

Abstract
Despite years of research investigating osteoblast differentiation, the mechanisms by which transcription factors regulate osteoblast maturation, bone formation, and bone homeostasis is still unclear. It has been reported that runt-related transcription factor 1 (Runx1) is expressed in osteoblast progenitors, pre-osteoblasts, and mature osteoblasts; yet, surprisingly, the exact function of RUNX1 in osteoblast maturation and bone formation remains unknown. Here, we generated and characterized a pre-osteoblast and differentiating chondrocyte-specific Runx1 conditional knockout mouse model to study RUNX1's function in bone formation. Runx1 ablation in osteoblast precursors and differentiating chondrocytes via osterix-Cre (Osx-Cre) resulted in an osteoporotic phenotype and decreased bone density in the long bones and skulls of Runx1f/fOsx-Cre mice compared with Runx1f/f and Osx-Cre mice. RUNX1 deficiency reduced the expression of SRY-box transcription factor 9 (SOX9), Indian hedgehog signaling molecule (IHH), Patched (PTC), and cyclin D1 in the growth plate, and also reduced the expression of osteocalcin (OCN), OSX, activating transcription factor 4 (ATF4), and RUNX2 in osteoblasts. ChIP assays and promoter activity mapping revealed that RUNX1 directly associates with the Runx2 gene promoter and up-regulates Runx2 expression. Furthermore, the ChIP data also showed that RUNX1 associates with the Ocn promoter. In conclusion, RUNX1 up-regulates the expression of Runx2 and multiple bone-specific genes, and plays an indispensable role in bone formation and homeostasis in both trabecular and cortical bone. We propose that stimulating Runx1 activity may be useful in therapeutic approaches for managing some bone diseases such as osteoporosis.
Keywords: Runt-related transcription factor 1 (Runx1), Runx2, osterix, osteocalcin, osteoblast, chondrocyte, bone homeostasis, osteoporosis, gene regulation, cell differentiation, bone marrow, cartilage, cell biology, osteoclast
Bone marrow mesenchymal stem cells are multipotent progenitors that give rise to osteoblasts, chondrocytes, and adipocytes upon specific stimulation for cell differentiation. Notably, the majority of bone disease conditions associated with bone loss, such as osteoporosis, result from impaired osteoblast function and disrupted bone homeostasis. Despite years of research in osteoblast lineage and osteoblast differentiation, the mechanisms by which transcription factors regulate bone homeostasis is unclear, even though an imbalance of bone homeostasis is significant to a number of diseases (1–3). A complete understanding of the mechanisms by which transcription factors control bone formation to maintain bone homeostasis is critical to developing therapies for the often-debilitating disorders of skeletal insufficiency (e.g. osteoporosis, periodontal disease, and tumor metastasis to bone).
The RUNX (Runt-related transcription factor) family is composed of RUNX1, RUNX2, and RUNX3, which play important roles in cell lineages (4). RUNX1 regulates hematopoietic stem cell differentiation into mature blood cells (5), and plays a major role in developing pain-transmitting neurons (6). RUNX1 has been reported to be involved in cartilage formation and fracture healing (7–9). In addition, others have explored the role of RUNX1 in the commitment and differentiation of chondroprogenitor cells into the chondrogenic lineage (9, 10). Although Runx1 has been reported to be expressed in osteoblast progenitors, pre-osteoblasts, and mature osteoblasts, the function of RUNX1 in osteoblasts has not yet been investigated (11). RUNX2 determines commitment to the osteoblastic lineage (12–15), and interacts with many co-regulators and transcription factors in the transcriptional regulation of its target genes (16). However, previous reports have shown that overexpression of Runx2 in cells of the osteoblastic lineage leads to an osteopenia phenotype due to negative regulation of osteoblast maturation (17). Thus, there is an urgent need to characterize transcription factor(s) that positively regulate osteoblast maturation and bone formation for healthy bone homeostasis to develop novel and efficient therapeutic approaches to treat bone diseases, including osteoporosis. RUNX1, a regulator of both Runx2 and bone genes, could facilitate the design of safer and novel therapeutic approaches for osteoporosis. Runx1 expression precedes Runx2, indicating its importance in cell lineage determination (11). Notably, it was previously reported that the overlapping expression of Runx1 and Runx2 supports cooperative induction of skeletal development (18). Furthermore, the Runx1 expression profile indicates that RUNX1 may have positive regulatory functions in osteoblasts that support bone formation for bone hemostasis. However, the function of RUNX1 in bone formation to maintain bone homeostasis, and how RUNX2 is regulated by RUNX1 is still largely unclear.
To investigate the role of RUNX1 in postnatal-skeletal development, we utilized Osterix-Cre (Osx-Cre) to specifically delete Runx1 in osteoblast precursors and differentiating chondrocytes (19, 20). We found that Runx1 deletion in osteoblast precursors and differentiating chondrocytes leads to a severe osteoporotic phenotype with a 50% reduction in both total bone volume and cortical bone, and a 40% reduction in trabecular bone. In this study, we have revealed that RUNX1 plays an indispensable role in postnatal skeletal development and bone homeostasis via directly associating with the Runx2 and Ocn promoters and regulating Runx2 and Ocn expression directly. Our results have revealed the important functions of RUNX1 in bone formation and the mechanisms underlying how RUNX1 maintains bone homeostasis, indicating that targeting RUNX1 may result in novel therapeutic approaches for degenerative bone diseases such as osteoporosis.
Results
Runx1f/fOsx-Cre mice exhibit shortened limbs, hypoplastic skeletons, and osteoporotic phenotype due to impaired bone mineralization
To investigate the role of RUNX1 in osteoblast development during postnatal skeletogenesis, we generated Runx1f/fOsx-Cre mice, deleting the Runx1 gene specifically in the osteoblast lineage. Runx1f/fOsx-Cre mice survived into adulthood, but the homozygote mice displayed severe skeletal defects characterized by shorter stature (Fig. 1A). PCR was used to confirm the genotypes of the mice (Fig. 1B). Through immunohistochemical (IHC) staining, we detected Runx1 expression in the trabecular bone of 14-week–old mice, which showed Runx1 was efficiently deleted in osteoblasts (Fig. 1C). X-ray analysis was performed to assess the bone density of Runx1f/fOsx-Cre and control mice (Fig. 1D). Radiographic analysis of 13-week–old Runx1f/fOsx-Cre and control skulls (Fig. 1D, Fig. S1C) and femurs (Fig. 1D, Fig. S1A) revealed a significant decrease in ossification and bone density, as well as mandibular defects in Runx1f/fOsx-Cre mice (Fig. 1D). Notably, Runx1f/fOsx-Cre mice had a significantly lower bone density in both the long bones and the calvarial area (Fig. 1D, Fig. S1A, white arrows). Micro-computed tomography (μCT) analysis of 14-week–old male and female Runx1f/fOsx-Cre femurs revealed a significant decrease in bone volume/tissue volume, trabecular number, trabecular thickness, and cortical bone compared with control (Fig. 1E). Both the cortical bone and total bone volume were reduced by 50% in the Runx1f/fOsx-Cre mice, whereas the trabecular bone was reduced by 40% (Fig. 1, E and F). These results demonstrated that Runx1 ablation in osteoblasts and chondrocyte precursors results in an osteoporotic phenotype characterized by short stature and impaired bone mineralization. Alizarin red and Alcian blue staining revealed that, except for the vertebrae and sternum, the bones in the newborn Runx1f/fOsx-Cre mice were severely underdeveloped (Fig. S2). Previous studies have indicated that Osx-cre mice have delayed calvarial ossification but no difference in the limbs (21, 22), which is consistent with our results (Fig. S2, H–K). Notably, we found that the newborn Runx1f/fOsx-Cre mice exhibited severe skull bone loss in the cranium and cranial base compared with controls (Fig. S2, A and B). Moreover, compared with Runx1f/f and Osx-Cre controls, Runx1f/fOsx-Cre mice exhibited shorter and underdeveloped forelimbs (Fig. S2C), clavicles (Fig. S2D), and hind limbs (Fig. S2E). Overall, the skeletons of the Runx1f/fOsx-Cre mice were severely underdeveloped and displayed an osteoporotic phenotype.
Figure 1.

Runx1f/fOsx-Cre mice have decreased bone mineralization and skeletal deformities. A, photographic images of 14-week–old (n = 16) Runx1f/fOsx-Cre (f/f/Δ) mice and WT (f/f) mice. B, PCR was used to determine Runx1 alleles (f/f, f/+, +/+, or deletion) and the presence of Cre. C, IHC staining of 14-week–old Runx1f/fOsx-Cre (f/f/Δ), and WT (f/f) mice tibias using RUNX1 antibody. D, x-ray analysis of 14-week-old male and female femurs and skulls, n = 19. E, μCT scans of femurs from 14-week-old male and female mice, n = 3. F, quantification of E. Results are expressed as mean ± S.D.; *, p < 0.05.
Runx1f/fOsx-Cre newborn femurs have impaired endochondral and intramembranous bone ossification and decreased osteoblast numbers
In further analyzing the growth retardation observed in the Runx1f/fOsx-Cre mice, we performed hematoxylin and eosin (H&E), Von Kossa, alkaline phosphatase (ALP), and tartrate-resistant acid phosphatase (TRAP) staining on femurs from newborn mice. H&E stain showed that newborn Runx1f/fOsx-Cre mice had decreased ossification and endochondral bone formation, as well as reduced cell density and a 50% decrease in osteoprogenitor cells in the periosteum (Fig. 2, A and F, red arrows). These osteoprogenitor cells differentiate into the first osteoblasts and produce a bone collar, which becomes the future cortical bone. Thus the 2-fold decrease in cortical bone width seen in Runx1f/fOsx-Cre may be due to the 50% decrease of the cells in the periosteum. ALP stain showed that osteoblast formation was significantly reduced in the Runx1f/fOsx-Cre mice compared with Osx-cre and WT mice, whereas there were significantly fewer ALP-positive cells within the periosteum (Fig. 2, B and E); we found a 3-fold decrease in ALP-positive cells in Runx1f/fOsx-Cre mice, which led to the reduction in both trabecular and cortical bone. Notably, the width of the cortical bone was reduced by 50% in Runx1f/fOsx-Cre mice (Fig. 2B). Von Kossa staining revealed a 50% decrease in osteoblast mineralization as well as a significantly shorter medullary cavity in newborn Runx1f/fOsx-Cre mice compared with Osx-cre and WT mice (Fig. 2, C, E, and F). Finally, TRAP staining confirmed that there were no significant differences in osteoclast numbers between WT and Runx1f/fOsx-Cre mice in both newborn and at the 3-week–old mice (Fig. 2, D and E, Fig. S3, A and B), demonstrating that the reduced bone density in Runx1f/fOsx-Cre mice was not due to an increase in osteoclast proliferation. Collectively, these results demonstrate that bone formation is impaired in Runx1f/fOsx-Cre mice, whereas osteoclast formation is not affected.
Figure 2.
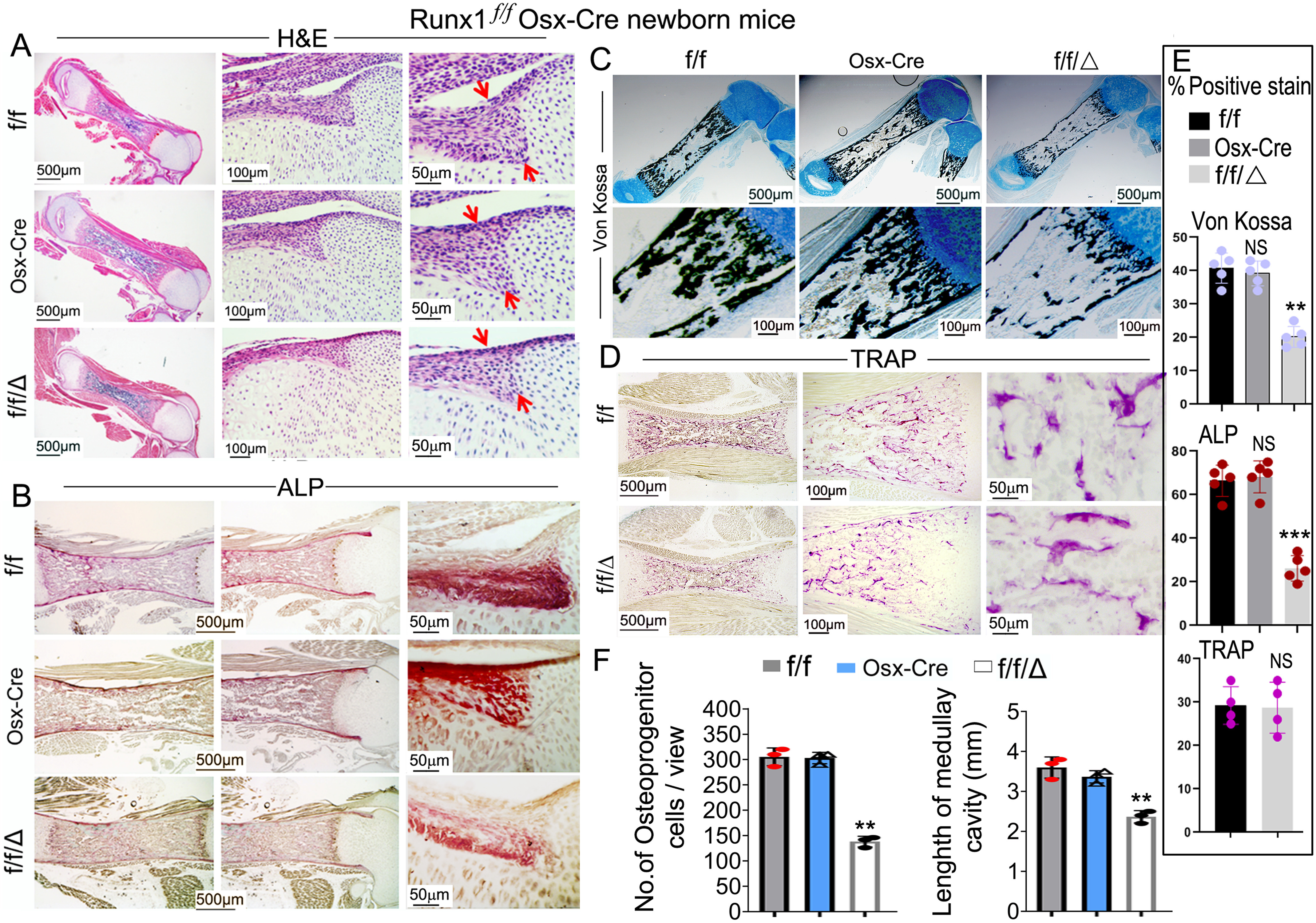
Endochondral bone ossification was significantly impaired and osteoblast numbers were largely decreased in Runx1f/fOsx-Cre newborn mice femurs, but not in Osx-Cre newborn mice femurs compared with WT mice femurs. A–D, newborn Runx1f/fOsx-Cre (f/f/Δ), Osx-Cre and WT (f/f) mice femurs were stained with (A) H&E stain, (B) ALP stain, (C) Von Kossa and Alcian blue stain, and newborn Runx1f/fOsx-Cre (f/f/Δ) and WT (f/f) mice femurs were stained with (D) TRAP stain. E, histomorphometry of WT (f/f), Osx-Cre and Runx1f/fOsx-Cre (f/f/Δ) mice (n = 4) showing ALP stain surface per bone surface (BS) area, Von Kossa stain surface per bone surface area, and TRAP stain surface per bone surface area. F, quantification of cells in the periosteum in A and length of the medullary cavity. Results are expressed as mean ± S.D. NS, not significant; **, p < 0.01; ***, p < 0.001.
RUNX1 deficiency affects the cortical and trabecular bone formation in the Runx1f/fOsx-Cre mice
Notably, we found that Runx2 expression in newborn Runx1f/fOsx-Cre mouse femurs decreased by 90% in the bone collar, and by 80% in the trabecular bone, whereas Runx2 expression in the hypertrophic zone was not significantly changed (Fig. 3, A and B). Notably, the width of the cortical bone was decreased by 50% in the newborn Runx1f/fOsx-Cre mouse femurs (Fig. 3B). To further study the effects of RUNX1 deficiency on bone homeostasis, we compared the mineral apposition rate in Runx1f/fOsx-Cre mice with their sex-matched littermate controls via double calcein labeling (Fig. S3E). The results demonstrated that the mineral apposition rate was decreased by 2-fold in the Runx1f/fOsx-Cre mice compared with the control, leading to the low bone density seen in the mutant mice. Furthermore, we found that the P1NP serum level (bone formation marker) was reduced by 2-fold in the Runx1f/fOsx-Cre mice, indicating that bone formation rate was affected by Runx1 CKO (Fig. 3C). Thus, RUNX1 plays an important role in up-regulating Runx2 expression in precursor cells and bone formation, and RUNX1 deficiency leads to significantly reduced cortical and trabecular bone.
Figure 3.

Runx2 expression in bone is significantly decreased in Runx1f/fOsx-Cre mice. A, IHC staining RUNX2 antibody of femur paraffin sections from P0 Runx1f/fOsx-Cre (f/f/Δ) and WT (f/f) mice. B, quantification of A. C, ELISA detection of P1NP (pg/ml) in 3-month–old mouse serum indicating bone formation is affected by gene CKO. Results are expressed as mean ± S.D., n = 3 in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RUNX1 deficiency affects the chondrocyte proliferation and maturation in the long bone of Runx1f/fOsx-Cre mice
Endochondral bone formation originates from cartilage templates, which are then replaced by bone (23). The delayed endochondral ossification observed in the Runx1f/fOsx-Cre mice prompted us to examine the impact of the Osx-Cre–mediated Runx1 deletion on the development of the growth plate. Safranin O staining of femurs revealed a significant decrease in relative length of the growth plate, proliferating zone, and hypertrophic zone over the total length of the bone in the mutant mice (Fig. 4, A and B). Furthermore, the proliferative zone in Runx1f/fOsx-Cre mice was shorter (Fig. 4A). We compared expression levels of Col2a1 and Col10a1 in the growth plate, which are specific collagens produced by proliferative and hypertrophic chondrocytes. We found that expression levels of both Col2a1 and Col10a1 were significantly decreased in the growth plate in Runx1f/fOsx-Cre mice (Fig. 4, C, D, and F). Upon staining to detect proliferating cell nuclear antigen (PCNA), we found that the expression was significantly decreased in both the proliferative and hypertrophic zones of Runx1f/fOsx-Cre mice (Fig. 4, E and F). These results indicate that the decreased length of the long bones in Runx1f/fOsx-Cre mice is due to reduced chondrocyte proliferation, which results in impaired growth plate development and trabecular bone formation.
Figure 4.
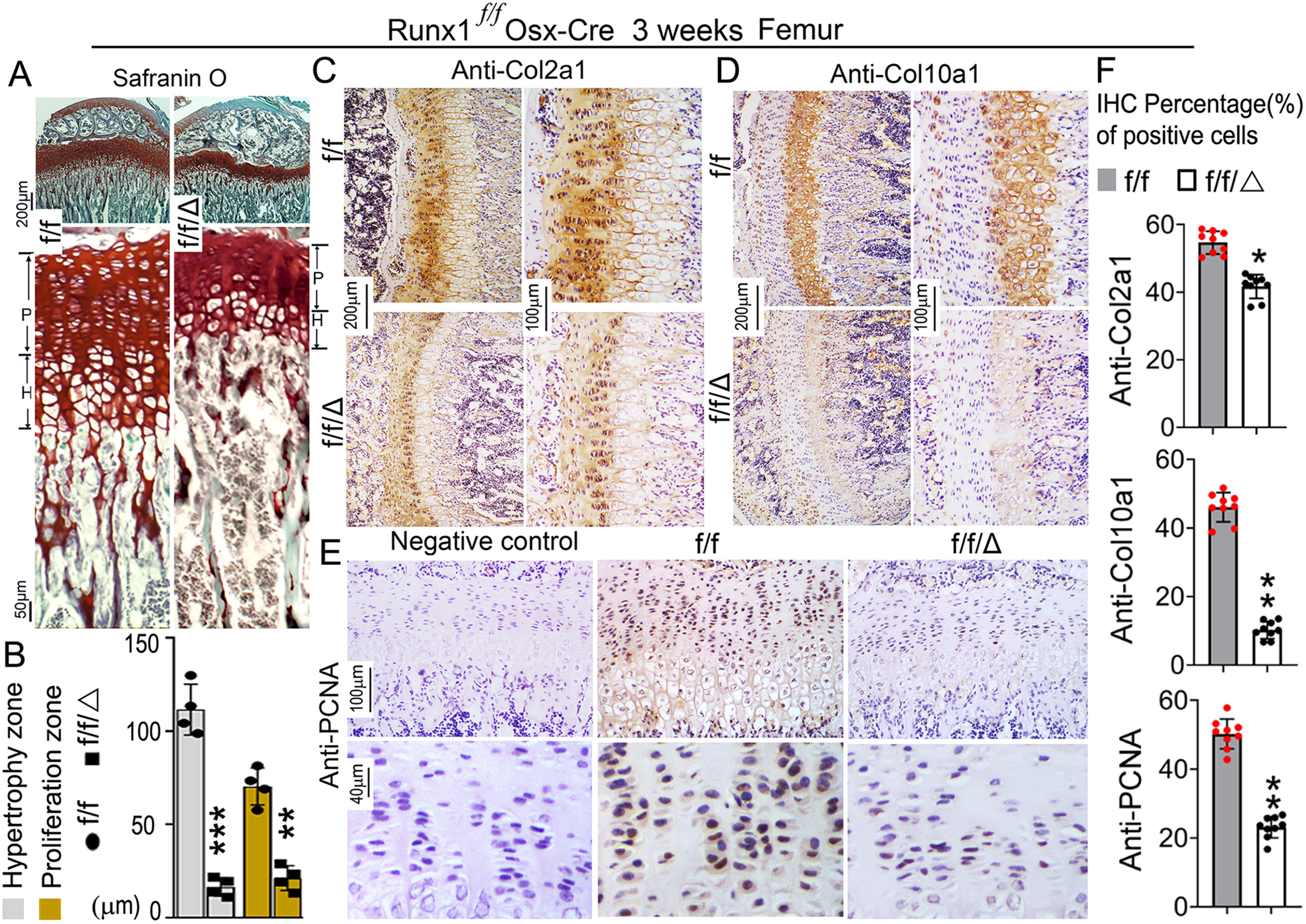
RUNX1 deficiency affects chondrocyte proliferation and maturation in Runx1f/fOsx-Cre mice. A, safranin O staining of tibia and comparison of growth plate development between 3-week–old Runx1f/fOsx-Cre and WT mice. B, quantification of hypertrophy zone and proliferation zone in A. C–E, IHC staining of tibia paraffin sections from 3-week–old Runx1f/fOsx-Cre (f/f/Δ) and WT (f/f) mice using (C) Col2a1 antibody, (D) Col10a1 antibody, and (E) PCNA antibody. F, quantification of immunostaining positive cells of anti-Col2a1, anti-Col10a1, and anti-PCNA in B-D. Results are expressed as mean ± S.D., n = 9 in each group. *, p < 0.05; **, p < 0.01.
Loss of Runx1 reduced the expression of Sox9, Ihh, cyclin D1, and Ptc in chondrocytes of Runx1f/fOsx-Cre mice, and impaired Ihh-Cyclin D1 signaling
Cyclin D1 is among the Cyclin/CDK protein complexes that regulate the cell cycle, and as a target of Indian hedgehog (Ihh) plays an important role in chondrocyte proliferation (24). To address the mechanism underlying the effects of RUNX1 on chondrocytes, we examined the expression levels of SRY-related high mobility group-Box gene 9 (Sox9), Cyclin D1, Ihh, and Patched (Ptc) by immunofluorescent (IF) staining of femur sections from newborn mice (Fig. 5, A–D). Compared with WT mice, the expression of a key transcription factor in the chondrocyte lineage, Sox9, was decreased in Runx1f/fOsx-Cre mice (Fig. 5, A and F). The expression of Ihh, which is important for the chondrocyte maturation, was reduced in Runx1f/fOsx-Cre mice (Fig. 5, B and F). The expression of the IHH target gene, Ptc, was also reduced in the growth plates in Runx1f/fOsx-Cre mice; Ptc expression was detected in the pre-hypertrophic zone in the growth plates of WT mice, but was greatly reduced in mutant mice (Fig. 5, C and F). Cyclin D1, a cell-cycle–regulating protein downstream of Ihh (24), was reduced in the proliferation zone of Runx1f/fOsx-Cre mice femurs (Fig. 5, D and F), which may induce the inhibition of chondrocyte proliferation. Thus, Runx1 plays an important role in pre-hypertrophic chondrocyte proliferation and differentiation. IF staining of newborn mice femurs showed that expression levels of both Osx and Ocn, genes critical to the skeletal formation, were decreased in Runx1f/fOsx-Cre mice (Fig. 5, E and F). In conclusion, we found that RUNX1 deficiency affects chondrocyte proliferation by inhibiting Ihh-cyclin D1 signaling. Furthermore, we found that RUNX1 deficiency reduces the expression of genes critical in skeletal formation.
Figure 5.

The expression of Sox9, Ihh, Cyclin D1, and Ptc in the growth plate, and the expression of Ocn in femurs were significantly decreased in Runx1f/fOsx-Cre mice compared with WT mice. A–D, IF staining of the growth plates of P0 Runx1f/fOsx-Cre mice compared with that of WT mice to detect (A) Sox9, (B) Ihh, (C) Ptc, and (D) Cyclin D1. E, IF staining with anti-Osterix, and anti-Ocn antibodies of femur frozen sections from P0 Runx1f/fOsx-Cre (f/f/Δ) and WT (f/f) mice. F, quantification of A–E. Results are expressed as mean ± S.D., n = 3 in each group.
RUNX1 deficiency in primary calvarial cells inhibits osteoblastogenesis
We investigated the impact of Runx1 deletion on osteoblastogenesis in vitro using calvarial cells from control and Runx1f/fOsx-Cre mice, which were maintained in the osteogenic medium for 14 and 21 days. Calvarial cells from Runx1f/fOsx-Cre mice after 14 days of culture showed reduced osteoblast formation, which was detected through ALP stain (Fig. 6A, Fig. S4A). The oil red O stain showed that adipocytes were increased in the Runx1f/fOsx-Cre mice after 14 days of culture (Fig. S4, B and C). The reduction in mineralization observed in Runx1f/fOsx-Cre mice was detected by Von Kossa staining after 21 days of culture (Fig. 6A, Fig. S4A). Through qPCR and Western blotting we analyzed the expression of several key factors that affect osteoblast differentiation and function in WT and Runx1f/fOsx-Cre mice. Western blotting at days 7 and 14 of culture showed that protein levels of key bone genes Runx1, Runx2, Osx, and Atf4 were all significantly reduced, whereas protein levels of CBFβ were unaffected (Fig. 6, B and C, Fig. S4D). AAV-mediated overexpression of Runx2 in Runx1f/fOsx-Cre cells partially rescued osteoblast differentiation (Fig. S5A). The protein levels of RUNX1, RUNX2, OSX, ATF4, and OCN were detected by Western blotting (Fig. S5, B and C). Consistently, qPCR showed that RUNX1 deficiency significantly reduced the expression Runx1, Runx2, Osx, Atf4, and Ocn (Fig. 6D). We performed genome-wide expression analysis from day 14 cultured osteoblasts in osteoblast differentiation medium to examine the transcriptional profile related with Runx1 knockdown. Our RNA-Seq analysis revealed significantly decreased expression levels of bone formation and homeostasis markers such as Runx2, Osx, Dmp1, Alp, and Ocn (Fig. 6E). We also found similar decreased signaling changes indicated by reduced Pth1r and Ihh expression (Fig. 6E). Our RNA-Seq data also revealed increased markers of adipogenesis in Runx1 knockdown osteoblasts through up-regulated expression of Pparg, Cebp/α, and Fabp4 (Fig. 6E). Taken together, these results demonstrate that Runx1 deletion impacts osteoblast differentiation by affecting the expression of genes critical to osteoblast differentiation at the mRNA and protein levels. These data also indicate that osteoblasts generated from Runx1f/fOsx-Cre mice were prevented from differentiating into mature osteoblasts.
Figure 6.

RUNX1 deficiency in primary calvarial cells cultured from Runx1f/fOsx-Cre mice inhibits osteoblastogenesis. A, calvarial cells from Runx1f/fOsx-Cre (f/f/Δ) and WT (f/f) newborn mice were cultured in osteogenic medium for 14 and 21 days, followed by ALP and Von Kossa stain, respectively. B, protein levels of Runx1, Runx2, Cbfβ, Osx, and Atf4 were analyzed by Western blotting analysis on days 7 and 14. GAPDH is shown as a control. C, quantification of B. D, qPCR analysis of mRNA expression levels of Runx1, Runx2, Osx, Atf4, ALP, Col1α1, Ocn, Opg, and RANKL in calvaria-derived osteoblasts from Runx1f/fOsx-Cre (f/f/Δ) and WT (f/f) mice. E, RNA-Seq analysis of the osteoblasts cultured for 14 days in osteoblast differentiation medium showed decreased osteoblast gene expression and increased adipocyte formation genes expression. Heat map analysis of differentially expressed genes with at least a 2-fold change and controlled by adjusted p value of 0.05, as comparing osteoblasts of WT and Runx1f/fOsx-Cre. In the heat map, red indicates up-regulation, whereas green indicates down-regulation. Select genes are listed separately. Results are expressed as mean ± S.D., n ≧ 3 in each group. N.S., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RUNX1 up-regulates Runx2 and Ocn expression by directly associating with their promoters
ChIP assay was performed to elucidate if RUNX1 binds to the Ocn and Runx2 promoters. We analyzed the respective promoter regions and designed primers accordingly. The RUNX1-binding sites in the Ocn promoter region (−4000/+200) were predicted (Fig. 7A). ChIP analysis was performed using the anti-RUNX1 antibody, and DNA was pulled down, amplified, and analyzed using primers. The ChIP input value using each primer represents the binding efficiency of an adjacent region around the location of the primer pair. We found that Ocn primer 2 resulted in the highest value, indicating that RUNX1-binding site 2 in the Ocn promoter region should be the most efficient (Fig. 7, B and C), and that RUNX1 potentially binds to the Ocn promoter around binding site 2 (Fig. 7A). We then examined the Runx2 promoter and found several potential RUNX1-binding sites in the Runx2 promoter region (−4000/+200) (Fig. 7D). We performed ChIP analysis using the anti-RUNX1 antibody, and DNA was pulled down, amplified, and analyzed using primers. We found that Runx2 primer 2 resulted in the highest value, indicating that RUNX1-binding site 4 in the Runx2 promoter region should be the most efficient (Fig. 7, E and F). This indicated that RUNX1 potentially binds to the Runx2 promoter around binding site 4 (Fig. 7D). The promoter luciferase assay showed that luciferase activity was highest when driven by the longest Runx2 promoter fragment (−2937/+80) and significantly lower (90% reduction) when driven by the other Runx2 promoter fragments (Fig. 7G). In conclusion, our data demonstrated that RUNX1 associates with the Ocn and Runx2 promoter regions and regulates their expression directly.
Figure 7.

RUNX1 regulates Runx2 expression by directly associating with its promoters. A, schematic display of the Ocn (–4000/+200) promoter region: TSS, predicted RUNX1-binding sites, and ChIP primers positions. B, ChIP analysis of RUNX1 binding to the Ocn promoter in WT calvaria-derived osteoblasts using primers as indicated on the x axis. Results are presented as ChIP/Input. C, agarose gel image using ChIP qPCR products in B. D, schematic display of Runx2 (–4000/+200) promoter region: TSS, predicted RUNX1-binding sites, and ChIP primers positions. E, ChIP analysis of RUNX1 binding to the Runx2 promoter in WT calvaria-derived osteoblast using primers as indicated on the x axis. Results are presented as ChIP/Input. F, agarose gel image using ChIP qPCR products in E. G, Runx2 promoter fragments were inserted into pG13-basic vector. C3H10T1/2 cells were transfected with pG13-Runx2 –126 bp, –465 bp, –1009 bp, –1409 bp, –1888 bp, and –2937 bp. Luciferase was detected at 48 h post-transfection and normalized to β-gal activity. Results are presented as mean ± S.D. with n = 3. N.S., not significant; *, p < 0.05; **, p < 0.01.
Discussion
In this study, we found that ablation of Runx1 reduced the expression of Sox9, Ihh, Ptc, and CyclinD1 in the growth plate, and impaired the proliferative and hypertrophic zones in the growth plate, whereas the trabecular bone was reduced by 40%, suggesting a central role of Runx1 in chondrocyte proliferation. Runx1 ablation also reduced the expression of Ocn, Osx, Atf4, and Runx2 in osteoblasts, and resulted in an osteoporotic phenotype in Runx1f/fOsx-Cre mice. Deficiency of RUNX1 results in a 2-fold decrease in osteoprogenitor cells in the periosteum, subsequently leading to the lack of bone collar in the newborn Runx1f/fOsx-Cre mice and 2-fold decrease in cortical bone width seen in 14-week–old Runx1f/fOsx-Cre mice. Interestingly, our data demonstrate that the expression levels of Runx2 and Osx are both decreased in the Runx1f/fOsx-Cre mice. The expression of osteocalcin, a mature osteoblast marker, was also decreased in the Runx1f/fOsx-Cre mice. Our data demonstrate that Runx1 plays a central role in promoting the commitment of osteoblast precursors into the osteoblast lineage. Our ChIP and luciferase assay show that RUNX1 can bind to the Runx2 promoter, and our ChIP assay showed that RUNX1 can bind to the Ocn promoter, indicating that RUNX1 can up-regulate Runx2 and Ocn expression directly.
Endochondral ossification and intramembranous ossification are two major modes of bone formation. Endochondral ossification is involved in the formation of long bones through a cartilage intermediate, whereas the intramembranous ossification directly forms the flat bone on the mesenchyme (25). Our results showed that the skull bone density was lower, the ALP and Von Kossa stain were decreased in the mutant mouse more than in the Osx-cre and Runx1f/f control mice, which indicated that the intramembranous ossification was affected because of the impaired osteoblast activity. As for the lone bone ossification, both chondrocyte and osteoblast were affected in the mutant mouse, so the deletion of Runx1 in both the chondrocyte and pre-osteoblast contributed to the endochondral ossification. Taken together, these results indicate that RUNX1 promotes osteoblast formation and differentiation to promote bone formation through up-regulating Runx2 and bone genes.
Previous reports have demonstrated that Runx1 is important for mesenchymal stem cell commitment to the early stages of chondrogenesis, and is required for chondrocyte lineage commitment and differentiation (9, 10, 26), whereas there may be a cross-regulation between Runx1 and Runx2 in chondrocytes (10). Runx1 was found expressed in vivo in osteoblasts at the site of bone formation and chondroblasts at sites of cartilage growth (11), and it was previously reported that RUNX1 dose-dependently regulates endochondral ossification during skeletal development and fracture healing (9). Yet the role of RUNX1 in bone formation and bone homeostasis has not been previously reported. RUNX2 regulates the expression of Ihh and Col10a1 in their respective layers in the growth plate (16). Our results show that the expression of Runx2 was not significantly changed in the hypertrophic zone in newborn mice, however, 3-week–old mice displayed an impaired hypertrophic zone, as well as significantly reduced expression of Col10a1. Through ChIP assays and promoter activity mapping, we demonstrated that Runx2 is directly up-regulated by RUNX1 at the transcriptional level. Furthermore, our ChIP data also showed that RUNX1 directly associates with the promoter of Ocn. Thus, RUNX1 regulates Runx2 expression directly to regulate chondrocyte and osteoblast differentiation and terminal maturation.
Lower bone mineral density and a 50% reduction in cortical thickness and 40% reduction in trabecular bone volume in the tibiae of Runx1f/fOsx-Cre mice, which may be due to decreased osteoblast numbers in the bone collar, suggest that RUNX1 is required as an endogenous regulator of skeletal size and bone architecture. Kimura et al. (7) previously reported that in Prx1 Runx1f/f mice, aside from delay in sternal development, no other skeletal elements of the mice showed any abnormalities, however, their analysis was limited to P0 to 3-week–old mice using Prx1 Cre. In our study, we examined mice from P0 to 14 weeks old to determine the role of Runx1 in maintaining bone homeostasis. Our data demonstrate that Runx1f/fOsx-Cre mice exhibited slower bone mineralization and bone formation rates, leading to lower bone density. These results suggest that RUNX1 is important for intramembranous and endochondral ossification. Interestingly, we found that Runx1f/fOsx-Cre mice also displayed a mandibular defect characterized by a smaller angle of the lower jaw. The growth plate is the specialized cartilaginous structure for the longitudinal growth of bones (27). Fibril-forming type II collagen is the major collagen type in the growth plate, whereas hypertrophic chondrocytes in the growth plate produce type X collagen (27). We found that COL2a1 and COL10a1, which are produced by the proliferative and hypertrophic chondrocytes, were decreased in the mutant mice. Runx1 is involved in chondrocyte proliferation and lineage determination (28), and whereas Runx1 is only expressed in the proliferative and resting chondrocytes, Runx2 is expressed primarily by hypertrophic chondrocytes of the growth plates (29). Thus, following conditional deletion of Runx1, proliferative and resting chondrocytes cannot differentiate into hypertrophic chondrocytes due to the loss of Runx2 regulation, leading to the significant reduction in trabecular bone observed in Runx1f/fOsx-Cre mice. Moreover, the expression of PCNA was decreased in Runx1f/fOsx-Cre mice. Thus, the shorter stature of the mice may be due to the inhibition of the chondrocyte differentiation and terminal maturation. Given the reduced numbers of proliferative and hypertrophic chondrocytes, osteoblasts, and reduced trabecular bone in Runx1f/fOsx-Cre mice, Runx1 may maintain trabecular bone formation through its regulation of Runx2, Sox9, Ihh, Ptc, and Cyclin D1 in hypertrophic chondrocytes.
Our data show that the expression levels of Sox9, Ihh, Ptc, and Cyclin D1, genes that are involved in chondrocyte differentiation, were decreased in Runx1f/fOsx-Cre mice, suggesting a potential role of Runx1 in pre-hypertrophic chondrocyte proliferation and differentiation. SOX9 is a transcription factor of the SRY-related high mobility group box family of proteins, which is the nuclear factor that is required for chondrogenesis (30, 31). Although Sox9 is dispensable for the initial formation of mesenchymal condensations, it necessary for the subsequent steps toward chondrocyte differentiation (32). A previous study showed that RUNX1 can directly regulate the expression of Sox9 (9). IHH produced by pre- and early hypertrophic chondrocytes leads to chondrocyte proliferation (33). Cyclin D1 plays an important role in chondrocyte proliferation and is a target of IHH (24). IHH, a major regulator of bone development, coordinating chondrocyte proliferation, chondrocyte differentiation, and osteoblast differentiation, also binds to membrane protein PTC (34–37). Thus, Runx1 regulates the early stages of the chondrocyte differentiation, whereas Runx2 is important for the chondrocyte maturation (16). Furthermore, Ihh signaling in mature osteoblasts controls bone resorption by regulating Rankl expression (38). Although it was previously believed that OSX functions exclusively as an osteoblast-specific transcription factor, our results and that of others (39–41) demonstrate that Osx also plays a role in postnatal chondrogenesis. Previous reports have demonstrated that Osx couples chondrogenesis and osteogenesis in postnatal condylar growth, and is essential for the coupling of terminal cartilage differentiation (39). Furthermore, Osx-Cre also targets olfactory glomerular cells and a subset of the gastric and intestinal epithelium in postnatal mice (40). Notably, Cheng et al. (41) demonstrated that haploinsufficiency of OSX in chondrocytes impairs skeletal growth in mice. Previous reports demonstrate that the Osx-Cre transgene itself has no effect on the trabecular bone parameters and minimal effect on the cortical parameters for 1-month–old mice, and as the mature mice, they display comparable cortical and trabecular bone parameters compared with the control mice (22, 42). At 6 weeks of age, Osx-Cre mice display reduced body weight and delayed cortical bone expansion and accrual, however, the delayed weight gain and cortical growth bone of Osx-Cre mice is overcome by 12 weeks of age, with no differences between Osx-Cre and WT mice (43). Thus, the phenotype observed in the Runx1f/fOsx-Cre mice was due to the deletion of Runx1, and not due to the Osx-Cre transgene itself.
Our in vitro mechanistic studies demonstrate that Runx1 conditional deletion in osteoblast precursors and differentiating chondrocytes has significant effects on genes critical for skeletal development such as Runx2, Osx, Atf4, and Ocn. These findings suggest that RUNX1 plays an indispensable role in postnatal skeletal development and bone homeostasis by up-regulating the expression of Runx2 and multiple bone genes. Our study shows that RUNX1 associates with the promoter regions of Runx2 and Ocn and highly up-regulates the expression of Runx2 at the transcriptional regulation level as shown by ChIP assay, qPCR analysis, and promoter reporter assay. Although we found multiple binding sites for RUNX1 on the Runx2 promoter, we found that RUNX1 potentially binds to the Runx2 promoter around binding site 4. Previous reports have shown that RUNX1 mediates epigenetic regulation to inhibit or facilitate multiple regulatory regions in the target genes (44–46). RUNX1 has been shown to interact with histone acetyltransferases, as well as components of the histone deacetylase complex (46). Thus, the interaction of RUNX1 with histone acetyltransferases acetylates chromatin-associated histones, which lead to chromatin conformational changes and transcriptional activation, could be responsible for the high binding efficiency of RUNX1 on the Runx2 promoter at binding site 4. However, the landscape of epigenetic alterations in many cell types is distinctively complex, thus future research is warranted to fully explore whether and how RUNX1 leads to epigenetic changes at genes central to osteoblastic lineage commitment.
Because Runx2 exhibits high homology with Runx1, it has been suspected that Runx1 may be responsible for some cases of CCD. Although no Runx1 mutation has yet been identified in classical CCD patients, our Runx1f/fOsx-Cre mouse models support the notion that search genetic alterations in the Runx1 gene may be responsible for CCD in those patients with no Runx2 mutation. Our results are in agreement with that of previous studies reporting that Runx2 deficiency causes an arrest in clavicular development (47). These findings provide great insight into the pathogenesis of CCD and the role of Runx1 in both postnatal skeletal and tooth development.
Overall, our genetic dissection approach revealed that RUNX1 plays an indispensable role in postnatal skeletal development and bone homeostasis by up-regulating the expression of Runx2 and multiple bone genes. Our study of Runx1 in the osteoblast lineage has demonstrated that Runx1 is required to promote chondrocyte and osteoblast differentiation to maintain bone homeostasis. Targeting RUNX1, a regulator of both Runx2 and bone genes, could facilitate the design of safer and novel therapeutic approaches for osteoporosis. Taken together, this work provides important insights into the role of RUNX1 in bone formation and the mechanisms underlying how RUNX1 maintains bone homeostasis. The insights resulting from this study may assist in the development of novel treatments for osteoporosis and other osteolytic diseases.
Experimental procedures
For more detailed descriptions, please refer to the Supporting Materials and Methods. For primer sequences please refer to Tables S1–S3.
Generation of Runx1f/fOsx-Cre mice
All animal experimentation was carried out according to the legal requirements of the Association for Assessment and Accreditation of the Laboratory Animal Care International and the University of Alabama at Birmingham Institutional Animal Care and Use Committee. Jackson Laboratory, strain name B6.129P2-Runx1tm1Tani/J, JAX No. 008772, were crossed with skeletal tissue cell (including osteoblast precursors, osteoblasts, chondrocytes, and odontoblasts)-specific Osx-Cre mice (19) (Tg(Sp7-tTA,tetO-EGFP/cre)1Amc, Mouse Genome Informatics) Their progeny were crossed with Runx1f/f mice to obtain Runx1f/fOsx-Cre mice. In our study, we only use one copy of Osx-Cre (Runx1f/f Osx-Cre/+) in the CKO mutation. Osx-Cre mice had no detectable bone phenotype and served as control groups along with Runx1f/f mice. Mice were bred in-house and euthanatized by CO2 asphyxiation. All mice were maintained under a 12-h light–dark cycle with ad libitum access to regular food and water at the University of Alabama Animal Facility. Both male and female mice of each strain were randomly selected into groups of five animals each. The investigators were not blinded during allocation, animal handling, and end point measurements. Genotyping by PCR was carried out as described (48).
Histomorphometric analysis
Histomorphometric samples were processed as un-decalcified hard-tissue sections as described (49, 50). Bone parameters were quantified via 6-μm sections obtained from mice.
Primary cell culture
Calvarial cells were isolated from newborn mice and seeded in cell culture dishes at a density of 3 × 103 cells/cm2 as described (51). After growing to confluence, cells were induced to differentiate into osteoblasts using osteogenic medium supplemented with 10% (v/v) FBS, 50 μg/ml of l-ascorbic acid (Sigma, A4544), and 5 mm β-glycerolphosphate (Sigma, G9891). Osteoblastogenesis was analyzed by ALP staining according to the manufacturer's manual (Sigma, A2356) on day 14. Osteoblast mineralization was examined by Von Kossa staining on day 21.
ChIP
Cells were derived from calvaria of newborn WT mice, and ChIP was performed as described (52). After immunoprecipitation using monoclonal anti-Runx1 antibody (ab23980; Abcam) and DNA extraction, quantitative PCR was performed using the primers in the promoter region of the mouse gene Runx2.
RNA-Sequencing analysis
Total mRNA was isolated using TRIzol reagent (Invitrogen Corp.) from the osteoblasts that were cultured for 14 days in osteoblasts differentiation medium following the manufacturer's protocol and submitted to Admera Health (South Plainsfield, NJ) who assessed sample quality with the Agilent Bioanalyzer and prepared the library using the NEBnext Ultra RNA-Poly(A) kit. Libraries were analyzed using Illumina next generation sequencing and relative quantification was provided by Admera Health.
Promoter analyses
Runx2 promoter sequences were analyzed for putative RUNX-binding sites with PROMO3.0 (RRID:SCR_016926) using version 8.3 of the TRANSFAC database. The promoter region (−) and (+) of the mouse Runx2 gene was amplified by PCR using Runx2 Bac clone (catalog No. CH29-571B2; CHORI). Then the promoter regions were inserted into the pGL3-basic vector to construct the pGL3-Runx2 promoter fragments. The insertions of the constructs were confirmed by sequencing. C3H10T1/2 cells were cultured in 24-well–plates, and were transiently transfected with a DNA mixture containing the pGL3-Runx2 construct (0.3 μg) and β-Gal-expressing plasmids (0.06 μg), with or without Runx1 expressing vector (pCMV-Sport6-Runx1, 0.3 μg) using Lipofectamine and Plus reagents. Luciferase was detected using Glo Luciferase Assay System (Promega) 48 h post-transfection as described (52). The β-Gal activity of the cell lysates was analyzed using β-Galactosidase Enzyme Assay System (E2000; Promega). The level of luciferase activity was normalized to the level of β-Gal activity.
Statistical analysis
All data are presented as the mean ± S.D. (n ≧ 6). Statistical significance was assessed using Student's t test. p values <0.05 were considered significant. Data are expressed as mean ± S.D., n ≧ 6, *, p < 0.05; **, p < 0.01; ***, p < 0.001. The results are representative of at least four individual experiments. The analyses of the data were performed with the SPSS 16.0 software (SPSS Incorporation, Chicago, IL, USA).
Data availability
The RNA-Seq data are available upon request from Yi-Ping Li, Department of Pathology, University of Alabama at Birmingham, E-mail: yipingli@uabmc.edu. All other data are contained within the manuscript.
Supplementary Material
Acknowledgments
We thank Abigail McVicar for excellent assistance with the manuscript, and Matthew McConnell for bioinformatics analysis assistance. We appreciate the assistance provided by the Center for Metabolic Bone Disease at the University of Alabama at Birmingham supported by Grant P30 AR046031. We are also grateful for the assistance from the Small Animal Phenotyping Core, Metabolism Core, and Neuroscience Molecular Detection Core Laboratory at the University of Alabama at Birmingham supported by Grant P30 NS0474666.
This article contains supporting information.
Author contributions—J. T., J. X., W. C., C. T., J. W., Y. W., X.-d. Z., H.-D. Z., and Y.-P. L. data curation; J. T., J. X., W. C., C. T., J. W., Y. W., X.-d. Z., H.-D. Z., and Y.-P. L. formal analysis; J. T., J. X., W. C., C. T., J. W., Y. W., X.-d. Z., H.-D. Z., and Y.-P. L. investigation; J. T., W. C., H.-D. Z., and Y.-P. L methodology; J. T., W. C., X.-d. Z., H-D. Z., and Y-P. L. writing-original draft; J. T., W. C., C. T., X.-d. Z., H.-D. Z., and Y.-P. L. writing-review and editing; J. X., W. C., C. T., Y. W., H.-D. Z., and Y.-P. L. visualization; W. C. and Y.-P. L. conceptualization; W. C., H.-D. Z., and Y.-P. L. supervision; W. C. and Y.-P. L. funding acquisition; W. C. and Y.-P. L. validation; Y.-P. L. resources; Y.-P. L. project administration.
Funding and additional information—This work was supported by National Institutes of Health Grants AR-075735, DE-028264 (to Y.-P. L.), AR-070135 and AG-056438 (to W. C.), and China Scholarship Council Grant 201606370184 (to J. T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- Runx1
- Runt-related transcription factor 1
- Osx
- Osterix
- Runx2
- Runt-related transcription factor 2
- Sox9
- SRY-related high-mobility group-box gene 9
- Ihh
- Indian hedgehog
- PTC
- Patched
- Ocn
- osteocalcin
- Atf4
- activating transcription factor 4
- qPCR
- quantitative polymerase chain reaction
- IHC
- immunohistochemical
- μCT
- micro-computed tomography
- H&E
- hematoxylin and eosin
- ALP
- alkaline phosphatase
- TRAP
- tartrate-resistant acid phosphatase
- CKO
- conditional knockout
- PCNA
- proliferating cell nuclear antigen
- IF
- immunofluorescent
- CCD
- cleidocranial dysplasia
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Nam M., Huh J. E., Kim M. S., Ryu D. H., Park J., Kim H. S., Lee S. Y., and Hwang G. S. (2018) Metabolic alterations in the bone tissues of aged osteoporotic mice. Sci. Rep. 8, 8127 10.1038/s41598-018-26322-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brommage R., and Ohlsson C. (2018) Translational studies provide insights for the etiology and treatment of cortical bone osteoporosis. Best Pract. Res. Clin. Endocrinol. Metab. 32, 329–340 10.1016/j.beem.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 3. Miyamoto T. (2018) Homeostasis and disorder of musculoskeletal system: pathogenesis of musculoskeletal diseases and strategies for their treatment. Clin. Calcium 28, 301–305 [PubMed] [Google Scholar]
- 4. Komori T. (2005) Regulation of skeletal development by the Runx family of transcription factors. J. Cell. Biochem. 95, 445–453 10.1002/jcb.20420 [DOI] [PubMed] [Google Scholar]
- 5. Okuda T., Nishimura M., Nakao M., and Fujita Y. (2001) RUNX1/AML1: a central player in hematopoiesis. Int. J. Hematol. 74, 252–257 10.1007/BF02982057 [DOI] [PubMed] [Google Scholar]
- 6. Chen C. L., Broom D. C., Liu Y., de Nooij J. C., Li Z., Cen C., Samad O. A., Jessell T. M., Woolf C. J., and Ma Q. (2006) Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron 49, 365–377 10.1016/j.neuron.2005.10.036 [DOI] [PubMed] [Google Scholar]
- 7. Kimura A., Inose H., Yano F., Fujita K., Ikeda T., Sato S., Iwasaki M., Jinno T., Ae K., Fukumoto S., Takeuchi Y., Itoh H., Imamura T., Kawaguchi H., Chung U. I., et al. (2010) Runx1 and Runx2 cooperate during sternal morphogenesis. Development 137, 1159–1167 10.1242/dev.045005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liakhovitskaia A., Lana-Elola E., Stamateris E., Rice D. P., van 't Hof R. J., and Medvinsky A. (2010) The essential requirement for Runx1 in the development of the sternum. Dev. Biol. 340, 539–546 10.1016/j.ydbio.2010.02.005 [DOI] [PubMed] [Google Scholar]
- 9. Soung do Y., Talebian L., Matheny C. J., Guzzo R., Speck M. E., Lieberman J. R., Speck N. A., and Drissi H. (2012) Runx1 dose-dependently regulates endochondral ossification during skeletal development and fracture healing. J. Bone Miner. Res. 27, 1585–1597 10.1002/jbmr.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Y., Belflower R. M., Dong Y. F., Schwarz E. M., O'Keefe R. J., and Drissi H. (2005) Runx1/AML1/Cbfa2 mediates onset of mesenchymal cell differentiation toward chondrogenesis. J. Bone Miner. Res. 20, 1624–1636 10.1359/JBMR.050516 [DOI] [PubMed] [Google Scholar]
- 11. Lian J. B., Balint E., Javed A., Drissi H., Vitti R., Quinlan E. J., Zhang L., Van Wijnen A. J., Stein J. L., Speck N., and Stein G. S. (2003) Runx1/AML1 hematopoietic transcription factor contributes to skeletal development in vivo. J. Cell. Physiol. 196, 301–311 10.1002/jcp.10316 [DOI] [PubMed] [Google Scholar]
- 12. Ducy P., Zhang R., Geoffroy V., Ridall A. L., and Karsenty G. (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89, 747–754 10.1016/S0092-8674(00)80257-3 [DOI] [PubMed] [Google Scholar]
- 13. Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R. T., Gao Y. H., Inada M., Sato M., Okamoto R., Kitamura Y., Yoshiki S., and Kishimoto T. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 10.1016/S0092-8674(00)80258-5 [DOI] [PubMed] [Google Scholar]
- 14. Otto F., Thornell A. P., Crompton T., Denzel A., Gilmour K. C., Rosewell I. R., Stamp G. W., Beddington R. S., Mundlos S., Olsen B. R., Selby P. B., and Owen M. J. (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771 10.1016/S0092-8674(00)80259-7 [DOI] [PubMed] [Google Scholar]
- 15. Soltanoff C. S., Chen W., Yang S., and Li Y.-P. (2009) Signaling networks that control the lineage commitment and differentiation of bone cells. Crit. Rev. Eukaryot. Gene Expr. 19, 1–46 10.1615/critreveukargeneexpr.v19.i1.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Komori T. (2018) Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 149, 313–323 10.1007/s00418-018-1640-6 [DOI] [PubMed] [Google Scholar]
- 17. Geoffroy V., Kneissel M., Fournier B., Boyde A., and Matthias P. (2002) High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol. Cell Biol. 22, 6222–6233 10.1128/mcb.22.17.6222-6233.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith N., Dong Y., Lian J. B., Pratap J., Kingsley P. D., van Wijnen A. J., Stein J. L., Schwarz E. M., O'Keefe R. J., Stein G. S., and Drissi M. H. (2005) Overlapping expression of Runx1(Cbfa2) and Runx2(Cbfa1) transcription factors supports cooperative induction of skeletal development. J. Cell. Physiol. 203, 133–143 10.1002/jcp.20210 [DOI] [PubMed] [Google Scholar]
- 19. Rodda S. J., and McMahon A. P. (2006) Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231–3244 10.1242/dev.02480 [DOI] [PubMed] [Google Scholar]
- 20. Chen S., Gluhak-Heinrich J., Wang Y. H., Wu Y. M., Chuang H. H., Chen L., Yuan G. H., Dong J., Gay I., and MacDougall M. (2009) Runx2, osx, and dspp in tooth development. J. Dent. Res. 88, 904–909 10.1177/0022034509342873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang W., and Olsen B. R. (2015) Skeletal defects in Osterix-Cre transgenic mice. Transgenic Res. 24, 167–172 10.1007/s11248-014-9828-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang L., Mishina Y., and Liu F. (2015) Osterix-Cre transgene causes craniofacial bone development defect. Calcif. Tissue Int. 96, 129–137 10.1007/s00223-014-9945-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Provot S., and Schipani E. (2005) Molecular mechanisms of endochondral bone development. Biochem. Biophys. Res. Commun. 328, 658–665 10.1016/j.bbrc.2004.11.068 [DOI] [PubMed] [Google Scholar]
- 24. Long F., Zhang X. M., Karp S., Yang Y., and McMahon A. P. (2001) Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128, 5099–5108 [DOI] [PubMed] [Google Scholar]
- 25. Berendsen A. D., and Olsen B. R. (2015) Bone development. Bone 80, 14–18 10.1016/j.bone.2015.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J., Wang X., Holz J. D., Rutkowski T., Wang Y., Zhu Z., and Dong Y. (2013) Runx1 is critical for PTH-induced onset of mesenchymal progenitor cell chondrogenic differentiation. PLoS ONE 8, e74255 10.1371/journal.pone.0074255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Myllyharju J. (2014) Extracellular matrix and developing growth plate. Curr. Osteoporos. Rep. 12, 439–445 10.1007/s11914-014-0232-1 [DOI] [PubMed] [Google Scholar]
- 28. Johnson K., Zhu S., Tremblay M. S., Payette J. N., Wang J., Bouchez L. C., Meeusen S., Althage A., Cho C. Y., Wu X., and Schultz P. G. (2012) A stem cell-based approach to cartilage repair. Science 336, 717–721 10.1126/science.1215157 [DOI] [PubMed] [Google Scholar]
- 29. Wu M., Li C., Zhu G., Wang Y., Jules J., Lu Y., McConnell M., Wang Y. J., Shao J. Z., Li Y. P., and Chen W. (2014) Deletion of core-binding factor beta (Cbfβ) in mesenchymal progenitor cells provides new insights into Cbfβ/Runxs complex function in cartilage and bone development. Bone 65, 49–59 10.1016/j.bone.2014.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Foster J. W., Dominguez-Steglich M. A., Guioli S., Kwok C., Weller P. A., Stevanovic M., Weissenbach J., Mansour S., Young I. D., and Goodfellow P. N. and (1994) Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 372, 525–530 10.1038/372525a0 [DOI] [PubMed] [Google Scholar]
- 31. Wagner T., Wirth J., Meyer J., Zabel B., Held M., Zimmer J., Pasantes J., Bricarelli F. D., Keutel J., Hustert E., Wolf U., Tommerup N., Schempp W., and Scherer G. (1994) Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 79, 1111–1120 10.1016/0092-8674(94)90041-8 [DOI] [PubMed] [Google Scholar]
- 32. Barna M., and Niswander L. (2007) Visualization of cartilage formation: insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Dev. Cell 12, 931–941 10.1016/j.devcel.2007.04.016 [DOI] [PubMed] [Google Scholar]
- 33. Long F., and Ornitz D. M. (2013) Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 5, a008334 10.1101/cshperspect.a008334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kronenberg H. M. (2003) Developmental regulation of the growth plate. Nature 423, 332–336 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- 35. Maeda Y., Nakamura E., Nguyen M. T., Suva L. J., Swain F. L., Razzaque M. S., Mackem S., and Lanske B. (2007) Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. U.S.A. 104, 6382–6387 10.1073/pnas.0608449104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Long F., Chung U. I., Ohba S., McMahon J., Kronenberg H. M., and McMahon A. P. (2004) Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development (Cambridge, Eng.) 131, 1309–1318 10.1242/dev.01006 [DOI] [PubMed] [Google Scholar]
- 37. Karp S. J., Schipani E., St-Jacques B., Hunzelman J., Kronenberg H., and McMahon A. P. (2000) Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development (Cambridge, Eng.) 127, 543–548 10631175 [Google Scholar]
- 38. Mak K. K., Bi Y., Wan C., Chuang P. T., Clemens T., Young M., and Yang Y. (2008) Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev. Cell 14, 674–688 10.1016/j.devcel.2008.02.003 [DOI] [PubMed] [Google Scholar]
- 39. Jing J., Hinton R. J., Jing Y., Liu Y., Zhou X., and Feng J. Q. (2014) Osterix couples chondrogenesis and osteogenesis in post-natal condylar growth. J. Dental Res. 93, 1014–1021 10.1177/0022034514549379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen J., Shi Y., Regan J., Karuppaiah K., Ornitz D. M., and Long F. (2014) Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PLoS ONE 9, e85161 10.1371/journal.pone.0085161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng S., Xing W., Zhou X., and Mohan S. (2013) Haploinsufficiency of osterix in chondrocytes impairs skeletal growth in mice. Physiol. Genomics 45, 917–923 10.1152/physiolgenomics.00111.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu F., Fang F., Yuan H., Yang D., Chen Y., Williams L., Goldstein S. A., Krebsbach P. H., and Guan J.-L. (2013) Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J. Bone Miner. Res. 28, 2414–2430 10.1002/jbmr.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davey R. A., Clarke M. V., Sastra S., Skinner J. P., Chiang C., Anderson P. H., and Zajac J. D. (2012) Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res. 21, 885–893 10.1007/s11248-011-9581-z [DOI] [PubMed] [Google Scholar]
- 44. Kitabayashi I., Aikawa Y., Nguyen L. A., Yokoyama A., and Ohki M. (2001) Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 20, 7184–7196 10.1093/emboj/20.24.7184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Allende-Vega N., Dayal S., Agarwala U., Sparks A., Bourdon J. C., and Saville M. K. (2013) p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 32, 1–14 10.1038/onc.2012.38 [DOI] [PubMed] [Google Scholar]
- 46. Koh C. P., Wang C. Q., Ng C. E., Ito Y., Araki M., Tergaonkar V., Huang G., and Osato M. (2013) RUNX1 meets MLL: epigenetic regulation of hematopoiesis by two leukemia genes. Leukemia 27, 1793–1802 10.1038/leu.2013.200 [DOI] [PubMed] [Google Scholar]
- 47. Cohen M. M., Jr (2013) Biology of RUNX2 and cleidocranial dysplasia. J. Craniofacial Surg. 24, 130–133 10.1097/SCS.0b013e3182636b7e [DOI] [PubMed] [Google Scholar]
- 48. Growney J. D., Shigematsu H., Li Z., Lee B. H., Adelsperger J., Rowan R., Curley D. P., Kutok J. L., Akashi K., Williams I. R., Speck N. A., and Gilliland D. G. (2005) Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 106, 494–504 10.1182/blood-2004-08-3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen W., Yang S., Abe Y., Li M., Wang Y., Shao J., Li E., and Li Y. P. (2007) Novel pycnodysostosis mouse model uncovers cathepsin K function as a potential regulator of osteoclast apoptosis and senescence. Hum. Mol. Genet. 16, 410–423 10.1093/hmg/ddl474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang S., and Li Y. P. (2007) RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation. Genes Dev. 21, 1803–1816 10.1101/gad.1544107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pockwinse S. M., Wilming L. G., Conlon D. M., Stein G. S., and Lian J. B. (1992) Expression of cell growth and bone specific genes at single cell resolution during development of bone tissue-like organization in primary osteoblast cultures. J. Cell. Biochem. 49, 310–323 10.1002/jcb.240490315 [DOI] [PubMed] [Google Scholar]
- 52. Chen W., Ma J., Zhu G., Jules J., Wu M., McConnell M., Tian F., Paulson C., Zhou X., Wang L., and Li Y. P. (2014) Cbfβ deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of Cbfβ required for skeletal development. Proc. Natl. Acad. Sci. U.S.A. 111, 8482–8487 10.1073/pnas.1310617111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq data are available upon request from Yi-Ping Li, Department of Pathology, University of Alabama at Birmingham, E-mail: yipingli@uabmc.edu. All other data are contained within the manuscript.