

Abstract
Metabolites are not only substrates in metabolic reactions, but also signaling molecules controlling a wide range of cellular processes. Discovery of the oncometabolite 2-hydroxyglutarate provides an important link between metabolic dysfunction and cancer, unveiling the signaling function of metabolites in regulating epigenetic and epitranscriptomic modifications, genome integrity, and signal transduction. It is now known that cancer cells remodel their metabolic network to support biogenesis, caused by or resulting in the dysregulation of various metabolites. Cancer cells can sense alterations in metabolic intermediates to better coordinate multiple biological processes and enhance cell metabolism. Recent studies have demonstrated that metabolite signaling is involved in the regulation of malignant transformation, cell proliferation, epithelial-to-mesenchymal transition, differentiation blockade, and cancer stemness. Additionally, intercellular metabolite signaling modulates inflammatory response and immunosurveillance in the tumor microenvironment. Here, we review recent advances in cancer-associated metabolite signaling. An in depth understanding of metabolite signaling will provide new opportunities for the development of therapeutic interventions that target cancer.
Keywords: tumor metabolism, signal transduction, cancer biology, tumor microenvironment, cancer stem cells, metabolite sensing, oncogenic signaling, 2HG, immunosurveillance, metabolic intermediate, cancer, metabolic regulation, signaling, metabolic disease, metabolomics, metabolite, oncometabolite, sensing
For decades, metabolites have been perceived as substrates of enzymes and/or regulators of their biosynthetic pathways (1–3). Recently, increasing evidence has suggested that metabolites are signaling molecules (4). Alterations in specific metabolites have been shown to modulate the activity of macromolecules. In this scenario, metabolites are emerging as signaling molecules that control biological processes, including epigenetic modification, signal transduction, and intercellular communication. Nutrients are digested into various intracellular and extracellular metabolites, which are produced via intermediary metabolism. Cells actively sense these metabolites to coordinate metabolic and nonmetabolic processes. These sensing and signaling mechanisms are core processes for the interaction between cellular metabolism and nonmetabolic processes. Notably, deregulation of metabolite signaling is implicated in numerous human diseases, including cancer. For example, 5′-adenosine monophosphate-activated protein kinase and mTOR complex 1 (mTORC1) are important energy/nutrient sensors that regulate energy production, protein synthesis, and autophagic processes to maintain metabolic homeostasis. Dysregulation of 5′-adenosine monophosphate-activated protein kinase and mTORC1 signaling leads to aberrant glucose and amino acids sensing in cancer cells, which have been reviewed elsewhere (5, 6).
Although metabolite signaling plays a fundamental role in interconnecting cellular metabolism with signaling events, the diversity of metabolites in their spatial structure and chemical properties precludes generic techniques that can be used to elucidate their biological function in cellular signaling. Consequently, the function of metabolites as signaling molecules has remained largely unexplored until recently. The discovery that the oncometabolite 2-hydroxyglutarate (2HG), generated by mutant isocitrate dehydrogenase 1 and 2 (IDH1/2), has functions beyond cell metabolism in cancer initiation and progression has placed renewed emphasis on this field. Besides 2HG, a growing number of metabolites have been shown to modulate various signaling pathways. Here, we review the recent progress in our understanding of how metabolite signaling interacts with diverse biological processes to regulate malignant transformation and remodel the tumor microenvironment.
Oncometabolite 2HG as a signaling molecule
To sustain malignant growth, cancer cells gauge nutrient availability to coordinate cellular metabolism. Deregulation of metabolic pathways, also known as metabolic reprogramming, is a key feature of cancer cells. Reprogrammed metabolic pathways reshape the cancer metabolome (i.e. the abundance of metabolites), which allows cancer cells to modulate oncogenic signaling with specific metabolites. The role of 2HG as a signaling molecule did not receive much attention until the discovery of cancer-driving mutations in IDHs. IDH1 and IDH2 mutations occur on specific residues in the catalytic center, conferring a new catalytic property to IDH, generating 2HG (7). 2HG is classified as an oncometabolite because mutated IDH1/2 can trigger malignant transformation, leading to the development of cancers such as myeloid leukemia, chondrosarcoma, and glioma (8). Additionally, 2HG can be generated by malate dehydrogenase, lactate dehydrogenase (LDH), and phosphoglycerate dehydrogenase through their catalytic promiscuity. 2-Hydroxyglutarate dehydrogenase counteracts these enzymes by clearing cellular 2HG at a low efficiency (9, 10).
2HG acts as a signaling metabolite that regulates a wide range of cellular processes (Fig. 1). It contributes to metabolic remodeling by inhibiting multiple metabolic enzymes, including ATP synthase (11), and by deregulating the tricarboxylic acid (TCA) cycle (12) in the mitochondria. In addition, 2HG disrupts redox metabolism, which is a key factor involved in tumor progression (1). 2HG acts as a structural analog of α-ketoglutarate (α-KG), an amino group acceptor in transamination reactions, resulting in suppression of branched-chain aminotransferase (BCAT) and impairment of GSH production to disrupt the redox balance in glioma (13). Furthermore, 2HG can also serve as a regulator of epigenetic and epitranscriptomic modifications. 2HG inhibits α-KG–dependent dioxygenases, including ten-eleven translocation (TET) enzymes, lysine demethylases (KDM), and fat mass and obesity–associated protein (FTO), leading to a global hypermethylation phenotype of DNA, histone, and RNA. Whereas transcriptional factor Wilms' tumor 1 (WT1) recruits TET2 to specific genetic loci to epigenetically direct myeloid differentiation, 2HG disrupts the TET2-WT1 signaling axis to promote the development of myeloid cancers (14, 15). In addition, 2HG suppresses histone demethylase KDM4C to enhance histone H3 Lys-9 methylation in glioma (16, 17). 2HG can also inhibit FTO activity to increase global N6-methyladenosine of RNA, further controlling the stability of mRNAs encoding myelocytomatosis oncogene (MYC)/CCAAT enhancer-binding protein α (CEBPA) transcripts (18).
Figure 1.
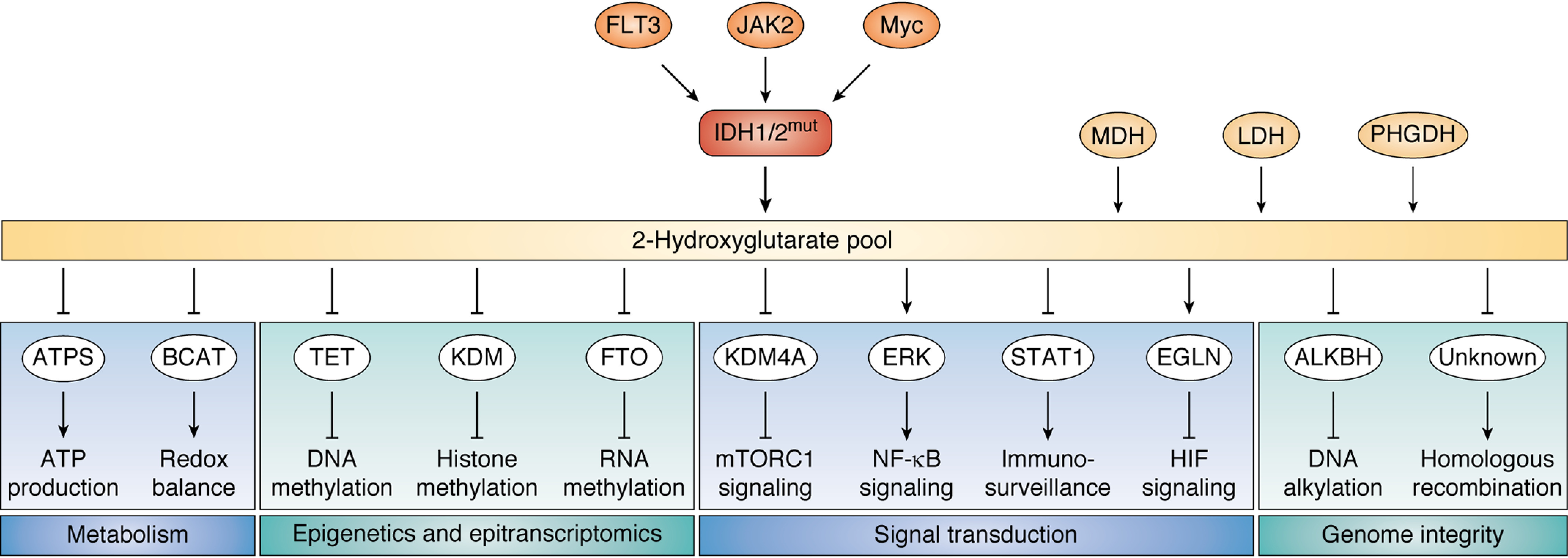
Oncometabolite 2-hydroxyglutarate-mediated signaling 2HG, produced by mutant isocitrate dehydrogenase 1/2 in cancer, modulates various cellular processes, including metabolic processes, epigenetic and epitranscriptomic modifications, signal transduction, and genome integrity maintenance. Tyrosine kinase FLT3 and JAK2, together with transcriptional factor Myc, regulates the 2HG-producing activity of mutant IDH. Malate dehydrogenase (MDH), lactate dehydrogenase (LDH), and phosphoglycerate dehydrogenase (PHGDH) are minor contributors of the 2HG pool. ATPS, ATP synthase; BCAT, branched-chain amino acids aminotransferase; TET, ten-eleven translocation methylcytosine dioxygenase; KDM, lysine-specific demethylase; FTO, fat mass and obesity-associated protein; KDM4A, lysine-specific demethylase 4A; ERK, extracellular signal–regulated kinase; STAT1, signal transducer and activator of transcription 1; EGLN, EGL-nine homolog enzyme; ALKBH, ALKB homolog enzymes.
2HG can also modulate oncogenic signaling pathways. After binding to KDM4A, 2HG destabilizes DEP domain–containing mTOR-interacting protein (DEPTOR) to activate mTORC1 signaling in brain tumors (11). By increasing the cellular reactive oxygen species level, 2HG also increases extracellular regulated protein kinase (ERK)-dependent phosphorylation of NF-κB, enhancing NF-κB's stability and transcriptional activity in bone marrow stromal cells. Specifically, 2HG signaling promotes the expression of cytokines, adhesion molecules, and cell-surface receptors that will provide stromal support for leukemia cell proliferation and chemoresistance (19). 2HG can also suppress signal transducer and activator of transcription 1 (STAT1) signaling and the production of CXC motif chemokine 10 (CXCL10), which in turn prevents T cell infiltration and suppresses immunosurveillance in glioma (20). Furthermore, 2HG activates EGL-nine (EGLN) prolyl hydroxylase, which mediates the degradation of hypoxia-inducible factor (HIF), thereby promoting the transformation of brain tumors expressing mutant IDH (21).
Finally, 2HG signaling leads to aberrant activity of the machinery that monitors genome integrity. 2HG suppresses ALKBH, a group of α-KG–dependent enzymes that remove alkylation damage (22). Interestingly, 2HG signaling also disrupts the activity of DNA repair enzymes through an unknown mechanism, resulting in deficient homologous recombination in glioma (23). Defective DNA repair in IDH-mutated cells opens a window for targeting glioma with DNA-damaging agents, as evidenced by the hypersensitivity of glioma cells to PARP inhibitor (23, 24). The direct targets of 2HG in homologous recombination remain poorly understood and potentially point to a new opportunity for sensitizing cancer cells to genotoxic agents. Notably, 2HG signaling converges with oncogenic signaling pathways to promote cancer progression. Oncogenes such as mutant FLT3, JAK2, and Myc up-regulate the 2HG-producing activity of mutant IDH to enhance 2HG signaling (25, 26), suggesting that inhibition of these cancer drivers, in collaboration with chemical inhibitors of mutated IDH, may be therapeutically beneficial.
Metabolic intermediates emerging as new players in oncogenic signaling
Cancer cells can sense and use the signals of a wide spectrum of metabolites to promote tumorigenesis and metastasis, including intermediate metabolites of central carbon metabolism, lipids, amino acids, and nucleotides (Fig. 2).
Figure 2.
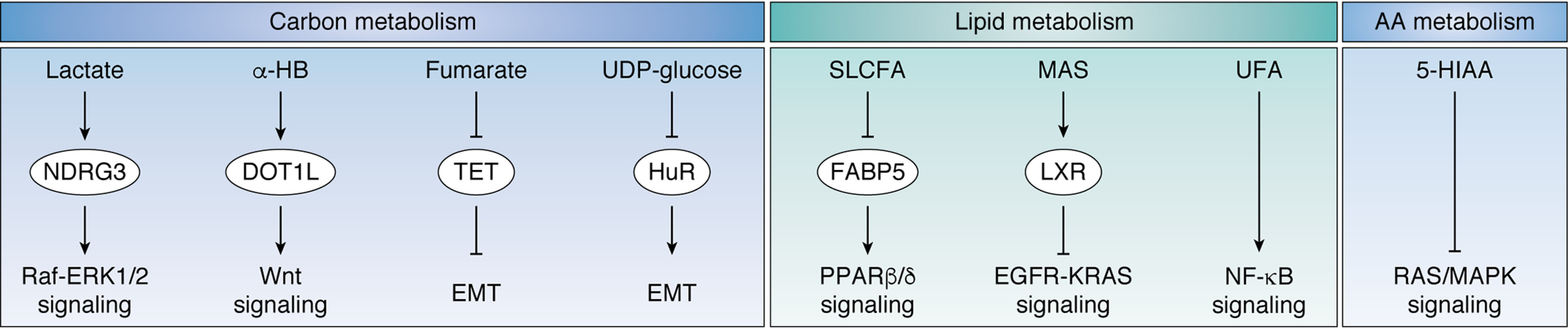
Metabolites modulate oncogenic signaling in cancer cells Metabolites from carbon metabolism, lipid metabolism, and amino acids metabolism modulate oncogenic signal transduction. Left, lactate binds to and stabilizes NDRG3 to enhance Raf-ERK1/2 signaling. α-HB increases the activity of DOT1L and enhances Wnt signaling by up-regulating histone methylation. Fumarate inhibits TET to increase genomic DNA methylation and promotes EMT. UDP-glucose suppresses Hu antigen R to inhibit EMT. Middle, SLCFA bind to FABP5 and suppresses PPAR β/δ signaling. MAS interact with LXR to inhibit EGFR-KRAS signaling. UFA suppress the transcriptional activity of NF-κB through an unknown mechanism. Right, 5-HIAA, a product in tryptophan catabolism, inhibits RAS/MAPK signaling. NDRG3, NDRG family member 3; α-HB, α-hydroxybutyrate; DOT1L, DOT1-like histone H3 methyltransferase; EMT, epithelial-mesenchymal transition; HuR, Hu antigen R; SLCFA, saturated long-chain fatty acids; FABP5, fatty acid-binding protein 5; MAS, meiosis-activating sterols; LXR, liver X receptor; UFA, unsaturated fatty acids; 5-HIAA, 5-hydroxyindoleacetic acid.
Signaling role of intermediates derived from central carbon metabolism
Central carbon metabolism consumes glucose and produces not only energy but also a variety of metabolic intermediates to satisfy the biosynthetic demands of cancer cells. Interestingly, these metabolic intermediates were found to be signaling molecules that control hypoxic signals and stress response. Specifically, glycolysis and the TCA cycle contribute to glucose degradation and ATP production (27), but they differ in oxygen dependence. Cells up-regulate glycolysis to enhance energy production when the surrounding environment lacks oxygen (28), whereas they activate the TCA cycle when oxygen is available. HIF1 signaling is a well-established oxygen-sensing pathway that enhances the transcription of glycolytic enzymes. Because the efficiency of transcription regulation is relatively slow, which requires synthesis of new mRNAs and proteins, cells have developed a rapid oxygen-sensing mechanism termed the lactate-induced hypoxic signal. Lactate accumulates when cells enter an anaerobic state, making lactate an indicator of oxygen supply. Cells sense the increase of lactate through N-Myc downstream-regulated 3 (NDRG3) protein and initiate a hypoxic response (29). Once bound to lactate, NDRG3 becomes more stable, resulting in the activation of Raf-ERK1/2 signaling and promotion of angiogenesis (29). In cancer cells, lactate accumulates even in the aerobic state, potentially leading to constitutive activation of lactate signaling. Interestingly, the enzyme responsible for lactate production, lactate dehydrogenase A (LDHA), is involved in a second mechanism by which metabolites impact signaling, although in this case its role relies on a noncanonical catalytic activity. Cancer cells are challenged by oxidative stress during transformation, in response to which LDHA translocates to the nucleus, where its noncanonical activity (α-ketobutyrate dehydrogenase) is increased. Nuclear LDHA produces α-hydroxybutyrate (α-HB) and epigenetically activates Wnt signaling by enhancing disruptor of telomeric silencing 1–like (DOT1L)-mediated histone H3 Lys-79 methylation. α-HB-DOT1L-Wnt signaling is critical in maintaining redox balance in human papillomavirus–induced cervical cancer (30). α-HB signaling potentially cooperates with Wnt/β-catenin signals to promote cell proliferation in several types of cancer.
The TCA cycle also produces a variety of signaling metabolites that may be of relevance to cancer biology (31, 32). Deletion of fumarate hydratase (FH), which is frequently observed in hereditary leiomyomatosis and renal cell cancer, leads to the accumulation of fumarate in human renal cancer cells. Fumarate is a structural analog to α-KG, and when in excess, it inhibits TET and suppresses demethylation of miR-200ba429, a metastasis repressor. Through this mechanism, fumarate up-regulates the expression of transcription factors that drive the epithelial-to-mesenchymal transition and promote cell transformation (33). Notably, a cell-permeable version of fumarate was shown to potentiate mitochondrial biogenesis (34), suggesting a link between fumarate signaling and cellular respiration.
Glucose is not only the fuel for energy production but also a precursor of the hexosamine pathway. UDP-glucose, a glucose derivative, is able to dissociate Hu antigen R from snail family transcriptional repressor 1 (SNAI1) mRNA and promote RNA decay. Lung cancer cells have been shown to overexpress UDP-glucose 6-dehydrogenase to oxidize UDP-glucose and stabilize SNAI1 mRNA, further supporting the processes of EMT and metastasis (35).
Cellular signaling of lipid synthesis and saturation
Lipids are critical sources of energy and the building blocks of plasma and intracellular membranes, but they have also been reported to regulate multiple signaling events. Cancer cells sense the abundance of long-chain fatty acids (LCFA) by fatty acid-binding protein 5 (FABP5). Interestingly, saturated and unsaturated LCFA release retinoic acid from FABP5 to activate retinoic acid–activated nuclear receptors. Specifically, saturated LCFA (SLCFA) bind FABP5 to down-regulate the transcriptional activity of peroxisome proliferator–activated receptor β/δ (PPAR β/δ) (36). Because PPAR β/δ plays a pro-proliferative role by enhancing the transcription of genes involved in cell growth and angiogenesis, diets enriched with SLCFA delay tumor growth in a mouse model of breast cancer (36).
Sterols are a specific subset of lipids, which can activate meiosis. Meiosis-activating sterols (MAS) can be sensed by the liver X receptor (LXR), which in turn will promote the transcription of the ABC transporters, including ABCA1 and ABCG1. This process results in the activation of the cholesterol pump and the reduction of intracellular cholesterol. Additionally, MAS can suppress the low-density lipoprotein receptor to inhibit cholesterol uptake. Therefore, LXR-mediated MAS signaling reduces cholesterol supply and suppresses malignant transformation driven by epidermal growth factor receptor (EGFR) and Kirsten rat sarcoma viral oncogene homolog (KRAS) in skin tumor cells (37).
The fluidity of cellular membranes may be a metabolic determinant of cell identity and is modulated by factors such as the saturation degree of lipids. This feature allows cells to be differentiated into different subsets. Cancer stem cells (CSCs) are thought to be the root of metastasis and relapse. These cells share metabolic features similar to those of normal stem cells; still, a better understanding of the metabolic determinants of stemness can help uncover metabolite signaling pathways that sustain CSCs (38–40). Of note, ovarian cancer stem cells have significantly higher levels of unsaturated lipids than non-CSCs, and the degree of unsaturation is closely linked to the stemness of these cells. Importantly, inhibition of desaturases efficiently blocks NF-κB signaling and suppresses tumor-initiating activity of ovarian CSCs (41). Therefore, desaturase may serve as a vital target in eradicating CSCs. Cholesterol is another structural component of membranes that controls membrane fluidity. Cholesterol metabolism is closely linked to intestinal stem cell proliferation. Lysophosphatidylcholine acyltransferase 3 (LPCAT3), a phospholipid remodeling protein, negatively regulates cholesterol production. The deletion of Lpcat3 induces accumulation of cholesterol and confers high proliferative capacity to intestinal stem cells, resulting in enhanced formation of intestinal tumors (42).
Cancer signaling of amino acids metabolism
Cancer cells consume high amounts of amino acids to sustain the fast turnover and need for proteins. Specifically, tumor cells have an increased requirement for glutamine, leading to glutamine depletion in the surrounding microenvironment. As a consequence, cancer cells sense glutamine insufficiency and seek alternative sources of amino acids during glutamine shortage. Pancreatic cancer cells sense glutamine insufficiency and activate EGFR and p21-activated kinase signaling to uptake extracellular proteins through macropinocytosis and support pancreatic cancer metabolism (43).
The intermediates of amino acids metabolism have been shown to intersect with cellular signaling. RAS/MAPK signaling plays a key role in promoting cell proliferation and survival. Activation of RAS/MAPK signaling has been frequently observed in various types of cancer. Interestingly, 5-hydroxyindoleacetic acid (5-HIAA), a product in tryptophan degradation, potentially interacts with membrane-bound receptors and effectively suppresses RAS/MAPK signaling (44). Therefore, 5-HIAA metabolism could potentially be targeted to suppress MAPK signaling and tumor growth. Additionally, dysfunction of the methionine salvage pathway has a signaling role in tumorigenesis. The methylthioadenosine phosphorylase (MTAP) gene from methionine metabolism is absent in certain cancers, such as glioblastoma. MTAP deficiency results in the accumulation of 5-methylthioadenosine (MTA). This nucleoside shares structural similarities with SAM, which is a methyl donor in methylation reactions. MTA inhibits protein arginine methyltransferase 5 (PRMT5), likely by occupying its SAM-binding domain, resulting in decreased methyltransferase activity and hypomethylation of downstream proteins (45, 46). Although the downstream biological events remain unclear, treatment with MTA decreases the viability of MTAP-deleted cancer cells.
Amino acids are also regulators of cancer stem cells. For example, branched-chain amino acids (BCAAs), which are essential for cell growth, seem to have an intrinsic link with both stem cell fate decision and cancer stemness. Low valine to isoleucine/leucine ratio, or BCAA imbalance, greatly slows the formation of hematopoietic stem cells (47). Importantly, branched-chain aminotransferase 1 (BCAT1), which is the cytosolic enzyme responsible for BCAA degradation, shows higher expression in leukemic stem cells. BCAT1 potentially links BCAAs abundance to differentiation programs and maintains the undifferentiated state of leukemia cells, as inhibition of BCAT1 promotes the differentiation of myeloid leukemia cells (48). BCAT1 transfers an amino group to α-KG and decreases intracellular α-KG level. Consequently, the activity of α-KG–dependent TET enzymes is suppressed to induce genomic DNA hypermethylation (49). Of note, BCAT2, the mitochondrial paralogue of BCAT1, is essential for the progression of pancreatic cancer (50). Nevertheless, the downstream targets of BCAA signaling and how BCAAs regulate cancer progression remain to be understood.
Nucleotide metabolism involved in cell fate decision
Cancer cells fulfill their high demands of DNA, RNA, and consequently nucleotides, at least in part, by up-regulating the de novo synthesis of ribose, purines, and pyrimidines. Pyrimidine can act as a signaling molecule that controls cell differentiation. In the case of acute myeloid leukemia (AML), leukemic cells are maintained at an undifferentiated state due to a differentiation barrier (51). High-throughput screenings have identified dihydroorotate dehydrogenase, an enzyme involved in de novo pyrimidine synthesis, as being responsible for this differentiation block. Because dihydroorotate dehydrogenase is a key enzyme in uridine synthesis, uridine metabolites may have a signaling function to control cell fate transition in AML. Elucidation of the uridine signaling pathway may help us to understand the metabolic basis of the differentiation barrier in AML (38).
Metabolite signaling in the tumor microenvironment
The tumor microenvironment is composed of cancer cells and cancer-associated fibroblasts as well as immune and pro-inflammatory cytokines (52). To sustain rapid proliferation, tumor cells compete for nutrients present in intercellular fluids and secrete metabolic waste to reshape the metabolic composition of the extracellular environment. Interestingly, the activity of tumor-associated cells is modulated by cancer metabolism to create an environment supportive of cancer progression (Fig. 3).
Figure 3.

Metabolite signaling in the tumor microenvironment Metabolite signaling in cancer-associated cells within the microenvironment. Lactate directly binds to MAVS to suppress RLR signaling and weakens cancer immunosurveillance. Kynurenine metabolism is activated in tumor-associating immune cells. Kynurenine binds to either transcription factor AHR or the cell-surface receptor GPR35 to mediate inflammatory signaling. In Th1 cells, glutamine-derived α-KG increases the expression of Tbet, through an unknown mechanism, and promotes Th1 cell differentiation. Bile acid triggers the release of CXCL16, which further acts on endothelial cells and NK cells to enhance antitumor immunosurveillance. AHR, aryl hydrocarbon receptor; GPR35, G protein–coupled receptor 35; Tbet, T-box transcription factor.
Glucose metabolism linked to cancer immunosurveillance
Tumor cells consume large amounts of glucose and deprive the tumor-infiltrating immune cells of glucose. As a result, glycolysis in these tumor-infiltrating T cells is disrupted, and phosphoenolpyruvate, a glycolytic intermediate, accumulates. Phosphoenolpyruvate is known to repress the activity of sarco-/endoplamic reticulum Ca2+-ATPase (SERCA), which pumps calcium from the cytoplasm into the endoplasmic reticulum, controlling cytoplasmic Ca2+ levels. A decrease in cytoplasmic Ca2+ deactivates the transcription factor nuclear factor of activated T cells (NFAT), to down-regulate the expression of target genes and suppress T cell immune response (53). This in turn weakens the effector function of CD4/CD8 T cells, which have tumor-eliminating effects. As such, glucose metabolism seems to be tightly linked to Ca2+ signaling within the tumor tissue. Furthermore, the glycolytic nature of most tumor cells results in lactate accumulation and acidification of the microenvironment. Lactate is a signaling metabolite that controls retinoic acid–inducible gene I–like receptor (RLR) signaling, which supports type I interferon production. Antiviral response RLR signaling is well-known to support type I interferon production and antiviral response. Importantly, RLR signaling is also involved in cancer immune surveillance. Lactate directly binds to the mitochondrial antiviral signaling protein (MAVS) and prevents its interaction with RLR. The consequent suppression of RLR signaling weakens immune surveillance and promotes cancer progression (54).
Amino acids metabolism implicated in tumor inflammation and T cell differentiation
Amino acids are important regulators of inflammation and anti-tumor response. Tryptophan catabolism generates kynurenine, a key signaling metabolite in tumor inflammation. Kynurenine can be sensed by both the aryl hydrocarbon receptor (AHR) and G protein–coupled receptor 35 (GPR35), which will suppress immunosurveillance. Kynurenine-AHR/GPR35 signaling plays a regulatory role in chronic inflammation and colonic tumorigenesis (55, 56).
T cells can differentiate into effector T cells that promote immune response against tumors, such as T helper type 1 (Th1) cells that are involved in the destruction of tumor cells, or into regulatory T cells that mediate immunosuppression. This cell differentiation process can be modulated by tumor metabolism. For instance, glutamine metabolism is a metabolic determinant of T cell differentiation as its product, α-KG, can direct T cell differentiation. Additionally, upon cytokine activation, naive CD4+ T cells can differentiate into Th1 cells in the presence of glutamine or into T regulatory (Treg) cells in the absence of this amino acid. α-KG increases the mRNA level of the transcription factor Tbet, which governs Th1 differentiation (57). Determination of whether α-KG directly binds to Tbet or epigenetically modulates Tbet expression requires further exploration. Furthermore, α-KG enhances mTORC1 signaling to direct naive T cells toward Th1 cell differentiation. Overall, cancer cells compete for glutamine present in the tumor microenvironment, a process that causes glutamine shortage for T cells. The consequent Treg phenotype will ultimately contribute to reduced immunosurveillance (58).
Intercellular signaling of fatty acids and cholesterol metabolism in the tumor microenvironment
Fatty acids and cholesterol are also involved in the regulation of inflammatory and immune responses. Exosomes serve as key vehicles connecting tumor cells with their adjacent cells. Notably, cancer-associated fibroblasts are important cellular components of the tumor microenvironment that are responsible for matrix deposition and signaling. Exosomes secreted by cancer-associated fibroblasts transport a wide spectrum of nutrients, such as amino acids, lipids, and TCA cycle metabolites, to fuel cancer cells during nutrient deprivation (59).
Exosome-mediated metabolite signaling is also involved in the development of nonalcoholic steatohepatitis (NASH), which is a high-risk factor for liver cancer. Clinically, NASH is featured by liver cell death and inflammation. Liver cells (hepatocytes) show enhanced release of extracellular vesicles when treated with high levels of palmitate and lysophosphatidylcholine (60). Interestingly, the secreted exosomes express ligands that can induce tumor necrosis factor signaling and hepatocyte cell death. Moreover, hepatocyte-derived exosomes interact with macrophages to up-regulate the expression of interleukin-1β and interleukin-6 and enhance hepatic inflammation (60). Because NASH potentially progresses to liver cancer, enhanced release of exosomes is potentially involved in the remodeling of the liver cancer microenvironment.
Interestingly, bile acids are also regulatory metabolites in the tumor microenvironment. Human liver cells produce bile acids as the end metabolites of cholesterol metabolism. The link between bile acids and colon cancer has been reported previously (61), with the signaling function of bile acids being intensively investigated in energy metabolism, inflammation, and cancer. Bile acids have been shown to act on receptors from both the nucleus and cell surface. Bile acids bind to farnesoid X receptor, vitamin D receptor, and G protein–coupled bile acid receptors, which further regulate lipid metabolism and energy production by altering the expression or activity of corresponding metabolic enzymes (62). In contrast, bile acids exert anti-tumor effects in liver cancer. In hepatic sinusoidal endothelial cells, bile acids trigger the release of chemokine CXCL16 to enhance anti-tumor immunosurveillance by increasing hepatic CXCR6+ natural killer cells (63).
Signaling of microbiota-derived metabolites
The microbiota is a rich source of metabolites within the human body and is also a vital regulator of metabolite signaling. Metabolites generated by microbiota are important signaling molecules in both normal cells and cancer cells in the colon. Several microbiota-produced metabolites, including taurine, histamine, and spermine, act as signaling molecules to suppress the NLRP6 inflammasome and inflammation-induced colorectal cancer (64). Specifically, indole is an important signaling metabolite in colon cancer. Indole associates with AHR in the gastrointestinal tract and promotes its nuclear translocation. The subsequent up-regulation of interleukin-6 transcription in human colon adenocarcinoma cells further promotes tumor inflammation (65).
Concluding remarks
Metabolite signaling is proving to be quite a fascinating topic, not only contributing to metabolic remodeling of cancer but also connecting cellular metabolism with signaling networks. While the signaling nature of more metabolites is being uncovered, how cells sense these metabolites and their physiological roles are being explored in depth. It is important to note that the abundance of metabolites is highly dynamic and largely depends on nutrient status and cell state. Metabolomic profiling of cancer cells under different nutritional status, or cancer cells from different stages, may help with the discovery of new signaling roles of metabolites. Moreover, metabolites are known to exist in a compartmentalized manner within cells (66). Therefore, the same metabolite may have distinct signaling roles in different cellular compartments.
Although we have gained rich knowledge of intracellular metabolite signaling, this process between different cell types and different organs remains largely unexplored. Moreover, tumor cells are known to induce systemic changes to promote cancer growth and metastasis. A clear understanding of intercellular and interorgan metabolite signaling would also help to elucidate complications in a tumor-bearing context, such as cachexia (extreme weight loss and muscle wasting).
The development of new tools or strategies would help to advance our current understanding of metabolite signaling in cancer. The signaling role of a metabolite is largely dependent on its interaction with macromolecules. Unfortunately, tools for high-throughput discovery of metabolite-binding proteins are still lacking, such as platforms that could allow the screening and discovery of metabolites acting on key signaling pathways in cancer.
Most importantly, therapeutic targeting of metabolite signaling holds great promise for clinical intervention in cancer. For example, chemical inhibitors of mutant IDH have been successful in the treatment of IDH-mutated leukemia, and we anticipate that ongoing discovery of functional roles of metabolites and their binding sites will provide other opportunities for the development of new targeted therapies of cancer.
Acknowledgments
We thank members of the Lei laboratory for discussion throughout this study.
Funding and additional information—This work was supported by MOST Grant 2019YFA0801703 (to Q.-Y. L.); Natural Science Foundation of China Grants 81790250, 81790253, and 91959202 (to Q.-Y. L.), 81872240 (to M. Y.), 81790251 and 81772946 (to Y.-P. W.), and 81802745 (to J. Q.); Innovation Program of the Shanghai Municipal Education Commission Grant N173606 (to Q.-Y. L.); and CAST Young Elite Scientist Sponsorship Program Grant 2018QNRC001 (to Y.-P. W.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- mTOR
- mechanistic target of rapamycin
- mTORC1
- mTOR complex 1
- BCAT
- branched-chain amino acids aminotransferase
- TET
- ten-eleven translocation methylcytosine dioxygenase
- KDM
- lysine-specific demethylase
- FTO
- fat mass and obesity–associated protein
- 2HG
- 2-hydroxyglutarate
- IDH
- isocitrate dehydrogenase
- TCA
- tricarboxylic acid
- α-KG
- α-ketoglutarate
- WT1
- Wilms' tumor 1
- HIF
- hypoxia-inducible factor
- LDHA
- lactate dehydrogenase A
- α-HB
- α-hydroxybutyrate
- DOT1L
- disruptor of telomeric silencing 1–like
- LCFA
- long-chain fatty acids
- SLCFA
- saturated LCFA
- PPAR
- peroxisome proliferator–activated receptor
- MAS
- meiosis-activating sterols
- LXR
- liver X receptor
- EGFR
- epidermal growth factor receptor
- KRAS
- Kirsten rat sarcoma viral oncogene homolog
- CSC
- cancer stem cell
- MAPK
- mitogen-activated protein kinase
- 5-HIAA
- 5-hydroxyindoleacetic acid
- BCAA
- branched-chain amino acids
- AML
- acute myeloid leukemia
- RLR
- retinoic acid–inducible gene I–like receptor
- AHR
- aryl hydrocarbon receptor
- Th1
- T helper type 1
- Treg
- T regulatory
- NASH
- nonalcoholic steatohepatitis.
References
- 1. Wang K., Jiang J., Lei Y., Zhou S., Wei Y., and Huang C. (2019) Targeting metabolic-redox circuits for cancer therapy. Trends Biochem. Sci. 44, 401–414 10.1016/j.tibs.2019.01.001 [DOI] [PubMed] [Google Scholar]
- 2. Wang Y., Bai C., Ruan Y., Liu M., Chu Q., Qiu L., Yang C., and Li B. (2019) Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 10, 201 10.1038/s41467-018-08033-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wegner A., Meiser J., Weindl D., and Hiller K. (2015) How metabolites modulate metabolic flux. Curr. Opin. Biotechnol. 34, 16–22 10.1016/j.copbio.2014.11.008 [DOI] [PubMed] [Google Scholar]
- 4. Wang Y. P., and Lei Q. Y. (2018) Metabolite sensing and signaling in cell metabolism. Signal Transduct. Target. Ther. 3, 30 10.1038/s41392-018-0024-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. González A., and Hall M. N. (2017) Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 36, 397–408 10.15252/embj.201696010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garcia D., and Shaw R. J. (2017) AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800 10.1016/j.molcel.2017.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Waitkus M. S., Diplas B. H., and Yan H. (2018) Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 34, 186–195 10.1016/j.ccell.2018.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fan J., Teng X., Liu L., Mattaini K. R., Looper R. E., Vander Heiden M. G., and Rabinowitz J. D. (2015) Human phosphoglycerate dehydrogenase produces the oncometabolite d-2-hydroxyglutarate. ACS Chem. Biol. 10, 510–516 10.1021/cb500683c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Intlekofer A. M., Dematteo R. G., Venneti S., Finley L. W., Lu C., Judkins A. R., Rustenburg A. S., Grinaway P. B., Chodera J. D., Cross J. R., and Thompson C. B. (2015) Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab. 22, 304–311 10.1016/j.cmet.2015.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fu X., Chin R. M., Vergnes L., Hwang H., Deng G., Xing Y., Pai M. Y., Li S., Ta L., Fazlollahi F., Chen C., Prins R. M., Teitell M. A., Nathanson D. A., Lai A., et al. (2015) 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 22, 508–515 10.1016/j.cmet.2015.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reitman Z. J., Jin G., Karoly E. D., Spasojevic I., Yang J., Kinzler K. W., He Y., Bigner D. D., Vogelstein B., and Yan H. (2011) Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. U. S. A. 108, 3270–3275 10.1073/pnas.1019393108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McBrayer S. K., Mayers J. R., DiNatale G. J., Shi D. D., Khanal J., Chakraborty A. A., Sarosiek K. A., Briggs K. J., Robbins A. K., Sewastianik T., Shareef S. J., Olenchock B. A., Parker S. J., Tateishi K., Spinelli J. B., et al. (2018) Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175, 101–116.e25 10.1016/j.cell.2018.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Figueroa M. E., Abdel-Wahab O., Lu C., Ward P. S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H. F., Tallman M. S., Sun Z., Wolniak K., Peeters J. K., Liu W., et al. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 10.1016/j.ccr.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y., Xiao M., Chen X., Chen L., Xu Y., Lv L., Wang P., Yang H., Ma S., Lin H., Jiao B., Ren R., Ye D., Guan K. L., and Xiong Y. (2015) WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 57, 662–673 10.1016/j.molcel.2014.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu C., Ward P. S., Kapoor G. S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C. R., Khanin R., Figueroa M. E., Melnick A., Wellen K. E., O'Rourke D. M., Berger S. L., Chan T. A., Levine R. L., et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chowdhury R., Yeoh K. K., Tian Y. M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K., Li X. S., Woon E. C., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D., et al. (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 10.1038/embor.2011.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Su R., Dong L., Li C., Nachtergaele S., Wunderlich M., Qing Y., Deng X., Wang Y., Weng X., Hu C., Yu M., Skibbe J., Dai Q., Zou D., Wu T., et al. (2018) R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172, 90–105.e123 10.1016/j.cell.2017.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J. Y., Lai Y. S., Tsai H. J., Kuo C. C., Yen B. L., Yeh S. P., Sun H. S., and Hung W. C. (2016) The oncometabolite R-2-hydroxyglutarate activates NF-κB–dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep. 6, 32428 10.1038/srep32428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kohanbash G., Carrera D. A., Shrivastav S., Ahn B. J., Jahan N., Mazor T., Chheda Z. S., Downey K. M., Watchmaker P. B., Beppler C., Warta R., Amankulor N. A., Herold-Mende C., Costello J. F., and Okada H. (2017) Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Invest. 127, 1425–1437 10.1172/JCI90644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koivunen P., Lee S., Duncan C. G., Lopez G., Lu G., Ramkissoon S., Losman J. A., Joensuu P., Bergmann U., Gross S., Travins J., Weiss S., Looper R., Ligon K. L., Verhaak R. G., et al. (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 10.1038/nature10898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen F., Bian K., Tang Q., Fedeles B. I., Singh V., Humulock Z. T., Essigmann J. M., and Li D. (2017) Oncometabolites d- and l-2-hydroxyglutarate inhibit the AlkB family DNA repair enzymes under physiological conditions. Chem. Res. Toxicol. 30, 1102–1110 10.1021/acs.chemrestox.7b00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sulkowski P. L., Corso C. D., Robinson N. D., Scanlon S. E., Purshouse K. R., Bai H., Liu Y., Sundaram R. K., Hegan D. C., Fons N. R., Breuer G. A., Song Y., Mishra-Gorur K., De Feyter H. M., de Graaf R. A., et al. (2017) 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 9, eaal2463 10.1126/scitranslmed.aal2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang P., Wu J., Ma S., Zhang L., Yao J., Hoadley K. A., Wilkerson M. D., Perou C. M., Guan K. L., Ye D., and Xiong Y. (2015) Oncometabolite d-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 13, 2353–2361 10.1016/j.celrep.2015.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen D., Xia S., Wang M., Lin R., Li Y., Mao H., Aguiar M., Famulare C. A., Shih A. H., Brennan C. W., Gao X., Pan Y., Liu S., Fan J., Jin L., et al. (2019) Mutant and wild-type isocitrate dehydrogenase 1 share enhancing mechanisms involving distinct tyrosine kinase cascades in cancer. Cancer Discov. 9, 756–777 10.1158/2159-8290.CD-18-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Terunuma A., Putluri N., Mishra P., Mathé E. A., Dorsey T. H., Yi M., Wallace T. A., Issaq H. J., Zhou M., Killian J. K., Stevenson H. S., Karoly E. D., Chan K., Samanta S., Prieto D., et al. (2014) MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 124, 398–412 10.1172/JCI71180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhong X. Y., Yuan X. M., Xu Y. Y., Yin M., Yan W. W., Zou S. W., Wei L. M., Lu H. J., Wang Y. P., and Lei Q. Y. (2018) CARM1 methylates GAPDH to regulate glucose metabolism and is suppressed in liver cancer. Cell Rep. 24, 3207–3223 10.1016/j.celrep.2018.08.066 [DOI] [PubMed] [Google Scholar]
- 28. Gao X., Lin S. H., Ren F., Li J. T., Chen J. J., Yao C. B., Yang H. B., Jiang S. X., Yan G. Q., Wang D., Wang Y., Liu Y., Cai Z., Xu Y. Y., Chen J., et al. (2016) Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 7, 11960 10.1038/ncomms11960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee D. C., Sohn H. A., Park Z. Y., Oh S., Kang Y. K., Lee K. M., Kang M., Jang Y. J., Yang S. J., Hong Y. K., Noh H., Kim J. A., Kim D. J., Bae K. H., Kim D. M., et al. (2015) A lactate-induced response to hypoxia. Cell 161, 595–609 10.1016/j.cell.2015.03.011 [DOI] [PubMed] [Google Scholar]
- 30. Liu Y., Guo J. Z., Liu Y., Wang K., Ding W., Wang H., Liu X., Zhou S., Lu X. C., Yang H. B., Xu C., Gao W., Zhou L., Wang Y. P., Hu W., et al. (2018) Nuclear lactate dehydrogenase A senses ROS to produce α-hydroxybutyrate for HPV-induced cervical tumor growth. Nat. Commun. 9, 4429 10.1038/s41467-018-06841-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan W. W., Liang Y. L., Zhang Q. X., Wang D., Lei M. Z., Qu J., He X. H., Lei Q. Y., and Wang Y. P. (2018) Arginine methylation of SIRT7 couples glucose sensing with mitochondria biogenesis. EMBO Rep. 19, e46377 10.15252/embr.201846377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang T., Cao Y., Zheng Q., Tu J., Zhou W., He J., Zhong J., Chen Y., Wang J., Cai R., Zuo Y., Wei B., Fan Q., Yang J., Wu Y., et al. (2019) SENP1-Sirt3 signaling controls mitochondrial protein acetylation and metabolism. Mol. Cell 75, 823–834.e5 10.1016/j.molcel.2019.06.008 [DOI] [PubMed] [Google Scholar]
- 33. Sciacovelli M., Goncalves E., Johnson T. I., Zecchini V. R., da Costa A. S., Gaude E., Drubbel A. V., Theobald S. J., Abbo S. R., Tran M. G., Rajeeve V., Cardaci S., Foster S., Yun H., Cutillas P., et al. (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 10.1038/nature19353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hayashi G., Jasoliya M., Sahdeo S., Saccà F., Pane C., Filla A., Marsili A., Puorro G., Lanzillo R., Brescia Morra V., and Cortopassi G. (2017) Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 26, 2864–2873 10.1093/hmg/ddx167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang X., Liu R., Zhu W., Chu H., Yu H., Wei P., Wu X., Zhu H., Gao H., Liang J., Li G., and Yang W. (2019) UDP-glucose accelerates SNAI1 mRNA decay and impairs lung cancer metastasis. Nature 571, 127–131 10.1038/s41586-019-1340-y [DOI] [PubMed] [Google Scholar]
- 36. Levi L., Wang Z., Doud M. K., Hazen S. L., and Noy N. (2015) Saturated fatty acids regulate retinoic acid signalling and suppress tumorigenesis by targeting fatty acid-binding protein 5. Nat. Commun. 6, 8794 10.1038/ncomms9794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gabitova L., Restifo D., Gorin A., Manocha K., Handorf E., Yang D. H., Cai K. Q., Klein-Szanto A. J., Cunningham D., Kratz L. E., Herman G. E., Golemis E. A., and Astsaturov I. (2015) Endogenous sterol metabolites regulate growth of EGFR/KRAS-dependent tumors via LXR. Cell Rep. 12, 1927–1938 10.1016/j.celrep.2015.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sykes D. B., Kfoury Y. S., Mercier F. E., Wawer M. J., Law J. M., Haynes M. K., Lewis T. A., Schajnovitz A., Jain E., Lee D., Meyer H., Pierce K. A., Tolliday N. J., Waller A., Ferrara S. J., et al. (2016) Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 167, 171–186.e15 10.1016/j.cell.2016.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kalaitzidis D., Lee D., Efeyan A., Kfoury Y., Nayyar N., Sykes D. B., Mercier F. E., Papazian A., Baryawno N., Victora G. D., Neuberg D., Sabatini D. M., and Scadden D. T. (2017) Amino acid-insensitive mTORC1 regulation enables nutritional stress resilience in hematopoietic stem cells. J. Clin. Invest. 127, 1405–1413 10.1172/JCI89452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Christian S., Merz C., Evans L., Gradl S., Seidel H., Friberg A., Eheim A., Lejeune P., Brzezinka K., Zimmermann K., Ferrara S., Meyer H., Lesche R., Stoeckigt D., Bauser M., et al. (2019) The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia 33, 2403–2415 10.1038/s41375-019-0461-5 [DOI] [PubMed] [Google Scholar]
- 41. Li J., Condello S., Thomes-Pepin J., Ma X., Xia Y., Hurley T. D., Matei D., and Cheng J. X. (2017) Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 20, 303–314.e5 10.1016/j.stem.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang B., Rong X., Palladino E. N. D., Wang J., Fogelman A. M., Martin M. G., Alrefai W. A., Ford D. A., and Tontonoz P. (2018) Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22, 206–220.e4 10.1016/j.stem.2017.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee S. W., Zhang Y., Jung M., Cruz N., Alas B., and Commisso C. (2019) EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev. Cell 50, 381–392.e5 10.1016/j.devcel.2019.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmid T., Snoek L. B., Fröhli E., van der Bent M. L., Kammenga J., and Hajnal A. (2015) Systemic regulation of RAS/MAPK signaling by the serotonin metabolite 5-HIAA. PLoS Genet. 11, e1005236 10.1371/journal.pgen.1005236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mavrakis K. J., McDonald E. R. 3rd, Schlabach M. R., Billy E., Hoffman G. R., deWeck A., Ruddy D. A., Venkatesan K., Yu J., McAllister G., Stump M., deBeaumont R., Ho S., Yue Y., Liu Y., et al. (2016) Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213 10.1126/science.aad5944 [DOI] [PubMed] [Google Scholar]
- 46. Kryukov G. V., Wilson F. H., Ruth J. R., Paulk J., Tsherniak A., Marlow S. E., Vazquez F., Weir B. A., Fitzgerald M. E., Tanaka M., Bielski C. M., Scott J. M., Dennis C., Cowley G. S., Boehm J. S., et al. (2016) MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218 10.1126/science.aad5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilkinson A. C., Morita M., Nakauchi H., and Yamazaki S. (2018) Branched-chain amino acid depletion conditions bone marrow for hematopoietic stem cell transplantation avoiding amino acid imbalance-associated toxicity. Exp. Hematol. 63, 12–16.e1 10.1016/j.exphem.2018.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hattori A., Tsunoda M., Konuma T., Kobayashi M., Nagy T., Glushka J., Tayyari F., McSkimming D., Kannan N., Tojo A., Edison A. S., and Ito T. (2017) Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 545, 500–504 10.1038/nature22314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Raffel S., Falcone M., Kneisel N., Hansson J., Wang W., Lutz C., Bullinger L., Poschet G., Nonnenmacher Y., Barnert A., Bahr C., Zeisberger P., Przybylla A., Sohn M., Tönjes M., et al. (2017) BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388 10.1038/nature24294 [DOI] [PubMed] [Google Scholar]
- 50. Li J. T., Yin M., Wang D., Wang J., Lei M. Z., Zhang Y., Liu Y., Zhang L., Zou S. W., Hu L. P., Zhang Z. G., Wang Y. P., Wen W. Y., Lu H. J., Chen Z. J., et al. (2020) BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 22, 167–174 10.1038/s41556-019-0455-6 [DOI] [PubMed] [Google Scholar]
- 51. Baryawno N., Przybylski D., Kowalczyk M. S., Kfoury Y., Severe N., Gustafsson K., Kokkaliaris K. D., Mercier F., Tabaka M., Hofree M., Dionne D., Papazian A., Lee D., Ashenberg O., Subramanian A., et al. (2019) A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell 177, 1915–1932.e16 10.1016/j.cell.2019.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang D., Wang Y., Shi Z., Liu J., Sun P., Hou X., Zhang J., Zhao S., Zhou B. P., and Mi J. (2015) Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 10, 1335–1348 10.1016/j.celrep.2015.02.006 [DOI] [PubMed] [Google Scholar]
- 53. Ho P. C., Bihuniak J. D., Macintyre A. N., Staron M., Liu X., Amezquita R., Tsui Y. C., Cui G., Micevic G., Perales J. C., Kleinstein S. H., Abel E. D., Insogna K. L., Feske S., Locasale J. W., et al. (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 10.1016/j.cell.2015.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang W., Wang G., Xu Z. G., Tu H., Hu F., Dai J., Chang Y., Chen Y., Lu Y., Zeng H., Cai Z., Han F., Xu C., Jin G., Sun L., et al. (2019) Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell 178, 176–189.e15 10.1016/j.cell.2019.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Opitz C. A., Litzenburger U. M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M., Jugold M., Guillemin G. J., Miller C. L., Lutz C., Radlwimmer B., et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 10.1038/nature10491 [DOI] [PubMed] [Google Scholar]
- 56. Cervenka I., Agudelo L. Z., and Ruas J. L. (2017) Kynurenines: tryptophan's metabolites in exercise, inflammation, and mental health. Science 357, eaaf9794 10.1126/science.aaf9794 [DOI] [PubMed] [Google Scholar]
- 57. Klysz D., Tai X., Robert P. A., Craveiro M., Cretenet G., Oburoglu L., Mongellaz C., Floess S., Fritz V., Matias M. I., Yong C., Surh N., Marie J. C., Huehn J., Zimmermann V., et al. (2015) Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 8, ra97 10.1126/scisignal.aab2610 [DOI] [PubMed] [Google Scholar]
- 58. Johnson M. O., Wolf M. M., Madden M. Z., Andrejeva G., Sugiura A., Contreras D. C., Maseda D., Liberti M. V., Paz K., Kishton R. J., Johnson M. E., de Cubas A. A., Wu P., Li G., Zhang Y., et al. (2018) Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795.e19 10.1016/j.cell.2018.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhao H., Yang L., Baddour J., Achreja A., Bernard V., Moss T., Marini J. C., Tudawe T., Seviour E. G., San Lucas F. A., Alvarez H., Gupta S., Maiti S. N., Cooper L., Peehl D., et al. (2016) Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 5, e10250 10.7554/eLife.10250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hirsova P., Ibrahim S. H., Krishnan A., Verma V. K., Bronk S. F., Werneburg N. W., Charlton M. R., Shah V. H., Malhi H., and Gores G. J. (2016) Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 150, 956–967 10.1053/j.gastro.2015.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reddy B. S., and Wynder E. L. (1977) Metabolic epidemiology of colon cancer: fecal bile acids and neutral sterols in colon cancer patients and patients with adenomatous polyps. Cancer 39, 2533–2539 [DOI] [PubMed] [Google Scholar]
- 62. Li T., and Chiang J. Y. (2014) Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 66, 948–983 10.1124/pr.113.008201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ma C., Han M., Heinrich B., Fu Q., Zhang Q., Sandhu M., Agdashian D., Terabe M., Berzofsky J. A., Fako V., Ritz T., Longerich T., Theriot C. M., McCulloch J. A., Roy S., et al. (2018) Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 10.1126/science.aan5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Levy M., Thaiss C. A., Zeevi D., Dohnalová L., Zilberman-Schapira G., Mahdi J. A., David E., Savidor A., Korem T., Herzig Y., Pevsner-Fischer M., Shapiro H., Christ A., Harmelin A., Halpern Z., et al. (2015) Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 10.1016/j.cell.2015.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hubbard T. D., Murray I. A., Bisson W. H., Lahoti T. S., Gowda K., Amin S. G., Patterson A. D., and Perdew G. H. (2015) Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 5, 12689 10.1038/srep12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Buescher J. M., Antoniewicz M. R., Boros L. G., Burgess S. C., Brunengraber H., Clish C. B., DeBerardinis R. J., Feron O., Frezza C., Ghesquiere B., Gottlieb E., Hiller K., Jones R. G., Kamphorst J. J., Kibbey R. G., et al. (2015) A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 34, 189–201 10.1016/j.copbio.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]