proteins with a transmembrane domain that transports Mg2+ and a cytosolic domain that binds PRLs (21). Binding is mediated by a conserved CNNM aspartic acid that acts as a pseudosubstrate and inserts into the PRL active site (13, 22, 23). There are four CNNM proteins in humans that bind with similar affinity to all three PRLs. PRL binding inhibits CNNM-associated Mg2+ efflux and acts to increase intracellular Mg2+ (4, 20, 24). CNNM mutations are associated with hereditary diseases in humans: Jalili syndrome (25, 26) and dominant hypomagnesemia (27). Studies in mice have identified roles in magnesium absorption, fertility, blood pressure, and tumorigenesis (4, 28–30). In Caenorhabditis elegans, loss of cnnm-1 and cnnm-3 cause a defect in gonadal development, which could be rescued by supplementing the media with Mg2+ (31). In Drosophila, PRL1 and the fly CNNM homolog, Uex, fulfill a neuroprotective function in response to CO2 stimulation (32).
A very large number of studies have established the importance of the PRL catalytic site for cellular functions and oncogenicity; however, none distinguish between the phosphatase activity and CNNM binding. Cysteine-to-serine or alanine mutations disrupt PRL inhibition of CNNMs (4) but they also abrogate phosphatase activity (12). Similarly, inhibitors that inhibit PRL cancer proliferation block both phosphatase activity and CNNM binding (33). Consequentially, the relative importance of PRL phosphatase activity versus CNNM binding has remained unknown. To address this, we designed PRL mutations that are specifically defective in one function and tested their activity in Mg2+ efflux assays and an animal model of tumor metastasis. The results demonstrate that phosphatase activity is dispensable and highlight the importance of the PRL-CNNM signaling axis in cancer.
Results
Mutagenesis of the PRL3 active site
Previous studies showed that substitution of the PRL3 catalytic cysteine by serine or alanine leads to loss of both phosphatase activity and CNNM binding (12, 22). In protein phosphatases, the active site cysteine is present as a negatively charged thiolate so we reasoned that substitution of cysteine by aspartic acid might preserve CNNM-binding. To assess this, the PRL3 C104D mutant was prepared and its binding affinity measured by isothermal titration calorimetry (Fig. 1). The substitution of the catalytic cysteine by aspartic acid retained full binding, whereas all other substitutions decreased binding affinity by 16-fold or more. The permissiveness of the binding site was very restricted: substitution by either asparagine, which is isosteric with aspartic acid, or glutamic acid, which is isoelectric, strongly decreased binding. To validate this remarkable result, we generated the same mutations in the other two PRL phosphatases. Both PRL2 C101D and PRL1 C104D showed near WT affinity in ITC experiments (Fig. 1B and Fig. S1). In contrast, mutations in the catalytic site (R101E) or the CNNM-binding site (R138E) blocked binding.
Figure 1.

CNNM-binding activity of PRL mutants. A, ITC thermograms of PRL3 binding to the CNNM3 CBS-pair GST fusion protein. The PRL3 C104D mutant shows WT affinity. Fitting parameters shown in Fig. S1. B, summary of binding affinities for PRL mutants binding to CNNM3 measured by ITC experiments (NB, no binding). PRL2 lacks three amino acids at its N terminus so that residue number of the catalytic cysteine differs. Affinities marked by † are reproduced from Ref. 22. C, co-immunoprecipitation analysis of COS7 cells transfected with CNNM4 and PRL3 shows the C104D mutant binds with WT affinity, whereas the C104E, R110E, and R138E mutants show no binding.
We next tested the ability of the PRL mutants to bind to CNNM proteins in cells. COS7 cells were transfected with Myc-tagged PRL3 mutant constructs and FLAG-tagged CNNM4. Proteins were subjected to co-immunoprecipitation analysis to investigate their interaction in cells. In agreement with the ITC results, only WT PRL3 and the C104D mutant associated with CNNM4 (Fig. 1C).
Having identified a mutant that retained CNNM-binding, we turned to assays of PRL3 phosphatase activity. We performed two types of assays to assess PRL3 enzymatic activity. In the first assay, we used a synthetic fluorogenic substrate, DiFMUP, to measure the rate of phosphate hydrolysis. The assays were conducted under single turnover conditions to detect the initial rate of catalysis (Fig. 2A). As expected, WT PRL3 showed good activity, whereas the C104D and other cysteine substitutions were completely inactive. In contrast, mutation of arginine 138, which is part of the CNNM-binding site but outside of the catalytic site, had little effect on phosphatase activity. The R138E mutant displayed nearly WT activity (Fig. 2C).
Figure 2.

Phosphatase activity of PRL mutants. A, in vitro phosphatase assay of PRL3 with fluorogenic substrate, DiFMUP. Only WT and the R138E mutant show activity. B, Phos-tag SDS-PAGE detection of PRL3 activity. Addition of a synthetic substrate, OMFP, leads to cysteine phosphorylation of catalytically active PRL3. C, specific phosphatase activity of PRL3 mutants.
As a second measure of phosphatase activity, we examined formation of the phosphocysteine intermediate upon incubation with a small molecule substrate, OMFP (22). As expected, Phos-tag SDS-PAGE analysis showed formation of the phosphocysteine intermediate upon incubation with a small molecule substrate by WT PRL3 and the R138E mutant, whereas the cysteine mutants were inactive (Fig. 2B). Thus, we have identified two PRL3 mutations, C104D and R138E, which are selectively defective in either phosphatase activity or CNNM binding.
Crystal structures of mutant PRL complexes
The requirement for the PRL catalytic cysteine in CNNM binding is poorly understood. It does not make direct contacts with CNNM but its mutation has a large effect on binding. Crystal structures of PRL-CNNM complexes show only minor conformational changes despite large differences in affinity (13, 22). Similarly, disulfide bond formation with an adjacent cysteine leads to a ∼500-fold loss of affinity with only modest structural changes (22, 23).
To understand the molecular basis for CNNM binding by the C104D mutant, we carried out structural studies using X-ray crystallography. We were able to obtain crystals of the cysteine-to-aspartic acid mutants of PRL1 and PRL2 in complexes with the CBS-pair domain of CNNM3 and CNNM2 (Fig. 3A). The mutant complexes adopted the same arrangement as the WT complex with binding mediated by an extended loop from CNNM3 that inserts into the PRL catalytic site (Fig. 3B and Fig. S2). Surprisingly, the substitution of cysteine by aspartic acid shifted the binding site residues by less than 0.5 Å.
Figure 3.
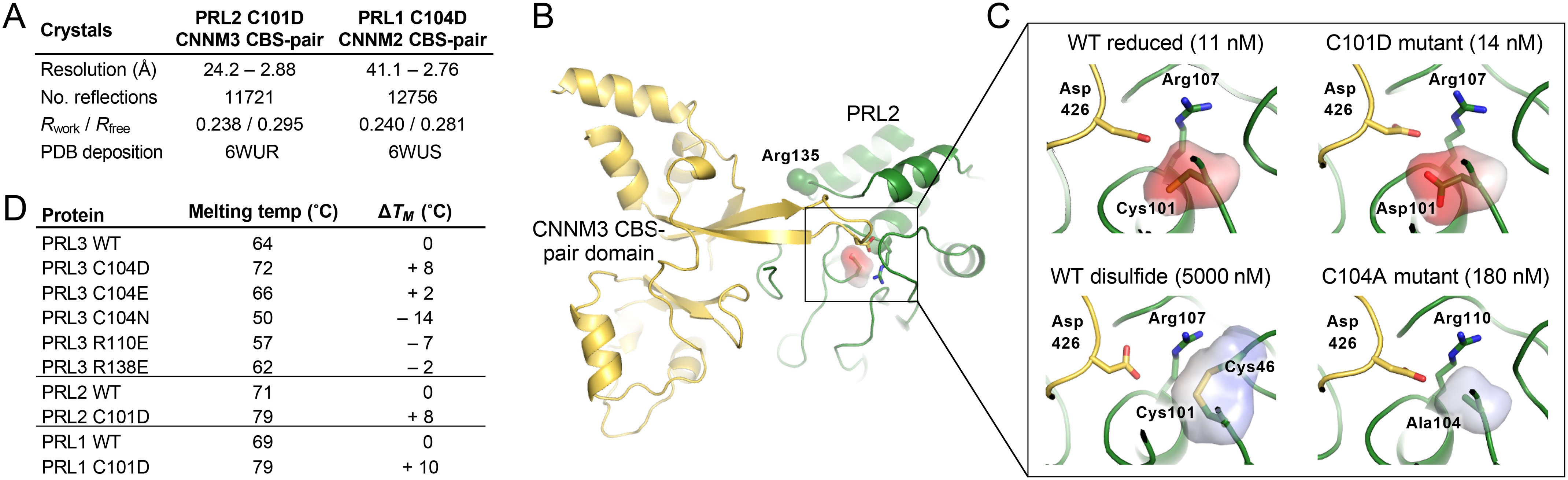
Structure of mutant PRL proteins with CNNM CBS-pair domains. A, structural statistics for the two crystal structures. B, overview of PRL2 C101D•CNNM3 CBS-pair complex. Binding is mediated by an extended β sheet that inserts into the PRL2 catalytic site with key contacts mediated by arginines 107 and 135 of PRL2 and aspartic acid 426 from CNNM3. PRL2 has a shorter N terminus than PRL3 so the numbering of key residues differs by three. C, comparison of PRL active site in four complexes. Full-binding affinity requires the negative charge of a cysteine thiolate or aspartic acid at position 101. PDB coordinates are PRL2 reduced (code 5K22), PRL2 C101D mutant (this work), PRL2 disulfide (code 5K23), and PRL3 C104A (code 5TSR). D, differential scanning fluorimetry shows the cysteine-to-aspartic acid mutations confer increased thermal stability.
Calculation of the partial charges in the PRL active site revealed the role of the catalytic cysteine in CNNM binding is primarily electrostatic (Fig. 3C). To act as a nucleophile, the pKa of the cysteine is shifted so it is deprotonated at neutral pH (10, 11). Measurements with Yersinia PTP show combined effects of the helix dipole and surrounding backbone amide groups shift the pKa by three pH units (34, 35). We used the computational tool H++ to estimate the pKa of the PRL2 active site cysteine. H++ uses atomic coordinates to calculate protonation states based on the AMBER force field with an implicit continuum solvent model (36). The active site cysteine of PRL2 was calculated to have pKa of 4.5 to 5. Thus, the C104D mutation preserves high affinity binding of CNNM proteins because it conserves the negative charge of the cysteine thiolate (Fig. 3C). This paradoxical result is likely due to polarization of other PRL residues in the CNNM-binding site by the negative charge.
The effects of mutations could also be readout as changes in the thermal stability of the PRL proteins. We used differential scanning fluorimetry (thermal shift assays), which measures binding of a hydrophobic dye to the denatured protein, to measure the melting temperatures of PRL1, PRL2, and PRL3 (Fig. 3D and Fig. S3). For all three, the cysteine-to-aspartic mutation increased the melting temperature by 8 °C or more, a large and highly significant change. The increase reflects the potential energy required to charge the cysteine. Strikingly, the PRL3 C104N mutation, which is the substitution of a single oxygen by nitrogen, led to a 22 °C decrease in stability relative to C104D. The importance of charge over steric fit was confirmed by the C104E mutation, which led to a small increase in stability of 2 °C. The R138E mutation that specifically inhibits CNNM binding had a negligible effect on protein stability.
Functional assay of mutant PRLs
We next turned to assessing the function of the PRL mutants in HEK293 cells transfected with Myc-tagged constructs. As controls, we analyzed cell extracts by immunoblotting to measure expression levels and immunofluorescence microscopy to confirm the mutants were properly localized. All the proteins expressed well (Fig. 4A) but the C104N mutant had a defect in its localization. WT PRL3 and other mutants mainly localized at the plasma membrane but the C104N mutant formed aggregates in the cytosol (Fig. 4B). Accordingly, the C104N mutant was omitted from further analysis.
Figure 4.

PRL phosphatase activity is not required for inhibition of CNNM-dependent Mg2+ efflux. A, immunoblotting analyses of HEK293 cells expressing PRL3 mutants. B, immunofluorescence images of HEK293 cells showing proper localization of transfected PRL3 mutants except C104N. The bar graph shows the co-localization index (Pearson's correlation coefficients) of actin filaments and PRL3, which was calculated by Coloc2 software in ImageJ (mean ± S.E., n = 10–12). The p values were determined by one-way ANOVA with Holm-Sidak post hoc tests. C, HEK293 cells were loaded with Magnesium Green and Mg2+ was removed from the extracellular medium at the indicated time. Cells transfected with CNNM4 showed a strong drop in fluorescence due to Mg2+ efflux. Co-transection with WT PRL3 and the C104D mutant inhibited Mg2+ efflux, whereas co-transfection with mutants defective in binding CNNM4 had no effect. The bar graph shows the relative fluorescence intensity after Mg2+ removal (at 5 min, mean ± S.E., n = 10). The p values were determined by one-way ANOVA with Holm-Sidak post hoc tests.
PRL3 binding to CNNM proteins inhibits their Mg2+ efflux activity but it is not known if the effect requires phosphatase activity (4, 13, 28). To address this, we tested the PRL3 mutants in a Mg2+ efflux assay. HEK293 cells were transfected with CNNM4 and intracellular Mg2+ levels were monitored using the fluorescent indicator Magnesium Green (Fig. 4C). In the absence of PRL3, the Mg2+ level dropped when Mg2+ was removed from the extracellular milieu. Co-expression of WT PRL3 and the C104D mutant blocked the Mg2+ efflux. Conversely, co-expression of mutants that prevent CNNM binding had no effect on Mg2+ efflux. This demonstrates that the inhibition of CNNM activity by PRL3 is not due to dephosphorylation of CNNM4 or any other substrate.
PRL oncogenicity
We next addressed the activities of the PRL3 mutants in a well-established experimental model of tumorigenesis. In this model, B16 mouse melanoma cells are injected into the tail vein of mice and the lung metastases were counted. Multiple groups have shown the importance of PRL3 in tumor implantation and outgrowth (4, 37–39). Funato and colleagues (4) showed that the C104S mutant is inactive in promoting metastases.
The new PRL mutants were tested to assess the relative importance of phosphatase activity versus CNNM binding. We verified the expression levels of the Myc-tagged PRL3 WT and mutant proteins by Western blotting prior to injection (Fig. 5A). Levels were very similar with the possible exception of the C104E mutant, which showed a slightly less protein. Following injection and outgrowth, mouse lungs were excised, and the number of tumor nodules were counted. B16 cells expressing WT PRL3 efficiently metastasized to the lung forming an average 60 nodules per animal, whereas the GFP-expressing control cells formed relatively few nodules (Fig. 5, B and C). Comparison of the C104D and WT PRL3 cell lines showed no significant difference in the number of lung nodules demonstrating that phosphatase activity is dispensable for metastasis. Conversely, the cell lines expressing the C104E and R138E mutant proteins, which are unable to bind CNNM proteins, were indistinguishable from the negative control. These results unambiguously show that PRL3 functions as a pseudophosphatase in this tumor model.
Figure 5.
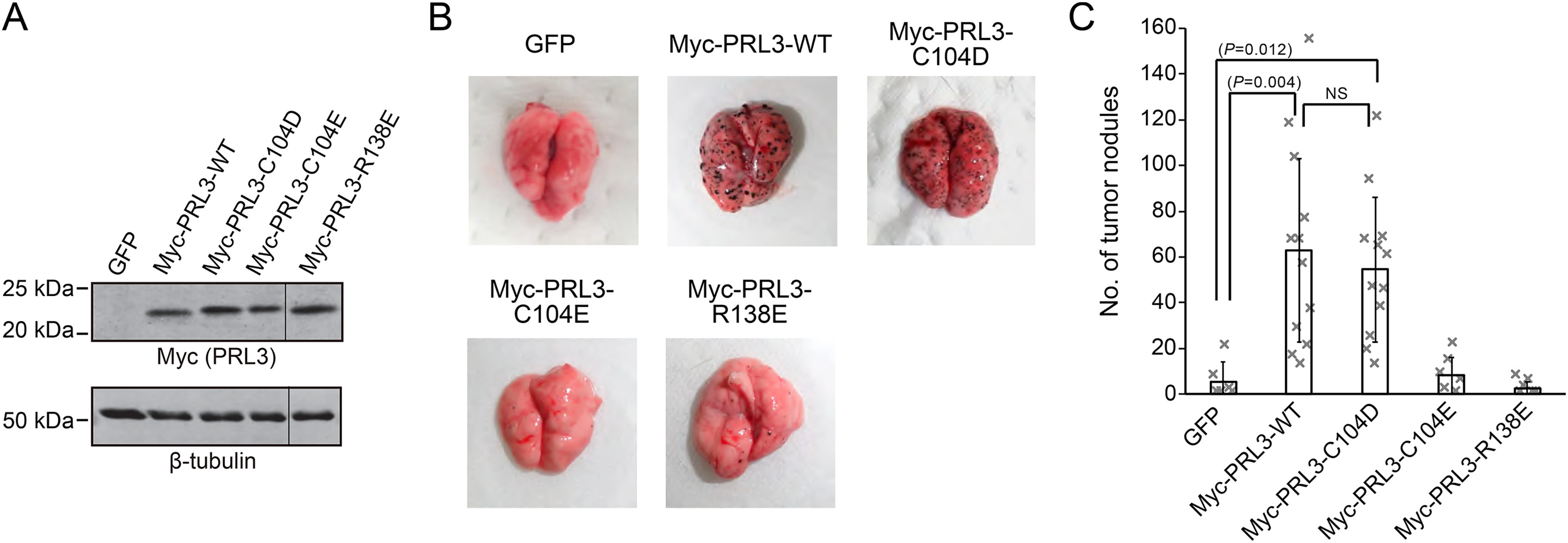
Tumor formation by PRL3 mutants. A, lysates of B16 mouse melanoma cells stably expressing the indicated constructs were subjected to immunoblotting analyses with Myc antibody to check expression levels of each PRL3 mutant. Staining for β-tubulin was used as a loading control. B, B16 cells were injected into mice via their tail veins. 3 weeks later, they were sacrificed, the lungs were removed, and tumor nodules were counted. C, number of tumor nodules per mouse. Stable expression of PRL3 WT and C104D mutant significantly augmented the number of tumor nodules. The catalytically active R138E, which is defective in CNNM binding, showed the same number of nodules as the GFP control. The bar graph shows the average ± S.D. (n = 6 for GFP, Myc-PRL3-C104E, and Myc-PRL3-R138E; n = 12 for Myc-PRL3-WT and Myc-PRL3-C104D).
PRL phosphorylation
Unlike true pseudophosphatases, which have no catalytic activity (40), PRLs are functional enzymes and formation of the phosphocysteine intermediate prevents CNNM binding (13). To address the question of whether PRLs are constitutively active in binding CNNMs, we examined levels of PRL phosphorylation in cultured cells and tissues. Extracts were analyzed by Phos-tag SDS-PAGE gels and Western blotting (Fig. 6A). As PRL3 expression is low and difficult to detect, we used a commercial antibody against PRL2 that also detects PRL1. Migration of phosphorylated proteins is retarded in Phos-tag gels and allowed us to assess the amount of phosphorylated and unphosphorylated protein. Phosphorylation on cysteine can be specifically recognized, as it is sensitive to boiling prior to gel electrophoresis (13).
Figure 6.

PRLs are cysteine phosphorylated in cells and tissues. A, Western blots of HeLa cell extracts show changes in PRL levels and phosphorylation in response to removal and re-addition of Mg2+ in the culture medium. Cells were cultured for the times shown without Mg2+ and after its re-addition. Total cell extracts were analyzed on SDS-PAGE gels with and without the Phos-tag reagent and blots were developed with an antibody that detects PRL1 and PRL2. Boiling prior to electrophoresis releases the phosphate from phosphocysteine. B, Western blots of mouse tissues show cysteine phosphorylation in all tissues. PRL1 and PRL2 from colon were roughly 50% phosphorylated whereas skeletal muscle PRLs were essentially 100% phosphorylated.
Analysis of an extract from HeLa cells grown with Mg2+ showed PRL1 and PRL2 were predominantly phosphorylated. Upon transfer to a growth medium without Mg2+, there was a slow loss of the phosphorylated PRLs and an accumulation of the unphosphorylated forms. After 24 h, the majority of PRL1 and PRL2 were unphosphorylated. Upon re-addition of Mg2+ to the culture medium, PRL1 and PRL2 were rapidly phosphorylated, revealing a burst of phosphatase activity. After 1 h, the large majority of PRL1/2 was phosphorylated, roughly in the same proportions as prior to Mg2+ withdrawal. In addition to changes in phosphorylation, Mg2+ deprivation led to a marked increase in PRL levels, which is visible in both the Phos-tag and regular SDS-PAGE gel. The up-regulation is the result of both transcriptional and translational regulatory mechanisms (41, 42).
We next examined PRL phosphorylation in mouse tissues (Fig. 6B). As previously reported for PRL1 (43), protein levels were high in the colon with lower amounts in heart, liver, and skeletal muscle. Comparison of boiled and unboiled samples and Phos-tag and regular SDS-PAGE gels showed that PRL1 and PRL2 were cysteine phosphorylated in all the tissues examined. The sample from the colon showed the most unphosphorylated protein; in the other tissues, the majority of PRL1 and PRL2 were phosphorylated. Taken together, these results show large changes in the proportion of PRLs bound to CNNMs in different culture conditions and tissues.
Uniqueness of PRL cysteine phosphorylation
The unusual kinetics of PRL phosphatase raised the question if other PTPs might similarly form stable phosphocysteine intermediates. To test this, we purified the catalytic domains of six phosphatases that have been previously reported to have very low activity (44). The domains were incubated with a synthetic substrate, OMFP, to generate the phosphocysteine intermediate and analyzed on Phos-tag gels with and without boiling to hydrolyze phosphocysteine (Fig. 7). Human PRL2 and PRL from a tardigrade species both showed formation of a slower migrating band upon incubation with OMFP. As previously observed, recombinant human PRL2 purified from Escherichia coli was partially phosphorylated prior to OMFP treatment (13). None of the other phosphatases showed the formation of a stable phosphocysteine intermediate. These results suggest that stable cysteine phosphorylation is a unique and remarkably conserved property of PRLs.
Figure 7.

Stable phosphorylation of cysteine is unique to PRLs. A, recombinant catalytic phosphatase domains of PTPs were analyzed on Coomassie-stained Phos-tag gels to detect cysteine phosphorylation. Proteins were dephosphorylated by boiling for 10 min in SDS-loading buffer or treated with OMFP at 37 °C for 1 h to generate the cysteine-phosphorylated form. Only human and tradigrade PRLs showed shifts consistent with cysteine phosphorylation. No phosphorylation was observed for the domains from PTPRN2 (also known as PTPR or IAR), PTPN14 (PTPD2), PTPRN (ICA3, ICA512 or PTP35), PTPRC (CD45), PTPRM (RPTPµ), and PTPN21 (PTPD1). Asterisks indicate contaminants in the preparations of PTPRN and PTPRC. B, SDS-PAGE of tradigrade PRL shows the OMFP-induced shift in A depends on the Phos-tag reagent.
Discussion
We have shown that PRL3 functions as a pseudophosphatase in the promotion of B16 tumorigenesis. PRL3 binds to CNNM proteins, which act as tumor suppressors by decreasing intracellular Mg2+ levels (4, 28). Mg2+ has long been suspected to play a central role in coordinating control of metabolism and growth in animal cells (45). Although Mg2+ does not undergo the dramatic, multifold changes in intracellular concentration that Ca2+ does, changes in Mg2+ levels have been reported in response to cellular stimulation and growth signals. Most Mg2+ is bound to phosphate-containing compounds: CNNM proteins themselves selectively bind Mg2+·ATP via their CBS-pair domain (24) and are important for regulating metabolism and preventing overproduction of reactive oxygen species (46). Compartmentalization of Mg2+ could increase the size of the concentration changes and allow coordinated control of different pathways. PRL and CNNM proteins may also act through other downstream effectors.
As PRL3 does have phosphatase activity, in some sense, it should be considered as pseudo-pseudophosphatase. Although dispensable for PRL3 inhibition of CNNM Mg2+ efflux and B16 tumorigenesis, it is unlikely that the catalytic activity of PRLs is without function. The PRL catalytic residues are conserved in all three isoforms and across metazoan species. The stability of the phosphocysteine form of tardigrade protein implies PRL cysteine phosphorylation has existed since the separation of protostomes and deuterostomes over 500 million years ago. PRL phosphorylation prevents CNNM binding and likely regulates CNNM function in cells (13). The tissue-specific differences and changes in response to Mg2+ availability argue for a conserved, physiological function of phosphatase activity, most likely as a mechanism for regulating the PRL-CNNM interaction.
B16 cells expressing the C104D mutant that cannot be phosphorylated might be expected to form more tumor nodules than cells expressing WT PRL3 (Fig. 5). We did not measure cysteine phosphorylation in the B16 cell lines but previous studies with exogenous expression of PRLs in HEK293 and COS7 cells showed high levels of PRL phosphorylation (13). One possible explanation for why the PRL3 C104D mutant does not act as a super oncogene is that the levels of WT PRL3 are high enough that the pool of unphosphorylated protein is sufficient to bind and inhibit CNNM proteins. Alternatively, some other aspect of migration, implantation, or growth may be limiting the number of tumor modules formed.
Although PRL3 has significant initial (burst) phosphatase activity (Fig. 2), the long lifetime of the catalytic intermediate makes it an extremely poor enzyme under steady-state conditions with a turnover rate on the order of once per hour (12, 13). As the second step in the catalytic cycle is rate-limiting, the catalytic throughput is not substrate dependent but depends on the rate of hydrolysis of phosphocysteine. Although a cellular factor, protein or small molecule, could in principle increase the rate of phosphate release, rapid dephosphorylation of PRL1 and PRL2 was not observed in cells (Fig. 6). In response to removal of Mg2+ from the culture medium, the levels of PRL phosphorylation decreased slowly, suggesting the absence of a mechanism for rapid dephosphorylation. In contrast, re-addition of Mg2+ led to rapid rephosphorylation. The substrates responsible for this rephosphorylation are not known but the observation of cysteine phosphorylation in PRL2 expressed in bacteria suggests broad substrate specificity (Fig. 7). Indeed, in vitro incubation of PRL2 with small phosphate-containing compounds leads to partial cysteine phosphorylation (22). As PRL phosphatase activity is inhibited when bound to CNNM (13), one possibility is that phosphorylation acts as a switch to prevent re-association of PRLs following release. The dissociated, phosphorylated forms could mediate proposed noncatalytic functions of PRLs in transforming growth factor β and SOX2 signaling (47).
There is a great deal of interest in identifying specific inhibitors of PRLs as therapeutics against metastatic cancer (18, 48, 49). The majority of efforts have focused on inhibiting PRL catalytic activity and identifying substrates. Among the more successful compounds, thienopyridone inhibitors have been developed that take advantage of the sensitivity of the PRL catalytic cysteine to oxidation (33, 50, 51). Other approaches have identified allosteric inhibitors that disrupt PRL trimerization (52) and PRL-specific antibodies (53). Intriguingly, an inhibitor of the low molecular weight phosphatase LMPTP (ACP1) was identified that specifically binds and stabilizes the cysteine-phosphorylated form of the protein (54). Thus, it may be possible to design small molecules that bind phosphorylated PRLs and alter the rate of phosphocysteine hydrolysis.
The identification of the C104D mutant should help these drug development efforts. The C104D mutant is insensitive to oxidation, which can lead to false-positives in high-throughput screens. PRL3 C104D binding to CNNMs is not inhibited by redox active compounds (33). Additionally, the C104D mutant cannot be phosphorylated so it is constitutively active in binding and inhibiting CNNMs.
More generally, cysteine-to-aspartic acid substitutions may be useful as a tool for studying other enzymes with active site cysteines. In cysteine proteases and protein phosphatases, cysteine has a shifted pKa to act as a nucleophile so aspartic acid is the amino acid most closely matched in size and charge. The cysteine-to-aspartic acid mutations may be useful for substrate trapping (55) and offer the additional advantage of increased thermal stability. Indeed, the phosphatase PTPRG contains a pseudophosphatase domain with a substitution of aspartic acid for the catalytic cysteine (56). Although ionized cysteines are a relatively small proportion of cysteines in proteins, the general guidance that serine and alanine are most likely to be neutral mutations needs to be qualified.
In conclusion, we have identified and characterized a PRL3 phosphatase mutant that does not have catalytic activity but is oncogenic in a well-established animal model. The reciprocal mutant that is unable to bind CNNM is not oncogenic. These results unequivocally establish that PRL3 acts as a pseudophosphatase independently of its phosphatase activity.
Experimental procedures
Expression and purification of proteins
Human phosphatases PRL3 (residues 1–169) and PRL2 (residues 1–167) were expressed and purified as His tag fusion proteins as described previously (13, 22). Mouse phosphatase PRL1 (residues 7–160) was codon-optimized for E. coli expression and inserted into the pET15b vector (Novagen Inc., Madison, WI) modified to contain noncleavable MHHHHHH tag fusion protein at the N terminus. Catalytic cysteine point mutants were generated with QuikChange Lighting Site-directed Mutagenesis Kit (Agilent Technologies) and confirmed by DNA sequencing and MS. Plasmids 38891 coding for PTPRN2(715-1010) and 38944 coding for PTPN14(886-1187) were obtained from Addgene (Cambridge, MA). DNAs coding for PTPRC(627-1228), PTPRM(867-1452), PTPN21(875-1174), PTPRN(681-979), and PRL homolog RvY_14574 from Ramazzottius varieornatus were chemically synthesized (Bio Basic Inc., Markham, Canada) and cloned into pET29a (EMD Biosciences). The CBS-pair domain of human CNNM3 was prepared as previously described (13). The murine CNNM2 CBS-pair domain (residues 430-580) was cloned into the pET SUMO vector to produce a protein fused with His tag and SUMO tag. The tags were removed using ULP-1 protease. The proteins were expressed in E. coli strain BL21 and purified using affinity chromatography. After His tag–based purification and tag cleavage for CBS domains, all proteins were additionally purified using Superdex-75 (GE Healthcare) size-exclusion column equilibrated with HPLC buffer containing 20 mm HEPES, 100 mm NaCl, 5 mm TCEP (tris(2-carboxyethyl)phosphine), pH 7.0. PRL concentrations were estimated by absorbance at 280 nm using an extinction coefficient of 19940 m−1 cm−1.
Isothermal titration calorimetry
ITC experiments were performed on a MicroCal VP-ITC titration calorimeter (Malvern Instruments Ltd). The syringe was loaded with 160 μm PRL phosphatase, whereas the sample cell contained 16 μm CBS-pair domain as a GST fusion. All experiments were carried out at 293 K with 19 injections of 15 µl with stirring at 310 rpm. Results were analyzed using ORIGIN software (MicroCal) and fitted to a binding model with a single set of identical sites.
Cell culture, transfection, and immunofluorescence analysis
COS7 and HEK293 cells were cultured in DMEM supplemented with 10% FBS and antibiotics. Expression plasmids were transfected in each cell with Lipofectamine 2000 (Invitrogen). Studies of PRL3 localization were performed as previously described for studies of CNNM4 localization (28) using a rabbit anti-Myc antibody (Santa Cruz) diluted in blocking buffer for Myc-PRL3 visualization and rhodamine-phalloidin (Invitrogen) for F-actin visualization. Cells were imaged with a FLUOVIEW FV1000 confocal scanning laser microscope (Olympus).
Co-immunoprecipitation and Western blotting
COS7 cells expressing the indicated constructs were washed with PBS and harvested with lysis buffer (20 mm Tris–HCl, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 2 mm EDTA, and 1 mm phenylmethylsulfonyl fluoride). The lysates were centrifuged and the supernatants were incubated with agarose bead-conjugated anti-FLAG antibody (Wako). The immunoprecipitated proteins were separated by SDS–PAGE and transferred to a polyvinylidene difluoride membrane. After blocking, membranes were incubated with primary antibodies, anti-Myc (Santa Cruz) or anti-FLAG (F1804, Sigma), and then with alkaline phosphatase-conjugated anti-rabbit IgG (Promega) for chromogenic detection with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium. For loading controls, anti-β-tubulin (T4026, Sigma) was used.
Phos-tag gels
For detection of protein phosphorylation, we used a Tris–Tricine SDS–PAGE system with 20–40 μm Phos-tag reagent (Wako Chemicals, Japan) and 40–80 μm MnCl2 was added before polymerization to the SDS-PAGE separating gel (12.5–17.5% acrylamide). For Western blots, the gel was soaked in transfer buffer with 1 mm EDTA to remove the Mn2+ ions before transfer.
Mg2+ efflux assays
Mg2+ imaging analyses with Magnesium Green were performed as described previously (13, 28). The cells were incubated with Mg2+ loading buffer (78.1 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 40 mm MgCl2, 5.5 mm glucose, and 5.5 mm HEPES–KOH, pH 7.4), including 2 μm Magnesium Green-AM (Invitrogen), and were viewed using a microscope (IX81; Olympus).
Phosphatase assays
Catalytic activity was measured with a fluorogenic substrate, 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) purchased from Molecular Probes, Thermo Fisher Scientific, as previously described (22). The reaction buffer contained 20 mm HEPES, 100 mm NaCl, 5 mm tris(2-carboxyethyl)phosphine. For measurements, the concentrations were 3 μm protein and 25 μm DiFMUP in a volume of 50 µl. The fluorescent product, DiFMU (6,8-difluoro-7-hydroxy-4-methylcoumarin), was detected using excitation at 360 nm and emission at 455 nm on a SpectraMax M5e (Molecular Devices, LLC) at room temperature. The concentration of DiFMU was estimated from the standard curve of coumarin from 0 to 200 μm.
PRL phosphorylation
Substrate-induced cysteine phosphorylation of PRLs was measured using Phos-tag SDS-PAGE as previously described (13). Purified PRL protein (50 μm) in 100 mm NaCl, 5 mm TCEP, 20 mm HEPES, pH 7.0, was incubated with or without 125 μm 3-O-methylfluorescein phosphate (OMFP) for 15 min at 30 °C and analyzed by Phos-tag SDS-PAGE. Gels were prepared by addition of 20 μm Phos-tag (Cedarlane Laboratory, CA) and 40 μm ZnCl2 prior to polymerization of a 15% acrylamide Tris–Tricine SDS–PAGE gel.
Crystallization and data collection
Crystals of the PRL2 C101D-CNNM3 CBS-pair domain complex were obtained using hanging drops at 293 K with 0.8-µl drops of a 1:1 mixture of the two proteins (10 mg/ml) in 20 mm HEPES, pH 7.0, 100 mm NaCl, 5 mm TCEP buffer equilibrated over 0.54 m sodium citrate, 0.1 m sodium acetate, pH 5.1. Crystals of the PRL1 C104D-CNNM2 CBS-pair domain complex were obtained using 0.8-µl drops of a 1:1 mixture of the proteins (15 mg/ml) equilibrated over 2.0 m sodium formate, 0.1 m sodium acetate, pH 5.3. The cryoprotection solutions contained crystallization conditions supplemented with 30% (v/v) glycerol. Diffraction data were collected at wavelength 0.977 Å on beamline F1 at the Cornell High-Energy Synchrotron Source (Table S1). Data processing and scaling were performed with HKL-2000 (57).
Structure solution and refinement
The starting phases for the structures were obtained using molecular replacement with the PRL2–CBS-pair complex (PDB entry 5K22) in Phaser (58). The resulting models were extended manually with the help of the program Coot (59) and improved by several cycles of refinement using PHENIX (60). The final models have good stereochemistry (Table S1). Figures were produced using PyMOL.
Electrostatics and pKa calculations
Side chain pKa and partial charges at pH 7 were calculated using H++ version 3.2 (36) with an internal dielectric constant of 5 and default parameters (e.g. 0.15 m salinity and implicit solvent). Atomic coordinates were obtained from PRL2 and PRL3 in complex with the CNNM3 CBS-pair domain. Electrostatic surfaces were calculated using the Adaptive Poisson-Boltzmann Solver (APBS) module in PyMOL (61).
Differential scanning fluorimetry
Each reaction contained 20 µl of solution with 50 μm PRL phosphatase, 5 µl of Protein Thermal ShiftTM buffer, 1× Protein Thermal ShiftTM dye (Protein Thermal Shift Dye kitTM, Life Technologies), HPLC buffer or phosphate buffer (0.1 m sodium phosphate, pH 6.8, 0.1 m sodium chloride, and 10 mm DTT). Samples were heated from 25 to 99 °C at a rate of 1 °C per minute and fluorescence signals were monitored by the StepOne Plus quantitative real-time PCR system (Life Technologies). Data were analyzed using Thermal Shift software (Life Technologies). The maximum change of fluorescence with respect to temperature was used to determine the Tm.
Tumor formation assay
Tumor formation experiments were performed as described previously (4). Briefly, one million B16 cells stably expressing Myc-PRL3 were injected into the tail veins of female C57BL/6J mice (Charles River Laboratories). Mice were euthanized 3 weeks after injection, the lungs were excised, and the black spherical B16 colonies on their surface larger than 0.1 mm in diameter were counted.
Mg2+ depletion experiments
HeLa cells were cultured in DMEM supplemented with 10% FBS and antibiotics. For Mg2+ depletion experiment, cells were cultured in Mg2+–free DMEM (Wisent) and dialyzed FBS (Wisent) with or without 1 mm MgSO4. Cells were lysed in RIPA buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) and 5 μg of total protein loaded per well on SDS-PAGE gels with and without the Phos-tag reagent. Proteins were transferred to a polyvinylidene difluoride membrane. The membrane was blocked and first incubated with mouse anti-PRL2 (Millipore), then with horseradish peroxidase-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch 115-035-062 1:5,000). Chemiluminescent detection on film was done using the Amersham Biosciences ECL Prime detection kit (GE Healthcare).
Cysteine phosphorylation in tissues
Frozen mouse tissues (gift of Philippe Gros) were homogenized and lysed in RIPA buffer. Total protein was estimated using Pierce BCA Protein assay kit and the amount for SDS-PAGE analysis adjusted to give roughly equal amounts of PRL staining: colon (10 μg/lane), heart (40 µg), liver (40 µg), and skeletal muscle (20 µg). Where indicated, samples were boiled for 10 min in loading buffer prior to electrophoresis. Western blotting was done as indicated for HeLa cell extracts.
Data availability
The atomic coordinates and structure factors (codes 6WUR and 6WUS) have been deposited in the Protein Data Bank.
Supplementary Material
Acknowledgments
We thank Nassima Fodil (Gros laboratory) and Sebastien Tabaries (Siegel laboratory) for assistance with the mouse tissues. Crystallographic data were acquired at the Canadian Light Source (CLS) and at the Macromolecular Diffraction (MacCHESS) facility at the Cornell High Energy Synchrotron Source (CHESS). CHESS is supported by National Science Foundation Award DMR-0225180 and National Institutes of Health, NCRR Grant RR-01646.
This article contains supporting information.
Author contributions—G. K. and Y. F. formal analysis; G. K., H. M., and K. G. supervision; G. K., Y. F., Y. S. C., Z. Z., and K. I. investigation; G. K., Y. F., and K. I. methodology; G. K., Y. F., H. M., and K. G. writing-original draft; G. K. and K. G. writing-review and editing; Y. F. and K. G. conceptualization; Y. F., Z. Z., K. I., and H. M. visualization; H. M. and K. G. funding acquisition; H. M. and K. G. project administration.
Funding and additional information—This work was supported by Natural Sciences and Engineering Research Council of Canada Grant RGPIN-2020-07195 (to K. G.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PRL
- phosphatase of regenerating liver
- PTP
- protein-tyrosine phosphatases
- CNNM
- CBS domain divalent metal cation transport mediator
- ITC
- isothermal calorimetry
- TCEP
- tris(2-carboxyethyl)phosphine
- GST
- glutathione S-transferase
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- DiFMUP
- 6,8-difluoro-4-methylumbelliferyl phosphate
- OMFP
- 3-O-methylfluorescein phosphate
- ANOVA
- analysis of variance.
References
- 1. Diamond R. H., Cressman D. E., Laz T. M., Abrams C. S., and Taub R. (1994) PRL-1, a unique nuclear protein tyrosine phosphatase, affects cell growth. Mol. Cell Biol. 14, 3752–3762 10.1128/mcb.14.6.3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saha S., Bardelli A., Buckhaults P., Velculescu V. E., Rago C., St Croix B., Romans K. E., Choti M. A., Lengauer C., Kinzler K. W., and Vogelstein B. (2001) A phosphatase associated with metastasis of colorectal cancer. Science 294, 1343–1346 10.1126/science.1065817 [DOI] [PubMed] [Google Scholar]
- 3. Zeng Q., Dong J. M., Guo K., Li J., Tan H. X., Koh V., Pallen C. J., Manser E., and Hong W. (2003) PRL-3 and PRL-1 promote cell migration, invasion, and metastasis. Cancer Res. 63, 2716–2722 [PubMed] [Google Scholar]
- 4. Funato Y., Yamazaki D., Mizukami S., Du L., Kikuchi K., and Miki H. (2014) Membrane protein CNNM4-dependent Mg2+ efflux suppresses tumor progression. J. Clin. Invest. 124, 5398–5410 10.1172/JCI76614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cates C. A., Michael R. L., Stayrook K. R., Harvey K. A., Burke Y. D., Randall S. K., Crowell P. L., and Crowell D. N. (1996) Prenylation of oncogenic human PTP(CAAX) protein tyrosine phosphatases. Cancer Lett. 110, 49–55 10.1016/S0304-3835(96)04459-X [DOI] [PubMed] [Google Scholar]
- 6. Zeng Q., Si X., Horstmann H., Xu Y., Hong W., and Pallen C. J. (2000) Prenylation-dependent association of protein-tyrosine phosphatases PRL-1, -2, and -3 with the plasma membrane and the early endosome. J. Biol. Chem. 275, 21444–21452 10.1074/jbc.M000453200 [DOI] [PubMed] [Google Scholar]
- 7. Zeng Q., Hong W., and Tan Y. H. (1998) Mouse PRL-2 and PRL-3, two potentially prenylated protein tyrosine phosphatases homologous to PRL-1. Biochem. Biophys. Res. Commun. 244, 421–427 10.1006/bbrc.1998.8291 [DOI] [PubMed] [Google Scholar]
- 8. Sun J. P., Wang W. Q., Yang H., Liu S., Liang F., Fedorov A. A., Almo S. C., and Zhang Z. Y. (2005) Structure and biochemical properties of PRL-1, a phosphatase implicated in cell growth, differentiation, and tumor invasion. Biochemistry 44, 12009–12021 10.1021/bi0509191 [DOI] [PubMed] [Google Scholar]
- 9. Jeong D. G., Kim S. J., Kim J. H., Son J. H., Park M. R., Lim S. M., Yoon T. S., and Ryu S. E. (2005) Trimeric structure of PRL-1 phosphatase reveals an active enzyme conformation and regulation mechanisms. J. Mol. Biol. 345, 401–413 10.1016/j.jmb.2004.10.061 [DOI] [PubMed] [Google Scholar]
- 10. Guan K. L., and Dixon J. E. (1991) Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem. 266, 17026–17030 [PubMed] [Google Scholar]
- 11. Zhang Z. Y., Wang Y., and Dixon J. E. (1994) Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 91, 1624–1627 10.1073/pnas.91.5.1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kozlov G., Cheng J., Ziomek E., Banville D., Gehring K., and Ekiel I. (2004) Structural insights into molecular function of the metastasis-associated phosphatase PRL-3. J. Biol. Chem. 279, 11882–11889 10.1074/jbc.M312905200 [DOI] [PubMed] [Google Scholar]
- 13. Gulerez I., Funato Y., Wu H., Yang M., Kozlov G., Miki H., and Gehring K. (2016) Phosphocysteine in the PRL-CNNM pathway mediates magnesium homeostasis. EMBO Rep. 17, 1890–1900 10.15252/embr.201643393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skinner A. L., Vartia A. A., Williams T. D., and Laurence J. S. (2009) Enzyme activity of phosphatase of regenerating liver is controlled by the redox environment and its C-terminal residues. Biochemistry 48, 4262–4272 10.1021/bi900241k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Funato Y., and Miki H. (2014) Reversible oxidation of PRL family protein-tyrosine phosphatases. Methods 65, 184–189 10.1016/j.ymeth.2013.06.032 [DOI] [PubMed] [Google Scholar]
- 16. Yu Z. H., and Zhang Z. Y. (2018) Regulatory mechanisms and novel therapeutic targeting strategies for protein-tyrosine phosphatases. Chem. Rev. 118, 1069–1091 10.1021/acs.chemrev.7b00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Al-Aidaroos A. Q., and Zeng Q. (2010) PRL-3 phosphatase and cancer metastasis. J. Cell. Biochem. 111, 1087–1098 10.1002/jcb.22913 [DOI] [PubMed] [Google Scholar]
- 18. Rubio T., and Kohn M. (2016) Regulatory mechanisms of phosphatase of regenerating liver (PRL)-3. Biochem. Soc. Transactions 44, 1305–1312 10.1042/BST20160146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duciel L., Monraz Gomez L. C., Kondratova M., Kuperstein I., and Saule S. (2019) The phosphatase PRL-3 is involved in key steps of cancer metastasis. J. Mol. Biol. 431, 3056–3067 10.1016/j.jmb.2019.06.008 [DOI] [PubMed] [Google Scholar]
- 20. Hardy S., Uetani N., Wong N., Kostantin E., Labbé D. P., Bégin L. R., Mes-Masson A., Miranda-Saavedra D., and Tremblay M. L. (2015) The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 34, 986–995 10.1038/onc.2014.33 [DOI] [PubMed] [Google Scholar]
- 21. Funato Y., and Miki H. (2019) Molecular function and biological importance of CNNM family Mg2+ transporters. J. Biochem. 165, 219–225 10.1093/jb/mvy095 [DOI] [PubMed] [Google Scholar]
- 22. Zhang H., Kozlov G., Li X., Wu H., Gulerez I., and Gehring K. (2017) PRL3 phosphatase active site is required for binding the putative magnesium transporter CNNM3. Sci. Rep. 7, 48 10.1038/s41598-017-04855-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giménez-Mascarell P., Oyenarte I., Hardy S., Breiderhoff T., Stuiver M., Kostantin E., Diercks T., Pey A. L., Ereño-Orbea J., Martínez-Chantar M. L., Khalaf-Nazzal R., Claverie-Martin F., Müller D., Tremblay M. L., and Martínez-Cruz L. A. (2017) Structural basis of the oncogenic interaction of phosphatase PRL-1 with the magnesium transporter CNNM2. J. Biol. Chem. 292, 786–801 10.1074/jbc.M116.759944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hirata Y., Funato Y., Takano Y., and Miki H. (2014) Mg2+-dependent interactions of ATP with the cystathionine-β-synthase (CBS) domains of a magnesium transporter. J. Biol. Chem. 289, 14731–14739 10.1074/jbc.M114.551176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polok B., Escher P., Ambresin A., Chouery E., Bolay S., Meunier I., Nan F., Hamel C., Munier F. L., Thilo B., Megarbane A., and Schorderet D. F. (2009) Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am. J. Hum. Genet. 84, 259–265 10.1016/j.ajhg.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parry D. A., Mighell A. J., El-Sayed W., Shore R. C., Jalili I. K., Dollfus H., Bloch-Zupan A., Carlos R., Carr I. M., Downey L. M., Blain K. M., Mansfield D. C., Shahrabi M., Heidari M., Aref P., et al. (2009) Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am. J. Hum. Genet. 84, 266–273 10.1016/j.ajhg.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stuiver M., Lainez S., Will C., Terryn S., Günzel D., Debaix H., Sommer K., Kopplin K., Thumfart J., Kampik N. B., Querfeld U., Willnow T. E., Němec V., Wagner C. A., Hoenderop J. G., et al. (2011) CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am. J. Hum. Genet. 88, 333–343 10.1016/j.ajhg.2011.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamazaki D., Funato Y., Miura J., Sato S., Toyosawa S., Furutani K., Kurachi Y., Omori Y., Furukawa T., Tsuda T., Kuwabata S., Mizukami S., Kikuchi K., and Miki H. (2013) Basolateral Mg2+ extrusion via CNNM4 mediates transcellular Mg2+ transport across epithelia: a mouse model. PLoS Genet. 9, e1003983 10.1371/journal.pgen.1003983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Funato Y., Yamazaki D., and Miki H. (2017) Renal function of cyclin M2 Mg2+ transporter maintains blood pressure. J. Hypertens. 35, 585–592 10.1097/HJH.0000000000001211 [DOI] [PubMed] [Google Scholar]
- 30. Yamazaki D., Miyata H., Funato Y., Fujihara Y., Ikawa M., and Miki H. (2016) The Mg2+ transporter CNNM4 regulates sperm Ca2+ homeostasis and is essential for reproduction. J. Cell Sci. 129, 1940–1949 10.1242/jcs.182220 [DOI] [PubMed] [Google Scholar]
- 31. Ishii T., Funato Y., Hashizume O., Yamazaki D., Hirata Y., Nishiwaki K., Kono N., Arai H., and Miki H. (2016) Mg2+ extrusion from intestinal epithelia by CNNM proteins is essential for gonadogenesis via AMPK-TORC1 signaling in Caenorhabditis elegans. PLoS Genet. 12, e1006276 10.1371/journal.pgen.1006276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo P., Xu X., Wang F., Yuan X., Tu Y., Zhang B., Zheng H., Yu D., Ge W., Gong Z., Yang X., and Xi Y. (2019) A novel neuroprotective role of phosphatase of regenerating liver-1 against CO2 stimulation in Drosophila. iScience 19, 291–302 10.1016/j.isci.2019.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z., Kozlov G., Chen Y. S., and Gehring K. (2019) Mechanism of thienopyridone and iminothienopyridinedione inhibition of protein phosphatases. MedChemComm 10, 791–799 10.1039/c9md00175a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Z. Y., and Dixon J. E. (1993) Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 32, 9340–9345 10.1021/bi00087a012 [DOI] [PubMed] [Google Scholar]
- 35. Peters G. H., Frimurer T. M., and Olsen O. H. (1998) Electrostatic evaluation of the signature motif (H/V)CX5R(S/T) in protein-tyrosine phosphatases. Biochemistry 37, 5383–5393 10.1021/bi971187i [DOI] [PubMed] [Google Scholar]
- 36. Anandakrishnan R., Aguilar B., and Onufriev A. V. (2012) H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541 10.1093/nar/gks375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim S. H., Kim Y., Kim M., Kim D. S., Lee S. C., Chi S. W., Lee D. H., Park S. G., Park B. C., Bae K. H., and Kang S. (2009) Comparative proteomic analysis of mouse melanoma cell line B16, a metastatic descendant B16F10, and B16 overexpressing the metastasis-associated tyrosine phosphatase PRL-3. Oncol. Res. 17, 601–612 10.3727/096504009789745494 [DOI] [PubMed] [Google Scholar]
- 38. Wu X., Zeng H., Zhang X., Zhao Y., Sha H., Ge X., Zhang M., Gao X., and Xu Q. (2004) Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am. J. Pathol. 164, 2039–2054 10.1016/S0002-9440(10)63763-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qian F., Li Y. P., Sheng X., Zhang Z. C., Song R., Dong W., Cao S. X., Hua Z. C., and Xu Q. (2007) PRL-3 siRNA inhibits the metastasis of B16-BL6 mouse melanoma cells in vitro and in vivo. Mol. Med. 13, 151–159 10.2119/2006-00076.Qian [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reiterer V., Eyers P. A., and Farhan H. (2014) Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol. 24, 489–505 10.1016/j.tcb.2014.03.008 [DOI] [PubMed] [Google Scholar]
- 41. Yoshida A., Funato Y., and Miki H. (2018) Phosphatase of regenerating liver maintains cellular magnesium homeostasis. Biochem. J. 475, 1129–1139 10.1042/BCJ20170756 [DOI] [PubMed] [Google Scholar]
- 42. Hardy S., Kostantin E., Wang S. J., Hristova T., Galicia-Vazquez G., Baranov P. V., Pelletier J., and Tremblay M. L. (2019) Magnesium-sensitive upstream ORF controls PRL phosphatase expression to mediate energy metabolism. Proc. Natl. Acad. Sci. U.S.A. 116, 2925–2934 10.1073/pnas.1815361116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Diamond R. H., Peters C., Jung S. P., Greenbaum L. E., Haber B. A., Silberg D. G., Traber P. G., and Taub R. (1996) Expression of PRL-1 nuclear PTPase is associated with proliferation in liver but with differentiation in intestine. Am. J. Physiol. 271, G121–G129 10.1152/ajpgi.1996.271.1.G121 [DOI] [PubMed] [Google Scholar]
- 44. Barr A. J., Ugochukwu E., Lee W. H., King O. N., Filippakopoulos P., Alfano I., Savitsky P., Burgess-Brown N. A., Muller S., and Knapp S. (2009) Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136, 352–363 10.1016/j.cell.2008.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rubin H. (1975) Central role for magnesium in coordinate control of metabolism and growth in animal cells. Proc. Natl. Acad. Sci. U.S.A. 72, 3551–3555 10.1073/pnas.72.9.3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hashizume O., Funato Y., Yamazaki D., and Miki H. (2020) Excessive Mg2+ impairs intestinal homeostasis by enhanced production of adenosine triphosphate and reactive oxygen species. Antioxid. Redox Signal. 33, 20–34 10.1089/ars.2019.7951 [DOI] [PubMed] [Google Scholar]
- 47. Sacchetti C., Bai Y., Stanford S. M., Di Benedetto P., Cipriani P., Santelli E., Piera-Velazquez S., Chernitskiy V., Kiosses W. B., Ceponis A., Kaestner K. H., Boin F., Jimenez S. A., Giacomelli R., Zhang Z. Y., et al. (2017) PTP4A1 promotes TGFβ signaling and fibrosis in systemic sclerosis. Nat. Commun. 8, 1060 10.1038/s41467-017-01168-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Frankson R., Yu Z. H., Bai Y., Li Q., Zhang R. Y., and Zhang Z. Y. (2017) Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 77, 5701–5705 10.1158/0008-5472.CAN-17-1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stanford S. M., and Bottini N. (2017) Targeting tyrosine phosphatases: time to end the stigma. Trends Pharmacol. Sci. 38, 524–540 10.1016/j.tips.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salamoun J. M., McQueeney K. E., Patil K., Geib S. J., Sharlow E. R., Lazo J. S., and Wipf P. (2016) Photooxygenation of an amino-thienopyridone yields a more potent PTP4A3 inhibitor. Org. Biomol. Chem. 14, 6398–6402 10.1039/c6ob00946h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lazo J. S., Blanco I. K., Tasker N. R., Rastelli E. J., Burnett J. C., Garrott S. R., Hart D. J., McCloud R. L., Hsu K. L., Wipf P., and Sharlow E. R. (2019) Next-generation cell-active inhibitors of the undrugged oncogenic PTP4A3 phosphatase. J. Pharmacol. Exp. Therap. 371, 652–662 10.1124/jpet.119.262188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bai Y., Yu Z. H., Liu S., Zhang L., Zhang R. Y., Zeng L. F., Zhang S., and Zhang Z. Y. (2016) Novel anticancer agents based on targeting the trimer interface of the PRL phosphatase. Cancer Res. 76, 4805–4815 10.1158/0008-5472.CAN-15-2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thura M., Al-Aidaroos A. Q., Gupta A., Chee C. E., Lee S. C., Hui K. M., Li J., Guan Y. K., Yong W. P., So J., Chng W. J., Ng C. H., Zhou J., Wang L. Z., Yuen J. S. P., et al. (2019) PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat. Commun. 10, 2484 10.1038/s41467-019-10127-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stanford S. M., Aleshin A. E., Zhang V., Ardecky R. J., Hedrick M. P., Zou J., Ganji S. R., Bliss M. R., Yamamoto F., Bobkov A. A., Kiselar J., Liu Y., Cadwell G. W., Khare S., Yu J., et al. (2017) Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol. 13, 624–632 10.1038/nchembio.2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xie L., Zhang Y. L., and Zhang Z. Y. (2002) Design and characterization of an improved protein tyrosine phosphatase substrate-trapping mutant. Biochemistry 41, 4032–4039 10.1021/bi015904r [DOI] [PubMed] [Google Scholar]
- 56. Chen M. J., Dixon J. E., and Manning G. (2017) Genomics and evolution of protein phosphatases. Sci. Signal. 10, eaag1796 10.1126/scisignal.aag1796 [DOI] [PubMed] [Google Scholar]
- 57. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 58. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 60. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors (codes 6WUR and 6WUS) have been deposited in the Protein Data Bank.