

Summary
Foxp3+ CD4+ regulatory T cells (Tregs) are an immune cell lineage endowed with immunosuppressive functionality in a wide array of contexts, including both anti‐pathogenic and anti‐self responses. In the past decades, our understanding of the functional diversity of circulating or lymphoid Tregs has grown exponentially. Only recently, the importance of Tregs residing within non‐lymphoid tissues, such as visceral adipose tissue, muscle, skin and intestine, has been recognized. Not only are Tregs critical for influencing the kinetics and strength of immune responses, but the regulation of non‐immune or parenchymal cells, also fall within the purview of tissue‐resident or infiltrating Tregs. This review focuses on providing a systematic and comprehensive comparison of the molecular maintenance, local adaptation and functional specializations of Treg populations operating within different tissues.
Keywords: inflammation, regulatory T cells, tissue regulatory T cells, tissue repair
Foxp3+ CD4+ regulatory T cells (Tregs) are an immune cell lineage endowed with immunosuppressive functionality. Only recently, the importance of Tregs residing within non‐lymphoid tissues, such as visceral adipose tissue, muscle, skin and intestine, has been recognized. Not only are Tregs critical for influencing the kinetics and strength of immune responses, but the regulation of non‐immune or parenchymal cells, also falls within the purview of tissue‐resident or infiltrating Tregs.
Abbreviations
- Areg
amphiregulin
- CAR‐Tregs
chimeric antigen receptor Tregs
- DTR
diphtheria toxin receptor
- EGFR
epidermal growth factor receptor
- Foxp3
Forkhead box P3 transcription factor
- HF
hair follicle
- HFSC
hair follicle stem cell
- LN
lymph node
- MHC
major histocompatibility complex
- PPARγ
peroxisome proliferator‐activated receptor
γ
- pTregs
peripherally derived Tregs
- TCR
T‐cell receptor
- Teffs
CD4+ T effector cells
- TGF‐β
transforming growth factor
β
- Tregs
regulatory T cells
- tTregs
thymic‐derived Tregs
- VAT
visceral adipose tissue
Introduction
The concept of T cells suppressing the immune system began in the early 1970s. 1 , 2 , 3 However, the field suffered scrutiny from the early 1980s to the 1990s, due to the lack of unique phenotypic markers and molecular mechanisms underlying immunosuppression (reviewed in refs 4, 5). Such a narrative began to change when Sakaguchi and colleagues identified a subset of thymic CD4+ T cells in murine lymph nodes (LN) expressing high levels of interleukin‐2 receptor‐α (IL‐2Rα; CD25), capable of suppressing autoimmunity. 6 Shortly after, Forkhead/winged‐helix transcription factor 3 (Foxp3) was recognized as a critical phenotypic and functional regulatory T cell (Treg) marker, which is mutated in ‘scurfy’ mice, 7 and also in human immunodysregulation polyendocrinopathy enteropathy X‐linked patients. 8 Foxp3 mutation in both scenarios results in an accelerated and fulminant systemic autoimmunity. These major breakthroughs led to the rebranding from ‘suppressor’ T cells to the present ‘regulatory’ T cells (i.e. Tregs), and the exponential bloom of our understanding in Treg development and functions (reviewed in ref. 9). We have recently come to appreciate populations of Tregs residing within non‐lymphoid tissues, collectively termed ‘tissue Tregs’. Here, we provide a comparison of how Tregs residing in tissue [such as visceral adipose tissue (VAT), intestine, skin or muscles] are specialized for orchestrating tissue homeostasis as regulators of both immune and non‐immune cells.
General definition and heterogeneity of Tregs
Besides CD25, Tregs may express other activation markers, such as co‐stimulatory/co‐inhibitory molecules [CTLA‐4, inducible T‐cell costimulator (ICOS), CD27, LAG‐3, LAP, CD69, PD‐1), tumour necrosis factor receptor superfamily (TNFRSF) members (OX40, GITR), cell adhesion‐related markers (CD49b, CD62L) and migratory receptors guiding to peripheral destinations (CD103, CCR4, CCR5, CCR6, CCR7). 10 In particular, Foxp3 is exclusively expressed in CD4+ CD25+ murine Treg populations, but at low levels in CD4+ CD25− effector T cells (Teffs) and largely absent in CD8+ T cells. 7 , 11 Importantly, Foxp3 is indispensable for Treg lineage development and immunosuppressive functions. 7 , 11 , 12 , 13 Hence, the general consensus is that the murine Treg lineage is defined as CD4+ CD25highFoxp3+. Yet, in human peripheral blood, not all Foxp3‐expressing cells correlate with high levels of CD25 expression (reviewed in ref. 10), nor immunosuppressive function. 14 The IL‐7Rα chain, CD127, is inversely correlated with Foxp3 expression, and CD4+ CD127low T cells in humans display similar suppressive capacity to CD4+ CD25high T cells in vitro. 15 Hence, human Tregs are more accurately defined as CD4+ CD25high CD127low/− Foxp3+.
While CD4+ CD25high CD127low/− Foxp3+ is the phenotypic backbone of Tregs, multiple flow cytometry‐based and RNA sequencing studies indicate that both mouse and human Tregs are highly heterogeneous (reviewed in ref. 16). One subdivision is based on Treg origin. CD25high CD4+ thymocytes initially undergo a positive selection process and develop into Treg precursors. This is triggered through T‐cell receptor (TCR) engagement with high‐affinity tissue‐specific self‐peptides on major histocompatibility complex type II (MHCII) presented by antigen‐presenting cells and medullary thymic epithelial cells. Stimulated by the cytokines IL‐2 or IL‐15, and in combination with transforming growth factor‐β (TGF‐β) signalling, Treg precursors differentiate into fully committed thymic Tregs (tTregs). 17 , 18 Peripherally derived Tregs (pTregs) can be generated from naive Foxp3− CD4+ T cells in secondary lymphoid organs and peripheral tissues, in response to various cytokines upon antigen exposure, including TGF‐β. 19 Furthermore, Tregs may also be subdivided according to their differentiation state (CD45RA+ naive, nTregs, or CD45RA− activated, aTregs), and their degree of activation (CD44low CD62Lhigh quiescent central, cTregs, or CD44highCD62Llow effector, eTregs) and by their expression of core T helper (Th) lineage‐defining transcription factors (T‐bet for Th1, Gata3 for Th2, and RoRγt for Th17) (reviewed in refs 16, 20).
Phenotypic dynamics of tissue Tregs
Unsurprisingly, murine Tregs share the core phenotypic backbone of their circulatory and lymphoid counterparts, regardless of their tissue of residence. For example, 63% of the VAT Treg transcriptome overlaps with that of LN Tregs, including the overexpression of hallmark activation markers, including Cd25, Gitr, Ctla4, Ox40 and Klrg1. 21 Indeed, genome‐wide chromatin accessibility profiling (ATAC‐seq) has indicated that VAT, muscle and colon Tregs share 79% of their open chromatin regions with their splenic counterparts, whereas only 3% are unique to these ‘pan‐tissue’ Tregs. 22 Re‐analysis of existing microarray data revealed that of ~2000 tissue Treg‐associated genes, 9% are shared among VAT, muscle and colon, including up‐regulation of the IL‐33 receptor (Ilrl1, also known as ST2) and amphiregulin (Areg) genes. 22 Another example is the peroxisome proliferator‐activated receptor γ gene (Pparγ), expressed in Tregs residing in VAT, muscle and neonatal liver. 22 , 23 Another single‐cell RNA sequence profiling also demonstrated the resemblance between ear skin Tregs and a subset of colon Tregs, sharing expression of Areg, Ilrl1 and Il10, but also Gata3, Rora and Tnfrsf4. 24 Additionally, inhibitors of DNA binding 3 (Id3) is also consistently absent among Tregs in skin, fat, colon and lung, but is abundant in circulating or lymphoid Tregs. 25
Human Tregs share <10% of overall mRNA expression with their mouse counterparts, such as Rora and Tnfrsf18 in skin, and Ikzf3 in colon. 24 Contrarily, multiple mRNA signatures, such as serine/threonine kinase (PIM1/2), cAMP‐related genes (PDE3B/4B/4D) or G‐protein signalling regulator (RGS1/2), are opposingly expressed between human and mouse tissue Tregs. 24 Whether this directly reflects species differences in function remains an open question. It may be reasonable to characterize tissue Tregs based on these commonly shared ‘pan‐tissue’ phenotypes, at least in mouse. Despite their phenotypic resemblance, certain genes remain uniquely expressed in each murine tissue Treg, which we will explore below.
Conventional anti‐inflammatory functions of Tregs
As its name suggests, Tregs regulate and suppress a variety of immune cell types (such as macrophages, dendritic cells, CD4+ Teffs and CD8+ T cells) (reviewed in ref. 26). Three general modes of suppression have been proposed: (i) cell‐to‐cell contact via Treg–cytotoxic T‐lymphocyte antigen 4 (CTLA‐4) regulation of CD28 co‐stimulation, an essential secondary signal for T‐cell activation. Mechanistically, CTLA‐4 physically removes and down‐regulates the ligands CD80/CD86 on target cells, through a process termed trans‐endocytosis; 27 (ii) Treg production of cytokines (IL‐10 and TGF‐β); and (iii) metabolic alteration, whereby CD25hi Tregs consume the T‐cell growth factor IL‐2, and restrict expansion of other T cells (reviewed in ref. 28).
The initial response towards tissue damage or infection is an acute inflammation. Upon removal of pathogens or damaged cells, Tregs dampen inflammation by secreting anti‐inflammatory cytokines, and regulating the recruitment/activation of other immune cells (reviewed in ref. 29). For example, Tregs directly suppress neutrophil recruitment via IL‐10, but also indirectly induce neutrophil apoptosis via TGF‐β. Hence, TGF‐β signalling not only functions as an inducer of the Treg lineage, but also as a facilitator of Treg‐mediated immunosuppression. Additionally, Tregs also promote macrophage polarization from a more inflammatory to anti‐inflammatory phenotype. Perhaps the best‐established suppressive function of Tregs is the regulation of pro‐inflammatory cytokines interferon‐γ (IFN‐γ) and tumour necrosis factor‐α (TNF‐α), as well as the abundance and activation of CD4+ and CD8+ T cells.
The major criterion for successful wound healing is the initiation of tissue remodelling to restore tissue integrity (reviewed in ref. 30). However, through inability to clear pathogens and/or dysregulation of the inflammatory process itself, tissues may transition to a chronic inflammatory and fibrosis‐associated phase. Fibrosis is broadly characterized by excessive extracellular matrix deposition, and eventually, tissue scarring. Although type 2 cytokines and macrophage‐derived TGF‐β promote fibrosis, Treg‐derived TGF‐β and IL‐10 play an opposing role (reviewed in ref. 31). In particular, the link between inflammation and fibrosis is best exemplified in the neutrophil‐ and macrophage‐deficient PU.1 null mice. This mutant lacks the major components of an inflammatory response. Yet, cutaneous wound closure kinetics are equivalent to wild‐type animals, with minimal tissue scarring. 32 It was later discovered that the knockdown of osteopontin (an inflammation‐dependent gene) improves collagen assembly, limits neutrophil, mast cell and macrophage recruitment, increases neovascularization, minimizes fibrosis and reduces scarring. 33 Taken together, the suppression of inflammation may minimize tissue fibrosis. It is, therefore, logical that tissue Tregs also play an essential role in suppressing fibrosis and promoting tissue repair, which we will explore thoroughly in this review. Unless otherwise stated, the majority of the data discussed below are derived from murine studies. We review our current knowledge on the phenotypes, origin and functions of four well characterized non‐lymphoid Treg populations, which reside in VAT, intestine, skin and skeletal muscle.
Visceral adipose tissue Tregs
Visceral adipose tissue refers to the white adipocytes localizing around various organs, which function primarily as reservoirs of energy storage (reviewed in ref. 34). In the steady‐state, murine VAT Tregs account for ~50% of CD4+ T cells. 21 Remarkably, VAT Tregs uniquely express peroxisome proliferator‐activated receptor γ (PPARγ), 35 a transcription factor usually restricted to adipocytes to drive adipose tissue development. Conditional knockout of PPARγ in Tregs results in a ~70% reduction of VAT Treg abundance, and the down‐regulation of the VAT Treg‐associated transcripts Ccr2, Gata3, Klrg1 and Cd69 (an early activation and tissue residency marker). 35 , 36 These findings indicate that PPARγ can act as a specific inducer and regulator of VAT Treg identity.
Thymic origin of VAT Tregs
Several lines of evidence indicate that VAT Tregs are likely of thymic origin. First, >90% of VAT Tregs express high levels of the thymic‐associated markers Helios and Nrp‐1, at comparable levels to splenic and LN Tregs. 37 Additionally, when pooled Teffs from 8‐week‐old CD45.1+ Foxp3iGFP reporter mice are transferred into 20‐week‐old congenic CD45.2+ Foxp3iGFP recipients, donor‐derived VAT Tregs are absent, suggesting that Teffs are unlikely to be the predominant source. 37
To better understand the origin of tissue Tregs, TCR sequencing analysis may be performed. In brief, the majority of T cells, including Tregs, express highly diverse TCRs. 38 Each TCR consists of a combination of α and β chains (αβ TCR), with each chain containing three complementary determining regions (CDR1–3). 39 The CDR3 region of a TCR is often in direct contact with the antigen, and so plays a defining role in the interaction with the peptide–MHC complex. Hence, CDR3 diversity is often reflective of T‐cell specificity, and in turn their clonality. If two T cells express identical CDR3 sequences, then they are likely derived from a clonally expanded T cell. Shared TCR sequences between Tregs and Teffs suggest the two populations may recognize the same antigens. Under the assumption that TCR repertoire remains unaltered during the transition from naive Teffs to pTregs, TCR similarity indirectly indicates whether a Treg population originates from Teffs. In VAT, the Treg TCR repertoire (specifically CDR3α and CDR3β sequences) resembles that of LN Tregs, but is distinct from VAT or LN Teffs. 37 The lack of CDR3 overlap between VAT Tregs and VAT Teffs indicates distinct antigen specificities. Thus, VAT Tregs are unlikely to originate from Teffs, suggesting minimal pTreg input. 37
Interestingly, thymectomy (surgical removal of the thymus) of C57BL/6 mice during neonatal life (postnatal week 3–4) or adulthood (postnatal week 13·5) does not alter VAT Treg accumulation, 37 suggesting that the VAT Tregs do not require constant replenishment from the thymus, at least beyond 3 weeks of life. A similar conclusion was drawn from Foxp3DTR mice, which harbour a diphtheria toxin receptor (DTR) transgene ahead of the Foxp3 locus. 40 The administration of diphtheria toxin permits systemic and specific deletion of all Foxp3‐expressing cells. Systemic Treg depletion at 8 or 13 weeks of age results in a failure of reconstructing the VAT Treg compartment, without influencing splenic Tregs. 37 Yet, Treg ablation before 4 weeks yields no change in Tregs within VAT or spleen. 37 Hence, VAT Tregs are probably seeded during the first 3 weeks of life. Once seeded, VAT Tregs may undergo clonal expansion, as suggested by the highly repetitive TCR sequencing results, and probably replenishes and maintains the VAT Treg compartment. 37
VAT Tregs suppress tissue inflammation and maintain energy homeostasis
The histopathology of obesity is defined by chronic low‐grade adipose tissue inflammation. Obesity is closely associated with the inability to respond to insulin, termed insulin resistance. In its severe form, obesity can further manifest to the pathology of type 2 diabetes, including elevated glucose and insulin in fasting individuals, lower insulin‐receptor activity, delayed blood glucose clearance (a poor glucose‐tolerance response), and increased glucose–insulin by‐products, as measured by the homeostatic model assessment of insulin resistance (reviewed in ref. 41, 42).
Obesity is associated with macrophage and CD8+ T‐cell accumulation in VAT, the production of pro‐inflammatory cytokines (TNF‐α and IL‐6), and a significant reduction of VAT Tregs. 21 , 35 , 43 , 44 , 45 A meta‐analysis of 91 human clinical studies has revealed that type 2 diabetes is associated with increased IL‐6 and TNF‐α, and reduced Treg abundance in human peripheral blood. 46 Indeed, murine VAT Tregs can suppress adipose tissue inflammation and potentially reduce diabetic pathology. Based on in vitro suppression assays, isolated murine VAT Tregs are immunosuppressive, and are not functionally different from splenic Tregs. 21 In healthy lean mice, VAT Tregs also express high levels of the anti‐inflammatory cytokine IL‐10, 21 , 35 , 47 further supporting their role as immunoregulators. Indeed, a number of in vivo studies describe VAT Treg suppression of other resident immune cells. First, Treg ablation leads to the induction of inflammatory mediators TNF‐α, IL‐6, RANTES and serum amyloid A‐3 within VAT. 21 Similarly, anti‐CD25‐mediated Treg depletion in diabetic leptin‐deficient db/db mice elevates the pro‐inflammatory cytokine transcripts, Ifng, Il6 and Tnfa, and down‐regulates the VAT Treg signature markers Gata3, Ccr2, Klrg1 and Cd69. 48 The loss of Tregs also results in pro‐inflammatory macrophage and monocyte accumulation, without affecting anti‐inflammatory monocytes, CD8+ T cells or B cells. 35 In contrast, oral administration of anti‐CD3 antibody and β‐glucosylceramide in ob/ob mice augments adipose‐resident Tregs, while reducing CD11b+ F4/80+ macrophage abundance and Tnfα transcript in adipose tissue, subsequently dampening inflammation. 49 These results provide clear evidence that VAT Tregs are essential to suppress adipose tissue inflammation.
Systemic Treg depletion in 10‐week‐old male Foxp3DTR mice also leads to elevated fasting insulin levels, accompanied by a reduction in epididymal fat and liver insulin‐receptor activity. 21 Similarly, Treg depletion in 6‐week‐old diabetic leptin‐deficient db/db mice also increases fasting glucose levels, lowers insulin sensitivity and elevates homeostatic model assessment of insulin resistance, relative to untreated db/db mice. 48 In contrast, Treg augmentation through IL‐2–IL‐2 antibody complexes or adoptive transfer of lymphoid Tregs improves glucose tolerance, 21 , 48 suggesting a protective role for VAT Tregs in insulin resistance.
The administration of pioglitazone, an insulin‐sensitizing PPARγ agonist, significantly increases VAT Treg numbers in high‐fat diet‐induced obese mice, and also lowers circulating glucose levels. 35 Indeed, metabolic improvement of pioglitazone is diminished in obese Foxp3cre PPARγ fl/fl mice, 35 indicating that PPARγ is a critical regulator. It remains to be confirmed whether pioglitazone treatment alone in obese mice is enough to restore VAT Treg abundance and aggravate obesity‐induced insulin resistance to a homeostatic level. This view has been challenged by Bapat et al., 36 where specific loss of VAT Tregs in Foxp3cre PPARγ fl/fl mice does not impact glucose metabolism in lean, young (12‐week‐old) or high‐fat diet‐induced obese (24‐week‐old) mice. Instead, specific loss of VAT Tregs leads to improved fasting glucose and insulin levels in aged (36‐week‐old) mice, 36 suggesting an opposing role for VAT Tregs in age‐associated insulin resistance. Nonetheless, there is an indisputable correlation between VAT Treg abundance and insulin sensitivity, and their essential role in maintaining metabolic homeostasis.
Despite preferential amphiregulin (Areg) expression in VAT Tregs, 21 , 35 , 43 , 47 this pathway remains to be explored in detail. Areg, an epidermal growth factor‐like molecule, is classified as a type‐2 associated cytokine, and has been associated with suppression of local inflammation and promotion of tissue repair (reviewed in ref. 50). The overexpression of Areg in obese db/db mice results in reduced perirenal and epididymal fat, and increased glucose and lipid metabolism‐related genes (including Pparγ coactivator 1α and Tnfa), implying that Areg is crucial for adipose tissue development. 51 Whether this directly translates to the function(s) of VAT Treg‐derived Areg remains an open question, as Areg is also expressed by other immune cells, including mast cells, basophils, eosinophils and type 2 innate lymphoid cells. 50
Intestinal Tregs
Within the small and large intestines, the majority of immune cells reside within the mucosa, the innermost layer of the intestinal wall (reviewed in ref. 52). The mucosal wall consists of an epithelial layer, lamina propria and a thin underlying muscle layer (known as muscularis mucosa). 53 Both small intestinal and colonic Tregs localize within the lamina propria of the intestinal mucosal wall, 54 , 55 contributing to ~35% and ~25% of residing CD4+ T cells, respectively. 56 However, Treg are most abundant in the colon, and lowest in the duodenum of the small intestine. 57
Similar to VAT, intestinal Tregs also adapt to their tissue environment. Of which, Rorc, a key Th17 lineage regulator encoding retinoic acid receptor‐related orphan receptor γt (RoRγt), is preferentially expressed in colonic Tregs, but not in other non‐lymphoid tissue or splenic Tregs. 58 Of total colonic Tregs, around 40% are RORγt+ Helios− and 30% are GATA3+, relative to >10% of Tregs in Peyer’s patches (intestinal lymphoid follicles) or in mesenteric LNs. 59 , 60 While ~30% of small intestinal Tregs express GATA3, only 15% are RORγt+ Helios−. 59 , 60 Notably, in both colonic and small intestinal Tregs, GATA3 and RORγt expression are mutually exclusive. 59 , 60 Overall, intestinal Tregs can be separated into three phenotypically distinct populations: GATA3+ Helios+, RORγt+ Helios− and RORγt− Helios−.
Mixed origins of intestinal Tregs
Given their phenotypic heterogeneity, intestinal Tregs appear to be of mixed origin. In specific pathogen‐free (SPF) mice, 70% of colonic Tregs are negative for Helios, 56 and 45% express low levels of Nrp‐1, in contrast to 11–19% of splenic or lymphoid Tregs, 61 suggesting a pTreg origin. Contrary to the stable high expression in splenic Tregs, the percentage of Helioshi or Nrp‐1hi colonic Tregs declines from 70% to 80% at 1 week of age to ~30% at 8–10 weeks, implying an increasing contribution of pTregs. 62 Although it is largely accepted that pTregs contribute to the establishment of intestinal Tregs, the majority of CDR3 TCR sequences of both colonic and small intestinal Tregs closely resemble that of thymic and peripheral LN Tregs. 63 This repertoire was largely distinct from intestinal Teffs in transgenic mice with limited TCR repertoires, 63 suggesting a tTreg origin. Minimal tTreg expansion was also observed when transferring Foxp3 gfp Rag1 −/− thymocytes retrovirally transfected with colonic Treg TCRs, into Rag1 −/− recipients (that lack T and B cells). 64 Furthermore, transferring LN Teffs to Treg‐depleted mice, Teffs can acquire a RORγt+ Helios− and Helios+ colonic Treg phenotype, depending on the level of nerve growth factor IB (otherwise known as Nur77). 65 Nur77 functions as an important regulator of cell survival, inflammation, but also as an early reporter for antigen‐specific signalling. 66 Taken together, both tTregs and pTregs contribute to the establishment of colonic Tregs.
Colonic Tregs are highly responsive towards microbiome colonization. The microbiome refers to the microorganisms (bacteria, archaea, protozoa, fungi or viruses) sharing a commensal, symbiotic or pathogenic relationship with their host of residence. Increasing evidence illustrates their vital role in regulating host metabolism, drug utilization, maintaining homeostasis, and ultimately host health (reviewed in ref. 67). In germ‐free C57BL/6 mice, only 10% of colonic CD4+ T cells are Tregs, in contrast to ~35% in SPF C57BL/6. 56 Treatment with Gram‐positive antibiotics (vancomycin) in SPF mice leads to an ~10% reduction in colonic Tregs, whereas no change was observed in SPF mice treated with Gram‐negative antibiotics (polymyxin B), indicating that Gram‐positive bacteria may promote colonic Treg establishment. 56 Using in vitro co‐cultivation of colonic Tregs and dendritic cells primed with autoclaved colonic contents, colonic Treg TCRs appear to engage microbiome‐derived antigens. 64 Unsurprisingly, colonic Treg activation during colonization is dependent on Toll‐like receptor signalling. Co‐deficiency of the Toll‐like receptor adaptor molecules (MyD88 and Ticam‐1) in germ‐free mice ablates colonic Treg accumulation during microbial colonization. 68 Conversely, Tregs residing in small intestinal lamina propria are unaffected by microbiome diversity, instead their induction appears to be largely driven by dietary antigens. 69 Hence, intestinal Treg establishment is probably facilitated by the environmental antigens present within organs, with microbiota as a key component.
Colonic Tregs suppress ongoing intestinal inflammation
Compromising colonic Treg function results in unresolved and ongoing inflammation, but their contribution to short‐term homeostatic maintenance appears less important. Deletion of Tregs for a period of 10 days in Foxp3DTR mice does not impact lymphocytic infiltration or proliferation in the colon. 70 Yet, IL‐10 receptor deficiency in Tregs (Foxp3cre Il10rafl/fl) manifests in severe colitis in aged mice (18–20 weeks), but not younger mice (8–10 weeks). 71 Similarly, patients with single point mutations in CTLA4 are associated with extensive gastrointestinal inflammation, indicating a major functional role for Tregs in controlling long‐term colon pathology. 72 , 73
Transforming growth factor‐β signalling is a key component in regulating T‐cell proliferation and activation. For example, TGFβ 1 deficiency in CD4+ T cells or in OX40+ T cells leads to severe colitis, accompanied by elevated IFN‐γ. 74 , 75 Yet, during steady‐state, deficiency of TGF‐β 1 or its receptor (TGF‐βR) in Tregs, does not impact IFN‐γ or IL‐17 production in small intestinal or colonic intraepithelial lymphocytes. 75 , 76 Similarly, colonic inflammation is absent in mice harbouring Treg conditional deletion of the TGF‐β activator (integrin α v β 8, Itgb8). 77 Hence, Treg‐derived TGF‐β signalling contributes minimally to the regulation of steady‐state colonic homeostasis.
Colitis can be artificially induced by transferring naive CD45RBhigh CD4+ T cells into immunodeficient Rag1−/− mice, characterized by colon thickening, and ultimately weight loss. 78 Co‐transfer of CD4+ CD25+ Tregs is protective against CD4+ Teff cell‐mediated inflammation. 78 However, co‐transferring TGF‐β 1‐deficient splenic Tregs induces an IFN‐γ‐mediated response and failure to resolve colitis. 79 Similarly, co‐transferring TGF‐βRI‐deficient Tregs leads to an increase in monocyte and neutrophil infiltration in the colon. 76 Likewise, a more severe colitis is observed when transferring Itgb8‐negative Tregs into inflamed Rag2−/− recipients. This exacerbated pathology was attributed to increased neutrophils, monocyte/macrophages, IFN‐γ‐expressing and IL‐17‐expressing CD4+ T cells within the colon. 77 Importantly, TGF‐βRI‐deficient Tregs fail to accumulate or be retained within Rag1−/− colon, explaining the uncontrolled inflammatory response. 76 In dextran sulphate sodium‐mediated colitis, the lack of Itgb8+ Tregs also exacerbates pathology, including the elevation of CD4+ T cells without altering Treg migration, activation or stability. 77 Taken together, colonic Tregs regulate ongoing inflammation via TGF‐β signalling, and may contribute minimally during the steady state.
Distinct intestinal Treg subpopulations appear to possess specialized functional roles in the suppression of ongoing inflammation. GATA3+ Helios+ Tregs function as major immunosuppressors during intestinal inflammation. Mice harbouring Gata3‐deficient Tregs (Foxp3IRES‐Cre Gata3fl/fl) do not display an inflammatory phenotype, nor changes in Treg abundance. 60 Yet, upon injury, colonic GATA3+ Helios+ Tregs express increased levels of IL‐10 and TGF‐β in an IL‐33‐dependent manner. 80 Co‐transfer of Gata3‐negative Tregs from OX40cre Gata3fl/fl, or Foxp3EGFP‐cre Gata‐3fl/fl mice, with naive T cells into Rag1−/− recipients induces severe colitis and weight loss, 60 , 81 suggesting that Tregs use Gata3 to limit tissue inflammation. The role of Gata3 was further confirmed in a competitive bone marrow chimera model, where CD45.2+ Gata3‐deficient and CD45.1+ CD45.2+ wild‐type cells were transferred into lethally irradiated CD45.1+ recipients. 60 While Teff accumulation is unaffected, Gata3 deficiency hinders Treg infiltration to inflamed intestine. 60 This was also true for Tregs within the spleen and mesenteric LNs. 60 Importantly, intestinal Gata3‐deficient Tregs express significantly higher levels of RORγt and IL‐17A relative to wild‐type cells, 60 indicating that Gata3 may also function as a restrictor of Th17 immunity. However, Gata3‐negative splenic Tregs also express higher Rorc and IL‐17a transcript. Although Gata3 is critical in facilitating intestinal Treg function, its regulatory role in suppressing Th17 immunity is unlikely to be intestinal Treg‐specific.
Ablation of RORγt+ colonic Tregs results in an up‐regulation of Teff‐derived IL‐17α and IFN‐γ, and the development of severe colitis. 59 RORγt+ Tregs also induce tolerance towards the pathogenic microbe Helicobacter hepaticus, via the cMaf transcription factor, which promotes IL‐10 secretion and restricts Th17 polarization. 82 However, it remains controversial as to what level human RORγt+ Tregs control intestinal inflammation. On the one hand, the abundance of human RORγt+ Tregs is comparable between healthy individuals and patients with inflammatory bowel disease. 59 Yet, more human RORγt+ Tregs were detected in dysplastic than non‐dysplastic histology from individuals with ulcerative colitis. 83
Intestinal Tregs facilitate tissue repair
Amphiregulin is also highly expressed in intestinal Tregs. 58 However, mutants lacking Areg‐expressing Tregs show no gross phenotypic pathology, nor changes in the frequency of colon‐resident Th1, Th2 or Th17 cells. 84 Although Treg‐derived Areg appears dispensable, Areg derived from other cell types may impact Treg control of colonic inflammation. Treg‐mediated suppression is less effective in Areg‐deficient Rag1−/− (Areg−/− Rag1−/−) mice relative to Rag1−/− controls. 85 In fact, sorted CD25+ Tregs from CD4cre Egfrfl/fl mice, which lack the Areg receptor epidermal growth factor receptor (EGFR), failed to suppress disease development, 85 indicating the importance of Areg in enhancing Treg immunosuppression. Additionally, Areg−/− mutants have significantly impaired intestinal epithelial regeneration after radiation exposure. 86 Similarly, tissue Tregs have also been associated with the maintenance of intestinal stem cells. 87 Whether Treg‐derived Areg is required to facilitate tissue repair remains currently unknown.
Skin Tregs
Both human and mouse skin are composed of epidermis and dermis, with hair follicles (HFs) interspersed between their junctions (reviewed in ref. 88). The majority of murine Tregs localize near HFs. 89 Similarly, the abundance of human Tregs is closely associated with hair density, with more skin Tregs in areas of high hair density (such as scalp or face). 90 In the steady state, skin Tregs contribute to an average of 50% of adult skin‐resident CD4+ T cells in mice, 91 and 20% in humans. 90 Of murine neonatal CD4+ cells, >80% are murine Tregs that express high levels of CTLA4 and ICOS, but their numbers are reduced to 50% in adulthood. 91 Murine skin Tregs are transcriptomically similar to colonic Tregs, but they uniquely express genes such as Dgat2 (related to lipid synthesis in skin). 24 Bulk RNA sequencing of dorsal skin and LN Tregs revealed preferential expression of Jagged1 (Jag1), a Notch signalling ligand, in skin Tregs. 92 In humans, the mitochondrial protein arginase 2 (Arg2) is also preferentially expressed in skin Tregs, relative to skin Teffs or circulating Tregs. 93 Using CRISPR‐Cas9‐mediated ARG2 deletion, the authors concluded that Tregs use this pathway to maintain a tissue‐specific signature. 93
Origin and residency of skin Tregs
Skin Tregs accumulate during a specific neonatal period (postnatal days 6–13) and are thought to originate from the thymus. 91 During this period, blockade of thymic or LN T‐cell egress, by the administration of sphingosine‐1‐phosphate receptor antagonist FTY720, results in ~10‐fold reduction in skin Treg numbers. This is also associated with an increase in thymic Tregs, 91 indicating that skin Tregs are potentially thymus‐derived. Until now, a comprehensive CDR3 sequencing comparing skin Tregs, skin Teffs and their counterparts in spleen or lymph nodes has not been performed. Hence, skin Treg origin remains inconclusive.
Further evidence comes from Kaede transgenic mice that harbour a photoactivatable fluorescence protein. The Kaede protein undergoes irreversible conversion from green to red fluorescence upon violet light exposure, allowing non‐invasive in vivo tracing of cell migration. 94 In inflamed ear skin, ~30% of Tregs have permanent residency, whereas 50% are migratory. 95 These migratory Tregs express higher levels of CTLA4 and Nrp1, but lower levels of CD25 and CD39 than those remaining in skin. 95 Given the increased expression of Nrp1, these migratory Tregs are probably of thymic origin. Whether this finding can extend our understanding of dorsal skin Treg migration during homeostasis remain unknown. In addition, Treg expression of FuT7, an enzyme facilitating E‐selectin and P‐selectin binding, is required for optimal Treg trafficking to both inflamed and non‐inflamed skin, 96 whereas their retention appears to require IL‐7, but not IL‐2. 97
Similar to the colon, the skin harbours a large quantity and diversity of microbial species. In germ‐free mice, there is a 20% reduction in neonatal skin Tregs, 98 but no differences are observed in adults. 99 Conversely, other T‐cell populations (such as CD4+ Foxp3− conventional T cells, CD8+ T cells, dermal γδ or dermal epithelial T cells) remain unaffected in neonatal skin, 98 indicating that the microbiome preferentially orchestrates skin Treg accumulation. The microbiome drives the production of hair follicle‐derived CCL20, the ligand for the skin‐homing receptor CCR6, and subsequently facilitates skin Treg migration into neonatal skin. 98 Successful accumulation of skin Tregs to neonatal skin, relative to adulthood, appears critical for the establishment of microbiome tolerance. 91 However, only specific strains can induce skin Treg tolerance. For example, neonatal colonization with the commensal Staphylococcus epidermidis, but not with the pathobiont Staphylococcus aureus, limits inflammation upon repeated encounter during adulthood. 100 The discrimination between commensals and pathobionts is attributed to the S. aureus‐derived α‐toxin and its regulation of IL‐1β production in skin. 100 Taken together, the timing of tissue Treg establishment and the nature of the microbiome plays a major role in instructing Treg‐mediated tolerance in skin.
Skin Tregs facilitate wound healing and control fibroblast activation
Similar to the colon, short‐term skin Treg deletion does not appear to impact immune homeostasis. Overt signs of cutaneous inflammation are absent during periods of acute Treg depletion in Foxp3DTR mice. These include epidermal hyperplasia, the abundance of other skin‐resident immune cells, and the production of the pro‐inflammatory cytokines IFN‐γ, TNF‐α and IL‐17. 92 , 101 However, Treg depletion up‐regulates the Th2 cytokines IL‐13 and IL‐4. 101 At the single‐cell level, Gata3 transcript is most abundant in murine skin Tregs, relative to other Th lineage transcription factors Rorc and Tbx21, suggesting that skin Tregs may be skewed towards Th2 regulation. 101 Conditional deletion of GATA3 in Tregs (Foxp3 YFP‐Cre Gata3 fl/fl) results in Type 2 associated skin inflammation, accompanied by a reduction in Treg abundance and activation. Treg‐specific deletion of GATA3 also manifests in polymorphonuclear cell infiltrate, as well as IL‐5 and IL‐13 producing CD8+ T cells in skin. 101 , 102 These findings imply that skin Tregs (or at least a subset thereof) maintain skin homeostasis via regulation of Th2 immunity.
In full‐thickness wounding of mouse dorsal skin, Treg numbers increase approximately 20‐fold, which is concurrent with increased expression of activation markers (Ctla4, Icos and Cd25) at days 3 and 7 post‐injury. 103 Consequentially, Treg ablation delays cutaneous wound closure, and is associated with the accumulation of IFN‐γ‐producing T cells and CD11b+ Ly‐6Chigh pro‐inflammatory macrophage in wounded skin, 103 demonstrating that Tregs are indispensable for resolving skin inflammation. Delayed wound closure was observed upon Treg‐specific deletion of the Areg receptor, EGFR, indicating a requirement for locally available Areg to sustain the kinetics of tissue repair. 103 Similarly, skin Tregs also facilitate repair of the injured epithelial barrier by suppression of IL‐17A, CXCL5, and subsequent neutrophil accumulation. 104 In turn, Tregs promote hair follicle stem cell (HFSC) activation, driving epidermal‐differentiation genes (such as filaggrin, keratin1) and restoration of the epidermal barrier. 104 Overall, skin Tregs promote cutaneous repair by facilitating early innate immunity.
Skin Tregs also play a major role in fibrotic disease. Using αSMA‐RFP/Foxp3DTR and female heterozygous Foxp3DTR mice, both acute (5 days) and chronic (4 weeks) steady‐state Treg depletion resulted in the accumulation of profibrogenic myofibroblasts, and up‐regulation of profibrotic genes (Col3a1, αSMA and Hsp47). 101 Chronic depletion also down‐regulates IL‐10 production and anti‐fibrosis genes (Mmp2a, Mmp8 and Bmp7), with increased dermal collagen density and dermal thickness, culminating in a fibrotic appearing pathology. 101 Severe fibrotic pathology was observed upon partial depletion of Tregs in αSMA‐RFP/ Foxp3DTR+/− treated with the dermal fibrosis‐inducing agent, bleomycin. 101 Hence, skin Tregs appear indispensable for suppressing or reducing steady‐state and injury‐induced fibrosis. These findings were largely recapitulated in bleomycin‐treated Foxp3creERT2 Gata3fl/fl mice, revealing Gata3+ Tregs as a core subset contributing to the control of Th2‐mediated skin fibrosis. 101
Interestingly, during both the acute and chronic Treg depletion models, TGF‐β transcripts (such asTgfbr1, Tgfb3 and Smad2) in dorsal skin are up‐regulated. 101 It is well‐established that TGF‐β signalling plays a critical role in maintaining the cutaneous barrier, including re‐epithelialization and the retention of memory T cells or Langerhans cells in skin (reviewed in ref. 105). In delayed‐type II hypersensitivity, integrin α v β 8 (Itgb8) in Foxp3+ Tregs is required to control ear skin inflammation and IFN‐γ‐producing CD4+ and CD8+ T cells. 77 However, during the steady state, cutaneous pathology was absent in mice lacking TGF‐βRI in Tregs. 76 Taken together, similar to intestinal Tregs, TGF‐β signalling also plays a direct role in skin Treg‐mediated suppression of ongoing inflammation. It remains unclear whether Treg‐derived TGF‐β impacts other immune populations, such as memory T cells and dendritic cells in skin.
Skin Tregs facilitate hair regeneration
In murine skin, HFs cycle between perpetual bouts of growth arrest (telogen) and activation (anagen) to form new hair shafts. This process is mediated by epithelial stem cells within the HF bulge region (HFSCs). 106 Depletion of Tregs during telogen significantly impairs anagen induction as evidenced by HFSC proliferation and differentiation defects, consequently impacting hair regeneration. 92 Yet, co‐depletion of Tregs with other immune effector cells, or neutralization of the IFN‐γ pathway, is unable to rescue HFSC activation. 92 This in turn suggests that skin Tregs may regulate HF regeneration outside their conventional role in suppressing skin inflammation. It was later revealed that the Notch ligand Jagged1 is preferentially expressed in skin Tregs, relative to skin‐draining LN Tregs. 92 Conditional deletion of Jag1 in Tregs, using Foxp3YFP‐cre Jag1fl/fl mutants, significantly impairs bulge HFSC proliferation, HFSC differentiation transcripts and, subsequently, anagen induction. 92 However, the mechanism(s) underlying the induction and maintenance of Jag1+ Tregs, and whether skin Tregs use the Notch signalling pathway to directly mediate HFSC function, remains unknown.
Skeletal muscle Tregs
Skeletal muscle is composed of multiple myofibres and connective tissues. The ability to activate quiescent satellite cells (muscle stem cells) on demand contributes to the remarkable capacity of skeletal muscle regeneration (reviewed in ref. 107). Unlike the previously discussed tissues, steady‐state skeletal muscle harbours a minimal quantity of Tregs, accounting for only 10% of CD4+ T cells, less than their counterparts in spleen. 58 , 108 Instead, Tregs are analysed in injured or inflamed muscle. Cardiotoxin‐induced muscle injury triggers rapid Treg accumulation, accounting for 40%–50% of CD4+ T cells at day 4 post‐injury. 58 , 108 Similarly, in both humans and experimental mouse models of Duchenne muscular dystrophy, muscle Treg numbers are significantly elevated. 109 Unsurprisingly, this is not exclusive to muscle Tregs, as the number of muscle Teffs share a similar pattern. 58 , 108 , 109
Unclear origin of injured muscle Tregs
The origin of Tregs in injured muscle appears complex to interpret. Although the number of muscle Tregs declines drastically after 1 week post‐injury, it remains eightfold higher than uninjured muscle. 58 , 108 There are potentially two possible origins of elevated Treg populations in injured muscle: (i) they represent infiltrating Tregs, which originate from other lymphatic organs, or (ii) they are derived from the expansion of a rare muscle‐resident Treg population. Upon successful tissue repair, Tregs migrate out of or do not survive in muscle. Experimental evidence for and against these ideas so far is conflicting. Administration of the T‐cell egress inhibitor FTY720, before cardiotoxin‐induced muscle injury, lowers Treg numbers, but not their percentage among CD4+ cells, nor total CD45+ cell abundance. 108 This indicates that the accumulation of muscle Tregs are likely dependent on T‐cell recruitment from other lymphatic organs. Yet, through in vivo tracing in Kaede mice, only a small fraction of Tregs in injured muscle traffic from cervical lymph nodes. 108 Although this does not exclude the possibility that muscle Tregs may be recruited from other lymphatic organs, at least a portion of muscle Tregs infiltrate upon injury. Furthermore, 84% of post‐injury muscle Tregs are highly proliferative, expressing higher levels of Ki67 and EdU relative to splenic Tregs or muscle/splenic Teffs. 58 , 109 Based on CDR3 sequencing of TCR‐α and TCR‐β chains, 20%–40% of muscle Tregs share the same TCR sequences, suggesting clonal expansion. 58 Importantly, the TCR sequences of muscle Tregs are distinct from muscle Teffs, 58 demonstrating muscle Tregs are unlikely to originate from pTregs. In a mutant strain with fixed TCR‐α and TCR‐β, TCR specificity drives muscle Treg phenotypic adaptation and accumulation in cardiotoxin‐injured muscle. 110 Hence, muscle Treg accumulation may be derived from the local proliferation of existing Tregs as well. However, the specific fraction in which existing muscle and infiltrating Tregs contribute to the accumulation of Tregs in injured muscle remains to be fully elucidated.
Muscle Tregs promote muscle repair
Under homeostasis, Tregs play a major role in facilitating macrophage transition from a pro‐inflammatory (CD11b+ Ly6chi) to anti‐inflammatory (CD11b+ Ly6clow) phenotype (reviewed in ref. 29). This transition is absent when Tregs are depleted post‐cardiotoxin injury. 58 Unsurprisingly, this is only observed in injured muscle, but not uninjured muscle, 58 suggesting that the functions of muscle Tregs are unlikely to be the same during the steady state, if any. Furthermore, muscle Tregs are also required to limit pro‐inflammatory IFN‐γ production, and the MHCII+ macrophage response. 109 , 111 This is accompanied by increased fibrosis, but also a reduction in regenerative nucleated myofibres, and attenuated expression of genes related to muscle repair (Mmp12, C1qa, Myog), immune response/inflammation (Cd8a, Ccl17, Arg1) and matrix protein (Col6a5), 58 suggesting a failure in muscle regeneration. Taken together, muscle Tregs, at least upon cardiotoxin‐induced injury, facilitate the shift of macrophage phenotype, suppressing fibrosis and mediating muscle repair.
The contribution of muscle Tregs to tissue repair appears to be Areg‐mediated. The administration of recombinant Areg in cardiotoxin‐injured Treg‐depleted mice up‐regulates transcripts associated with muscle function (Myl2, Myl3 and Me1), and down‐regulates muscle development/differentiation (Adam12 and Myog) and fibrosis‐related transcripts (Col1a1, Col2a1, Col3a1 and Col4a1). 58 Hence, Areg may maintain satellite cell ‘stemness’ and fibrosis suppression, at least at the molecular level. In vitro culture of wild‐type satellite cells with Areg enhances colony‐forming efficiency and myogenic differentiation, 58 suggesting that Areg can directly impact muscle stem cells. However, this remains to be shown in vivo.
Similarly, infection with the protozoan parasite Toxoplasma gondii augments muscle Treg abundance and Areg production, with subsequent loss of tissue function. 112 These findings indicate the induction of muscle Tregs, as well as Treg‐derived Areg, are probably regulated by muscle injury, irrespective of the nature of injury. However, in this model, Treg depletion did not impact the absolute number of pro‐ and anti‐inflammatory macrophages. 112 Instead, there is an increase in centrally nucleated muscle fibres. In summary, it appears that Tregs hinder, rather than promote, muscle regeneration upon T. gondii infection. 112 Of note, in fibrosis‐resistant ADAM17 PTC knockout mice, administration of recombinant Areg induces kidney fibrosis in response to post‐ischaemia reperfusion injury. 113 These studies collectively imply that the role(s) played by muscle Tregs may be dependent on the injury stimuli.
Duchenne muscular dystrophy pathogenesis is largely characterized by chronic muscle inflammation and eventual loss of structural tissue integrity. 58 , 109 Relative to control muscle, Treg numbers (irrespective of anatomical location), their activation and IL‐10 production, are elevated in diseased muscle. 58 , 109 Anti‐CD25‐mediated Treg depletion in an experimental model leads to increased IFN‐γ expression, without influencing IL‐10 expression. The absence of Tregs also down‐regulates genes implicated in muscle homeostasis, but up‐regulates muscle‐reparative and fibrosis‐promoting genes, such as osteopontin (Spp1) and connective‐tissue growth factor (Ctgf). 58 Conversely, augmentation of muscle Tregs through IL‐2–IL‐2 complexes attenuates muscle damage. 58 Taken together, muscle Tregs regulate muscle fibro‐pathology and regeneration in response to both acute and chronic injury.
Future directions
In summary, we have presented the current advancements in the phenotypic diversity and functions of tissue Tregs (Fig. 1). However, many areas remain underexplored. Below, we highlight the most pertinent.
Figure 1.
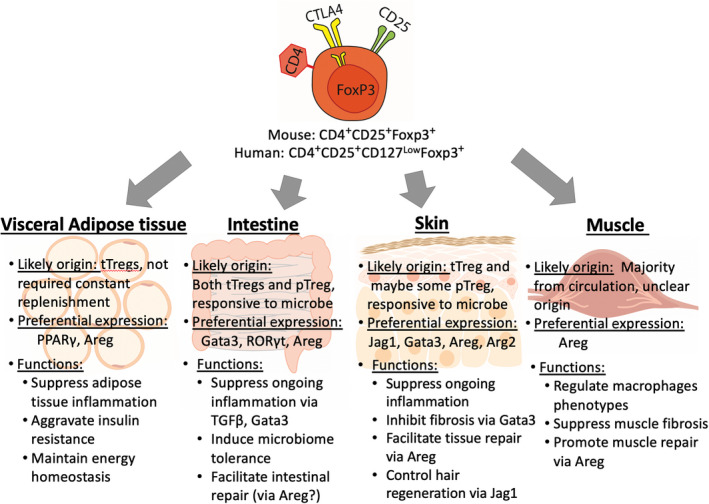
Regulatory T cells (Tregs) residing within four major tissue sites. The majority of visceral adipose tissue Tregs, marked by their preferential expression of peroxisome proliferator‐activated receptor γ (PPARγ), are mainly thymically derived. Contrarily, intestinal Tregs are highly heterogeneous, in both preferential expression and origin. Some evidence has suggested skin Tregs are of thymic origin, and accumulate during neonatal life. Uniquely, the majority of muscle Tregs are derived from the circulation, but retain a preferential transcriptome relative to lymphoid Tregs. These four tissue Treg populations serve as important regulators of tissue inflammation and fibrosis, as well as playing a major role in facilitating tissue repair.
A potential Areg–TGF‐β axis in facilitating Treg‐mediated tissue repair
In this review we have focused on Tregs in four organs with the most experimental data, but it is important to note that Tregs also reside in other tissues, and equally play a central role in maintaining homeostasis. For example, Treg depletion can aggravate lung fibrosis in Aspergillus fungus‐exposed mice, accompanied by an increased infiltration of inflammatory cells, in particular CD103lo GATA3hi T cells. 114 During PR8‐OTI influenza virus infection, Treg‐derived Areg preserves lung tissue integrity and blood oxygen saturation. 84 Similarly, after ischaemic stroke, Treg‐derived Areg was required to regulate neurotoxic astrogliosis . 115 Despite the unanimous role of Areg in multiple tissue Treg populations, the exact molecular mechanisms of how Tregs use this pathway for tissue regeneration is not fully understood. One possible interconnection may be with the TGF‐β pathway. After acute lung and liver injury, macrophage‐derived Areg locally activates TGF‐β on mesenchymal stromal cells (pericytes), by induction of integrin‐α v complexes. 116 These pericytes then differentiate into collagen‐producing myofibroblasts, promoting re‐vascularization and tissue restoration. 116 It is well‐established that TGF‐β, along with IL‐15 and T‐box transcription factors, plays a major role in the survival and development of tissue‐resident CD8+ CD103+ memory T cells, 117 as well as Treg development and function. 74 In addition, skin Treg abundance is significantly reduced in Foxp3cre EGFRfl/fl mice, suggesting a dependency on locally available Areg. 103 As discussed, Treg‐derived Areg is critical for the control of intestinal and skin inflammation. Hence, it is plausible that Areg, in co‐operation with TGF‐β, may indirectly enable the self‐regulation of tissue Treg survival and function.
Interconnection between Tregs at different tissue sites
An active and exciting area of investigation is whether the functions of Tregs residing in different tissues interconnect with each other. Emerging physiological and pathological evidence has repeatedly demonstrated an intimate, bidirectional association between skin and intestinal immune homeostasis (reviewed in ref. 118). Indeed, Treg subtypes are spatially localized in their unique niches but appear to be influenced by similar external cues. For instance, the repertoire and exposure to commensal and pathogenic microbiota is a major contributor to the regulation of both skin and colonic Treg abundance. More recently, it has been hypothesized that the gut microbiome can also influence skin Treg abundance and function(s), through the gut–skin axis. Several studies have suggested that oral consumption of specific microbial strains can influence cutaneous inflammation and wound healing processes (reviewed in ref. 118). For example, oral consumption of Lactobacillus reuteri accelerates healing of excision‐induced wounds in murine dorsal skin. 119 Adoptive transfer of lymphoid Tregs from L. reuteri‐infected mice into Rag2−/− recipients promotes the localization of Tregs proximal to skin wounded sites, resulting in accelerated healing. 119 Contrarily, anti‐CD25‐mediated Treg depletion abolishes the improved response observed in the L. reuteri‐infected group, 119 suggesting that L. reuteri‐mediated wound healing is facilitated by resident tissue Tregs, or a subset thereof. Similarly, oral administration of Lactobacillus casei enhances skin Treg abundance, IL‐10 production and delayed skin inflammation. 120 The gut–skin axis remains to yet be fully explored, but it is an exciting field that may reveal a potential cross‐link between immunity in different organs, and whether this influences global tissue homeostasis.
Current Treg immunotherapies
In recent years, increasing research has focused on the utilization of Tregs for immunotherapy. Traditionally, inflammation may be controlled by immunosuppressants (in the forms of drug metabolizing enzymes, hormone‐based or small molecules) in autoimmune diseases and for the prevention of transplantation rejection. One example is Azathioprine, which is commonly used to inhibit lymphocyte proliferation through inhibition of purine synthesis to delay graft rejection and as a treatment for rheumatoid arthritis or inflammatory bowel disease (reviewed in ref. 121). Besides the limited efficacy, adverse toxicity and undesirable secondary effects, the long‐term use of immunosuppressants can be detrimental overall. 122 , 123 There are currently three major Treg‐targeting approaches. (i) using known Treg co‐stimulatory pathways to suppress T‐cell activation. For example, CTLA4‐Ig (a fusion CTLA4 protein with human immunoglobulin) blocks the CD28/B7 co‐stimulation signal, and has shown high efficacy in rheumatoid arthritis and type 1 diabetes; 124 , 125 (ii) inducing in vivo expansion of Tregs and/or enhancing their functions with IL‐2, rapamycin or anti‐CD3. Low‐dose IL‐2 has been effective in chronic graft‐versus‐host disease to promote Treg proliferation and thymic export, rescue IL‐7 and IL‐15 levels, while showing a minimal effect on Teffs; 126 and (iii) using adoptive transfer of ex vivo stimulated and expanded Tregs from patients’ peripheral blood mononuclear cells to ameliorate type I diabetes or delay solid organ transplant rejection. However, specificity remains a major obstacle in these therapies (reviewed in ref. 127, 128).
Early evidence of Tregs with defined antigen specificity (chimeric antigen receptor Tregs, CAR‐Tregs) appear promising. Although not clinically approved, multiple pre‐clinical studies have indicated the potential of CAR‐Tregs to induce immunological tolerance (reviewed in ref. 129). For example, transfer of HLA‐A2‐specific CAR‐Tregs can prevent HLA‐A2+ human skin graft rejection and promote graft survival in humanized mice. 130 , 131 One construct of CAR‐Tregs has proved beneficial over polyclonal Tregs, while limiting bystander cytotoxicity, 131 suggesting that antigen specificity can improve therapeutic outcomes. A recent study has also shed light on the importance of Treg TCR specificity for muscle Treg accumulation, phenotype and function. 110 Hence, elucidating the nature of antigens recognized by Tregs within tissue sites may be essential in designing therapies to induce immune tolerance.
However, CAR‐Treg therapies are limited by antigen selection, in vivo persistence and potential off‐target cytotoxicity (reviewed in ref. 129). Homing to target tissue is possibly the biggest hurdle. One possibility will be to engineer expression of tissue‐homing chemokine receptors (such as CCR6 for skin and CCR9 for intestine), creating an artificial means by which Tregs can infiltrate target tissues. It has been shown that Treg sequential migration to inflamed allografts helps to shape Treg phenotypes and immunosuppressive function within the tissue, 132 proposing that Treg flux may be associated with their phenotypic diversity. Fully elucidating the tissue Treg phenotypes, and how this translates to each tissue Treg function(s) will be a critical piece of the puzzle in developing tissue‐specific CAR‐Treg therapy.
Targeting tissue Tregs in situ
Furthermore, as highlighted in this review, immunosuppression is not the sole function of tissue Tregs. Conventional antigen recognition through the TCR may be one of many pathways to initiate tissue Treg‐mediated repair. In the lung, Treg‐mediated tissue repair functions in a TCR‐independent fashion and instead relies on IL‐18 or IL‐33 signalling. 84 Interleukin‐33 is also required for the induction of IL‐13‐producing ST2+ Tregs during acute lung injury. 133 In this scenario, rather than employing Tregs to suppress undesired target cells, a mechanism to activate Treg functions in a specific manner will be necessary. One potential method is the use of bi‐specific antibodies, which can bind to two unique antigens simultaneously. It may be possible to redirect the proximity between a given cell type with a second target cell within tissues. One clinically approved construct is blinatumomab, with anti‐CD3 targeting CD8+ T cells and anti‐CD19 against tumour cells. The link‐crossing between these two cell types ultimately leads to tumour eradication (reviewed in ref. 134). Hypothetically, one could engineer a bi‐specific antibody with a tissue ligand and Treg‐tissue‐specific expressed receptor to cross‐link Tregs with a parenchymal, or even stem cell. By targeting two receptors that are uniquely expressed in the tissue, there is potential to increase spatial co‐localization compared with monospecific antibodies. This in turn may reduce the risk of systemic Treg activation and improve efficacy.
Concluding remarks
In summary, we have discussed the experimental evidence that Tregs residing in multiple tissue compartments share core phenotypic signatures and carry out immunosuppression. However, Tregs adapt to their respective environment, and express tissue‐specific factors that confer distinct roles in tissue repair. To clinically exploit these cells will require a more in‐depth understanding of the factors that regulate and maintain tissue Treg heterogeneity. Given that Tregs exert the majority of their functions in tissue sites, it is imperative the Treg community focuses on augmenting specific tissue‐resident subsets for the treatment of tissue regenerative and inflammatory disorders.
Disclosures
The authors have no competing interests to declare.
Acknowledgements
We apologise to our colleagues for the many articles that could not be quoted because of space limitation.
References
- 1. Gershon RK, Cohen P, Hencin R, Liebhaber SA. Suppressor T cells. J Immunol 1972; 108:586–90. [PubMed] [Google Scholar]
- 2. Rammensee HG, Nagy ZA, Klein J. Suppression of cell‐mediated lymphocytotoxicity against minor histocompatibility antigens mediated by Lyt‐1+Lyt–2+ T cells of stimulator‐strain origin. Eur J Immunol 1982; 12:930–4. [DOI] [PubMed] [Google Scholar]
- 3. Baxevanis CN, Ishii N, Nagy ZA, Klein J. Role of the Ek molecule in the generation of suppressor T cells in response to LDHB. Scand J Immunol 1982; 16:25–31. [DOI] [PubMed] [Google Scholar]
- 4. Sakaguchi S, Wing K, Miyara M. Regulatory T cells – a brief history and perspective. Eur J Immunol 2007; 37(Suppl 1):S116–23. [DOI] [PubMed] [Google Scholar]
- 5. Germain RN. Special regulatory T‐cell review: a rose by any other name: from suppressor T cells to Tregs, approbation to unbridled enthusiasm. Immunology 2008; 123:20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self‐tolerance maintained by activated T cells expressing IL‐2 receptor alpha‐chains (CD25). Breakdown of a single mechanism of self‐tolerance causes various autoimmune diseases. J Immunol 1995; 155:1151–64. [PubMed] [Google Scholar]
- 7. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4:330–6. [DOI] [PubMed] [Google Scholar]
- 8. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova J‐L, Buist N et al X‐linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001; 27:18–20. [DOI] [PubMed] [Google Scholar]
- 9. Josefowicz SZ, Lu L‐F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30:531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X, Oppenheim JJ. Resolving the identity myth: key markers of functional CD4+FoxP3+ regulatory T cells. Int Immunopharmacol 2011; 11:1489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hori S, Nomura T, Sakaguchi S. Control of regulatory T Cell development by the transcription factor Foxp3. Science. 2003; 299:1057–61. [DOI] [PubMed] [Google Scholar]
- 12. Khattri R, Cox T, Yasayko S‐A, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 2003; 4:337–42. [DOI] [PubMed] [Google Scholar]
- 13. Williams LM, Rudensky AY. Maintenance of the Foxp3‐dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol 2007; 8:277–84. [DOI] [PubMed] [Google Scholar]
- 14. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A et al Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009; 30:899–911. [DOI] [PubMed] [Google Scholar]
- 15. Liu W, Putnam AL, Xu‐Yu Z, Szot GL, Lee MR, Zhu S et al CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 2006; 203:1701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol 2020; 10; 3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF‐β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol 2008; 9:632–40. [DOI] [PubMed] [Google Scholar]
- 18. Lio C‐WJ, Hsieh C‐S. A two‐step process for thymic regulatory T cell development. Immunity 2008; 28:100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 2009; 30:626–35. [DOI] [PubMed] [Google Scholar]
- 20. Fang D, Zhu J. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J Exp Med 2017; 214:1861–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A et al Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009; 15:930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DiSpirito JR, Zemmour D, Ramanan D, Cho J, Zilionis R, Klein AM et al Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol 2018; 3:eaat5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li M, Zhao W, Wang Y, Jin L, Jin G, Sun X et al A wave of Foxp3+ regulatory T cell accumulation in the neonatal liver plays unique roles in maintaining self‐tolerance. Cell Mol Immunol 2020; 17:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN et al Single‐cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity 2019; 50:493–504.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sullivan JM, Höllbacher B, Campbell DJ. Cutting edge: dynamic expression of Id3 defines the stepwise differentiation of tissue‐resident regulatory T cells. J Immunol 2019; 202:31–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shevach EM. Foxp3+ T regulatory cells: still many unanswered questions—a perspective after 20 years of study. Front Immunol 2018; 9; 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM et al Trans‐endocytosis of CD80 and CD86: a molecular basis for the cell‐extrinsic function of CTLA‐4. Science 2011; 332:600–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg‐mediated T cell suppression. Front Immunol 2012; 3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J, Tan J, Martino MM, Lui KO. Regulatory T‐cells: potential regulator of tissue repair and regeneration. Front Immunol 2018; 9:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci. 2009; 122:3209–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin P, D’Souza D, Martin J, Grose R, Cooper L, Maki R et al Wound healing in the PU.1 Null Mouse–Tissue repair is not dependent on inflammatory cells. Curr Biol 2003; 13:1122–8. [DOI] [PubMed] [Google Scholar]
- 33. Mori R, Shaw TJ, Martin P. Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med 2008; 205:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell 2014; 156:20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE et al PPAR‐γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012; 486:549–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A et al Depletion of fat‐resident Treg cells prevents age‐associated insulin resistance. Nature 2015; 528:137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM et al Antigen‐ and cytokine‐driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab 2015; 21:543–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O et al Comprehensive assessment of T‐cell receptor beta‐chain diversity in αβ T cells. Blood 2009; 114:4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 2006; 24:419–66. [DOI] [PubMed] [Google Scholar]
- 40. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 2006; 8:191–7. [DOI] [PubMed] [Google Scholar]
- 41. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007; 132:2169–80. [DOI] [PubMed] [Google Scholar]
- 42. Singh B, Saxena A. Surrogate markers of insulin resistance: a review. World J Diabetes 2010; 1:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D. Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARγ effects. Proc Natl Acad Sci USA 2015; 112:482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V et al Visceral adipose inflammation in obesity is associated with critical alterations in T regulatory cell numbers. PLoS One 2011; 6:e16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mathis D. Immunological goings‐on in visceral adipose tissue. Cell Metab 2013; 17:851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qiao Y‐C, Shen J, He L, Hong X‐Z, Tian F, Pan Y‐H et al Changes of regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: a systematic review and meta‐analysis. J Diabetes Res. 2016; 2016:3694957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han JM, Wu D, Denroche HC, Yao Y, Verchere CB, Levings MK. IL‐33 reverses an obesity‐induced deficit in visceral adipose tissue ST2+ T regulatory cells and ameliorates adipose tissue inflammation and insulin resistance. J Immunol 2015; 194:4777–83. [DOI] [PubMed] [Google Scholar]
- 48. Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U et al Potential role of regulatory T cells in reversing obesity‐linked insulin resistance and diabetic nephropathy. Diabetes 2011; 60:2954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ilan Y, Maron R, Tukpah A‐M, Maioli TU, Murugaiyan G, Yang K et al Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci USA. 2010; 107:9765–9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zaiss DMW, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015; 42:216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang B, Kumoto T, Arima T, Nakamura M, Sanada Y, Kumrungsee T et al Transgenic mice specifically expressing amphiregulin in white adipose tissue showed less adipose tissue mass. Genes Cells 2018; 23:136–45. [DOI] [PubMed] [Google Scholar]
- 52. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014; 14:667–685. [DOI] [PubMed] [Google Scholar]
- 53. Collins J, Badireddy M. Anatomy, Abdomen and Pelvis, Small Intestine. Treasure Island: StatPearls Publishing, 2019. [PubMed] [Google Scholar]
- 54. Makita S, Kanai T, Nemoto Y, Totsuka T, Okamoto R, Tsuchiya K et al Intestinal lamina propria retaining CD4+CD25+ regulatory T cells is a suppressive site of intestinal inflammation. J Immunol 2007;178:4937–46. [DOI] [PubMed] [Google Scholar]
- 55. Guo Z, Jang MH, Otani K, Bai Z, Umemoto E, Matsumoto M et al CD4+CD25+ regulatory T cells in the small intestinal lamina propria show an effector/memory phenotype. Int Immunol 2008; 20:307–15. [DOI] [PubMed] [Google Scholar]
- 56. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y et al Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011; 331:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Denning TL, Norris BA, Medina‐Contreras O, Manicassamy S, Geem D, Madan R et al Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol 2011; 187:733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y et al A special population of regulatory T cells potentiates muscle repair. Cell 2013; 155:1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sefik E, Geva‐Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM et al Mucosal Immunology. Individual intestinal symbionts induce a distinct population of RORγ + regulatory T cells. Science 2015; 349:993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH et al GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Investig 2011; 121:4503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN et al Neuropilin 1 is expressed on thymus‐derived natural regulatory T cells, but not mucosa‐generated induced Foxp3+ T reg cells. J Exp Med. 2012; 209:1723–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nutsch K, Chai JN, Ai TL, Russler‐Germain E, Feehley T, Nagler CR et al Rapid and efficient generation of regulatory T cells to commensal antigens in the periphery. Cell Rep 2016; 17:206–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL et al Thymus‐derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 2013; 497:258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio C‐W, Santacruz N et al Peripheral education of the immune system by colonic commensal microbiota. Nature 2011; 478:250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pratama A, Schnell A, Mathis D, Benoist C. Developmental and cellular age direct conversion of CD4+ T cells into RORγ + or Helios+ colon Treg cells. J Exp Med 2019; 217:e20190428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ashouri JF, Weiss A. Endogenous Nur77 is a specific indicator of antigen receptor signaling in human T and B cells. J Immunol 2017; 198:657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pryor R, Martinez‐Martinez D, Quintaneiro L, Cabreiro F. The role of the microbiome in drug response. Annu Rev Pharmacol Toxicol 2019; 60:417–35. [DOI] [PubMed] [Google Scholar]
- 68. Geuking MB, Cahenzli J, Lawson MAE, Ng DCK, Slack E, Hapfelmeier S et al Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011; 34:794–806. [DOI] [PubMed] [Google Scholar]
- 69. Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG et al Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 2016; 351:858–63. [DOI] [PubMed] [Google Scholar]
- 70. Boehm F, Martin M, Kesselring R, Schiechl G, Geissler EK, Schlitt H‐J et al Deletion of Foxp3+ regulatory T cells in genetically targeted mice supports development of intestinal inflammation. BMC Gastroenterol 2012; 12:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich J‐M et al Interleukin‐10 signaling in regulatory T cells is required for suppression of Th17 cell‐mediated inflammation. Immunity 2011; 34:566–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT et al Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014; 345:1623–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A et al Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 2014; 20:1410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li MO, Sanjabi S, Flavell RA. Transforming growth factor‐beta controls development, homeostasis, and tolerance of T cells by regulatory T cell‐dependent and ‐independent mechanisms. Immunity 2006; 25:455–71. [DOI] [PubMed] [Google Scholar]
- 75. Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor‐β1 promotes in vivo Th17 cell differentiation. Immunity 2011; 34:396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle‐Murray T, Jin W et al Transforming growth factor‐β signaling in regulatory T cells controls T helper‐17 cells and tissue‐specific immune responses. Immunity 2017; 46:660–74. [DOI] [PubMed] [Google Scholar]
- 77. Worthington JJ, Kelly A, Smedley C, Bauché D, Campbell S, Marie JC et al Integrin αvβ8‐mediated TGF‐β activation by effector regulatory T cells is essential for suppression of T‐cell‐mediated inflammation. Immunity 2015; 42:903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol 2003; 170:3939–43. [DOI] [PubMed] [Google Scholar]
- 79. Li MO, Wan YY, Flavell RA. T Cell‐produced transforming growth factor‐β1 controls T cell tolerance and regulates Th1‐ and Th17‐cell differentiation. Immunity 2007; 26:579–91. [DOI] [PubMed] [Google Scholar]
- 80. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA et al The alarmin IL‐33 promotes regulatory T‐cell function in the intestine. Nature 2014; 513:564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA‐3 for the function of regulatory T cells. Immunity 2011; 35:337–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ et al C‐MAF‐dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018; 554:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rizzo A, Di Giovangiulio M, Stolfi C, Franzè E, Fehling H‐J, Carsetti R et al RORγt‐expressing Tregs drive the growth of colitis‐associated colorectal cancer by controlling IL6 in dendritic cells. Cancer Immunol Res 2018; 6:1082–92. [DOI] [PubMed] [Google Scholar]
- 84. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S et al A distinct function of regulatory T cells in tissue protection. Cell 2015; 162:1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zaiss DMW, van Loosdregt J, Gorlani A, Bekker CPJ, Gröne A, Sibilia M et al Amphiregulin enhances regulatory T cell‐suppressive function via the epidermal growth factor receptor. Immunity 2013; 38:275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shao J, Sheng H. Amphiregulin promotes intestinal epithelial regeneration: roles of intestinal subepithelial myofibroblasts. Endocrinology 2010; 151:3728–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A et al T Helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 2018; 175:1307–20.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zomer HD, Trentin AG. Skin wound healing in humans and mice: challenges in translational research. J Dermatol Sci 2018; 90:3–12. [DOI] [PubMed] [Google Scholar]
- 89. Ali N, Rosenblum MD. Regulatory T cells in skin. Immunology 2017; 152:372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW et al Memory regulatory T cells reside in human skin. J Clin Invest 2014; 124:1027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Scharschmidt TC, Vasquez KS, Truong H‐A, Gearty SV, Pauli ML, Nosbaum A et al A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity 2015; 43:1011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K et al Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell 2017; 169:1119–29.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lowe MM, Boothby I, Clancy S, Ahn RS, Liao W, Nguyen DN et al Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI insight 2019; 4:129756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A et al Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci USA 2008; 105:10871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ikebuchi R, Fujimoto M, Nakanishi Y, Okuyama H, Moriya T, Kusumoto Y et al Functional phenotypic diversity of regulatory T cells remaining in inflamed skin. Front Immunol 2019; 10; 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dudda JC, Perdue N, Bachtanian E, Campbell DJ. Foxp3+ regulatory T cells maintain immune homeostasis in the skin. J Exp Med 2008; 205:1559–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gratz IK, Truong H‐A, Yang SH‐Y, Maurano MM, Lee K, Abbas AK et al Edge Cutting: memory regulatory t cells require IL‐7 and not IL‐2 for their maintenance in peripheral tissues. J Immunol 2013; 190:4483–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong H‐A et al Commensal microbes and hair follicle morphogenesis coordinately drive Treg migration into neonatal skin. Cell Host Microbe 2017; 21:467–77.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W et al Compartmentalized control of skin immunity by resident commensals. Science 2012; 337:1115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Leech JM, Dhariwala MO, Lowe MM, Chu K, Merana GR, Cornuot C et al Toxin‐triggered interleukin‐1 receptor signaling enables early‐life discrimination of pathogenic versus commensal skin bacteria. Cell Host Microbe. 2019; 26:795–809.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kalekar LA, Cohen JN, Prevel N, Sandoval PM, Mathur AN, Moreau JM et al Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci Immunol 2019; 4:eaaw2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Harrison OJ, Linehan JL, Shih H‐Y, Bouladoux N, Han S‐J, Smelkinson M et al Commensal‐specific T cell plasticity promotes rapid tissue adaptation to injury. Science 2019; 363:eaat6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nosbaum A, Prevel N, Truong H‐A, Mehta P, Ettinger M, Scharschmidt TC et al Cutting edge: regulatory T cells facilitate cutaneous wound healing. J Immunol 2016;196:2010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mathur AN, Zirak B, Boothby IC, Tan M, Cohen JN, Mauro TM et al Treg‐cell control of a CXCL5‐IL‐17 inflammatory axis promotes hair‐follicle‐stem‐cell differentiation during skin‐barrier repair. Immunity 2019; 50:655–67.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. McEntee CP, Gunaltay S, Travis MA. Regulation of barrier immunity and homeostasis by integrin‐mediated transforming growth factor β activation. Immunology 2000; 160:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Müller‐Röver S, Handjiski B, Van Der Veen C, Eichmüller S, Foitzik K, McKay IA et al A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Invest Dermatol 2001; 117:3–15. [DOI] [PubMed] [Google Scholar]
- 107. Morgan JE, Partridge TA. Muscle satellite cells. Int J Biochem Cell Biol 2003; 35:1151–6. [DOI] [PubMed] [Google Scholar]
- 108. Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ et al Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin‐33‐dependent accumulation of regulatory T cells. Immunity 2016; 44:355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG et al Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med 2014; 6:258ra142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Cho J, Kuswanto W, Benoist C, Mathis D. T cell receptor specificity drives accumulation of a reparative population of regulatory T cells within acutely injured skeletal muscle. Proc Natl Acad Sci USA 2019; 116:26727 –33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Panduro M, Benoist C, Mathis D. Treg cells limit IFN‐γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc Natl Acad Sci USA 2018; 115: E2585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Jin RM, Blair SJ, Warunek J, Heffner RR, Blader IJ, Wohlfert EA. Regulatory T cells promote myositis and muscle damage in Toxoplasma gondii infection. J Immunol 2016; 198:352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kefaloyianni E, Keerthi Raja MR, Schumacher J, Muthu ML, Krishnadoss V, Waikar SS et al Proximal tubule‐derived amphiregulin amplifies and integrates profibrotic EGF receptor signals in kidney fibrosis. J Am Soc Nephrol 2019; 30:2370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ichikawa T, Hirahara K, Kokubo K, Kiuchi M, Aoki A, Morimoto Y et al CD103hi Treg cells constrain lung fibrosis induced by CD103lo tissue‐resident pathogenic CD4 T cells. Nat Immunol 2019; 20:1469–80. [DOI] [PubMed] [Google Scholar]
- 115. Ito M, Komai K, Mise‐Omata S, Iizuka‐Koga M, Noguchi Y, Kondo T et al Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019; 565:246–50. [DOI] [PubMed] [Google Scholar]
- 116. Minutti CM, Modak RV, Macdonald F, Li F, Smyth DJ, Dorward DA et al A macrophage–pericyte axis directs tissue restoration via amphiregulin‐induced transforming growth factor β activation. Immunity 2019; 50:645–54.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mackay LK, Wynne‐Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM et al T‐box Transcription factors combine with the cytokines TGF‐β and IL‐15 to control tissue‐resident memory T cell fate. Immunity 2015; 43:1101–11. [DOI] [PubMed] [Google Scholar]
- 118. Salem I, Ramser A, Isham N, Ghannoum MA. The gut microbiome as a major regulator of the gut–skin axis. Front Microbiol 2018; 9:1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Poutahidis T, Kearney SM, Levkovich T, Qi P, Varian BJ, Lakritz JR et al Microbial symbionts accelerate wound healing via the neuropeptide hormone oxytocin. PLoS One 2013; 8:e78898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hacini‐Rachinel F, Gheit H, Le Luduec J‐B, Dif F, Nancey S, Kaiserlian D. Oral probiotic control skin inflammation by acting on both effector and regulatory T cells. PLoS One 2009; 4:e4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Evans WE. Pharmacogenetics of thiopurine S‐methyltransferase and thiopurine therapy. Ther Drug Monit 2004; 26:186–91. [DOI] [PubMed] [Google Scholar]
- 122. Vial T, Descotes J. Immunosuppressive drugs and cancer. Toxicology 2003; 185:229–40. [DOI] [PubMed] [Google Scholar]
- 123. Singh JA, Hossain A, Kotb A, Wells G. Risk of serious infections with immunosuppressive drugs and glucocorticoids for lupus nephritis: a systematic review and network meta‐analysis. BMC Med 2016; 14:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Maxwell LJ, Singh JA. Abatacept for rheumatoid arthritis: a Cochrane systematic review. J Rheumatol. 2010; 37:234–45. [DOI] [PubMed] [Google Scholar]
- 125. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R et al Co‐stimulation modulation with abatacept in patients with recent‐onset type 1 diabetes: a randomised, double‐blind, placebo‐controlled trial. Lancet 2011; 378:412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y et al Low‐dose interleukin‐2 therapy restores regulatory T cell homeostasis in patients with chronic graft‐versus‐host disease. Sci Transl Med 2013; 5:179ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Raffin C, Vo LT, Bluestone JA. Treg cell‐based therapies: challenges and perspectives. Nat Rev Immunol 2020; 20:158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rosenblum MD, Gratz IK, Paw JS, Abbas AK. Treating human autoimmunity: current practice and future prospects. Sci Transl Med 2012; 4:125sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhang Q, Lu W, Liang C‐L, Chen Y, Liu H, Qiu F et al Chimeric Antigen Receptor (CAR) Treg: a promising approach to inducing immunological tolerance. Front Immunolo 2018; 9:2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Noyan F, Zimmermann K, Hardtke‐Wolenski M, Knoefel A, Schulde E, Geffers R et al Prevention of allograft rejection by use of regulatory T cells with an MHC‐specific chimeric antigen receptor. Am J Transplant 2017; 17:917–30. [DOI] [PubMed] [Google Scholar]
- 131. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MAA, Hannen RF, Cooper D et al Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant 2017; 17:931–43. [DOI] [PubMed] [Google Scholar]
- 132. Zhang N, Schröppel B, Lal G, Jakubzick C, Mao X, Chen D et al Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity 2009; 30:458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Liu Q, Dwyer GK, Zhao Y, Li H, Mathews LR, Chakka AB et al IL‐33‐mediated IL‐13 secretion by ST2+ Tregs controls inflammation after lung injury. JCI insight 2019; 4:123919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther 2018; 12:195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]