

Abstract
The phenotypes of each breast cancer subtype are defined by their transcriptomes. However, the transcription factors that regulate differential patterns of gene expression that contribute to specific disease outcomes are not well understood. Here, using gene silencing and overexpression approaches, RNA-Seq, and splicing analysis, we report that the transcription factor B-cell leukemia/lymphoma 11A (BCL11A) is highly expressed in triple-negative breast cancer (TNBC) and drives metastatic disease. Moreover, BCL11A promotes cancer cell invasion by suppressing the expression of muscleblind-like splicing regulator 1 (MBNL1), a splicing regulator that suppresses metastasis. This ultimately increases the levels of an alternatively spliced isoform of integrin-α6 (ITGA6), which is associated with worse patient outcomes. These results suggest that BCL11A sustains TNBC cell invasion and metastatic growth by repressing MBNL1-directed splicing of ITGA6. Our findings also indicate that BCL11A lies at the interface of transcription and splicing and promotes aggressive TNBC phenotypes.
Keywords: breast cancer, metastasis, invasion, transcription, alternative splicing, B-cell leukemia/lymphoma 11A (BCL11A), muscleblind-like splicing regulator 1 (MBNL1), integrin-α6 (ITGA6), gene regulation, gene transcription
Breast cancer subtypes are defined by unique transcriptomes that are reflective of their developmental lineage and specify distinct phenotypes. The luminal breast epithelial lineage highly expresses estrogen receptor (ER), FOXA1, and GATA3, and these factors drive more differentiated breast cancer phenotypes that are associated with good patient prognoses (1, 2). In contrast, triple-negative breast cancers (TNBC), while heterogeneous, are generally more primitive and are devoid of ER, progesterone receptor (PR), and HER2 receptors. These tumors convey the poorest prognoses because of the high incidence of metastatic recurrence, with a median 5-year survival rate of ∼68% depending on the patient population (3). Once metastasis has occurred, patient prognosis is very poor, with a median overall survival of only 13 months (4). Thus, there is a significant need to understand the molecular mechanisms that drive metastatic disease in TNBC patients.
Development of metastases requires tumor cells to disengage their cellular adhesions, allowing them to dissociate from the primary tumor, migrate, and invade the basement membrane, providing vascular access. After extravasation, tumor cells invade secondary sites and reinitiate growth pathways. Only a small percentage of cells that are shed from the primary tumor possess these invasive and tumor-initiating properties and, hence, are termed tumor-initiating cells (TIC) (5, 6). TIC are more migratory and invasive than most cells within the tumor bulk and exhibit a molecular profile that significantly overlaps that of cells that have undergone epithelial-to-mesenchymal transition (7). The rapid rate of metastatic recurrence in TNBC is thought to be due, in part, to the high proportion of TIC within these tumors compared with luminal breast cancers (7–9). Several factors expressed in TNBC have been found to drive the TIC phenotype such as SOX9, SOX10, ZEB, and SNAIL (7, 10, 11). However, it is likely that additional, lineage-associated factors also contribute to metastatic progression of this disease.
The zinc finger transcription factor, BCL11A, was initially identified because of a chromosomal translocation site in chronic B-cell lymphocytic leukemia (12, 13). However, BCL11A has been more recently identified as being preferentially upregulated in TNBC compared to all other breast cancer subtypes and is critical for the maintenance of normal and malignant mammary epithelial stem/progenitor populations (14). Most commonly characterized as a transcriptional repressor, BCL11A has been implicated as a member of multiple corepressor complexes, including SWI/SNF, NURD, and RBB4/7 as well as DNMT1 (15–19). In the mouse mammary gland, BCL11A is one of 22 genes defining an embryonic mammary signature that is silenced in adult epithelia and reactivated in mouse and human TNBC (20). BCL11A silencing also reduces the TIC population in TNBC xenografts, and ectopic BCL11A expression in nontransformed immortalized mammary epithelial cells can promote xenograft tumor formation (14, 21). Whereas these studies have implicated BCL11A as a key driver of stem cell fate in the mammary epithelium that controls primary tumor growth, it is unknown whether this factor also contributes to metastatic progression. The potential for BCL11A to contribute to the metastatic cascade is supported by it being a component of an 11-gene expression signature that is prognostic for breast cancer metastasis to the bone (22) and a recent report demonstrating its importance in in vitro migration and invasion assays (23). In addition to a lack of clarity regarding its role in metastasis, the downstream transcriptional targets of BCL11A and the factors that preclude its expression in luminal tumors remain to be discovered. Here, we define a new pathway involving BCL11A that controls TNBC metastasis. Elucidation of the BCL11A-regulated transcriptome, followed by genetic modulation studies, further revealed that BCL11A is necessary for sustained expression of extracellular matrix and adhesion genes as well as the RNA splicing regulator, muscleblind-like protein 1 (MBNL1), and its downstream targets.
Results
BCL11A sustains optimal invasion and metastatic outgrowth of TNBC cells
Basal breast cancer is the most abundant subtype of TNBC. Consistent with prior reports, we found that BCL11A expression is highly upregulated in basal breast cancer compared with the more differentiated luminal tumors in a publicly available data set (24) (Fig. 1A, left). Further confirming its differential expression, the level of BCL11A is also significantly higher in TNBC than in all other breast cancer subtypes in the TCGA data set (Fig. 1A, right) (14, 24, 25). BCL11A has been reported to be necessary for maintaining TNBC stem cell properties (14). Because increased stem cell activity can promote metastatic disease in various cancers, we postulated that BCL11A is essential for breast cancer metastasis. To test this, we first assessed the impact of altering BCL11A levels on in vitro surrogates of metastatic behaviors in cell lines representing two TNBC subtypes: MDA-MB-231 (mesenchymal-like or claudin-low subtype of TNBC) and MDA-MB-468 cells (basal-like 1) (26, 27). Transiently silencing BCL11A (Fig. 1B) had no impact on cell migration for either cell line (Fig. 1C). However, invasion was significantly reduced with the loss of BCL11A expression compared with the nonsilencing control (Fig. 1D), in alignment with a previous report (23). Similar to previous reports (14, 26), no changes in cell growth occurred with short-term suppression of BCL11A expression (Fig. S1, A and B), indicating that the observed change in invasion was not simply a result of decreased cell number. Together, these data indicate that sustained BCL11A expression is necessary to maintain the invasive phenotype of TNBC cells and may ultimately control their metastatic potential.
Figure 1.
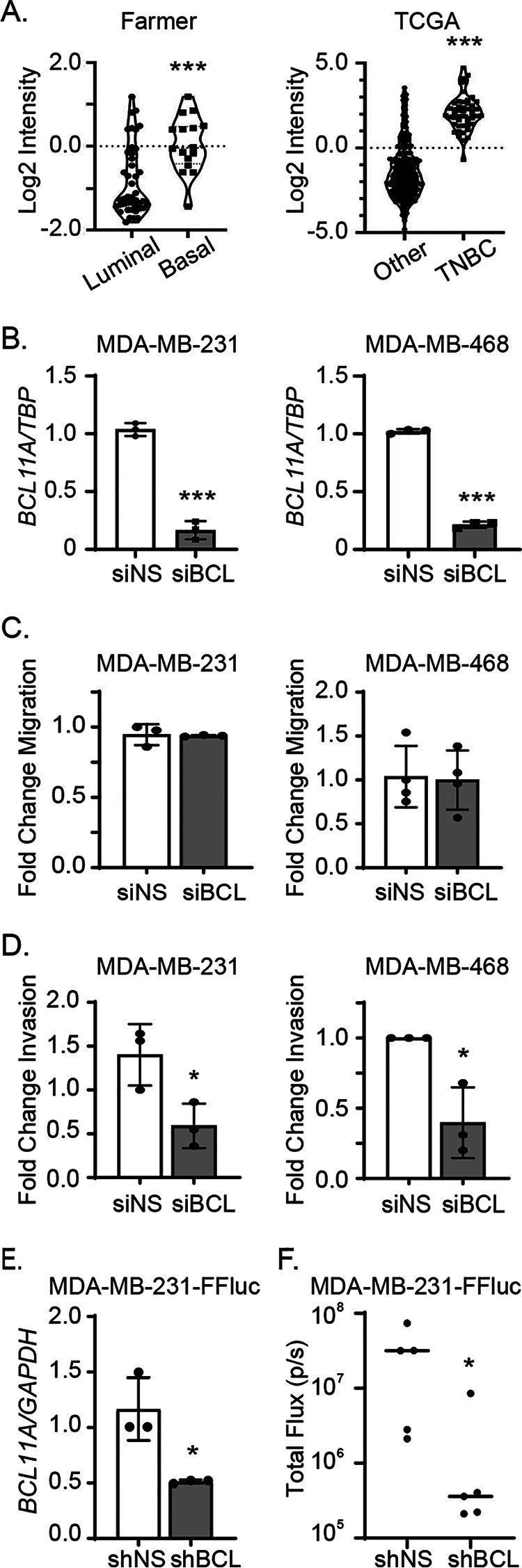
BCL11A sustains optimal metastatic outgrowth of TNBC cells. A, BCL11A expression in basal-like (n = 16) versus luminal (n = 43) invasive breast cancers from the Farmer data set (23) (left) and TNBC (n = 49) versus all other subtypes (n = 300) in the TCGA data set (38) (right). B, BCL11A expression in MDA-MB-231 or MDA-MB-468 cells was measured by qPCR 48 h after transient silencing. C and D, fold change of migration (C) or invasion (D) after 48 h of BCL11A silencing in the MDA-MB-231 or MDA-MB-468 cell lines. E, BCL11A expression measured by qPCR in the MDA-MB-231-FFluc cell line 2–4 passages after stable transduction with control (shNS) or BCL11A-targeted shRNA (shBCL)-expressing lentiviruses. F, bioluminescence quantitation of mice 18 days after intracardiac injection of shNS or shBCL11A stable cell lines (n = 5/group). Significance of differences was calculated in panels A–E using an unpaired t test and in panel F using a Mann-Whitney test. *, p < 0.05; ***, p < 0.001.
To directly assess the impact of BCL11A on metastatic outgrowth, we transduced luciferase-expressing MDA-MB-231 cells (MDA-MB-231-FFluc) with lentiviral vectors expressing an shRNA targeting BCL11A or a nonsilencing control. Upon confirming BCL11A silencing (Fig. 1E), these cells were injected into the left ventricles of the hearts of immunocompromised mice to assess their ability to colonize the body. After 18 days, BCL11A-silenced cells were significantly compromised in their ability to establish metastatic disease compared with control cells (Fig. 1F). Combined, these data suggest that sustained expression of BCL11A is necessary for invasion and metastatic outgrowth in models of TNBC.
BCL11A enhances metastatic outgrowth of TNBC cells
To determine whether BCL11A is also sufficient to induce metastasis within the context of TNBC, we overexpressed BCL11A in a cell line of low metastatic potential. Compared with the MDA-MB-231 cell line, SUM149PT cells (BSL2 TNBC subtype) have a lower incidence of cancer stem cells and are less invasive in vitro and in vivo (26, 28–30). Thus, we stably overexpressed BCL11A (xL isoform [31]) in luciferase-labeled SUM149PT cells (SUM149PT-FFluc) and assessed its impact on metastatic behaviors. Similar to BCL11A-silenced cells, overexpression of BCL11A had no impact on cell growth (Fig. 2, A and B). However, BCL11A overexpression significantly increased metastatic outgrowth in the lungs 31 days after intracardiac injection compared with cells transduced with an empty vector (Fig. 2C). Whereas no macroscopic metastases were observed in control mice, 4/9 mice injected with BCL11A-overexpressing cells had visible macrometastatic lesions (Fig. 2D and Fig. S2).
Figure 2.
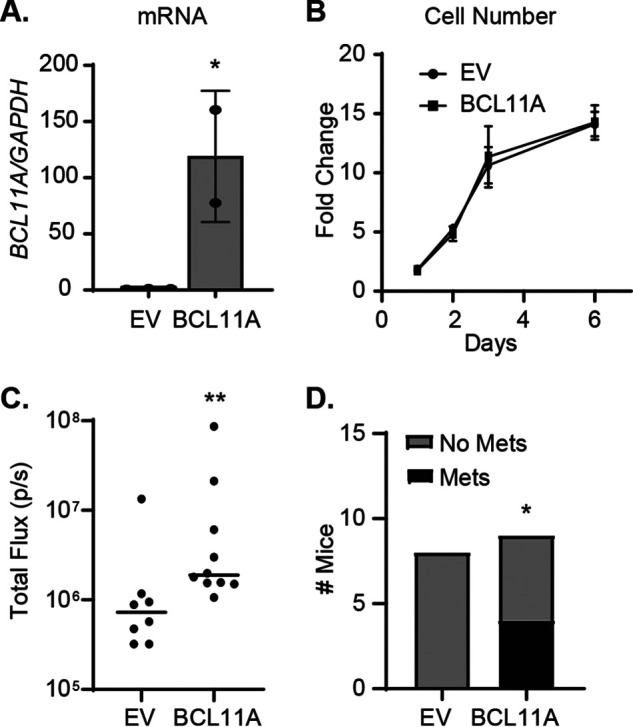
BCL11A enhances metastatic outgrowth of TNBC cells. A, stable overexpression of BCL11A mRNA (compared with empty vector [EV]) in SUM149PT-FFluc cells was measured by qPCR. B, cell growth was assessed over 6 days using the Cyto-X colorimetric cell counting reagent. C, quantitation of bioluminescence 31 days after intracardiac injection of stable cell lines (EV = 8 mice/group and BCL11A = 9 mice/group). D, number of mice with macrometastases. Significance of differences was calculated in panel A using an unpaired t test, in panel C using a Mann-Whitney test, and in panel D using chi-square test. *, p < 0.05; **, p < 0.01.
BCL11A governs the spliced transcriptome of TNBC cells
Given the function of BCL11A as a transcriptional regulator, we expected that its impact on invasion and metastatic outgrowth would be mediated by changes in target gene expression. Therefore, we used RNA-Seq to identify candidate BCL11A target genes. To identify early proximal targets of BCL11A, rather than downstream changes, that corresponded to invasion changes in vitro, MDA-MB-231 cells were transiently transfected with siRNAs against BCL11A or a nonsilencing control (Fig. 3A), and changes in RNA expression were evaluated. This revealed 183 and 395 genes that were up- or downregulated, respectively (Table S1), with transient BCL11A silencing. The top 25 most significantly changed genes are shown in Fig. 3B. The downregulated gene set was most strongly enriched in pathways involved in extracellular matrix organization and integrins in angiogenesis (top 4 enriched pathways are shown in Fig. 3C, left), which was consistent with the loss of invasion and metastatic behaviors that occurs with BCL11A silencing. In contrast, the upregulated gene set was enriched in cell cycle and mitosis-associated genes (top 4 enriched pathways shown in Fig. 3C, right). This was surprising because we, and others, have observed an absence of changes in cell number following BCL11A silencing (Fig. 1 and Fig. S1) (14). Notably, Bcl11a-null mice die shortly after birth, and we were unable to generate homozygous BCL11A-null clones in the MDA-MB-231 cell line using CRISPR/Cas9, suggesting that there is a critical threshold or time dependence for BCL11A expression that is required for long-term cell viability/growth. Regardless, the downregulated gene set identified several potential candidates that may collaborate to modulate metastatic behaviors.
Figure 3.

BCL11A controls the spliced transcriptome of TNBC cells. MDA-MB-231 cells were transiently transfected with SMARTpool siRNA targeting BCL11A or a nonsilencing control (siNS) for 48 h, and changes in the transcriptome were analyzed by RNA-Seq. A, confirmation of BCL11A silencing using qPCR. B, heatmap of the 25 most significantly altered genes (±2-fold change and adjusted p < 0.01; RPKM > 2). C, pathway analysis of differentially expressed genes using ConsensusPathDb (total expressed genes in MDA-MB-231 as a comparative background list). D, RNA-Seq reads were mapped and measured for differential splicing of cassette exons (cass), tandem exons spliced in a coordinated or mutually exclusive manner (taca or mutx, respectively), differences in 5′ and 3′ splice site (SS) selection (alt5 or alt3, respectively), and changes in intron retention (iret). The numbers of AS genes are in parentheses. Shown is a violin plot of statistically significant splicing changes (p < 0.05; FDR < 0.05). Gray lines are positioned at ΔPSI of ±10%. E, volcano plot of all differentially expressed genes with adjusted p value of <0.01, and dotted lines indicate genes with fold change of <2. Red dot, MBNL1.
In addition to changes in absolute expression of various genes, we also observed a striking change in the spliced isoforms for many genes. To determine whether BCL11A is associated with splicing in human basal breast cancers, we interrogated publicly available gene expression datasets to identify the set of genes that are most positively correlated with BCL11A (25, 32). Evaluation of these genes for functionality revealed that the highest ranked gene ontologies (GO) were “mRNA splicing” (GO 0000398; p = 2 × 10−4) and “mRNA splice site selections” (GO 0006376; p = 9.44 × 10−4), suggesting that BCL11A is a major regulator of the spliced transcriptome in basal breast cancers. Analysis of the RNA-Seq data set described here revealed 415 significant alternative splicing events, including 256 alternatively spliced cassette (cass) exons occurring in response to BCL11A silencing (Fig. 3D). We identified cassette exons for which the change in the percent exons spliced in (ΔPSI) was ≥10. This yielded 107 differentially spliced cassette exons that occur when BCL11A is repressed (Table S2). In alignment with the group of genes whose expression is downregulated with BCL11A silencing, genes that were alternatively spliced were also enriched in pathways controlling cell adhesion (adjusted p < 0.0421), as well as other ontologies. These data suggest that BCL11A controls the transcriptome, particularly that associated with cell adhesion, through at least two processes: gene expression and mRNA splicing.
Given that BCL11A-correlated genes are associated with mRNA splicing functions in basal breast cancer, our observation that BCL11A silencing results in a significant shift in splicing events in MDA-MB-231 cells, and that these splicing events are also enriched in pathways governing extracellular matrix (ECM) remodeling, we interrogated the RNA-Seq data set for genes that encode splicing factors that are differentially expressed in response to BCL11A silencing. One of the genes whose expression was most significantly induced was the RNA-splicing regulator muscleblind-like protein 1 (MBNL1), an established suppressor of TNBC invasion and metastasis (Fig. 3E).
MBNL1 is a key target of BCL11A that controls the TNBC spliced transcriptome and invasion
MBNL1 promotes cellular differentiation by repressing embryonic stem and iPS cell-specific alternative splicing programs (33–35). Thus, we proposed that BCL11A regulation of MBNL1 contributes to the alternative splicing events that occur when BCL11A expression is altered. We first confirmed that BCL11A represses the expression of MBNL1 in independent samples of MDA-MB-231 and MDA-MB-468 cells. As expected, transient silencing of BCL11A increased MBNL1 mRNA and protein expression in both cell lines (Fig. 4, A and B, and Fig. S3A). MBNL1 was also upregulated in MDA-MB-231-FFLuc cells that had undergone stable silencing of BCL11A using 5 independent shRNAs (Fig. S3B). Conversely, MBNL1 protein expression was reduced in SUM149PT cells upon transient overexpression of BCL11A (Fig. S3C). These data confirm that BCL11A is both necessary and sufficient to suppress MBNL1 expression in TNBC. To determine whether BCL11A directly binds to the MBNL1 gene to repress its expression, we examined publicly available genome-wide ChIP-Seq data for BCL11A in human lymphoblastoid (GM12878) and embryonic stem cell lines (h1-hESC) available in the Encyclopedia of DNA Elements (ENCODE) (36, 37). This analysis revealed multiple BCL11A binding sites in the MBNL1 gene and suggests that BCL11A is a general repressor of MBNL1 gene expression in multiple cell lineages.
Figure 4.

MBNL1 is a key target of BCL11A that controls the TNBC spliced transcriptome and invasion. A, MBNL1 mRNA expression was assessed by qPCR. B, MBNL1 protein was quantified by western blotting following transfection with BCL11A or nonspecific control siRNA in MDA-MB-231 or MDA-MB-468 cells. Both bands correspond to MBNL1 protein, as determined by using siRNA to MBNL1. C, MBNL1, BCL11A, or both were transiently silenced in MDA-MB-231 cells, and the relative mRNA expression of BCL11A (C) and MBNL1 (D) was evaluated by qPCR. E, fold change in invasion was assessed after BCL11A and MBNL1 silencing in the MDA-MB-231 cells. F, fold change in viable cells 4 days after BCL11A and MBNL1 silencing was determined using the Cyto-X colorimetric assay. G, disease-specific (DSS) survival was determined using the METABRIC data set (39). Samples (all subtypes) were divided into BCL11A high and MBNL1 low (n = 148), BCL11A low + MBNL1 high (n = 345), or intermediate (n = 1478). For panels A–F, significance of differences was calculated using an unpaired t test: *, p < 0.05; ***, p < 0.01; ***, p < 0.001; n.s., not signficant. For panel G, significance of difference between the survival curves was calculated using a log-rank test: green versus red line, p = 0.0002; red versus black line, p = 0.04; green versus black line, p = 0.004.
We then determined if the changes in the spliced transcriptome that occur with BCL11A silencing could be ascribed to alterations in established MBNL1 splicing events. To accomplish this, we intersected the list of alternatively spliced exons in the BCL11A RNA-Seq data set with a list of MBNL1-binding sites reported in a publically available HITS-CLIP data set from the MDA-MB-231 cell line (38). This revealed that MBNL1 directly binds to the majority (92 of 107, Table S2) of cassette exons that are alternatively spliced in response to BCL11A silencing.
MBNL1 suppresses invasion and metastasis in mouse models of TNBC (38). To determine whether the loss of invasion that occurs with BCL11A silencing is because of an induction of MBNL1 expression, we used RNAi to impede this induction in BCL11A-silenced cells. As expected, transient BCL11A silencing in MDA-MB-231 cells (Fig. 4C) caused an increase in MBNL1 mRNA and protein (Fig. 4D and Fig. S3D), but MBNL1 silencing alone had no impact on BCL11A expression (Fig. 4C and Fig. S3D). Likewise, BCL11A silencing decreased invasion of MDA-MB-231 cells, but MBNL1 silencing alone was insufficient to alter invasion (Fig. 4E). Most importantly, blocking the induction of MBNL1 that occurs in the context of BCL11A silencing restored the invasive capacity of MDA-MB-231 cells (Fig. 4E). In contrast to the changes in invasion, no differences in cell viability were observed following the silencing of BCL11A, MBNL1, or their combination (Fig. 4F). Nearly identical results were observed in the MDA-MB-468 cell line (Fig. S3, E–H), indicating that BCL11A modulation of invasion depends upon its regulation of MBNL1 in multiple models of TNBC.
To determine whether combined BCL11A and MBNL1 expression is associated with breast cancer patient outcomes, we interrogated the METABRIC gene expression data set. Primary breast cancers with combined high BCL11A and low MBNL1 expression (BCL11A high + MBNL1 low) were prognostic of significantly shorter disease-free survival than those with intermediate expression of both genes (intermediate) or to those that had low BCL11A and elevated MBNL1 expression (Fig. 4G) (39). Moreover, patients whose tumors expressed low MBNL1 and low BCL11A had intermediate outcomes, suggesting low MBNL1 expression alone does not drive aggressive TNBC phenotypes (Fig. S3I). Together, these data indicate that the suppression of MBNL1 is required for BCL11A to promote TNBC invasiveness and may contribute to worse patient outcomes.
BCL11A suppression of MBNL1 leads to an accumulation of the stem cell-driving alternative splice form of ITGA6 (ITGA6B)
The specific targets of MBNL1 that control various stages of metastasis are not well understood. Because MBNL1 suppression is an integral step in BCL11A-induced invasion, we sought to identify direct targets of MBNL1 whose alternative splicing may contribute to BCL11A-induced invasion. We first used RNA-Seq coupled with MBNL1 silencing, alone and in the context of siBCL11A (Fig. 5A and Fig. S4), to discover alternatively spliced exons controlled by both BCL11A and MBNL1. We identified 107, 110, and 146 cassette exons with altered splicing patterns following transient transfection with siBCL11A, siMBNL1, or the combination of siBCL11A and siMBNL1 (Fig. 5A).
Figure 5.

BCL11A increases expression of the ITGA6B stem-like splice form. A, alternatively spliced (AS) cassette exons identified using RNA-Seq of MDA-MB-231 cells after transiently silencing BCL11A, MBNL1, or both were intersected with HITS-CLIP data for MBNL1-bound transcripts (37). AS events induced by BCL11A silencing that were then reversed by cosuppression of MBNL1 were selected as candidates for further analysis. B, top, CLIP reads from MBNL1-binding libraries; library 1, red; library 2, blue. Middle, diagram of AS ITGA6 isoforms ± exon 27 (red outline). Bottom, corresponding RNA-Seq tracks for each siRNA condition and conservation across species (Cons, blue). C, top, representative agarose gel image examining changes in the expression of ITGA6 exon 27 AS isoforms following silencing of BCL11A, MBNL1, or both in MDA-MB-231 cells using RT-PCR. Bottom, quantitation of ITGA6 AS in 3 independent experiments, each performed in either duplicate or triplicate. D, top, representative western blot assessing changes in the levels of the ITGA6 stem-like splice form (ITGA6B) that occur with changes in BCL11A, MBNL1, or both. Bottom, quantitation of protein expression of the ITGA6B splice form from 3 independent experiments, each performed in either singlet or duplicate. E, PSI for ITGA6 exon 27 was obtained from the TCGA data set for normal breast, n = 114 or invasive BRCA, n = 1081. Significance of differences was calculated for panels C–E using an unpaired t test. F, ITGA6 exon 27 expression in basal breast cancers from the TCGA data set were stratified into groups with high expression (upper 15th percentile, n = 25) and low expression (remaining 85th percentile, n = 24) and overall survival compared by log-rank test, p = 0.005, HR = 0.148. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine which alternatively spliced mRNAs are direct targets of MBNL1, we reexamined the publicly available HITS-CLIP data of biologically reproducible MBNL1-RNA interactions identified in the MDA-MB-231 cell line (38). In all cases, the majority of the affected genes were previously shown to bind MBNL1 within the affected exon and/or the flanking intron sequences (86 to 88%) (Fig. S5). We next compared the three datasets (siBCL11A, siMBNL1, or both) and identified 14 cassette exons that were differentially spliced under all conditions and could bind to MBNL1 in close proximity (Fig. 5A). We interrogated each of these to discover which exons were impacted in opposite fashion with BCL11A silencing versus MBNL1 silencing alone or in combination with BCL11A loss. This resulted in two candidate genes, kinesin family member 13A (KIF13A) and integrin subunit alpha 6 (ITGA6), with alternative mRNA splicing patterns that were sensitive to MBNL1 levels in the MDA-MB-231 cell line. In BCL11A-silenced cells (elevated MBNL1), KIF13A displayed increased inclusion of exon 28, a 39-bp cassette encoding 13 amino acids that is located between the 2 noncontinuous coiled-coil domains (CC2 and CC3) in the motor protein stalk. These are proposed to function as points of dimerization in the Kinesin-3 family of monomeric proteins. Silencing of MBNL1 (alone or in combination with BCL11A silencing) reversed the effects of siBCL11A and further reduced splicing of exon 28 compared with the siRNA control (Fig. S6). Similarly, ITGA6 displayed increased inclusion of exon 27 with BCL11A silencing that was reversed and further reduced by MBNL1 loss compared with the control (Fig. 5B). Differential splicing of ITGA6 exon 27 generates protein isoforms with distinct C-terminal sequences (35 or 53 amino acids, exon 27 included or excluded, respectively) that are predicted to affect ITGA6 intracellular signaling (40).
Whereas KIF13A is a microtubule-based motor protein, its function and splicing in normal breast biology or breast cancer has not been reported. In contrast, ITGA6 encodes an integral cell surface protein that is a well-established biomarker and regulator of cancer stem cell biology, particularly in the breast (41–44). Notably, prior studies have shown that alternative splicing of the ITGA6 cytoplasmic tail dictates the fate of breast cancer stem cells and tumorigenicity of TNBC (44). Ectopic expression of ITGA6 lacking exon 27 (ITGA6B) promotes tumor formation and mesenchymal/stem-like phenotypes, whereas the contrary is true for the epithelium-specific splice form in which exon 27 is incorporated (ITGA6A) (44). Thus, inclusion of exon 27 promotes a more differentiated epithelial cell state, whereas reduction in exon 27 expression drives stem-like properties and tumor formation.
Using semiquantitative RT-PCR, we confirmed that the percentage of ITGA6 transcripts with inclusion of exon 27 (ITGA6A PSI) is elevated with the loss of BCL11A, and that this was reversed (ITGA6B) with combined silencing of MBNL1 and BCL11A in both MDA-MB-231 (Fig. 5C) and MDA-MB-468 cells (Fig. S7, A and B). Moreover, the loss of BCL11A resulted in differential protein expression with a decrease in the levels of the stem cell-associated ITGA6B (Fig. 5D and Fig. S7, C and D). These data indicate that BCL11A, through its suppression of MBNL1 expression, causes the differential splicing of ITGA6, resulting in the accumulation of the ITGA6B protein isoform that lacks exon 27 and promotes stem cell fate in TNBC cells. To assess the clinical significance of ITGA6 differential splicing, we queried the TCGA data set (25) and found that invasive breast cancers have a significantly lower inclusion of exon 27 than normal breast tissue (Fig. 5E). Most importantly, reduced expression of ITGA6 exon 27 is associated with worse overall survival of patients with basal breast cancer (Fig. 5F). These data support the postulate that BCL11A suppression of MBNL1 promotes accumulation of the ITGA6B stem cell-associated splice form and drives metastatic outgrowth of TNBC (Fig. 6).
Figure 6.

BCL11A drives invasion and metastatic outgrowth by suppressing MBNL1 and is associated with expression of the stem-like splice form of ITGA6 (ITGA6B). Reduced expression relieves repression of MBNL1 and promotes alternative splicing of ITGA6. Inclusion of exon 27 is associated with reduced TNBC invasion in vitro, reduced metastatic outgrowth in mice, and extended overall survival of patients with basal breast cancer.
Discussion
Here, we report that the transcriptional repressor BCL11A is a key driver of invasion of TNBC cell lines in vitro and metastatic seeding and outgrowth in xenografted mouse models of this disease. We also report that BCL11A governs both transcriptional and posttranscriptional programs known to promote tissue remodeling, extracellular matrix signaling, and alternative splicing (35, 45–48). Together, these data extend the function of BCL11A beyond its roles in breast epithelial stem cells and primary tumor formation identified by Khaled et al. (14), nominating it as a potential drug target in the metastatic setting. Supporting its critical role and therapeutic potential in breast cancer cells, we were unable to fully disrupt the BCL11A gene using CRISPR/Cas9, even though 4 heterozygous null lines were generated. Thus, we anticipate that complete ablation of BCL11A expression is incompatible with extended TNBC cell viability. Khaled et al. demonstrated tumor volume changes with altered BCL11A (14), supporting a role for BCL11A in cancer stem cell self-renewal. Mechanistic studies utilizing multicellular 3D cultures are needed to discern the function of BCL11A in cell cycle and self-renewal in a context that recapitulates components of the tumor microenvironment. RNA-Seq analysis of stably silenced cells cultured in this environment would aid in the identification of BCL11A transcriptional targets driving proliferation and self-renewal. As a transcription factor, BCL11A would traditionally be considered undruggable, although PROTAC strategies (49) may be effective. An alternative approach is to identify its downstream targets or upstream regulators with the goal of discovering additional therapeutically targetable intermediates.
BCL11A is a well-established regulator of differentiation, including formation of hematopoietic stem cells, repression of fetal hemoglobin expression, stimulating neuronal polarity and migration, and establishment of the epidermal permeability barrier (50–54). The specific function of BCL11A in each of these contexts likely involves its regulation of a series of global and cell-specific target genes. Here, we used a transient silencing paradigm to identify the BCL11A-regulated transcriptome in TNBC that is changed within 48 h. Because BCL11A does not impact proliferation within this window, we could leverage the timing of this study to identify changes that were more associated with the invasion phenotype. This revealed that BCL11A controls the expression of many genes involved in remodeling of cellular adhesions and the extracellular matrix. Given the mechanistic similarities in neuronal migration and cancer cell migration/invasion, it is likely that at least some of these BCL11A effector genes have overlapping functions between these two processes.
Although BCL11A controls the gene expression for many proteins involved in ECM remodeling, and these factors themselves are associated with metastatic progression, the suppression of MBNL1 is essential for maximal BCL11A-induced invasion and metastatic outgrowth. While we did not examine the possibility that dysregulation of ECM components by BCL11A would also play a core role in invasion and metastatic outgrowth, it is likely that BCL11A regulation of multiple pathways, including splicing and the ECM, together contribute to its ability to regulate these processes. Importantly, we also found that BCL11A broadly controls alternative splicing events, and many of these are the downstream consequence of changes in the expression of MBNL1, a context-dependent enhancer and suppressor of alternative splicing. Extending this finding, we postulate that MBNL1 also is a key intermediate for at least some of the differentiation processes controlled by BCL11A. Notably, recent studies have shown that cytoplasmic accumulation of MBNL1 promotes neurite morphogenesis (48), a process that is also controlled by BCL11A. Although the data presented here demonstrate that BCL11A represses the expression of the MBNL1 gene, it is unclear if this is direct or indirect. We could not identify BCL11A binding to the MBNL1 gene using 6 sites identified in lymphoblastoid cells in ENCODE (data not shown). This could indicate that BCL11A does not directly bind to and regulate MBNL1, or it could reflect the use of different BCL11A binding sites in TNBC cells compared with lymphoblastoid cells. Whole-genome BCL11A ChIP-Seq analyses in TNBC cells will be necessary to definitively identify the cohort of genes directly bound and regulated by BCL11A in this cell lineage.
Targeting MBNL1 for therapeutic benefit in TNBC or any cancer will likely prove difficult. However, we more specifically found that BCL11A dictates the splicing pattern of ITGA6. Global ITGA6 protein expression is independently prognostic of adverse outcomes in ER-negative breast cancer (41) but is only detectable in a minority of breast cancer cells, presumably cancer stem cells. When combined with low EpCam expression, ITGA6 has also been widely used to isolate a stem cell subpopulation within normal and malignant breast epithelial cells (43, 55–58). Our data suggest that BCL11A stimulates alternative splicing of ITGA6 and provides another level of regulation for this integrin. BCL11A induces the exclusion of the terminal exon of ITGA6 to form ITGA6B protein, a variant associated with stem cell phenotypes (44). In contrast, when BCL11A levels are low, the epithelial isoform (ITGA6A) is expressed. ITGA6B has a functional role in breast cancer cells where prior studies have demonstrated that its increased expression enhances the formation of colonies in soft agar and mammospheres over several passages. Moreover, ectopic expression of ITGA6B increases tumor formation in xenografted mice (44). ITGA6B is also elevated in tissues that are enriched in stem cells, such as embryonic mouse kidney, undifferentiated endoderm, and, most notably, embryonic stem cells (59–61). Mechanistically, loss of the terminal exon of ITGA6 results in an altered amino acid sequence at the cytoplasmic tail, which is a critical site for integrin signaling. The PDZ-binding motif in the cytoplasmic tail shifts from the canonical SDA amino acid sequence to an atypical SYS motif, and a previous study suggests that these differing sequences confer differential binding affinities to downstream effector proteins (62; reviewed in reference 40). Given the impact of BCL11A on ITGA6 splicing, we propose that altering the expression of BCL11A has profound effects on the integrin signaling pathways in TNBC stem cells. Further analysis of clinical samples will be necessary to determine the interplay between BCL11A protein expression and ITGA6 splicing in TNBC.
It is now well established that cancers undergo changes in splicing behaviors (63, 64), yet mechanistic studies elucidating which splicing changes contribute to invasion and metastasis have lagged behind. This is because of the complexity of RNA splicing events driven by RNA binding proteins that both cooperate with and compete for transcript binding (65). This includes MBNL1 and RBFOX, which cooperate to promote alternative splicing events. Relevant to this particular study, ITGA6 splicing is not uniquely controlled by MBNL1. ESRP1 (epithelial splicing protein 1) controls ITGA6 splicing in the nontransformed mammary epithelial cell line, MCF10A, and its activity is regulated by VEGF (vascular endothelial growth factor) signaling (44). Notably, our assessment of the BCL11A-regulated transcriptome in TNBC failed to reveal any changes in the expression of ESRP1 and RBFOX. However, a modest change in the expression of PTPB3 (log2 fold change = −0.7, adjusted p = 3 × 10−10), a gene encoding another splicing protein, was observed. Additional studies focusing on PTPB3-driven splicing changes may uncover an even greater transcript complexity driven by BCL11A than that reported here. It is also important to note that MBNL1 regulates additional targets beyond ITGA6 that also control invasion and metastasis of TNBC. Fish et al. reported that TACC1 (transforming acidic coiled-coil-containing protein 1) and DBNL (drebrin-like protein) are also direct targets of MBNL1 that can modulate invasion and metastatic colonization of MDA-MB-231 cells (38). Surprisingly, we did not detect alternative splicing of these genes with transient BCL11A silencing, and we could not detect differences in steady-state protein expression (data not shown). It is possible that the alternative splicing of TACC1 and DBNL is an adaptive process that occurs with sustained MBNL1 manipulation, whereas our studies involved transient silencing. Clearly, additional studies are needed to identify the full spectrum of alternative splicing events that drive metastatic progression of cancers, including breast cancer.
In summary, prior studies identified BCL11A as a highly expressed gene in TNBC that is critical for sustaining the tumor-initiating cell population. The studies reported here reveal that BCL11A is also a regulator of metastatic progression, the key determinant in breast cancer patient survival. Although BCL11A is a transcription factor and generally considered difficult to target for therapeutic benefit, suppressing BCL11A expression using lentiviral shRNA delivery and genetic deletion of BCL11A using CRISPR-Cas9 editing are both in clinical trials for the treatment of sickle cell disease and beta-thalassemia. Whereas these methods will prove challenging for the treatment of solid tumors, elucidating pathways that regulate BCL11A expression as well as those that mediate its downstream effects may prove beneficial for improving patient outcomes. This work begins to unravel the complex network of gene regulation governed by BCL11A that contributes to the aggressive nature of TNBC.
Materials and methods
Cell culture and reagents
MDA-MB-231, MDA-MB-468, T-47D, and ZR-75-1 cells were purchased from the ATCC and grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. T-47D cells were additionally supplemented with 0.2 units/ml insulin. Cell lines were authenticated in 2018 by STR profiling (Genetica DNA Laboratories, Cincinnati, OH). MDA-MB-231-FFluc cells were a kind gift from Drs. Jenny Parvani and Mark Jackson, and stable expression of the luciferase gene was achieved with Blasticidin selection. SUM149PT cells were purchased in 2016 (BioIVT, Westbury, NY) and grown in Ham's F-12 medium supplemented with 5% FBS, HEPES (10 mm), hydrocortisone (1 μg/ml), insulin (5 μg/ml), and 1% penicillin–streptomycin. All cell lines were maintained in a humidified tissue culture incubator at 37 °C with 5% CO2, tested for mycoplasma using the Mycoplasma detection kit-quick test (Bimake), and were mycoplasma free.
Gene silencing and overexpression
a. RNAi
For transient gene silencing, MDA-MB-231 and MDA-MB-468 cells were seeded into 6-well plates to achieve ∼60% confluency the following day. SMARTpool siRNAs targeting BCL11A (Dharmacon, catalog no. L-006996), MBNL1 (Dharmacon, catalog no. L-014136), or a nontargeting siRNA directed to firefly luciferase mRNA (U47296) (Dharmacon, catalog no. D-001210-02-50) were transfected in Opti-MEM medium (Invitrogen, catalog no. 3195088) at a final concentration of 100 nM for 6 h. For stable gene silencing, transduction of BCL11A-targeted shRNA lentiviral particles (TRCN000003349, Sigma-Aldrich) was conducted with puromycin selection. Four additional shRNA clones were individually transduced to confirm changes in MBNL1 expression (TRCN0000033453, TRCN0000430666, TRCN00000359207, and TRCN0000359144, Sigma-Aldrich).
b. Overexpression
BCL11A-xL-pCDNA3 expression plasmid was a kind gift from Dr. Haley Tucker (University of Texas, Austin). BCL11A-xL-pcDNA3 or empty vector (pcDNA3) was transfected into SUM149PT cells using Lipofectamine 2000. A stable cell line was achieved using neomycin (Genetecin, 200 μg/ml) selection.
In vivo studies
All in vivo experiments were performed with approval from the Institutional Animal Care and Use Committee at Case Western Reserve University, which is certified by the American Association of Accreditation for Laboratory Animal Care. For both MDA-MB-231-FFluc and SUM149PT-FFluc studies, 200,000 cells in a total volume of 100 μl were injected into the left ventricle of NOD Scid-γ female mice that were anesthetized with 2% isofluorane. Mice were imaged for bioluminescence signal as an indicator of metastatic outgrowth using a Xenogen IVIS system. Mice were administered 150 mg/kg luciferin-D via intraperitoneal injection and imaged after 10 min. Bioluminescence was quantified as total flux (p/s) per mouse.
RNA isolation and real-time qPCR
RNA was isolated using either Invitrogen TRIzol reagent (Life Technologies, Carlsbad, CA, USA) and DNase (Ambion DNA-free kit; Life Technologies) or an RNeasy plus minikit (Qiagen). Complementary DNA was generated using SuperScript II reverse transcriptase with random hexamers (Life Technologies) per the manufacturer's protocol. Quantitative real-time PCR (qPCR) was performed on a StepOnePlus real-time PCR system using TaqMan gene expression assays (Applied Biosystems, Foster City, CA, USA) for human BCL11A (Hs01093197) or MBNL1 (Hs01582594).
Cell viability/counting
Cells were transfected with siRNAs and viable cells seeded the following morning in quadruplicate in 96-well plates at either 3000 cells/well (MDA-MB-231 and SUM149PT cells) or 6000 cells/well (MDA-MB-468 cells). Cyto-X (CELL Applications, Inc.) was used to determine the relative number of cells/well each day by reading the optical density at 450 nm. Duplicate wells were used to validate gene silencing using qPCR.
Western blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer and diluted in Laemmli buffer plus β-mercaptoethanol. After boiling, 40–80 μg of protein lysate and molecular weight marker (LI-COR, catalog no. 928-60000) were fractionated in a 4–20% Tris-glycine gel (Invitrogen, catalog no. XP04200BOX). Proteins were then transferred to Immobilon-FL PVDF membrane (catalog no. IPFL00010) by wet transfer. Blots were stained using REVERT total protein stain (P/N, 926-11011) for ∼5 min and then washed with 6.7% (v/v) glacial acetic acid, 30% (v/v) methanol in water twice for 30 s. After total protein was detected using a LI-COR Odyssey Fc, blots were incubated in a destaining solution (0.1% [w/v] sodium hydroxide, 30% [v/v] methanol in water). Membranes were blocked using 5% nonfat milk in TBS with 0.05% Tween-20 for 1 h and were probed with anti-BCL11A (Abcam, catalog no. Ab19487; lot GR3201086-3, 1:200), anti-MBNL1 (Millipore, catalog no. 2883433, 1:1000), or anti-ITGA6b (stem-ITGA6, Millipore, catalog no. MAB1358, 1:250 diluted in LI-COR buffer) antibodies overnight. Blots were incubated for 1 h with fluorophore-bound secondary antibody (IRDye 800CW goat anti-mouse IgM, 1:10,000) or HRP-bound secondary (1:5000). Bound antibody was detected using either chemiluminescence (SignalFire ECL reagent, Cell Signaling Technology, Danvers, MA, USA) or the Odyssey Fc (OFC-1279) for fluorescent secondary antibodies. Western blots were imaged using a LI-COR Odyssey Fc and quantified using Image Studio v5.2 compared with total protein. MBNL1 exists as two isoforms (66); thus, we included both in our quantitation of MBNL1 expression. Quantitation was performed on Western blots from 3 independent experiments.
Migration and invasion
Cells were transiently transfected as described above with 100 nM SMARTpool siRNA for the following targets: BCL11A, MBNL1, nontargeting control (siNS), or a combination of siBCL11A and siMBNL1, each at 50 nM. After 48 h, cells were plated for migration assays in modified Boyden chambers (transwell permeable supports, catalog no. 3422, Corning Life Sciences, Tewksbury, MA, USA) or invasion assays in transwell inserts coated with Matrigel (BioCoat Matrigel invasion chamber, catalog no. 354480, Corning Life Sciences). The upper chamber contained cells in serum-free media, and the bottom chamber contained 10% serum. Cells were allowed to migrate or invade for 16–24 h. Filters were fixed and stained with Diff-Quik. The number of migrated or invaded cells per filter was calculated by averaging the number of cells per 10× magnification field in five independent fields. Three independent experiments were performed with technical replicates, in triplicate for migration assays and in duplicate for invasion assays.
RNA-Seq
MDA-MB-231 cells were transiently transfected with SMARTpool siRNAs as indicated above. Total RNA was isolated 48 h later using the RNeasy Plus minikit (Qiagen) and evaluated using a Qubit fluorometer (Invitrogen) for quantitation and Agilent 2100 Bioanalyzer to assess quality using a cutoff of RNA integrty number > 7.0 to select samples for further analysis. For library preparation, the Illumina TruSeq stranded total RNA kit with Ribo Zero Gold for rRNA removal was used. The Ribo Zero kit was used to remove rRNA from 150 ng of total RNA. Illumina TruSeq libraries were tagged with unique adapter indexes. Final libraries were validated on the Agilent 2100 Bioanalyzer, quantified via qPCR (KAPA Biosystems Illumina Library Quantification Kit), and pooled at equimolar ratios. Pooled libraries were diluted, denatured, and loaded onto the Illumina HiSeq 2500 using a paired-end Rapid Run flow cell.
Reads were mapped to the hg19 genome with Olego (67). The Quantas pipeline was used to infer transcript structure and quantify alternative splicing and gene expression (68). Genes that expressed at an RPKM of >2 in one group and had a fold change in expression of 2 or greater and adjusted p value of <0.01 were considered significantly differentially expressed. Significant splicing changes were defined as those with a false discovery rate (FDR) of <0.05, adjusted p value of <0.05, and average RPKM across biological replicates of >1 under both conditions. Cassette (cass) exons were collapsed and filtered for events where ΔPSI ≥ 10 (107, 110, and 146 significant cass exons in siNS versus siBCL11A, siMBNL1, and dual silencing, respectively).
Pathway analysis was performed using the ConsensusPathDb database (69, 70) using overrepresentation analysis and all expressed genes in the MDA-MB-231 cell line as a background list. Significance was determined using the hypergeometric test and p values corrected for multiple testing using the FDR method (expressed as a q value). The top 4 most strongly (most significantly) enriched pathways were reported. Basal breast cancer genes that were correlated with BCL11A in a meta-analysis of the TCGA (25), GSE81538 and GSE96058 (32), were identified and assessed for enriched gene ontology (GO) pathways using bc-GenExMiner (71).
Splicing analysis and HITS-CLIP processing
Publicly available HITS-CLIP data for MBNL1 included SRR3082411.sra and RR3082412.sra. Raw SRA files were converted to fastq format using the fastqdump tool. After trimming the 3′ linker (RL3, 15 nt), reads were mapped to the hg19 genome using Bowtie2, and PCR duplicates were removed. To identify high-confidence MBNL1 binding sites relative to alternatively spliced cassette exons, biologically reproducible binding sites (BR2, reads contributed from both CLIP libraries) were intersected with coordinates flanking the significantly spliced exon to include the upstream and downstream constitutive exons. CLIP reads mapping to these regions were then collapsed and filtered for binding events that occur within 500-bp regions flanking the alternative exon splice sites as well as 500-bp regions up- and downstream of the constitutive exons.
Splicing validation
ITGA6 alternative splice products were identified using relative RT-PCR as described above. The following primer pair was used to amplify a region around ITGA6 exon 27: 5′-CTAACGGAGTCTCACAACTC-3′ and 5′-ACTCTGAAATCAGTCCTCAG-3′. PCR products were resolved on a 1% agarose gel and identified with ethidium bromide. Quantitation of the PSI was determined using either Image J or Image Studio Software (LI-COR).
Kaplan-Meier analyses
Data from METABRIC (EGAS00000000083) were accessed via Oncomine v4.5. The population was stratified by the upper 10th percentile (high) versus the remainder (low) expression for BCL11A (ILMN_1752899) and by upper 25th percentile (high) versus the remainder (low) for MBNL1 (ILMN_1807304) expression. All remaining samples with intermediate expression of BCL11A and MBNL1 were designated intermediate. Kaplan-Meier curves were generated for the resulting BCL11A/MBNL1 expression groups, and disease-specific survival for these groups was compared by the log-rank test. Splicing data for ITGA6 exon 27 in the TCGA data set were downloaded from TCGASpliceSeq and combined with PAM50 classification and clinical outcomes in the same data set. The population was partitioned into the upper 15% ITGA6 exon 27 expressing (high) and lower 85% expressing (low) tumors and assessed for patient overall survival. Significance of differences was determined by the log-rank test.
Statistical analyses
All data are expressed as means ± standard deviations. Statistical analyses were performed using one-way analyses of variance followed by unpaired Student's t test, Fisher's exact test, Mann-Whitney test, or chi-square tests in GraphPad Prism where indicated. Log-rank tests were used to calculate significant differences in survival curves, and a minimum p value of <0.05 was considered statistically significant. Assays were performed in at least 3 independent (biological) experiments with technical duplicates or triplicates.
Data availability
The RNA-Seq data were deposited in the Gene Expression Omnibus, series GSE149435. All other data are present within the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Drs. Jenny Parvani and Mark Jackson for their kind gift of the MDA-MB-231-FFluc cells and to Dr. Haley Tucker for her generous gift of the BCL11A expression plasmid. We are also grateful to Dr. Lindsey Anstine for her thoughtful review of our manuscript.
This article contains supporting information.
Author contributions—D. D. S., P. Y., G. B., and R. A. K. conceptualization; D. D. S., M. M. H., N. N. I., B. M. W., S. S., S. T. S., and D. D. L. data curation; D. D. S., N. N. I., K. L. W.-B., S. S., S. T. S., and D. D. L. formal analysis; D. D. S., M. M. H., N. N. I., S. S., and S. T. S. investigation; D. D. S., M. M. H., N. N. I., B. M. W., K. L. W.-B., S. S., and D. D. L. methodology; D. D. S., M. M. H., N. N. I., B. M. W., K. L. W.-B., P. Y., G. B., S. S., S. T. S., V. V., and D. D. L. writing-original draft; D. D. S., M. M. H., N. N. I., B. M. W., K. L. W.-B., P. Y., G. B., S. S., S. T. S., V. V., D. D. L., and R. A. K. writing-review and editing; N. N. I. and D. D. L. validation; G. B. and R. A. K. funding acquisition; S. S., S. T. S., and D. D. L. visualization; V. V., D. D. L., and R. A. K. supervision; R. A. K. project administration.
Funding and additional information—This work was supported by the Department of Defense, W81XWH-14-1-0354, and NIH R01 CA206505 to R. A. K. and the Clinical and Translational Science Collaborative of Cleveland, KL2 TR000440, and University Hospitals Department of Medicine Team Science Award 2014 to G. B. This research was also supported by the Genomics Core Facility of the CWRU School of Medicine's Genetics and Genome Sciences Department. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ER
- estrogen receptor
- TNBC
- triple-negative breast cancer
- PR
- progesterone receptor
- TIC
- tumor-initiating cells
- PSI
- percent exons spliced in
- FDR
- false discovery rate
- cass
- cassette
- GO
- gene ontology
- ECM
- extracellular matrix
- RPKM
- reads per kilobase of transcript, per million mapped reads.
References
- 1. Fallahpour S., Navaneelan T., De P., and Borgo A. (2017) Breast cancer survival by molecular subtype: a population-based analysis of cancer registry data. CMAJ Open 5, E734–E739 10.9778/cmajo.20170030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haque R., Ahmed S. A., Inzhakova G., Shi J., Avila C., Polikoff J., Bernstein L., Enger S. M., and Press M. F. (2012) Impact of breast cancer subtypes and treatment on survival: an analysis spanning two decades. Cancer Epidemiol. Biomarkers Prev. 21, 1848–1855 10.1158/1055-9965.EPI-12-0474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ovcaricek T., Frkovic S., Matos E., Mozina B., and Borstnar S. (2011) Triple negative breast cancer–prognostic factors and survival. Radiol. Oncol. 45, 46 10.2478/v10019-010-0054-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kassam F., Enright K., Dent R., Dranitsaris G., Myers J., Flynn C., Fralick M., Kumar R., and Clemons M. (2009) Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design. Clin. Breast Cancer 9, 29–33 10.3816/CBC.2009.n.005 [DOI] [PubMed] [Google Scholar]
- 5. Luzzi K. J., MacDonald I. C., Schmidt E. E., Kerkvliet N., Morris V. L., Chambers A. F., and Groom A. C. (1998) Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 153, 865–873 10.1016/S0002-9440(10)65628-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cameron M. D., Schmidt E. E., Kerkvliet N., Nadkarni K. V., Morris V. L., Groom A. C., Chambers A. F., and MacDonald I. C. (2000) Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 60, 2541–2546 [PubMed] [Google Scholar]
- 7. Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., Brooks M., Reinhard F., Zhang C. C., Shipitsin M., Campbell L. L., Polyak K., Brisken C., Yang J., and Weinberg R. A. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Idowu M. O., Kmieciak M., Dumur C., Burton R. S., Grimes M. M., Powers C. N., and Manjili M. H. (2012) CD44(+)/CD24(-/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Human Pathol. 43, 364–373 10.1016/j.humpath.2011.05.005 [DOI] [PubMed] [Google Scholar]
- 9. Croker A. K., Goodale D., Chu J., Postenka C., Hedley B. D., Hess D. A., and Allan A. L. (2009) High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 13, 2236–2252 10.1111/j.1582-4934.2008.00455.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo W., Keckesova Z., Donaher J. L., Shibue T., Tischler V., Reinhardt F., Itzkovitz S., Noske A., Zurrer-Hardi U., Bell G., Tam W. L., Mani S. A., van Oudenaarden A., and Weinberg R. A. (2012) Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028 10.1016/j.cell.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parvani J. G., and Schiemann W. P. (2013) Sox4, EMT programs, and the metastatic progression of breast cancers: mastering the masters of EMT. Breast Cancer Res. 15, R72 10.1186/bcr3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Satterwhite E., Sonoki T., Willis T. G., Harder L., Nowak R., Arriola E. L., Liu H., Price H. P., Gesk S., Steinemann D., Schlegelberger B., Oscier D. G., Siebert R., Tucker P. W., and Dyer M. J. (2001) The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood 98, 3413–3420 10.1182/blood.v98.12.3413 [DOI] [PubMed] [Google Scholar]
- 13. Yin B., Delwel R., Valk P. J., Wallace M. R., Loh M. L., Shannon K. M., and Largaespada D. A. (2009) A retroviral mutagenesis screen reveals strong cooperation between Bcl11a overexpression and loss of the Nf1 tumor suppressor gene. Blood 113, 1075–1085 10.1182/blood-2008-03-144436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khaled W. T., Choon Lee S., Stingl J., Chen X., Raza Ali H., Rueda O. M., Hadi F., Wang J., Yu Y., Chin S.-F., Stratton M., Futreal A., Jenkins N. A., Aparicio S., Copeland N. G., et al. (2015) BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat. Commun. 6, 5987–5987 10.1038/ncomms6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kadoch C., Hargreaves D. C., Hodges C., Elias L., Ho L., Ranish J., and Crabtree G. R. (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 10.1038/ng.2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang H. T., Chen S. M., Pan L. B., Yao J., and Ma H. T. (2015) Loss of function of SWI/SNF chromatin remodeling genes leads to genome instability of human lung cancer. Oncol. Rep. 33, 283–291 10.3892/or.2014.3584 [DOI] [PubMed] [Google Scholar]
- 17. Xu J., Bauer D. E., Kerenyi M. A., Vo T. D., Hou S., Hsu Y. J., Yao H., Trowbridge J. J., Mandel G., and Orkin S. H. (2013) Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci. U S A 110, 6518–6523 10.1073/pnas.1303976110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moody R. R., Lo M.-C., Meagher J. L., Lin C.-C., Stevers N. O., Tinsley S. L., Jung I., Matvekas A., Stuckey J. A., and Sun D. (2018) Probing the interaction between the histone methyltransferase/deacetylase subunit RBBP4/7 and the transcription factor BCL11A in epigenetic complexes. J. Biol. Chem. 293, 2125–2136 10.1074/jbc.M117.811463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen F., Luo N., Hu Y., Li X., and Zhang K. (2018) MiR-137 suppresses triple-negative breast cancer stemness and tumorigenesis by perturbing BCL11A-DNMT1 interaction. Cell. Physiol. Biochem. 47, 2147–2158 10.1159/000491526 [DOI] [PubMed] [Google Scholar]
- 20. Zvelebil M., Oliemuller E., Gao Q., Wansbury O., Mackay A., Kendrick H., Smalley M. J., Reis-Filho J. S., and Howard B. A. (2013) Embryonic mammary signature subsets are activated in Brca1-/- and basal-like breast cancers. Breast Cancer Res. 15, R25 10.1186/bcr3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu L., Pan R., Zhou D., Ye G., and Tan W. (2019) BCL11A enhances stemness and promotes progression by activating Wnt/beta-catenin signaling in breast cancer. Cancer Manag. Res. 11, 2997–3007 10.2147/CMAR.S199368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hayashi N., Manyam G. C., Gonzalez-Angulo A. M., Niikura N., Yamauchi H., Nakamura S., Hortobagyi G. N., Baggerly K. A., and Ueno N. T. (2014) Reverse-phase protein array for prediction of patients at low risk of developing bone metastasis from breast cancer. Oncologist 19, 909–914 10.1634/theoncologist.2014-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang X., Xu Y., Xu K., Chen Y., Xiao X., and Guan X. (2020) BCL11A confers cell invasion and migration in androgen receptor-positive triple-negative breast cancer. Oncol. Lett. 19, 2916–2924 10.3892/ol.2020.11383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Farmer P., Bonnefoi H., Becette V., Tubiana-Hulin M., Fumoleau P., Larsimont D., MacGrogan G., Bergh J., Cameron D., Goldstein D., Duss S., Nicoulaz A.-L., Brisken C., Fiche M., Delorenzi M., et al. (2005) Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 24, 4660–4671 10.1038/sj.onc.1208561 [DOI] [PubMed] [Google Scholar]
- 25. Cancer Genome Atlas Network. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lehmann B. D., Bauer J. A., Chen X., Sanders M. E., Chakravarthy A. B., Shyr Y., and Pietenpol J. A. (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herschkowitz J. I., Simin K., Weigman V. J., Mikaelian I., Usary J., Hu Z., Rasmussen K. E., Jones L. P., Assefnia S., Chandrasekharan S., Backlund M. G., Yin Y., Khramtsov A. I., Bastein R., Quackenbush J., et al. (2007) Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 8, R76 10.1186/gb-2007-8-5-r76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chu J. E., Xia Y., Chin-Yee B., Goodale D., Croker A. K., and Allan A. L. (2014) Lung-derived factors mediate breast cancer cell migration through CD44 receptor-ligand interactions in a novel ex vivo system for analysis of organ-specific soluble proteins. Neoplasia 16, 180–191. 10.1593/neo.132076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nieman M. T., Prudoff R. S., Johnson K. R., and Wheelock M. J. (1999) N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell. Biol. 147, 631–644 10.1083/jcb.147.3.631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bartholomeusz C., Xie X., Pitner M. K., Kondo K., Dadbin A., Lee J., Saso H., Smith P. D., Dalby K. N., and Ueno N. T. (2015) MEK inhibitor selumetinib (AZD6244; ARRY-142886) prevents lung metastasis in a triple-negative breast cancer xenograft model. Mol. Cancer. Ther. 14, 2773–2781 10.1158/1535-7163.MCT-15-0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu H., Ippolito G. C., Wall J. K., Niu T., Probst L., Lee B. S., Pulford K., Banham A. H., Stockwin L., Shaffer A. L., Staudt L. M., Das C., Dyer M. J., and Tucker P. W. (2006) Functional studies of BCL11A: characterization of the conserved BCL11A-XL splice variant and its interaction with BCL6 in nuclear paraspeckles of germinal center B cells. Mol. Cancer 5, 18 10.1186/1476-4598-5-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brueffer C., Vallon-Christersson J., Grabau D., Ehinger A., Häkkinen J., Hegardt C., Malina J., Chen Y., Bendahl P.-O., Manjer J., Malmberg M., Larsson C., Loman N., Rydén L., Borg Å., et al. (2018) Clinical value of RNA sequencing–based classifiers for prediction of the five conventional breast cancer biomarkers: a report from the population-based multicenter Sweden Cancerome Analysis Network—Breast Initiative. JCO Precision Oncol. 2018, 1–18 10.1200/PO.17.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han H., Irimia M., Ross P. J., Sung H.-K., Alipanahi B., David L., Golipour A., Gabut M., Michael I. P., Nachman E. N., Wang E., Trcka D., Thompson T., O'Hanlon D., Slobodeniuc V., et al. (2013) MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498, 241–245 10.1038/nature12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davis J., Salomonis N., Ghearing N., Lin S.-C. J., Kwong J. Q., Mohan A., Swanson M. S., and Molkentin J. D. (2015) MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat. Commun. 6, 10084 10.1038/ncomms10084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Venables J. P., Lapasset L., Gadea G., Fort P., Klinck R., Irimia M., Vignal E., Thibault P., Prinos P., Chabot B., Abou Elela S., Roux P., Lemaitre J.-M., and Tazi J. (2013) MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat. Commun. 4, 2480 10.1038/ncomms3480 [DOI] [PubMed] [Google Scholar]
- 36. The E. P. C., Dunham I., Kundaje A., Aldred S. F., Collins P. J., Davis C. A., Doyle F., Epstein C. B., Frietze S., Harrow J., Kaul R., Khatun J., Lajoie B. R., Landt S. G., Lee B.-K., et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davis C. A., Hitz B. C., Sloan C. A., Chan E. T., Davidson J. M., Gabdank I., Hilton J. A., Jain K., Baymuradov U. K., Narayanan A. K., Onate K. C., Graham K., Miyasato S. R., Dreszer T. R., Strattan J. S., et al. (2018) The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 10.1093/nar/gkx1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fish L., Pencheva N., Goodarzi H., Tran H., Yoshida M., and Tavazoie S. F. (2016) Muscleblind-like 1 suppresses breast cancer metastatic colonization and stabilizes metastasis suppressor transcripts. Genes Dev. 30, 386–398 10.1101/gad.270645.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Curtis C., Shah S. P., Chin S. F., Turashvili G., Rueda O. M., Dunning M. J., Speed D., Lynch A. G., Samarajiwa S., Yuan Y., Graf S., Ha G., Haffari G., Bashashati A., Russell R., METABRIC Group. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 10.1038/nature10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou Z., Qu J., He L., Peng H., Chen P., and Zhou Y. (2018) α6-Integrin alternative splicing: distinct cytoplasmic variants in stem cell fate specification and niche interaction. Stem Cell Res. Ther. 9, 122–122 10.1186/s13287-018-0868-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ali H. R., Dawson S.-J., Blows F. M., Provenzano E., Pharoah P. D., and Caldas C. J. B. C. R. (2011) Cancer stem cell markers in breast cancer: pathological, clinical and prognostic significance. Breast Cancer Res. 13, R118 10.1186/bcr3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cariati M., Naderi A., Brown J. P., Smalley M. J., Pinder S. E., Caldas C., and Purushotham A. D. (2008) Alpha-6 integrin is necessary for the tumourigenicity of a stem cell-like subpopulation within the MCF7 breast cancer cell line. Int. J. Cancer 122, 298–304 10.1002/ijc.23103 [DOI] [PubMed] [Google Scholar]
- 43. Brooks D. L. P., Schwab L. P., Krutilina R., Parke D. N., Sethuraman A., Hoogewijs D., Schörg A., Gotwald L., Fan M., Wenger R. H., and Seagroves T. N. (2016) ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol. Cancer 15, 26 10.1186/s12943-016-0510-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goel H. L., Gritsko T., Pursell B., Chang C., Shultz L. D., Greiner D. L., Norum J. H., Toftgard R., Shaw L. M., and Mercurio A. M. (2014) Regulated splicing of the α6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. Cell Rep. 7, 747–761 10.1016/j.celrep.2014.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu P., Takai K., Weaver V. M., and Werb Z. (2011) Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 3, a005058–a005058 10.1101/cshperspect.a005058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jin H., and Varner J. (2004) Integrins: roles in cancer development and as treatment targets. Br. J. Cancer 90, 561–565 10.1038/sj.bjc.6601576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tabaglio T., Low D. H., Teo W. K. L., Goy P. A., Cywoniuk P., Wollmann H., Ho J., Tan D., Aw J., Pavesi A., Sobczak K., Wee D. K. B., and Guccione E. (2018) MBNL1 alternative splicing isoforms play opposing roles in cancer. Life Sci. Alliance 1, e201800157 10.26508/lsa.201800157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang P.-Y., Chang K.-T., Lin Y.-M., Kuo T.-Y., and Wang G.-S. (2018) Ubiquitination of MBNL1 is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep. 22, 2294–2306 10.1016/j.celrep.2018.02.025 [DOI] [PubMed] [Google Scholar]
- 49. Sun X., Wang J., Yao X., Zheng W., Mao Y., Lan T., Wang L., Sun Y., Zhang X., Zhao Q., Zhao J., Xiao R.-P., Zhang X., Ji G., and Rao Y. (2019) A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov. 5, 10 10.1038/s41421-018-0079-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu P., Keller J. R., Ortiz M., Tessarollo L., Rachel R. A., Nakamura T., Jenkins N. A., and Copeland N. G. (2003) Bcl11a is essential for normal lymphoid development. Nat. Immunol. 4, 525–532 10.1038/ni925 [DOI] [PubMed] [Google Scholar]
- 51. Sankaran V. G., Menne T. F., Xu J., Akie T. E., Lettre G., Van Handel B., Mikkola H. K. A., Hirschhorn J. N., Cantor A. B., and Orkin S. H. (2008) Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842 10.1126/science.1165409 [DOI] [PubMed] [Google Scholar]
- 52. John A., Brylka H., Wiegreffe C., Simon R., Liu P., Jüttner R., Crenshaw E. B., Luyten F. P., Jenkins N. A., Copeland N. G., Birchmeier C., and Britsch S. (2012) Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development 139, 1831–1841 10.1242/dev.072850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wiegreffe C., Simon R., Peschkes K., Kling C., Strehle M., Cheng J., Srivatsa S., Liu P., Jenkins N. A., Copeland N. G., Tarabykin V., and Britsch S. (2015) Bcl11a (Ctip1) controls migration of cortical projection neurons through regulation of Sema3c. Neuron 87, 311–325 10.1016/j.neuron.2015.06.023 [DOI] [PubMed] [Google Scholar]
- 54. Li S., Teegarden A., Bauer E. M., Choi J., Messaddeq N., Hendrix D. A., Ganguli-Indra G., Leid M., and Indra A. K. (2017) Transcription factor CTIP1/BCL11A regulates epidermal differentiation and lipid metabolism during skin development. Sci. Rep. 7, 13427–13427 10.1038/s41598-017-13347-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vieira A. F., Ricardo S., Ablett M. P., Dionísio M. R., Mendes N., Albergaria A., Farnie G., Gerhard R., Cameselle-Teijeiro J. F., Seruca R., Schmitt F., Clarke R. B., and Paredes J. (2012) P-cadherin is coexpressed with CD44 and CD49f and mediates stem cell properties in basal-like breast cancer. Stem Cells 30, 854–864 10.1002/stem.1075 [DOI] [PubMed] [Google Scholar]
- 56. Shehata M., Teschendorff A., Sharp G., Novcic N., Russell I. A., Avril S., Prater M., Eirew P., Caldas C., Watson C. J., and Stingl J. (2012) Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 14, R134 10.1186/bcr3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eirew P., Stingl J., Raouf A., Turashvili G., Aparicio S., Emerman J. T., and Eaves C. J. (2008) A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat. Med. 14, 1384–1389 10.1038/nm.1791 [DOI] [PubMed] [Google Scholar]
- 58. To K., Fotovati A., Reipas K. M., Law J. H., Hu K., Wang J., Astanehe A., Davies A. H., Lee L., Stratford A. L., Raouf A., Johnson P., Berquin I. M., Royer H.-D., Eaves C. J., et al. (2010) Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 70, 2840–2851 10.1158/0008-5472.CAN-09-3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cooper H. M., Tamura R. N., and Quaranta V. (1991) The major laminin receptor of mouse embryonic stem cells is a novel isoform of the alpha 6 beta 1 integrin. J. Cell Biol. 115, 843–850 10.1083/jcb.115.3.843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jiang R., and Grabel L. B. (1995) Function and differential regulation of the α6 integrin isoforms during parietal endoderm differentiation. Exp. Cell Res. 217, 195–204 10.1006/excr.1995.1079 [DOI] [PubMed] [Google Scholar]
- 61. Morini M., Piccini D., De Santanna A., Levi G., Barbieri O., and Astigiano S. (1999) Localization and expression of integrin subunits in the embryoid bodies of F9 teratocarcinoma cells. Exp. Cell Res. 247, 114–122 10.1006/excr.1998.4344 [DOI] [PubMed] [Google Scholar]
- 62. El Mourabit H., Poinat P., Koster J., Sondermann H., Wixler V., Wegener E., Laplantine E., Geerts D., Georges-Labouesse E., Sonnenberg A., and Aumailley M. (2002) The PDZ domain of TIP-2/GIPC interacts with the C-terminus of the integrin α5 and α6 subunits. Matrix Biol. 21, 207–214 10.1016/S0945-053X(01)00198-6 [DOI] [PubMed] [Google Scholar]
- 63. El Marabti E., and Younis I. (2018) The cancer spliceome: reprograming of alternative splicing in cancer. Front. Mol. Biosci. 5, 80–80 10.3389/fmolb.2018.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sveen A., Kilpinen S., Ruusulehto A., Lothe R. A., and Skotheim R. I. (2016) Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35, 2413–2427 10.1038/onc.2015.318 [DOI] [PubMed] [Google Scholar]
- 65. Dassi E. (2017) Handshakes and fights: the regulatory interplay of RNA-binding proteins. Front. Mol. Biosci. 4, 67 10.3389/fmolb.2017.00067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Terenzi F., and Ladd A. N. (2010) Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol. 7, 43–55 10.4161/rna.7.1.10401 [DOI] [PubMed] [Google Scholar]
- 67. Wu J., Anczuków O., Krainer A. R., Zhang M. Q., and Zhang C. (2013) OLego: fast and sensitive mapping of spliced mRNA-Seq reads using small seeds. Nucleic Acids Res. 41, 5149–5163 10.1093/nar/gkt216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yan Q., Weyn-Vanhentenryck S. M., Wu J., Sloan S. A., Zhang Y., Chen K., Wu J. Q., Barres B. A., and Zhang C. (2015) Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc. Natl. Acad. Sci. U S A 112, 3445–3450 10.1073/pnas.1502849112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kamburov A., Pentchev K., Galicka H., Wierling C., Lehrach H., and Herwig R. (2011) ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 39, D712–D717 10.1093/nar/gkq1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kamburov A., Wierling C., Lehrach H., and Herwig R. (2009) ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res. 37, D623–D628 10.1093/nar/gkn698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jézéquel P., Campone M., Gouraud W., Guérin-Charbonnel C., Leux C., Ricolleau G., and Campion L. (2012) bc-GenExMiner: an easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res. Treatment 131, 765–775 10.1007/s10549-011-1457-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq data were deposited in the Gene Expression Omnibus, series GSE149435. All other data are present within the manuscript.