

Key Points
Question
What is the association between MUC16 mutation and response to immune checkpoint inhibitors (ICIs) in solid tumors?
Findings
In this cohort study of 3 groups of patients, including The Cancer Genome Atlas cohort with 10 195 patients across 30 solid tumor types, 56 patients with non–small cell lung cancer, and 145 patients with melanoma, MUC16 mutation was associated with greater response rates, prolonged overall survival, and genomic factors associated with ICI response, including tumor mutational burden and programmed cell death ligand–1 expression.
Meaning
These findings suggest that MUC16 mutation may be associated with superior response to ICIs in patients with solid tumors.
This cohort study examines whether MUC16 mutation is associated with response to treatment with immune checkpoint inhibitors and outcomes among patients with solid tumors, non–small cell lung cancer, and melanoma.
Abstract
Importance
As the third most frequently mutated gene in cancers, the association between MUC16 mutation and response to immune checkpoint inhibitors (ICIs) in solid tumors remains unclear.
Objective
To examine whether MUC16 mutation is associated with genomic factors in ICI response in solid tumors and with outcomes in ICI-treated patients.
Design, Setting, and Participants
This cohort study used multidimensional genomic data of 10 195 patients from The Cancer Genome Atlas (TCGA) across 30 solid tumor types, 56 patients from a non–small cell lung cancer (NSCLC) cohort, and 145 patients from a melanoma cohort. Genomic factors associated with ICI response covered tumor mutational burden, neoantigens, immune-related gene signatures, and tumor immune microenvironment. Both NSCLC and melanoma cohorts included ICI-treated patients. The TCGA cohort was used to examine the association of MUC16 mutation with genomic factors. Two ICI-treated cohorts were used to explore the significance of outcomes associated with MUC16 mutation, using Kaplan-Meier curves and Cox models with adjusting for potential confounders. Gene set enrichment analysis was used to identify MUC16 mutation–associated biological processes. Data were obtained from October 1 through October 10, 2019, and were analyzed from October 11 through December 31, 2019.
Main Outcomes and Measures
Genomic factors associated with ICI response, overall survival, and clinical response.
Results
Of the 10 195 patients, 4821 (47.6%) were men (median [interquartile range {IQR}] age, 60 [50-70] years). MUC16 was mutated in 2006 of 10 195 patients (19.68%). In this pan-cancer data set, patients with MUC16 mutation had higher tumor mutational burden (median [IQR], 230 [93-595] mutations vs 48 [25-92] mutations; difference, 182 mutations; 95% CI, 164-199 mutations; P < .001) and neoantigen load (median [IQR], 179 [74-394.5] neoantigens vs 48 [24-89] neoantigens; difference, 131 antigens; 95% CI, 116.5-145 neoantigens; P < .001) than those without mutations. The tumor immune microenvironment with dual-positive CD8A and PD-L1 was overrepresented in MUC16-mutated tumors compared with wild-type ones (43.8% vs 32.4%; odds ratio, 1.63; 95% CI, 1.46-1.80; P < .001). Of the 40 immune-related genes, 37 (92.5%) exhibited differential expression between 2 states. MUC16 mutation was associated with improved overall survival in both the NSCLC (hazard ratio, 0.34; 95% CI, 0.12-0.99; P = .04) and melanoma (hazard ratio, 0.57; 95% CI, 0.36-0.90; P = .02) cohorts. The improvement persisted after adjusting for age, sex, and dominant mutational signatures in the melanoma cohort (hazard ratio, 0.57; 95% CI, 0.33-0.96; P = .04). MUC16 mutation was associated with greater response rates in the NSCLC cohort (odds ratio, 4.03; 95% CI, 1.06-16.43; P = .03) and the melanoma cohort (odds ratio, 3.38; 95% CI, 1.07-14.25; P = .03). Gene set enrichment analysis revealed that gene sets regarding cell proliferation and immune response were enriched in MUC16-mutated tumors (false discovery rate, <.001).
Conclusions and Relevance
MUC16 mutation appears to be associated with reported genomic factors associated with response to and improved outcomes for ICI treatment in solid tumors. It may hold promise as a marker for guiding immunotherapeutic responsiveness.
Introduction
Immune checkpoint inhibitor (ICI)–based immunotherapy, which mainly targets cytotoxic T lymphocyte–associated protein 4 (CTLA-4), programmed cell death 1 (PD-1), and its ligand (PD-L1), has shown remarkable clinical benefits to patients with advanced-stage cancers.1 Early studies2 suggest that ICI response may be associated with PD-L1 protein levels, tumor mutational burden, neoantigens, tumor-infiltrating lymphocytes (TILs), T cell receptor clonality, and transcriptional signatures.
As the third most frequently mutated gene according to The Cancer Genome Atlas (TCGA), MUC16 (OMIM 606154) encodes a membrane-spanning mucin, comprising a tandem repeat region sandwiched between N-terminal and C-terminal domains.3,4 Tumor antigen 125, residing in the tandem repeat region, is a common clinical biomarker to monitor epithelial ovarian cancer.3 A recent study5 reported that MUC16 mutation may be associated with elevated tumor mutational burden and superior overall survival (OS) in patients with gastric adenocarcinoma. However, a comprehensive analysis of the relationship of MUC16 mutation with ICI response across a large set of solid tumors is lacking. Here, we explored the associations of MUC16 mutation with ICI response based on multidimensional data from multiple solid tumors.
Methods
Multidimensional Data Collection
This study was exempted from approval by an institutional review board and from the need for informed consent because its data were gathered from publicly available data sets that have received approval, in accordance with 45 CFR §46. This study follows the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Somatic mutations, neoantigens, and gene expression data for TCGA samples from multiple solid tumor types were gathered. Specifically, somatic mutations of 10 195 samples and mRNA expression profiles of 9850 samples for 30 solid tumor types were downloaded from the TCGA Pan-Cancer Atlas project deposited in the cBioPortal database.6,7 These somatic mutations were uniformly called by the Multi-Center Mutation Calling in Multiple Cancers working group to adjust for variance and batch effects introduced by the differences in DNA extraction, hybridization-capture, sequencing, and analytical methods over time, which enables robust cross-tumor-type analyses.8 The mRNA expression levels were quantified using RNA-Seq by Expectation-Maximization algorithm9; batch-corrected for sequencers (Illumina GAII and HiSeq), sequencing centers (University of North Carolina and British Columbia Cancer Agency), and a plate effect identified in prostate adenocarcinoma; and normalized using the upper quartile method by Hoadley et al.10 Neoantigens of 5935 samples across 19 solid tumor types were obtained from The Cancer Immunome Atlas.11 Abundance data of 64 immune and stromal cell types in the tumor microenvironment for 9358 TCGA tumor samples were inferred using xCell12 and downloaded from the xCell website. xCell is a gene signatures–based method that uses a compensation technique to reduce spillover effects between closely related cell types. Overlapping associations of somatic mutations data with mRNA expression, neoantigens, and cell abundance data are shown in eFigure 1 in the Supplement.
Clinical Cohorts Treated With ICIs
Uniformly analyzed clinical cohorts would help to more robustly evaluate biomarkers of response to ICI therapy.13 Thus, we obtained uniformly processed somatic mutations and clinical information of pretreatment tumors from patients with metastatic lung cancer or melanoma receiving ICI therapies. Briefly, all these patients were gathered from 5 published cohorts.13,14,15,16,17 To avoid the bias introduced by different studies, all raw whole-exome sequencing data (BAM files) and clinical annotations were processed through standardized quality control and mutation calling pipelines as described by Miao et al.13 We excluded 1 patient with small cell lung cancer from the lung cancer cohort with 57 patients and thus obtained 56 patients with non–small cell lung cancer (NSCLC) treated with anti-PD-1/PD-L1 as our NSCLC cohort. The melanoma cohort initially consisted of 151 patients treated with anti-CTLA-4, anti-PD-1, anti-PD-L1, or a combination of these therapies. Given the substantial differences in response rates and survival among patients treated with anti-CTLA-4, anti-PD-1, and anti-CTLA-4 plus anti-PD-1,18 we focused on 145 patients treated with anti-CTLA-4 alone to avoid potential bias introduced by different therapy classes.
With regard to the evaluation of clinical response to ICI therapy, several response definitions,15,19,20 which varied mostly in their assignment of patients with stable disease (SD) by Response Evaluation Criteria in Solid Tumors version 1.1,21 were reported. Given the evolving viewpoints on classifying clinical response to ICI therapy,22 a conservative method was applied: objective response was defined as complete response (CR) or partial response (PR) by Response Evaluation Criteria in Solid Tumors, and no response was defined as progressive disease (PD) by Response Evaluation Criteria in Solid Tumors.13
MUC16 Mutation and Tumor Mutational Burden
All nonsynonymous somatic mutations in MUC16, including missense, nonsense, nonstop, frame-shift insertion and deletion, in-frame insertion and deletion, and splice site mutations, were considered. Tumor mutational burden was calculated as the total count of nonsynonymous mutations in coding sequence using maftools.23
Association of MUC16 Mutation With Immune-Related Genes Expression Profile and Tumor Immune Microenvironment
First, we collected 40 immune-related genes, which were classified into 3 categories: immune checkpoint, T-effector and interferon-γ gene signature, and T cell receptor (eTable 1 in the Supplement). These genes were retrieved from previous reports.24,25,26 We next extracted normalized expression levels of these genes from 9580 samples with both somatic mutations and expression data available. The expression levels were log2-transformed after adding an offset of 1 to avoid taking log of 0 before analysis. In addition, we stratified all samples into 4 tumor immune microenvironment (TIME) types on the basis of median expression levels of CD8A (OMIM 186910) and PD-L1 (OMIM 605402) according to the previous report,27 which defined the positive as gene expression above the median level. On the basis of 8755 samples with both somatic mutations and cell abundances data, we further evaluated the extent of immune cells infiltration into TIME by generating a composite immune score as the sum of abundances for B cells, CD4+ T cells, CD8+ T cells, dendritic cells, eosinophils, macrophages, monocytes, mast cells, neutrophils, and natural killer cells, as described elsewhere.12
Survival Analysis
A Kaplan-Meier estimator of OS was used to construct the survival curves. The log-rank test was performed to compare OS of patients stratified by MUC16 mutational status, and hazard ratios (HRs) with 95% CIs are provided. For the advanced melanoma cohort, complete clinical characteristics available included age at start of treatment and sex; 5 dominant COSMIC mutational signatures28 (signature 7, exposure to UV light; signature 5, unknown environmental exposure; signature 11, prior chemotherapeutic treatment with alkylating agents; signature 1, age-related spontaneous deamination of 5-methyl-cytosine; and signature 6, defective DNA mismatch repair) were also derived from the previous study,13 where mutational burden was not associated with ICI response after adjusting these dominant signatures, suggesting that it is a surrogate for an underlying mutagenic biological process. In melanoma, a large proportion of somatic mutations are known to arise from exposure to UV light.29 Finally, a Cox proportional hazards model was fitted for OS, controlling for potential confounding factors (age, sex, and mutational signatures, except signature 1) and ruling out multicollinearity. Multicollinearity among variables was determined by variance inflation factor calculated using the rms package in R statistical software version 3.5.2 (R Project for Statistical Computing). All survival analyses were performed using the survival package in R statistical software.
Gene Set Enrichment Analysis
Gene set enrichment analysis (GSEA) was performed using the GSEA Desktop Application (version 3.0).30,31 Specifically, genes with 0 count in more than 40% of samples were first removed, and then log2-transformed fold changes for expression of qualified genes in MUC16-mutated vs wild-type states were calculated as a ranking metric for GSEAPreranked to perform GSEA against 50 hallmark gene sets annotated by the Molecular Signatures Database (version 6.2).32 This analysis involved 100 000 random permutations for gene set and weighted enrichment statistic. The normalized enrichment score was used as the magnitude of enrichment. The proportion of false-positives was controlled by calculating the false discovery rate (FDR). The FDR estimates the probability that a gene set with a given normalized enrichment score represents a false-positive finding. A significantly enriched gene set was expected at FDR < .001.
Statistical Analysis
The Mann-Whitney U test was used to determine the association between MUC16 mutation and a continuous variable. Fisher exact test and χ2 test were used to compare the frequencies of clinical response to ICIs and TIME types in MUC16-mutated vs wild-type tumors, respectively. The difference in mean mRNA expression of each immune-related gene in MUC16-mutated vs wild-type patients for different cancer types was hierarchically clustered with the pheatmap package in R statistical software, using Euclidean distance as a distance metric for samples, and ward.D2 clustering method. Statistical significance was expected at 2-tailed P < .05 unless otherwise specified. Statistical analyses were done in R statistical software version 3.5.2 (R Project for Statistical Computing). Data were obtained from October 1 through October 10, 2019, and were analyzed from October 11 through December 31, 2019.
Results
MUC16 Mutation Associated With Elevated Tumor Mutational Burden and Neoantigen Load
Of the 10 195 patients with solid tumor from the TCGA Pan-Cancer Atlas project, 4821 (47.6%) were men, and the median (interquartile range [IQR]) age was 60 (50-70) years. The mutational frequency of MUC16 was 19.68% (2006 of 10 195 patients). The frequency within each solid tumor type is summarized in eTable 2 in the Supplement, with the highest frequency, 73.86%, in skin cutaneous melanoma, followed by 42.76% in lung adenocarcinoma, and 38.84% in lung squamous cell carcinoma.
To investigate the association of MUC16 mutation with established genomic factors associated with ICI response, we began with tumor mutational burden, because it has been the most widely reproduced biomarker for ICI therapy.33 We found that, in this pan-cancer data set, patients with MUC16 mutations exhibited significantly higher tumor mutational burden than those without them (median [IQR], 230 [93-595] mutations vs 48 [25-92] mutations; difference, 182 mutations; 95% CI, 164-199 mutations; P < .001, Mann-Whitney U test) (Figure 1A). This finding persisted within the majority of solid tumor types (25 of 30 tumor types) (eFigure 2 in the Supplement). We next examined the association of MUC16 mutation with neoantigen load and observed that neoantigen load was significantly increased in patients with MUC16-mutated tumors vs those with wild-type tumors (median [IQR], 179 [74-394.5] neoantigens vs 48 [24-89] neoantigens; difference, 131 antigens; 95% CI, 116.5-145 neoantigens; P < .001, Mann-Whitney U test) (Figure 1B). Twelve of 19 solid tumor types had significantly increased load (eFigure 3 in the Supplement).
Figure 1. Association of MUC16 Mutation With Tumor Mutational Burden and Neoantigen Load in Patients With Solid Tumor.

A and B, Graphs show comparison of tumor mutational burden (A) and neoantigen load (B) between MUC16-mutated and wild-type solid tumors from The Cancer Genome Atlas pan-cancer data set. Box plots show the median, first, and third quartiles; error bars extend to 1.5 times the interquartile range; and outlier data are shown as dots. Mann-Whitney U test was used for both comparisons.
MUC16 Mutation Associated With Immune-Related Gene Signatures and Immune Infiltration
It has been proposed that 4 different types of TIMEs exist by measuring TILs recruitment, as evaluated by CD8A and PD-L1 expression. Tumors that fit into TIME I, defined by positive CD8A and PD-L1 expression, most likely receive benefit from anti-PD1/PD-L1 therapies.27,34 Thus, we categorized tumors into 4 groups according to median values of CD8A and PD-L1 expression. We found that the proportion of TIME I (CD8A and PD-L1 positive) was significantly higher in MUC16-mutated tumors compared with wild-type ones (43.8% vs 32.4%; odds ratio, 1.63; 95% CI, 1.46-1.80; P < .001, χ2 test) (Figure 2A). To confirm the association between MUC16 mutation and TIME, we generated a composite immune score based on immune cell types in innate and adaptive immune responses and observed a significantly elevated immune score in MUC16-mutated tumors compared with wild-type tumors (median [IQR] score, 0.17 [0.11-0.29] vs 0.14 [0.09-0.24]; difference, 0.03; 95% CI, 0.02-0.04; P < .001, Mann-Whitney U test) (Figure 2B), suggesting greater extent of immune cells infiltration into TIME. Interestingly, our analysis showed a significant positive correlation between CD8A expression and the composite immune score (Pearson correlation coefficient, 0.64; P < .001).
Figure 2. Association of MUC16 Mutation With Immune Infiltration and Immune-Related Gene Signatures.
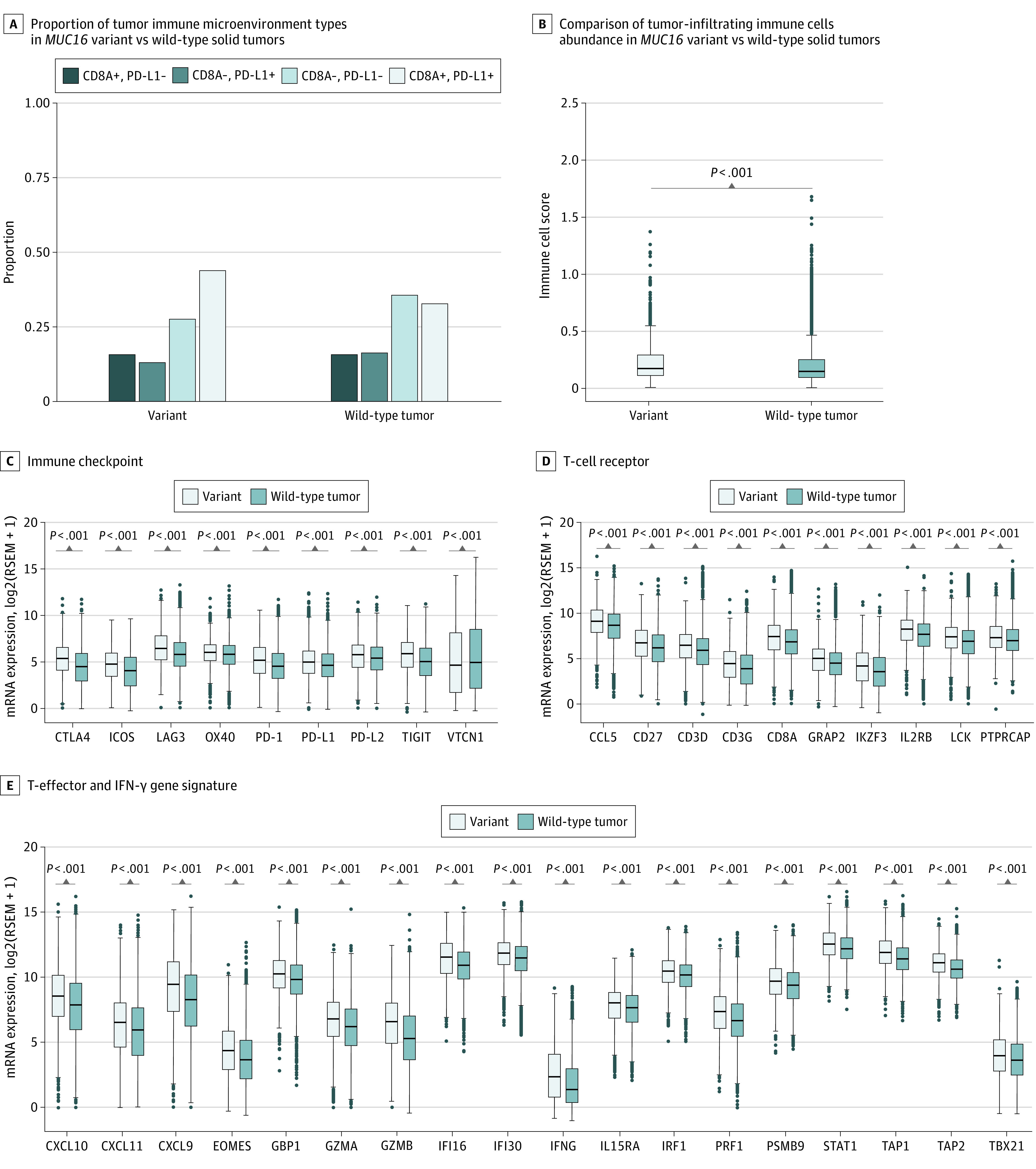
A, Proportion of tumor immune microenvironment (TIME) types in MUC16-mutated vs wild-type solid tumors. TIME I, 43.8% vs 32.4%; TIME II, 28.1% vs 36.3%; TIME III, 12.4% vs 16.1%; TIME IV, 15.7% vs 15.3%. B, Comparison of tumor-infiltrating immune cells abundance in MUC16-mutated vs wild-type solid tumors. C, D, and E, Differentially expressed genes classified into 3 categories, immune checkpoint (C), T-cell receptor (D), and T-effector and interferon-gamma gene signature (E). Box plots show the median, first, and third quartiles; error bars extend to 1.5 times the interquartile range; and outlier data are shown as dots. The χ2 test was used in A, and the Mann-Whitney U test was used in B, C, D, and E.
Given the link between MUC16 mutation and TIME, we further investigated whether MUC16 mutation was associated with immune-related gene signatures, including T-effector and interferon-γ gene signature, which has previously been associated with activated T cells, immune cytolytic activity, and interferon-γ expression.35,36 Of the 40 immune-related genes, 36 (90.0%) demonstrated higher expression levels in MUC16-mutated tumors in comparison with wild-type ones, but 1 gene (2.5%), VTCN1 (OMIM 608162), showed decreased expression, implying a potential negative population for anti-VTCN1 immunotherapy (median [IQR] expression, 4.6 [1.7 to 8.1] vs 4.9 [2.1 to 8.5; difference, −0.3; 95% CI, −0.6 to 0.04; Bonferroni-corrected P < .05, Mann-Whitney U test) (Figure 2C, 2D, and 2E). The differences in mean expression levels of these genes in MUC16-mutated vs wild-type tumors from the respective cancer types are presented in eFigure 4 in the Supplement.
MUC16 Mutation Associated With Favorable Outcomes for ICI Treatment
We further examined the association of MUC16 mutation with outcomes in 2 ICI-treated cohorts, comprising 1 NSCLC cohort with 56 patients (median [IQR] age, 61 [57-66] years; 24 men [43%]) and 1 melanoma cohort with 145 patients (median [IQR] age, 61 [47-71] years; 98 men [67.6%]). As in the pan-cancer analyses, MUC16 mutation was significantly associated with elevated tumor mutational burden (median mutational burden, NSCLC cohort, 325.0 vs 125.5 mutations; difference, 199.5 mutations; 95% CI, 49.0-290.0 mutations; P < .001, Mann-Whitney U test; melanoma cohort, 449.5 vs 82.0 mutations; difference, 367.5 mutations; 95% CI, 262.0-490.0 mutations; P < .001, Mann-Whitney U test) and neoantigen load (median neoantigen load, NSCLC cohort, 1038.0 vs 372.5 neoantigens; difference, 665.5 neoantigens; 95% CI, 100.9-920.7 neoantigens; P = .002, Mann-Whitney U test; melanoma cohort, 1404.5 vs 230.0; difference, 1174.5 neoantigens; 95% CI, 936.6-1614.5 neoantigens; P < .001, Mann-Whitney U test) in both cohorts (eFigure 5A and 5B and eFigure 6A and 6B in the Supplement). In Kaplan-Meier curves analysis for the NSCLC cohort, MUC16 mutation was significantly associated with prolonged OS (median, not reached vs 11.3 months; hazard ratio, 0.34; 95% CI, 0.12-0.99; P = .04, log-rank test) (Figure 3A). There were 18 deaths in the NSCLC cohort.
Figure 3. Overall Survival of Patients With MUC16-Mutated vs Wild-Type Tumors Treated With Immune Checkpoint Inhibitors.
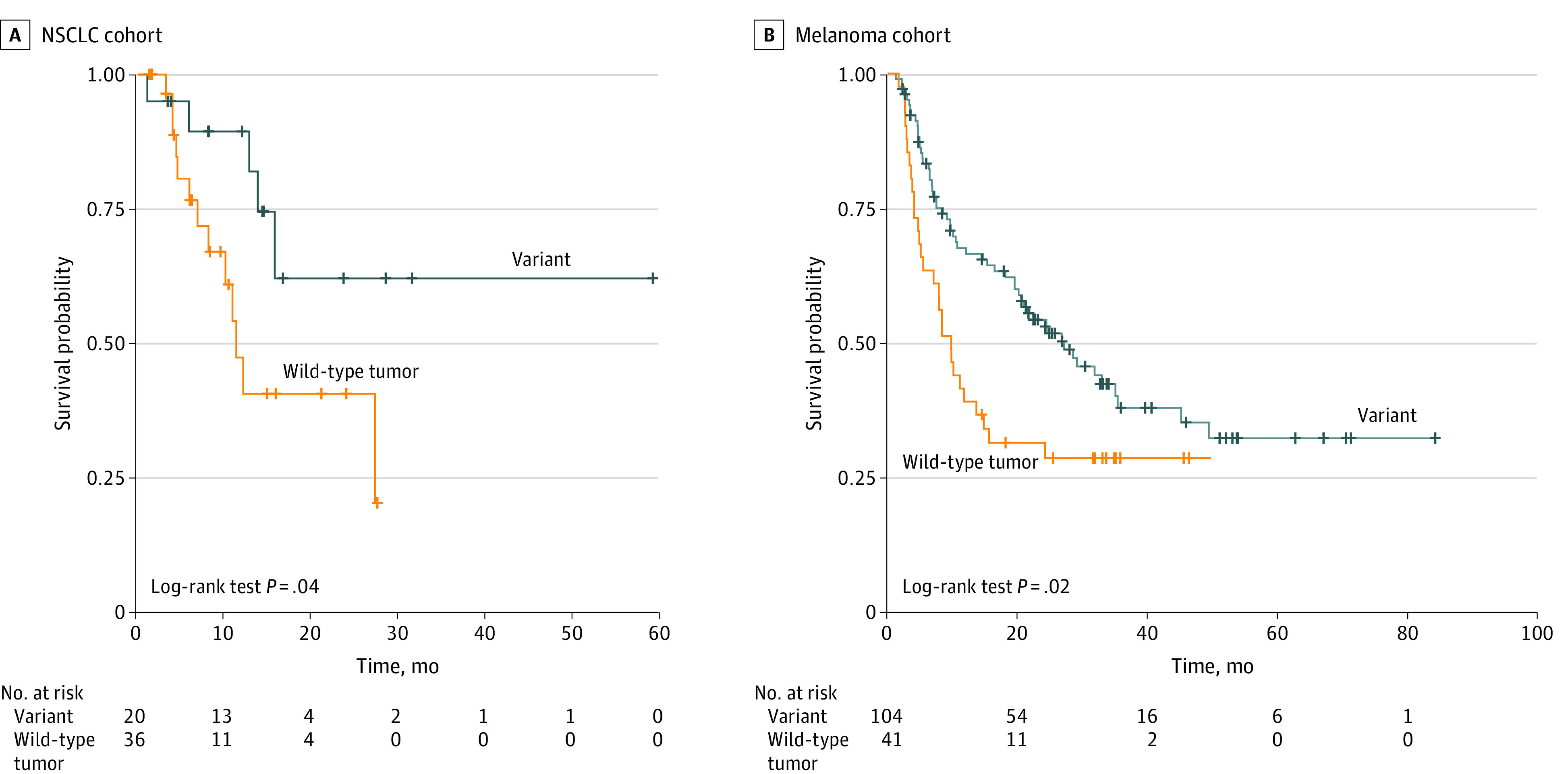
A, Kaplan-Meier curves of overall survival in anti-PD-1/PD-L1–treated patients with non–small cell lung cancer (NSCLC). The median overall survival was 11.3 months (95% CI, 10.1 months to not available) in patients with wild-type tumors and was not reached in patients with MUC16-mutated tumors. B, Kaplan-Meier curves of overall survival in anti-CTLA4–treated patients with melanoma. The median overall survival was 27.03 months (95% CI, 20.07-45.00 months) vs 9.73 months (95% CI, 7.03-15.50 months) in patients with MUC16-mutated and wild-type tumors, respectively.
We next confirmed the association between MUC16 mutation and OS in the melanoma cohort. As shown in Figure 3B, significant OS improvement was also observed in patients with melanoma with MUC16 mutation (median, 27.03 vs 9.73 months; hazard ratio, 0.57; 95% CI, 0.36-0.90; P = .02, log-rank test). We used linear regression model to analyze the association of tumor mutational burden with neoantigen load and observed that each nonsynonymous mutation produced, on average, 2.79 estimated neoantigens, with a strong correlation between them (R2 = 0.98; P < 2 × 10−16) (eFigure 7 in the Supplement), making it difficult to extricate the association of each burden on response from the other. On the basis of the mutational signature activity in each patient with melanoma (eFigure 8 in the Supplement), we observed significant associations of UV light–related signature 7 (median contribution, 0.71 vs 0.15; difference, 0.56; 95% CI, 0.3-0.68; P < .001, Mann-Whitney U test) and alkylating agents-related signature 11 (median contribution, 0.21 vs 0.11; difference, 0.10; 95% CI, 0.02-0.19; P < .001, Mann-Whitney U test) with MUC16 mutation (eFigure 9 in the Supplement). To eliminate the possibility that the association was skewed by confounders, a multivariable Cox model was adopted to adjust for age, sex, and dominant mutational signatures. No multicollinearity was detected in this model, whereas high variance inflation factor was observed for tumor mutational burden when including it (variance inflation factor = 5.24) (eTable 3 in the Supplement). The significant association persisted after controlling these factors (hazard ratio, 0.57; 95% CI, 0.33-0.96; P = .04) (Figure 4).
Figure 4. Forest Plot of Association Between MUC16 Mutation and Overall Survival in Melanoma Cohort.

Data are adjusted for age, sex, and dominant mutational signatures. The vertical line represents hazard ratio (HR) of 1.0. Square data markers reflect estimated HRs. Error bars indicate 95% CIs.
We also examined the likelihood of response to ICI therapy stratified by MUC16 mutational status and found that MUC16 mutation was enriched in patients with CR or PR for both the NSCLC cohort (10 of 20 patients with MUC16 mutation [50%] vs 7 of 36 patients with wild-type tumor [19%]; odds ratio, 4.03; 95% CI, 1.06-16.43; P = .03, Fisher exact test) and melanoma cohort (28 of 104 patients with MUC16 mutation [27%] vs 4 of 41 patients with wild-type tumor [10%]; odds ratio, 3.38; 95% CI, 1.07-14.25; P = .03, Fisher exact test) (Figure 5A and 5B).
Figure 5. Comparison of Response Rates to Immune Checkpoint Therapy Between Patients With MUC16-Mutated and Wild-Type Tumors.

A and B, Likelihood of response stratified by MUC16 mutational status in anti-PD-1/PD-L1–treated patients with non–small cell lung cancer (NSCLC) (A) and anti-CTLA4–treated patients with melanoma (B). CR/PR indicates complete response or partial response; PD, progressive disease; SD, stable disease.
Enriched Biological Processes in MUC16-Mutated Tumors
To identify biological processes associated with MUC16 mutational status, we conducted GSEA on 50 hallmark gene sets, representing major biological processes, for 9580 patients with and without MUC16 mutations. Our analysis yielded 8 gene sets whose expression was significantly upregulated in patients with MUC16 mutations (FDR < .001) (eFigure 10 in the Supplement). Upon inspecting these gene sets, we were able to assign them to 3 biological themes involved in cell proliferation (E2F targets, G2/M checkpoint, MYC targets variant 1, and MYC targets variant 2), immune response (interferon-α response, interferon-γ response, and allograft rejection), and mTORC1 signaling.
Discussion
Our analysis of the TCGA pan-cancer data set across multiple solid tumor types demonstrated that MUC16 mutation was associated with factors previously associated with response to ICI therapy. For instance, patients with MUC16 mutations exhibited higher tumor mutational burden and neoantigen load, compared with patients with wild-type tumor, indicating increased tumor immunogenicity. In a recent study5 of gastric adenocarcinoma, MUC16 mutation was associated with elevated tumor mutational burden. On the basis of a simplistic and pragmatic framework of stratifying tumors, we classified all patients into 4 different TIMEs (I, PD-L1 positive with TILs driving adaptive immune resistance; II, PD-L1 negative with no TIL indicating immune ignorance; III, PD-L1 positive with no TIL indicating intrinsic induction; and IV, PD-L1 negative with TILs indicating the role of other suppressors in promoting immune tolerance)34 and found a higher proportion of TIME I in patients with MUC16 mutation. Notably, TIME I was reported to be associated with a high mutational burden, abundant neoantigen, PD-L1 amplification, and infection with an oncogenic virus, representing an immunoresponsive microenvironment to anti-PD1/PD-L1 therapies.2 This finding is further supported by MUC16-mutated tumors characterized by upregulated expression of T-effector and interferon-γ gene signature, a hallmark of preexisting immunity associated with pronounced benefit from checkpoint blockade.36 Furthermore, the greater abundance of immune cells observed in the microenvironment of MUC16-mutated tumors might more directly demonstrate the association of MUC16 mutation with TIME. An additional hallmark of MUC16-mutated tumors is the augmented expression of multiple inhibitory checkpoints, such as PD-L1, PD-1, CTLA4, LAG3, and others, suggesting that there exists potential adaptive immune resistance to anti-PD-1/PD-L1 therapies and that additional inhibitory pathways beyond the PD-1/PD-L1 axis might be targetable.37 Taken together, these findings support the hypothesis that MUC16 mutation is associated with high immunogenicity and a responsive TIME with PD-L1–dependent or PD-L1–independent adaptive immune resistance.38 Consistent with the hypothesis, to our knowledge, we first confirmed the association of MUC16 mutation with superior outcomes in cohorts receiving ICI treatment. MUC16 mutation was associated with improved OS and response rates in anti-PD-1/PD-L1–treated patients with NSCLC, and this finding was verified in the independent melanoma cohort treated with anti-CTLA-4.
MUC16 has previously been implicated in tumor cell proliferation, metastasis, and modulation of the innate immune response by direct suppression of natural killer cell function.3,4 Also, our GSEA analysis revealed that 8 biological processes regarding cell proliferation, immune response, and mTORC1 signaling were significantly upregulated in MUC16-mutated tumors, providing biological insights concerning the link between MUC16 mutation and ICI response.
Limitations
Our study has several limitations. First, although MUC16-mutated solid tumors were characterized by alteration of pathways involved in cell proliferation, immune response, and mTORC1 signaling, mechanistic underpinnings of MUC16 mutation relevant to ICI response remain elusive and merit further experimental work. Second, the sample size of both ICI-treated cohorts was limited. The limited number of deaths (18 deaths) in the NSCLC cohort restricted the ability to controlling confounders. Additional and larger clinical studies are required. Despite these limitations, this observed association between MUC16 mutation and ICI response in this pan-cancer study represents a further step toward the development of factors associated with outcomes for ICI therapy in solid tumors.
Conclusions
MUC16 mutation is associated with established genomic factors associated with response and better outcomes for ICI therapy in solid tumors. It may serve as a prognostic stratification factor to help optimize the application of ICI-based immunotherapy.
eTable 1. Summary of the 40 Immune-Related Genes
eTable 2. Prevalence of MUC16 Mutations Across 30 Solid Tumor Types
eTable 3. Assessment of Multicollinearity for the Cox Model With and Without Tumor Mutational Burden in Melanoma Cohort
eFigure 1. Venn Diagrams of Number of Samples With Each Type of Data Available
eFigure 2. Correlation of MUC16 Mutation With Tumor Mutational Burden Across 30 Solid Tumor Types
eFigure 3. Correlation of MUC16 Mutation With Neoantigen Load Across 19 Solid Tumor Types
eFigure 4. Unsupervised Hierarchical Clustering of 37 Differentially Expressed Immune-Related Genes Across 30 Solid Tumor Types
eFigure 5. Correlation of MUC16 Mutation With Tumor Mutational Burden and Neoantigen Load in Non-small Cell Lung Cancer Cohort
eFigure 6. Correlation of MUC16 Mutation With Tumor Mutational Burden and Neoantigen Load in Melanoma Cohort
eFigure 7. Relationship Between Tumor Mutational Burden and Neoantigen Load in Melanoma Cohort
eFigure 8. Mutational Signature Activity in Melanoma Cohort
eFigure 9. Correlation of MUC16 Mutation With Ultraviolet Light or Alkylating Agents-Related Signatures
eFigure 10. Gene Set Enrichment Analysis Plots of Significantly Enriched Gene Sets
References
- 1.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133-150. doi: 10.1038/s41568-019-0116-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nat Med. 2019;25(3):389-402. doi: 10.1038/s41591-019-0382-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Felder M, Kapur A, Gonzalez-Bosquet J, et al. . MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. 2014;13:129. doi: 10.1186/1476-4598-13-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aithal A, Rauth S, Kshirsagar P, et al. . MUC16 as a novel target for cancer therapy. Expert Opin Ther Targets. 2018;22(8):675-686. doi: 10.1080/14728222.2018.1498845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, Pasche B, Zhang W, Chen K. Association of MUC16 mutation with tumor mutation load and outcomes in patients with gastric cancer. JAMA Oncol. 2018;4(12):1691-1698. doi: 10.1001/jamaoncol.2018.2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerami E, Gao J, Dogrusoz U, et al. . The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401-404. doi: 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao J, Aksoy BA, Dogrusoz U, et al. . Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellrott K, Bailey MH, Saksena G, et al. ; MC3 Working Group; Cancer Genome Atlas Research Network . Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018;6(3):271-281.e7. doi: 10.1016/j.cels.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoadley KA, Yau C, Hinoue T, et al. ; Cancer Genome Atlas Network . Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173(2):291-304.e6. doi: 10.1016/j.cell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charoentong P, Finotello F, Angelova M, et al. . Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18(1):248-262. doi: 10.1016/j.celrep.2016.12.019 [DOI] [PubMed] [Google Scholar]
- 12.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi: 10.1186/s13059-017-1349-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miao D, Margolis CA, Vokes NI, et al. . Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet. 2018;50(9):1271-1281. doi: 10.1038/s41588-018-0200-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder A, Makarov V, Merghoub T, et al. . Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189-2199. doi: 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Allen EM, Miao D, Schilling B, et al. . Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207-211. doi: 10.1126/science.aad0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizvi NA, Hellmann MD, Snyder A, et al. . Cancer immunology: mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124-128. doi: 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garofalo A, Sholl L, Reardon B, et al. . The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8(1):79. doi: 10.1186/s13073-016-0333-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. . Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23-34. doi: 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hugo W, Zaretsky JM, Sun L, et al. . Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165(1):35-44. doi: 10.1016/j.cell.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roh W, Chen P-L, Reuben A, et al. . Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9(379):eaah3560. doi: 10.1126/scitranslmed.aah3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenhauer EA, Therasse P, Bogaerts J, et al. . New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247. doi: 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 22.Wolchok JD, Hoos A, O’Day S, et al. . Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412-7420. doi: 10.1158/1078-0432.CCR-09-1624 [DOI] [PubMed] [Google Scholar]
- 23.Mayakonda A, Lin D-C, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28(11):1747-1756. doi: 10.1101/gr.239244.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong ZY, Zhong WZ, Zhang XC, et al. . Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012-3024. doi: 10.1158/1078-0432.CCR-16-2554 [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Zhao J, Wang G, et al. . Comutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Cancer Res. 2018;78(22):6486-6496. doi: 10.1158/0008-5472.CAN-18-1814 [DOI] [PubMed] [Google Scholar]
- 26.Gao Q, Zhu H, Dong L, et al. . Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell. 2019;179(2):561-577.e22. doi: 10.1016/j.cell.2019.08.052 [DOI] [PubMed] [Google Scholar]
- 27.Ock C-Y, Keam B, Kim S, et al. . Pan-cancer immunogenomic perspective on the tumor microenvironment based on PD-L1 and CD8 T-cell infiltration. Clin Cancer Res. 2016;22(9):2261-2270. doi: 10.1158/1078-0432.CCR-15-2834 [DOI] [PubMed] [Google Scholar]
- 28.Catalogue of Somatic Mutations in Cancer Mutational signatures version 2. Published March 2015. Accessed July 15, 2020. https://cancer.sanger.ac.uk/cosmic/signatures_v2
- 29.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. ; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain . Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415-421. doi: 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mootha VK, Lindgren CM, Eriksson K-F, et al. . PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267-273. doi: 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417-425. doi: 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14(11):655-668. doi: 10.1038/nrclinonc.2017.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teng MWL, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015;75(11):2139-2145. doi: 10.1158/0008-5472.CAN-15-0255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1-2):48-61. doi: 10.1016/j.cell.2014.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fehrenbacher L, Spira A, Ballinger M, et al. ; POPLAR Study Group . Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837-1846. doi: 10.1016/S0140-6736(16)00587-0 [DOI] [PubMed] [Google Scholar]
- 37.Koyama S, Akbay EA, Li YY, et al. . Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benci JL, Xu B, Qiu Y, et al. . Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167(6):1540-1554.e12. doi: 10.1016/j.cell.2016.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Summary of the 40 Immune-Related Genes
eTable 2. Prevalence of MUC16 Mutations Across 30 Solid Tumor Types
eTable 3. Assessment of Multicollinearity for the Cox Model With and Without Tumor Mutational Burden in Melanoma Cohort
eFigure 1. Venn Diagrams of Number of Samples With Each Type of Data Available
eFigure 2. Correlation of MUC16 Mutation With Tumor Mutational Burden Across 30 Solid Tumor Types
eFigure 3. Correlation of MUC16 Mutation With Neoantigen Load Across 19 Solid Tumor Types
eFigure 4. Unsupervised Hierarchical Clustering of 37 Differentially Expressed Immune-Related Genes Across 30 Solid Tumor Types
eFigure 5. Correlation of MUC16 Mutation With Tumor Mutational Burden and Neoantigen Load in Non-small Cell Lung Cancer Cohort
eFigure 6. Correlation of MUC16 Mutation With Tumor Mutational Burden and Neoantigen Load in Melanoma Cohort
eFigure 7. Relationship Between Tumor Mutational Burden and Neoantigen Load in Melanoma Cohort
eFigure 8. Mutational Signature Activity in Melanoma Cohort
eFigure 9. Correlation of MUC16 Mutation With Ultraviolet Light or Alkylating Agents-Related Signatures
eFigure 10. Gene Set Enrichment Analysis Plots of Significantly Enriched Gene Sets