

Abstract
Cleavage of influenza virus hemagglutinin (HA) by host proteases is essential for virus infectivity. HA of most influenza A and B (IAV/IBV) viruses is cleaved at a monobasic motif by trypsin-like proteases. Previous studies have reported that transmembrane serine protease 2 (TMPRSS2) is essential for activation of H7N9 and H1N1pdm IAV in mice but that H3N2 IAV and IBV activation is independent of TMPRSS2 and carried out by as-yet-undetermined protease(s). Here, to identify additional H3 IAV- and IBV-activating proteases, we used RNA-Seq to investigate the protease repertoire of murine lower airway tissues, primary type II alveolar epithelial cells (AECIIs), and the mouse lung cell line MLE-15. Among 13 candidates identified, TMPRSS4, TMPRSS13, hepsin, and prostasin activated H3 and IBV HA in vitro. IBV activation and replication was reduced in AECIIs from Tmprss2/Tmprss4-deficient mice compared with WT or Tmprss2-deficient mice, indicating that murine TMPRSS4 is involved in IBV activation. Multicycle replication of H3N2 IAV and IBV in AECIIs of Tmprss2/Tmprss4-deficient mice varied in sensitivity to protease inhibitors, indicating that different, but overlapping, sets of murine proteases facilitate H3 and IBV HA cleavages. Interestingly, human hepsin and prostasin orthologs did not activate H3, but they did activate IBV HA in vitro. Our results indicate that TMPRSS4 is an IBV-activating protease in murine AECIIs and suggest that TMPRSS13, hepsin, and prostasin cleave H3 and IBV HA in mice. They further show that hepsin and prostasin orthologs might contribute to the differences observed in TMPRSS2-independent activation of H3 in murine and human airways.
Keywords: infectious disease, serine protease, trypsin, virus, mouse, airway proteases, hemagglutinin, influenza virus, cleavage, protease gene expression, TMPRSS2, TMPRSS4, hepsin, prostasin, trypsin-like protease, host-pathogen interactions, mouse lung proteases
Influenza is a highly contagious respiratory illness. Of the four genera of influenza viruses (A, B, C, and D), influenza A and B viruses (IAV/IBV)2 pose a continuous threat to public health and are responsible for seasonal epidemics that may result in 3–5 million cases of severe respiratory illness and 290,000 to 650,000 deaths annually (World Health Organization, November 2018). The natural reservoir of IAV is wild aquatic birds, from where they are transmitted to a wide range of mammalian and avian hosts, including humans, pigs, and poultry (1). Based on antigenic criteria of the two viral surface glycoproteins hemagglutinin (HA) and neuraminidase (NA), 16 HA subtypes (H1–H16) and nine NA subtypes (N1–N9) have been identified in avian species, and two more HA subtypes (H17 and H18) and NA subtypes (N10 and N11) have been found in bats. In contrast, humans are the primary host for IBV. Currently, two antigenic lineages of IBV, Victoria and Yamagata, co-circulate with IAV of subtype H1N1 and H3N2 in the human population (2). In addition to seasonal influenza, the recurrent transmission of avian IAV to other host species provides the basis for the emergence of new influenza viruses for which there is little or no preexisting immunity in the human population and may provoke an influenza pandemic. Four influenza pandemics occurred in the 20th and 21st century, with the 1918 influenza pandemic (H1N1, Spanish flu) being the most devastating influenza pandemic in recent history with an estimated 50 million deaths worldwide. Since their emergence in 1997 and 2013, respectively, human infections with avian H5N1 and H7N9 viruses have caused over 860 and 1500 confirmed cases of severe illness with mortality rates of 53 and 39% (World Health Organization Influenza (Seasonal) Fact Sheet; https://www.who.int/news-room/fact-sheets/detail/influenza (accessed September 24, 2019)).3
IAV and IBV belong to the family Orthomyxoviridae and are enveloped viruses with a negative-sense, single-stranded RNA genome that consists of eight segments. Influenza virus infection is initiated by the major surface glycoprotein HA through binding to sialic acid–containing receptors and fusion of the viral lipid envelope and the endosomal membrane following receptor-mediated endocytosis to release the viral genome into the host cell. HA is synthesized as a fusion-incompetent precursor protein, HA0, in the infected cell and requires cleavage by a host cell protease into the subunits HA1 and HA2, which remain covalently linked by a disulfide bond. Cleavage of HA is a prerequisite for conformational changes at low pH in the endosome that trigger membrane fusion activity, and it is essential for virus infectivity (reviewed in Ref. 4). Most influenza viruses, including human IAV and IBV and low pathogenic avian IAV, possess a monobasic HA cleavage site composed of a single arginine (rarely lysine) residue. HA with a monobasic cleavage site is activated by trypsin-like proteases present in the airways of mammalian hosts and respiratory and intestinal tissues of avian species, respectively, that remained unknown for a long time (reviewed in Ref. 5). In 2006, we identified the type II transmembrane serine proteases (TTSP) transmembrane serine protease 2 (TMPRSS2) and human airway trypsin-like protease (HAT, also designated as TMPRSS11D) as the first human proteases activating IAV HA with a monobasic cleavage site in vitro (6). Thereafter, a number of human TTSPs have been shown to activate IAV HA and more recently IBV HA with a monobasic cleavage site in vitro (7–11). In addition, human kallikrein 1 (KLK1) (also known as tissue kallikrein) and the kallikrein-related peptidases KLK5 and KLK12 were shown to cleave IAV HA, but not IBV HA with a monobasic cleavage site in vitro (11–13). Further studies demonstrated that TMPRSS2 (also designated as epitheliasin in mice) is essential for activation and spread, and consequently pathogenesis, of H1N1pdm, H7N9, and H10 IAV in mice (14–18). Intriguingly, TMPRSS2-deficient mice were protected from pathogenesis and lethal outcome of infection. In contrast, proteolytic activation and pathogenesis of certain H3N2 IAV strains and IBV was shown to be independent of TMPRSS2 in mice, indicating that an additional yet undetermined protease(s) supports activation of H3 and IBV HA (14–16, 19). TMPRSS4 was demonstrated to be involved in H3N2 activation in vivo. Tmprss2−/−Tmprss4−/− double-knockout mice showed reduced body weight loss and mortality upon H3N2 infection compared with WT or single-knockout mice (17). However, H3N2 virus was still proteolytically activated in the double-knockout mice, indicating that at least one more H3-cleaving protease is present in murine lung. Together, these previous findings have made clear that HA with a monobasic cleavage site can differ in its sensitivity to host cell proteases in mice in vivo.
Recently, we identified TMPRSS2 as the major activating protease of H1N1pdm, H7N9, and also H3N2 IAV in primary human bronchial epithelial cells (HBECs) and alveolar epithelial cells type II (AECIIs) as well as in Calu-3 human airway cells (20). Proteolytic activation of IBV was found to be independent of TMPRSS2 in Calu-3 cells and primary HBECs, consistent with results previously observed in mice (19, 20). Interestingly, TMPRSS2 was crucial for IBV activation in primary human AECIIs, indicating that expression of IBV-activating proteases differs along the respiratory tract and that appropriate proteases in addition to TMPRSS2 are present in human bronchial cells, but obviously not in AECIIs (20). In summary, these data showed that IBV can be activated by a broader range of host cell proteases in the airways of both mice and humans, whereas activation of H3N2 is supported by proteases in addition to TMPRSS2 only in mice. The proteases responsible for IBV activation in Calu-3 cells and HBECs remained undetermined. Furthermore, it remained unknown whether IBV is activated by orthologous or different proteases in human and murine airway cells.
In this study, by comprehensive RNA-Seq, we examined the repertoire of protease-specific mRNAs present in trachea, bronchi, and lungs of mice to investigate expression and distribution of trypsin-like proteases in the murine lower airways. In addition, we compared the protease transcriptome of primary murine AECIIs and the mouse lung cell line MLE-15, which do and do not support cleavage of HA with a monobasic cleavage site, respectively, to identify H3/IBV HA-activating proteases in addition to TMPRSS2 in murine lung. Potential protease candidates were then analyzed for cleavage of IBV HA and H3 in vitro. We demonstrate that TMPRSS4 contributes to IBV activation in primary murine AECIIs and identify TMPRSS13, hepsin, and prostasin as further potential H3/IBV HA–cleaving proteases in murine lung. Moreover, our data show that hepsin and prostasin orthologues might contribute to the differences observed for TMPRSS2-independent activation of H3 in murine and human cells.
Results
Transcriptome analysis of the murine lower respiratory tract reveals differences in the expression and distribution of trypsin-like serine proteases
First, we analyzed the protease repertoire of murine lower airways to screen for further HA-activating protease candidates and to further examine their distribution along the murine respiratory tract. This was achieved by using RNA-Seq of mRNA species present in trachea, bronchi, and lungs of naive mice (C57BL/6J background). Detected genes expressed in total were 18,086 in trachea, 17,871 in bronchi, and 17,683 in lungs, representing approximately 72.3, 71.5, and 70.7% of the murine genome (∼25,000 genes (21)), respectively (Fig. 1A). Transcripts were prefiltered to those that yielded at least one FPKM (fragments per kilobase of exon model per million reads mapped) value of ≥0.3 and a tag count ≥50 out of three replicates to exclude insufficiently covered genes from the analysis. Similar numbers of genes for proteases and homologs expressed in trachea, bronchi, and lungs with 468, 465, and 461, respectively, were detected (Fig. 1A), representing approximately 69.6, 69.2, and 68.6% of all murine protease genes (total of 672; Mammalian Degradome Database), respectively. Of these, 68 (trachea and bronchi) and 66 (lungs) expressed serine protease genes were detected (Fig. 1A), which represent 29.6 and 28.7% of all murine serine proteases (total of 230; Mammalian Degradome Database). Among the detected serine proteases, we were particularly interested in serine proteases with trypsin-like activity that have a substrate preference for an arginine residue in the P1 position. Therefore, detected serine proteases were analyzed with the UniProt (22) and MEROPS (23) databases. Finally, 31 expressed trypsin-like serine proteases were selected in trachea and 28 in bronchi, whereas only 25 were present in lungs (Fig. 1A). Expression levels of these trypsin-like protease candidates are shown in Fig. 1B; depicted are mean values of three biological replicates (mean and individual expression values with corresponding S.D. values are included in Table S1). The expression profile of trypsin-like serine proteases differed between examined tissues. Tmprss2 expression was found to be solid and rather constant in trachea, bronchi, and lungs, respectively. A number of human TTSPs, including TMPRSS4, TMPRSS13, and matriptase as well as the KLK members KLK1, KLK5, and KLK12, have been shown to cleave IAV and recently IBV HA with a monobasic cleavage site in vitro (reviewed in Refs. 9, 11, and 22). Therefore, we first focused on expression of these protease genes in murine lower airways. The expression profile of TTSP members Corin, Hpn/hepsin, Tmprss11a, Tmprss11g, Tmprss13, Tmprss3, Tmprss4, Tmprss5, and Tmprss6 was less strong and varied between tissues, with four TTSPs (Tmprss11a, Tmprss11g, Tmprss3, and Tmprss6) being not expressed in lung tissue. However, expression of Tmprss4 increased from trachea to lung, whereas the opposite was found for St14/matriptase. We did not detect expression of Klk5 and Klk12 in murine trachea, bronchi, and lungs. Low expression of Klk1 was detected in lungs, and even lower gene expression values were found in trachea and bronchi. Klk11 was expressed in trachea and bronchi and, to a lower extent, in lungs, whereas Klk8 was expressed at higher levels in lung tissue compared with trachea and bronchi. Expression of Klk14 was detected only in lung. Strong expression of Cfb/complement factor B (CFB), Plat/tissue-type plasminogen activator (tPA), and Prss8/prostasin was detected in all three tissues. Expression of Ltf/lactotransferrin was extremely high compared with all other serine proteases in trachea and bronchi, but much lower in lung.
Figure 1.

Protease transcriptome analysis of the murine lower respiratory tract by RNA-Seq. A, numbers of detected protease genes in trachea, bronchi, and lungs of mice. Depicted are all detected genes with at least one FPKM value of ≥0.3 and a tag count ≥50 out of three biological replicates (a), all detected genes coding for proteases and homologs according to the gene ontology classification and genes annotated as hydrolases (EC 3.4) (b), all detected genes coding for proteases that are classified as serine proteases (EC 3.4.21) (c), and all detected genes coding for serine proteases with substrate preference for an arginine residue in P1 based on the UniProt and MEROPS databases (d). B, heatmap of detected proteases with trypsin-like activity in trachea, bronchi, and lungs of mice with at least one FPKM value of ≥0.3 and a tag count ≥50 out of three biological replicates. Shown are mean values with expression levels color-coded (blue, low expression; red, high expression).
In summary, analysis of the protease transcriptome of the murine lower airways revealed 34 proteases possessing a trypsin-like activity expressed, although some of them at very low levels. Moreover, the data show that these serine proteases can vary in their distribution in respiratory tissues and that a slightly reduced number are expressed in lungs compared with trachea and bronchi of mice.
Immortalized murine lung cell line MLE-15 does not promote proteolytic activation of H3N2 IAV and IBV
Comprehensive transcriptomic profiling of the murine lower respiratory tract revealed 68 protease candidates in trachea and bronchi and 66 in lungs; of those, 31 (trachea), 28 (bronchi), and 25 (lung) remained as potential HA-cleaving protease candidates due to their trypsin-like substrate specificity. To further reduce the number of protease candidates, we wished to compare the protease repertoire of murine lung cells that do support HA cleavage with murine lung cells that do not support HA cleavage and, thus, most likely lack expression of relevant proteases. Recently, we demonstrated that primary murine AECIIs support proteolytic activation of IAV and IBV with a monobasic HA cleavage site and express H3– and IBV HA–cleaving proteases in addition to TMPRSS2 (20). To confirm our observations, multicycle replication and HA cleavage of Aichi/H3N2/PR8 and Malaysia/B in murine AECIIs were examined in the absence and presence of exogenous trypsin. Primary murine AECIIs were inoculated with Aichi/H3N2/PR8 and Malaysia/B at low multiplicity of infection (MOI) and incubated in the absence or presence of trypsin (0.2 or 0.4 μg/ml) for 72 h. At the indicated time points, virus titers in cell culture supernatants were determined by a plaque assay. Both viruses replicated efficiently in primary AECIIs in the absence of trypsin as shown in Fig. 2A. Trypsin treatment caused no increase in viral titers of Aichi/H3N2/PR8 over time. Replication of Malaysia/B was marginally increased in the presence of 0.2 and 0.4 μg/ml at 48 h postinfection (p.i.) and at 72 h p.i. in the presence of 0.4 μg/ml trypsin. Moreover, cell lysates were analyzed for HA cleavage by SDS-PAGE and Western blotting at 72 h p.i. using HA-specific antibodies. As expected, cleavage of HA0 of Aichi/H3N2/PR8 and Malaysia/B into HA1 and HA2 (not detected by the antibodies) was observed in primary AECIIs in the absence of trypsin (Fig. 2B). Cleavage of H3 was enhanced in the presence of 0.2 and 0.4 μg/ml trypsin. Cleavage of B HA was also enhanced by trypsin treatment in a dose-dependent manner. Interestingly, enhanced HA cleavage did not result in significantly higher virus titers for both viruses in primary AECIIs. In sum, the data show that primary AECIIs express proteases that support cleavage of HA with a monobasic cleavage site.
Figure 2.

Multicycle replication and HA cleavage analysis of IAV and IBV with a monobasic cleavage site in primary AECIIs and MLE-15 cells in the absence and presence of exogenous trypsin. A, primary AECIIs were infected with Aichi/H3N2/PR8 or Malaysia/B at a low MOI of 0.004 and incubated in the absence (w/o) or presence of trypsin (0.2 or 0.4 μg/ml) for 72 h. Viral titers were determined as pfu/ml by a plaque assay at the indicated time points postinfection. Results are mean values ± S.D. (error bars) of two independent experiments. B, analysis of HA cleavage in AECIIs at 72 h p.i. Cell lysates were subjected to reducing SDS-PAGE and Western blot analysis with HA-specific antibodies. β-Actin was used as a loading control. C, MLE-15 mouse lung epithelial cells were infected with Aichi/H3N2/PR8 and Malaysia/B at a MOI of 0.01 and 0.1, respectively, and incubated in the absence (w/o) or presence of trypsin (0.2 or 0.4 μg/ml) for 72 h. Viral titers were determined by plaque assays at the indicated time points postinfection. Results are mean values ± S.D. of three independent experiments. Log-transformed data were analyzed by one-way ANOVA for each time point followed by Tukey's multiple-comparison test according to the number of parameters and groups being compared. Obtained p values were corrected for multiple-hypothesis testing using Benjamini–Hochberg correction. Statistical significance is indicated for w/o versus 0.2 μg/ml and w/o versus 0.4 μg/ml. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****) were considered significant; p > 0.05 (ns) was considered nonsignificant. D, analysis of HA cleavage in MLE-15 cells. Cells were infected with Aichi/H3N2/PR8 or Malaysia/B with a high MOI of 1 and incubated with or without (w/o) trypsin treatment for 24 h. Cell lysates were subjected to reducing SDS-PAGE and Western blot analysis with HA-specific antibodies. Uninfected cells were used as mock control. β-Actin served as loading control.
In search of a murine cell line that does not support cleavage of HA with a monobasic cleavage site, we examined proteolytic activation and multicycle replication of Aichi/H3N2/PR8 and Malaysia/B in the immortalized mouse lung epithelial cell line MLE-15 (25). Confluent MLE-15 monolayers were infected with Aichi/H3N2/PR8 or Malaysia/B at a MOI of 0.01–0.1 and incubated in the absence or presence of trypsin (0.2 or 0.4 μg/ml) for 72 h. At the indicated time points, virus titer in cell culture supernatants was determined by plaque assay. As shown in Fig. 2C, both viruses were not able to replicate in MLE-15 cells in the absence of trypsin. In contrast, trypsin treatment supported efficient replication of Aichi/H3N2/PR8 in MLE-15 cells and caused significant increase in virus titer in a dose-dependent manner. Virus titer of Malaysia/B was increased 10-fold at 16 h p.i. only by the high amount of trypsin (0.4 μg/ml) treatment, whereas the effect was marginal at 0.2 μg/ml. However, the virus was not able to replicate efficiently in these cells, indicating that MLE-15 cells are not permissive for Malaysia/B for so far unknown reasons. To analyze HA cleavage in MLE-15 cells, cells were infected with Aichi/H3N2/PR8 or Malaysia/B at a high MOI of 1 and incubated with or without exogenous trypsin for 24 h. Cell lysates were subjected to SDS-PAGE and Western blotting using HA-specific antibodies. Infection with Aichi/H3N2/PR8 resulted in detection of only uncleaved precursor HA0 in the absence of trypsin in MLE-15 cells, whereas cleavage of HA0 was observed in trypsin-treated cells (Fig. 2D). The data indicate that MLE-15 cells most likely lack expression of an appropriate HA-cleaving protease, and compensation of this lack by exogenous trypsin supports proteolytic activation and multiplication of Aichi/H3N2/PR8 in the cells. Cleavage of HA of Malaysia/B in MLE-15 cells was also strongly enhanced by trypsin treatment (Fig. 2D). However, a low amount of Malaysia/B HA1 was also detected in untreated MLE-15 cells, suggesting that MLE-15 cells express low levels of an appropriate IBV HA–cleaving protease.
Together, the data suggest that MLE-15 cells lack expression of (enzymatically active) H3-cleaving proteases and sufficient amounts of IBV HA–cleaving proteases. Therefore, MLE-15 cells were considered as a suitable testing system for a murine lung cell line that does not support cleavage of HA with a monobasic cleavage site. Hence, proteases expressed in AECIIs but not in MLE-15 cells or expressed at much higher levels in AECIIs compared with MLE-15 cells are potential HA-cleaving candidates, whereas proteases present at higher levels in MLE-15 cells compared with AECIIs or only in MLE-15 cells represent negative candidates.
Comparative expression profiling of trypsin-like serine protease mRNAs in MLE-15 cells and primary murine AECIIs
Comparative transcriptome analysis of proteases with trypsin-like specificity expressed in primary murine AECIIs and MLE-15 cells was performed by RNA-Seq as described above. The expression profile of serine proteases with trypsin-like substrate specificity in MLE-15 cells and AECIIs is depicted in a heatmap containing mean values of three biological replicates (Fig. 3A) (mean and individual expression values with corresponding standard deviations are included in Table S2). Overall, 22 listed proteases were detected in at least one condition. Nine proteases (Hgfac, Klk11, Klk8, Ltf, Prss57, Tmprss13, Tmprss2, Prss27, and Prss32) were found to be expressed only in AECIIs and not in MLE-15 cells. However, gene expression values of Klk11, Prss27, and Prss32 were very low, and the proteases were therefore discarded as promising candidates. The expression profile of Tmprss2 exemplifies here our model as a protease involved in HA cleavage with barely detectable expression in MLE-15 cells compared with robust expression levels in AECII. Two protease genes were detected only in MLE-15 cells (Klk14 and Prss12/neurotrypsin) and therefore considered as negative candidates. Eleven protease genes were found to be expressed in both cell culture models (C2, Cfb, Hpn, Plat, Plau, Proc, Prss22, Prss36, St14, Tmprss4, and Prss8). The number of protease candidates present in both cell models was then further reduced to six protease genes (Cfb, Hpn, Plau, Prss22, St14, Tmprss4) based on higher expression levels in AECIIs compared with MLE-15 (with a log2 -fold change (log2FC) of ≥2.0 in AECIIs compared with MLE-15) (Fig. 3B and Table S3). In sum, 11 proteases present in AECIIs only (Hgfac/hepatocyte growth factor activator (HGFA), Klk8, Ltf, Prss57/neutrophil serine protease 4 (NSP4), and Tmprss13) or expressed at higher levels in AECIIs versus MLE-15 cells (Cfb, Hpn/hepsin, Plau/urokinase-type plasminogen activator (uPA), Prss22/tryptase ϵ, St14/matriptase, and Tmprss4) were identified as potential H3/IBV HA–cleaving protease candidates (Fig. 3C). We furthermore included Plat/tPA and Prss8/prostasin as candidates in our further studies, although both proteases were expressed at slightly higher levels in MLE-15 cells compared with AECII. However, both proteases were available as expression plasmids to us; were found to be expressed in murine trachea, bronchi, and lung; and have not been analyzed for HA cleavage in more detail so far. Thus, altogether 13 potential H3/IBV HA–cleaving protease candidates were identified by comparison of the protease transcriptome of MLE-15 versus primary murine AECIIs (Fig. 3C). Hereinafter, all 13 protease candidates were analyzed for cleavage of H3 and IBV HA upon co-expression in cell culture.
Figure 3.

Comparative expression profiling of trypsin-like serine protease mRNAs in MLE-15 cells and murine AECIIs by RNA-Seq. A, heatmap of detected protease genes in MLE-15 cells and AECIIs are mean values of three replicates with at least one FPKM value of ≥0.3 and a tag count of ≥50. Expression levels are color-coded (blue, low expression; red, high expression). B, differential expression analysis of AECIIs compared with MLE-15 cells in log2 -fold change (log2FC), with the threshold of log2FC ≥ 2 representing promising protease candidates. C, identified putative promising protease candidates with respective protein names and abbreviations used in this study.
Malaysia/B HA is activated by murine TMPRSS4, TMPRSS13, hepsin, and prostasin, but not matriptase
First, we investigated whether murine TTSP members TMPRSS4, TMPRSS13, hepsin, and matriptase as well as the glycosylphosphatidylinositol-anchored protease prostasin are able to process the HA of Malaysia/B. HA cleavage was examined by transient co-expression of murine proteases and HA of Malaysia/B in human embryonic kidney 293 (HEK293) cells for 48 h and subsequent analysis of cell lysates by SDS-PAGE and Western blotting. Murine TMPRSS2 served as positive control. As expected, HA of Malaysia/B was not cleaved by an endogenous protease in HEK293 cells, and only the precursor protein HA0 was detected (Fig. 4A). Co-expression of TMPRSS2 supported moderate cleavage of HA0 into HA1 and HA2 (the latter was not detected by the antibody). Cleavage of Malaysia/B HA was also observed upon co-expression of TMPRSS4, TMPRSS13, prostasin, and hepsin, although at a low level for hepsin. In contrast, no cleavage of HA was observed upon co-expression of HA and matriptase (Fig. 4A).
Figure 4.

Malaysia/B HA is proteolytically activated by murine membrane-anchored proteases TMPRSS4, TMPRSS13, hepsin, and prostasin, but not matriptase. A, examination of HA cleavage. HEK293 cells were co-transfected with plasmids encoding Malaysia/B HA and either TMPRSS2, TMPRSS4, TMPRSS13, prostasin, hepsin, or matriptase. Co-transfection of HA-expressing plasmid and empty vector (w/o) and transfection of empty vector only (ev) were used as control. Cell lysates were subjected to SDS-PAGE and Western blot analysis at 48 h post-transfection using HA-specific antibodies. β-Actin served as loading control. B, proteolytic activation and multicycle replication analysis. MDCK cells were transfected with protease-encoding plasmids for 24 h and then infected with Malaysia/B at a MOI 0.01. At 24 h p.i., cells were fixed and immunostained against the viral NP to examine virus spread. Transfection of cells with empty vector (ev) was used as negative control. Data are representatives of at least three independent experiments. C, cleavage of H9 by matriptase. HEK293 cells were co-transfected with plasmids encoding H9 of A/quail/Shantou/782/00 (H9N2) and either TMPRSS2 or matriptase. Co-transfection of H9-expressing plasmid and empty vector (w/o) or transfection of empty vector only (ev) were used as control. Cell lysates were analyzed by SDS-PAGE and Western blotting at 48 h post-transfection using H9-specific antibodies. β-Actin served as loading control. D, MDCK cells were transfected with protease-encoding plasmids for 24 h and then infected with H9N2 at an MOI of 0.03 for 24 h to allow multicycle replication. Cells were fixed and immunostained against NP. Transfection of cells with empty vector (ev) was used as negative control. Data are representative of three independent experiments.
We next investigated whether cleavage of HA by the different proteases activates the fusogenic potential of HA and supports proteolytic activation and multicycle replication of Malaysia/B. For that purpose, MDCK cells were transiently transfected with protease-encoding plasmids, incubated for 24 h, and subsequently infected with Malaysia/B at a MOI of 0.01 and incubated for further 24 h. Cells were fixed and immunostained against the viral nucleoprotein (NP). As shown in Fig. 4B, MDCK cells transfected with empty vector did not support multicycle replication of Malaysia/B, and no spread of infection was visible. In contrast, foci of infection were observed in TMPRSS2-expressing cells at 24 h p.i. due to proteolytic activation of HA of progeny virus. Efficient virus multicycle replication and spread were also visible in MDCK cells expressing TMPRSS4, TMPRSS13, hepsin, and prostasin. No virus spread was observed in matriptase-expressing cells in congruence with the lack of HA cleavage by matriptase.
Expression of matriptase was confirmed by SDS-PAGE and Western blot analysis of the cell lysates using antibodies against the C-terminal FLAG epitope, and both the zymogen and the catalytic domain of the two-chain form were detected (Fig. S1), suggesting that matriptase is expressed as mature protease in the cells. Furthermore, co-expression of murine matriptase and H9 of A/quail/Shantou/782/00 (H9N2) that has been shown to be proteolytically activated by matriptase in vitro (26) was analyzed in HEK293 cells. As shown in Fig. 4C, H9 was cleaved upon co-expression of murine matriptase, although less efficiently compared with cleavage by TMPRSS2, which was used as a positive control. Small foci of infection of H9N2 virus were observed in MDCK cells with transient expression of murine matriptase, indicating proteolytic activation of H9N2 virus in these cells (Fig. 4D). The data show that murine matriptase is expressed as enzymatically active protease in HEK293 and MDCK cells, and thus, lack of Malaysia/B HA cleavage by matriptase was not due to lack of matriptase activity. Taken together, our data show that Malaysia/B HA can be activated by murine membrane-anchored proteases hepsin, prostasin, TMPRSS4, and TMPRSS13 in addition to TMPRSS2 in vitro, but not by matriptase.
Murine proteases CFB, HGFA, lactotransferrin (LTF), tPA, uPA, tryptase ϵ, NSP4, and rat kallikrein-related peptidase 8 (KLK8) do not cleave HA of Malaysia/B
Next, we investigated whether the soluble protease candidates CFB, HGFA, LTF, tPA, uPA, tryptase ϵ (also designated as brain-specific serine protease 4 (BSSP-4) or serine protease 22 (PRSS22)), NSP4 (also designated as serine protease 57 (PRSS57)), and KLK8, are able to activate Malaysia/B HA. Respective protease-expressing plasmids were purchased to investigate HA cleavage by co-expression in cell culture. CFB, LTF, NSP4, tPA, uPA, and HGFA were expressed with a C-terminal Myc-DDK tag to facilitate protein detection. Murine KLK8 was not available during our study, but we tested HA cleavage by recombinant rat KLK8 instead. To examine protease expression in HEK293 cells, the cells were transiently transfected with protease-encoding plasmids for 48 h, and subsequently cell lysates and concentrated cell supernatants were subjected to SDS-PAGE and immunoblotting using FLAG-specific antibodies that recognize the DDK tag. Expression of CFB, LTF, NSP4, tPA, and uPA was detected in both cell lysates and cell supernatants (Fig. S1A). tPA and uPA were detected as zymogen and mature forms by immunoblotting, suggesting that these proteases are expressed as active enzymes in HEK293 cells. LTF was present as 80-kDa protein in the supernatant and as lower-molecular-weight forms of 50, 38, and 36 kDa, most likely representing the zymogen and the mature form following autocatalytic activation as described for bovine LTF (26). CFB with C-terminal Myc-DDK tag was detected as protein of ∼100 kDa, probably the zymogen. NSP4-Myc-DDK was detected as a double band in cell lysate and supernatant, which may represent the zymogen and the mature form lacking the propeptide due to processing by endogenous dipeptidyl peptidase 1 (DPP1) in HEK293 cells (27, 29) (HPA, RRID:SCR_006710, gene name: CTSC (accessed January 3, 2020)).
Next, we examined whether proteases released into the supernatant were able to cleave Malaysia/B HA. HEK293 cells were transiently transfected with HA-encoding plasmid for 48 h and then harvested and incubated with protease-containing supernatants (Fig. 5A, left) for 2 h at 37 °C. In addition, to analyze HA cleavage by rKLK8, HA-expressing HEK293 cells were incubated with soluble, recombinant rKLK8 at 48 h post-transfection (Fig. 5A, right). Subsequently, HA-expressing cells were subjected to SDS-PAGE and Western blot analysis. Treatment of HA-expressing cells with buffer (w/o) or trypsin served as controls. As shown in Fig. 5A, none of the supernatants nor rKLK8 was able to support cleavage of HA0 of Malaysia/B, whereas HA was efficiently cleaved by trypsin. Trypsin treatment resulted in decreased β-actin protein intensity, most likely due to nonspecific protein degeneration and cell lysis. To investigate whether the soluble proteases possess enzymatic activity, the cell supernatants were incubated with fluorogenic peptides. Thereby, high levels of enzymatic activity were measured for tPA and uPA using methylsulfonyl (Mes)-dArg-Gly-Arg-AMC and rKLK8 using Boc-Val-Pro-Arg-AMC as substrates (Fig. S1B), whereas only low, if any, protease activity was measured for cell supernatants containing CFB, LTF, and NSP4 (data not shown). Finally, we examined multicycle Malaysia/B replication in MDCK cells with transient expression of tPA, uPA, CFB, LTF, and NSP4 as a different cell-based assay (Fig. 5B). MDCK cells were transfected with protease-encoding plasmids and then infected with Malaysia/B at a low MOI for 24 h. Infected cells were stained against viral NP. Murine TMPRSS2 was used as control. No viral spread was visible in MDCK cells expressing tPA, uPA, LTF, NSP4, or CFB, whereas foci of infection due to proteolytic activation of progeny virus were observed in TMPRSS2-expressing cells.
Figure 5.

Malaysia/B HA is not activated by murine tPA, uPA, LTF, NSP4, CFB, tryptase ϵ, and HGFA. A, examination of HA cleavage by soluble proteases present in cell supernatants. HEK293 cells with transient expression of Malaysia/B HA were incubated with cleared protease containing HEK293 cell supernatants as described under “Experimental procedures” (left) or recombinant rKLK8 (right). Treatment of HA-expressing cells with buffer (w/o) or trypsin was used as control. Cell lysates were analyzed for HA cleavage by immunoblotting. β-Actin was used as loading control. B, MDCK cells with transient protease expression were infected with Malaysia/B at a low MOI of 0.01 and incubated for 24 h to allow multicycle viral replication. Cells transfected with empty vector (ev) or murine TMPRSS2-expressing plasmid were used as control. Virus spread was visualized by immunostaining of infected cells against NP. C, expression analysis of tryptase ϵ_DDDDK mutant in HEK293 cells with or without enterokinase treatment. Supernatants of cells transfected with empty vector (ev) or tryptase ϵ_DDDDK-encoding plasmid were concentrated (5×) at 48 h post-transfection and analyzed by SDS-PAGE and Western blotting using tryptase ϵ–specific antibodies. Zymogen and mature form are indicated by filled and open arrowheads, respectively. D, examination of HA cleavage by tryptase ϵ. HEK293 cells expressing Malaysia/B HA were incubated with tryptase ϵ_DDDDK mutant–containing cell supernatants treated with or without enterokinase (10 IU). Treatment of HA-expressing cells with trypsin was used as control. Cell lysates were analyzed for HA cleavage. E, expression analysis of HGFA in HEK293 supernatants with and without matriptase treatment. At 48 h post-transfection with empty vector (ev) or HGFA-encoding plasmid cell supernatants were concentrated (5×), treated with or without matriptase (5.0 μg/ml) for 1 h at 37 °C, and analyzed by immunoblotting using a FLAG-specific antibody. Zymogen and mature form are indicated by filled and open arrowheads, respectively. F, examination of HA cleavage by HGFA. HEK293 cells co-transfected with plasmids encoding Malaysia/B HA and either empty vector (w/o) or HGFA-encoding plasmid were incubated with exogenous matriptase or trypsin (0.5 μg/ml each) or remained untreated for 24 h. Cell lysates were analyzed for HA cleavage by Western blotting.
In sum, the data show that tPA, uPA, and rKLK8 do not cleave HA of Malaysia/B. For LTF, CFB, and NSP4 it remains to be confirmed whether they were expressed as enzymatically active proteases and whether the observed lack of HA cleavage is due to being unable to cleave HA rather than to a lack of enzymatic activity under the experimental settings used here.
Further, we examined whether tryptase ϵ is able to cleave IBV HA. Tryptase ϵ was released into the supernatant of HEK293 cells upon transient expression, but did not show any enzymatic activity against various fluorogenic peptides tested (data not shown). Therefore, we generated a mutant tryptase ϵ variant (tryptase ϵ_DDDDK) with Arg-49 substituted by Asp-Asp-Asp-Asp-Lys (DDDDK), the cleavage motif for enterokinase, comparable with what was previously described (30). Treatment of tryptase ϵ_DDDDK containing HEK293 supernatants with enterokinase led to partial conversion of the full-length zymogen (∼40 kDa) to its mature form (∼33 kDa) (Fig. 5C). This was further confirmed by measuring increased activity in enterokinase-treated tryptase ϵ_DDDDK supernatants compared with untreated supernatants or supernatants of mock-transfected cells (Fig. S1C). Incubation of Malaysia/B HA-expressing HEK293 cells with tryptase ϵ_DDDDK–containing supernatants did not support cleavage of HA independent of enterokinase treatment, indicating that tryptase ϵ was not able to cleave IBV HA (Fig. 5D).
HGFA was also released in the supernatant of HGFA-expressing HEK293 cells (Fig. 5E), but without measurable proteolytic activity (data not shown). Treatment of HGFA-containing cell supernatants with recombinant soluble matriptase caused partial conversion of the full-length HGFA zymogen (∼71 kDa) into the mature form (∼34 kDa) (Fig. 5E). Unfortunately, specific activity of matriptase-activated HGFA could not be measured directly due to strong background activity of matriptase cleaving various of the tested peptide substrates (data not shown). To analyze cleavage of Malaysia/B HA by HGFA, the protease was co-expressed with HA in HEK293 cells in the absence or presence of exogenous matriptase. Only uncleaved HA0 was detected by co-expression of HGFA both in the presence and absence of matriptase by Western blot analysis (Fig. 5F), indicating that HGFA does not cleave Malaysia/B HA. In contrast, HA cleavage was observed in control cells treated with trypsin.
In summary, our data show that the murine protease candidates CFB, HGFA, LTF, tPA, uPA, tryptase ϵ, and NSP4 as well as rat KLK8 do not support proteolytic activation of IBV HA under the experimental settings applied in this study.
IAV H3 is activated by murine TMPRSS13, hepsin, and prostasin
Infection of mice deficient for TMPRSS2 and TMPRSS4 revealed that both proteases contribute to H3 cleavage, but that at least one additional H3-cleaving protease is present in murine airways (17). Therefore, in addition to analysis of Malaysia/B HA cleavage by the protease candidates, we conducted similar in vitro experiments with H3 of A/HongKong/1/68 (H3N2). H3 was co-expressed with murine TMPRSS4, TMPRSS13, prostasin, and hepsin in HEK293 cells, and lysates were analyzed for HA expression and cleavage by SDS-PAGE and immunoblotting with H3-specific antibodies at 48 h post-transfection. As expected, only uncleaved H3 was detected in HEK293 in the absence of expression of an appropriate protease, whereas cleavage of H3 was observed by co-expression of murine TMPRSS2, TMPRSS4, prostasin, and, to a lesser extent, by hepsin (Fig. 6A). Upon co-expression of H3 and TMPRSS13, a HA cleavage product of lower molecular weight was detected in addition to HA1, indicating that TMPRSS13 cleaves H3 at different sites. To examine whether cleavage of H3 by these proteases supports proteolytic activation, MDCK cells were transfected with protease-encoding plasmids and then infected with Aichi/H3N2/PR8 at a low MOI for 24 h. Cells were fixed and immunostained against NP. Efficient spread of infection was visible in cells expressing murine TMPRSS13, hepsin, and prostasin similar to virus foci in TMPRSS2- or TMPRSS4-expressing cells that served as positive control (Fig. 6B).
Figure 6.
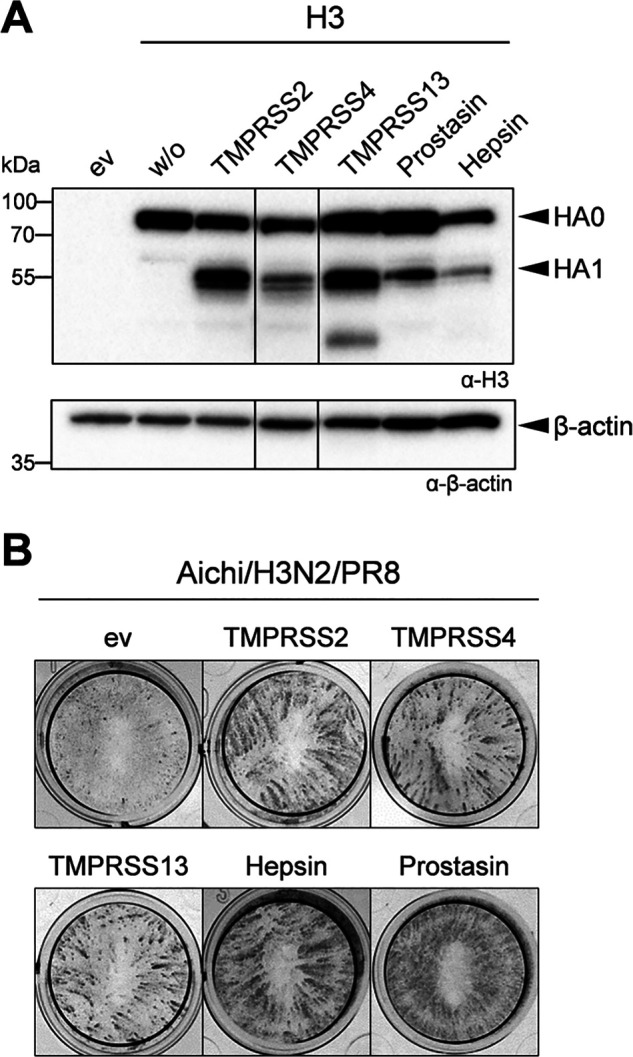
IAV H3 is activated by murine TMPRSS13, prostasin and hepsin. A, HEK293 cells were co-transfected with H3- and protease-encoding plasmids for 48 h. Transfection with empty vector (ev) was used as control. Cell lysates were subjected to SDS-PAGE and Western blot analysis using a H3-specific antibody. β-Actin served as loading control. B, MDCK cells were transfected with expression plasmids of murine proteases and then infected with Aichi/H3N2/PR8 at a MOI of 0.01 for 24 h. Foci of infection was analyzed by immunostaining against viral NP. Data shown are representative of three independent experiments.
The murine protease candidates CFB, HGFA, LTF, matriptase, NSP4, tPA, uPA, and tryptase ϵ and rat KLK8 were not able to cleave H3 or to promote multicycle viral replication of Aichi/H3N2/PR8 in MDCK cells (data not shown), similar to the results obtained with Malaysia/B. The results for cleavage activation of H3 and IBV HA by the different protease candidates are summarized in Table 1. Together, our data show that H3 can be activated by murine proteases TMPRSS13, hepsin, and prostasin in addition to TMPRSS2 and TMPRSS4.
Table 1.
Cleavage of IBV HA and H3 by protease candidates and confirmed protease activity
| Protease | Cleavage of B-HA | Cleavage H3 | Protease activity |
|---|---|---|---|
| CFB | − | − | − |
| HGFA | − | − | (+)a |
| Hepsin | + | + | +b,c |
| LTF | − | − | (+)a |
| Matriptase | − | − | (+)a |
| NSP4 | − | − | (+)a |
| Prostasin | + | + | +b,c |
| rKLK8 | − | − | +b |
| TMPRSS13 | + | + | +b,c |
| TMPRSS4 | + | + | +b,c |
| tPA | − | − | +a,b |
| Tryptase ϵ | − | − | +a,b |
| uPA | − | − | +a,b |
| hHepsin | + | − | +b |
| hProstasin | + | − | +b |
a Protease activity determined by detection of both zymogen and mature forms by immunoblotting.
b Protease activity determined by enzymatic activity measurements.
c Protease activity determined by HA cleavage upon co-expression.
Human prostasin and hepsin activate Malaysia/B HA but not H3
As described above, murine hepsin and prostasin were able to activate H3 and IBV HA. Interestingly, human prostasin and hepsin have been shown to be unable to activate H3 in previous studies by us and others (10, 11, 31). Here, we confirmed these observations. Co-expression of H3 with human prostasin did not result in cleavage of HA0 (Fig. 7A). A minor band for HA1 was detected upon co-expression of H3 and human hepsin as described before (11). Moreover, no spread of Aichi/H3N2/PR8 was observed in MDCK cells transiently expressing human prostasin or hepsin (Fig. 7B). Expression of human TMPRSS2 was used as a positive control, and clear foci of infection were visible in TMPRSS2-expressing cells. Interestingly, both human prostasin and hepsin supported proteolytic activation of Malaysia/B in MDCK cells upon co-expression (Fig. 7B). Thus, prostasin and hepsin orthologues from human and mouse seem to differ in their substrate specificity regarding cleavage of H3 but not cleavage of IBV HA.
Figure 7.
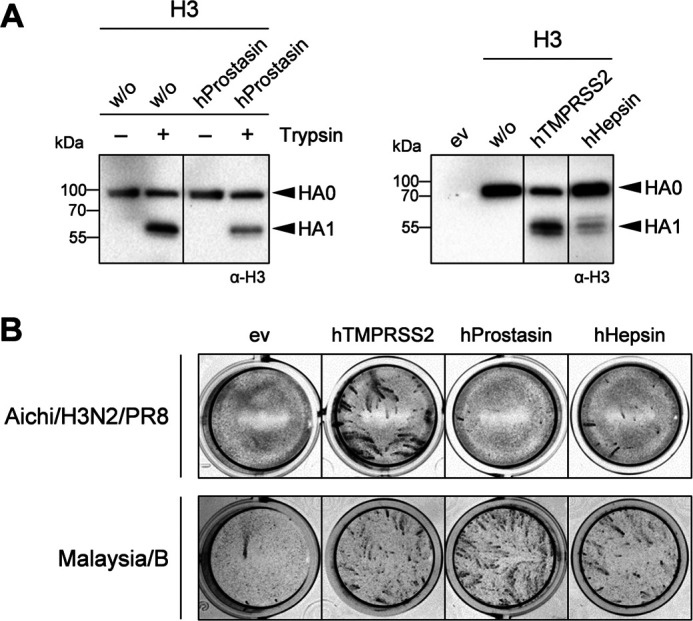
Human hepsin and prostasin do not activate H3 but rather IBV HA. A, HA cleavage analysis of H3 and Malaysia/B HA by human hepsin and prostasin. A, HEK293 cells were co-transfected with plasmids encoding H3 and either human prostasin or hepsin or TMPRSS2 (as control) for 24 h and incubated in the absence or presence of trypsin (0.5 μg/ml) for a further 24 h. Cell lysates were analyzed for HA cleavage by SDS-PAGE and immunoblotting using H3-specific antibody. B, examination of proteolytic activation of H3 and IBV HA by human prostasin and hepsin. MDCK cells were transfected with protease-encoding plasmids or empty vector (ev) for 24 h, followed by infection with Aichi/H3N2/PR8 or Malaysia/B at an MOI of 0.01 for 24 h. Virus spread was visualized by immunostaining of infected cells against NP. Data shown are representative of three independent experiments.
TMPRSS4 is involved in proteolytic activation of Malaysia/B in primary murine AECIIs
We next analyzed replication of Malaysia/B in primary AECIIs of Tmprss2−/−Tmprss4−/− double-knockout mice to examine the role of TMPRSS4 in IBV activation. Primary AECIIs of WT mice and Tmprss2−/− single-knockout mice were used as controls. In addition, replication of Anhui/H7/PR8, which is not able to replicate in AECIIs of Tmprss2−/− mice due to lack of HA cleavage (20), and Aichi/H3N2/PR8, which is proteolytically activated independent of TMPRSS2 and TMPRSS4 in mice (17), was examined (Fig. S2). Primary murine AECIIs of Tmprss2−/−, Tmprss2−/−Tmprss4−/−, or WT mice were inoculated with Malaysia/B, Anhui/H7/PR8, or Aichi/H3N2/PR8 at a MOI of 0.01 and incubated for 72 h. Virus replication was determined by plaque assay at different time points p.i., and cell lysates were analyzed for HA cleavage by SDS-PAGE and Western blotting at 72 h p.i. As described previously (20), replication of Anhui/H7/PR8 was strongly reduced in AECIIs of Tmprss2−/− mice, and a similar blockage of virus replication was observed in AECIIs of Tmprss2−/−Tmprss4−/− mice (Fig. S2A). In contrast, efficient replication of Anhui/H7/PR8 and cleavage of H7 was observed in AECIIs of WT mice. The amount of β-actin detected by Western blot analysis was strongly decreased in AECII lysates of WT mice, consistent with the strong cytopathic effect observed upon Anhui/H7/PR8 replication in the cells. Replication of Aichi/H3N2/PR8 was slightly reduced in AECIIs of Tmprss2−/− mice and significantly suppressed in AECIIs of Tmprss2−/−Tmprss4−/− double-knockout mice (Fig. S2A). Furthermore, also cleavage of HA of Aichi/H3N2/PR8 was reduced in AECIIs of Tmprss2-Tmprss4-deficient mice (Fig. S2B). Malaysia/B replicated to similar titers in AECIIs of both Tmprss2−/− and WT mice, but virus titers were reduced about 10-fold in AECIIs of Tmprss2−/−Tmprss4−/− double-knockout mice (Fig. 8A). As shown in Fig. 8B, Malaysia/B HA was still cleaved in AECIIs of Tmprss2-Tmprss4-deficient mice, although less efficiently compared with AECIIs of Tmprss2−/− and WT mice. The data suggest that TMPRSS4 contributes to proteolytic activation of Malaysia/B HA in mice in a scenario similar to H3N2. Proteolytic activation of Malaysia/B and Aichi/H3N2/PR8 in primary AECIIs of Tmprss2-Tmprss4-deficient mice, however, suggested that both H3 and IBV HA can be activated by at least three or more proteases in murine airways.
Figure 8.
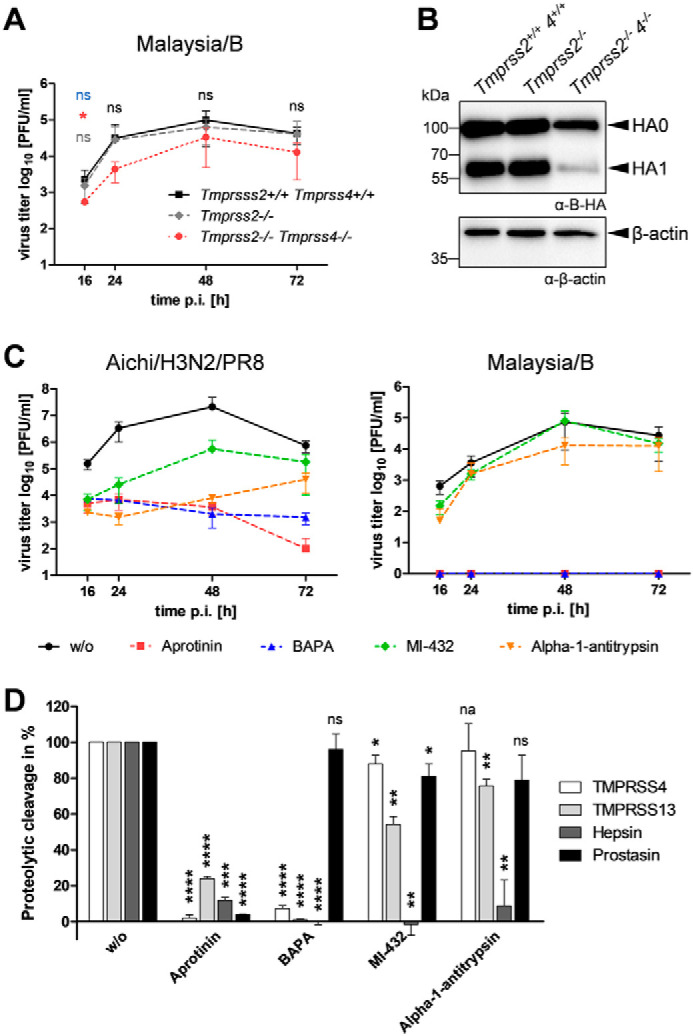
Virus replication in primary murine AECIIs of Tmprss2−/−Tmprss4−/− double-knockout mice in the presence of protease inhibitors. A, replication and proteolytic activation of Malaysia/B in AECIIs of Tmprss2−/−, Tmprss2−/−Tmprss4−/−, and WT mice. Primary AECIIs were infected with Malaysia/B at an MOI of 0.01 and incubated for 72 h. Virus titers were determined by a plaque assay at the indicated time points. Data are mean values ± S.D. (error bars) (n = 3). Log-transformed data were analyzed by one-way ANOVA followed by Tukey's multiple-comparison test according to the number of parameters and groups being compared. Obtained p values were corrected for multiple-hypothesis testing using Benjamini–Hochberg correction. Statistical significance is color-coded (gray, WT versus Tmprss2−/− mice; red, WT versus Tmprss2−/−Tmprss4−/− mice; blue, Tmprss2−/− versus Tmprss2−/−Tmprss4−/− mice). p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****) were considered significant; p > 0.05 (ns) was considered nonsignificant. No significance between all groups is indicated in black letters. B, at 72 h p.i., cell lysates were analyzed by SDS-PAGE and Western blotting using antibodies against HA. β-Actin served as loading control. C, virus growth kinetics of Aichi/H3N2/PR8 and Malaysia/B in AECIIs of Tmprss2-Tmprss4-deficient mice in the presence of protease inhibitors. AECIIs were infected with virus at a low MOI of 0.01 and incubated in the presence or absence of aprotinin, BAPA, or MI-432 (50 μm each) or α1-antitrypsin (0.5 mg/ml) for 72 h. Virus replication was analyzed by plaque titration at the indicated time points. Shown are the mean values ± S.D. of 2–3 independent experiments. D, inhibition of cell surface–expressed murine proteases by protease inhibitors. HEK293 cells were transfected with protease-expressing plasmids for 48 h. Cells were then treated with aprotinin, BAPA, or MI-432 (each 50 μm) or α1-antitrypsin (0.5 mg/ml) or remained untreated for 5 min and further supplemented with 50 μm fluorogenic peptide substrate Mes-dArg-Gly-Arg-AMC for 2 h to assay protease activity at the cell surface. Nonspecific substrate turnover of empty vector–transfected cells was subtracted, and remaining protease activity (as a percentage) in the presence of inhibitor was standardized to untreated controls. Data shown are representative of 2–3 independent experiments and were analyzed by one-sample t tests, and obtained p values were corrected for multiple testing using Benjamini–Hochberg correction. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****) were considered significant; ns, no significance; na, not applicable.
Suppression of virus replication in murine AECIIs by protease inhibitors suggests that H3 and IBV HA are activated by different but overlapping sets of proteases in mice
To investigate whether the same set of proteases supports proteolytic activation of IBV and H3N2 in AECIIs of Tmprss2-Tmprss4-deficient mice, we examined multicycle replication analysis of Aichi/H3N2/PR8 and Malaysia/B in these cells in the presence of different serine protease inhibitors. The broad-range serine protease inhibitor aprotinin from bovine lung has been shown to efficiently inhibit activation and multiplication of H3N2 IAV and IBV in embryonated chicken eggs and H3N2 in human airway epithelial cells and in lungs of infected mice (32, 33). Here, aprotinin treatment strongly inhibited replication of Aichi/H3N2/PR8 and blocked multiplication of Malaysia/B in AECIIs of Tmprss2−/−Tmprss4−/− double-knockout mice (Fig. 8C). The peptidomimetic inhibitor benzylsulfonyl-d-Arg-Pro-4-amidinobenzylamide (BAPA) inhibits proteolytic activation of IAV and IBV in Calu-3 human airway epithelial cells (34). BAPA treatment strongly suppressed Aichi/H3N2/PR8 replication by 3 log -fold reduction of viral titer and completely blocked multicycle replication of Malaysia/B in AECIIs of Tmprss2-Tmprss4-deficient mice. The sulfonylated 3-amindinophenylalanylamide–derived small-molecule inhibitor MI-432, which has been shown to inhibit TMPRSS2 in cell culture (35), reduced replication of Aichi/H3N2/PR8 about 100-fold but only marginally reduced Malaysia/B replication. Interestingly, the human lung protease inhibitor α1-antitrypsin strongly reduced titers of Aichi/H3N2/PR8 3–4 log-fold, whereas replication of Malaysia/B was suppressed only 10-fold by α1-antitrypsin treatment (Fig. 8C). Together, we observed differences in the sensitivity of H3– and IBV HA–activating proteases present in murine AECIIs for MI-432 and α1-antitrypsin, but not aprotinin and BAPA. The data suggest either that different but partially overlapping sets of proteases are involved in proteolytic activation of Malaysia/B and Aichi/H3N2/PR8 in AECIIs of Tmprss2−/−Tmprss4−/− mice or that Malaysia/B is activated by a broader range of proteases compared with Aichi/H3N2/PR8 in the cells.
Finally, we examined the sensitivity of murine TMPRSS13, hepsin, and prostasin, which were identified as potential HA-cleaving candidates in murine AECIIs above, to aprotinin, BAPA, MI-432, and α1-antitrypsin. We furthermore included TMPRSS4. For that purpose, HEK293 cells were transiently transfected with protease-encoding plasmids. At 48 h post-transfection, cells were preincubated with inhibitor and further supplemented with the fluorogenic peptide substrate Mes-d-Arg-Gly-Arg-AMC to assay protease activity at the cell surface. Aprotinin almost completely inhibited the enzymatic activity of TMPRSS4 and prostasin at the cell surface and significantly reduced the activity of hepsin and TMPRSS13, by about 85 and 70%, respectively (Fig. 8D). BAPA strongly inhibited the enzymatic activity of TMPRSS4, TMPRSS13, and hepsin but did not inhibit prostasin activity at the cell surface. MI-432 completely inhibited hepsin activity but showed only marginal inhibition of TMPRSS4 and prostasin activity. Activity of TMPRSS13 was reduced by about 45% by MI-432. α1-Antitrypsin treatment also strongly suppressed activity of hepsin but had only little if any effect on the activities of TMPRSS4, TMPRSS13, and prostasin. Hepsin was very sensitive to all inhibitors and was the only protease inhibited by α1-antitrypsin. Hence, hepsin might be involved in H3 activation in murine AECIIs.
In conclusion, comprehensive protease transcriptome profiling of the murine lower airways, HA cleavage analysis, and protease inhibitor studies in primary murine AECIIs identified TMPRSS4, TMPRSS13, hepsin, and prostasin as H3– and IBV HA–cleaving proteases in murine lung. In addition, our data indicate that activation of H3 and IBV HA is caused by different but overlapping sets of proteases in mice. Moreover, this study shows that human hepsin and prostasin did not cleave H3, but rather IBV HA, suggesting that hepsin and/or prostasin orthologues might contribute to the differences observed in TMPRSS2-independent activation of H3 in murine versus human airway cells.
Discussion
Influenza continues to be a global public health problem despite the availability of vaccines against seasonal influenza, highlighting the need for novel treatment approaches. Protection of TMPRSS2-deficient mice from H7N9, H1N1pdm, and H10 IAV pathogenesis demonstrated that prevention of HA cleavage provides a promising therapeutic approach for influenza treatment in humans and identified TMPRSS2 as drug target for the development of potent protease inhibitors (14–16, 18). Proteolytic activation of IBV and certain H3N2 strains by yet-undetermined proteases in TMPRSS2-deficient mice, however, revealed that influenza viruses can differ in their sensitivity to host proteases in vivo and that further proteases may be involved. More recent studies suggested that activation of H3 in mice might be due to a rather mouse-specific protease or a protease not present as active enzyme in human airway cells (20). In contrast, activation of IBV in primary human bronchial cells and Calu-3 cells was supported by proteases in addition to TMPRSS2 (20). It remained unknown which proteases activate IBV in human bronchial cells and Calu-3 cells and whether orthologous proteases facilitate IBV activation in humans and mice.
In this present study, we examined the repertoire of trypsin-like serine proteases in murine lower airways to identify further H3– and IBV HA–cleaving proteases in mice. By comprehensive transcriptome profiling, we detected 31 proteases with trypsin-like specificity in murine trachea, 28 in bronchi, and 25 in lung, indicating a slight decrease in the number of serine proteases with trypsin-like activity from murine trachea to lung. 21 of these 25 lung proteases were expressed in all three tissues. One protease, Klk14, was expressed only in the lung, although gene expression values were very low. In our study, expression values of many serine protease genes were very low compared with other genes, and only 20–30% of the murine serine protease genes were detected by the transcriptome analysis. However, RNA-Seq data showed high quality and reliable gene expression values, suggesting that the data are robust and reflect the tissue-specific expression of serine proteases. Serine protease expression in different tissues is not well-characterized. Among TTSPs, Hpn/hepsin, St14/matriptase, Tmprss2, and Tmprss4 showed the most robust expression levels in all three tissues. Expression value for Tmprss11a, Tmprss11d (also designated as HAT in humans), and Tmprss11g was very low or not detected at all. This is in agreement with previous studies on distribution of TTSP members in human and murine tissues, which showed that most HAT/DESC subfamily members are expressed in upper airways, trachea, and bronchi, but not in lung (15, 24, 36). Thus, the slight decrease in expression of trypsin-like serine proteases from trachea to lung observed here is mainly due to a decrease in the number of TTSP genes expressed. It may be interesting to determine the repertoire of serine proteases with trypsin-like specificity in murine upper airways, such as the nasal and the pharyngeal epithelium providing the initial site of influenza virus infection.
Comparative transcriptome analysis of serine proteases with trypsin-like activity expressed in primary murine AECIIs and the murine lung cell line MLE-15 identified 13 protease candidates, which were then tested for HA activation in vitro and thereby narrowed down to the four candidates hepsin, prostasin, TMPRSS4, and TMPRSS13. Knockout of TMPRSS4 expression in addition to TMPRSS2 in mice reduced cleavage of IBV HA and suppressed virus titers 10-fold in primary AECIIs, demonstrating that TMPRSS4 contributes to IBV activation in mice. Nevertheless, the data show that at least one more IBV HA–cleaving protease is present in murine AECIIs similar to what has been observed for H3 in mice (17). Our data suggest that hepsin, prostasin, and/or TMPRSS13 are involved.
The remaining nine protease candidates were shown to be incapable of cleaving IBV HA and H3 upon co-expression. Expression of uPA, tPA, and tryptase ϵ_DDDDK as enzymatically active proteases in cell supernatants and enzymatic activity of recombinant rat KLK8 were validated by protease activity assays using fluorogenic peptides. In contrast, only poor if any enzymatic activity was measured for transient expressed HGFA, LTF, matriptase, and NSP4 released in the cell supernatant or present on the surface of HEK293 cells. However, by Western blot analysis, we detected both zymogen and cleaved forms of HGFA, LTF, matriptase, and NSP4 in cell lysates and/or supernatants, suggesting that these candidates were expressed as mature proteases in HEK293 cells. Thus, low levels of fluorogenic peptide cleavage measured for HGFA, LTF, matriptase, and NSP4 might be due to the experimentally settings (buffer, pH) and/or missing co-factors in the protease activity assay rather than lack of enzymatic activity of the transient expressed proteases. In contrast, CFB was not able to cleave IBV HA and H3 upon co-expression, and we did not detect enzymatic activity or zymogen conversion in our study. Therefore, we cannot fully exclude the possibility that lack of IBV HA and H3 cleavage was due to lack of CFB activity, and it finally remains to be examined whether or not CFB can cleave HA with a monobasic cleavage site.
It should be noted that we cannot exclude the possibility that cell types other than AECIIs present in murine lung express HA-cleaving proteases different from the proteases identified here that may contribute to TMPRSS2-independent activation of H3 and/or IBV HA in mice. Immune cells or other cell types might release appropriate HA-cleaving proteases that have not been examined in our comparative transcriptional profiling. Comparison of protease genes detected in murine lung versus AECIIs here indicates that factor X (F10), granzyme A (Gzma), Klk1, Klk14, and Prss12/neurotrypsin are expressed in murine lung tissue but not in AECII. Co-expression of murine KLK1 with IBV HA and H3 in HEK239 and MDCK cells revealed that KLK1 is not able to cleave HA.4 Murine factor X, granzyme A, KLK14, and neurotrypsin have not been tested for HA cleavage so far. Previous studies using human proteases showed that factor Xa (activated form of factor X) does not cleave IAV HA with a monobasic cleavage site, including H3, and that KLK14 is not able to activate IBV HA in vitro (11, 37).
We found that multicycle replication of Aichi/H3N2/PR8 and Malaysia/B in murine AECIIs of Tmprss2−/−Tmprss4−/− mice varied in sensitivity to protease inhibitors. Multicycle replication of Aichi/H3N2/PR8 was strongly suppressed by the small-molecule nonpeptidic inhibitor MI-432 and the serpin α1-antitrypsin, whereas viral titer of Malaysia/B was only marginally reduced by MI-432 treatment and 10-fold reduced by α1-antitrypsin. We found that cell-surface activity of transient expressed hepsin was very sensitive to inhibition by MI-432 and α1-antitrypsin. In contrast, α1-antitrypsin had only a minor effect on enzymatic activity of TMPRSS4, TMPRSS13, and prostasin. MI-432 reduced the activity of TMPRSS13 to 50% but hardly inhibited prostasin and TMPRSS4. Thus, the data suggest that hepsin supports proteolytic activation and replication of Aichi/H3N2/PR8 in AECIIs of Tmprss2−/−Tmprss4−/− mice. The data further indicate that hepsin is either not involved in IBV activation in murine AECIIs or that other proteases are also present in the cells, which facilitate cleavage of IBV HA, but not H3, and are less sensitive to MI-432 and α1-antitrypsin. The substrate-analogue peptidic inhibitor BAPA efficiently inhibited multicycle replication of both Aichi/H3N2/PR8 and Malaysia/B in AECIIs of Tmprss2−/−Tmprss4−/− mice. BAPA strongly inhibited the activity of TMPRSS4, hepsin, and TMPRSS13 but only slightly inhibited prostasin. However, the data do not exclude prostasin as H3– or IBV HA–cleaving protease in mice. In contrast to TMPRSS2, TMPRSS4, TMPRSS13, and hepsin, which all undergo autocatalytic activation, prostasin requires activation by matriptase. Thus, inhibition of Aichi/H3N2/PR8 and Malaysia/B multicycle replication by BAPA in murine AECIIs could be due to prevention of prostasin zymogen activation by a BAPA-sensitive protease. Together, our data indicate that cleavage of H3 and IBV HA in murine lung is facilitated by different but partially overlapping sets of proteases and that IBV HA is activated by a broader number of proteases. Moreover, the data suggest that H3 cleavage in murine airways is due to hepsin in addition to TMPRSS2 and TMPRSS4, whereas prostasin and/or TMPRSS13 may play a minor role. In contrast, activation of IBV HA might be supported by all five proteases.
Interestingly, we observed that human hepsin and prostasin did not cleave H3 but rather IBV HA upon co-expression, revealing that orthologous proteases in humans and mice can differ in their capability to cleave HA. Similarly, human KLK5 was recently shown to cleave IAV H3, whereas murine KLK5 was not able to support H3 cleavage (38). Notably, activation of H3 by murine but not human hepsin and/or prostasin might explain why activation of H3 is independent of TMPRSS2 in murine airways but requires TMPRSS2 activity in human airway cells (14–16, 20). Moreover, the data suggest that hepsin and/or prostasin may support IBV activation independent of TMPRSS2 in primary human bronchial cells and human Calu-3 airway cells.
Currently available measures for the treatment of influenza infections in humans are inhibitors of the viral NA (oseltamivir, zanamivir, peramivir). Recently, two inhibitors of the viral polymerase complex, the cap-dependent polymerase acid endonuclease inhibitor baloxavir marboxil and the viral polymerase inhibitor favipiravir/T-705, have been approved for influenza treatment in Japan, and baloxavir marboxil has also been approved in the United States. The first class of influenza antivirals, the ion channel protein M2 blockers amantadine and rimantadine, are no longer recommended, due to the widespread drug resistance in circulating IAVs. Development of drug resistance against antivirals is a major concern in influenza control. Targeting host cell factors instead of viral proteins has the advantage of reducing or even preventing the emergence of drug-resistant viruses. The development of drug resistance against protease inhibitors due to mutations at the HA cleavage site is very unlikely. The amino acid sequence at the HA cleavage site differs among IAV HA subtypes but is highly conserved for each IAV subtype and among IBV strains. In agreement with that, by using an HA cleavage site mutant IAV strictly depending on elastase as a live vaccine approach, Stech et al. (39) did not obtain revertants by sequential passaging of the virus in mouse lungs. Furthermore, efficient inhibition of HA cleavage of progeny virus may restrict virus replication to a single cycle and further reduce the emergence of drug resistance.
Major concerns in targeting host cell factors are side effects or toxicity due to inhibition of their physiological function(s). The physiological role of TMPRSS2 and TMPRSS4, respectively, is still not known. Tmprss2−/− or Tmprss4−/− mice exhibit no adverse phenotype, indicating functional redundancy or compensation of the function by other cellular proteases (40, 41). Thus, short-time inhibition of TMPRSS2 or TMPRSS4 activity during an acute influenza infection seems feasible. Prostasin (also designated as channel-activating protease 1 (CAP1)) is a glycosylphosphatidylinositol-anchored protease expressed in a variety of epithelial tissues with high expression levels in prostate, kidney, bronchus, and lung. Prostasin activates the epithelial sodium channel (ENaC) by cleavage of the γ subunit and plays important roles in epithelial development and tissue homeostasis (36, 42–44). Paradoxically, prostasin has been shown to require neither zymogen activation nor enzymatic activity to execute its important functions in epidermal development, including activation of the ENaC (45–47). Other functions of prostasin, however, such as matriptase zymogen activation in the placenta, were found to require its enzymatic activity (47). TMPRSS13 (also known as MSPL (mosaic serine protease large-form)) is present in various human tissues (48). Its physiological role is not well-understood. TMPRSS13-deficient mice display impaired epidermal barrier formation, however, by a mechanism independent of profilaggrin processing and tight junction formation (49). Hepsin (also known as TMPRSS1) is abundant in the liver and expressed at low levels in other tissues, including the lung (reviewed in Refs. 24 and 38). Guipponi et al. (50) found that hepsin-deficient mice exhibit profound hearing loss and inner ear effects. The underlying mechanism is still not clear. Moreover, hepsin has been shown to be expressed in many tumor types and to promote tumor growth in animal models (51). The physiological roles of these membrane-bound serine proteases in the airways still needs to be investigated. Moreover, it remains to be determined whether short-term inhibition of these enzymes during an acute influenza infection would be feasible with respect to side effects. Notably, it has long been known that inhibition of HA cleavage by the broad-range serine protease inhibitor aprotinin from bovine lung efficiently suppresses influenza virus replication and spread in cell cultures and mice and reduces symptoms of disease. Aerosolized aprotinin markedly reduced the duration of symptoms without causing side effects in influenza patients (reviewed in Ref. 35). Here, aprotinin strongly inhibited cell-surface activity of transient expressed TMPRSS4, prostasin, and hepsin and reduced activity of TMPRSS13 to ∼30%. TMPRSS2 has been shown to be sensitive to inhibition by aprotinin too (52). Ideally, protease inhibitors should be administered through inhalation to specifically target proteases in the airways and should be preferably used in combination with antiviral drugs. This may allow the use of lower concentrations of protease inhibitors and thereby the maintenance of low levels of cleavage of physiological substrates while strongly preventing HA activation. Combinations of the NA inhibitor oseltamivir and peptidomimetic inhibitors of TMPRSS2 or furin synergistically and efficiently blocked replication of H1N1pdm and highly pathogenic avian H5N1 or H7N1 IAV, respectively, in cell culture at remarkably lower concentrations for each compound than single-drug treatment (34, 53).
The major role of TMPRSS2 in activation of H1N1pdm, H3N2, and H7N9 IAV in primary human airway cells and its obviously redundant physiological function represent a rare opportunity for the development of host factor–directed influenza drugs. Thus, a highly selective TMPRSS2 inhibitor may efficiently prevent IAV activation without causing considerable side effects. However, more recent studies, including data presented here, indicate that for treatment of IBV in humans, a broad-range serine protease inhibitor represents the most promising strategy. It should be noted that IBV activation in primary human AECIIs required TMPRSS2 activity, and it still remains to be investigated to what extent TMPRSS2 is involved in IBV activation in human airways. Thus, a TMPRSS2 inhibitor may also cause significant reduction in IBV activation in humans. Future studies on the development of protease inhibitors for influenza treatment should therefore consider both approaches. In addition, the differences in proteolytic activation of H3 and IBV by human and murine proteases exemplify limitations in using the mouse model to examine the efficacy of protease inhibitors in preventing influenza virus activation and spread. In this respect, it may be interesting to determine IAV- and IBV-activating proteases in the ferret model.
In conclusion, our study identifies H3– and IBV HA–cleaving proteases in addition to TMPRSS2 in mice that may also be involved in IBV activation in human airways and therefore are potential targets for host factor–directed drug development for influenza treatment.
Experimental procedures
Animals
Mice were bred and housed in the animal facility of the Philipps-University Marburg under specific pathogen-free conditions. All protocols involving mice have been approved by the Commission on Animal Protection and Experimentation at the Philipps-University Marburg. Homozygous deficient mice for TMPRSS2 (Tmprss2−/−,Tmprss2tm1Tsyk strain, mixed C57BL/6J-129 background) were kindly provided by Peter Nelson (Fred Hutchinson Cancer Research Center, Seattle) (40). Homozygous double-knockout mice for TMPRSS2 and TMPRSS4 (Tmprss2−/−Tmprss4−/−; Tmprss2tm1Tsyk Tmprss4tm1.1Hum strain, congenic B6.129S1 background) have been described previously (17). Tmprss4tm1.1Hum were kindly provided by Edith Hummler (Department of Pharmacology and Toxicology, University of Lausanne). WT control mice (C57BL/6J background) of approximately the same age were commercially obtained from Charles River Laboratories (Wilmington, MA) and housed short-term in the animal facility of the Philipps-University Marburg.
Cells and viruses
All cell growth and incubations were carried out at 37 °C and 5% CO2. HEK293 cells, Madin–Darby canine kidney cell lines MDCK(H) and MDCK(II), and MLE-15 mouse lung epithelial cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 5% glutamine, and 5% penicillin/streptomycin (DMEM/FCS). Infection experiments of cells were performed in DMEM supplemented with 0.1% BSA (Sigma–Aldrich), 5% glutamine, and 5% penicillin/streptomycin (DMEM/BSA). Preparation of primary AECIIs was conducted according to the protocol described previously (20). AECII identity was confirmed by immunofluorescence staining against prosurfactant protein C as an airway marker for type II pneumocytes. Transient expression of specific recombinant proteins was performed in DMEM/BSA or FreeStyle 293 Expression Medium (Gibco) as described below.
Influenza viruses used here were recombinant A/Anhui/H7/PR8, consisting of HA of A/Anhui/1/13 (H7N9) and seven genes of A/PR8/34 (H1N1) and recombinant Aichi/H3N2/PR8 (HA and NA of A/Aichi/2/68 (H3N2) and six genes of PR8) described previously (20); A/quail/Shantou/782/00 (H9N2) (designated as H9N2); and B/Malaysia/2506/2004 (Malaysia/B). IAVs were propagated in MDCK(II) cells in DMEM/BSA containing 1 μg/ml tosyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma). IBVs were grown in the allantoic cavity of 11-day-old embryonated chicken eggs. Cell supernatants and allantoic fluid were cleared from cell debris by low-speed centrifugation and stored at −80 °C.
Plasmids
Expression plasmids encoding viral HAs used here were pCAGGS-H3, encoding HA of A/HongKong/1/68 (H3N2) (6), and pHW2000-H9, encoding HA of A/quail/Shantou/782/00 (H9N2) (54). The cDNA of the HA gene of B/Malaysia/2506/2004 was cloned from viral RNA by RT-PCR using HA-specific primers and subsequently subcloned into pCAGGS expression plasmid using EcoRI and NotI restriction sites. pCMV6-Entry expression plasmids encoding murine proteases with a C-terminal Myc-DDK tag were obtained from OriGene Technologies: CFB (MR210521), HGFA (MR216475), hepsin (MR219750), LTF (MR210170), tPA (MR208868), uPA (MR225747), tryptase ϵ (MR204321), NSP4 (MR217041), prostasin (MR223227), matriptase (MR222240), TMPRSS4 (MR206946), and TMPRSS13 (MR220731). pCAGGS plasmids encoding human or murine TMPRSS2 with C-terminal FLAG epitope have been described previously (6, 15). pCAGGS encoding human prostasin was described previously (31). pcDNA6.2/C-emGFP-HPN encoding human hepsin (Human ORFeome Collaboration, Clone ID: 100004749) was generously provided by the Juha Klefström laboratory (University of Helsinki). The cDNA encoding tryptase ϵ_DDDDK mutant with substitution of Arg-49 by the enterokinase cleavage site motif DDDDK was synthesized by GeneArt Gene Synthesis (Thermo Fisher Scientific) based on the cDNA sequence of the murine Prss22 gene (OriGene Technologies, MR204321) and was cloned into the pCMV6-Entry plasmid using HindIII and NotI restriction sites.
Antibodies, proteases, and protease inhibitors
Antibodies used in this study were a polyclonal rabbit antibody against HA1 of A/TW/3446/02 (H3N2) (GeneTex, GTX127363), a polyclonal rabbit antibody against HA1 of B/Brisbane/60/2008 (Sino Biological, 40016-T38), a polyclonal rabbit antibody against HA of A/Anhui/1/13 (H7N9) (Sino Biological, 40103RP02), a polyclonal rabbit antibody against HA of H9N2 (Thermo Fisher Scientific, PA5–81658), a monoclonal mouse antibody against β-actin (Abcam, ab6276), a monoclonal mouse antibody against IBV NP (Thermo Fisher Scientific, B017 (B35G)), a monoclonal mouse antibody against IAV NP (Abcam, ab43821), a polyclonal rabbit antibody against FLAG tag (Sigma–Aldrich), and a polyclonal goat antibody against mouse tryptase ϵ (Thermo Fisher Scientific, PA5–47245). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Agilent DAKO.
TPCK-treated trypsin (Sigma–Aldrich) was stored as 1 mg/ml stock solutions in dH2O. Recombinant rat kallikrein-related peptidase 8 (rKLK8) was kindly provided by Viktor Magdolen (School of Medicine, Technical University of Munich).
Broad-range serine protease inhibitor aprotinin (Bachem) was dissolved in dH2O. Human α1-proteinase inhibitor (also referred to as α1-antitrypsin) purchased from Talecris, stored as 25 mg/ml stock solution at −80 °C, was kindly provided by Andreas Rembert Koczulla (Department of Pulmonary Rehabilitation, Philipps-University Marburg). Peptidomimetic inhibitors BAPA (34, 55) and 3-amidinophenylalanine–derived inhibitor MI-432 (28, 54) were dissolved in dH2O. Inhibitor stock solutions were stored at −20 °C.
RNA isolation from murine tissues and cells
For isolation of total RNA from murine airway tissues, 8–16-week-old WT mice (C57BL/6 background) were used. Animals were sacrificed by cervical dislocation. The respiratory tract of mice was prepared under the sterile bench, washed in sterile PBS, and sectioned into trachea, bronchi, and lungs. Tissues were separately transferred into fresh tubes, shock-frozen in liquid nitrogen, and stored at −80 °C until further use. 20–40 mg of lungs, ≤20 mg of bronchi, and ≤10 mg of trachea tissues were used for each RNA extraction. Homogenization of lung tissue was performed with a precooled mortar/pestle and liquid nitrogen. The corresponding lysate was further homogenized using a syringe and needle (0.8 mm). Homogenization of bronchi and trachea tissues were performed with Lysing Matrix H (MP Biomedicals) and a Retsch MM200 homogenizer at 30 Hz for 2.5 min, three times with a 1-min incubation on ice in between. Total RNA isolation from homogenized tissues as well as MLE-15 cells and primary murine AECIIs was performed using the RNeasy kit (Qiagen), applying the DNase digestion step according to the manufacturer's protocol. Isolated RNA was stored at −80 °C until further use.
RNA-Seq and bioinformatic analysis for gene expression profiling
The quality of total RNA samples was assessed with Experion RNA StdSens Chips (Bio-Rad), and library preparation was performed with the TruSeq Stranded mRNA Library Prep Kit (Illumina) according to the manufacturer's instructions. Sample sequencing was performed on an Illumina HiSeq 1500 using mRNA derived from three biological replicates, for each organ as well as cell population. The reads obtained by RNA-Seq were aligned with STAR (2.4.1.a) against Ensembl Genome Mus musculus release 89 (mm10). Prior to expression profiling, transcripts were prefiltered to those that yielded at least one FPKM value of ≥0.3 and a tag count ≥50 out of three replicates to exclude insufficiently covered genes from the analysis. Subsequently, differential expression was assessed using DEseq2 (version 1.16.1) (3). Protease and protease inhibitor genes were filtered based on gene ontology classification and according to the enzyme classification (EC) as hydrolases (EC 3.4). Serine protease genes were filtered with reference to EC 3.4.21 classified genes as well as UniProt (22) and MEROPS (23) database search to filter for proteases with trypsin-like activity. RNA-Seq data have been deposited in the EBI ArrayExpress archive (accession numbers E-MTAB-8571 and E-MTAB-8573).
Infection of murine cells and multicycle viral replication
MLE-15 cells were seeded in 12-well plates and grown to >90% of confluence. For virus growth kinetics, cells were gently washed with PBS and infected with Aichi/H3N2/PR8 and Malaysia/B at an MOI of 0.001 and 0.01, respectively, in DMEM/BSA for 1 h at 37 °C. The inoculum was removed, and cells were washed with PBS and incubated in fresh DMEM/BSA with or without TPCK-treated trypsin at final concentrations of 0.2 or 0.4 μg/ml. At 16, 24, 48, and 72 h p.i., viral titers in supernatant were determined by plaque assay on MDCK(II) cells with Avicel overlay as described previously (31). Cell lysates were subjected to SDS-PAGE and Western blot analysis as described below.
Primary AECIIs of Tmprss2−/−, Tmprss2−/−Tmprss4−/−, or WT mice were prepared as described previously (20) and seeded into membrane supports (12-mm Transwell culture inserts, 0.4-μm pore size, Costar), coated with 0.05 mg of collagen type I from calf skin (Sigma–Aldrich) per well, and cultivated with DMEM/FCS for 24 h at 37 °C and 5% CO2. Cells were washed and incubated under air-liquid interface conditions (medium in the basal but not apical chamber) for a further 24 h. Cells were then washed and inoculated apically with Malaysia/B, Aichi/H3N2/PR8, or Anhui/H7/PR8 at a MOI of 0.01 in DMEM/BSA for 1 h. The inoculum was removed, and cells were washed and incubated with fresh DMEM/BSA in the apical and basal chamber for 72 h. At 16, 24, 48, and 72 h p.i., virus replication was quantified by plaque assay as described above. Cell lysates were analyzed for HA cleavage by SDS-PAGE and Western blotting as described below.
To analyze virus replication in primary AECIIs in the presence of protease inhibitors, the cells were cultivated and seeded as described above and then infected with Malaysia/B or Aichi/H3N2/PR8 at an MOI of 0.01 in DMEM/BSA for 1 h. The inoculum was removed, and cells were washed with PBS and then incubated with fresh DMEM/BSA in both apical and basal chambers. The apical medium was supplemented with 50 μm inhibitor (aprotinin, BAPA, MI-432) or 0.5 mg/ml inhibitor (α1-antitrypsin). At 16, 24, 48, and 72 h p.i., virus replication was determined by a plaque assay.
Analysis of HA cleavage by co-expression with proteases or incubation of HA with protease-containing cell supernatants
Transfection of cells was performed using the transfection reagent Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's protocol.
For co-expression of HA with protease, HEK293 cells were seeded in 12-well plates, co-transfected with HA-encoding and protease-encoding plasmids (ratio 160:1), and incubated for 48 h in DMEM/BSA. To examine HA cleavage by cell supernatants of transient protease-expressing cells, HEK293 cells in 6-well plates were transfected with HA-encoding plasmid and incubated in DMEM/BSA for 24–48 h. Separately, HEK293 cells seeded in 24-well plates were transfected with protease expression plasmids and incubated in FreeStyle 293 Expression Medium for 48 h. Then cell supernatants were collected and cleared by centrifugation. HA-expressing cells were harvested, washed with buffer (100 mm NaCl, 50 mm Tris-HCl, 0.005% Tween 20, pH 8.0), and divided into fresh tubes. Cell pellets were resuspended in buffer (negative control, w/o), rKLK8 (diluted 1:4), 0.5 μg/ml TPCK-treated trypsin in buffer (positive control), or cleared cell supernatant followed by incubation at 37 °C for 2 h. Cells were then subjected to SDS-PAGE and Western blot analysis as described below.
Transient protease expression, zymogen activation, and concentration of cell supernatants
Transient expression of proteases and proteolytic activity measurements were performed using HEK293 cells. Cells were seeded in 96-/24-/12-well plates and transfected with protease-encoding plasmids using Lipofectamine 2000 as described above. Cells were incubated in DMEM/BSA (TMPRSS4, TMPRSS13, hepsin, HGFA, and prostasin) or FreeStyle 293 Expression Medium (CFB, LTF, NSP4, tPA, uPA, and tryptase ϵ) for 48 h. Cell lysates were prepared as described below and subjected to SDS-PAGE and Western blot analysis. Protease-containing cell supernatants were cleared from cell debris by centrifugation (4000 × g, 10 min, 4 °C) and concentrated (5–10 times) with centrifugal filter units (Sartorius Stedim). HGFA containing concentrated (5×) cell supernatant was treated with 5.0 μg/ml recombinant matriptase (54) for 1 h at 37 °C for zymogen activation. Tryptase ϵ_DDDDK mutant containing cell supernatant was treated with 10 IU bovine PRSS7 (GenScript) for 24 h for zymogen activation. Supernatants were then concentrated (5×) and subjected to SDS-PAGE and Western blot analysis or analyzed for enzymatic activity as described below.
SDS-PAGE and Western blot analysis
Cells were washed, lysed in CelLytic M Cell Lysis Reagent (Sigma-Aldrich) supplemented with protease inhibitor mixture (Sigma–Aldrich) for 30 min on ice, and cleared from cell debris by centrifugation (8000 × g, 5 min, 4 °C). Lysates were supplemented with reducing SDS-PAGE sample buffer, heated at 95 °C for 10 min, and centrifuged at 10,000 × g for 1 min. Cell supernatants were cleared from cell debris and concentrated by ultrafiltration as described above. Concentrated supernatants were supplemented with reducing SDS-PAGE sample buffer, heated at 95 °C for 10 min, and centrifuged at 10,000 × g for 1 min prior to SDS-PAGE. Electrophoresis was carried out with discontinuous Tris/glycine-based SDS-polyacrylamide gels (12%). Proteins were transferred to polyvinylidene fluoride membranes and detected by specific primary antibodies and HRP-conjugated secondary antibodies using SuperSignal West Femto or Dura chemiluminescent Western blotting substrates (Thermo Fisher Scientific). Chemiluminescence was detected with the ChemiDoc XRS+ system and Image Lab software (Bio-Rad).
Multicycle virus replication in transient protease-expressing cells
To analyze multicycle replication and virus spread in MDCK(H) cells, the cells were seeded in 24-well plates and transfected with protease-encoding plasmids as described above. At 24 h post-transfection, cells were infected with virus at a low MOI of 0.01 for 1 h. Inoculum was removed, and cells were washed and further incubated for 24 h in DMEM/BSA. At 24 h p.i., cells were immunostained against the viral NP as described previously (6). Briefly, cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.3% Triton X-100 in PBS. Cells were incubated with IAV NP– or IBV NP–specific antibodies and HRP-conjugated secondary antibodies and stained using the peroxidase substrate TrueBlue (KPL). Images were taken with the ChemiDoc XRS+ system and Image Lab software (Bio-Rad).
Protease activity at the cell surface and in cell supernatants
Enzymatic activity of the proteases expressed at the cell surface or released in the cell supernatant was assessed with the synthetic AMC-conjugated peptide Mes-dArg-Gly-Arg-AMC. To assay protease activity on the cell surface, HEK293 cells were seeded in 96-well plates, transfected with protease-encoding plasmid or empty vector as a control as described above, and incubated in DMEM/BSA for 48 h. Then cells were washed and incubated with 50 μl of fresh DMEM/BSA supplemented with 50 μm inhibitor (aprotinin, BAPA, MI-432) or 0.5 mg/ml α1-antitrypsin or equal volumes of dH2O for 5 min at room temperature. Subsequently, 50 μl of 100 μm fluorogenic peptide substrate in DMEM/BSA was added to each well, and cells were incubated for 2 h at room temperature. Hydrolysis of the peptide was monitored by the fluorescence intensity of released AMC using a PerkinElmer Life Sciences LS55 luminescence spectrometer with wavelengths set at 350 nm for excitation and 460 nm for emission. Fluorescence intensity of empty vector–transfected cells treated with inhibitors were subtracted as background for nonspecific substrate turnover. To assay protease activity in cell culture supernatants, HEK293 cells were seeded in 12-/24-well plates, transfected with protease-encoding plasmids or empty plasmid as control, and incubated in FreeStyle 293 Expression medium for 48 h. Subsequently, supernatants were cleared from cell debris and concentrated by ultrafiltration as described above. 40 μl of the supernatant were incubated with aprotinin, BAPA, or MI-432 (50 μm each) or 0.5 mg/ml α1-antitrypsin or equal volumes of dH2O for 5 min at room temperature and then mixed with 40 μl of 100 μm fluorogenic peptide substrate diluted in reaction buffer (100 mm NaCl, 50 mm Tris-HCl, 0.005% Tween 20, pH 8.0). Fluorescence intensity of AMC was measured over a period of 30 min using a kinetic mode. The steady-state rates were calculated from the linear slopes of the progress curves. Supernatants of cells transfected with empty vector were subtracted as background of nonspecific substrate turnover. The enzymatic activity of rKLK8 was measured with the fluorogenic substrate tert-butyloxycarbonyl-Val-Pro-Arg-AMC in buffer (100 mm NaCl, 50 mm Tris-HCl, 0.005% Tween 20, pH 8.0) over a period of 30 min using a kinetic mode.
Statistical analysis
Virus titers of growth kinetics were log-transformed prior to statistical evaluation as a variance-stabilizing transformation. Analysis of variance (ANOVA) assumptions were checked where applicable using the Levene test (test of heteroscedascity) and the Shapiro–Wilk test (test of normality). Statistical significance between the conditions was analyzed by one-way ANOVA for different time points followed by Tukey's multiple-comparison test according to the number of parameters and groups being compared. Obtained p values were corrected by Benjamini–Hochberg correction. p values below a threshold of 0.05 were considered significant, and different significance levels were indicated as follows: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, p > 0.05 (nonsignificant). Inhibition of protease activities was analyzed by one-sample t tests on relative proteolytic activity values normalized to 100% (w/o) proteolytic activity of untreated cells. Obtained p values were corrected for multiple testing using Benjamini–Hochberg correction. The assumption of normality was tested using the Shapiro–Wilk test (test of normality). The resulting p values are given as described above.
Data availability
The gene expression data of trachea, bronchi, and lungs of mice have been deposited in ArrayExpress with the accession code E-MTAB-8573. The gene expression data of primary murine AECIIs and the murine lung cells MLE-15 have been deposited in ArrayExpress with the accession code E-MTAB-8571. Data supporting the findings of this study are available within the article and its supporting materials or, if stated otherwise, available from the corresponding author (Eva Böttcher-Friebertshäuser, friebertshaeuser@staff.uni-marburg.de) upon reasonable request.
Author contributions
A. H. and E. B.-F. conceptualization; A. H., M. M., and E. B.-F. formal analysis; A. H., L. B., S. P., K. S., T. Steinmetzer, T. Stiewe, A. N., and E. B.-F. investigation; A. H. and E. B.-F. writing-original draft; M. M. data curation; S. P., K. S., T. Steinmetzer, and E. B.-F. funding acquisition; A. N. and E. B.-F. supervision; E. B.-F. project administration.
Supplementary Material
Acknowledgments
We thank Guido Schemken and Uta Eule for technical assistance and support. We thank Viktor Magdolen, Edith Hummler, Andreas Rembert Koczulla, Peter Nelson, and Juha Klefström for support in generating this work. We also thank Hans-Dieter Klenk for critical reading of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the collaborative research center SFB 1021 (to E. B.-F. and S. P.), by the Von-Behring Röntgen Stiftung (to E. B.-F.), by the LOEWE Center DRUID (to E. B.-F. and T. S.), by intramural grants from the Helmholtz-Association (Program Infection and Immunity) (to K. S.), and by the German Ministry for Education and Research (BMBF)-funded German Center for Infectious Diseases (DZIF) partner site Giessen (to S. P.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Tables S1–S3, Figs. S1 and S2, and supporting materials.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
A. Harbig, and E. Böttcher-Friebertshäuser, unpublished data.
- IAV
- influenza A virus
- IBV
- influenza B virus
- AMC
- 7-amino-4-methyl coumarin
- AECII
- alveolar epithelial cell type II
- BAPA
- benzylsulfonyl-d-Arg-Pro-4-amidinobenzylamide
- CFB
- complement factor B
- ENaC
- epithelial sodium channel
- FPKM
- fragments per kilobase of exon model per million mapped reads
- HA
- hemagglutinin
- HAT
- human airway tryptase
- HBEC
- human bronchial epithelial cell
- HGFA
- hepatocyte growth factor activator
- KLK
- kallikrein-related peptidase
- LTF
- lactotransferrin
- MDCK
- Madin–Darby canine kidney
- MOI
- multiplicity of infection
- NA
- neuraminidase
- PRSS
- serine protease
- TMPRSS
- transmembrane protease serine S1 member
- tPA
- tissue-type plasminogen activator
- TTSP
- type II transmembrane serine protease
- uPA
- urokinase-type plasminogen activator
- p.i.
- postinfection
- NP
- nucleoprotein
- NSP4
- neutrophil serine protease 4
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- TPCK
- tosyl phenylalanyl chloromethyl ketone
- HRP
- horseradish peroxidase
- dH2O
- distilled H2O
- Mes
- methylsulfonyl
- w/o
- without.
References
- 1. Yoon S.-W., Webby R. J., and Webster R. G. (2014) Evolution and Ecology of Influenza A Viruses, pp. 359–375, Springer, Cham, Switzerland: [DOI] [PubMed] [Google Scholar]
- 2. Koutsakos M., Nguyen T. H., Barclay W. S., and Kedzierska K. (2016) Knowns and unknowns of influenza B viruses. Future Microbiol. 11, 119–135 10.2217/fmb.15.120 [DOI] [PubMed] [Google Scholar]
- 3. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steinhauer D. A. (1999) Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258, 1–20 10.1006/viro.1999.9716 [DOI] [PubMed] [Google Scholar]
- 5. Böttcher-Friebertshäuser E., Garten W., Matrosovich M., and Klenk H. D. (2014) The hemagglutinin: a determinant of pathogenicity. Curr. Top. Microbiol. Immunol. 385, 3–34 10.1007/82_2014_384 [DOI] [PubMed] [Google Scholar]
- 6. Böttcher E., Matrosovich T., Beyerle M., Klenk H.-D., Garten W., and Matrosovich M. (2006) Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 80, 9896–9898 10.1128/JVI.01118-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaipan C., Kobasa D., Bertram S., Glowacka I., Steffen I., Tsegaye T. S., Takeda M., Bugge T. H., Kim S., Park Y., Marzi A., and Pöhlmann S. (2009) Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83, 3200–3211 10.1128/JVI.02205-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hamilton B. S., Gludish D. W. J., and Whittaker G. R. (2012) Cleavage activation of the human-adapted influenza virus subtypes by matriptase reveals both subtype and strain specificities. J. Virol. 86, 10579–10586 10.1128/JVI.00306-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beaulieu A., Gravel É., Cloutier A., Marois I., Colombo É., Désilets A., Verreault C., Leduc R., Marsault É., and Richter M. V. (2013) Matriptase proteolytically activates influenza virus and promotes multicycle replication in the human airway epithelium. J. Virol. 87, 4237–4251 10.1128/JVI.03005-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zmora P., Blazejewska P., Moldenhauer A.-S., Welsch K., Nehlmeier I., Wu Q., Schneider H., Pöhlmann S., and Bertram S. (2014) DESC1 and MSPL activate influenza A viruses and emerging coronaviruses for host cell entry. J. Virol. 88, 12087–12097 10.1128/JVI.01427-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laporte M., Stevaert A., Raeymaekers V., Boogaerts T., Nehlmeier I., Chiu W., Benkheil M., Vanaudenaerde B., Pöhlmann S., and Naesens L. (2019) Hemagglutinin cleavability, acid-stability and temperature dependence optimize influenza B virus for replication in human airways. J. Virol. 94, e01430–19 10.1128/JVI.01430-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hamilton B. S., and Whittaker G. R. (2013) Cleavage activation of human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J. Biol. Chem. 288, 17399–17407 10.1074/jbc.M112.440362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leu C.-H., Yang M.-L., Chung N.-H., Huang Y.-J., Su Y.-C., Chen Y.-C., Lin C.-C., Shieh G.-S., Chang M.-Y., Wang S.-W., Chang Y., Chao J., Chao L., Wu C.-L., and Shiau A.-L. (2015) Kallistatin ameliorates influenza virus pathogenesis by inhibition of kallikrein-related peptidase 1-mediated cleavage of viral hemagglutinin. Antimicrob. Agents Chemother. 59, 5619–5630 10.1128/AAC.00065-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hatesuer B., Bertram S., Mehnert N., Bahgat M. M., Nelson P. S., Pöhlman S., and Schughart K. (2013) Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 9, e1003774 10.1371/journal.ppat.1003774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tarnow C., Engels G., Arendt A., Schwalm F., Sediri H., Preuss A., Nelson P. S., Garten W., Klenk H.-D., Gabriel G., and Böttcher-Friebertshäuser E. (2014) TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 influenza A virus in mice. J. Virol. 88, 4744–4751 10.1128/JVI.03799-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakai K., Ami Y., Tahara M., Kubota T., Anraku M., Abe M., Nakajima N., Sekizuka T., Shirato K., Suzaki Y., Ainai A., Nakatsu Y., Kanou K., Nakamura K., Suzuki T., et al. (2014) The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J. Virol. 88, 5608–5616 10.1128/JVI.03677-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kühn N., Bergmann S., Kösterke N., Lambertz R. L. O., Keppner A., van den Brand J. M. A., Pöhlmann S., Weiss S., Hummler E., Hatesuer B., and Schughart K. (2016) The proteolytic activation of (H3N2) influenza A virus hemagglutinin is facilitated by different type II transmembrane serine proteases. J. Virol. 90, 4298–4307 10.1128/JVI.02693-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lambertz R. L. O., Gerhauser I., Nehlmeier I., Leist S. R., Kollmus H., Pöhlmann S., and Schughart K. (2019) Tmprss2 knock-out mice are resistant to H10 influenza A virus pathogenesis. J. Gen. Virol. 100, 1073–1078 10.1099/jgv.0.001274 [DOI] [PubMed] [Google Scholar]
- 19. Sakai K., Ami Y., Nakajima N., Nakajima K., Kitazawa M., Anraku M., Takayama I., Sangsriratanakul N., Komura M., Sato Y., Asanuma H., Takashita E., Komase K., Takehara K., Tashiro M., et al. (2016) TMPRSS2 independency for haemagglutinin cleavage in vivo differentiates influenza B virus from influenza A virus. Sci. Rep. 6, 29430 10.1038/srep29430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Limburg H., Harbig A., Bestle D., Stein D. A., Moulton H. M., Jaeger J., Janga H., Hardes K., Koepke J., Schulte L., Koczulla A. R., Schmeck B., Klenk H.-D., and Böttcher-Friebertshäuser E. (2019) TMPRSS2 is the major activating protease of influenza A virus in primary human airway cells and influenza B virus in human type II pneumocytes. J. Virol. 93, e00649–19 10.1128/JVI.00649-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guénet J. L. (2005) The mouse genome. Genome Res. 15, 1729–1740 10.1101/gr.3728305 [DOI] [PubMed] [Google Scholar]
- 22. UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rawlings N. D., Barrett A. J., Thomas P. D., Huang X., Bateman A., and Finn R. D. (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632 10.1093/nar/gkx1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Böttcher-Friebertshäuser E. (2018) Membrane-anchored serine proteases: host cell factors in proteolytic activation of viral glycoproteins. in Activation of Viruses by Host Proteases, pp. 153–203, Springer International Publishing, Cham, Switzerland [Google Scholar]
- 25. Wikenheiser K. A., Vorbroker D. K., Rice W. R., Clark J. C., Bachurski C. J., Oie H. K., and Whitsett J. A. (1993) Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 90, 11029–11033 10.1073/pnas.90.23.11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Massucci M. T., Giansanti F., Di Nino G., Turacchio M., Giardi M. F., Botti D., Ippoliti R., De Giulio B., Siciliano R. A., Donnarumma G., Valenti P., Bocedi A., Polticelli F., Ascenzi P., and Antonini G. (2004) Proteolytic activity of bovine lactoferrin. BioMetals 17, 249–255 10.1023/B:BIOM.0000027700.90780.45 [DOI] [PubMed] [Google Scholar]
- 27. Perera N. C., Schilling O., Kittel H., Back W., Kremmer E., and Jenne D. E. (2012) NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 6229–6234 10.1073/pnas.1200470109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hammami M., Rühmann E., Maurer E., Heine A., Gütschow M., Klebe G., and Steinmetzer T. (2012) New 3-amidinophenylalanine-derived inhibitors of matriptase. Medchemcomm 3, 807 10.1039/c2md20074k [DOI] [Google Scholar]
- 29. Thul P. J., Åkesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., Alm T., Asplund A., Björk L., Breckels L. M., Bäckström A., Danielsson F., Fagerberg L., Fall J., Gatto L., et al. (2017) A subcellular map of the human proteome. Science 356, eaal3321 10.1126/science.aal3321 [DOI] [PubMed] [Google Scholar]
- 30. Wong G. W., Yasuda S., Madhusudhan M. S., Li L., Yang Y., Krilis S. A., Šali A., and Stevens R. L. (2001) Human tryptase ϵ (PRSS22), a new member of the chromosome 16p13.3 family of human serine proteases expressed in airway epithelial cells. J. Biol. Chem. 276, 49169–49182 10.1074/jbc.M108677200 [DOI] [PubMed] [Google Scholar]
- 31. Böttcher-Friebertshäuser E., Freuer C., Sielaff F., Schmidt S., Eickmann M., Uhlendorff J., Steinmetzer T., Klenk H.-D., and Garten W. (2010) Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 84, 5605–5614 10.1128/JVI.00140-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ovcharenko A. V., and Zhirnov O. P. (1994) Aprotinin aerosol treatment of influenza and paramyxovirus bronchopneumonia of mice. Antiviral Res. 23, 107–118 10.1016/0166-3542(94)90038-8 [DOI] [PubMed] [Google Scholar]
- 33. Zhirnov O. P., Klenk H. D., and Wright P. F. (2011) Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res. 92, 27–36 10.1016/j.antiviral.2011.07.014 [DOI] [PubMed] [Google Scholar]
- 34. Böttcher-Friebertshäuser E., Lu Y., Meyer D., Sielaff F., Steinmetzer T., Klenk H. D., and Garten W. (2012) Hemagglutinin activating host cell proteases provide promising drug targets for the treatment of influenza A and B virus infections. Vaccine 30, 7374–7380 10.1016/j.vaccine.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 35. Meyer D., Sielaff F., Hammami M., Böttcher-Friebertshäuser E., Garten W., and Steinmetzer T. (2013) Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 452, 331–343 10.1042/BJ20130101 [DOI] [PubMed] [Google Scholar]
- 36. Szabo R., and Bugge T. H. (2008) Type II transmembrane serine proteases in development and disease. Int. J. Biochem. Cell Biol. 40, 1297–1316 10.1016/j.biocel.2007.11.013 [DOI] [PubMed] [Google Scholar]
- 37. Scheiblauer H., Reinacher M., Tashiro M., and Rott R. (1992) Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J. Infect. Dis. 166, 783–791 10.1093/infdis/166.4.783 [DOI] [PubMed] [Google Scholar]
- 38. Magnen M., Elsässer B. M., Zbodakova O., Kasparek P., Gueugnon F., Petit-Courty A., Sedlacek R., Goettig P., and Courty Y. (2018) Kallikrein-related peptidase 5 and seasonal influenza viruses, limitations of the experimental models for activating proteases. Biol. Chem. 399, 1053–1064 10.1515/hsz-2017-0340 [DOI] [PubMed] [Google Scholar]
- 39. Stech J., Garn H., Wegmann M., Wagner R., and Klenk H. D. (2005) A new approach to an influenza live vaccine: modification of the cleavage site of hemagglutinin. Nat. Med. 11, 683–689 10.1038/nm1256 [DOI] [PubMed] [Google Scholar]
- 40. Kim T. S., Heinlein C., Hackman R. C., and Nelson P. S. (2006) Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell Biol. 26, 965–975 10.1128/MCB.26.3.965-975.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keppner A., Andreasen D., Mérillat A. M., Bapst J., Ansermet C., Wang Q., Maillard M., Malsure S., Nobile A., and Hummler E. (2015) Epithelial sodium channel-mediated sodium transport is not dependent on the membrane-bound serine protease CAP2/Tmprss4. PLoS ONE 10, e0135224 10.1371/journal.pone.0135224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leyvraz C., Charles R.-P., Rubera I., Guitard M., Rotman S., Breiden B., Sandhoff K., and Hummler E. (2005) The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J. Cell Biol. 170, 487–496 10.1083/jcb.200501038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vuagniaux G., Vallet V., Jaeger N. F., Pfister C., Bens M., Farman N., Courtois-Coutry N., Vandewalle A., Rossier B. C., and Hummler E. (2000) Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J. Am. Soc. Nephrol. 11, 828–834 [DOI] [PubMed] [Google Scholar]
- 44. Netzel-Arnett S., Currie B. M., Szabo R., Lin C.-Y., Chen L.-M., Chai K. X., Antalis T. M., Bugge T. H., and List K. (2006) Evidence for a matriptase-prostasin proteolytic cascade regulating terminal epidermal differentiation. J. Biol. Chem. 281, 32941–32945 10.1074/jbc.C600208200 [DOI] [PubMed] [Google Scholar]
- 45. Andreasen D., Vuagniaux G., Fowler-Jaeger N., Hummler E., and Rossier B. C. (2006) Activation of epithelial sodium channels by mouse channel activating proteases (mCAP) expressed in Xenopus oocytes requires catalytic activity of mCAP3 and mCAP2 but not mCAP1. J. Am. Soc. Nephrol. 17, 968–976 10.1681/ASN.2005060637 [DOI] [PubMed] [Google Scholar]
- 46. Peters D. E., Szabo R., Friis S., Shylo N. A., Uzzun Sales K., Holmbeck K., and Bugge T. H. (2014) The membrane-anchored serine protease prostasin (CAP1/PRSS8) supports epidermal development and postnatal homeostasis independent of its enzymatic activity. J. Biol. Chem. 289, 14740–14749 10.1074/jbc.M113.541318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Szabo R., Lantsman T., Peters D. E., and Bugge T. H. (2016) Delineation of proteolytic and non-proteolytic functions of the membrane-anchored serine protease prostasin. Development 143, 2818–2828 10.1242/dev.137968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kido H., Okumura Y., Takahashi E., Pan H.-Y., Wang S., Chida J., Le T. Q., and Yano M. (2008) Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J. Mol. Genet. Med. 3, 167–175 [PMC free article] [PubMed] [Google Scholar]
- 49. Madsen D. H., Szabo R., Molinolo A. A., and Bugge T. H. (2014) TMPRSS13 deficiency impairs stratum corneum formation and epidermal barrier acquisition. Biochem. J. 461, 487–495 10.1042/BJ20140337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guipponi M., Tan J., Cannon P. Z. F., Donley L., Crewther P., Clarke M., Wu Q., Shepherd R. K., and Scott H. S. (2007) Mice deficient for the type II transmembrane serine protease, TMPRSS1/hepsin, exhibit profound hearing loss. Am. J. Pathol. 171, 608–616 10.2353/ajpath.2007.070068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tanabe L. M., and List K. (2017) The role of type II transmembrane serine protease-mediated signaling in cancer. FEBS J. 284, 1421–1436 10.1111/febs.13971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Böttcher E., Freuer C., Steinmetzer T., Klenk H. D., and Garten W. (2009) MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 27, 6324–6329 10.1016/j.vaccine.2009.03.029 [DOI] [PubMed] [Google Scholar]
- 53. Lu Y., Hardes K., Dahms S. O., Böttcher-Friebertshäuser E., Steinmetzer T., Than M. E., Klenk H.-D., and Garten W. (2015) Peptidomimetic furin inhibitor MI-701 in combination with oseltamivir and ribavirin efficiently blocks propagation of highly pathogenic avian influenza viruses and delays high level oseltamivir resistance in MDCK cells. Antiviral Res. 120, 89–100 10.1016/j.antiviral.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 54. Baron J., Tarnow C., Mayoli-Nüssle D., Schilling E., Meyer D., Hammami M., Schwalm F., Steinmetzer T., Guan Y., Garten W., Klenk H.-D., and Böttcher-Friebertshäuser E. (2013) Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A viruses. J. Virol. 87, 1811–1820 10.1128/JVI.02320-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sisay M. T., Steinmetzer T., Stirnberg M., Maurer E., Hammami M., Bajorath J., and Gütschow M. (2010) Identification of the First Low-Molecular-Weight Inhibitors of Matriptase-2. J. Med. Chem. 53, 5523–5535 10.1021/jm100183e [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene expression data of trachea, bronchi, and lungs of mice have been deposited in ArrayExpress with the accession code E-MTAB-8573. The gene expression data of primary murine AECIIs and the murine lung cells MLE-15 have been deposited in ArrayExpress with the accession code E-MTAB-8571. Data supporting the findings of this study are available within the article and its supporting materials or, if stated otherwise, available from the corresponding author (Eva Böttcher-Friebertshäuser, friebertshaeuser@staff.uni-marburg.de) upon reasonable request.