

Abstract
Cardiolipin (CL) is the signature phospholipid of mitochondrial membranes, where it is synthesized locally and plays an important role in mitochondrial bioenergetics. Previous studies in the yeast model have indicated that CL is required for optimal iron homeostasis, which is disrupted by a mechanism not yet determined in the yeast CL mutant, crd1Δ. This finding has implications for the severe genetic disorder, Barth syndrome (BTHS), in which CL metabolism is perturbed because of mutations in the CL-remodeling enzyme, tafazzin. Here, we investigate the effects of tafazzin deficiency on iron homeostasis in the mouse myoblast model of BTHS tafazzin knockout (TAZ-KO) cells. Similarly to CL-deficient yeast cells, TAZ-KO cells exhibited elevated sensitivity to iron, as well as to H2O2, which was alleviated by the iron chelator deferoxamine. TAZ-KO cells exhibited increased expression of the iron exporter ferroportin and decreased expression of the iron importer transferrin receptor, likely reflecting a regulatory response to elevated mitochondrial iron. Reduced activities of mitochondrial iron–sulfur cluster enzymes suggested that the mechanism underlying perturbation of iron homeostasis was defective iron–sulfur biogenesis. We observed decreased levels of Yfh1/frataxin, an essential component of the iron–sulfur biogenesis machinery, in mitochondria from TAZ-KO mouse cells and in CL-deleted yeast crd1Δ cells, indicating that the role of CL in iron–sulfur biogenesis is highly conserved. Yeast crd1Δ cells exhibited decreased processing of the Yfh1 precursor upon import, which likely contributes to the iron homeostasis defects. Implications for understanding the pathogenesis of BTHS are discussed.
Keywords: cardiolipin, iron–sulfur protein, iron metabolism, frataxin, Barth syndrome, Friedreich ataxia, mitochondria, phospholipid, metal homeostasis, tafazzin, iron, protein import
As the signature lipid of the mitochondrial membrane, cardiolipin (CL) is essential for optimal mitochondrial function and bioenergetics (1–6). The de novo synthesis of CL occurs in the inner mitochondrial membrane (7) and is followed by a remodeling process in which saturated fatty acyl chains are replaced by unsaturated fatty acids (8, 9). Perturbation of CL synthesis leads to defects in mitochondrial bioenergetics, resulting in decreased respiration, reduced mitochondrial membrane potential, and decreased ATP synthesis (3, 10–12). CL is tightly associated with a wide range of proteins in the mitochondrial inner membrane (3, 10) and is essential for the stability of respiratory chain supercomplexes (13–15). The severe genetic disorder Barth syndrome (BTHS) results from loss of function of tafazzin, the transacylase that remodels CL, highlighting the significance of this lipid (16). Patients with BTHS suffer from cardio- and skeletal myopathy, neutropenia, and growth retardation (17) and exhibit biochemical and bioenergetic defects at the cellular level that are similar to those observed in CL-deficient yeast cells (18–20). However, the clinical presentation of the disorder in BTHS individuals is highly variable, ranging from neonatal death to lack of clinical symptoms, suggesting that physiological modifiers strongly influence the phenotype of CL deficiency (21). The mechanism linking the pathology in BTHS to CL deficiency is not known.
A previous study indicated that yeast cells lacking CL exhibit decreased iron–sulfur biosynthesis, resulting in perturbation of iron homeostasis (22). Iron is essential for cellular functions in all eukaryotes, from yeast to humans (23). A deficiency in iron causes anemia and even death, whereas iron overload can be toxic as a result of free radical damage (24). Therefore, appropriate intracellular iron levels must be maintained by the regulation of iron transport, storage, and regulatory proteins (25). In mammalian cells, transferrin receptor 1 (TFR1) is responsible for the uptake of extracellular iron by binding to the plasma iron transport protein, transferrin (25). The transmembrane protein ferroportin-1 (FPN1) transports intracellular iron to the outside of the cell (26). Iron is utilized in the formation of iron–sulfur clusters, which are essential co-factors for many enzymatic reactions, including those of electron transfer, enzyme catalysis, and regulatory processes (27). Iron–sulfur biosynthesis is a complex and highly conserved process. The protein Isu provides the primary scaffold for iron–sulfur cluster assembly (28). Cysteine desulfurase (Nfs1) removes sulfur from cysteine and delivers it to Isu (29), a step that requires interaction with accessory protein Isd11 (30). It has been proposed that imported iron is carried by Yfh1/frataxin to Isu (31). After the incorporation of sulfur and iron, the cluster assembly reaction requires reducing equivalents, which are provided by ferredoxin reductase (Arh1) and ferredoxin (Yah1) (32, 33). The iron–sulfur cluster is then delivered to apoproteins by interacting with the Hsp70 chaperone, Ssq1, as well as several other proteins (34).
Frataxin plays a key role in iron–sulfur biogenesis (35) and is highly conserved from prokaryotes to eukaryotes (36–38). It is encoded in the nucleus, is targeted to the mitochondria (39), and acts as a gatekeeper for the formation of iron–sulfur clusters. Several functions of frataxin have been proposed, including regulation of iron homeostasis, storage of iron to decrease toxic levels of free mitochondrial iron, and control of Nfs1 activity (40). Although the exact role of frataxin is unclear, its interaction with Isu1, Nfs1, and Isd11 suggests that it is a key player in regulating iron–sulfur biogenesis (41). Frataxin deficiency in humans causes the severe genetic disorder, Friedreich ataxia (42, 43), which is characterized by progressive neurodegeneration and hypertrophic cardiomyopathy (44). Interestingly, the yeast frataxin mutant yfh1Δ exhibits phenotypes similar to those of CL-deficient crd1Δ cells, including accumulation of mitochondrial iron (45), decreased activity of aconitase (46), and hypersensitivity to oxidative stress (46). In the current study, we show that CL deficiency impairs the second processing step of imported Yfh1 precursor and that CL-deficient cells have decreased levels of mature frataxin and concomitant perturbation of iron homeostasis. Elucidating the role of CL in maintaining iron homeostasis may identify specific mitochondrial defects that contribute to the pathology in BTHS.
Results
Impaired iron homeostasis and iron–sulfur deficiencies in TAZ-KO cells
We reported previously that CL-deficient yeast cells exhibit defective iron–sulfur biogenesis, as reflected in decreased activities of mitochondrial and cytoplasmic enzymes requiring iron–sulfur co-factors and in altered iron homeostasis (22). To determine whether the role of CL in these essential processes is conserved, iron–sulfur biogenesis and iron homeostasis were analyzed in the CL-deficient mouse cell line, TAZ-KO, in which the tafazzin gene is disrupted (47). TAZ-KO cells exhibit the prototypical biochemical phenotypes of BTHS, including decreased levels of total CL and unsaturated CL species and increased monolysocardiolipin (47). Iron–sulfur–requiring enzymes were assayed in TAZ-KO cells, as shown in Table 1. Activities of aconitase, NADH dehydrogenase, succinate dehydrogenase, and ubiquinol–cytochrome c reductase were decreased in TAZ-KO mitochondria to 42–57% of WT. Levels of these mitochondrial proteins were not decreased in the mutant mitochondria (Fig. 1), suggesting that the defect was in catalytic activity. Although previous studies indicated that CL is required for activity of cytochrome c oxidase (48, 49), which does not require an iron–sulfur co-factor, complex IV activity was not affected in TAZ-KO cells.
Table 1.
Decreased iron–sulfur enzyme activities in TAZ-KO mitochondria. The enzyme activities were assayed as described under “Experimental procedures.” The data shown are means ± S.D. (n = 3)
| Activity in TAZ-KO mitochondria relative to WT | |
|---|---|
| % | |
| Aconitase | 42.3 ± 4.7 |
| Complex I (NADH dehydrogenase) | 56.7 ± 8.6 |
| Complex II (succinate dehydrogenase) | 49.2 ± 7.5 |
| Complex III (ubiquinol–cytochrome c oxidoreductase) | 52.3 ± 4.1 |
| Complex IV (cytochrome c oxidase) | 98.7 ± 7.2 |
Figure 1.

Protein levels of iron–sulfur enzymes are unaltered in TAZ-KO cells. Western blotting analysis of mitochondrial protein (30 μg of total protein) purified from mouse cells grown to 100% confluency. ACO2, aconitase 2; SDHA, succinate dehydrogenase subunit a; UQCRC1, ubiquinol–cytochrome c oxidoreductase core 1 subunit; NDUFB6, NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 6. The signal intensities of protein bands and surrounding background were scanned and quantified using ImageJ. The background-subtracted value for each protein band was normalized to that of NDUFB6 and quantified relative to WT.
A decrease in iron–sulfur biogenesis is expected to lead to impaired iron homeostasis, which would be reflected in altered transport of iron (50). As discussed above, TFR1 and FPN1 mediate iron transport in mammalian cells (51). TFR1 transfers extracellular iron into cells by endocytosis (25), and FPN1 exports iron (26). In TAZ-KO cells, the TFR1 mRNA and protein levels are decreased, whereas the levels of FPN1 are increased (Fig. 2), as expected if iron levels are elevated. In agreement with this, an 18% increase in mitochondrial iron was observed in TAZ-KO cells relative to WT (data not shown), similar to what was observed in yeast crd1Δ cells, which lack CL (22), and in cells with a deficiency in iron–sulfur cluster synthesis or export of iron–sulfur co-factors (52–57). Fibroblasts obtained from Friedreich ataxia patients exhibit a greater (40%) increase in mitochondrial iron (58). Increased mitochondrial iron levels lead to hypersensitivity to oxidative stress, which results in sensitivity to H2O2 (28, 45, 53) or iron supplementation (46, 59–61). Consistent with this, TAZ-KO cells exhibited significantly greater sensitivity than WT cells to H2O2 (Fig. 3A) and to iron (Fig. 3B). To test whether increased sensitivity of TAZ-KO cells to H2O2 was due to elevated iron, the effect on viability of the iron chelator deferoxamine (DFO) was determined. Growth of TAZ-KO cells treated with DFO was less sensitive to H2O2-induced death than untreated TAZ-KO cells (Fig. 3C). Taken together, these findings are consistent with impaired iron homeostasis and defective iron–sulfur biogenesis in TAZ-KO cells and indicate that the requirement for CL in these processes is conserved from yeast to mammals.
Figure 2.

TAZ-KO cells exhibit altered expression of genes for iron transport. A, mRNA levels of iron transport genes TFR1 and FPN1 from WT and TAZ-KO cells were quantified by qPCR. The values are reported as fold change in expression in mutant relative to WT. Expression was normalized to the mRNA levels of the internal control GAPDH. The data shown are means ± S.D. (n = 3). *, p < 0.05. B, Western blotting analysis of FPN1 and TFR1 from total extracts of mouse cells grown to 100% confluency. ACTIN is included as a loading control. The signal intensities of protein bands and surrounding background were scanned and quantified using ImageJ. The background-subtracted value for each protein band was normalized to that of ACTIN and quantified relative to WT.
Figure 3.

Sensitivity of TAZ-KO cells to oxidants is rescued by DFO. The cells were seeded equally in 96-well plates and incubated for 3 h until they attached to the plate. 20 μm H2O2 (A) or 50 mm FeCl3 (B) was added, and the cells were incubated for the indicated times, after which cell viability was measured by MTT assay, as described under “Experimental Procedures.” C, TAZ-KO cells were either mock-treated (CTRL) or exposed to 200 μm DFO for 3 h prior to the addition of 20 μm H2O2. The data shown are means ± S.D. (n = 3). *, p < 0.05.
Impaired processing of imported Yfh1 precursor in yeast crd1Δ cells
Previous studies in yeast cells indicated that CL is required for the optimal import of some mitochondrial proteins (11, 62). Therefore, we speculated that altered iron–sulfur biogenesis in CL-deficient cells may result from defective import of proteins required for iron–sulfur biogenesis. In addition, Yfh1 maturation may be perturbed, because the biogenesis of Yfh1 involves two processing steps of the Yfh1 precursor by the mitochondrial processing peptidase upon import into mitochondria (63, 64). To analyze whether biogenesis of Yfh1 depends on CL, we imported radiolabeled Yfh1 precursor into isolated WT and crd1Δ mitochondria. The intermediate and mature forms were only detected in the presence of a membrane potential, confirming that the Yfh1 precursor was imported into WT and crd1Δ mitochondria (Fig. 4A). Interestingly, the second processing step of the Yfh1 precursor was impaired, whereas the first processing was only mildly decreased (Fig. 4A). In contrast, the import of other proteins (Isu1p and Yah1p) involved in iron–sulfur biosynthesis was largely unaffected or only mildly reduced (Nfs1p) (Fig. 4B). The decreased import rates of Yfh1p and Nfs1p are likely due to the reduced membrane potential (Figs. 4C) (10, 60), which drives protein translocation into the matrix and inner membrane. Consistent with the impaired processing of Yfh1, crd1Δ exhibited decreased Yfh1 protein levels but unchanged Nfs1 and Isu1 levels (Fig. 5A). Decreased Yfh1 in crd1Δ was not the result of reduced gene expression, because YFH1 transcript levels were comparable in WT and CL mutant cells (Fig. 5B).
Figure 4.

CL promotes processing of the Yfh1 precursor. Mitochondria (Mito.) were isolated from WT and crd1Δ cells and incubated with 35S-labeled precursors Yfh1 (top panel of A), or 35S-labeled Isu1, Yah1, or Nfs1 (B) in the presence or absence of a membrane potential (Δψ). Following import, the samples were treated with proteinase K, which removes portions of the nonimported precursor proteins. The import reactions were analyzed by SDS-PAGE and autoradiography. A, lower panels, quantification of the intermediate and mature forms of Yfh1 in four independent import experiments with the corresponding standard error of the mean. As control, the longest import time point of Yfh1 into WT mitochondria was set to 100%. C, the membrane potential of WT and crd1Δ mitochondria was measured by quenching of the fluorescence of DiSC3. The reaction was stopped by addition of the AVO mix.
Figure 5.

Decreased Yfh1 in yeast crd1Δ cells. A, Western blotting analysis of Nfs1, Yfh1, and Isu1 proteins from mitochondrial extracts of WT, crd1Δ, and yfh1Δ cells grown in SD galactose at 36 °C. Por1 was used as a loading control. The signal intensities of protein bands and surrounding background were scanned and quantified using ImageJ. The background-subtracted value for each protein band was normalized to that of Por1 and quantified relative to WT. The data shown are means ± S.D. (n = 3). *, p < 0.05. B, mRNA levels of YFH1 were quantified by qPCR from WT and crd1Δ yeast cells grown in SD galactose at 30 and 36 °C. The data shown are means ± S.D. (n = 3).
Yfh1 was previously shown to associate with the mitochondrial inner membrane (39), which is enriched in CL. Therefore, the possibility was tested that CL physically interacts with Yfh1. As shown in the protein lipid overlay assay (Fig. 6), purified recombinant Yfh1 displayed a high affinity for CL. Yfh1 did not bind to phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol 4,5-bisphosphate, or phosphatidylinositol (3,4,5)-trisphosphate. Comparatively weaker binding to phosphatidic acid was also observed. These findings suggest that binding to CL may be required for optimal processing of Yfh1.
Figure 6.
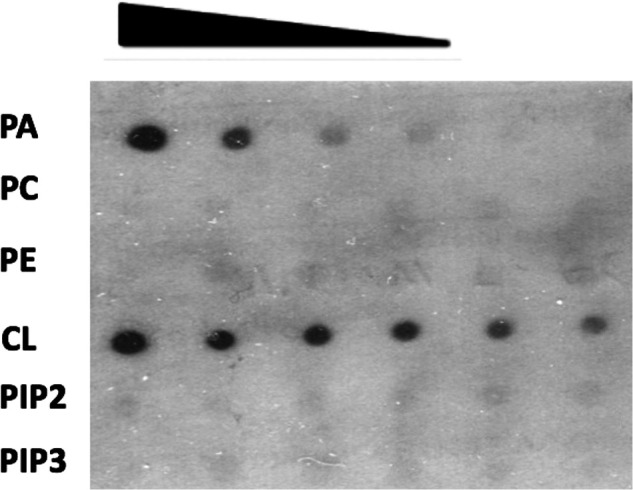
CL physically interacts with Yfh1. Yfh1 protein was purified from E. coli cells expressing a His-tagged Yfh1 gene on the pET28a vector. Serial dilutions of the indicated lipids were spotted on a nitrocellulose membrane, which was incubated overnight in buffer containing 30 μg of Yfh1 protein. Interactions between Yfh1 and lipids were determined by immunoblotting with antibody against the His tag. PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; CL, cardiolipin; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate.
Decreased frataxin in TAZ-KO cells
Because perturbation of iron homeostasis and iron–sulfur biogenesis is conserved in CL-deficient mammalian cells, we considered the possibility that in mammalian cells, as in yeast, the underlying defect is in mitochondrial frataxin. In agreement with this, mitochondria from TAZ-KO cells exhibited decreased levels of mature frataxin, consistent with defective mitochondrial processing of the protein (Fig. 7A). Taken together, these experiments suggest that CL is required for optimal processing of Yfh1/frataxin and that perturbation of CL synthesis leads to defects in iron–sulfur biogenesis and iron homeostasis as a result of Yfh1/frataxin deficiency.
Figure 7.

Decreased frataxin in CL-deficient mammalian cells. A, Western blotting analysis of frataxin from WT and TAZ-KO mitochondria. NDUFB6 is included as a loading control. The signal intensities of protein bands and surrounding background were scanned and quantified using ImageJ. The background-subtracted value for each protein band was normalized to that of NDUFB6 and quantified relative to WT. The data shown are means ± S.D. (n = 2). *, p < 0.05. B, mRNA levels of frataxin from WT and TAZ-KO cells were quantified by qPCR. The values are reported as fold change in expression in mutant relative to WT. Expression was normalized to the mRNA levels of the internal control Gapdh. The data shown are means ± S.D. (n = 3).
Discussion
In this study, we demonstrate for the first time that CL-deficient cells exhibit defective processing of imported Yfh1 precursor. Yfh1/frataxin is a unique protein with respect to processing in both yeast and mammalian cells, because the precursor undergoes two cleavages by the mitochondrial processing peptidase (MPP) to form the mature protein (63). Impaired second-step processing (Fig. 4) is the likely mechanism underlying reduced levels of mature Yfh1/frataxin in CL-deficient yeast and mouse mitochondria (Figs. 5 and 7). These findings suggest the model shown in Fig. 8. Accordingly, following import into mitochondria, processing of the Yfh1/frataxin precursor to the intermediate by the MPP occurs similarly in CL mutant and WT cells. However, optimal processing of the intermediate to the mature form, which is deficient in the mutant (Fig. 4), requires CL. A possible explanation for this defect is that CL is required to maintain the proper conformation of Yfh1/frataxin to expose its second cleavage site to MPP to facilitate the processing step. This is supported by the observation that Yfh1/frataxin binds to CL in vitro (Fig. 6) and has been shown to associate with the inner membrane (37). Gordon et al. (65) reported that cells deficient in the mature form of Yfh1 failed to exhibit any defects, suggesting that the intermediate can functionally compensate for the deficiencies in the yfh1Δ mutant. However, iron–sulfur defects were clearly observed in both crd1Δ yeast and TAZ-KO mouse cells, which have abundant levels of the intermediate form of Yfh1/frataxin. Therefore, it is likely that Yfh1 does not fold properly in CL-deficient mitochondria, leading to a defect in its processing and potentially contributing to the iron homeostasis defects.
Figure 8.

Model: CL is essential for the optimal processing of frataxin. Frataxin precursor is synthesized in the cytoplasm and imported into mitochondria, where it is processed twice by the MPP. The model proposes that CL is required for maintaining an optimal conformation of Yfh1/frataxin for processing from the intermediate to the mature protein. Improperly folded Yfh1/frataxin in CL-deficient mitochondria results in defective processing, potentially contributing to the iron homeostasis defects.
In the current study, the Yfh1/frataxin deficiency in crd1Δ could account for defective iron–sulfur biogenesis, resulting in decreased activity of enzymes requiring iron–sulfur co-factors (Table 1) and perturbation of iron homeostasis (Figs. 2 and 3). The phenotypes observed in the crd1Δ cells are similar to those of the yfh1Δ mutant, including accumulation of mitochondrial iron (45), decreased activities of iron–sulfur enzymes (46), and hypersensitivity to oxidative stress (46). These findings are consistent with defective iron–sulfur biogenesis in both mutants. Yfh1/frataxin, which is essential for iron–sulfur biogenesis, functions as a chaperone that delivers iron to the scaffold assembly protein, Isu1 (66). Direct interaction between CL and frataxin, as suggested by the experiment in Fig. 6, may facilitate processing of the protein by the MPP. The current studies do not distinguish between decreased total and unsaturated CL and increased monolyso-CL as the cause of frataxin deficiency in TAZ-KO cells. However, the observation that mature Yfh1 is decreased in yeast cells that completely lack CL suggests that CL interaction promotes frataxin maturation.
The findings in this report may have implications for understanding the pathology in BTHS, which is characterized by wide disparities in clinical phenotypes among patients, strongly suggesting that physiological modifiers exacerbate the outcome of altered CL metabolism (21). In this context, iron homeostasis and the robustness of iron–sulfur biogenesis may be factors that modify the clinical phenotype of BTHS. Interestingly, the iron chelator DFO, which has been proposed as a treatment to counteract oxidant sensitivity in Friedreich ataxia (58), partially rescued decreased growth of TAZ-KO cells (Fig. 3C).
Elegant studies in other laboratories have established that many of the proteins required for iron–sulfur biogenesis are highly conserved from bacteria to humans (67). In this light, it is not surprising that model organisms have been invaluable in elucidating the complex mechanisms underlying this process. The striking finding in the current study, utilizing yeast and mammalian models of CL deficiency, is that the signature mitochondrial lipid CL plays a pivotal and highly conserved role in the process, linking lipid biosynthesis to iron–sulfur biogenesis for the first time.
Experimental procedures
Yeast strains and growth conditions
The Saccharomyces cerevisiae strains used in this work are listed in Table 2. Complete synthetic medium (CSM) contained adenine (20.25 mg/liter), arginine (20 mg/liter), histidine (20 mg/liter), leucine (60 mg/liter), lysine (200 mg/liter), methionine (20 mg/liter), threonine (300 mg/liter), tryptophan (20 mg/liter), uracil (20 mg/liter), yeast nitrogen base without amino acids, and glucose (2%). CSM-lactate medium contained 2% lactate as carbon source in place of glucose. Complex medium (YPD) contained yeast extract (1%), peptone (2%), and glucose (2%). For import studies, yeast cells were grown overnight in YPD precultures at 30 °C. Subsequently, the cells were transferred to CSM containing 2% (w/v) galactose as carbon source. The cells were grown at 30 °C until they reached the stationary phase, followed by a shift to 35 °C for 2 h.
Table 2.
Yeast strains used in this study
Mouse cells and growth conditions
The C2C12 tafazzin knockout mouse cell line, TAZ-KO, has been described previously (47). Growth medium consisted of Dulbecco's modified Eagle's Medium (Gibco) containing 10% fetal bovine serum (Hyclone), 2 mm glutamine (Gibco), penicillin (100 units/ml), and streptomycin (100 μg/ml) (Invitrogen). The cells were grown at 37 °C in a humidified incubator with 5% CO2.
MTT cell proliferation assay
3000 cells were suspended in 100 μl of growth medium and seeded into 96-well plates. Viable cells were measured in triplicate using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Fisher) after 3, 24, and 48 h. In brief, 10 μl of 5 mg/ml MTT were added to each well. As a negative control, MTT was applied to wells lacking cells. The plates were incubated for the indicated times at 37 °C, after which the medium was carefully removed, and 150 μl of DMSO was added to dissolve the MTT product. The plates were covered with foil and incubated for 10 min at 37 °C. Samples from each well were mixed well with a pipette, and absorbance was read at 570 nm.
Mitochondria isolation
The cell pellets were washed with cold PBS and suspended in mitochondrial isolation buffer (280 mm sucrose, 0.25 mm EDTA, 20 mm Tris-HCl, pH 7.2). Mouse cells were collected and manually homogenized with a glass homogenizer. Yeast cell extracts were collected by disrupting the cell wall with glass beads. Mitochondria were isolated by differential centrifugation. Cell debris was removed by centrifugation at 800 rpm for 5 min. Mitochondria were subsequently collected by centrifugation at 11,500 rpm for 10 min. Protein concentration was determined using a DC protein assay kit (Bio-Rad). For protein import studies, mitochondria were isolated by differential centrifugation (68). In brief, yeast cells were grown to an early logarithmic growth. Subsequently, the cells were harvested by centrifugation (5,500 × g, 8 min, 20 °C), incubated with DTT buffer (10 mm DTT, 100 mm Tris, H2SO4 pH 9.4) and treated with Zymolyase 20T from Arthrobacter luteus (Nacalai Tesque, Kyoto, Japan) to digest the cell wall. We opened the plasma membrane with a glass potter in homogenization buffer (0.6 m sorbitol, 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm PMSF) at 4 °C. After removal of cell debris (2,500 × g, 5 min, 4 °C), mitochondria were isolated (17,000 × g, 15 min, 4 °C), resuspended in S.E. buffer (250 mm sucrose, 1 mm EDTA, 10 mm MOPS/KOH, pH 7.2) in 10 mg/ml protein concentration and stored in aliquots at −80 °C until use.
Immunoblotting
Mammalian or yeast cell extracts corresponding to 30 μg of protein were analyzed by SDS-PAGE using a 10% gel. Immunoblotting was performed using primary antibodies as indicated and corresponding secondary antibodies conjugated to horseradish peroxidase. Immunoreactivity was visualized using enhanced chemiluminescence substrate (Thermo).
Enzyme assays
NADH dehydrogenase activity was assayed by measuring electron transfer from NADH to coenzyme Q1. The absorbance of each sample was measured at 340 nm to account for rotenone-insensitive NADH oxidation. Succinate dehydrogenase activity was measured spectrophotometrically at 600 nm as described, with some modifications (68). The 1-ml incubation volume contained 1 g/liter BSA, 80 mm potassium phosphate, 2 mm EDTA, 10 mm succinate, 0.2 mm ATP, 0.3 mm potassium cyanide, 80 μm dichloroindophenol, 1 μm antimycin A, 50 μm decylubiquinone, and 3 μm rotenone, pH 7.8. Antimycin A and rotenone were dissolved in ethanol. Decylubiquinone was dissolved in DMSO. Succinate dehydrogenase from purified mitochondria was measured in the incubation buffer at 1-min intervals for 5 min with absorptivity at 600 nm. Ubiquinol–cytochrome c oxidoreductase activity of isolated mitochondria was measured spectrophotometrically at 550 nm by supplying decylubiquinol and cytochrome c and following the rate of reduction of cytochrome c, while inhibiting cytochrome c oxidase with 2 mm KCN. Aconitase activity was assayed by the aconitase–isocitrate dehydrogenase–coupled assay in which NADPH formation was monitored at A340 (69). Cytochrome c oxidase was assayed by monitoring oxygen consumption. The cells were solubilized in buffer (10 mm HEPES, pH 7.4, 40 mm KCl, 1% Tween 20, 2 mm EGTA, 1 mm sodium vanadate, 10 mm KF, 1 mm PMSF, 1 μm oligomycin) and sonicated for 5 s. Total protein concentration was determined using the DC protein assay kit (Bio-Rad) with BSA as the standard. Oxygen consumption was analyzed in a closed 200-μl chamber equipped with a micro Clark-type oxygen electrode type (Oxygraph Plus System, Hansatech Instruments) at 25 °C. 30 μm cytochrome c (from bovine heart, Sigma) was added after a baseline measurement. Oxygen consumption was recorded on a computer and analyzed with Oxygraph software. The turnover number was defined as nmol of oxygen consumed per minute per milligram of protein.
qPCR analysis
The cells were harvested, and total RNA was isolated using the RNeasy plus mini kit (Qiagen). cDNAs were synthesized with transcriptor first-strand cDNA synthesis kit (Roche Diagnostics), and quantitative PCRs were performed in a 25-μl volume using BrilliantTM SYBR® Green qPCR Master Mix (Stratagene) in 96-well plates. Duplicates were included for each reaction. The primers used for qPCR are listed in Table 3. The RNA level of the gene of interest was normalized to levels of GAPDH, which served as an internal control. PCRs were initiated at 95 °C for 10 min for denaturation followed by 40 cycles consisting of 30 s at 95 °C and 60 s at 55 °C.
Table 3.
Primers used for qPCR analysis
| Gene | Prime | Sequence |
|---|---|---|
| Tfr1 | Forward | TCCTGTCGCCCTATGTATCT |
| Reverse | CAGCAGCTCTTGAGATTGTTTG | |
| Fpn1 | Forward | CGGTCTTTGGTCCTTTGATTTG |
| Reverse | GCAGAAGGTCAAGAAGGTAGTT | |
| Gapdh | Forward | AGTCTTTTGGGTGGCGGTCA |
| Reverse | ACATTGACGCTGGTGCCAAG | |
| Frataxin | Forward | ACCTCCACTACCTCCAGATT |
| Reverse | TCTTCCGCCAGTCTTTCATAC | |
| YFH1 | Forward | GACGAGACAGCGTATGAAAGA |
| Reverse | ATCGTAGTCCTCCAGGGTATAG | |
| ACT1 | Forward | TCCGGTGATGGTGTTACTCA |
| Reverse | GGCCAAATCGATTCTCAAAA |
Import of precursor proteins into mitochondria
Precursor proteins were synthesized in rabbit reticulocyte lysates in the presence of [35S]methionine (70). Import into isolated mitochondria was performed in import buffer (3% [w/v] BSA, 250 mm sucrose, 80 mm KCl, 5 mm methionine, 5 mm MgCl2, 2 mm KH2PO4, 10 mm MOPS-KPH (pH 7.2), 2 mm NADH, 4 mm ATP, 20 mm creatine phosphate, 0.1 mg/ml creatine kinase) at 24 °C. The import reaction was stopped by the addition of AVO (8 μm antimycin A, 1 μm valinomycin, 20 μm oligomycin). In control reactions, AVO was added before the import experiment to dissipate ΔΨ. After the import reaction, mitochondria were treated for 15 min on ice with proteinase K (15–30 µg/ml) to remove the nonimported precursor proteins. Subsequently, the protease was inactivated by incubation with PMSF for 10 min on ice (final concentration, 4 mm).
Membrane potential measurement
The mitochondrial membrane potential was determined by quenching of the fluorescence dye DiSC3 (15). DiSC3 is taken up by mitochondria in a membrane potential–dependent manner, which leads to quenching of the fluorescence signal in the reaction mix. For this assay, isolated mitochondria were incubated with DiSC3 in membrane potential buffer (0.6 m sorbit, 0.1% (w/v) BSA, 10 mm MgCl2, 0.5 mm EDTA, 20 mm KPi, pH 7.2) supplemented with 5 mm succinate and 5 mm malate for the indicated time points. The reaction was terminated by the addition of the AVO mix.
Protein lipid overlay assay
PET28a-Yfh1 was constructed by inserting the YFH1 gene carrying a polyhistidine tag (His tag) at the C terminus into a PET28a plasmid. PET28a-Yfh1 was transformed into Escherichia coli BL21 (DE3) pLysS cells for isopropyl β-d-thiogalactopyranoside–induced overexpression. The cells were lysed with a French press, and Yfh1-His protein was purified from extracts using a His-Bind purification kit (Millipore). Protein lipid overlay assays were performed according to the protocol of Dowler et al. (71) with modification. In brief, lyophilized lipids were dissolved in a 1:1 solution of methanol and chloroform to make 1 mm stocks. Lipids were diluted to concentrations ranging from 500 to 5 μm in a 2:1:0.8 solution of methanol:chloroform:water. Then 1-μl lipid samples from each dilution were spotted on nitrocellulose membranes. After drying for 1 h at room temperature, the membranes were incubated in blocking buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl2, 0.1% Tween 20 and 2 mg/ml BSA) with gentle rocking for 1 h at room temperature. The membranes were incubated overnight at 4 °C with gentle rocking in 10 ml of blocking buffer containing 25 μg of purified Yfh1-His protein. Yfh1-His protein bound to lipid on the membrane was detected by immunoblotting using primary antibodies against the His tag and corresponding secondary antibodies.
Data availability
All data are contained within the article.
Author contributions—Y. L., V. A. P., and M. L. G. conceptualization; Y. L. data curation; Y. L., W. L., A. G., L. B., Z. L., J. J., J. L., C. Y., M. H., and T. B. investigation; Y. L., T. B., and M. L. G. writing-original draft; Y. L., T. B., and M. L. G. writing-review and editing; M. L. G. funding acquisition; M. L. G. project administration.
Funding and additional information—This work was supported by grants from the National Institutes of Health Grant HL 117880 and by funds from the Barth Syndrome Foundation (to M. L. G.). T. B. was supported by funds from the Deutsche Forschungsgemeinschaft, Germany's Excellence Strategy Grant CIBSS-EXC 2189 under Project 390939984 and Grant BE 4679/2-2. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- CL
- cardiolipin
- BTHS
- Barth syndrome
- TAZ-KO
- tafazzin knockout C2C12 cells
- DFO
- deferoxamine
- MPP
- mitochondrial processing peptidase
- qPCR
- quantitative PCR
- TFR
- transferrin receptor
- FPN
- ferroportin
- CSM
- complete synthetic medium
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PMSF
- phenylmethylsulfonyl fluoride.
References
- 1. Ren M., Phoon C. K., and Schlame M. (2014) Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 55, 1–16 10.1016/j.plipres.2014.04.001 [DOI] [PubMed] [Google Scholar]
- 2. Paradies G., Paradies V., De Benedictis V., Ruggiero F. M., and Petrosillo G. (2014) Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 1837, 408–417 10.1016/j.bbabio.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 3. Joshi A. S., Zhou J., Gohil V. M., Chen S., and Greenberg M. L. (2009) Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793, 212–218 10.1016/j.bbamcr.2008.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raja V., and Greenberg M. L. (2014) The functions of cardiolipin in cellular metabolism: potential modifiers of the Barth syndrome phenotype. Chem. Phys. Lipids 179, 49–56 10.1016/j.chemphyslip.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shen Z., Ye C., McCain K., and Greenberg M. L. (2015) The role of cardiolipin in cardiovascular health. Biomed. Res. Int. 2015, 891707 10.1155/2015/891707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mårtensson C. U., Doan K. N., and Becker T. (2017) Effects of lipids on mitochondrial functions. Biochim. Biophys. Acta 1862, 102–113 10.1016/j.bbalip.2016.06.015 [DOI] [PubMed] [Google Scholar]
- 7. Mejia E. M., Nguyen H., and Hatch G. M. (2014) Mammalian cardiolipin biosynthesis. Chem. Phys. Lipids 179, 11–16 10.1016/j.chemphyslip.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 8. Beranek A., Rechberger G., Knauer H., Wolinski H., Kohlwein S. D., and Leber R. (2009) Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578 10.1074/jbc.M805511200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu Y., Kelley R. I., Blanck T. J., and Schlame M. (2003) Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 278, 51380–51385 10.1074/jbc.M307382200 [DOI] [PubMed] [Google Scholar]
- 10. Planas-Iglesias J., Dwarakanath H., Mohammadyani D., Yanamala N., Kagan V. E., and Klein-Seetharaman J. (2015) Cardiolipin interactions with proteins. Biophys. J. 109, 1282–1294 10.1016/j.bpj.2015.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., and Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 10.1074/jbc.M909868199 [DOI] [PubMed] [Google Scholar]
- 12. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., and Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 10.1083/jcb.200801152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang M., Mileykovskaya E., and Dowhan W. (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556 10.1074/jbc.C200551200 [DOI] [PubMed] [Google Scholar]
- 14. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., and Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 10.1074/jbc.M308366200 [DOI] [PubMed] [Google Scholar]
- 15. Böttinger L., Horvath S. E., Kleinschroth T., Hunte C., Daum G., Pfanner N., and Becker T. (2012) Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J. Mol. Biol. 423, 677–686 10.1016/j.jmb.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., and Toniolo D. (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389 10.1038/ng0496-385 [DOI] [PubMed] [Google Scholar]
- 17. Clarke S. L., Bowron A., Gonzalez I. L., Groves S. J., Newbury-Ecob R., Clayton N., Martin R. P., Tsai-Goodman B., Garratt V., Ashworth M., Bowen V. M., McCurdy K. R., Damin M. K., Spencer C. T., Toth M. J., et al. (2013) Barth syndrome. Orphanet J. Rare Dis. 8, 23 10.1186/1750-1172-8-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brandner K., Mick D. U., Frazier A. E., Taylor R. D., Meisinger C., and Rehling P. (2005) Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol. Biol. Cell 16, 5202–5214 10.1091/mbc.e05-03-0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McKenzie M., Lazarou M., Thorburn D. R., and Ryan M. T. (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469 10.1016/j.jmb.2006.06.057 [DOI] [PubMed] [Google Scholar]
- 20. Gu Z., Valianpour F., Chen S., Vaz F. M., Hakkaart G. A., Wanders R. J., and Greenberg M. L. (2004) Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51, 149–158 10.1046/j.1365-2958.2003.03802.x [DOI] [PubMed] [Google Scholar]
- 21. Schlame M., and Ren M. (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 10.1016/j.febslet.2006.07.022 [DOI] [PubMed] [Google Scholar]
- 22. Patil V. A., Fox J. L., Gohil V. M., Winge D. R., and Greenberg M. L. (2013) Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J. Biol. Chem. 288, 1696–1705 10.1074/jbc.M112.428938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lasocki S., Gaillard T., and Rineau E. (2014) Iron is essential for living! Crit. Care 18, 678 10.1186/s13054-014-0678-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hider R. C., and Kong X. (2013) Iron: effect of overload and deficiency. Met. Ions Life Sci. 13, 229–294 10.1007/978-94-007-7500-8_8 [DOI] [PubMed] [Google Scholar]
- 25. Hentze M. W., Muckenthaler M. U., and Andrews N. C. (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117, 285–297 10.1016/S0092-8674(04)00343-5 [DOI] [PubMed] [Google Scholar]
- 26. Donovan A., Lima C. A., Pinkus J. L., Pinkus G. S., Zon L. I., Robine S., and Andrews N. C. (2005) The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1, 191–200 10.1016/j.cmet.2005.01.003 [DOI] [PubMed] [Google Scholar]
- 27. Beinert H., Holm R. H., and Münck E. (1997) Iron–sulfur clusters: nature's modular, multipurpose structures. Science 277, 653–659 10.1126/science.277.5326.653 [DOI] [PubMed] [Google Scholar]
- 28. Schilke B., Voisine C., Beinert H., and Craig E. (1999) Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 96, 10206–10211 10.1073/pnas.96.18.10206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Biederbick A., Stehling O., Rösser R., Niggemeyer B., Nakai Y., Elsässer H. P., and Lill R. (2006) Role of human mitochondrial Nfs1 in cytosolic iron–sulfur protein biogenesis and iron regulation. Mol. Cell. Biol. 26, 5675–5687 10.1128/MCB.00112-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mühlenhoff U., Gerber J., Richhardt N., and Lill R. (2003) Components involved in assembly and dislocation of iron–sulfur clusters on the scaffold protein Isu1p. EMBO J. 22, 4815–4825 10.1093/emboj/cdg446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y., Lyver E. R., Knight S. A., Pain D., Lesuisse E., and Dancis A. (2006) Mrs3p, Mrs4p, and frataxin provide iron for Fe–S cluster synthesis in mitochondria. J. Biol. Chem. 281, 22493–22502 10.1074/jbc.M604246200 [DOI] [PubMed] [Google Scholar]
- 32. Shi Y., Ghosh M., Kovtunovych G., Crooks D. R., and Rouault T. A. (2012) Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron–sulfur cluster biogenesis. Biochim. Biophys. Acta 1823, 484–492 10.1016/j.bbamcr.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alves R., Herrero E., and Sorribas A. (2004) Predictive reconstruction of the mitochondrial iron–sulfur cluster assembly metabolism: II. Role of glutaredoxin Grx5. Proteins 57, 481–492 10.1002/prot.20228 [DOI] [PubMed] [Google Scholar]
- 34. Lill R., and Mühlenhoff U. (2008) Maturation of iron–sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 77, 669–700 10.1146/annurev.biochem.76.052705.162653 [DOI] [PubMed] [Google Scholar]
- 35. Stemmler T. L., Lesuisse E., Pain D., and Dancis A. (2010) Frataxin and mitochondrial FeS cluster biogenesis. J. Biol. Chem. 285, 26737–26743 10.1074/jbc.R110.118679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dhe-Paganon S., Shigeta R., Chi Y. I., Ristow M., and Shoelson S. E. (2000) Crystal structure of human frataxin. J. Biol. Chem. 275, 30753–30756 10.1074/jbc.C000407200 [DOI] [PubMed] [Google Scholar]
- 37. He Y., Alam S. L., Proteasa S. V., Zhang Y., Lesuisse E., Dancis A., and Stemmler T. L. (2004) Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry 43, 16254–16262 10.1021/bi0488193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roche B., Aussel L., Ezraty B., Mandin P., Py B., and Barras F. (2013) Iron/sulfur proteins biogenesis in prokaryotes: formation, regulation and diversity. Biochim. Biophys. Acta 1827, 455–469 10.1016/j.bbabio.2012.12.010 [DOI] [PubMed] [Google Scholar]
- 39. Campuzano V., Montermini L., Lutz Y., Cova L., Hindelang C., Jiralerspong S., Trottier Y., Kish S. J., Faucheux B., Trouillas P., Authier F. J., Dürr A., Mandel J. L., Vescovi A., Pandolfo M., et al. (1997) Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780 10.1093/hmg/6.11.1771 [DOI] [PubMed] [Google Scholar]
- 40. Adinolfi S., Iannuzzi C., Prischi F., Pastore C., Iametti S., Martin S. R., Bonomi F., and Pastore A. (2009) Bacterial frataxin CyaY is the gatekeeper of iron–sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 16, 390–396 10.1038/nsmb.1579 [DOI] [PubMed] [Google Scholar]
- 41. Gerber J., Mühlenhoff U., and Lill R. (2003) An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 4, 906–911 10.1038/sj.embor.embor918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chamberlain S., Shaw J., Rowland A., Wallis J., South S., Nakamura Y., von Gabain A., Farrall M., and Williamson R. (1988) Mapping of mutation causing Friedreich's ataxia to human chromosome 9. Nature 334, 248–250 10.1038/334248a0 [DOI] [PubMed] [Google Scholar]
- 43. Pandolfo M., and Pastore A. (2009) The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 256, 9–17 10.1007/s00415-009-1003-2 [DOI] [PubMed] [Google Scholar]
- 44. Harding A. E. (1983) Classification of the hereditary ataxias and paraplegias. Lancet 1, 1151–1155 10.1016/S0140-6736(83)92879-9 [DOI] [PubMed] [Google Scholar]
- 45. Babcock M., de Silva D., Oaks R., Davis-Kaplan S., Jiralerspong S., Montermini L., Pandolfo M., and Kaplan J. (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276, 1709–1712 10.1126/science.276.5319.1709 [DOI] [PubMed] [Google Scholar]
- 46. Foury F. (1999) Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Lett. 456, 281–284 10.1016/S0014-5793(99)00961-8 [DOI] [PubMed] [Google Scholar]
- 47. Lou W., Reynolds C. A., Li Y., Liu J., Hüttemann M., Schlame M., Stevenson D., Strathdee D., and Greenberg M. L. (2018) Loss of tafazzin results in decreased myoblast differentiation in C2C12 cells: a myoblast model of Barth syndrome and cardiolipin deficiency. Biochim. Biophys. Acta 1863, 857–865 10.1016/j.bbalip.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Paradies G., Petrosillo G., and Ruggiero F. M. (1997) Cardiolipin-dependent decrease of cytochrome c oxidase activity in heart mitochondria from hypothyroid rats. Biochim. Biophys. Acta 1319, 5–8 10.1016/S0005-2728(97)00012-1 [DOI] [PubMed] [Google Scholar]
- 49. Robinson N. C. (1993) Functional binding of cardiolipin to cytochrome c oxidase. J. Bioenerg. Biomembr. 25, 153–163 10.1007/BF00762857 [DOI] [PubMed] [Google Scholar]
- 50. Huang M. L., Becker E. M., Whitnall M., Suryo Rahmanto Y., Ponka P., and Richardson D. R. (2009) Elucidation of the mechanism of mitochondrial iron loading in Friedreich's ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. U.S.A. 106, 16381–16386 10.1073/pnas.0906784106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang J., and Pantopoulos K. (2011) Regulation of cellular iron metabolism. Biochem. J. 434, 365–381 10.1042/BJ20101825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hausmann A., Samans B., Lill R., and Mühlenhoff U. (2008) Cellular and mitochondrial remodeling upon defects in iron–sulfur protein biogenesis. J. Biol. Chem. 283, 8318–8330 10.1074/jbc.M705570200 [DOI] [PubMed] [Google Scholar]
- 53. Mühlenhoff U., Richhardt N., Ristow M., Kispal G., and Lill R. (2002) The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 11, 2025–2036 10.1093/hmg/11.17.2025 [DOI] [PubMed] [Google Scholar]
- 54. Li J., Kogan M., Knight S. A., Pain D., and Dancis A. (1999) Yeast mitochondrial protein, Nfs1p, coordinately regulates cluster proteins, cellular iron uptake, and iron distribution. J. Biol. Chem. 274, 33025–33034 10.1074/jbc.274.46.33025 [DOI] [PubMed] [Google Scholar]
- 55. Garland S. A., Hoff K., Vickery L. E., and Culotta V. C. (1999) Saccharomyces cerevisiae ISU1 and ISU2: members of a well-conserved gene family for iron–sulfur cluster assembly. J. Mol. Biol. 294, 897–907 10.1006/jmbi.1999.3294 [DOI] [PubMed] [Google Scholar]
- 56. Knight S. A., Sepuri N. B., Pain D., and Dancis A. (1998) Mt-Hsp70 homolog, Ssc2p, required for maturation of yeast frataxin and mitochondrial iron homeostasis. J. Biol. Chem. 273, 18389–18393 10.1074/jbc.273.29.18389 [DOI] [PubMed] [Google Scholar]
- 57. Kispal G., Csere P., Guiard B., and Lill R. (1997) The ABC transporter Atm1p is required for mitochondrial iron homeostasis. FEBS Lett. 418, 346–350 10.1016/S0014-5793(97)01414-2 [DOI] [PubMed] [Google Scholar]
- 58. Wong A., Yang J., Cavadini P., Gellera C., Lonnerdal B., Taroni F., and Cortopassi G. (1999) The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 8, 425–430 10.1093/hmg/8.3.425 [DOI] [PubMed] [Google Scholar]
- 59. Ramazzotti A., Vanmansart V., and Foury F. (2004) Mitochondrial functional interactions between frataxin and Isu1p, the iron–sulfur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett. 557, 215–220 10.1016/S0014-5793(03)01498-4 [DOI] [PubMed] [Google Scholar]
- 60. Foury F., Pastore A., and Trincal M. (2007) Acidic residues of yeast frataxin have an essential role in Fe–S cluster assembly. EMBO Rep. 8, 194–199 10.1038/sj.embor.7400881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leidgens S., De Smet S., and Foury F. (2010) Frataxin interacts with Isu1 through a conserved tryptophan in its beta-sheet. Hum. Mol. Genet. 19, 276–286 10.1093/hmg/ddp495 [DOI] [PubMed] [Google Scholar]
- 62. Gebert N., Joshi A. S., Kutik S., Becker T., McKenzie M., Guan X. L., Mooga V. P., Stroud D. A., Kulkarni G., Wenk M. R., Rehling P., Meisinger C., Ryan M. T., Wiedemann N., Greenberg M. L., et al. (2009) Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139 10.1016/j.cub.2009.10.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Branda S. S., Cavadini P., Adamec J., Kalousek F., Taroni F., and Isaya G. (1999) Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase. J. Biol. Chem. 274, 22763–22769 10.1074/jbc.274.32.22763 [DOI] [PubMed] [Google Scholar]
- 64. Voisine C., Schilke B., Ohlson M., Beinert H., Marszalek J., and Craig E. A. (2000) Role of the mitochondrial Hsp70s, Ssc1 and Ssq1, in the maturation of Yfh1. Mol. Cell. Biol. 20, 3677–3684 10.1128/.20.10.3677-3684.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gordon D. M., Kogan M., Knight S. A., Dancis A., and Pain D. (2001) Distinct roles for two N-terminal cleaved domains in mitochondrial import of the yeast frataxin homolog, Yfh1p. Hum. Mol. Genet. 10, 259–269 10.1093/hmg/10.3.259 [DOI] [PubMed] [Google Scholar]
- 66. Wang T., and Craig E. A. (2008) Binding of yeast frataxin to the scaffold for Fe–S cluster biogenesis, Isu. J. Biol. Chem. 283, 12674–12679 10.1074/jbc.M800399200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rouault T. A. (2012) Biogenesis of iron–sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model Mech. 5, 155–164 10.1242/dmm.009019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rustin P., Chretien D., Bourgeron T., Gérard B., Rötig A., Saudubray J. M., and Munnich A. (1994) Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 228, 35–51 10.1016/0009-8981(94)90055-8 [DOI] [PubMed] [Google Scholar]
- 69. Pierik A. J., Netz D. J., and Lill R. (2009) Analysis of iron–sulfur protein maturation in eukaryotes. Nat. Protoc. 4, 753–766 10.1038/nprot.2009.39 [DOI] [PubMed] [Google Scholar]
- 70. Priesnitz C., Pfanner N., and Becker T. (2020) Studying protein import into mitochondria. Methods Cell Biol. 155, 45–79 10.1016/bs.mcb.2019.11.006 10.1016/bs.mcb.2019.11.006 [DOI] [PubMed] [Google Scholar]
- 71. Dowler S., Kular G., and Alessi D. R. (2002) Protein lipid overlay assay. Sci. STKE 2002, pl6 10.1126/stke.2002.129.pl6 [DOI] [PubMed] [Google Scholar]
- 72. Jiang F., Rizavi H. S., and Greenberg M. L. (1997) Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol. Microbiol. 26, 481–491 10.1046/j.1365-2958.1997.5841950.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within the article.