

Abstract
Directed migration of endothelial cells (ECs) is an important process during both physiological and pathological angiogenesis. The binding of vascular endothelial growth factor (VEGF) to VEGF receptor-2 (VEGFR-2) on the EC surface is necessary for directed migration of these cells. Here, we used TAXIScan, an optically accessible real-time horizontal cell dynamics assay approach, and demonstrate that reactive oxygen species (ROS)-producing NADPH oxidase 4 (NOX4), which is abundantly expressed in ECs, mediates VEGF/VEGFR-2-dependent directed migration. We noted that a continuous supply of endoplasmic reticulum (ER)-retained VEGFR-2 to the plasma membrane is required to maintain VEGFR-2 at the cell surface. siRNA-mediated NOX4 silencing decreased the ER-retained form of VEGFR-2, resulting in decreased cell surface expression levels of the receptor. We also found that ER-localized NOX4 interacts with ER-retained VEGFR-2 and thereby stabilizes this ER-retained form at the protein level in the ER. We conclude that NOX4 contributes to the directed migration of ECs by maintaining VEGFR-2 levels at their surface.
Keywords: NADPH oxidase, Nox4, vascular endothelial growth factor receptor 2 (VEGFR-2), endothelial cell migration, angiogenesis, capillary formation, reactive oxygen species (ROS), redox signaling, endoplasmic reticulum (ER)
Angiogenesis is the formation of new capillary blood vessels to supply oxygen and nutrients to tissues (1) and is fundamental to the process of recovery from tissue ischemia in adults (2). Pathological angiogenesis occurs in an uncontrolled manner in retinopathy of prematurity (3) and diabetic retinopathy (4), resulting in excessive and abnormal vascular patterns. Abnormal angiogenesis is essential for tumor growth, invasion, and metastasis (5). Both physiological and pathological angiogenesis require the migration, differentiation, and proliferation of the endothelial cells (ECs) that line blood vessels to form capillary-like structures (6). ECs are activated by vascular endothelial growth factor (VEGF), which is the most potent and primary endothelium-specific angiogenic growth factor in both physiological and pathological angiogenesis (6). The binding of VEGF to VEGF receptor-2 (VEGFR-2) at the surface of ECs activates a tyrosine kinase pathway that stimulates angiogenesis (7–9). VEGF/VEGFR-2 signaling is required for physiological angiogenesis for tissue ischemia but is also actively engaged in retinopathy and tumor progression by pathological angiogenesis (10).
Reactive oxygen species (ROS) play a role in angiogenesis (11). The ROS-producing NADPH oxidase (Nox) family is abundantly expressed in the vasculature and is involved in angiogenesis (12–14). The Nox family includes seven members: Nox1–5 and Duox 1–2 (15–20). Nox4 is abundantly expressed in ECs (21), and the global genetic deletion of Nox4 attenuates angiogenesis in response to ischemia after femoral artery ligation (22). Transgenic mice overexpressing endothelial-specific WT Nox4 showed accelerated recovery from hind limb ischemia (23, 24). Use of a mouse model overexpressing WT Nox4 showed that Nox4 plays a positive role in cardiac adaptation to pressure overload (25). Furthermore, deletion of Nox4 in an oxygen-induced retinopathy mouse model prevented abnormal angiogenesis (26). Endothelial Nox4-derived ROS was shown to play a role in the insulin-induced pathological angiogenic response (27). Nox4 was also shown to mediate tumor angiogenesis (28). Thus, Nox4 is involved in physiological and pathological angiogenesis.
The directed migration of ECs is an important process that occurs during angiogenesis. VEGF/VEGFR-2 signaling is required for directed migration (29) and is frequently affected by ROS (11). The present study investigated whether Nox4 is involved in VEGF/VEGFR-2-dependent directed migration using TAXIScan, which visualizes the migration of cells using various parameters. The siRNA-mediated silencing of Nox4 leads to decreased cell surface levels of VEGFR-2, resulting in decreased velocity and directionality of ECs. The maintenance of surface VEGFR-2 levels depends on the continuous supply of the endoplasmic reticulum (ER)-retained form of the receptor to the plasma membrane, and Nox4 is involved in stabilization of the ER-retained form at the protein level in the ER. Thus, Nox4 regulates the directed migration of ECs by maintaining surface VEGFR-2 levels.
Results
Regulation of EC migration by Nox4
We first investigated the migratory capacity of EA.hy926 cells, which are cells obtained from a cultured cell line derived from human umbilical vein endothelial cells and possess many of the unique characteristics of ECs (30, 31), by assessing directed migration toward fetal bovine serum (FBS). Cells were tracked using TAXIScan, which is a real-time assay device for imaging cell migration (Fig. 1A) (32), and collected images of migrating cells at 0, 100, 200, and 300 min (Fig. 1B). Two-dimensional trajectories using various concentrations of FBS are shown in Fig. 1C. We calculated the mean values of directionality and velocity of individual cells during migration and expressed these as velocity–directionality (VD) plots (33, 34) (Fig. 1D). The mean migration directionality and velocity toward the FBS gradient increased in a dose-dependent manner from 0% to 5% (Fig. 1E).
Figure 1.

Migration of ECs in TAXIScan. A, schematic illustration of the experiment. See Experimental procedures for details. B, images of ECs migrating toward FBS in TAXIScan channels. The concentration of injected FBS and their gradient are indicated schematically on the left axis. The distance between the start line (bottom) and finish line (top) of each channel is 260 μm. Scale bar, 100 μm. C, trajectory patterns of ECs migrating toward FBS. The images of migrating cells in TAXIScan channels were filmed, and their paths were traced. Each point shows the direction and distance of each cell in 5 min, from which the mean values of directionality and velocity were calculated. Traces in different colors indicate an independent cell migration. D, velocity–directionality (VD) plots of EC migration. The methods used to calculate each value are described in Experimental procedures. Representative VD plots obtained from 0% FBS and each concentration in panel B are shown. E, statistical analysis of the data of panel D. Each graph represents the mean ± S.D. of pooled data of migrating cells (n ≥ 30) from three independent experiments: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Statistical analysis used the Tukey–Kramer test. The data are representative of results from three independent experiments.
Previous studies have demonstrated that Nox4 promotes angiogenesis (22–24), which requires directed migration. To investigate whether Nox and Nox-derived ROS are involved in FBS-dependent cell migration, we treated EA.hy926 cells with the Nox inhibitor diphenyleneiodonium (DPI) or the antioxidant N-acetylcysteine (NAC). The H2O2 released by the treated cells was measured by a sensitive fluorimetric assay; the assay is based on the oxidation of homovanillic acid (HVA) into a fluorescent dimer, which is catalyzed by horseradish peroxidase (HRP) (Fig. 2A). H2O2 (0.2 μm) was added from a standard H2O2 solution to the HVA-HRP detection system, and then any corresponding increase in fluorescence was checked (Fig. 2B). The H2O2-dependent relative fluorescence intensity was attenuated to background level by the addition of 6,000 units/ml catalase, which decomposes H2O2 to water and oxygen. The H2O2 generated by the EA.hy926 cells increased fluorescence intensity, which was decreased by DPI treatment. The cell viability was determined by propidium iodide (PI) exclusion using flow cytometry and was unaffected by the DPI or NAC treatment (Fig. 2C). The mean migration directionality and velocity toward the FBS of DPI- or NAC-treated cells were significantly lower than those of control cells (Fig. 2, D and E). Thus, endothelial Nox activity and ROS are required for cell migration.
Figure 2.

Directed migration of DPI or NAC-treated ECs. A, schematic illustration of the experiment. See also the Experimental procedures for details. B, catalase-inhibitable H2O2 production by EA.hy926 cells. The panel shows the H2O2 production by EA.hy926 cells with or without DPI treatment. C, assay of cell viability. Viable cells were assessed as those that excluded PI staining (PI-negative). D and E, cell migration assay. Left, images of ECs migrating toward FBS in TAXIScan channels. The concentration of injected FBS was 1%. EA.hy926 cells were exposed to 3 μm DPI with or without 1 mm NAC for 48 h (added every 12 h). The distance between the start line (bottom) and finish line (top) of each channel was 260 μm. Scale bar, 100 μm. The bar graphs show the statistical analysis. Each graph represents the mean ± S.D. of pooled data of migrating cells (n ≥ 30) from three independent experiments. ***, p < 0.001. Statistical analysis used the Tukey-Kramer test. The data are representative of results from three independent experiments.
Nox4 is abundantly expressed in the endothelial cells (21). To investigate whether Nox4 is involved in directed migration, we treated EA.hy926 cells with siRNA targeting Nox4, resulting in an almost complete depletion of Nox4 (Fig. 3, A and B). Specificity of the rabbit polyclonal anti-Nox4 antibody was confirmed by immunoblot analysis of HeLa cells transfected with FLAG–Nox1–5 (a FLAG tag inserted at the N terminus of Nox1–Nox5) expression vectors (Fig. 3C). Nox4 knockdown markedly decreased H2O2 levels in EA.hy926 cells (Fig. 3D). Nox2 knockdown (Fig. 3E) did not affect the H2O2 generation by EA.hy926 cells (Fig. 3D). These results suggest that Nox4 is a major source of H2O2 produced in EA.hy926 cells. Transfection with Nox4- or Nox2-targeted siRNA did not affect the cell viability (Fig. 3F). We examined the migration of Nox4- or Nox2-knockdown EA.hy926 cells and found that the mean migration directionality and velocity of Nox4-silenced cells was significantly lower than those of control cells (Fig. 3, G and H). We obtained the same results using another siRNA sequence (Fig. S1). Treatment of EAhy.926 cells with Nox2-specific siRNA did not affect the directionality or velocity of cell migration (Fig. 3, G and H).
Figure 3.

Directed migration of Nox4 knocked down ECs. A, Nox4 mRNA levels. The bar graph represents the relative density of the bands normalized to β-actin (n = 3). B, Nox4 protein levels with or without Nox4-specific siRNA. Immunoblotting showing control and Nox4 siRNA-treated cell samples. The bar graph represents the relative density of the bands normalized to β-tubulin (n = 3). C, specificity of the rabbit polyclonal anti-Nox4 antibody used in this study. Protein levels expressed in FLAG-tagged Nox-transfected HeLa cells were analyzed by immunoblotting using the indicated antibodies. The positions of the marker proteins are indicated in kDa. D, H2O2 production by EA.hy926 cells transfected with siRNA. ¶, the same data set as that in Fig. 2B. E, Nox2 mRNA levels with or without Nox2-specific siRNA. F, assay of cell viability. Viable cells were assessed as those that excluded PI staining (PI negative). G, images of ECs migrating toward FBS in TAXIScan channels. The concentration of injected FBS was 1%. The distance between the start line (bottom) and finish line (top) of each channel was 260 μm. Scale bar, 100 μm. H, statistical analysis of VD plots. Each graph represents the mean ± S.D. of pooled data of migrating cells (n ≥ 30) from three independent experiments. ***, p < 0.001. Statistical analysis used the Tukey-Kramer test. The data are representative of results from three independent experiments.
Maintenance of cell surface VEGFR-2 levels by Nox4
Several studies have identified the mechanisms of Nox-derived ROS-mediated angiogenesis driven in a VEGF/VEGFR-2 signaling-dependent manner (11). VEGF/VEGFR-2 signaling is required for the directed migration of ECs (29, 35, 36). Because Nox4 knockdown cells showed reduced directional migration, we investigated the expression pattern of VEGFR-2. Transfection with Nox4-targeted siRNA reduced VEGFR-2 protein expression by around 70% compared with that of cells transfected with control siRNA (Fig. 4A). A second Nox4-specific siRNA also blocked VEGFR-2 protein expression compared with control RNA, whereas Nox2-specific siRNA did not decrease VEGFR-2 protein expression (Fig. S2). However, Nox4 silencing did not affect VEGFR-2 mRNA levels (Fig. 4B), demonstrating that the link between Nox4 and VEGFR-2 expression is not transcriptionally controlled. To examine whether the decreased amount of VEGFR-2 protein seen with depletion of Nox4 affected the cell surface levels of VEGFR-2 protein, we performed a cell surface biotinylation assay to analyze the effect of Nox4 knockdown on levels of VEGFR-2 at the plasma membrane. The results showed that Nox4 knockdown markedly reduced the cell surface levels of VEGFR-2 (Fig. 4C). The reduction of surface VEGFR-2 levels induced by Nox4 knockdown attenuated the migration of ECs toward human recombinant VEGF-A injected as a ligand (Fig. 4, D and E).
Figure 4.

Maintenance of surface VEGFR-2 levels by Nox4. A, Nox4 and VEGFR-2 protein levels. Protein expression levels were analyzed by immunoblotting using the indicated antibodies. The bar diagram represents the relative density of the bands normalized to β-tubulin (n = 3). B, Nox4 and VEGFR-2 mRNA levels. The bar diagram represents the relative density of the bands normalized to β-actin (n = 3). C, cell surface VEGFR-2 levels. EA.hy926 cells were treated with Nox4 or control siRNA for 72 h. Cell surface biotinylation experiments were performed as described in Experimental procedures. Protein levels were analyzed by immunoblotting using anti-VEGFR-2. The positions of the marker proteins are indicated in kDa. D, images of ECs migrating toward FBS in TAXIScan channels. The concentration of injected human recombinant VEGF-A was 500 pg/ml. The distance between the start line (bottom) and finish line (top) of each channel was 260 μm. Scale bar, 100 μm. E, statistical analysis of VD plots. Each graph represents the mean ± S.D. of pooled data of migrating cells (n ≥ 30) from three independent experiments. ***, p < 0.001. Statistical analysis used the Tukey-Kramer test. The data are representative of results from three independent experiments.
Stabilization of VEGFR-2 by Nox4 in the ER
Immunoblotting using the anti-VEGFR-2 antibody showed that VEGFR-2 existed as a doublet, with the higher-molecular-weight form being the predominant species (Fig. 5A, lane 1). The higher-molecular-weight band was endoglycosidase H (Endo H) resistant, whereas the lower-molecular-weight band was Endo H sensitive (Fig. 5B, lane 3). Furthermore, the Endo H-resistant species was sensitive to treatment with peptide:N-glycosidase F (PNGase F) (Fig. 5B, lane 2). Thus, in ECs, VEGFR-2 was found at both the cell surface (Endo H-resistant form) and the ER (Endo H-sensitive form), which is responsible for membrane protein maturation.
Figure 5.

Stabilization of ER-retained VEGFR-2 stabilized by Nox4. A, VEGFR-2 protein levels. Proteins were resolved using SDS-PAGE (7.5% gel). The bar diagram represents the relative density of the lower-molecular-weight bands normalized to that of the highest (n = 3). B, N-glycosylation of VEGFR-2. Nox4 knockdown or control EA.hy926 cells were treated with 10 µg/ml BFA for 3 h. Cell lysates were extracted using Triton X-100 and digested with PNGase F or Endo H. C, effect of BFA on the surface VEGFR-2 level. EA.hy926 cells were exposed to 10 µg/ml BFA for the indicated times. D, cell surface VEGFR-2 levels. EA.hy926 cells were treated with or without 10 µg/ml BFA for 3 h. Cell surface biotinylation experiments were performed as described in Experimental procedures. E, stability of ER-retained VEGFR-2 in Nox4 knockdown cells. Upper, Nox4 knockdown or control EA.hy926 cells were exposed to 10 µg/ml BFA for 3 h. Middle, cells were then treated with or without 10 µg/ml CHX for the indicated times. Protein levels were analyzed by immunoblotting with anti-VEGFR-2. The protein band density was quantitated and plotted on a graph, with the value obtained for untreated cells set as 100% (n = 3). The data are representative of results from three independent experiments.
The binding of VEGF leads to rapid internalization of VEGFR-2 (37, 38), followed by the uptake of VEGFR-2 by lysosomes for degradation (39). To investigate the role of ER-retained VEGFR-2 in the maintenance of surface VEGFR-2 levels, we examined the effects of brefeldin A (BFA), which inhibits protein transport from the ER to the Golgi apparatus, by inhibiting coat protein complex I (40). VEGFR-2 carrying complex-type glycan (Endo H resistant) was reduced in a time-dependent manner (Fig. 5C). Consistent with this, BFA treatment decreased the cell surface VEGFR-2 levels (Fig. 5D). These data suggest that the maintenance of cell surface VEGFR-2 depends on the continuous transport of VEGFR-2 from the ER.
Interestingly, the Endo H-sensitive band was decreased in Nox4 knockdown cells (Fig. 5A, lane 2, and B, lanes 7–9), suggesting that VEGFR-2 protein is influenced by Nox4 expression in the ER. To test this hypothesis, we investigated the effects of Nox4 on the stability of ER-retained VEGFR-2. Nox4-silenced or control EA.hy926 cells were preincubated with BFA to retain VEGFR-2 in the ER. Cells were then treated with cycloheximide (CHX) to inhibit the de novo synthesis of VEGFR-2 in a time course experiment (0, 1, 3, 6, and 7.5 h), and cell lysates were analyzed by immunoblotting. In Nox4 knockdown cells, the VEGFR-2 protein levels decreased to ∼40% after 7.5 h of exposure to CHX (Fig. 5E), indicating that existing VEGFR-2 protein is easily degraded in the absence of Nox4 protein.
H2O2 diffuses easily between extra- and intracellular environments (41). Indeed, ER-localized Nox4-derived H2O2 was trapped by extracellular catalase in the HVA-HRP detection system (Fig. 2, A and B). We investigated whether removal of extracellular H2O2 affects cell migration. Addition of catalase had no effect on FBS- or VEGF-A-dependent cell migration (Fig. 6, A and B, and Fig. S3). VEGFR-2 protein expression levels were decreased in DPI-treated cells (Fig. 6C, lane 2). The addition of exogenous H2O2 did not restore the expression level of VEGFR-2 (Fig. 6C, lane 3). These results indicate that the stabilizing effect of ER-retained VEGFR-2 by Nox4 required the production of H2O2 in the closed space between Nox4 and VEGFR-2.
Figure 6.
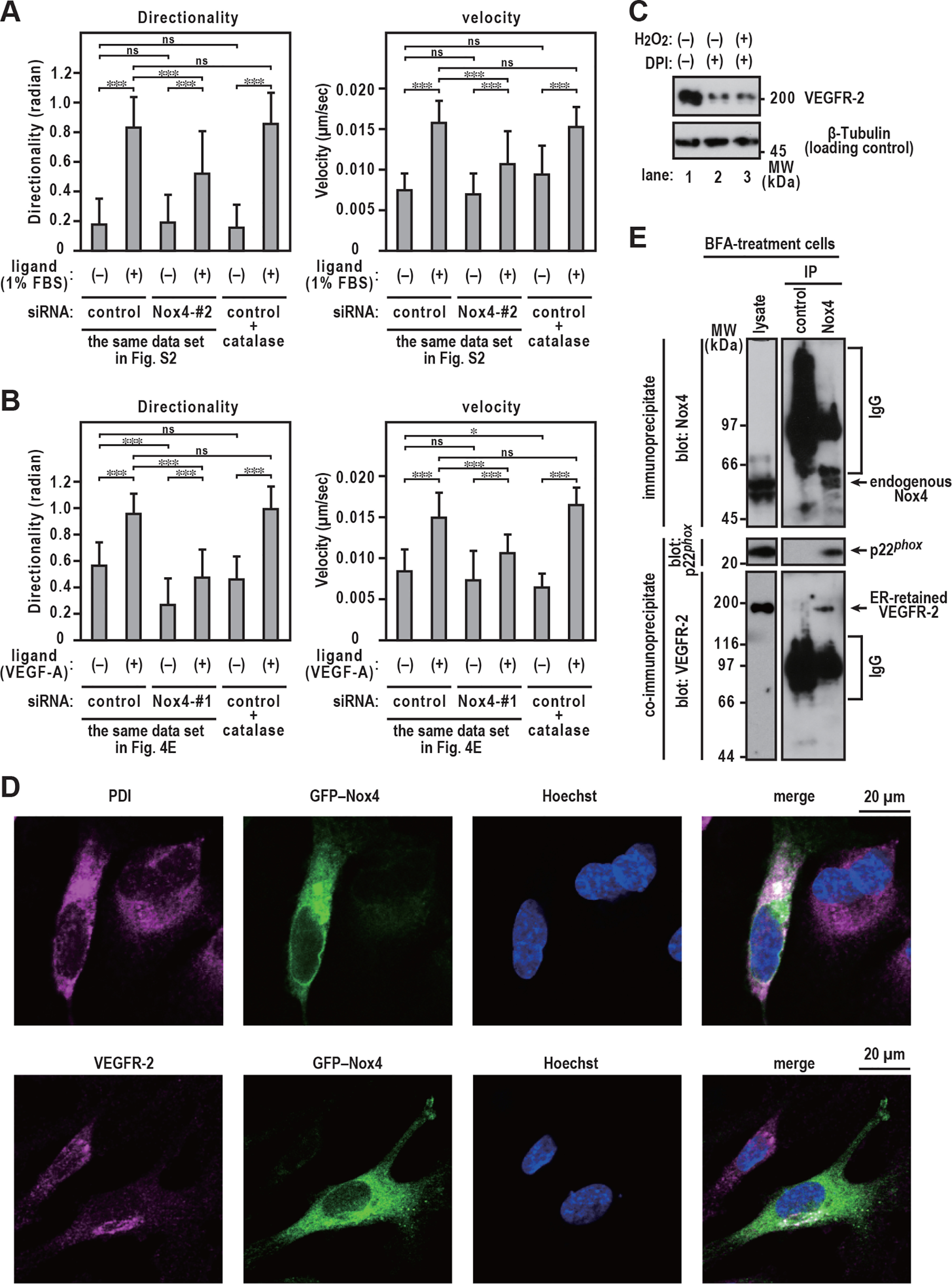
Interaction of Nox4 with VEGFR-2. A and B, statistical analysis of VD plots. EA.hy926 cells were exposed to 6,000 units/ml catalase for 48 h (added every 12 h). Each graph represents the mean ± S.D. of pooled data of migrating cells (n ≥ 30) from three independent experiments. ***, p < 0.001. Statistical analysis used the Tukey-Kramer test. C, VEGFR-2 protein levels. Proteins were resolved with SDS-PAGE (7.5% gels). EA.hy926 cells were exposed to 3 μm DPI with or without 1 μm H2O2 for 48 h (added every 12 h). D, distribution of GFP–Nox4 and VEGFR-2 in EA.hy926 cells. EA.hy926 cells were transfected with the expression plasmid for GFP–Nox4. After fixation, the immunofluorescence signals were observed by confocal microscopy; anti-PDI was used to detect the ER marker PDI (magenta), anti-VEGFR-2 (magenta) was used to detect VEGFR-2, and Hoechst staining was used to detect cell nuclei (blue). Scale bars, 20 μm. E, interaction of endogenous Nox4 with VEGFR-2. EA.hy926 cells were exposed to 10 µg/ml BFA for 3 h. Cell lysates were extracted with Triton X-100. Immunoprecipitation experiments were performed as described in Experimental procedures. The positions of the marker proteins are indicated in kDa. The data are representative of results from three independent experiments.
To determine the effect of Nox4 on the stabilization of ER-retained VEGFR-2, we investigated whether Nox4 physically interacts with VEGFR-2. Confocal imaging showed that GFP–Nox4 was predominantly present in the compartments of the ER (Fig. 6D, upper), which is consistent with previous studies (42–45). Interestingly, GFP–Nox4 was partially colocalized with VEGFR-2 (Fig. 6D, lower). To demonstrate the binding of endogenous Nox4 to ER-retained VEGFR-2, cell lysates derived from BFA-treated cells, which did not include cell surface-localized VEGFR-2 protein (Fig. 5, B–D), were subjected to a coimmunoprecipitation assay. Immunoprecipitation of endogenous Nox4 using an anti-Nox4 antibody led to an expected coprecipitation of endogenous p22phox (Fig. 6E), a partner protein that forms a complex in the membrane with Nox4 (46, 47). Under the same experimental conditions, we also observed that ER-retained VEGFR-2 was coimmunoprecipitated with Nox4 (Fig. 6E), demonstrating that Nox4 interacts with the ER-retained form in the ER compartments.
Discussion
Nox frequently affects VEGF/VEGFR-2 signaling, which is required for physiological and pathological angiogenesis. Nox-derived ROS can mediate VEGF-stimulated VEGFR-2 dimerization and autophosphorylation of VEGFR-2, leading to the activation of downstream signaling pathways (11, 12, 48). VEGF/VEGFR-2 signaling can also activate Nox, leading to increased ROS production, indicating a positive feedback loop between Nox4 and VEGF/VEGFR-2 signaling. VEGF binding stimulates rapid surface VEGFR-2 internalization (37, 38) and degradation (39). In the present study, we demonstrated that Nox4 contributes to the stabilization of ER-retained VEGFR-2, which is responsible for increasing the amount of surface VEGFR-2, and enhances the directed migration of ECs, which is an important process during angiogenesis.
Previous studies have shown that Nox4 promotes endothelial angiogenesis in a mouse hind limb ischemia model (22–24). Nox4 plays a positive role in cardiac adaptation to pressure overload (25), whereas overexpression of Nox4 and excess production of ROS are associated with undesirable angiogenesis, indicating a pathogenic role for Nox4 (49, 50). Nox4 was reported to induce angiogenesis in retinopathy (26, 27) and during tumor development (28), and it has also been shown to enhance the velocity of EC migration (24, 51). The present study demonstrated that Nox4 controls the directionality of EC migration. Thus, Nox4 is clearly involved in physiological and pathological angiogenesis via Nox4-mediated directed migration.
Previous studies have demonstrated that the overexpression of Nox4 results in increased VEGFR-2 protein in ECs (23). Consistent with this observation, RNAi inhibition of Nox4 protein led to the downregulation of VEGFR-2 at the protein level (Fig. 4). A surface biotinylation assay (Fig. 4) showed that the amount of VEGFR-2 on the plasma membrane was substantially reduced in response to Nox4 knockdown. VEGFR-2 was reported to be required for the directed migration of ECs (29). Our results suggest that the decrease in the directed migration of Nox4 knockdown cells is because of decreased levels of VEGFR-2.
We used immunoblot analysis to detect a 200-kDa doublet band of VEGFR-2 in EAhy.926 cells (Fig. 5). Previous studies have reported the detection of a double band using the anti-VEGFR-2 antibody in human umbilical vein ECs (52). A surface biotinylation assay (Fig. 5) showed that VEGFR-2 was present on the cell surface as an Endo H-resistant form (high molecular weight) but not as an Endo H-sensitive form (low molecular weight). These findings are consistent with those of previous studies that reported that the higher-molecular-weight form is the major form of VEGFR-2 on the cell surface (52, 53). Thus, in ECs, VEGFR-2 is found at both the cell surface and the ER, which is responsible for the maturation of membrane protein.
BFA treatment blocks the transport of protein from the ER to the Golgi apparatus, leading to the rapid degradation of cell surface VEGFR-2 (with complex N-glycan) within 3 h (Fig. 5). A continuous supply of the ER-retained form of VEGFR-2 to the plasma membrane appears to be important for maintaining the cell surface receptor level. Surface VEGFR-2 levels are maintained by various molecules. Myoferlin, a member of the ferlin family, prevents VEGFR-2 polyubiquitination and proteasomal degradation, resulting in increased levels of surface VEGFR-2 (54). The chaperone protein PDCL3 protects ER-retained VEGFR-2 from misfolding and aggregation (55). Ring finger protein 121 is an ER ubiquitin E3 ligase that recognizes ER-retained VEGFR-2 and promotes the ubiquitination of VEGFR-2 (56), suggesting its potential in regulating the maturation of VEGFR-2 by controlling its exit from the ER. We also demonstrated that Nox4 contributes to the stabilization of ER-retained VEGFR-2 (Fig. 5), resulting in increased amounts of surface VEGFR-2 (Fig. 4).
As Nox4 was reported to be localized in the ER (42, 43), we believe that it may be involved in the stabilization of ER-retained VEGFR-2 at the protein level. Indeed, Nox4 expression affected the stability of VEGFR-2 in the ER at the protein level (Fig. 5). Nox4 is known to bind to calnexin (57) and protein disulfide isomerase (PDI) (42) in the ER. Calnexin transiently associates with newly synthesized N-linked glycoproteins in the ER (58). PDI catalyzes disulfide oxidation, reduction, and isomerization, thereby playing an important role in protein folding (59–61). These proteins comprise an ER chaperone system that ensures the proper folding and quality control of newly synthesized proteins. VEGFR-2 is a glycoprotein with multiple disulfide bonds. Thus, Nox4 may form a complex with VEGFR-2 as well as chaperone proteins that facilitate protein folding in the ER. In conclusion, we found that Nox4 enhances the directed migration of ECs by maintaining surface VEGFR-2 levels via stabilization of VEGFR-2 in the ER.
Experimental procedures
Materials
Chemicals, reagents, and materials
All general ultrapure-grade reagents were purchased from Nacalai Tesque (Kyoto, Japan), Wako Pure Chemicals Industries (Tokyo, Japan), or Sigma–Aldrich Japan (Tokyo, Japan), unless otherwise stated. Primers were purchased from Eurofins Genomics (Tokyo, Japan).
Plasmids and cDNA
pcDNA3.1-hNox1 was a gift from Botond Bánfi and Karl-Heinz Krause (Addgene plasmid 58344; http://n2t.net/addgene:58344; RRID: Addgene_58344). pcDNA3.1-hNox3 was a gift from Botond Bánfi (Addgene plasmid 58341; http://n2t.net/addgene:58341; RRID: Addgene_58341). pcDNA3.1-hNox4 was a gift from Karl-Heinz Krause (Addgene plasmid 69352; http://n2t.net/addgene:69352; RRID: Addgene_69352). pcDNA3.1-hNox5 was a gift from Karl-Heinz Krause (Addgene plasmid 69354; http://n2t.net/addgene:69354; RRID: Addgene_69354). FLAG-HA-pcDNA3.1 was a gift from Adam Antebi (Addgene plasmid 52535; http://n2t.net/addgene:52535; RRID: Addgene_52535). Human Nox2 cDNA was prepared as previously described (62).
Cells and cell culture
HeLa cells were obtained from RIKEN Cell Bank (Tsukuba, Japan), and EA.hy926 cells were purchased from the ATCC. Both cell lines were maintained in Dulbecco's modified Eagle's medium (Sigma–Aldrich) supplemented with 10% (v/v) FBS (Sigma–Aldrich) at 37 °C in a humidified 5% (v/v) CO2 atmosphere.
Migration of ECs
Directed migration experiments were performed using a TAXIScan (ECI, Tokyo, Japan), as previously described (32, 34, 63). Briefly, we used silicon chips with a 10-μm-high terrace, which forms a 10-μm-deep microchannel. After filling the space at the top end of the holes with assay buffer, 1 μl of EC suspension (2 × 106 cells/ml) was injected into one of the two compartments, and the cells were aligned along the start line on the edge of the channel. After starting to record the images, 1 μl of attractant/stimulus solution (0.1%–5% FBS or 500 pg/ml VEGF-A) was injected into the compartment opposite that containing the cells. Cellular migration at 37 °C was recorded using a camera focused on the migration path every 5 min for 5 h. The migration profile was then analyzed using TAXIScan analyzer 2 software (33). The concentration of VEGF-A injected as a ligand during the assay did not affect the protein levels of Nox4 and VEGFR-2 (Fig. S4).
Assay of H2O2 production
Production of H2O2 by EA.hy926 cells was assayed using HVA (Nacalai Tesque) and HRP (TOYOBO), according to the methods by Ruch et al. (64) and Martyn et al. (65), with minor modifications. Briefly, EA.hy926 cells were harvested by incubation with trypsin–EDTA for 1 min at 37 °C. 1 ml HVA solution (100 μm HVA, 4 units/ml HRP in PBS [137 mm NaCl, 2.7 mm KCl, 8.1 mm Na2HPO4, and 1.5 mm KH2PO4, pH 7.4]) with or without 6,000U/ml catalase was added to each tube, and 1 × 105 cells were incubated for 30 min at 37 °C; 150 μl of HVA stop buffer (0.1 M glycine–0.1 M NaOH, pH 12) was then added per 1 ml of HVA solution. The HVA oxidation product, as a measure of the amount of H2O2, was determined fluorimetrically using a fluorescence spectrophotometer F-2700 (Hitachi); excitation was at 312 nm, and emission was measured at 420 nm.
Estimation of cell viability
Cell viability was assessed using the annexin V apoptosis detection kit (BD Pharmingen, San Diego, CA, USA), with minor modifications. Briefly, siRNA- or reagent-treated EA.hy926 cells were washed by ice-cold PBS and then resuspended in PBS at a concentration of 1 × 106 cells/ml. 100 μl of suspension was transferred to a 5-ml culture tube, mixed with 5 μl of PI, and incubated for 15 min at 25 °C in the dark. After incubation, 400 μl of PBS was added to each tube. All samples were analyzed with the BD FACSCANTO II and Diva software (Becton Dickinson Franklin Lakes, NJ, USA).
RNAi knockdown of Nox2 and Nox4
The following 25-nucleotide modified synthetic double-stranded siRNAs targeting Nox2 and Nox4 (Stealth RNAi) were purchased from Invitrogen: Nox4 siRNA number 1, 5′-ACAGUGAAGACUUUGUUGAACUGAA-3' (sense) and 5′-UUCAGUUCAACAAAGUCUUCACUGU-3′ (antisense); Nox4 siRNA number 2, 5′-AUCUGGUGGAGGUAGUGAUACUCUG-3′ (sense) and 5′-CAGAGUAUCACUACCUCCACCAGAU-3′ (antisense); and Nox2 siRNA number 1, 5′-CCGAGUCAAUAAUUCUGAUCCUUAU-3′ (sense) and 5′-AUAAGGAUCAGAAUUAUUGACUCGG-3′ (antisense). Low- or high-GC duplex of stealth RNAi negative-control duplexes (Invitrogen) was used as a negative control. EA.hy926 cells (1.9 × 104 cells/cm2) were cultured in 15- or 6-cm dishes or six-well plates and then transfected with 50–100 pmol RNA using Lipofectamine RNAiMAX (ThermoFisher Scientific) in 50 μl of Opti-MEM (ThermoFisher Scientific) according to the manufacturer's instructions. Transfected cells were cultured for 72 h and used to measure EC migration, total protein, surface VEGFR-2 levels, and mRNA levels.
Estimation of protein levels
EA.hy926 and HeLa cells were lysed using lysis buffer (20 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100). Cell lysates were subjected to SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore), and probed using anti-Nox4 polyclonal antibody (no. GTX121929, GeneTex), anti-DYKDDDDK tag (FLAG) mouse mAb (no. 014-22383, Wako Pure Chemical Industries), anti-β-tubulin mouse mAb (no. 014-25041, Wako Pure Chemical Industries), or anti-VEGFR-2 rabbit mAb (no. 2479, BD Transduction Laboratories). Blots were developed using ImmunoStar Zeta or ImmunoStar LD (Wako Pure Chemical Industries), and band intensities were quantified using NIH Image J software.
Protein expression in HeLa cells
Sequences encoding peptide epitopes (FLAG–Gly–Gly–Gly–FLAG–Gly–Gly–Gly) recognized using commercially available monoclonal antibodies were inserted into FLAG-HA-pcDNA3.1. A modified vector, termed pcDNA3.1–N-FLAG, allows a FLAG tag to be inserted at the N terminus of the protein. Human cDNA encoding full-length Nox1–Nox5 was subcloned into pcDNA3.1–N-FLAG for expression as a FLAG-tagged protein, as previously described (62). All constructs were sequenced to confirm their identities using an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Warrington, UK) and a BigDye Terminator cycle sequencing kit version 3.1 (Applied Biosystems, Foster City, CA, USA). Plasmids were purified using a QIAfilter plasmid kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Transfections were performed using the TransIT-X 2 dynamic delivery system (Mirus Bio) according to the manufacturer's instructions.
RT-PCR
Total RNA was extracted and purified from EA.hy926 cells (1 × 105 cells) using an RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Total RNA (0.5 µg) was reverse transcribed into cDNA using random primers and Ready-To-Go You-Prime first-strand beads (GE Healthcare) according to the manufacturer's instructions. The following primers were used: Nox4, 5′-CACAGAAGGTTCCAAGCAGGA-3′ and 5′-AGTCAGGTCTGTTCTCTTGCC-3′ (65); β-actin, 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′ and 5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′ (66); and VEGFR-2, 5′-GGAAGACCAAGAAAAGACATTGCG-3′ and 5′-GGTTGACCACATTGAGATGGTGAC-3′ (66); and Nox2, 5′-CATGTTTCTGTATCTCTGTGA-3′ and 5′-GTGAGGTAGATGTTGTAGCT-3′ (44). PCR products were analyzed using 2% agarose gel electrophoresis.
Glycosidase treatment
EA.hy926 cells were lysed using a solution of 20 mm Tris-HCl, pH 7.4, and 150 mm NaCl containing 1% (w/v) Triton X-100, and lysates were centrifuged for 10 min at 20,000 × g. The supernatant was digested using peptide:N-glycosidase F (PNGase F; New England Biolabs) or endoglycosidase H (Endo H; New England Biolabs), as previously described (67). The treated proteins were then separated using 7.5% SDS-PAGE, followed by immunoblot analysis with an anti-VEGFR-2 antibody.
Cell surface biotinylation assay
Cell surface biotinylation experiments were performed as previously described (67). EA.hy926 cells (8 × 106 in a 15-cm dish) were cultured with or without 10 µg/ml BFA for 3 h at 37 °C in a humidified 5% (v/v) CO2 atmosphere. Monolayer cultures were washed three times with PBS to remove contaminating FBS and other soluble proteins and then incubated with or without the membrane-impermeable biotinylation reagent 0.5 mm EZ-link Sulfo-NHS-SS-biotin (ThermoFisher Scientific) for 2.5 min at room temperature. The reaction was quenched with 50 mm glycine, and then the cells were lysed using lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100). Lysates were incubated at 4 °C for 1 h and then centrifuged for 20 min at 20,000 × g. The resulting supernatants were precipitated using streptavidin–Sepharose (GE Healthcare Biosciences). The precipitants were analyzed by immunoblotting with an anti-VEGFR-2 antibody.
Protein stabilization
Nox4 knockdown or control EA.hy926 cells (6 × 105 in six-well plates) were treated with 10 µg/ml BFA for 3 h at 37 °C in a humidified 5% (v/v) CO2 atmosphere. Monolayer cultures were exposed to 10 µg/ml CHX for 0, 1, 3, 6, and 7.5 h and then washed using PBS. The cells were lysed using lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100). Cell lysates were incubated at 4 °C for 1 h and then centrifuged for 20 min at 20,000 × g before subjecting them to SDS-PAGE followed by immunoblot analysis using the anti-VEGFR-2 antibody.
Immunofluorescent microscopy
pEGFP-C1-Nox4 plasmid transfection was performed using the TransIT-X 2 Dynamic Delivery System (Mirus Bio) according to the manufacturer's instructions. To stain VEGFR-2 and GFP–Nox4, EA.hy926 cells grown on glass coverslips were fixed for 15 min in 4% formaldehyde at room temperature and permeabilized for 60 min with 0.3% Triton X-100 in PBS with 5% BSA. To stain PDI (ER marker) and GFP–Nox4, EA.hy926 cells were fixed for 15 min in 4% formaldehyde at room temperature and then for 10 min in ice-cold 100% methanol at −20°C, followed by permeabilization for 60 min in 0.3% Triton X-100 in PBS with 5% BSA. The samples were incubated overnight at 4 °C with the indicated primary antibodies in PBS with 1% BSA and 0.3% Triton X-100 and subsequently incubated for 1–2 h at room temperature with secondary antibodies in PBS with 1% BSA and 0.3% Triton X-100. Immunofluorescence analysis was performed using Alexa Fluor 647-labeled goat anti-rabbit antibody (no. ab150083, Abcam) as a secondary antibody. Nuclei were stained with Hoechst 33342 (no. 346-07951, Dojindo). Images were captured at room temperature with an LSM700 confocal microscope (Carl Zeiss) and analyzed using ZEN (Carl Zeiss) and Fiji/ImageJ (version 2.0; National Institutes of Health).
Immunoprecipitation assay
Monolayer cultures of EA.hy926 cells (8 × 106 in a 15-cm dish) were treated with 10 µg/ml BFA for 3 h at 37 °C in a humidified 5% (v/v) CO2 atmosphere and then washed three times with PBS. Cells were lysed using lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100). Cell lysates were incubated at 4 °C for 1 h and then centrifuged for 20 min at 20,000 × g. Proteins in the lysates of EA.hy926 cells were precipitated with the anti-Nox4 antibody or control IgG in the presence of protein G–Sepharose (GE Healthcare Biosciences). Precipitants were incubated with SDS sample buffer for 1 h at 4 °C and centrifuged for 3 min at 10,000 × g. Supernatants (including the immunoprecipitated and coimmunoprecipitated proteins) were analyzed by immunoblotting using the anti-Nox4 and anti-VEGFR-2 antibodies.
Statistical analysis
Data were expressed as means ± standard deviations (S.D.). Between-group comparisons were performed with t test and the Tukey-Kramer multiple-comparison of means test. Statistical analyses were performed with EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria). Normal distribution of the velocity and directionality values was confirmed by the Kolmogorov-Smirnov test.
Data availability
All data are contained within the manuscript and supporting information.
Supplementary Material
Acknowledgments
We are grateful to Masumi Itadani (Kawasaki Medical School, Japan) for technical assistance and to the Central Research Institute of Kawasaki Medical School for technical support.
This article contains supporting information.
Author contributions—K. M., S. O., A. Y., and F. K. conceptualization; K. M. and S. O. data curation; K. M., S. O., A. Y., C. K., M. K., T. K., M. Tamura, and F. K. formal analysis; K. M., S. O., C. K., M. K., T. K., M. Tamura, and F. K. funding acquisition; K. M., S. O., and C. K. investigation; K. M., S. O., A. Y., C. K., M. K., M. Tamura, M. Taura, and F. K. methodology; K. M., S. O., and M. Taura writing-original draft; K. M. and S. O. project administration; K. M., S. O., A. Y., M. K., T. K., M. Tamura, and F. K. writing-review and editing; M. Taura validation.
Funding and additional information—This study was supported in part by JSPS KAKENHI grant numbers JP17K08637 (to K. M.), JP18K07804 (to F. K.), and JP19K07676 (to A. Y.), in part by the Wesco Scientific Promotion Foundation (to K. M. and A. Y.), in part by the Ryobi Teien Memory Foundation (to K. M. and A. Y.), and in part by Research Project Grants (nos. R01S-003 (to K. M.), H30Y-002 (to S. O.), R01B-081 (to F. K.), and R01B-093 (to A. Y.)) from Kawasaki Medical School.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ECs
- endothelial cells
- VEGF
- vascular endothelial growth factor
- ROS
- reactive oxygen species
- ER
- endoplasmic reticulum
- DPI
- diphenyleneiodonium
- NAC
- N-acetylcysteine
- HVA
- homovanillic acid
- PI
- propidium iodide
- Endo H
- endoglycosidase H
- BFA
- brefeldin A
- CHX
- cycloheximide
- VD
- velocity–directionality
- PNGase F
- peptide:N-glycosidase F.
References
- 1. Folkman J., and Shing Y. (1992) Angiogenesis. J. Biol. Chem. 267, 10931–10934 [PubMed] [Google Scholar]
- 2. Couffinhal T., Silver M., Zheng L. P., Kearney M., Witzenbichler B., and Isner J. M. (1998) Mouse model of angiogenesis. Am. J. Pathol. 152, 1667–1679 [PMC free article] [PubMed] [Google Scholar]
- 3. Sapieha P., Joyal J. S., Rivera J. C., Kermorvant-Duchemin E., Sennlaub F., Hardy P., Lachapelle P., and Chemtob S. (2010) Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 120, 3022–3032 10.1172/JCI42142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crawford T. N., Alfaro D. V. 3rd, Kerrison J. B., and Jablon E. P. (2009) Diabetic retinopathy and angiogenesis. Curr. Diabetes. Rev 5, 8–13. 10.2174/157339909787314149 [DOI] [PubMed] [Google Scholar]
- 5. Hicklin D. J., and Ellis L. M. (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 23, 1011–1027 10.1200/JCO.2005.06.081 [DOI] [PubMed] [Google Scholar]
- 6. Carmeliet P. (2003) Angiogenesis in health and disease. Nat. Med. 9, 653–660 10.1038/nm0603-653 [DOI] [PubMed] [Google Scholar]
- 7. Ferrara N. (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev. 25, 581–611 10.1210/er.2003-0027 [DOI] [PubMed] [Google Scholar]
- 8. Moens S., Goveia J., Stapor P. C., Cantelmo A. R., and Carmeliet P. (2014) The multifaceted activity of VEGF in angiogenesis—implications for therapy responses. Cytokine. Growth. Factor. Rev. 25, 473–482 10.1016/j.cytogfr.2014.07.009 [DOI] [PubMed] [Google Scholar]
- 9. Holash J., Maisonpierre P. C., Compton D., Boland P., Alexander C. R., Zagzag D., Yancopoulos G. D., and Wiegand S. J. (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284, 1994–1998 10.1126/science.284.5422.1994 [DOI] [PubMed] [Google Scholar]
- 10. De Bock K., Cauwenberghs S., and Carmeliet P. (2011) Vessel abnormalization: another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr. Opin. Genet. Dev. 21, 73–79 10.1016/j.gde.2010.10.008 [DOI] [PubMed] [Google Scholar]
- 11. Kim Y. W., and Byzova T. V. (2014) Oxidative stress in angiogenesis and vascular disease. Blood 123, 625–631 10.1182/blood-2013-09-512749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ushio-Fukai M. (2006) Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 71, 226–235 10.1016/j.cardiores.2006.04.015 [DOI] [PubMed] [Google Scholar]
- 13. Takac I., Schröder K., and Brandes R. P. (2012) The Nox family of NADPH oxidases: friend or foe of the vascular system?. Curr. Hypertens. Rep. 14, 70–78 10.1007/s11906-011-0238-3 [DOI] [PubMed] [Google Scholar]
- 14. Zhang M., and Shah A. M. (2014) ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 102, 249–257 10.1093/cvr/cvu050 [DOI] [PubMed] [Google Scholar]
- 15. Brandes R. P., Weissmann N., and Schröder K. (2014) Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 76, 208–226 10.1016/j.freeradbiomed.2014.07.046 [DOI] [PubMed] [Google Scholar]
- 16. Lambeth J. D., and Neish A. S. (2014) Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu. Rev. Pathol. 9, 119–145 10.1146/annurev-pathol-012513-104651 [DOI] [PubMed] [Google Scholar]
- 17. Leto T. L., Morand S., Hurt D., and Ueyama T. (2009) Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox. Signal. 11, 2607–2619 10.1089/ars.2009.2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nauseef W. M. (2014) Detection of superoxide anion and hydrogen peroxide production by cellular NADPH oxidases. Biochim. Biophys. Acta 1840, 757–767 10.1016/j.bbagen.2013.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nayernia Z., Jaquet V., and Krause K.-H. (2014) New insights on NOX enzymes in the central nervous system. Antioxid. Redox. Signal. 20, 2815–2837 10.1089/ars.2013.5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sumimoto H., Minakami R., and Miyano K. (2019) Soluble regulatory proteins for activation of NOX family NADPH oxidases. Methods Mol. Biol 121–137. 1982. [DOI] [PubMed] [Google Scholar]
- 21. Kuroda J., Nakagawa K., Yamasaki T., Nakamura K., Takeya R., Kuribayashi F., Imajoh-Ohmi S., Igarashi K., Shibata Y., Sueishi K., and Sumimoto H. (2005) The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes. Cells. 10, 1139–1151 10.1111/j.1365-2443.2005.00907.x [DOI] [PubMed] [Google Scholar]
- 22. Schröder K., Zhang M., Benkhoff S., Mieth A., Pliquett R., Kosowski J., Kruse C., Luedike P., Michaelis U. R., Weissmann N., Dimmeler S., Shah A. M., and Brandes R. P. (2012) Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 110, 1217–1225 10.1161/CIRCRESAHA.112.267054 [DOI] [PubMed] [Google Scholar]
- 23. Chen L., Xiao J., Kuroda J., Ago T., Sadoshima J., Cohen R. A., and Tong X. (2014) Both hydrogen peroxide and transforming growth factor beta 1 contribute to endothelial Nox4 mediated angiogenesis in endothelial Nox4 transgenic mouse lines. Biochim. Biophys. Acta 1842, 2489–2499 10.1016/j.bbadis.2014.10.007 [DOI] [PubMed] [Google Scholar]
- 24. Craige S. M., Chen K., Pei Y., Li C., Huang X., Chen C., Shibata R., Sato K., Walsh K., and Keaney J. F. Jr (2011) NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 124, 731–740 10.1161/CIRCULATIONAHA.111.030775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang M., Brewer A. C., Schröder K., Santos C. X., Grieve D. J., Wang M., Anilkumar N., Yu B., Dong X., Walker S. J., Brandes R. P., and Shah A. M. (2010) NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. U S A 107, 18121–18126 10.1073/pnas.1009700107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vogel J., Kruse C., Zhang M., and Schröder K. (2015) Nox4 supports proper capillary growth in exercise and retina neo-vascularization. J. Physiol. 593, 2145–2154 10.1113/jphysiol.2014.284901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meng D., Mei A., Liu J., Kang X., Shi X., Qian R., and Chen S. (2012) NADPH oxidase 4 mediates insulin-stimulated HIF-1α and VEGF expression, and angiogenesis in vitro. PLoS ONE 7, e48393 10.1371/journal.pone.0048393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Helfinger V., Henke N., Harenkamp S., Walter M., Epah J., Penski C., Mittelbronn M., and Schröder K. (2016) The NADPH Oxidase Nox4 mediates tumour angiogenesis. Acta Physiol. 216, 435–446 10.1111/apha.12625 [DOI] [PubMed] [Google Scholar]
- 29. Bernatchez P. N., Soker S., and Sirois M. G. (1999) Vascular endothelial growth factor effect on endothelial cell proliferation, migration, and platelet-activating factor synthesis is Flk-1-dependent. J. Biol. Chem. 274, 31047–31054 10.1074/jbc.274.43.31047 [DOI] [PubMed] [Google Scholar]
- 30. Edgell C. J., McDonald C. C., and Graham J. B. (1983) Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc. Natl. Acad. Sci. U S A 80, 3734–3737 10.1073/pnas.80.12.3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hseu Y. C., Chen S. C., Lin W. H., Hung D. Z., Lin M. K., Kuo Y. H., Wang M. T., Cho H. J., Wang L., and Yang H. L. (2011) Toona sinensis (leaf extracts) inhibit vascular endothelial growth factor (VEGF)-induced angiogenesis in vascular endothelial cells. J. Ethnopharmacol. 134, 111–121 10.1016/j.jep.2010.11.058 [DOI] [PubMed] [Google Scholar]
- 32. Kanegasaki S., Nomura Y., Nitta N., Akiyama S., Tamatani T., Goshoh Y., Yoshida T., Sato T., and Kikuchi Y. (2003) A novel optical assay system for the quantitative measurement of chemotaxis. J. Immunol. Methods 282, 1–11 10.1016/j.jim.2003.07.008 [DOI] [PubMed] [Google Scholar]
- 33. Nitta N., Tsuchiya T., Yamauchi A., Tamatani T., and Kanegasaki S. (2007) Quantitative analysis of eosinophil chemotaxis tracked using a novel optical device–TAXIScan. J. Immunol. Methods 320, 155–163 10.1016/j.jim.2006.12.010 [DOI] [PubMed] [Google Scholar]
- 34. Yamauchi A., Degawa-Yamauchi M., Kuribayashi F., Kanegasaki S., and Tsuchiya T. (2014) Systematic single cell analysis of migration and morphological changes of human neutrophils over stimulus concentration gradients. J. Immunol. Methods 404, 59–70 10.1016/j.jim.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 35. Witzenbichler B., Maisonpierre P. C., Jones P., Yancopoulos G. D., and Isner J. M. (1998) Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie2. J. Biol. Chem. 273, 18514–18521 10.1074/jbc.273.29.18514 [DOI] [PubMed] [Google Scholar]
- 36. Herzog B., Pellet-Many C., Britton G., Hartzoulakis B., and Zachary I. C. (2011) VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Biol. Cell 22, 2766–2776 10.1091/mbc.E09-12-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lampugnani M. G., Orsenigo F., Gagliani M. C., Tacchetti C., and Dejana E. (2006) Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 174, 593–604 10.1083/jcb.200602080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Santos S. C., Miguel C., Domingues I., Calado A., Zhu Z., Wu Y., and Dias S. (2007) VEGF and VEGFR-2 (KDR) internalization is required for endothelial recovery during wound healing. Exp. Cell Res. 313, 1561–1574 10.1016/j.yexcr.2007.02.020 [DOI] [PubMed] [Google Scholar]
- 39. Ewan L. C., Jopling H. M., Jia H., Mittar S., Bagherzadeh A., Howell G. J., Walker J. H., Zachary I. C., and Ponnambalam S. (2006) Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic 7, 1270–1282 10.1111/j.1600-0854.2006.00462.x [DOI] [PubMed] [Google Scholar]
- 40. Fujiwara T., Oda K., Yokota S., Takatsuki A., and Ikehara Y. (1988) Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol.Chem. 263, 18545–18552 [PubMed] [Google Scholar]
- 41. Forman H. J., Maiorino M., and Ursini F. (2010) Signaling functions of reactive oxygen species. Biochemistry 49, 835–842 10.1021/bi9020378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janiszewski M., Lopes L. R., Carmo A. O., Pedro M. A., Brandes R. P., Santos C. X., and Laurindo F. R. (2005) Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 280, 40813–40819 10.1074/jbc.M509255200 [DOI] [PubMed] [Google Scholar]
- 43. Lee H. Y., Zeeshan H. M. A., Kim H. R., and Chae H. J. (2017) Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic. Biol. Med. 113, 26–35 10.1016/j.freeradbiomed.2017.09.010 [DOI] [PubMed] [Google Scholar]
- 44. Petry A., Djordjevic T., Weitnauer M., Kietzmann T., Hess J., and Görlach A. (2006) NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid. Redox Signal. 8, 1473–1484 10.1089/ars.2006.8.1473 [DOI] [PubMed] [Google Scholar]
- 45. Chen K., Kirber M. T., Xiao H., Yang Y., and Keaney J. F. Jr (2008) Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 181, 1129–1139 10.1083/jcb.200709049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ambasta R. K., Kumar P., Griendling K. K., Schmidt H. H., Busse R., and Brandes R. P. (2004) Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 279, 45935–45941 10.1074/jbc.M406486200 [DOI] [PubMed] [Google Scholar]
- 47. von Löhneysen K., Noack D., Jesaitis A. J., Dinauer M. C., and Knaus U. G. (2008) Mutational analysis reveals distinct features of the Nox4-p22phox complex. J. Biol. Chem. 283, 35273–35282 10.1074/jbc.M804200200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kleikers P. W., Wingler K., Hermans J. J., Diebold I., Altenhöfer S., Radermacher K. A., Janssen B., Görlach A., and Schmidt H. H. (2012) NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 90, 1391–1406 10.1007/s00109-012-0963-3 [DOI] [PubMed] [Google Scholar]
- 49. Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M. D., and Sadoshima J. (2010) NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. U S A 107, 15565–15570 10.1073/pnas.1002178107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sirokmány G., Donkó Á., and Geiszt M. (2016) Nox/Duox family of NADPH oxidases: lessons from knockout mouse models. Trends Pharmacol. Sci. 37, 318–327 10.1016/j.tips.2016.01.006 [DOI] [PubMed] [Google Scholar]
- 51. Datla S. R., Peshavariya H., Dusting G. J., Mahadev K., Goldstein B. J., and Jiang F. (2007) Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 27, 2319–2324 10.1161/ATVBAHA.107.149450 [DOI] [PubMed] [Google Scholar]
- 52. Esser S., Lampugnani M. G., Corada M., Dejana E., and Risau W. (1998) Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 111, 1853–1865 [DOI] [PubMed] [Google Scholar]
- 53. Takahashi T., and Shibuya M. (1997) The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene 14, 2079–2089 10.1038/sj.onc.1201047 [DOI] [PubMed] [Google Scholar]
- 54. Bernatchez P. N., Acevedo L., Fernandez-Hernando C., Murata T., Chalouni C., Kim J., Erdjument-Bromage H., Shah V., Gratton J. P., McNally E. M., Tempst P., and Sessa W. C. (2007) Myoferlin regulates vascular endothelial growth factor receptor-2 stability and function. J. Biol. Chem. 282, 30745–30753 10.1074/jbc.M704798200 [DOI] [PubMed] [Google Scholar]
- 55. Srinivasan S., Chitalia V., Meyer R. D., Hartsough E., Mehta M., Harrold I., Anderson N., Feng H., Smith L. E., Jiang Y., Costello C. E., and Rahimi N. (2015) Hypoxia-induced expression of phosducin-like 3 regulates expression of VEGFR-2 and promotes angiogenesis. Angiogenesis 18, 449–462 10.1007/s10456-015-9468-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maghsoudlou A., Meyer R. D., Rezazadeh K., Arafa E., Pudney J., Hartsough E., and Rahimi N. (2016) RNF121 inhibits angiogenic growth factor signaling by restricting cell surface expression of VEGFR-2. Traffic 17, 289–300 10.1111/tra.12353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Prior K. K., Wittig I., Leisegang M. S., Groenendyk J., Weissmann N., Michalak M., Jansen-Dürr P., Shah A. M., and Brandes R. P. (2016) The endoplasmic reticulum chaperone calnexin is a NADPH oxidase NOX4 interacting protein. J. Biol. Chem. 291, 7045–7059 10.1074/jbc.M115.710772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Williams D. B. (1995) The Merck Frosst Award Lecture 1994/La conference Merck Frosst 1994. Calnexin: a molecular chaperone with a taste for carbohydrate. Biochem. Cell Biol. 73, 123–132 [DOI] [PubMed] [Google Scholar]
- 59. Oka O. B., and Bulleid N. J. (2013) Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2425–2429 10.1016/j.bbamcr.2013.02.007 [DOI] [PubMed] [Google Scholar]
- 60. Okumura M., Kadokura H., and Inaba K. (2015) Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic. Biol. Med. 83, 314–322 10.1016/j.freeradbiomed.2015.02.010 [DOI] [PubMed] [Google Scholar]
- 61. Zito E. (2015) ERO1: a protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 83, 299–304 10.1016/j.freeradbiomed.2015.01.011 [DOI] [PubMed] [Google Scholar]
- 62. Kawai C., Yamauchi A., and Kuribayashi F. (2018) Monoclonal antibody 7D5 recognizes the R147 epitope on the gp91phox, phagocyte flavocytochrome b558 large subunit. Microbiol. Immunol. 62, 269–280 10.1111/1348-0421.12584 [DOI] [PubMed] [Google Scholar]
- 63. Yamauchi A., Yamamura M., Katase N., Itadani M., Okada N., Kobiki K., Nakamura M., Yamaguchi Y., and Kuribayashi F. (2017) Evaluation of pancreatic cancer cell migration with multiple parameters in vitro by using an optical real-time cell mobility assay device. BMC Cancer 17, 234 10.1186/s12885-017-3218-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruch W., Cooper P. H., and Baggiolini M. (1983) Assay of H2O2 production by macrophages and neutrophils with homovanillic acid and horse-radish peroxidase. J. Immunol. Methods 63, 347–357 10.1016/S0022-1759(83)80008-8 [DOI] [PubMed] [Google Scholar]
- 65. Martyn K. D., Frederick L. M., von Loehneysen K., Dinauer M. C., and Knaus U. G. (2006) Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 18, 69–82 10.1016/j.cellsig.2005.03.023 [DOI] [PubMed] [Google Scholar]
- 66. Morini M., Benelli R., Giunciuglio D., Carlone S., Arena G., Noonan D. M., and Albini A. (2000) Kaposi's sarcoma cells of different etiologic origins respond to HIV-Tat through the Flk-1/KDR (VEGFR-2): relevance in AIDS-KS pathology. Biochem. Biophys. Res. Commun. 273, 267–271 10.1006/bbrc.2000.2941 [DOI] [PubMed] [Google Scholar]
- 67. Miyano K., and Sumimoto H. (2014) N-Linked glycosylation of the superoxide-producing NADPH oxidase Nox1. Biochem. Biophys. Res. Commun 443, 1060–1065 10.1016/j.bbrc.2013.12.086 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript and supporting information.