

Summary
Some human antibodies may paradoxically inhibit complement activation on bacteria and enhance pathogen survival in humans. This property was also claimed for IgG antibodies reacting with terminal galactose‐α‐1,3‐galactose (Galα3Gal; IgG anti‐αGal), a naturally occurring and abundant antibody in human plasma that targets numerous different pathogens. To reinvestigate these effects, we used IgG anti‐αGal affinity isolated from a pool of normal human IgG and human hypogammaglobulinaemia serum as a complement source. Flow cytometry was performed to examine antibody binding and complement deposition on pig erythrocytes, Escherichia coli O86 and Streptococcus pneumoniae serotype 9V. Specific nanobodies were used to block the effect of single complement factors and to delineate the complement pathways involved. IgG anti‐αGal was capable of activating the classical complement pathway on all the tested target cells. The degree of activation was exponentially related to the density of bound antibody on E. coli O86 and pig erythrocytes, but more linearly on S. pneumoniae 9V. The alternative pathway of complement amplified complement deposition. Deposited C3 fragments covered the activating IgG anti‐αGal, obstructing its detection and highlighting this as a likely general caveat in studies of antibody density and complement deposition. The inherent capacity for complement activation by the purified carbohydrate reactive IgG anti‐αGal was similar to that of normal human IgG. We propose that the previously reported complement inhibition by IgG anti‐αGal relates to suboptimal assay configurations, in contrast to the complement activating property of the antibodies demonstrated in this paper.
Keywords: alpha‐galactosyl epitope, antibodies, antigens/peptides/epitopes, complement, human
The naturally occurring human antibody IgG anti‐αGal is claimed to diminish complement attacks on invading pathogens. Aided by novel nanobody‐based specific complement inhibitors, we reinvestigated complement activation by the antibody and found that its binding dose‐dependently increased complement deposition on cells by initiating the classical pathway and receiving pivotal amplification from the alternative pathway. We propose that the previously reported complement inhibition by IgG anti‐αGal relates to suboptimal assay configurations.
![]()
Abbreviations
- AP
alternative pathway
- CP
classical pathway
- EcO86
Escherichia coli serotype O86
- ED50
half‐maximal effective antibody density
- EDTA
ethylenediaminetetraacetic acid
- Galα3Gal
galactose‐
α
‐1,3‐galactose
- hRBC
human ABO blood type O red blood cells
- HSA
human serum albumin
- Ig
immunoglobulin
- HHS
human hypogammaglobulinaemic serum (used as complement source)
- IgG anti‐αGal
human IgG reacting with terminal Galα3Gal
- nhIgG
normal human IgG
- NHS
normal human serum
- P
doubling‐percentage
- pRBC
pig red blood cells
- SD
standard deviation
- Sp9V
Streptococcus pneumoniae serotype 9V
- T½
half‐time until plateau phase (in one‐phase association model)
- y∞
Signal at plateau phase (in one‐phase association model)
- 95% CI
95% confidence intervals
Introduction
In general, antibodies confer protection from invading pathogens. However, some IgG antibodies cause undesirable effects by enhancing pathogen survival in humans. The classic example is dengue virus, which exploits reactive IgG to gain access to host cells. 1 More controversially, various other IgG antibodies are proposed to promote infections by extracellular bacteria by diminishing complement attacks. 2 , 3 , 4 , 5 , 6 Protection against invading extracellular bacteria is the main function of IgG and complement, as emphasized by the predisposition to infections by such pathogens in individuals who are deficient in IgG and complement. 7
IgG antibody against terminal galactose‐α‐1,3‐galactose (Galα3Gal) (IgG anti‐αGal) is a naturally occurring antibody that is proposed to enhance bacterial infections by conveying protection from complement. 4 This is of particular interest as IgG anti‐αGal is abundant in essentially all humans (comprising on average 0·1% of the total IgG 8 , 9 , 10 ) and reacts with a wide range of extracellular bacterial pathogens. 4 , 11 , 12 In this light, clarification of the role of IgG anti‐αGal in complement activation is imperative.
IgG anti‐αGal occurs naturally in humans and is also present in other Catarrhini (great apes and Old World monkeys). Catarrhini are the only mammals that do not possess terminal Galα3Gal carbohydrate. 13 They may produce anti‐αGal antibodies after exposure to commensal bacteria carrying the disaccharide. 14 Humans can generate anti‐αGal antibodies of all immunoglobulin classes. IgM anti‐αGal can bind αGal‐presenting tissue in models of xenotransplantation, which activates the complement system. 15 , 16 However, naturally occurring IgG anti‐αGal is claimed to lack the potential for complement activation. 4 , 8 , 15 , 17 , 18 , 19 Hamadeh et al. 4 reported that IgG anti‐αGal reacts with most Gram‐negative bacteria isolated from persons with sepsis. The authors proposed that the antibody paradoxically promotes pathogen survival based on findings that it protected a Serratia strain from complement‐mediated killing by an undefined mechanism. 4 Other groups also reported that IgG anti‐αGal did not activate the classical pathway (CP) of complement 8 , 15 , 17 , 18 , 19 and seemed to inhibit complement activation by occupying binding sites for potent complement activators. 8 , 18 IgG anti‐αGal is mainly IgG2, 8 , 12 which was proposed to explain the antibody’s claimed attenuation of complement activation. 8 Historically, IgG2 was believed to activate the CP very poorly compared with IgG1 and IgG3. 20 , 21 , 22 However, later studies have established IgG2 antibodies as potent activators of the CP, as reviewed elsewhere. 23 , 24 Only human IgG4 cannot activate the CP, as it cannot fix C1q. 25 Hence, the claimed inability of IgG anti‐αGal to activate the CP is not sufficiently explained by the predominance of IgG2. Furthermore, other IgG antibodies also reportedly inhibit the CP. Examples are an IgG2 antibody against O‐antigens in the lipopolysaccharide of Pseudomonas aeruginosa 6 and an IgG3 antibody against peptides on the surface of Neisseria meningitides. 5 These observations indicate that the ‘shielding phenomenon’ is not restricted to certain IgG subclasses or only to antibodies to carbohydrate antigens.
Here, we aimed to reinvestigate the apparent inability of some IgG antibodies to activate the CP. IgG anti‐αGal was selected as a model antibody.
Material and methods
Human sera
Blood was collected at the Department of Clinical Immunology, Aarhus University Hospital, Denmark. A pool of normal human serum (NHS) was prepared from 10 random blood donors. From each donor, a 4‐ml blood sample was collected in test tubes containing silica as a clot activator (BD Vacutainer, ref. 369032; BD, Franklin Lakes, NJ). After 45 min at room temperature, the tubes were centrifuged (2000 g, 5 min). Serum was collected, pooled, split into aliquots and stored at −80° until use. Human hypogammaglobulinaemia serum (HHS) was prepared from the blood obtained from an adult patient diagnosed with hypogammaglobulinaemia. The HHS contained low IgG (< 0·8 g/l), low IgA (< 0·16 g/l), and low IgM (0·10 g/l) levels, and normal complement activity of all three pathways (Euro Diagnostica complement assay; Svar Life Science AB, Malmö, Sweden).
Affinity purification of IgG anti‐αGal
Highly purified IgG anti‐αGal was prepared as previously described. 12 In brief, normal human IgG pool (nhIgG; Beriglobulin, CSL Behring, King of Prussia, PA) was diluted to 30 g/l and passed through a column containing Galα3Gal‐derivatized beads. The preparation of the Galα3Gal‐derivatized beads is described in the Supplementary material (Appendix S1). The beads were washed extensively before bound antibodies were eluted with glycine buffer (0·1 m, pH 2·5). To remove column‐reactive antibodies, the eluate was neutralized (pH 7·4) and passed over a column containing uncoupled beads. The final IgG anti‐αGal preparation was the eluate of a second positive selection using the Galα3Gal‐derivatized beads that was performed as described above.
Antibody biotinylation
A solution of antibody (1 mg/ml) was dialysed twice (2 and 18 hr) against 4 l of phosphate buffered saline (PBS), pH 7·4 and then against 4 l PBS, pH 8·5 for 3 hr. N‐Hydroxysuccinimido‐biotin (Sigma‐Aldrich, St Louis, MO) was added (170 µl of 1 mg/ml in dimethylsulphoxide per mg IgG) and reacted for 4 hr before termination of the reaction by three rounds of dialysis against Tris‐buffered saline [TBS; 10 mm Tris–HCl, 140 mm NaCl, 0·09% (weight/volume; w/v) NaN3, pH 7·4] overnight and then twice for 1.5 hr each time.
Red blood cells
Venous ethylenediaminetetraacetic acid (EDTA) ‐stabilized blood was obtained from anonymized human blood donors attending the blood bank at Aarhus University Hospital in accordance with Danish legislation, and from pigs undergoing experimental surgery at the Institute of Clinical Medicine, Aarhus University, Denmark. Blood cells from 3 ml of blood were washed twice in 50 ml PBS by centrifugation (200 g, 5 min), resuspended in 45 ml PBS containing 2% (w/v) glucose and 0·25% (v/v) glutaraldehyde, and rotated gently end‐over‐end for 1 hr at room temperature. Ethanolamine (2·5 ml, 1 m, pH 8) was added and the tube was rotated slowly for 15 min to inactivate the residual aldehyde groups. Stabilized RBCs were washed by centrifugation (200 g, 5 min) in TBS and then in PBS before resuspension in PBS with 0·1% (w/v) human serum albumin (HSA; CSL Behring) and 0·09% (w/v) sodium azide.
Microorganisms
Escherichia coli serotype O86 (EcO86) strain ATCC 12701 was obtained from the American Type Culture Collection (Manassas, VA). Streptococcus pneumoniae serotype 9V (Sp9V) strain (SK2017) was obtained from the Kilian collection, the bacterial culture collection at the Department of Biomedicine, Aarhus University. Bacteria were cultured in 50 ml Todd–Hewitt broth overnight (35°, 5% CO2), collected by centrifugation (2000 g, 30 min) and resuspended in PBS with 1% (v/v) formaldehyde. The following day, the fixed bacteria were washed twice in 50 ml PBS and resuspended in 10 ml TBS to block residual aldehyde groups. The next day, bacteria were centrifuged and the supernatant was discarded before resuspension of the collected cells in TBS. Bacteria were stored at 4° until use.
Complement deposition on cells and inhibition of complement activation
Primary incubation of cells was performed in RPMI‐1640 with 0·1% (w/v) HSA. HHS (the complement source), antibody and/or complement inhibitor were included as described below. The target cell concentration was 10 000/µl. Antibodies were purified IgG anti‐αGal, rituximab (monoclonal, chimeric mouse/human anti‐hCD20; Roche, Basel, Switzerland) as the irrelevant specificity control or nhIgG. The nhIgG preparations were Beriglobulin (same batch as used for IgG anti‐αGal purification), Privigen (CSL Behring) or Subcuvia (Baxter, Deerfield, IL). Complement inhibitors were 10 mm EDTA, eculizumab (monoclonal IgG against human C5; Alexion Pharmaceuticals, Boston, MA) or single‐domain antibodies (nanobodies) against complement factors. EDTA was used to chelate divalent cations and abrogate complement activation. Eculizumab inhibits cleavage of C5. 26 Nanobodies were derived from heavy‐chain antibodies from llamas, selected by phage display, expressed in bacteria and purified as described previously. 27 Four nanobodies were used: (i) C1qNb75 binds to the globular head of C1q, blocking C1q docking to immunoglobulin Fc domains; 28 (ii) hC3Nb1 binds C3, selectively blocking the C3 convertase of the alternative pathway (AP);, 27 (iii) hC3Nb2 binds C3, blocking both the CP and AP C3 convertases; 29 and (iv) KRA152, which was used as control. KRA152 binds an intracellular yeast kinase (i.e. an irrelevant reactivity). Nanobodies and eculizumab were used in concentrations to approximate a twofold higher molar concentration than expected of the targeted epitopes in the particular experiment. To do this, the following serum concentrations of each of the three complement factors were assumed: (i) C1q, 220 nm (mass concentration 100 mg/l, 30 MW 460 000 31 ), but as a C1q molecule carries six globular heads, the epitope concentration for C1qNb75 was sixfold higher (i.e. 1·3 µm); (ii) C3, 6·3 µm (mass concentration 1·2 g/l, 32 MW 190 000 31 ); and (iii) C5, 390 nm (75 mg/l, 32 MW 190 000 31 ). In experiments incorporating 10% HHS, concentrations of the complement inhibitors were as follows: C1qNb75, 260 nm; anti‐C3 nanobodies, 1·3 µm; and eculizumab, 78 nm. The concentrations of the inhibitors were 10‐fold lower in experiments with 1% HHS. The concentration of the control nanobody (KRA152) was equal to the highest concentration of an active nanobody (C1qNb75, hC3Nb1 or hC3Nb2) in the respective experiments. All constituents in each primary incubation were mixed in an Eppendorf tube at 0° to a total volume of 20 µl. The mixture was then placed in a water bath at 37° for 2 hr or as described in the text. The primary incubation was terminated by the addition of 1 ml PBS and mixing. Cells were then washed by centrifugation at 200 g (RBCs) or 2500 g (bacteria) for 10 min. An additional wash was applied for bacteria (all experiments) and for RBCs as described in the text. The supernatants were discarded and the cells were examined using flow cytometry.
Flow cytometry
After the final wash, the antibody‐ and complement‐treated cells (see above) were resuspended in 20 µl PBS containing 0·1% (w/v) HSA and 1 : 300 dilutions of stock solutions of secondary antibody for detection. Detection antibodies were: (i) anti‐IgG, fluorescein‐isothiocyanate‐labelled polyclonal rabbit F(ab')2 anti‐human IgG (F0315; Dako, Glostrup, Denmark); (ii) anti‐IgM, biotinylated (in‐house) polyclonal rabbit anti‐human IgM (Rockland, Gilbertsville, PA); (iii) anti‐C3c, fluorescein‐isothiocyanate‐labelled polyclonal rabbit IgG anti‐human C3c (product # PA1‐29718; Invitrogen, Carlsbad, CA) or biotinylated (in‐house) polyclonal rabbit anti‐human C3c (Q0368, Dako); and (iv) ‘anti‐C4c’, biotinylated (in‐house) polyclonal rabbit anti‐human C4c (A0065, Dako). The anti‐C3c antibodies detect deposited C3b and iC3b fragments (as the C3c sequence is present in these fragments 31 ). The anti‐C4c antibody detects deposited C4b and iC4b fragments (as the C4c sequence is present in these fragments). A maximum of two detection antibodies with different labels were included in each experiment. Incubations with detection antibodies were performed in the dark at room temperature for 30 min. When biotin‐labelled antibodies were used, cells were washed in PBS/HSA by centrifugation before the addition of 20 µl Streptavidin allophycocyanin‐eFluor780 (eBioscience/ThermoFisher Scientific, Waltham, MA) in PBS/HSA (1/300, v/v). After mixing, the samples were incubated in the dark at room temperature for an additional 30 min. Labelled cells were resuspended in 100 µl PBS (flow buffer) and analysed on a NovoCyte Quanteon (ACEA Biosciences, Inc., San Diego, CA). The threshold for the exclusion of noise was set on the forward‐scatter height signal based on analysis of the flow buffer. From each tube, 30 µl was injected at a rate of 35 µl/min. The event rate was under 1000/s. Mixing (1500 rpm, 10 seconds) and rinsing were performed between each tube. The median fluorescence intensity (MFI) of each experiment was subtracted from the background fluorescence and expressed relative to the mean of a reference experiment as stated in the text. Background fluorescence was defined as 99·9% of the lowest MFI measured in associated experiments (99·9% was used instead of 100% to allow calculations of geometric means). Concentrations of residual cell‐reactive antibody in the HHS (i.e. the complement source) relative to concentrations in NHS were estimated from measured MFIs, which were converted to measures of concentration using standard curves. Standard curves were established in parallel for each assay and setup from the MFIs determined for twofold serial dilution of NHS incubated with the particular cells.
Complement deposition measured on microtitre plates
Purified IgG anti‐αGal or Beriglobulin (same batch as used for IgG anti‐αGal purification) was coated on plastic surfaces and the deposition of C4b from NHS was measured as follows. Serial dilutions of each IgG preparation were prepared with 0·1 m carbonate buffer (pH 9·4) in Tween‐20 blocked Eppendorf tubes. The maximal IgG concentration was 10 mg/l, followed by seven 2·5‐fold dilutions. Aliquots of the serial dilutions (100 µl) were applied to wells on microtitre plates (FluoroNunc; Thermo Scientific Nunc A/S, Denmark), and the IgG was allowed to coat the well surfaces overnight in a humid chamber at 4°. The wells were emptied and residual binding sites were blocked with TBS/Tween/HSA [TBS with 0·05% (v/v) Tween‐20 and 1 g/l HSA]. Each step in the assay was followed by three wash cycles in TBS/Tween performed using an automated plate washer. To each well of the microtitre plate was added 100 µl of Veronal buffer (4 mm barbital, 145 mm NaCl, 2 mm CaCl2, 1 mm MgCl2, pH 7·5) with either 1% NHS (for measurement of deposited C4b) or no NHS (for measurement of coated IgG). The microtitre plates were incubated for 1·5 hr at 37°C. Each well then received 100 µl of 1 mg/l biotinylated rabbit polyclonal anti‐human C4c or anti‐IgG (both from Dako) in TBS/Tween/HSA followed by incubation for 1 hr at room temperature. This was followed by the addition of 100 µl TBS/Tween containing 25 µm EDTA and 0·1% (v/v) europium‐labelled streptavidin (product 1244‐360; PerkinElmer, Waltham, MA). The plates were kept at room temperature for 1 hr. Finally, 200 µl enhancement solution (PerkinElmer) was loaded into each well, and the plates were shaken for 5 min before the measurement of time‐resolved fluorescence using a VICTOR X5 apparatus (PerkinElmer). In experiments with nanobodies, purified IgG anti‐αGal was coated at 10 mg/l as described above, and samples contained 1% NHS alone or containing 10 mg/l of the specified nanobody.
Statistical analysis
Quantification of antibody reacting with target cells was performed as follows. Estimations were performed for log10‐transformed data using standard curve equations approximated by a third‐order polynomial approximation in Microsoft excel spreadsheets (excel 2016; Microsoft Corporation, Redmond, WA). Concentrations were retransformed to a linear scale for presentation in figures.
Curve fitting
Curve fitting for graphical presentations was made using the graphpad prism 6·07 program (GraphPad Software, Inc., San Diego, CA) as follows:
Exponential model: ; doubling‐percentage; P = ln(2)/k.
One‐phase association model: ; plateau phase, ; half‐time, T ½ = ln(2)/k.
Sigmoid model: ; antibody density required for half‐maximal deposition, ED50; Hill slope, n.
Ethics
Human blood was obtained from excess blood donated by anonymized voluntary blood donors at the blood bank, Department of Clinical Immunology, Aarhus University Hospital, Denmark, in accordance with Danish legislation. HHS was collected from excess material from clinical tests of a patient referred for laboratory investigations for immunodeficiency. All methods were carried out in accordance with the relevant guidelines and regulations. The studies and inclusion of the human material were approved by the Danish Data Protection Agency (reference number 1‐16‐02‐40‐12/2007‐58‐0010) and the Ethics Committee in Central Denmark Region (reference number 1‐10‐72‐127‐12).
Results
IgG anti‐αGal activates the complement CP on pig red blood cells
We addressed the ability of IgG anti‐αGal to activate complement on RBCs from pigs (pRBCs). A single pRBC presents approximately 6 × 104 Galα3Gal epitopes on the surface. 13 Affinity‐purified IgG anti‐αGal efficiently binds pRBCs. 12 We applied IgG anti‐αGal at a concentration of 20 mg/l and 1% HHS as a complement source to investigate the deposition of complement fragments on pRBCs by flow cytometry. IgG anti‐αGal markedly triggered deposition of C3 and C4 fragments compared with HHS alone (Fig. 1). HHS caused negligible complement deposition (Fig. 1), although it did contain small amounts of anti‐pRBC antibody (see Supplementary material, Fig. S1). To clarify whether complement activation by IgG anti‐αGal proceeded through the CP, we tested the effect of including the C1qNb75 nanobody, which binds the globular heads of C1q and inhibits C1q docking to immunoglobulin. 28 C1qNb75 abrogated deposition to levels that could not be distinguished from the background (i.e. a control without added HHS) (Fig. 1). The control nanobody with irrelevant reactivity showed no effect.
Figure 1.
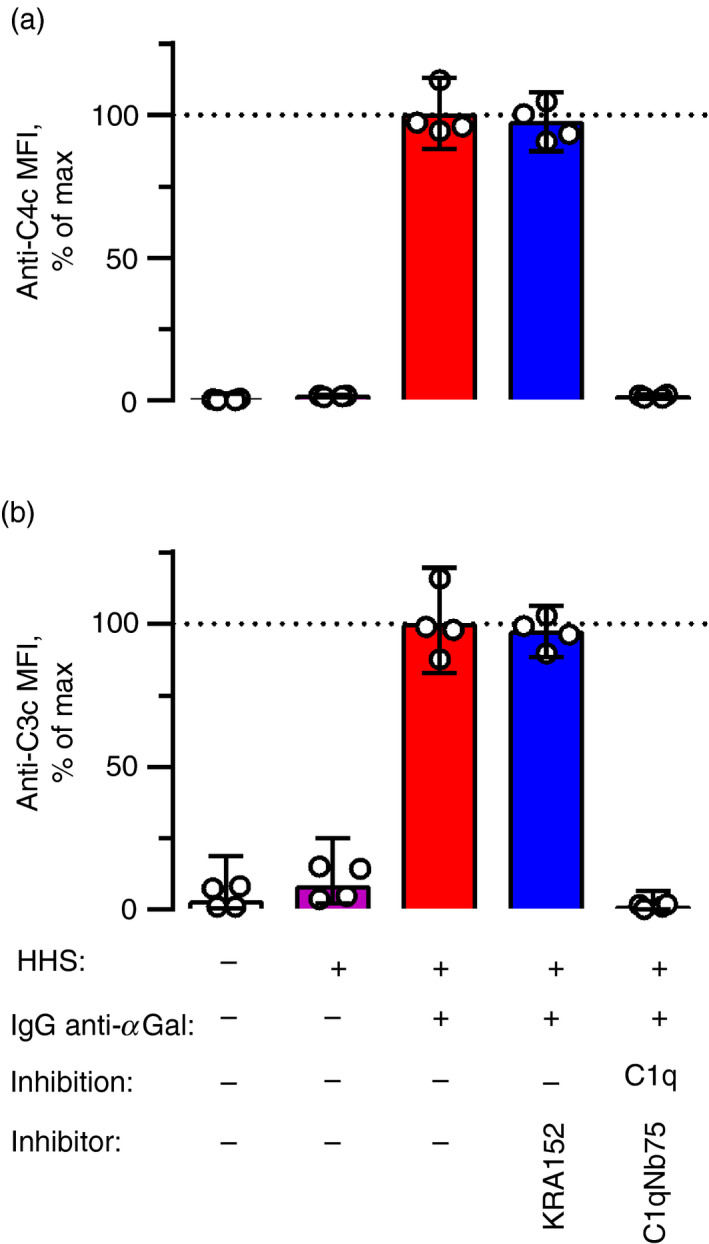
IgG anti‐αGal activates the classical pathway on pig red blood cells (pRBCs). Complement deposition on fixed pRBCs measured by flow cytometry using detection antibodies against C4c (which recognize C4b and iC4b fragments) (a) or C3c (which recognize C3b and iC3b fragments) (b). The pRBCs were incubated for 2 hr at 37° in 1% hypogammaglobulinaemia human serum (HHS) as the complement source supplemented with IgG anti‐αGal at 20 mg/l and inhibitors as indicated. Inhibitors were C1qNb75 or KRA152 nanobody. C1qNb75 targets the globular heads of C1q, thereby blocking C1q docking to immunoglobulin and inhibiting the classical pathway of complement. KRA152 possess irrelevant specificity and was included as a control. MFI, median fluorescence intensity. The columns represent the mean of four separate experiments (each represented by a dot) and error bars are 95% confidence intervals.
As controls, we tested complement deposition on cells lacking Galα3Gal (human RBCs of ABO blood group type O) in otherwise identical experiments. As expected, no complement deposition was achieved (see Supplementary material, Fig. S2). Moreover, an IgG1 antibody of irrelevant reactivity (monoclonal anti‐CD20) failed to promote complement deposition on pRBCs (see Supplementary material, Fig. S2).
The collective results demonstrate that human IgG anti‐αGal activates complement on pRBCs and that the process is initiated via the CP.
Variations in incubation time and complement concentration
We addressed the effect of incubation time from 0 to 180 min and the importance of complement concentration (1%, 4% or 10% HHS). Again, we supplemented HHS with IgG anti‐αGal to a final concentration of 20 mg/l. Complement deposition increased with prolonged incubation time, whereas the effect of HHS was more complex (Fig. 2). Detection of complement fragments never decreased, emphasizing that the speed of degradation to fragments that are not recognized by the detection antibodies never exceeded the generation of fragments that are recognized by the detection antibodies. Hence, such degradation was considered negligible and was ignored in the experiments below. We applied a one‐phase association model to extrapolate the plateau levels of detected fragments (i.e. maximal achievable level of detected fragment) and estimated the half‐time to reaching such plateaus. In this model, increasing HHS concentration entailed higher plateaus of detected C3 fragments (Fig. 2). However, the opposite was observed for the C4 fragments. Increasing HHS concentration entailed lower plateaus of the detected C4 fragments (Fig. 2). For both types of complement fragments, the half‐time to a plateau was inversely related to HHS concentration, emphasizing that their deposition speed increased with HHS concentration. For a given HHS concentration, the half‐time to the plateau was similar for the two types of fragments, supporting the idea that C4b deposition is rate limiting for the subsequent C3 fragment deposition that occurs almost instantaneously once the C4bC2a enzyme complex has been generated.
Figure 2.
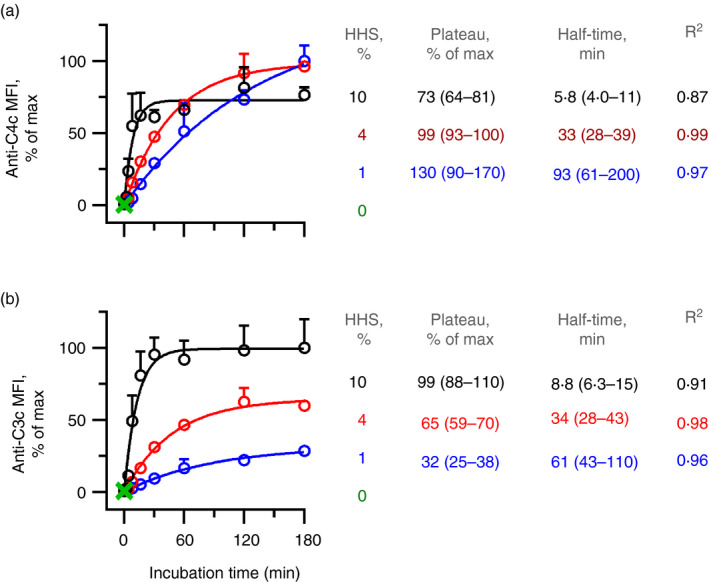
Complement deposition increases with time and complement concentration. Complement deposition on pig red blood cells (pRBCs) was measured by flow cytometry using detection antibodies against C4c (a) or C3c (b). The pRBCs were incubated for various times at 37° in increasing concentrations (1%, 4% or 10%) of hypogammaglobulinaemia human serum (HHS) as the complement source with IgG anti‐αGal at 20 mg/l. The data are expressed as the mean and standard deviation of two independent experiments. Curve fitting was performed using one‐phase association modelling.
Deposited complement products conceal the initiating IgG anti‐αGal
The detection of C4 fragments decreased when HHS concentration increased (i.e. increasing C4 concentration). We speculated that more efficient deposition of fragments downstream of C4 (at higher concentrations of complement) concealed the C4 fragments and possibly also the initiating antibodies to which the detection antibodies could bind. Such steric hindrance would be a general flaw in studies of antibody binding and complement deposition. Hence, it was important to examine this possibility. We challenged the complement deposition of IgG anti‐αGal on pRBCs using four different inhibitors: C1qNb75, hC3Nb2 (nanobody that binds C3, blocking C3 cleavage by both CP and AP 27 ), eculizumab (monoclonal IgG that binds C5, blocking C5 cleavage 26 ) and EDTA (chelates divalent cations, inhibiting complement activation in general). These experiments were conducted in 10% HHS and IgG anti‐αGal at 20 mg/l.
We found that essentially no C3 fragments were deposited on pRBCs in experiments containing either C1qNb75, hC3Nb2 or EDTA (Fig. 3a). The effect of C1qNb75 illustrates that spontaneous activation of AP was negligible in this setup. To test if AP supplied amplification after activation by CP, we used the hC3Nb1 anti‐C3 nanobody, which preferentially inhibits C3 cleavage by AP. HC3Nb1 inhibited half of the signal from the deposited C3 fragments (see Supplementary material, Fig. S3), supporting the idea that considerable amplification was provided by AP. Some C3 fragment deposition was evident in experiments without added IgG anti‐αGal (Fig. 3a). Such deposition was quenched by the addition of C1qNb75 (see Supplementary material, Fig. S3), demonstrating that the low levels of endogenous anti‐pRBC antibodies in the HHS were responsible for this background activation. Notably, the addition of eculizumab had no effect on the detection of deposited C3 fragments (Fig. 3a), suggesting that C5b‐9 complexes did not conceal C3 fragments. However, no controls were included to assess the inhibition of C5‐cleavage or C5b‐9 complex integration.
Figure 3.
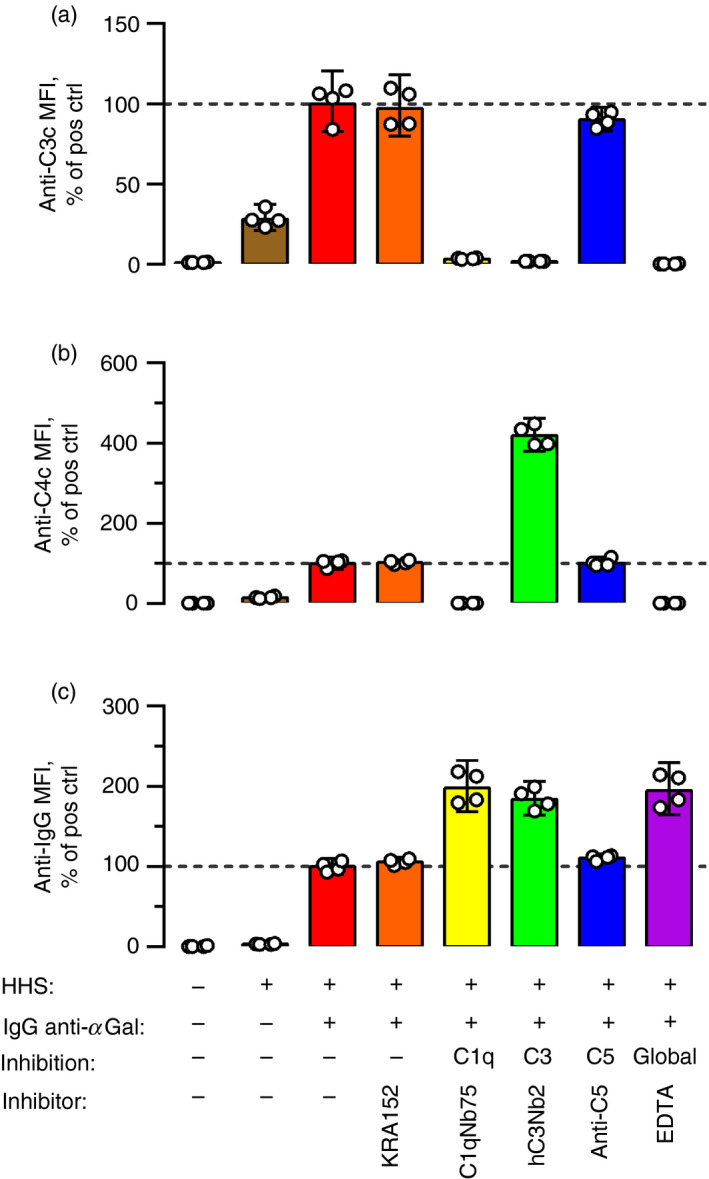
Deposited C3 fragments conceal proximal complement factors and initiating antibodies. Complement deposition and antibody binding on pig red blood cells (pRBCs) were measured by flow cytometry using detection antibodies against C3c (a), C4c (b), or IgG (c). The pRBCs were incubated for 2 hr at 37° in 10% hypogammaglobulinaemia human serum (HHS) as the complement source supplemented with IgG anti‐αGal at 20 mg/l and complement inhibitors as indicated. KRA152 (control nanobody) at 90 mg/l; C1qNb75 (inhibiting C1q docking) at 12 mg/l; hC3Nb2 (binds C3 and blocks the action of the C3 convertases of both classical and alternative pathway) at 90 mg/l, anti‐C5 (eculizumab, binds C5 and blocks the action of the C5 convertases of both classical and alternative pathway) at 50 mg/l, and EDTA at 10 mm. ‘% of pos ctrl’ on the y‐axis denotes experiments containing HHS and IgG anti‐αGal, but no inhibitor. The columns represent the mean of four separate experiments (each represented by a dot). The error bars are 95% confidence intervals. The figure shows that inhibition of C3 cleavage increased the detection of C4b and IgG. IgG detection was also increased by global complement inhibition and inhibition of C1q docking.
In accordance with our concern that C3 fragments could hinder the detection of other proteins, markedly more C4 fragments were detected when C3 fragment generation was blocked using hC3Nb2 (Fig. 3b). As controls, both C1qNb75 and EDTA abrogated deposition of C4 fragments. The addition of eculizumab did not influence the level of detected C4 fragments, further indicating that C5b‐9 complexes do not conceal the upstream complement components.
Moreover, detection of bound IgG anti‐αGal was increased by the inclusion of any of the complement inhibitors, except eculizumab (Fig. 3c). Complement factors upstream of C3 fragments (i.e. C1‐complex and fragments of C2 and C4) did not block detection of antibodies, as C1qNb75 (and EDTA) did not increase the level of detected antibody compared with experiments containing hC3Nb2 (Fig. 3c). These findings emphasize that C3 fragments also concealed the detection of the complement activating antibodies bound on pRBCs.
Based on these results, we inferred that accurate assessment of the level of complement activating antibodies requires inhibition of C3 fragment deposition.
Level of bound IgG anti‐αGal versus complement deposition on pRBCs
To further study the concealment of the initiating antibody by complement, we investigated the level of IgG anti‐αGal detected on pRBCs for different complement concentrations (1%, 4% or 10% HHS). Experiments were performed using various IgG anti‐αGal concentrations in the absence or presence of EDTA as a complement inhibitor. EDTA did not influence IgG anti‐αGal binding on pRBCs (see Supplementary material, Fig. S4). In 1% HHS without EDTA, the level of detected IgG anti‐αGal increased in a concentration‐dependent manner (Fig. 4a). This pattern was not observed for 4% or 10% HHS lacking EDTA. In the latter experiments, the detected antibody level increased with increasing IgG anti‐αGal concentration until approximately 10 mg/l, whereupon the detected antibody plateaued and then started to decrease. The complement deposition signal started to increase around the peak signal of the detected antibody and did not drop when more IgG anti‐αGal was added (see Supplementary material, Fig. S5A–C). The concurrence of these observations suggests that increasing the density of deposited C3 fragments masks the detection of a substantial part of the complement activating antibody. In agreement with this finding, abolition of complement fixation by inclusion of EDTA showed a positive association between the detection of bound antibody and the concentration of IgG anti‐αGal for all HHS concentrations (Fig. 4b). These findings underscore that significant complement deposition masks the initiating antibodies for binding by the detection antibody, which is why accurate measurement of cell‐bound antibody necessitates inhibition of complement.
Figure 4.
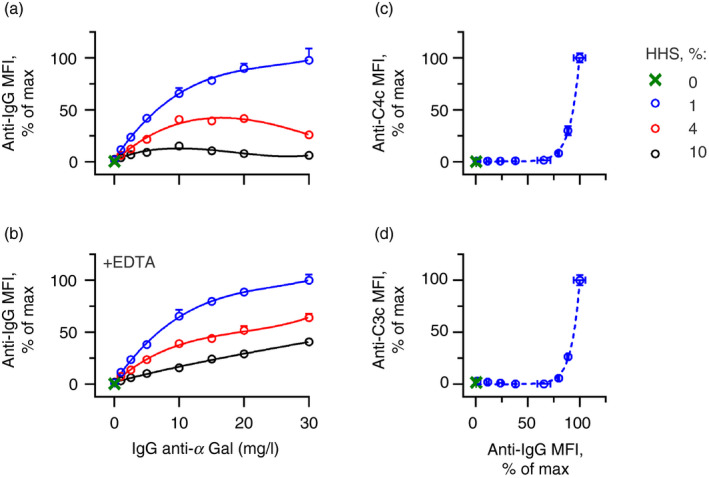
Relation of IgG anti‐αGal density and complement deposition on pig red blood cells (pRBCs). Complement deposition and antibody binding on pRBCs were measured by flow cytometry using detection antibodies against IgG (a and b), C4c (c), or C3c (d). (a) Antibody binding on pRBCs as a function of IgG anti‐αGal concentration are shown for different concentrations of hypogammaglobulinaemia human serum (HHS) as the complement source. (b) As in the previous panel (conducted in parallel) except that the experiments contained 10 mm EDTA. The data are expressed as the mean and standard deviation of two independent experiments. Curve fittings were performed in a third‐order polynomial model. (c and d) Complement deposition as a function of the level of bound IgG anti‐αGal for experiments performed with 1% HHS [antibody binding data from panel (b), blue curve]. Curve fitting was done in an exponential model ((c) P = 6·2 [95% CI 5·8–6·8), R 2 = 1·0; (d) P = 5·6 (95% CI 5·2–6·2), R 2 = 1·0].
The data in Fig. 4(b) also reveal that the level of detected antibody for a given concentration of added IgG anti‐αGal was inversely related to the HHS concentration. As these experiments contained EDTA, this observation is not explained by increased masking of bound IgG at higher complement concentrations. It is more likely that it relates to the small amounts of unbound endogenous IgG present in HHS. Such unbound IgG will adsorb some of the reagents used to detect IgG anti‐αGal on pRBCs. As the concentration of the detection reagent was constant, less reagent was probably available to detect IgG anti‐αGal on pRBCs at higher HHS concentrations. An additional washing step of the pRBCs before the addition of the reagent for IgG detection eliminated the influence of HHS concentration (see Supplementary material, Fig. S6). This interaction does not alter the conclusion that complement inhibition is required for the reliable detection of bound antibodies. In addition, free immunoglobulin must be avoided during detection. In the rest of the presented experiments, cells were washed twice before addition of detection antibodies.
The relationship between deposited complement fragments as a function of the actual level of bound antibody was then evaluated. Cell‐bound antibodies were detected in parallel experiments containing EDTA, and complement deposition was detected in experiments without EDTA. An exponential relationship was evident between antibody binding and complement deposition in 1% HHS (Fig. 4c,d). Complement deposition was negligible at low densities of IgG anti‐αGal but rapidly accelerated when densities exceeded that associated with an IgG anti‐αGal concentration of approximately 15 mg/l (Fig. 4c,d). In 4% and 10% HHS, the relationship followed a sigmoid curve (see Supplementary material, Fig. S7A–D), probably reflecting that complement deposition approximated the maximum possible level. The threshold of bound IgG anti‐αGal required to trigger complement deposition was inversely related to HHS concentration. The antibody density required to achieve half‐maximal complement deposition was approximately 90% of the maximum antibody density (IgG anti‐αGal of approximately 20 mg/l) in 1% HHS (Fig. 4c,d), 70% (IgG anti‐αGal of approximately 15 mg/l) in 4% HHS (see Supplementary material, Fig. S7A and B), and 40% (IgG anti‐αGal of approximately 10 mg/l) in 10% HHS (see Supplementary material, Fig. S7C and D).
The collective results indicate that increasing the density of IgG anti‐αGal does not activate CP on pRBCs before exceeding a critical threshold density, after which complement deposition increased exponentially.
IgG anti‐αGal activates the complement CP on microorganisms
We investigated the ability of IgG anti‐αGal to activate complement on each of the two bacterial strains. The Gram‐negative bacterium EcO86 is often used as a model bacterium for IgG anti‐αGal reactivity. 12 , 14 , 33 As IgG anti‐αGal react more frequently with Gram‐positive bacterial pathogens, 12 we also included Sp9V, which was identified as highly reactive in preliminary screening of 91 pneumococcal serotypes (data being prepared for publication).
We investigated complement deposition in 1% HHS as a function of bacterial‐bound IgG anti‐αGal. For both bacteria, positive associations were observed (Fig. 5a), strongly suggesting that bound antibody initiates complement deposition on the bacteria. However, the relationships differed markedly between the two bacteria. For EcO86, the relation was exponential (R 2 = 0·99), as observed above for pRBCs. Complement deposition started at an IgG anti‐αGal density achieved at approximately 2·5 mg/l of the applied antibody. A direct comparison with the experiments with pRBCs is not justified, because cell sizes and surface epitope density differ markedly between pRBCs and the bacteria. Instead, we compared the relative increments in bound antibody required to double the signal of deposited complement (doubling‐percentage). The doubling‐percentage was approximately fourfold lower for EcO86 (1·5%; 95% CI 1·3%–1·8%) (Fig. 5a) than for pRBCs (5·6%; 95% CI 5·2%–6·2%, Fig. 4d). These findings indicated that IgG anti‐αGal more efficiently initiated the deposition of complement on EcO86. As the two bacteria are similar in size, a direct comparison is more informative. Although Sp9V bound much more IgG anti‐αGal per cell than did EcO86, complement deposition was clearly lower for Sp9V (Fig. 5a). In contrast to EcO86, Sp9V displayed a linear relationship between antibody binding and complement deposition (R 2 = 0·95). Direct comparison was further facilitated by the fact that the two bacteria displayed similar levels of autofluorescence and that HHS alone contributed to negligible IgG binding and complement deposition (see Supplementary material, Fig. S8).
Figure 5.
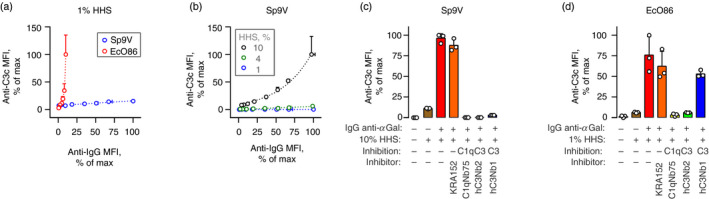
IgG anti‐αGal activates the classical pathway on bacteria. (a) Complement deposition on bacteria as function of the level of IgG anti‐αGal bound on the bacteria measured by flow cytometry (‘anti‐IgG MFI’). Bacteria, Streptococcus pneumonia serotype 9V (Sp9V) or Escherichia coli O86 (EcO86), were incubated in 1% hypogammaglobulinaemia human serum (HHS) as the complement source supplemented with IgG anti‐αGal at 0, 0·1, 1, 2·5, 5, 10, 15, 20 or 30 mg/l. The data are expressed as the mean and standard deviation of two independent experiments. Curve fittings were performed using a linear regression model for Sp9V (R 2 = 0·94) and an exponential model for EcO86 (doubling‐percentage, P = 1·5% (95% CI 1·3%–1·8%), R 2 = 0·99). P represents the increase in IgG binding signal associated with doubling of the complement deposition signal. (b) As in the previous panel but experiments were conducted for Sp9V in 1%, 4% or 10% HHS. Curve fitting was performed using linear regression models for 1% HHS (slope 0·00; 95% CI 0·0017–0·0028; R 2 = 0·95) and 4% HHS (slope 0·052; 95% CI 0·040–0·064; R 2 = 0·95) but in an exponential model for 10% HHS (P = 29% (95% CI 26%–32%), R 2 = 0·99). (c) Sp9V, IgG anti‐αGal at 20 mg/l and 10% HHS were incubated at 37° with or without nanobodies: KRA152 (irrelevant specificity), C1qNb75 (inhibits C1q docking to Ig), hC3Nb2 (inhibits C3 cleavage by CP and AP) and hC3Nb1 (inhibits C3 cleavage by AP). Complement deposition on bacteria was detected by flow cytometry. The data are expressed as the mean (bars) of three separate experiments (each represented by a dot) and standard deviation (error bars). (d) As in the previous panel, but for EcO86 in 1% HHS.
Taken together, these findings demonstrate that IgG anti‐αGal is able to activate complement on bacteria, but the relationship to the density of bound antibodies differs widely between strains and bacterial species.
We examined complement deposition by IgG anti‐αGal on Sp9V using higher concentrations of HHS, that is, with more available complement. In 4% HHS, a nearly linear relationship between IgG anti‐αGal binding and deposition of complement was also observed (R 2 = 0·95) (Fig. 5b). The slope was greater than that observed with 1% HHS (Fig. 5b), so we concluded that complement deposition was more effective in 4% HHS. Complement deposition was clearly more efficient in 10% HHS (Fig. 5b) where an exponential model provided close approximation (R 2 = 0·99). Complement deposition on Sp9V was markedly less efficient than on pRBCs and EcO86. For Sp9V in 10% HHS, the doubling‐percentage was 29% (95% CI 26%–32%), which was 20‐fold higher than for EcO86 (Fig. 5a) and fivefold higher than for pRBCs (Fig. 4d), despite the latter experiments being performed in only 1% HHS. Hence, IgG anti‐αGal activates complement on Sp9V but is much less efficient than on EcO86 and pRBCs.
Complement deposition on the bacteria was investigated using specific anti‐complement factor nanobodies. For Sp9V in 10% HHS, blockade of C1q binding abolished deposition (reading < 0·4% of the uninhibited experiment) (Fig. 5c), proving that activation required the CP. However, AP provided pivotal amplification as hC3Nb1 (which preferentially inhibits C3 cleavage by AP) and produced nearly as efficient an inhibition as hC3Nb2 (which inhibits C3 cleavage by both AP and CP). The control nanobody having irrelevant specificity caused no inhibition. We conducted similar experiments for EcO86, but in 1% HHS, and observed similar findings, except that selective blockade of AP C3 convertase was markedly less efficient than the simultaneous inhibition of both convertases (Fig. 5d). We suspect that this result reflects that the dilution of HHS to 1% compromises AP function. However, experiments in 10% HHS were not feasible for EcO86 because HHS alone produced a very high level of background complement deposition (see Supplementary material, Fig. S9), driven spontaneously by the AP or by endogenous anti‐EcO86 antibodies in HHS (see Supplementary material, Fig. S10).
The collective findings supported the conclusion that IgG anti‐αGal can activate the CP on microorganisms and recruit amplification from the AP.
IgG anti‐αGal activates complement on cells more effectively than antibodies in nhIgG
We compared complement deposition on target cells by IgG anti‐αGal with that elicited by the source of the antibody, that is, the preparation of nhIgG used for the affinity isolation of the IgG anti‐αGal. This nhIgG is comprised of approximately 0·1% IgG anti‐αGal 12 and contains many antibodies that are unlikely to react with the target cells. Consistent with this suggestion, far lower concentrations of IgG anti‐αGal than nhIgG were required to achieve a given level of cell‐bound IgG on Sp9V, EcO86 and pRBCs (Fig. 6a–c). In addition, the relationship between IgG binding and IgG concentration was less steep for nhIgG (and was most extreme with pRBCs). This could indicate that cell binding of IgG anti‐αGal in the nhIgG is inhibited by other antibodies present in the nhIgG. In support of this inhibition, binding of the affinity isolated IgG anti‐αGal to pRBCs was inhibited by the addition of nhIgG (see Supplementary material, Fig. S11 and S12). Hence, we chose to compare complement activation as a function of bound antibody. With Sp9V in 10% HHS, the two IgG preparations displayed similar ranges of cell binding (Fig. 6a,d). Increasing the density of purified IgG anti‐αGal on Sp9V was accompanied by a linear increase in complement deposition (Fig. 6d). In contrast, increasing the density of nhIgG on Sp9V resulted in an initial increase in complement deposition that quickly plateaued and finally decreased (Fig. 6d). With EcO86 in 4% HHS, increasing densities of purified IgG anti‐αGal produced complement deposition in a sigmoid manner, whereas increasing densities of nhIgG produced no complement deposition (Fig. 6e). With pRBCs in 4% HHS, increasing densities of purified IgG anti‐αGal produced complement deposition in an exponential manner, whereas nhIgG produced no complement deposition (Fig. 6f).
Figure 6.
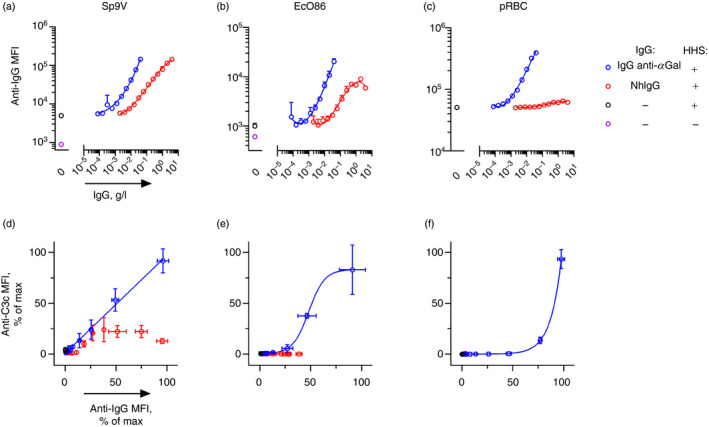
Binding and complement activation on cells by IgG anti‐αGal compared with normal human IgG. (a–c) Cell‐bound IgG as a function of IgG concentration examined by flow cytometry. IgG used in the experiments was either purified IgG anti‐αGal or was the source material for the purified IgG anti‐αGal (normal human IgG, nhIgG). Experiments contained 10 mm EDTA and hypogammaglobulinaemia human serum (HHS) (concentration in (a), 0% (−) or 10% (+); and in (b and c), 0% (−) or 4% (+)). The data are expressed as the mean and standard deviation of two independent experiments. Curve fitting was done by third‐order polynomial approximation. (d–f) Deposited C3 fragments as a function of cell‐bound IgG were examined by flow cytometry. Complement deposition was assayed in parallel and equivalent experiments as in panels (a–c) but without EDTA. The data for cell‐bound IgG were from panels (a–c). Curve fittings for IgG anti‐αGal‐associated data were performed in a linear model (d, R 2 = 1·00), a sigmoid model (e, R 2 = 1·00) and an exponential model (f, R 2 = 1·00).
We conclude that purified IgG anti‐αGal more efficiently activates complement on target cells than nhIgG. Moreover, nhIgG may inhibit the action of purified IgG anti‐αGal.
Inherent activation of the CP is similar for nhIgG and purified IgG anti‐αGal
We compared the inherent ability of the complement activation of IgG anti‐αGal and its source material, nhIgG. The two IgG preparations were coated directly onto microtitre wells. The amount of bound IgG was detected with anti‐IgG antibodies and complement deposition from NHS onto the surfaces was compared using anti‐C4c antibody. For both preparations, deposition was exponentially related to the amount of coated IgG (Fig. 7). The nhIgG supported deposition at slightly lower densities than IgG anti‐αGal (see the leftward curve of nhIgG in Fig. 7). On the other hand, IgG anti‐αGal more efficiently caused deposition, once deposition commenced, as doubling of deposited complement required 6% (95% CI 5%–7%) more IgG signal from the IgG anti‐αGal coat, but 9% (95% CI 7%–14%) more IgG signal from the nhIgG coat.
Figure 7.
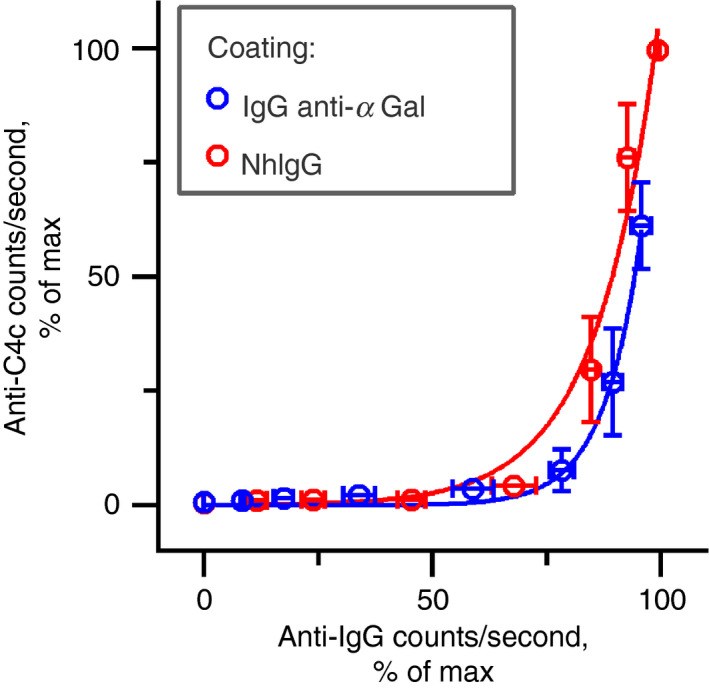
IgG anti‐αGal and normal human IgG display similar inherent ability to activate the classical complement pathway. Complement deposition as a function of IgG density determined by solid‐phase immunoassay. Microtitre wells were coated with purified IgG anti‐αGal or nhIgG (normal human IgG used as the starting material for purification of the IgG anti‐αGal). The complement source [1% normal human serum (NHS)] was added, the plate was incubated at 37°, and the deposited complement was measured with anti‐C4c. The level of coated IgG was measured with anti‐IgG in parallel experiments that were identical, except for the lack of NHS. The data are expressed as the mean with 95% confidence intervals of four independent experiments.
As a control of the assay, the two antibody preparations did not show different potentials for coating (see Supplementary material, Fig. S13). Furthermore, addition of C1qNb75 to experiments on the IgG anti‐αGal coat abrogated complement deposition, indicating that activation progressed through CP (see Supplementary material, Fig. S14).
We conclude that IgG anti‐αGal and nhIgG deposited in microtitre wells did not differ importantly in their ability to activate CP despite their difference in IgG subclass composition, with the first being 76% IgG2 and the second being 71% IgG1. 12
Discussion
Antibody binding on invading pathogens is essential for the killing of many intruding pathogens by activating complement and enhancing phagocytosis. Nevertheless, several human IgG antibodies have been reported to attenuate complement deposition on virulent bacteria and thereby have been proposed to be deleterious to host defences. 2 , 3 , 4 , 5 , 6 We investigated complement activation by IgG anti‐αGal, which is claimed to enhance infection. 4
We used antibodies and complement of human origin, which are obviously important to use in studies of human immunology. The antibody source was IgG anti‐αGal purified from therapeutic grade nhIgG. This has recently been characterized. 12 The complement source was serum from a person with hypogammaglobulinaemia. The serum was not totally devoid of endogenous immunoglobulin, which limited its utility in some assays because of the background signal. An alternative strategy would have been to deplete a serum for immunoglobulin. However, we refrained from doing this because complement components are likely to be depleted concomitantly, leading to impaired complement activity. 34
In contrast to the conclusion of earlier studies, 4 , 8 , 15 , 17 , 18 , 19 we found that IgG anti‐αGal is fully capable of activating the classical complement pathway on pRBCs as well as on bacteria. It is of interest to speculate why other studies observed complement inhibition by IgG anti‐αGal. 4 , 8 , 18 Hamadeh et al. reported that IgG anti‐αGal did not activate CP and moreover inhibited AP‐mediated killing of a Serratia strain. 4 The authors did not provide the mechanisms. We observed that IgG anti‐αGal efficiently activated the CP and showed no signs of inhibiting the AP. In contrast, we found that the AP played an important role in amplifying CP activation by IgG anti‐αGal. From the current understanding of AP activation, it is difficult to imagine how antibodies bound to a cell should protect the cell from AP activation (e.g. the antibody itself may well be substrate for C3b deposition). IgG anti‐αGal may inhibit CP by occupying binding sites of potent activators. 8 , 18 We found that increasing the density of cell‐bound IgG anti‐αGal may not activate the CP before reaching threshold levels, after which complement deposition increases exponentially with IgG density. The same applied for nhIgG and IgG anti‐αGal directly coated onto plastic surfaces. We expect this relationship to reflect that several IgG molecules must be in very close proximity to bind the hexavalent C1q, which has low functional affinity for single IgG molecules, 35 , 36 but at least 1000 times higher functional affinity for multiple clustered IgG molecules. 36 , 37 This is because the binding strength between one IgG molecule and one of the C1q globular heads is low, 38 whereas several closely positioned IgG molecules can provide a pattern that allows the C1q molecule to deploy several of its six globular heads and establish multivalent binding. 39 This markedly increases the binding strength and allows CP activation. However, this does not apply to all C1q fixing molecules. For instance, a single molecule of IgM can fix C1q. 40 IgG anti‐αGal at densities below the threshold for CP activation may occupy binding sites for IgM antibodies and thereby suppress complement deposition. We propose that all IgG antibodies that are displayed at configurations incompatible with proper C1q docking (i.e. scattered locations and a mutual distance that is too long) have the potential to inhibit the action of single molecule complement activators. The mechanism may explain the past findings of complement inhibition by IgG antibodies.
Preparations of nhIgG inhibited the binding of purified IgG anti‐αGal to the studied cells. It may be that the inhibition was caused by the anti‐idiotype antibodies present in nhIgG. Pools of nhIgG contain a mixture of many anti‐idiotypic antibodies, which may form dimers of IgG. The proportion of such dimers increases with the number of individuals contributing to the pool, with up to 40% of the IgG in pools for therapeutic use. 41 , 42 It is conceivable that the IgG anti‐αGal fraction is efficiently released from the anti‐idiotypic antibodies during the purification procedures with extreme antigen excess. This could remove the anti‐idiotypic antibodies that react with IgG anti‐αGal and would explain the markedly better binding of the affinity isolated preparation. Moreover, for similar densities of cell‐bound IgG, nhIgG seemed to cause less complement activation than IgG anti‐αGal. However, as the potential inhibitory effects of nhIgG are not essential to the present study, we did not examine the mechanisms any further.
Our experimental design incorporated only a limited part of the human immune defence ‐ antibody and complement why inference to the importance of IgG anti‐αGal in vivo requires caution. Keeping this in mind, our observations indicated that complement activation may require higher concentrations of IgG anti‐αGal than the average concentration found in the plasma of healthy humans (approximately 10 mg/l). 8 , 9 , 10 However, we also observed that the density of cell‐bound IgG anti‐αGal required for CP activation decreased when the complement concentration was increased. We anticipate that 10‐ to 100‐fold higher complement concentrations in human plasma than those used in our experiments support CP activation at lower levels of IgG anti‐αGal. Data from humans are warranted because IgG anti‐αGal and complement obviously do not work in isolation in vivo but operate in concert with other anti‐bacterial antibodies as well as many other defence mechanisms.
Interestingly, although IgG anti‐αGal bound to the Sp9V strain markedly better than the EcO86 strain, this difference did not translate into more efficient complement deposition. Possibly, some configurations of bound antibody may be suboptimal for C1q docking despite being dense. This may be related to the surface structures of the microorganisms and/or possession of complement‐inhibiting molecules. Moreover, we also found that the nature of the relationship between antibody binding and complement deposition differed markedly among the bacteria. The relationship was exponential for EcO86 (and pRBCs as well as on plastic surfaces), but more linear with Sp9V. Exponential relationships may indicate an abrupt ability to fix C1q when the IgG density increases enough to allow sufficiently strong binding of C1 complexes. The linear relationship was more surprising. It will be interesting to determine the mechanisms for the differences.
Our results provide at least three additional unexpected insights. First, IgG anti‐αGal, despite being mostly IgG2 (76% IgG2 12 ), possessed an inherent ability to activate complement that was very similar to that of nhIgG, which mostly contains IgG1 (71% IgG1 12 ). These data may challenge the notion that IgG2 is a less efficient complement activator compared with IgG1. In agreement with our results, recent findings by others have also suggested that IgG1 and IgG2 may not differ significantly in their propensity to activate complement. 43 In further support of important CP activation by IgG2 antibodies, individuals with CP deficiencies are particularly prone to infections with encapsulated bacteria, mainly S. pneumoniae, 44 and human antibodies to such pathogens are mainly IgG2. 45 , 46 Second, the present results reveal that C3 fragments deposited on cells can conceal the detection of upstream components, such as C4b, and the initiating antibodies. This phenomenon is well‐recognized in bead‐based assays used to quantify antibodies to human leucocyte antigens. 47 However, our findings emphasize that this phenomenon also applies to cells. Hence, the level of bound antibody should be determined in separate, parallel experiments with added complement inhibitors, such as EDTA, or one of the complement reactive nanobodies that were used. Third, our finding that IgG anti‐αGal binds Sp9V, which does not carry the Galα3Gal moiety in its capsular polysaccharide, 48 supports the notion that the antibody can bind additional microbial epitopes beyond terminal Galα3Gal. Indeed, IgG anti‐αGal has been reported to bind additional antigens, although of non‐microbial origin, 49 , 50 but the actual polyspecificity for microbial epitopes remains unknown.
In conclusion, the carbohydrate reactive IgG anti‐αGal can activate the complement CP on target cells. This insight is important as IgG anti‐αGal is naturally occurring, abundant in humans, 8 , 10 and targets a wide range of common pathogens. 4 , 12 The contribution of IgG anti‐αGal to human immunity awaits future studies.
Disclosure
The authors declare no competing interests.
Author contributions
JMBJ designed the study, performed the experiments, analysed the data, and wrote the first draft. NSL, RKJ, GRA, JCJ, USS and ST provided resources, and reviewed and edited the manuscript. All authors approved the final manuscript.
Supporting information
Appendix S1. Preparation of the Galα3Gal‐derivatized beads.
Figure S1. Antibody level against pig red blood cells in human hypogammaglobulinaemic serum relative to normal human serum.
Figure S2. No complement deposition was initiated by irrelevant antibody on pig red blood cells or IgG anti‐αGal on human red blood cells that are devoid of Galα3Gal.
Figure S3. Alternative pathway amplifies classical pathway activation but is not spontaneously active.
Figure S4. Effect of EDTA upon anti‐αGal binding to pig red blood cells.
Figure S5. Relations between IgG anti‐αGal concentration, IgG binding, and complement deposition on pig red blood cells.
Figure S6. Importance of number of washes upon anti‐αGal detection on pig red blood cells.
Figure S7. Relation between IgG anti‐αGal density and complement deposition on pig red blood cells.
Figure S8. Anti‐bacterial antibodies in human hypogammaglobulinaemic serum.
Figure S9. Complement deposition on Escherichia coli O86 from human hypogammaglobulinaemic serum.
Figure S10. Content of anti‐bacterial antibodies in human hypogammaglobulinaemic serum compared with normal human serum.
Figure S11. Normal human IgG inhibits binding of purified IgG anti‐αGal to pig red blood cells.
Figure S12. Comparison of normal human IgG pools inhibitory effect on pig red blood cell binding of purified IgG anti‐αGal.
Figure S13. Coating of antibodies in microtitre wells.
Figure S14. Complement activation by IgG anti‐αGal on plastic surface progress through classical pathway.
Acknowledgements
This work was supported by an Aarhus University grant to JMBJ, Bloddonorernes Forskningsfond, and Forskningstræningspuljen, Region Midt.
Senior author: Steffen Thiel
References
- 1. Halstead SB, O'Rourke EJ. Dengue viruses and mononuclear phagocytes. I. infection enhancement by non‐neutralizing antibody. J Exp Med 1977; 146:201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guttman RM, Waisbren BA. Bacterial blocking activity of specific IgG in chronic Pseudomonas aeruginosa infection. Clin Exp Immunol 1975; 19:121–30. [PMC free article] [PubMed] [Google Scholar]
- 3. Rice PA, Vayo HE, Tam MR, Blake MS. Immunoglobulin G antibodies directed against protein III block killing of serum‐resistant Neisseria gonorrhoeae by immune serum. J Exp Med 1986; 164:1735–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hamadeh RM, Jarvis GA, Galili U, Mandrell RE, Zhou P, Griffiss JM. Human natural anti‐gal IgG regulates alternative complement pathway activation on bacterial surfaces. J Clin Invest 1992; 89:1223–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ray TD, Lewis LA, Gulati S, Rice PA, Ram S. Novel blocking human IgG directed against the pentapeptide repeat motifs of Neisseria meningitidis Lip/H.8 and laz lipoproteins. J Immunol 2011; 186:4881–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wells TJ, Whitters D, Sevastsyanovich YR, Heath JN, Pravin J, Goodall M et al Increased severity of respiratory infections associated with elevated anti‐LPS IgG2 which inhibits serum bactericidal killing. J Exp Med 2014; 211:1893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM et al Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol 2015; 136:205. e1–78. [DOI] [PubMed] [Google Scholar]
- 8. Yu PB, Holzknecht ZE, Bruno D, Parker W, Platt JL. Modulation of natural IgM binding and complement activation by natural IgG antibodies: a role for IgG anti‐gal α1‐3Gal antibodies. J Immunol 1996; 157:5163–8. [PubMed] [Google Scholar]
- 9. Barreau N, Blancho G, Boulet C, Martineau A, Vusio P, Liaigre J et al Natural anti‐gal antibodies constitute 0.2% of intravenous immunoglobulin and are equally retained on a synthetic disaccharide column or on an immobilized natural glycoprotein. Transplant Proc 2000; 32:882–3. [DOI] [PubMed] [Google Scholar]
- 10. Bernth‐Jensen JM, Moller BK, Jensenius JC, Thiel S. Biological variation of anti‐αGal‐antibodies studied by a novel time‐resolved ImmunoFluorometric assay. J Immunol Methods 2011; 373:26–35. [DOI] [PubMed] [Google Scholar]
- 11. Wetter LA, Hamadeh RM, Griffiss JM, Oesterle A, Aagaard B, Way LW. Differences in outer membrane characteristics between gallstone‐associated bacteria and normal bacterial flora. Lancet 1994; 343:444–8. [DOI] [PubMed] [Google Scholar]
- 12. Bernth Jensen JM, Petersen MS, Ellerman‐Eriksen S, Moller BK, Jensenius JC, Sorensen UBS et al Abundant human anti‐Galα3Gal antibodies display broad pathogen reactivity. Sci Rep 2020; 10:4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Galili U, Clark MR, Shohet SB, Buehler J, Macher BA. Evolutionary relationship between the natural anti‐gal antibody and the gal α 1–3Gal epitope in primates. Proc Natl Acad Sci U S A 1987; 84:1369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffiss JM. Interaction between human natural anti‐α‐galactosyl immunoglobulin G and bacteria of the human flora. Infect Immun 1988; 56:1730–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parker W, Bruno D, Holzknecht ZE, Platt JL. Characterization and affinity isolation of xenoreactive human natural antibodies. J Immunol 1994; 153:3791–803. [PubMed] [Google Scholar]
- 16. Collins BH, Cotterell AH, McCurry KR, Alvarado CG, Magee JC, Parker W et al Cardiac xenografts between primate species provide evidence for the importance of the α‐galactosyl determinant in hyperacute rejection. J Immunol 1995; 154:5500–10. [PubMed] [Google Scholar]
- 17. Yu PB, Parker W, Everett ML, Fox IJ, Platt JL. Immunochemical properties of anti‐Gal α 1–3Gal antibodies after sensitization with xenogeneic tissues. J Clin Immunol 1999; 19:116–26. [DOI] [PubMed] [Google Scholar]
- 18. Magee JC, Collins BH, Harland RC, Lindman BJ, Bollinger RR, Frank MM et al Immunoglobulin prevents complement‐mediated hyperacute rejection in swine‐to‐primate xenotransplantation. J Clin Invest 1995; 96:2404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Minanov OP, Itescu S, Neethling FA, Morgenthau AS, Kwiatkowski P, Cooper DK et al Anti‐GaL IgG antibodies in sera of newborn humans and baboons and its significance in pig xenotransplantation. Transplantation 1997; 63:182–6. [DOI] [PubMed] [Google Scholar]
- 20. Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R et al Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med 1987; 166:1351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bindon CI, Hale G, Bruggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med 1988; 168:127–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lucisano Valim YM, Lachmann PJ. The effect of antibody isotype and antigenic epitope density on the complement‐fixing activity of immune complexes: A systematic study using chimaeric anti‐NIP antibodies with human fc regions. Clin Exp Immunol 1991; 84:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014; 5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Irani V, Guy AJ, Andrew D, Beeson JG, Ramsland PA, Richards JS. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol Immunol 2015; 67:171–82. [DOI] [PubMed] [Google Scholar]
- 25. Davies AM, Sutton BJ. Human IgG4: A structural perspective. Immunol Rev 2015; 268:139–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schatz‐Jakobsen JA, Zhang Y, Johnson K, Neill A, Sheridan D, Andersen GR. Structural basis for eculizumab‐mediated inhibition of the complement terminal pathway. J Immunol 2016; 197:337–44. [DOI] [PubMed] [Google Scholar]
- 27. Jensen RK, Pihl R, Gadeberg TAF, Jensen JK, Andersen KR, Thiel S et al A potent complement factor C3‐specific nanobody inhibiting multiple functions in the alternative pathway of human and murine complement. J Biol Chem 2018; 293:6269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laursen NS, Pedersen DV, Gytz H, Zarantonello A, Bernth Jensen JM, Hansen AG et al Functional and structural characterization of a potent C1q inhibitor targeting the classical pathway of the complement system. Front Immunol 2020. 10.3389/fimmu.2020.01504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pedersen H, Jensen RK, Hansen AG, Gadeberg TAF, Thiel S, Laursen NS et al A C3 specific nanobody that blocks all three activation pathways in the human and murine complement system. J Biol Chem 2020; 295(26):8746–8758. 10.1074/jbc.RA119.012339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dillon SP, D'Souza A, Kurien BT, Scofield RH. Systemic lupus erythematosus and C1q: A quantitative ELISA for determining C1q levels in serum. Biotechnol J 2009; 4:1210–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agarwal Aea . The complement FactsBook, 2nd edn. London: Academic Press, 2018: 514. [Google Scholar]
- 32. Kohler PF, Muller‐Eberhard HJ. Immunochemical quantitation of the third, fourth and fifth components of human complement: concentrations in the serum of healthy adults. J Immunol 1967; 99:1211–6. [PubMed] [Google Scholar]
- 33. Posekany KJ, Pittman HK, Bradfield JF, Haisch CE, Verbanac KM. Induction of cytolytic anti‐gal antibodies in α‐1,3‐galactosyltransferase gene knockout mice by oral inoculation with Escherichia coli O86:B7 bacteria. Infect Immun 2002; 70:6215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mutti M, Ramoni K, Nagy G, Nagy E, Szijártó V. A new tool for complement research: In vitro reconstituted human classical complement pathway. Front Immunol 2018; 9:2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sledge CR, Bing DH. Binding properties of the human complement protein Clq. J Biol Chem 1973; 248:2818. [PubMed] [Google Scholar]
- 36. Hughes‐Jones NC, Gardner B. The reaction between the complement subcomponent C1q. IgG complexes and polyionic molecules. Immunology 1978; 34:459–63. [PMC free article] [PubMed] [Google Scholar]
- 37. Lin TY, Fletcher DS. Interaction of human Clq with insoluble immunoglobulin aggregates. Immunochemistry 1978; 15:107–17. [DOI] [PubMed] [Google Scholar]
- 38. Hughes‐Jones NC, Gardner B. Reaction between the isolated globular sub‐units of the complement component C1q and IgG‐complexes. Mol Immunol 1979; 16:697–701. [DOI] [PubMed] [Google Scholar]
- 39. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA et al Complement is activated by IgG hexamers assembled at the cell surface. Science 2014; 343:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Borsos T, Rapp HJ. Complement fixation on cell surfaces by 19S and 7S antibodies. Science 1965; 150:505–6. [DOI] [PubMed] [Google Scholar]
- 41. Tankersley DL, Preston MS, Finlayson JS. Immunoglobulin G dimer: an idiotype–anti‐idiotype complex. Mol Immunol 1988; 25:41–8. [DOI] [PubMed] [Google Scholar]
- 42. Roux KH, Tankersley DL. A view of the human idiotypic repertoire. electron microscopic and immunologic analyses of spontaneous idiotype‐anti‐idiotype dimers in pooled human IgG. J Immunol 1990; 144:1387–95. [PubMed] [Google Scholar]
- 43. Nishiya CT, Boxx GM, Robison K, Itatani C, Kozel TR, Zhang MX. Influence of IgG subclass on human antimannan antibody‐mediated resistance to hematogenously disseminated candidiasis in mice. Infect Immun 2015; 84:386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skattum L, van Deuren M, van der Poll T, Truedsson L. Complement deficiency states and associated infections. Mol Immunol 2011; 48:1643–55. [DOI] [PubMed] [Google Scholar]
- 45. Siber GR, Schur PH, Aisenberg AC, Weitzman SA, Schiffman G. Correlation between serum IgG‐2 concentrations and the antibody response to bacterial polysaccharide antigens. N Engl J Med 1980; 303:178–82. [DOI] [PubMed] [Google Scholar]
- 46. Barrett DJ, Ayoub EM. IgG2 subclass restriction of antibody to pneumococcal polysaccharides. Clin Exp Immunol 1986; 63:127–34. [PMC free article] [PubMed] [Google Scholar]
- 47. Schwaiger E, Wahrmann M, Bond G, Eskandary F, Bohmig GA. Complement component C3 activation: The leading cause of the prozone phenomenon affecting HLA antibody detection on single‐antigen beads. Transplantation 2014; 97:1279–85. [DOI] [PubMed] [Google Scholar]
- 48. Calix JJ, Saad JS, Brady AM, Nahm MH. Structural characterization of Streptococcus pneumoniae serotype 9A capsule polysaccharide reveals role of glycosyl 6‐O‐acetyltransferase wcjE in serotype 9V capsule biosynthesis and immunogenicity. J Biol Chem 2012; 287:13996–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sandrin MS, Vaughan HA, Xing PX, McKenzie IF. Natural human anti‐gal α(1,3)gal antibodies react with human mucin peptides. Glycoconj J 1997; 14:97–105. [DOI] [PubMed] [Google Scholar]
- 50. Satapathy AK, Ravindran B. Naturally occurring α‐galactosyl antibodies in human sera display polyreactivity. Immunol Lett 1999; 69:347–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Preparation of the Galα3Gal‐derivatized beads.
Figure S1. Antibody level against pig red blood cells in human hypogammaglobulinaemic serum relative to normal human serum.
Figure S2. No complement deposition was initiated by irrelevant antibody on pig red blood cells or IgG anti‐αGal on human red blood cells that are devoid of Galα3Gal.
Figure S3. Alternative pathway amplifies classical pathway activation but is not spontaneously active.
Figure S4. Effect of EDTA upon anti‐αGal binding to pig red blood cells.
Figure S5. Relations between IgG anti‐αGal concentration, IgG binding, and complement deposition on pig red blood cells.
Figure S6. Importance of number of washes upon anti‐αGal detection on pig red blood cells.
Figure S7. Relation between IgG anti‐αGal density and complement deposition on pig red blood cells.
Figure S8. Anti‐bacterial antibodies in human hypogammaglobulinaemic serum.
Figure S9. Complement deposition on Escherichia coli O86 from human hypogammaglobulinaemic serum.
Figure S10. Content of anti‐bacterial antibodies in human hypogammaglobulinaemic serum compared with normal human serum.
Figure S11. Normal human IgG inhibits binding of purified IgG anti‐αGal to pig red blood cells.
Figure S12. Comparison of normal human IgG pools inhibitory effect on pig red blood cell binding of purified IgG anti‐αGal.
Figure S13. Coating of antibodies in microtitre wells.
Figure S14. Complement activation by IgG anti‐αGal on plastic surface progress through classical pathway.