

Abstract
Cysteamine dioxygenase (ADO) has been reported to exhibit two distinct biological functions with a nonheme iron center. It catalyzes oxidation of both cysteamine in sulfur metabolism and N-terminal cysteine-containing proteins or peptides, such as regulator of G protein signaling 5 (RGS5). It thereby preserves oxygen homeostasis in a variety of physiological processes. However, little is known about its catalytic center and how it interacts with these two types of primary substrates in addition to O2. Here, using electron paramagnetic resonance (EPR), Mössbauer, and UV-visible spectroscopies, we explored the binding mode of cysteamine and RGS5 to human and mouse ADO proteins in their physiologically relevant ferrous form. This characterization revealed that in the presence of nitric oxide as a spin probe and oxygen surrogate, both the small molecule and the peptide substrates coordinate the iron center with their free thiols in a monodentate binding mode, in sharp contrast to binding behaviors observed in other thiol dioxygenases. We observed a substrate-bound B-type dinitrosyl iron center complex in ADO, suggesting the possibility of dioxygen binding to the iron ion in a side-on mode. Moreover, we observed substrate-mediated reduction of the iron center from ferric to the ferrous oxidation state. Subsequent MS analysis indicated corresponding disulfide formation of the substrates, suggesting that the presence of the substrate could reactivate ADO to defend against oxidative stress. The findings of this work contribute to the understanding of the substrate interaction in ADO and fill a gap in our knowledge of the substrate specificity of thiol dioxygenases.
Keywords: thiol regulation, EPR spectroscopy, nitric oxide, oxygen activation, oxygen sensing, cysteamine dioxygenase (ADO), cysteine dioxygenase (CDO), sulfur metabolism, thiol dioxygenase, thiol, electron paramagnetic resonance (EPR), dioxygenase, metalloenzyme, sulfur, oxygen binding, thiol regulation
Thiol dioxygenases are a group of nonheme, ferrous enzymes that incorporate two oxygen atoms from molecular oxygen into the thiol groups of their corresponding substrates (1). Among this group of proteins, cysteamine dioxygenase (ADO) (2), cysteine dioxygenase (CDO) (3–6), 3-mercaptopropionate dioxygenase (3MDO) (7–9), and mercaptosuccinate dioxygenase (MSDO) (10) are found to catalyze the oxidation of small molecules, as shown in Fig. 1A. To date, ADO and CDO are the only two known thiol dioxygenases in humans, and they are directly involved in cysteine metabolism and the biosynthesis of hypotaurine and taurine (1). Dysfunction of these thiol dioxygenases is associated with oxidative stress, autoimmune and neurodegenerative diseases (11–18).
Figure 1.

Reactions catalyzed by thiol dioxygenases and superposition of human ADO and CDO structures. A, thiol dioxygenases catalyze the oxidation of thiol-bearing small molecules. Cysteamine is the only small molecule without a carboxylate group among all the substrates. B, thiol dioxygenases oxidize selective peptides containing cysteine at the N terminus. C, superposed structures of a predicted model of ADO (green) and a ternary complex of CDO (white, PDB code 6N43). The substrate of CDO, cysteine, extensively interacts with the second sphere residues, including Cys93-Tyr157 cross-link, Arg60, and Tyr58. Cys220-Tyr222 cross-link of ADO locates at a position opposite that of CDO.
Additionally, some thiol dioxygenases oxidize N-terminal cysteine to cysteine sulfinic acid in polypeptides to regulate protein stability (Fig. 1B). For example, plant cysteine oxidase (PCO) promotes the N-terminal thiol dioxygenation of VII ethylene response factors in normoxia to precede arginylation, resulting in proteasomal degradation (19). Recently, this type of posttranslational modification mediated by thiol dioxygenases was reported in ADO (20). Analogous to PCO in plants, ADO has been proposed to function as a conserved oxygen sensor in animals, regulating the degradation of N-cysteine proteins, including regulator of G signaling 5 (RGS5), and, thus, transduces cellular responses to hypoxia. RGS5 belongs to the R4 subfamily of RGS proteins that are known to negatively regulate the signaling of G protein-coupled receptors (21). Enriched in cardiovascular tissues, RGS5 is related to pericyte maturation, vascular functions, and hypoxia-induced apoptosis (22–24). As an essential modulator, RGS5 is also one of the most effective substrates of human ADO, with kcat of 16.9 s−1 and KM of 71.5 μm (20). In contrast, CDO does not possess such dioxygenation activity on N-cysteine peptides. Overall, ADO is not only part of the thiol metabolism cycle, regulating through its dioxygenase activity of cysteamine, but also has functions which extend to oxygen homeostasis and signal transduction in mammals. The biological significance of ADO is increasingly appreciated; thus, exploring its catalytic center and especially how it responds to small-molecule and protein substrates is needed.
A protein-derived Cys-Tyr cofactor is autocatalytically generated in mammalian ADO and CDO to amplify their enzymatic activities when the substrate level rises (25–27). This substrate-initiated enzyme posttranslational modification and activity amplification is likely a mechanism for maintaining the physiologically significant, free thiol-containing substances at a proper cellular level. The active-site geometry and substrate binding mode of CDO in the ternary complex with a mature Cys-Tyr cofactor is revealed by the X-ray crystal structure with nitric oxide (•NO) as an O2 surrogate (28). The Cys-Tyr cofactor is formed by Cys93 and Tyr157 through a thioether bond located ∼5 Å from the iron center in CDO (Fig. 1C). Its presence increases the catalytic rate by 10-fold (25). The substrate cysteine binds to the iron center in a bidentate fashion with both its amine and thiol groups. Together with an end-on bound •NO, the iron center forms octahedral coordination. This active-site architecture in the complex resembles the persulfenate species that was structurally characterized in CDO, but its catalytic competence remains to be established (29). The Cys-Tyr cofactor of CDO not only interacts with •NO but also forms hydrogen bonds with both the amine and carboxylate groups of cysteine. In addition to the cross-linked Tyr157, the carboxylate group of the substrate is also recognized by other second-sphere residues, such as Tyr58 and Arg60 (Fig. 1C). Such extensive interactions assist the correct positioning of cysteine in the process of enzyme-substrate (ES) complex formation.
Although ADO is a sibling enzyme of CDO catalyzing a similar reaction, the structural features and substrate binding of ADO are much less understood because of the absence of a crystal structure. However, it is important to characterize how the ADO active site handles both small-molecule and protein or peptide substrates, a feature that is so far unique among thiol dioxygenases. Our previous study identified the presence of a Cys-Tyr cofactor in ADO through noncanonical amino acid incorporation via the genetic code expansion strategy, followed by high-resolution mass spectrometric analysis (27). In contrast with the cofactor of CDO, which is formed by two residues distant from each other (63 residues apart) leading to a global structural change (30), the Cys-Tyr cofactor in ADO is formed by two adjacent residues, Cys220 and Tyr222, and the structures of cross-linked and uncross-linked proteins have little global difference that is undetectable by low-resolution techniques such as SDS-PAGE (27). Understandably, a predicted structure of ADO indicates a different location of the Cys-Tyr cofactor relative to the iron center of CDO (Fig. 1C). Moreover, amino acid sequence alignment suggests residues responsible for substrate recognition in CDO are mostly missing in ADO, except for Tyr58 (Fig. S1). Therefore, although some structural and mechanistic similarity is generally anticipated between ADO and CDO because of the catalytic reactions they promote, the substrate-binding mode of ADO cannot be simply extrapolated because of intrinsic differences.
Here, we conducted an EPR-centered spectroscopic study of ADO from both human and mouse origin. The two proteins share 85.6% amino acid sequence identity (Fig. S1). We have found that the substrate of the ADO, either cysteamine or the N terminus of RGS5, is capable of reducing the iron center by forming a disulfide product if the enzyme is in the ferric oxidation state. The spectroscopic data also reveal a new binding mode of the substrate, monodentate coordination through the thiol group. This study fills the gap in the substrate specificity of the thiol dioxygenases for promoting a reaction without a free carboxylate group.
Results
Enzyme reactivation through chemical reduction of the ferric iron center by the primary substrate
Considering that cysteamine, as well as the cysteine-containing peptide substrates, contains a free thiol, we tested if these ADO substrates can reduce the ferric form of the enzyme to the ferrous state. X-band continuous-wave EPR spectroscopy was used to monitor the change of the oxidation state of the iron center upon incubation with cysteamine or the N terminus of RGS5. After exposure to air overnight at 4 °C, the as-purified ADO became oxidized, and the EPR spectrum showed a notable species with a central g value of 4.30. This g = 4.30 feature originates from the middle doublet of a rhombic (E/D = ∼1/3), high-spin (S = 5/2) nonheme ferric iron coordinated by protein ligands (Fig. 2A, black trace). A few minor EPR signals were observed at g = 9.31, 7.25, and 5.82. The g = 9.31 feature can be attributed to the overlapping resonances within the lowest- and highest-lying doublets of the S = 5/2 spin system, whereas other minor features are likely because of inhomogeneity of the iron center that will be discussed later. After anaerobically mixing with the substrate cysteamine and incubating for 1 min, the major ferric EPR signal of the oxidized ADO was diminished, and its resonance at g = 4.30 broadened slightly (Fig. 2A). Additionally, new spectral features at g = 9.22 and 4.73 arose, concomitant with the disappearance of the minor species observed in the spectrum of FeIII-ADO. The noticeable shift of the g value and the appearance of new resonance after cysteamine addition are tentatively attributed to substrate ligation to the ferric iron center. Similar EPR features have been observed in samples of as-isolated desulfoferrodoxin (36) and neelaredoxin (37), two nonheme iron-binding proteins coordinated by cysteinyl-sulfur ligands. Hence, based on literature precedence, the thiol moiety of cysteamine is expected to participate in iron coordination, although at this stage it is not definitive. After 3- and 6-min substrate incubations, the high-spin ferric signal at g = 4.30 continuously decreased, along with the resonances at g = 9.22 and 4.73. After a 10-min incubation, only a minor fraction of g = 4.30 remained, and all other resonances were no longer detectable. This experiment confirmed that cysteamine can reduce ferric ADO to the EPR silent, ferrous state.
Figure 2.

Continuous-wave EPR spectra of time-resolved incubation of oxidized human ADO with cysteamine (A) and N terminus (B) of RGS5. Oxidized FeIII-ADO exhibited a major high-spin (S = 5/2) signal centered at g = 4.30 with multiple low-field resonances (inset, amplified 6 times) (black trace). FeIII-ADO was incubated with 30 mm cysteamine (blue traces) and RGS5 peptide (red traces) for 1, 3, 6, and 10 min. The EPR signal intensity is present in arbitrary units. Spectra were recorded at 10 K with 0.2 mW microwave power.
Next, the same anaerobic incubation was done with the peptide substrate. The first 14 amino acids of the Met-excised N terminus of RGS5 was chemically synthesized with a sequence of CKGLAALPHSCLER by following a previous study (20). A similar phenomenon was observed when the N terminus of RGS5 was incubated with ADO (Fig. 2B). After incubation with the peptide substrate for 1 min, the high-spin ferric signal centered at g = 4.30 was converted to a new signal at g = 4.42 and 4.26 with a decreased ferric population; small resonances at g = 9.13 and 5.02 were present. These new signals have different g values compared with the samples incubated with cysteamine, which suggests the electronic structure of the ferric iron center differs upon cysteamine or peptide ligation, though both substrates are able to reduce FeIII-ADO. At longer incubation times of 3 and 6 min, the primary ferric signal, as well as the minor resonances, continuously decreased. After a 10-min incubation, only a small fraction of the major ferric species remained oxidized, and the minor resonances were nearly absent. Together, the above results revealed that both the cysteamine and N-terminal cysteine-containing peptide could readily reduce the ferric center of ADO through their corresponding thiol groups, implicating thiolate ligation in both cases.
It was previously unknown that the primary substrate of ADO can reduce its ferric ion to the catalytically active ferrous form. To verify this finding, especially that the disappearance of the nonheme ferric ion EPR signal upon substrate binding was because of the change of the metal oxidation state rather than the relaxation caused spectral line broadening of the EPR signal, a separate set of EPR experiments was conducted on mouse ADO (Fig. 3A). The mouse ADO lacks 12 amino acids that are present in the N terminus of the human protein. However, this region is not directly related to the active site (Fig. S1). The mouse protein can tolerate chemical oxidation. Thus, ferricyanide was employed to oxidize as-isolated ADO for 5 h. After removing excess oxidant by desalting and buffer exchange, the oxidized ADO (Fig. 3B) was made anaerobic and mixed with 50 mm cysteamine under O2-free conditions. A putative cysteamine-bound high-spin ferric species was shown concomitantly with a reduction of the EPR signal (Fig. 3, C and D). Finally, the high-spin ferric EPR signal of the oxidized ADO decreased slowly and completely disappeared after 20 min of cysteamine incubation (Fig. 3E). Exposure of the substrate-reduced ADO to nitric oxide (•NO) under O2-free conditions led to the formation of an Fe(II)-nitrosyl complex (Fig. 3F), whose electronic structure will be further characterized below. Together, the results from human ADO and mouse ADO unambiguously demonstrate that the substrate of the enzyme is capable of reducing the catalytically inactive ferric center to the enzymatically active ferrous state.
Figure 3.
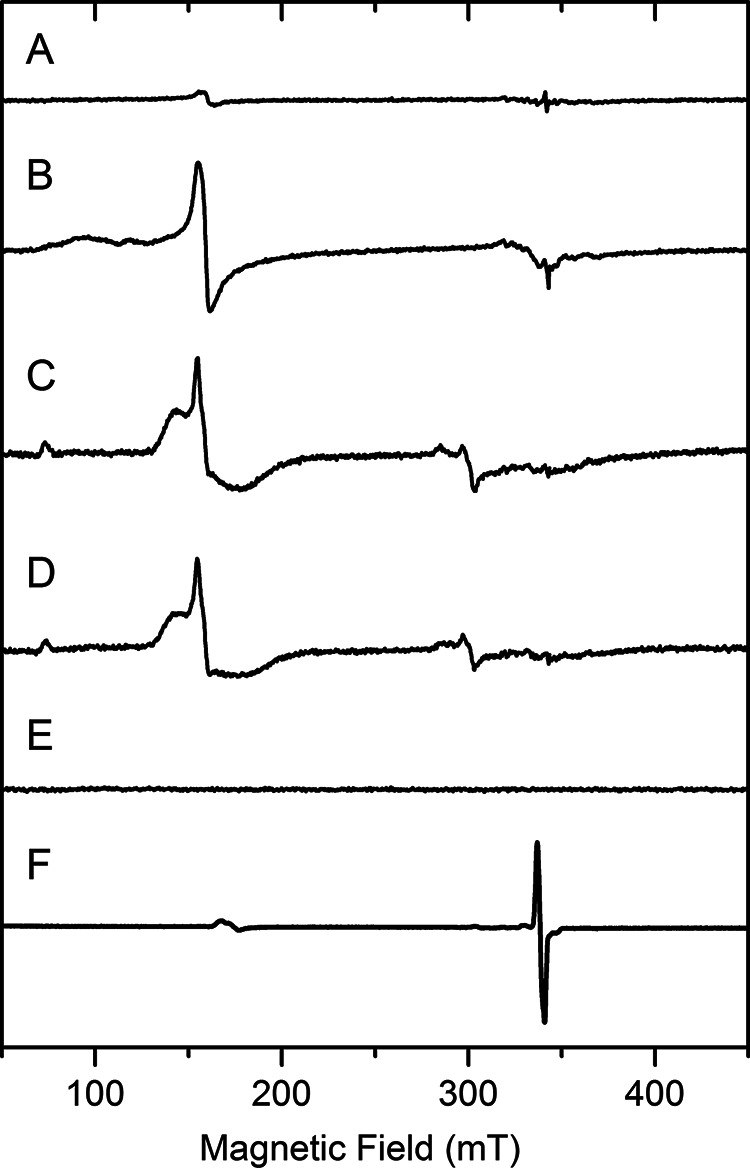
EPR spectra demonstrating cysteamine reduction of the ferric center of mouse ADO. These are freshly prepared, as-isolated mouse ADO (A), after oxidation by ferricyanide and desalting (B), incubation with 50 mm cysteamine for 1.5 min (C), 5 min after cysteamine incubation (D), 20 min after cysteamine incubation (E), and addition of •NO (F). The signal intensity is presented at an arbitrary scale to put each spectrum in approximately the same height for easy comparison. Spectra were recorded at 10 K with 1.0 mW microwave power and 0.5 mT modulation.
Characterization of the ADO iron center by Mössbauer spectroscopy
To elaborate the substrate-induced iron reduction and analyze the iron inhomogeneity in the ligand-free ADO by an independent technique, we executed a Mössbauer spectroscopic characterization of the iron center of as-isolated mouse ADO. The concentrated, as-isolated ADO protein was analyzed by Mössbauer spectroscopy at 4.2 K. For resting ADO, the observed zero-field Mössbauer spectrum was fitted to three species. As shown in Fig. 4A, ∼23% of the total iron was assigned to a ferric doublet (red trace, δ = 0.27 mm/s; ΔEQ = 0.47 mm/s), and the remaining 77% was in the ferrous state. The 77% ferrous iron was composed of two quadrupole doublets, FeIIA [purple trace, δA = 1.16 mm/s; ΔEQ(A)= 2.45 mm/s] and FeIIB [green trace, δB = 1.26 mm/s; ΔEQ(B) = 2.93 mm/s], with the ratio 1:2.2. The quadrupole doublets and isomer shift values of FeIIA and FeIIB suggest a high-spin ferrous iron coordinated by N- and O-atom ligands, forming penta- or hexa-coordination. All thiol dioxygenases known so far possess a 3-His motif as the first sphere metal coordination in a cupin fold (1), and this is also expected to be the case for ADO (27). Hence, the iron center presumably contains two or three solvent-derived ligands. The multiple species observed also suggest that the geometry of the iron center is inhomogeneous. The Mössbauer spectrum is largely in accord with the results reported for other thiol dioxygenases with the 3-His coordination motif (9, 31, 38).
Figure 4.

Mössbauer spectra of 57Fe-enriched ADO. A, as-isolated ADO. Spectrum fitted with three components, FeIII (red), FeIIA (purple), and FeIIB (green). B, ADO with the addition of 30 mm cysteamine. The spectrum was fitted to three components, FeIII (red), FeIIA (purple), FeIIc (blue). C, the overlay of the subtracted spectra of the active enzyme alone (green line) and ES complex (blue line). All spectra were collected at 4.2 K with 50 mT applied field parallel to γ-radiation.
To pinpoint how the iron center responds to the addition of cysteamine, the binary complex was characterized (Fig. 4B). The spectral fitting indicates that there was still one ferric and two ferrous species present in ADO upon substrate incubation. The quadrupole splitting and isomer shift of the ferric species (red trace) had no measurable change; however, its population decreased to 14%. The FeIIA species (purple trace) also maintained the same δ and ΔEQ values but with a reduced population. Notably, the FeIIB species in the resting ADO (green trace) completely disappeared, concomitant with the appearance of a new, predominant ferrous species, termed FeIIC [blue trace, δC = 1.19 mm/s, ΔEQ(C) = 2.85 mm/s] that comprises 72% of the iron content. The Mössbauer parameters are summarized in Table 1. This observation suggests that FeIIB is directly responsible for the substrate binding and reduction, and the newly evolved FeIIC is the binary ES complex. Moreover, the addition of cysteamine induces homogeneity of the iron species by converting the inactive species, FeIII and FeIIA, to the active form, FeIIB.
Table 1.
Mössbauer parameters of as-isolated and cysteamine-incubated ADO
| ADO and Fe species | δ (mm/s) | ΔE (mm/s) | ΓLa (mm/s) | ΓR (mm/s) | % of total iron |
|---|---|---|---|---|---|
| As-isolated | |||||
| FeIII | 0.27 | 0.47 | 0.51 | 0.51 | 23 |
| FeIIA | 1.16 | 2.45 | 0.34 | 0.31 | 24 |
| FeIIB | 1.26 | 2.93 | 0.34 | 0.34 | 53 |
| Cysteamine incubated | |||||
| FeIII | 0.27 | 0.47 | 0.40 | 0.40 | 14 |
| FeIIA | 1.16 | 2.45 | 0.31 | 0.31 | 14 |
| FeIIC | 1.19 | 2.85 | 0.40 | 0.43 | 72 |
a Spectra were fitted using Voigt line shape.
By subtracting the fitted signals of FeIII and FeIIA from the experimental spectra, the spectra of active enzyme alone (green line) and ES complex (blue line) are shown in Fig. 4C. Comparing the subtracted spectra reveals a change to both isomer shift and quadrupole splitting of the iron center upon substrate binding. The decrease of the δ value, from 1.26 mm/s of the substrate-free enzyme to 1.19 mm/s of the ES complex, agrees with sulfur atom coordination to the ferrous center (9, 38, 39).
Detection of by-products in the substrate-mediated enzyme reactivation
Based on the Mössbauer and EPR analyses, the reduction of FeIII-ADO occurred through the interaction between the thiol of the substrate and the iron center. The reactivation of ADO from the ferric form to the catalytically active ferrous form is at the expense of substrate oxidation. The process forming disulfide product could proceed through the following reaction (Equation 1):
| (Eq. 1) |
To investigate whether a fraction of the substrate forms R-S-S-R, cysteamine was anaerobically incubated with the oxidized mouse ADO at room temperature for one hour. Cysteamine is known to be readily oxidized to cystamine, which is a dimerized product with a disulfide bridge. Thus, commercially purchased cysteamine unavoidably contains cystamine impurity. A control sample was prepared with cysteamine in the absence of ADO. Samples were then filtered and analyzed by LC-MS. As shown in Fig. S2, although a portion of cysteamine was already dimerized and eluted as cystamine in the control, the amount of cystamine had a noticeable increase after anaerobic incubation with the oxidized enzyme. This shows that the oxidized product is indeed the substrate dimer with a disulfide bond.
The most appealing conclusion was, however, derived from the experiment with the peptide substrate. The N-terminal peptide contains 14 amino acids (CKGLAALPHSCLER), which should have a larger steric hindrance than cysteamine. Although we did observe the N-terminal RGS5 peptide could readily reduce the iron center and reactivate the ferric form of ADO to the catalytically active form, whether or not the dimeric peptide can still be formed as the product was in question. We next executed the experiment with the RGS5 peptide and human ADO. Similarly, RGS5 (5 mm) was anaerobically incubated with oxidized ADO for one hour and analyzed using high-resolution MS. The control of peptide alone generated a spectrum of pure RGS5 peptide with peaks carrying different charges (Fig. 5A). The m/z values of 375.1975, 499.9274, and 749.3871 represent the intact peptide at +4, +3, and +2 charge states, respectively, with a mass error of less than 1.1 ppm. The heavier species shown at m/z of 999.1814 corresponds to a dimer of two nonspecifically interacted peptide molecules at the +3 charge state. After incubation with the oxidized enzyme, the mass spectrum of the peptide appeared differently (Fig. 5B). The peaks with m/z of 374.6933, 499.2552, and 748.3788 are from a modified peptide at +4, +3, and +2 charge states, respectively (marked in red). The loss of 2 Da compared with the intact peptide indicates the modified peptide has two fewer protons (mass error less than 0.3 ppm). Considering there is another cysteine residue present in the peptide besides the N-terminal cysteine, most likely an intramolecular disulfide bond was formed between these two cysteines. Additionally, peaks with m/z of 599.3061 and 998.1714 were also found in the spectrum (marked in blue), corresponding to a dimerized peptide with a two-proton loss (mass error less than 2.9 ppm), which is presumably an intermolecular disulfide product formed between two N-terminal cysteines, though the population of the intramolecular disulfide peptide was overwhelming compared with the intermolecular disulfide peptide. It is worth mentioning that a nonspecific dimer of the intramolecular disulfide peptide was also observed but with relatively low intensity, and its m/z peaks partially overlapped ones of the intermolecular cross-linked dimer. The presence of both intra- and intermolecular disulfide peptides suggests that after activation in the active site of ADO, the thiol group of the peptide substrate diffuses out to the solution and reacts with another free thiol to form the disulfide product. It is more favored when encountering the thiol within the same peptide chain. Because the RGS5 peptide contained cross-linked dimer in controls only after ADO reactivation, these results provide strong support for Equation 1. Notably, other than disulfide products, the highly reactive substrate thiol radicals also randomly reacted with surface cysteine residues to form a disulfide bond, as we observed by intact protein MS. For such a reason, substrate or product quantitation was not further pursued to investigate the precise stoichiometry of the reduction.
Figure 5.

MS analysis of RGS5 peptide before (A) and after (B) incubation with oxidized human ADO. Charge states and m/z values of RGS5 N terminus, intramolecular cross-linked peptide, and intermolecular cross-linked peptide are marked in black, red, and blue, respectively. The putative structures and theoretical molecular weights are shown on the right.
Cysteamine alters the binding behavior of nitric oxide to the nonheme iron center of ADO
In the resting state, nonheme iron-dependent thiol dioxygenases contain a mononuclear ferrous iron coordinated by three histidine residues in their active sites. Unlike heme-dependent iron proteins, which have unique UV-visible, EPR, and other spectroscopic features, these nonheme iron centers lack representative spectroscopic characteristics to monitor the process of substrate binding and reaction turnovers. Historically, in spectroscopic studies, using •NO as a spin probe and an oxygen surrogate to form an EPR-visible nitrosyl complex is a common method to investigate how primary substrate and dioxygen interact with an EPR-silent ferrous center.
Initially, we sought to test the binding behavior of •NO to FeII-ADO. The reduced FeII-ADO protein was completely EPR silent (Fig. 6A, black trace). Addition of 0.1 equivalent (equiv) of •NO releasing agent to the ferrous enzyme resulted in the formation of a high-spin (S = 3/2) resonance with g values of gmax = 4.10, gmid = 3.95, and gmin = 2.00 (gray trace). The g value features correspond to an E/D value of ∼0.01 based on the S = 3/2 rhombogram. This nearly axial EPR signal corresponds to the nonheme ferrous iron center bound to one equivalent of •NO generating {Fe(NO)}7, which can also be described as an antiferromagnetic coupling between a high-spin FeIII (S = 5/2) and a nitroxylate anion (NO–, S = 1) based on the Feltham–Enemark theorem (40). This high-spin signal is similar to the {Fe(NO)}7 species of 3MDO in the presence of its substrate (8). Additional •NO releasing agent (0.5 and 2.5 equiv) was then added to the enzyme, resulting in an increase in the signal intensity of the {Fe(NO)}7 species (light gray and red traces). The high-spin signal reached a plateau after the addition of 2.5 equiv of •NO releasing agent, concomitant with the formation of a minor low-spin axial signal with g⊥ = 2.039 and g//= 2.015. The line shape and g values of this new S = 1/2 signal are reminiscent of a dinitrosyl species found in synthetic and protein-based samples (41–43). However, the dinitrosyl species did not increase much, even when excess •NO was added to the sample, and its population remains insignificant, accounting for 3% of the total FeII-ADO (Fig. S3 and S4), indicating that FeII-ADO tends to bind only 1 equiv of •NO in the absence of substrate. The trace amount of dinitrosyl species only forms when excess •NO is present, and the mononitrosyl species is predominant in substrate-free ADO.
Figure 6.

EPR spectra of •NO addition to ADO in the absence (A) and presence (B and C) of cysteamine. •NO was released by 0 (black), 0.1 (gray), 0.5 (light gray), and 2.5 (red) equiv of •NO releasing agent (1 equiv of agent produces approximately 1.5 equiv of •NO). A, in the absence of substrate, changes occur in the low-field (high-spin) region with increasing •NO. Spectra were taken at 10 K with 50 mW microwave power. B, in the presence of substrate, changes occur in both high- and low-spin regions with increasing •NO. Spectra were taken at 10 K, 1 mW. C, in the presence of substrate, changes occur in the g = 2 region with increasing •NO. Spectra were taken at 50 K with 0.05 mW microwave power.
Next, we tested whether the presence of cysteamine would alter the binding behavior of •NO to the ferrous center. •NO was introduced into FeII-ADO precomplexed with cysteamine. The ES complex was completely EPR silent (Fig. 6B, black trace). Once a small amount of •NO releasing agent (0.1 equiv) was added, interestingly, no EPR signal was observed in the high-spin region; rather, a low-spin (S = 1/2) signal was observed with g values of g⊥ = 2.040 and g//= 2.014 (Fig. 6B). This signal is very similar to the signal from dinitrosyl species in the aforementioned substrate-free ADO samples; however, the g values are slightly different. These two signals also responded very differently upon •NO titration. This new low-spin signal produced in the presence of a substrate is more sensitive to the population of •NO (Fig. 6C). Compared with similar signals reported in the literature, it is best explained as a substrate-ligated dinitrosyl iron center (DNIC), i.e. a B-type {Fe(NO)2}9 species that has been reported in other nonheme iron-binding proteins (Table S1) (42–46). The addition of more •NO into the sample increased the DNIC signal, but no {Fe(NO)}7 (S = 3/2) signal was observed throughout the addition of •NO to the ES complex. These experiments show that ligation of the substrate to the iron center increases the binding affinity of •NO and alters the binding stoichiometry of •NO to the Fe. The same DNIC EPR spectra were obtained from cysteamine-reduced ADO samples, as observed from the ascorbate-reduced ADO samples upon exposure to •NO and the addition of cysteamine (Fig. 3F and Fig. S5). Notably, such a dinitrosyl iron complex was not described in previous EPR studies of other thiol dioxygenases. Addition of •NO to cysteine-bound CDO mainly gave rise to a low-spin (S = 1/2), {Fe(NO)}7 complex (47), whereas a high-spin (S = 3/2), {Fe(NO)}7 complex was generated in the case of substrate-bound 3MDO (8).
In this work, ADO samples incubated with •NO generated either mononitrosyl (S = 3/2) or dinitrosyl (S = 1/2) complexes, depending on the presence of the substrate. These complexes responded differently to temperatures and microwave power. Therefore, EPR spectra were recorded under different conditions according to the power saturation profile (Fig. S6).
Cysteamine monodentate coordinates to ADO through thiolate ligation
To further investigate the observation that cysteamine alters the binding behavior of •NO to the iron center, cysteamine was titrated into ADO preincubated with excess •NO. As shown in Fig. 7, A and B, titration of cysteamine gradually converted the high-spin {Fe(NO)}7 complex into the low-spin {Fe(NO)2}9, which is consistent with the stoichiometric change of •NO from one to two. Additionally, the newly produced DNIC EPR signal at g⊥ = 2.040, g//= 2.014 continuously increased upon titration of cysteamine (Fig. 7B). The population of DNIC was further quantitated using a Cu-EDTA standard. As shown in Fig. S4, in the presence of saturating cysteamine (30 mm), the majority of iron centers were converted to DNIC, accounting for 58% of the total iron content. Under the circumstances, there is only one coordination site of iron available for substrate binding, and the line shape and g values suggest monodentate thiolate ligation to the iron center (44, 48).
Figure 7.

The ferrous nitrosyl complex EPR spectra obtained through addition of cysteamine or analogues to human ADO precomplexed with excess •NO. A and B, high-spin (A) and low-spin (B) features of enzyme-nitrosyl complex after addition of 0 (black), 0.2 (gray), 1 (light gray), and 5 (red) mm cysteamine. High-spin and low-spin signals were recorded at 10 K with 50 mW microwave power and at 50 K with 0.05 mW microwave power, respectively. C and D, full scans (C) and low-spin regions (D) of the enzyme-nitrosyl complex after the addition of 30 mm cysteamine (black), β-ME (red), and ethanolamine (blue). Structures of cysteamine, β-ME, and ethanolamine are shown in black, red, and blue, respectively. Full scans and low-spin signals were recorded at 10 K with 1 mW microwave power and at 50 K with 0.05 mW microwave power, respectively.
With the understanding that cysteamine coordinates to the iron center of ADO in a monodentate fashion, we next attempted to determine whether it is indeed the thiol rather than the amine moiety of cysteamine that is responsible for ligation. Substrate analogues ethanolamine and β-mercaptoethanol (β-ME) were added to ADO precomplexed with •NO. Both analogues are comprised of a functionalized ethane chain; ethanolamine replaces the thiol of cysteamine with a hydroxyl, and β-ME replaces the amine with a hydroxyl (Fig. 7C). Substitution of a specific functional group allows for a detailed comparison of the binding mode of the organic substrate through EPR spectroscopy. If the substrate ligates in a bidentate fashion with both amine and thiol groups, the EPR spectra of ethanolamine and β-ME should be distinguishable from the spectrum of cysteamine. In contrast, if the substrate coordinates in a monodentate fashion, the EPR spectrum of cysteamine should have spectroscopic features similar to those of either ethanolamine or β-ME based on which group ligates to iron. Similarity with ethanolamine or β-ME represents amine or thiol coordination, respectively.
Substrate-free ADO exposed to •NO had a predominant high-spin (S = 3/2) {Fe(NO)}7 and a minor low-spin (S = 1/2) dinitrosyl species (Fig. 6A and Fig. S3). The addition of 30 mm ethanolamine did not cause obvious change to the EPR spectrum, and the high-spin {Fe(NO)}7 remained as the majority of the iron signal (Fig. 7C, blue trace). A trace amount of dinitrosyl species (4% of total iron) at g⊥ = 2.039, g//= 2.015 was seen in the spectrum of ethanolamine sample because of the excess of •NO, regardless of the addition of organic substrate (Fig. 7D, blue trace). This result indicates that ethanolamine has no specific binding to the metal center of ADO. Alternatively, in the presence of β-ME, the low-field region became nearly featureless, whereas the prominent signal was a low-spin (S = 1/2) species (Fig. 7C, red trace). The low-spin dinitrosyl species, which accounts for 56% of the total iron, has spectral features nearly identical to those of the {Fe(NO)2}9 complex observed when cysteamine was present (Fig. 7D, black and red traces). The high-spin {Fe(NO)}7 species without any organic substrate showed near-complete conversion to the {Fe(NO)2}9 complex. The observation of high-spin to low-spin conversion upon addition of β-ME indicates that it coordinates to the iron center in a manner similar to that of cysteamine and that the iron center binds two equiv of •NO as well. The addition of 30 mm cysteamine and β-ME yielded a comparable population of the {Fe(NO)2}9 species at saturating concentrations. Therefore, the replacement of the amine with a hydroxyl has a negligible impact on the coordination geometry and electronic structure of the iron center. Altogether, these data indicate that cysteamine coordinates to the iron center of ADO with its thiol group in a monodentate fashion.
RGS5 peptide coordinates to ADO through the N-terminal thiol
Like cysteamine, the reported N-terminal cysteine-containing protein substrates of ADO have a free thiol and amine. However, we wondered if the peptide chain binds to the iron in the same monodentate manner. Thus, the N terminus of RGS5 peptide was titrated into the enzyme-nitrosyl complex, and the changes were monitored by EPR spectroscopy. As shown in Fig. 8A, with no substrate present, the enzyme-nitrosyl complex had a pronounced high-spin (S = 3/2) {Fe(NO)}7 signal (black trace). Under the experimental conditions (10 K, microwave power of 50 mW), an additional positive absorptive peak was observed at g = 1.98, which was assigned to excess free •NO in solution (49). Upon titration of the peptide substrate, the high-spin signal gradually decreased, consistent with the observation from the samples that underwent cysteamine addition (Fig. 8A). Additionally, the EPR signal of free •NO decreased and completely disappeared upon substrate addition at 5 mm concentration (Fig. 8A, red trace). The decrease of free •NO signal suggests that the free •NO in solution was consumed to form other nitrosyl species.
Figure 8.

EPR and absorbance spectra of RGS5 peptide addition to the ferrous form of human ADO precomplexed with excess •NO. A, ADO-nitrosyl complex after addition of 0 (black), 0.2 (gray), 1 (light gray), and 5 (red) mm RGS5 peptide. Spectra were taken at 10 K with 50 mW microwave power. B, the g = 2 region of enzyme-nitrosyl complexes after addition of 0 (black), 0.2 (gray), 1 (light gray) and 5 (red) mm RGS5 peptide. An axial signal (*, amplified 9 times in the inset) and a broad signal (#) were observed. Spectra were taken at 50 K with 0.05 mW microwave power. C, UV-visible absorption spectra of FeII-ADO (black), ADO-nitrosyl complex (green), ADO complexed with •NO and cysteamine (blue), and ADO complexed with •NO and RGS5 peptide (red).
Next, the low-spin region of the above samples was analyzed at 50 K. The initial enzyme-nitrosyl complex had a small dinitrosyl signal at g⊥ = 2.039, g//= 2.015 (Fig. 8B, black trace). As expected, after titrating the peptide substrate, the major resonance of the low-spin (S = 3/2), {Fe(NO)2}9 complex shifted to g⊥ = 2.040, g//= 2.014 and intensified with increasing concentration of the peptide. In the presence of 5 mm RGS5 peptide, the majority of the iron center was converted to DNIC, which accounts for 61% of the total iron content (Fig. S4). This observation mirrors the phenomenon caused by cysteamine, which suggests the iron center was coordinated by a thiol group and two •NO molecules, i.e. the peptide monodentate coordinates to the iron center via thiolate ligation. The peptide substrate alters the binding stoichiometry of •NO from one to two equivalents, as cysteamine does. Based on the EPR spectra, the DNIC {Fe(NO)2}9 signal was more responsive upon peptide addition than cysteamine addition, especially at lower concentrations (<1 mm) (Fig. S4). This suggests the iron center of ADO has a higher affinity to the peptide substrate than cysteamine, so that the peptide substrate converts the mononitrosyl complex to the ternary catalytic mimic, thiol-bound {Fe(NO)2}9 complex more efficiently. This interpretation is consistent with the reported Km values of 71.5 and 3400 μm for the peptide and cysteamine, respectively (20, 27).
Other than the predominant dinitrosyl resonances at g⊥ = 2.040, g//= 2.014, two new signals were seen in the low-spin region of peptide samples that were absent from the samples of cysteamine. An axial signal arose at g⊥ = 2.244, g//= 2.131 (Fig. 8B, marked by an asterisk), and a broad signal centered at g = 2.032 (marked by the pound sign) overlapped the major resonance of thiol monodentate-bound, {Fe(NO)2}9 complex. Both signals were very similar to the signals shown in CDO nitrosyl studies, and the g = 2.032 signal corresponds to a characteristic, cysteine bidentate-bound, ferrous-nitrosyl {Fe(NO)}7 complex (28, 47). Such an observation suggests that at high concentrations of a peptide substrate (at mm level), the amine of the peptide interacts with the iron center, forming bidentate coordination, but its population is smaller than that of the predominant, thiol monodentate coordination.
As a nonheme iron-dependent enzyme, the ferrous form of human ADO at the resting state is expected to be colorless and uncharacteristic above 280 nm by UV-visible spectroscopy. However, the concentrated protein (200 μm) showed a pale green color, and its absorbance spectrum exhibited a small peak with a λmax at 405 nm (Fig. 8C, black trace). A similar spectral feature is also present in mouse ADO, with a λmax at 409 nm (Fig. S6). This absorbance feature was only observable in concentrated protein samples and consistently showed in samples after multistep purification and from both human and mouse ADO expression systems. The origin of this absorbance feature remains unresolved. It may result from a small molecule tightly bound to a small fraction of protein during protein expression and cell lysis. Anaerobic addition of •NO gave rise to new, broad peaks centered at 340 and 406 nm (green trace), which resemble the features observed in the nitrosyl samples of other nonheme iron enzymes, including isopenicillin N synthase (IPNS) which catalyzes oxidative thioether formation of a thiol-containing substrate (39). Such absorption bands are attributed to charge transfer transitions associated with the {Fe(NO)}7 complex. The addition of cysteamine resulted in a distinct peak with a λmax at 397 nm, but the peaks observed in the enzyme-nitrosyl complex were no longer present (blue trace). The addition of RGS5 peptide also generated a new peak but with a different λmax at 390 nm and more intensity than that of the cysteamine-bound nitrosyl complex (red trace). The absorbance bands centered at about 400 nm are suggestive of thiol-containing mononuclear DNICs that have been previously documented (50, 51). Therefore, our UV-visible spectroscopic results again confirm the formation of ADO-based dinitrosyl complexes in the presence of substrates. The substrate-bound nitrosyl complex of IPNS had absorbance features at 508 and 720 nm that are quite different from the ADO case, because DNIC was absent in the former. Together with the EPR and absorption spectra, the peptide substrate exhibits binding behavior similar to that of ADO as cysteamine but with higher affinity. Moreover, the thiol-bound, mononuclear DNIC has been independently characterized by both spectroscopies.
Discussion
Among the thiol dioxygenases, CDO has been unambiguously characterized to bind its substrate with bidentate coordination. Without an ES complex structure, the substrate-binding mode of 3MDO is controversial because of the inconsistency between spectroscopic data and a computational study (8, 38, 52), whereas the substrate binding of ADO and MSDO remains undefined. Studies on 3MDO and MSDO consistently suggest an arginine residue (Arg168 in 3MDO from Pseudomonas aeruginosa and Arg66 in MSDO from Variovorax paradoxus strain B4) plays a crucial role in their catalysis, likely interacting through a highly specific salt bridge to the carboxylate group of substrates, which correlates to the function of Arg60 in human CDO (9, 52, 53). Additionally, SbzM and OvoA are two nonheme iron enzymes related to cysteine oxidation but are not yet classified as thiol dioxygenases because of their atypical reactions and different metal ligands. SbzM catalyzes the two-step oxidation and decarboxylation of cysteine (54), whereas OvoA promotes either dioxygenation or dimerization of cysteine as modulated by histidine (55). The substrate binding of neither enzyme has been spectroscopically characterized. As the only thiol substrate without a carboxylate group among these native thiol substrates for this enzyme family (Fig. 1A), lacking the assistance from such an arginine, cysteamine is likely to bind the enzyme in a different manner than the other thiol dioxygenases. Whether cysteamine binds in a bidentate fashion with both amine and thiol coordinated or as a monodentate ligand with either its thiol or amine was an open question before this study. Likewise, substrates of N-terminal cysteine-containing peptides such as RGS5 contain a free thiol and amine as well but lack a carboxylate group (Fig. 1B). Determining the specific binding of these polypeptide substrates in the active site of ADO is of great importance to further understand this biologically significant enzyme.
Incubation with either cysteamine or the N terminus peptide of RGS5 with FeIII-ADO gave rise to respective EPR signals, which suggests the reduction is achieved through direct ligation between substrates and the enzyme. Similar EPR signals have been detected in FeIII-CDO with the incubation of cysteine, but no cysteine-mediated reduction was reported (56). Because the thiol groups of N-terminal cysteine and free cysteine have similar redox potentials and the iron centers of ADO and CDO have identical coordination (3-His motif), it is interesting that the substrate-mediated reduction was only found in the ADO system. A previous EPR study of CDO pointed out that the amine group of l-cysteine is likely to coordinate to the iron center prior to the thiol, and the thiol coordination is relatively transient in the absence of •NO or O2 (47), which is also supported by X-ray absorption and crystallographic data (57, 58). A plausible explanation is that the initial coordination of amine causes a strong d-orbital splitting of iron, resulting in a distinct coordination geometry that hinders further reduction by the thiol. This may explain that in the ADO case, thiol monodentate ligation enables a facile reduction of the metal center.
Mössbauer spectra show that ∼75% of the iron center remained as ferrous iron in as-isolated ADO in the concentrated samples. Although two major ferrous species were observed, FeIIA and FeIIB, only FeIIB is directly related to substrate binding and subsequent catalytic activity. However, there is an interconversion between these two species triggered by substrate addition. The mechanism for ADO preserving such an inhomogenous iron center is unclear, but it seems to be a common feature in some nonheme iron dioxygenases besides thiol dioxygenases (9, 31, 59, 60).
Substrate incubation with oxidized ADO leads to the finding that the active site of ADO could be readily reduced by the substrate thiol group and, hence, reactivate the enzyme if it were to become oxidized. As a result, substrates form disulfide by-products, for example, cysteamine generates cystamine and RGS5 peptide produces either intra- or intermolecular disulfide cross-links. Other peptides whose dioxygenation may be catalyzed by ADO could have different disulfide products based on the location of cysteine, because the formation of a disulfide bond is a nonenzymatic outcome that occurs after the activated thiol is liberated from the active site. Importantly, the substrate-mediated reduction could have a profound impact on oxygen homeostasis. ADO regulates N-terminal cysteine-containing proteins with an extremely high Km for oxygen (>500 μm) (20), which implies that the activity of ADO keeps a linear relationship with the oxygen concentration within the physiological range. The reduction effect of the primary substrates keeps the enzyme ready in an active form to respond as an oxygen sensor, even when facing oxidative stress. Such a substrate-induced reactivation was also reported in a life-essential enzyme, tryptophan 2,3-dioxygenase, that generates the tryptophan dimer and mono-oxygenated tryptophan as the by-products (61). The finding from ADO is another example of how metalloenzymes maintain their catalytic activity to defend against an oxidizing environment, such as that in cancer cells. We have previously shown that when the primary substrate and O2 are both available, ADO autocatalytically generates a protein-bound Cys-Tyr cofactor by the iron center that functions as a catalytic amplifier to more rapidly reduce the level of the free-thiol-bearing substrate (27). Thus, the elevated level of the substrate can reactivate ADO and bring its catalysis up to speed to respond to the rise of thiol levels.
EPR results with nitrosyl complexes of ADO have striking differences from other nonheme iron-dependent enzymes that oxidize thiol-containing substrates, including CDO, 3MDO, and IPNS (Table 2). In the absence of substrate cysteine, the CDO-nitrosyl complex exhibited an S = 3/2 and mononitrosyl {Fe(NO)}7 EPR spectrum with features similar to those of the enzyme-nitrosyl complex of ADO. The substrate bidentate coordination of CDO promotes the generation of a broad S = 1/2, substrate-bound {Fe(NO)}7 signal. A DNIC was also seen in CDO as a minor species (∼5% of the total iron), which was not a substrate-bound enzyme-nitrosyl complex (47). In the absence of a substrate, 3MDO was unable to bind •NO, and its substrate coordination mode is unclear. However, the substrate-bound nitrosyl complex gave rise to an S = 3/2, {Fe(NO)}7 species. This high-spin species showed an EPR spectrum very similar to that of the substrate-free nitrosyl complexes of ADO and CDO, which implies a unique substrate binding mode for 3MDO that differs from both ADO and CDO. IPNS is a well-documented enzyme that catalyzes double-ring closure of a tripeptide substrate that monodentate binds to the iron center via a thiol group (62). Its nitrosyl complex in the absence of the substrate was reported to have EPR features similar to those of ADO and most nonheme iron-dependent proteins (39). In contrast to the low-spin (S = 1/2), {Fe(NO)2}9 species of ADO, the ES-nitrosyl complex of IPNS became more rhombic than substrate-free nitrosyl complex but remained as a high-spin (S = 3/2), {Fe(NO)}7 species. This difference may be because of a different set of first-sphere metal ligand (2-His-2-carboxylate motif in IPNS). In general, ADO exhibits a distinct DNIC among these thiol-oxidizing enzymes.
Table 2.
EPR and UV-visible absorbance spectroscopic parameters of nitrosyl complexes formed in nonheme iron enzymes catalyzing thiol oxidatione
| Protein | Protein ligand | Substrate coordination | Enzyme-nitrosyl complex | ES-nitrosyl complex |
|---|---|---|---|---|
| ADOa | 3-His | Monodentate, via thiol | {Fe(NO)}7, S = 3/2, g = 4.10, 3.95, 2.00; 340, 406 nm | {Fe(NO)2}9, S = 1/2, g = 2.04, 2.01; 390 or 397 nm |
| CDOb | 3-His | Bidentate, via thiol and amine | {Fe(NO)}7, S = 3/2, g ∼ 4, 4, 2 | {Fe(NO)}7, S = 1/2, g = 2.07, 2.02, 1.98 |
| 3MDOc | 3-His | Unclear, thiol involved | Not applicable | {Fe(NO)}7, S = 3/2, g = 4.06, 3.96, 2.00 |
| IPNSd | 2-His-2-carboxylate | Monodentate, via thiol | {Fe(NO)}7, S = 3/2, g = 4.09, 3.95, 2.00; 340, 430, 600 nm | {Fe(NO)}7, S = 3/2, g = 4.22, 3.81, 1.99; 508, 720 nm |
The unique phenomenon of DNIC in ADO involves the binding of its primary substrate, either cysteamine or N-cysteine-containing peptide. DNIC could be generated by mononuclear iron or the [2Fe-2S] or [4Fe-4S] cluster, whereas the mononuclear DNIC is generally more important in terms of biological events (63). As summarized in Table S1, the EPR spectra of DNICs reported in the literature can be classified into A-type and B-type based on anisotropy of resonance, as first proposed by Chasteen and coworkers (42). The DNIC observed in ADO is a B-type mononuclear DNIC with thiol-containing ligands that is commonly described by the formula {Fe(NO)2}9, with two proposed models as [Fe3+(NO−)2] and [Fe+(NO)2] (64). The former contains a high-spin (S = 5/2) d5-configuration of the iron atom and two nitroxyl ligands each at triplet state (S = 1), whereas the latter is a high-spin (S = 3/2) d7-configuration of the iron atom bound to two neutral nitric oxide molecules (S = 1/2). An alternative model for DNIC is [Fe+(NO+)2] with the formula {Fe(NO)2}7, which involves the transfer of electron density from sulfur atoms to iron and nitrosonium ligands (64). The third model is feasible where a soluble thiol-bound DNIC is concerned, especially if the DNIC could act as a •NO donor and trigger the S-nitrosation of the thiol. Further investigation is required to identify the model that best represents the structure and character of the DNIC present in ADO.
Presumably, the ES-nitrosyl complex mimics the ternary structure related to the catalytic reaction. The observation of a DNIC in ADO in the presence of primary substrate leads to the interpretation that both oxygen atoms of molecular oxygen could spontaneously bind to the iron center, forming an octahedral ternary complex after monodentate thiolate ligation. If so, our findings here add a new, side-on oxygen binding model to the thiol dioxygenase family, because previously binding modes were exclusively proposed as end-on (5, 8, 28, 54, 58, 65). A new oxygen binding mode potentially represents a new oxygen activation mechanism during thiol oxidation and Cys-Tyr cross-link biogenesis in thiol dioxygenases. Our finding that cysteamine and N-cysteine peptide bind ADO through the thiol group in a monodentate fashion also supports the idea that the amine group does not participate in either catalysis or cofactor biogenesis. Moreover, a previous study showed that •NO affects the N-terminal cysteine oxidation and the stability of RGS proteins (66), a process that is regulated by ADO (20). The ADO-nitrosyl complexes presented here may provide a direction for investigating how the N-degron pathway acts as an •NO sensor. Hence, even if the ES-nitrosyl complex is not directly relevant to the catalytic mechanism, the DNIC formation in ADO certainly opens a new avenue for correlating ADO with •NO-related physiological study.
Concluding remarks
Thiol dioxygenases are a family of 3-His coordinated ferrous enzymes that activate molecular oxygen and transform thiol substrates to corresponding sulfinates. Their biological significance is in regulating sulfur metabolism and thiol homeostasis in various organisms. Although they share some structural and catalytic features, those enzymes specifically handle different thiol-containing substrates, ranging from small molecules to N-terminal cysteine-containing peptides. As the only enzyme catalyzing the oxidation of substrates without a carboxylate moiety, the iron center of ADO and its interaction with cysteamine and RGS5 peptide were investigated here via biophysical and bioanalytical techniques, including EPR, Mössbauer, UV-visible spectroscopies, and MS. The results suggest that ADO preserves an inhomogeneous ferrous center in the resting state, whereas only one form is directly responsible for substrate binding and subsequent catalysis. Other forms could be readily converted to the active form in the presence of primary substrates. We also found a substrate-mediated reduction of the iron center generating disulfide by-products. These two features compose a substrate-induced reactivation mechanism that enables ADO to function as an oxygen sensor under physiological conditions. •NO, as an oxygen mimic and spin probe, was incubated anaerobically with ADO to examine further the binding behavior of primary substrates and substrate analogues. Our data suggest that substrates could alter the binding stoichiometry of •NO from one equivalent to two, generating a thiolate-bound, low-spin, B-type DNIC signal that was not previously observed in other thiol dioxygenases or oxidases. Such an observation indicates an oxygen side-on binding mode distinct from those of other thiol dioxygenases. The absence of an amine in substrate coordination also implies that the thiol dioxygenase-promoted oxidation and cofactor biogenesis do not require the involvement of an amine. Altogether, our work provides the first systematic investigation of the ferrous center and substrate binding of ADO, and it contributes to the overall understanding of the substrate specificity in thiol dioxygenases.
Materials and methods
Chemicals and protein preparation
Cysteamine (95%), ethanolamine (99%), and 2-mercaptoethanol (99%) were purchased from Acros Organics. Peptide substrate with the sequence of CKGLAALPHSCLER (>95%) was synthesized by Biomatik, which corresponds to the first 14 amino acids of the Met-excised N terminus of RGS5 as reported previously (20).
Cloning, overexpression, and purification of His6-tagged, full-length human ADO were described previously (27). The untagged protein was obtained through the cleavage of His tag by tobacco etch virus protease. The tagged and untagged proteins showed identical activity (kcat of 1.2 s−1 and Km of 3.4 mm cysteamine) (27) and spectral features in this study.
The mouse ADO expression plasmid was a generous gift from Dr. Martha H. Stipanuk (2). Most of the spectroscopic work, except for Mössbauer characterization, was carried out on both the mouse and human proteins. No difference was observed unless otherwise stated. The results presented were mostly from the human version of the protein. Cell culture was grown in a baffled flask with Luria-Bertani medium at 220 rpm, 37 °C until optical density at 600 nm reached 0.8 absorbance units. Isopropyl-β-thiogalactoside and ferrous ammonium sulfate were added to a final concentration of 600 and 50 μm, respectively, before the temperature was lowered to 28 °C. After 12 h of further incubation, the cells were harvested by centrifugation and lysed using a Microfluidizer LM20 cell disruptor. The enzyme was separated from the cell lysis using nickel-nitrilotriacetic acid resin and was then desalted into 100 mm Tris-HCl at pH 8.0 and 50 mm NaCl for future use. Protein concentration was determined by UV-visible absorbance at 280 nm (ε280 = 24,785 cm−1 m−1) (27).
ADO binds its iron ion tightly. However, the iron ion in the protein could be easily reduced by reducing agents such as ascorbate. Hence, the ferrous form of ADO was prepared by supplying the iron source during protein expression and then reducing chemically. The iron occupancy of purified ADO was determined as ∼80% using a ferrozine assay (31).
Enzyme reduction by substrate
The oxidized FeIII-ADO was prepared by exposing the as-purified protein to air at 4 °C in 100 mm Tris-HCl at pH 8.0, 50 mm NaCl overnight. An alternative method to generate oxidized ADO was to incubate the protein with potassium ferric cyanide (10 mm) for 5 h before desalting. Proteins prepared by either air or chemical oxidation showed identical spectroscopic features, but human ADO was less tolerant of chemical reduction. The enzyme then was concentrated to 200 μm and degassed under vacuum. A stock solution of cysteamine or RGS5 peptide (300 mm) was prepared in the same buffer and added to the enzyme to a final concentration of 30 mm under anaerobic conditions. Samples were incubated in an anaerobic chamber, transferred to quartz EPR tubes, and then frozen in liquid nitrogen. Sample incubation and handling were controlled within 1, 3, 6, and 10 min. Spectra were recorded on a Bruker E560 X-band spectrometer equipped with a cryogen-free 4 K temperature system together with an SHQE high-Q resonator (9.4 GHz). Experimental conditions were set at 100-kHz modulation frequency, 1-mT modulation amplitude, 0.2-mW microwave power, and a temperature of 10 K. Each spectrum is the average of 4 scans.
MS
For RGS5-mediated reduction, the peptide (5 mm) was anaerobically incubated with oxidized ADO (200 μm) in NH4HCO3 (50 mm) at pH 7.0. The control sample was 5 mm peptide alone in the buffer. Both samples were incubated at room temperature for 1 h before removal of the enzyme using centrifugal filters. The filtrate was then analyzed by high-resolution MS. Spectra were collected on a maXis plus quadrupole-TOF mass spectrometer equipped with an electrospray ionization source (Bruker Daltonics) and operated in the positive ionization mode. Samples were introduced via a syringe pump at a constant flow rate of 3 μl/min. Important source parameters are summarized as follows: capillary voltage, 3,500 V; end plate offset, –500 V; nebulizer gas pressure, 0.4 bar; dry gas flow rate, 4.0 liters/min; source temperature, 200 °C; collision energy, 5.0 eV. Mass spectra were averages of 1 min of scans collected at a rate of 1 scan per second in the range 50 ≤ m/z ≤ 1500. For cysteamine-mediated reduction, samples were prepared similarly but with 10 mm cysteamine anaerobically. The filtrate was injected into LC-MS for reverse-phase separation after the addition of 50 mm ethanolamine as an internal standard. 50% CH3CN with 0.1% HCOOH was used as the mobile phase. Mass selected for the scan was set at m/z values of 62, 87, and 153 for positive mode.
Preparation and EPR measurement of nitrosyl complexes
O2-free FeII-ADO was freshly prepared to generate nitrosyl complexes. Sodium ascorbate (100 μm) was included to keep the protein in a reduced environment. Nitrosyl samples were prepared by the addition of •NO-releasing agent, DEA-NONOate (≥98%, Cayman Chemical), to FeII-ADO (200 μm) and incubating for 20 min under anaerobic conditions. The amount of •NO in each prepared sample was estimated through titration of •NO releasing agent to the enzyme solution (1.5 mol of •NO can be liberated per mol of DEA NONOate). For substrate or analog titration, small molecules or RGS5 peptide were added to the enzyme-nitrosyl complex to a concentration of 0.2, 1, 5, or 30 mm. For •NO addition experiments, cysteamine (30 mm) was complexed with FeII-ADO (200 μm) prior to the addition of •NO-releasing agent. The samples were transferred to quartz EPR tubes and slowly frozen in liquid nitrogen. EPR spectra were recorded on a Bruker E560 X-band spectrometer equipped with a cryogen-free 4 K temperature system as described elsewhere (32–34), with either a dual mode (9.6 GHz, for small molecules) or an SHQE high-Q resonator (9.4 GHz, for peptide) at 100-kHz modulation frequency, 0.6-mT modulation amplitude. High-spin signals were measured at 10 K with a microwave power of 50 mW and one scan for each spectrum, and low-spin signals were measured at a higher temperature, 50 K, with a lower microwave power of 0.05 mW and an average of four scans for each spectrum. To visualize both features of high- and low-spin EPR signals in the same spectrum, the data acquisition conditions were set to a temperature at 10 K and microwave power of 1.0 mW.
UV-visible absorbance spectroscopy
The preparation of nitrosyl complexes for UV-visible absorbance spectroscopic measurement was identical to the procedure described in the EPR measurement of nitrosyl complexes. 200 μm O2-free FeII-ADO was used in each sample. Excess •NO was precomplexed with enzyme prior to the addition of 10 mm cysteamine and RGS5 peptide. All spectra were recorded in a 1-cm, anaerobic quartz cuvette (SpectrEcology) using a Lambda 25 spectrometer (Perkin Elmer).
Mössbauer spectroscopy
To generate 57Fe-incorporated ADO with high metal occupancy, 57Fe stock solution was supplied to the metal-depleted LB medium during cell culture. The stock solution was prepared by dissolving 57Fe powder (95.4%, Science Engineering & Education Co.) in 2.5 M H2SO4 under anaerobic conditions, which was then anaerobically incubated at 60 °C with continuous stirring until completely dissolved. The stock solution of the pH was drop adjusted by sodium hydroxide, as described previously (35). A final concentration of 80 μm 57Fe was added for cell growth. Expression and purification of ADO were done as aforementioned, and the protein was finally desalted into a 100 mm Tris-HCl, pH 8.0, 25 mm NaCl, and 5% glycerol buffer. The protein was then concentrated to 2–3 mm. A 500-μl aliquot of the concentrated protein was transferred into a Mössbauer cup and frozen in liquid nitrogen. Mössbauer spectra were recorded in a weak-field spectrometer equipped with a Janis 8DT variable-temperature cryostat as described previously (35). The zero velocity of the spectra refers to the centroid of a room-temperature spectrum of a metallic iron foil. For samples of the enzyme-substrate complex, cysteamine was added to a final concentration of 30 mm under anaerobic conditions. All spectra were collected at 4.2 K with 50-mT applied field parallel to γ-radiation. Isomer shifts are reported relative to Fe metal at 298 K. Least square fitting of the spectra was performed with the WMOSS software package (WEB Research, Edina, MN).
Data availability
All of the data are described in Tables 1 and 2 and Fig. 1–8, as well as in Table S1 and Fig. S1–S6.
Supplementary Material
Acknowledgments
This article is dedicated to Dr. Sunil G. Naik, FRSC, a former postdoctoral associate of this team who succumbed to H1N1 virus infection in June 2017. We thank Dr. Martha Stipanuk of Cornell University for the gift of plasmid of mouse ADO and Dr. Boi Hanh (Vincent) Huynh of Emory University for access to Mössbauer instrumentation. We are grateful to Dr. Jiasong Li for helpful discussion.
This article contains supporting information.
Author contributions—Y. W., I. D., Y. C., S. G. N., and W. P. G. data curation; Y. W., I. D., Y. C., S. G. N., W. P. G., and A. L. formal analysis; Y. W., I. D., Y. C., S. G. N., and W. P. G. investigation; Y. W., I. D., Y. C., S. G. N., and W. P. G. visualization; Y. W. writing-original draft; I. D. and A. L. conceptualization; I. D., Y. C., S. G. N., W. P. G., and A. L. validation; Y. W., I. D., and A. L. writing-review and editing; W. P. G. methodology; A. L. supervision; A. L. funding acquisition; A. L. project administration.
Funding and additional information—This work was supported by National Science Foundation award CHE-1808637 and in part by National Institutes of Health grant GM108988. The MS facility used in this work was sponsored by the National Institutes of Health grant G12MD007591. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ADO
- cysteamine dioxygenase
- EPR
- electron paramagnetic resonance
- CDO
- cysteine dioxygenase
- 3MDO
- 3-mercaptopropionate dioxygenase
- MSDO
- mercaptosuccinate dioxygenase
- β-ME
- β-mercaptoethanol
- IPNS
- isopenicillin N synthase
- DNIC
- dinitrosyl iron center.
References
- 1. Stipanuk M. H., Simmons C. R., Karplus P. A., and Dominy J. E. Jr. (2011) Thiol dioxygenases: unique families of cupin proteins. Amino Acids 41, 91–102 10.1007/s00726-010-0518-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dominy J. E. Jr., Simmons C. R., Hirschberger L. L., Hwang J., Coloso R. M., and Stipanuk M. H. (2007) Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J. Biol. Chem. 282, 25189–25198 10.1074/jbc.M703089200 [DOI] [PubMed] [Google Scholar]
- 3. Simmons C. R., Hao Q., and Stipanuk M. H. (2005) Preparation, crystallization and X-ray diffraction analysis to 1.5 Å resolution of rat cysteine dioxygenase, a mononuclear iron enzyme responsible for cysteine thiol oxidation. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 1013–1016 10.1107/S1744309105033737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dominy J. E., Simmons C. R., Karplus P. A., Gehring A. M., and Stipanuk M. H. (2006) Identification and characterization of bacterial cysteine dioxygenases: a new route of cysteine degradation for eubacteria. J. Bacteriol. 188, 5561–5569 10.1128/JB.00291-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McCoy J. G., Bailey L. J., Bitto E., Bingman C. A., Aceti D. J., Fox B. G., and Phillips G. N. Jr. (2006) Structure and mechanism of mouse cysteine dioxygenase. Proc. Natl. Acad. Sci. U S A 103, 3084–3089. 10.1073/pnas.0509262103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simmons C. R., Liu Q., Huang Q. Q., Hao Q., Begley T. P., Karplus P. A., and Stipanuk M. H. (2006) Crystal structure of mammalian cysteine dioxygenase–a novel mononuclear iron center for cysteine thiol oxidation. J. Biol. Chem. 281, 18723–18733 10.1074/jbc.M601555200 [DOI] [PubMed] [Google Scholar]
- 7. Bruland N., Wubbeler J. H., and Steinbuchel A. (2009) 3-Mercaptopropionate dioxygenase, a cysteine dioxygenase homologue, catalyzes the initial step of 3-mercaptopropionate catabolism in the 3,3-thiodipropionic acid-degrading bacterium variovorax paradoxus. J. Biol. Chem. 284, 660–672 10.1074/jbc.M806762200 [DOI] [PubMed] [Google Scholar]
- 8. Pierce B. S., Subedi B. P., Sardar S., and Crowell J. K. (2015) The “Gln-Type” thiol dioxygenase from Azotobacter vinelandii is a 3-mercaptopropionic acid dioxygenase. Biochemistry 54, 7477–7490 10.1021/acs.biochem.5b00636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tchesnokov E. P., Fellner M., Siakkou E., Kleffmann T., Martin L. W., Aloi S., Lamont I. L., Wilbanks S. M., and Jameson G. N. (2015) The cysteine dioxygenase homologue from Pseudomonas aeruginosa is a 3-mercaptopropionate dioxygenase. J. Biol. Chem. 290, 24424–24437 10.1074/jbc.M114.635672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brandt U., Schurmann M., and Steinbuchel A. (2014) Mercaptosuccinate dioxygenase, a cysteine dioxygenase homologue, from Variovorax paradoxus strain B4 is the key enzyme of mercaptosuccinate degradation. J. Biol. Chem. 289, 30800–30809 10.1074/jbc.M114.579730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paul B. D., and Snyder S. H. (2019) Therapeutic applications of cysteamine and cystamine in neurodegenerative and neuropsychiatric diseases. Front. Neurol. 10, 1315 10.3389/fneur.2019.01315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sarkar B., Kulharia M., and Mantha A. K. (2017) Understanding human thiol dioxygenase enzymes: structure to function, and biology to pathology. Int. J. Exp. Pathol. 98, 52–66 10.1111/iep.12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McBean G. J., Aslan M., Griffiths H. R., and Torrao R. C. (2015) Thiol redox homeostasis in neurodegenerative disease. Redox Biol. 5, 186–194 10.1016/j.redox.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Donnelly E. T., McClure N., and Lewis S. E. (2000) Glutathione and hypotaurine in vitro: effects on human sperm motility, DNA integrity and production of reactive oxygen species. Mutagenesis 15, 61–68 10.1093/mutage/15.1.61 [DOI] [PubMed] [Google Scholar]
- 15. Bousquet M., Gibrat C., Ouellet M., Rouillard C., Calon F., and Cicchetti F. (2010) Cystamine metabolism and brain transport properties: clinical implications for neurodegenerative diseases. J. Neurochem. 114, 1651–1658 10.1111/j.1471-4159.2010.06874.x [DOI] [PubMed] [Google Scholar]
- 16. Hara K., Nakamura M., Haranishi Y., Terada T., Kataoka K., and Sata T. (2012) Antinociceptive effect of intrathecal administration of hypotaurine in rat models of inflammatory and neuropathic pain. Amino Acids 43, 397–404 10.1007/s00726-011-1094-9 [DOI] [PubMed] [Google Scholar]
- 17. Cisbani G., Drouin-Ouellet J., Gibrat C., Saint-Pierre M., Lagace M., Badrinarayanan S., Lavallee-Bourget M. H., Charest J., Chabrat A., Boivin L., Lebel M., Bousquet M., Levesque M., and Cicchetti F. (2015) Cystamine/cysteamine rescues the dopaminergic system and shows neurorestorative properties in an animal model of Parkinson's disease. Neurobiol. Dis. 82, 430–444 10.1016/j.nbd.2015.07.012 [DOI] [PubMed] [Google Scholar]
- 18. Nishimura T., Duereh M., Sugita Y., Yoshida Y., Higuchi K., Tomi M., and Nakashima E. (2015) Protective effect of hypotaurine against oxidative stress-induced cytotoxicity in rat placental trophoblasts. Placenta 36, 693–698 10.1016/j.placenta.2015.02.014 [DOI] [PubMed] [Google Scholar]
- 19. White M. D., Klecker M., Hopkinson R. J., Weits D. A., Mueller C., Naumann C., O'Neill R., Wickens J., Yang J., Brooks-Bartlett J. C., Garman E. F., Grossmann T. N., Dissmeyer N., and Flashman E. (2017) Plant cysteine oxidases are dioxygenases that directly enable arginyl transferase-catalysed arginylation of N-end rule targets. Nat. Commun. 8, 14690 10.1038/ncomms14690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masson N., Keeley T. P., Giuntoli B., White M. D., Puerta M. L., Perata P., Hopkinson R. J., Flashman E., Licausi F., and Ratcliffe P. J. (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 365, 65–69 10.1126/science.aaw0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Vries L., Zheng B., Fischer T., Elenko E., and Farquhar M. G. (2000) The regulator of G protein signaling family. Annu. Rev. Pharmacol. Toxicol. 40, 235–271 10.1146/annurev.pharmtox.40.1.235 [DOI] [PubMed] [Google Scholar]
- 22. Hamzah J., Jugold M., Kiessling F., Rigby P., Manzur M., Marti H. H., Rabie T., Kaden S., Grone H. J., Hammerling G. J., Arnold B., and Ganss R. (2008) Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 453, 410–414 10.1038/nature06868 [DOI] [PubMed] [Google Scholar]
- 23. Jin Y., An X., Ye Z., Cully B., Wu J., and Li J. (2009) RGS5, a hypoxia-inducible apoptotic stimulator in endothelial cells. J. Biol. Chem. 284, 23436–23443 10.1074/jbc.M109.032664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silini A., Ghilardi C., Figini S., Sangalli F., Fruscio R., Dahse R., Pedley R. B., Giavazzi R., and Bani M. (2012) Regulator of G-protein signaling 5 (RGS5) protein: a novel marker of cancer vasculature elicited and sustained by the tumor's proangiogenic microenvironment. Cell. Mol. Life Sci. 69, 1167–1178 10.1007/s00018-011-0862-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stipanuk M. H., Ueki I., Dominy J. E. Jr., Simmons C. R., and Hirschberger L. L. (2009) Cysteine dioxygenase: a robust system for regulation of cellular cysteine levels. Amino Acids 37, 55–63 10.1007/s00726-008-0202-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dominy J. E. Jr., Hwang J., Guo S., Hirschberger L. L., Zhang S., and Stipanuk M. H. (2008) Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel posttranslational regulation of activity. J. Biol. Chem. 283, 12188–12201 10.1074/jbc.M800044200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y., Griffith W. P., Li J., Koto T., Wherritt D. J., Fritz E., and Liu A. (2018) Cofactor biogenesis in cysteamine dioxygenase: C-F bond cleavage with genetically incorporated unnatural tyrosine. Angew. Chem. Int. Ed. 57, 8149–8153 10.1002/anie.201803907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J., Koto T., Davis I., and Liu A. (2019) Probing the Cys-Tyr cofactor biogenesis in cysteine dioxygenase by the genetic incorporation of fluorotyrosine. Biochemistry 58, 2218–2227 10.1021/acs.biochem.9b00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Simmons C. R., Krishnamoorthy K., Granett S. L., Schuller D. J., Dominy J. E. Jr., Begley T. P., Stipanuk M. H., and Karplus P. A. (2008) A putative Fe2+-bound persulfenate intermediate in cysteine dioxygenase. Biochemistry 47, 11390–11392 10.1021/bi801546n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li J., Griffith W. P., Davis I., Shin I., Wang J., Li F., Wang Y., Wherritt D. J., and Liu A. (2018) Cleavage of a carbon-fluorine bond by an engineered cysteine dioxygenase. Nat. Chem. Biol. 14, 853–860 10.1038/s41589-018-0085-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tchesnokov E. P., Wilbanks S. M., and Jameson G. N. (2012) A strongly bound high-spin iron(II) coordinates cysteine and homocysteine in cysteine dioxygenase. Biochemistry 51, 257–264 10.1021/bi201597w [DOI] [PubMed] [Google Scholar]
- 32. Nguyen R. C., Yang Y., Wang Y. F., Davis I., and Liu A. (2020) Substrate-assisted hydroxylation and O-demethylation in the peroxidase-like cytochrome P450 enzyme CYP121. ACS Catal. 10, 1628–1639 10.1021/acscatal.9b04596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dornevil K., Davis I., Fielding A. J., Terrell J. R., Ma L., and Liu A. (2017) Cross-linking of dicyclotyrosine by the cytochrome P450 enzyme CYP121 from Mycobacterium tuberculosis proceeds through a catalytic shunt pathway. J. Biol. Chem. 292, 13645–13657 10.1074/jbc.M117.794099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fielding A. J., Dornevil K., Ma L., Davis I., and Liu A. (2017) Probing ligand exchange in the P450 enzyme CYP121 from Mycobacterium tuberculosis: dynamic equilibrium of the distal heme ligand as a function of pH and temperature. J. Am. Chem. Soc. 139, 17484–17499 10.1021/jacs.7b08911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y., Naik S. G., Krzystek J., Shin S., Nelson W. H., Xue S., Yang J. J., Davidson V. L., and Liu A. (2012) Role of calcium in metalloenzymes: effects of calcium removal on the axial ligation geometry and magnetic properties of the catalytic diheme center in MauG. Biochemistry 51, 1586–1597 10.1021/bi201575f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tavares P., Ravi N., Moura J. J., LeGall J., Huang Y. H., Crouse B. R., Johnson M. K., Huynh B. H., and Moura I. (1994) Spectroscopic properties of desulfoferrodoxin from Desulfovibrio desulfuricans (ATCC 27774). J. Biol. Chem. 269, 10504–10510 [PubMed] [Google Scholar]
- 37. Jovanovic T., Ascenso C., Hazlett K. R., Sikkink R., Krebs C., Litwiller R., Benson L. M., Moura I., Moura J. J., Radolf J. D., Huynh B. H., Naylor S., and Rusnak F. (2000) Neelaredoxin, an iron-binding protein from the syphilis spirochete, Treponema pallidum, is a superoxide reductase. J. Biol. Chem. 275, 28439–28448 10.1074/jbc.M003314200 [DOI] [PubMed] [Google Scholar]
- 38. Sardar S., Weitz A., Hendrich M. P., and Pierce B. S. (2019) Outer-sphere tyrosine 159 within the 3-mercaptopropionic acid dioxygenase S-H-Y motif gates substrate-coordination denticity at the non-heme iron active site. Biochemistry 58, 5135–5150 10.1021/acs.biochem.9b00674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen V. J., Orville A. M., Harpel M. R., Frolik C. A., Surerus K. K., Münck E., and Lipscomb J. D. (1989) Spectroscopic studies of isopenicillin N synthase. A mononuclear nonheme Fe2+ oxidase with metal coordination sites for small molecules and substrate. J. Biol. Chem. 264, 21677–21681 [PubMed] [Google Scholar]
- 40. Enemark J. H., and Feltham R. D. (1974) Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord. Chem. Rev. 13, 339–406 10.1016/S0010-8545(00)80259-3 [DOI] [Google Scholar]
- 41. Tsai M. L., Tsou C. C., and Liaw W. F. (2015) Dinitrosyl iron complexes (DNICs): from biomimetic synthesis and spectroscopic characterization toward unveiling the biological and catalytic roles of DNICs. Acc. Chem. Res. 48, 1184–1193 10.1021/ar500459j [DOI] [PubMed] [Google Scholar]
- 42. Lee M., Arosio P., Cozzi A., and Chasteen N. D. (1994) Identification of the EPR-active iron-nitrosyl complexes in mammalian ferritins. Biochemistry 33, 3679–3687 10.1021/bi00178a026 [DOI] [PubMed] [Google Scholar]
- 43. Kozlov A. V., Yegorov D., Vladimirov Y. A., and Azizova O. A. (1992) Intracellular free iron in liver tissue and liver homogenate: studies with electron paramagnetic resonance on the formation of paramagnetic complexes with desferal and nitric oxide. Free Radic. Biol. Med. 13, 9–16 10.1016/0891-5849(92)90159-E [DOI] [PubMed] [Google Scholar]
- 44. Vanin A. F., Serezhenkov V. A., Mikoyan V. D., and Genkin M. V. (1998) The 2.03 signal as an indicator of dinitrosyl-iron complexes with thiol-containing ligands. Nitric Oxide 2, 224–234 10.1006/niox.1998.0180 [DOI] [PubMed] [Google Scholar]
- 45. Le Brun N. E., Andrews S. C., Moore G. R., and Thomson A. J. (1997) Interaction of nitric oxide with non-haem iron sites of Escherichia coli bacterioferritin: reduction of nitric oxide to nitrous oxide and oxidation of iron(II) to iron(III). Biochem. J. 326, 173–179 10.1042/bj3260173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kennedy M. C., Antholine W. E., and Beinert H. (1997) An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J. Biol. Chem. 272, 20340–20347 10.1074/jbc.272.33.20340 [DOI] [PubMed] [Google Scholar]
- 47. Pierce B. S., Gardner J. D., Bailey L. J., Brunold T. C., and Fox B. G. (2007) Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: electronic structure of the active site and mechanistic implications. Biochemistry 46, 8569–8578 10.1021/bi700662d [DOI] [PubMed] [Google Scholar]
- 48. Vanin A. F., Borodulin R. R., and Mikoyan V. D. (2017) Dinitrosyl iron complexes with natural thiol-containing ligands in aqueous solutions: synthesis and some physico-chemical characteristics (a methodological review). Nitric Oxide 66, 1–9 10.1016/j.niox.2017.02.005 [DOI] [PubMed] [Google Scholar]
- 49. Hendrich M. P., Upadhyay A. K., Riga J., Arciero D. M., and Hooper A. B. (2002) Spectroscopic characterization of the NO adduct of hydroxylamine oxidoreductase. Biochemistry 41, 4603–4611 10.1021/bi011332z [DOI] [PubMed] [Google Scholar]
- 50. Lo Bello M., Nuccetelli M., Caccuri A. M., Stella L., Parker M. W., Rossjohn J., McKinstry W. J., Mozzi A. F., Federici G., Polizio F., Pedersen J. Z., and Ricci G. (2001) Human glutathione transferase P1-1 and nitric oxide carriers; a new role for an old enzyme. J. Biol. Chem. 276, 42138–42145 10.1074/jbc.M102344200 [DOI] [PubMed] [Google Scholar]
- 51. Borodulin R. R., Kubrina L. N., Shvydkiy V. O., Lakomkin V. L., and Vanin A. F. (2013) A simple protocol for the synthesis of dinitrosyl iron complexes with glutathione: EPR, optical, chromatographic and biological characterization of reaction products. Nitric Oxide 35, 110–115 10.1016/j.niox.2013.08.007 [DOI] [PubMed] [Google Scholar]
- 52. Aloi S., Davies C. G., Karplus P. A., Wilbanks S. M., and Jameson G. N. L. (2019) Substrate specificity in thiol dioxygenases. Biochemistry 58, 2398–2407 10.1021/acs.biochem.9b00079 [DOI] [PubMed] [Google Scholar]
- 53. Brandt U., Galant G., Meinert-Berning C., and Steinbuchel A. (2019) Functional analysis of active amino acid residues of the mercaptosuccinate dioxygenase of Variovorax paradoxus B4. Enzyme Microb. Technol. 120, 61–68 10.1016/j.enzmictec.2018.09.007 [DOI] [PubMed] [Google Scholar]
- 54. Hu Z., Awakawa T., Ma Z., and Abe I. (2019) Aminoacyl sulfonamide assembly in SB-203208 biosynthesis. Nat. Commun. 10, 184 10.1038/s41467-018-08093-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Song H., Her A. S., Raso F., Zhen Z., Huo Y., and Liu P. (2014) Cysteine oxidation reactions catalyzed by a mononuclear non-heme iron enzyme (OvoA) in ovothiol biosynthesis. Org. Lett. 16, 2122–2125 10.1021/ol5005438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Crawford J. A., Li W., and Pierce B. S. (2011) Single turnover of substrate-bound ferric cysteine dioxygenase with superoxide anion: enzymatic reactivation, product formation, and a transient intermediate. Biochemistry 50, 10241–10253 10.1021/bi2011724 [DOI] [PubMed] [Google Scholar]
- 57. Chai S. C., Jerkins A. A., Banik J. J., Shalev I., Pinkham J. L., Uden P. C., and Maroney M. J. (2005) Heterologous expression, purification, and characterization of recombinant rat cysteine dioxygenase. J. Biol. Chem. 280, 9865–9869 10.1074/jbc.M413733200 [DOI] [PubMed] [Google Scholar]
- 58. Ye S., Wu X., Wei L., Tang D., Sun P., Bartlam M., and Rao Z. (2007) An insight into the mechanism of human cysteine dioxygenase. Key roles of the thioether-bonded tyrosine-cysteine cofactor. J. Biol. Chem. 282, 3391–3402 10.1074/jbc.M609337200 [DOI] [PubMed] [Google Scholar]
- 59. Arciero D. M., Lipscomb J. D., Huynh B. H., Kent T. A., and Münck E. (1983) EPR and Mössbauer studies of protocatechuate 4,5-dioxygenase. Characterization of a new Fe2+ environment. J. Biol. Chem. 258, 14981–14991 [PubMed] [Google Scholar]
- 60. Price J. C., Barr E. W., Tirupati B., Bollinger J. M. Jr., and Krebs C. (2003) The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508 10.1021/bi030011f [DOI] [PubMed] [Google Scholar]
- 61. Fu R., Gupta R., Geng J., Dornevil K., Wang S., Zhang Y., Hendrich M. P., and Liu A. (2011) Enzyme reactivation by hydrogen peroxide in heme-based tryptophan dioxygenase. J. Biol. Chem. 286, 26541–26554 10.1074/jbc.M111.253237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roach P. L., Clifton I. J., Hensgens C. M., Shibata N., Schofield C. J., Hajdu J., and Baldwin J. E. (1997) Structure of isopenicillin N synthase complexed with substrate and the mechanism of penicillin formation. Nature 387, 827–830 10.1038/42990 [DOI] [PubMed] [Google Scholar]
- 63. Tinberg C. E., Tonzetich Z. J., Wang H., Do L. H., Yoda Y., Cramer S. P., and Lippard S. J. (2010) Characterization of iron dinitrosyl species formed in the reaction of nitric oxide with a biological Rieske center. J. Am. Chem. Soc. 132, 18168–18176 10.1021/ja106290p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vanin A. F., and Burbaev D. (2011) Electronic and spatial structures of water-soluble dinitrosyl iron complexes with thiol-containing ligands underlying their ability to act as nitric oxide and nitrosonium ion donors. J. Biophys. 2011, 878236 10.1155/2011/878236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Joseph C. A., and Maroney M. J. (2007) Cysteine dioxygenase: structure and mechanism. Chem. Commun. 3338–3349 10.1039/b702158e [DOI] [PubMed] [Google Scholar]
- 66. Hu R. G., Sheng J., Qi X., Xu Z., Takahashi T. T., and Varshavsky A. (2005) The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 437, 981–986 10.1038/nature04027 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data are described in Tables 1 and 2 and Fig. 1–8, as well as in Table S1 and Fig. S1–S6.