

Abstract
Up to 95% of novel interventions demonstrating significant effects at the bench fail to translate to the bedside. In recent years, the windfalls of “big data” have afforded investigators more substrate for research than ever before. However, issues with translation have persisted: although countless biomarkers for diagnostic and therapeutic targeting have been proposed, few of these generalize effectively. We assert that inadequate heterogeneity in datasets used for discovery and validation causes their nonrepresentativeness of the diversity observed in real-world patient populations. This nonrepresentativeness is contrasted with advantages rendered by the solicitation and utilization of data heterogeneity for multisystemic disease modeling. Accordingly, we propose the potential benefits of models premised on heterogeneity to promote the Institute for Healthcare Improvement’s Triple Aim. In an era of personalized medicine, these models can confer higher quality clinical care for individuals, increased access to effective care across all populations, and lower costs for the health care system.
Keywords: medical Informatics, health equity, health care disparities, population health, quality improvement, precision medicine
Background
Philosopher Karl Popper commented in 1934 that “non-reproducible single occurrences are of no significance to science” [1]. Yet, 85 years since this statement was made, science remains inundated with nonreproducible single occurrences. John Ioannidis famously wrote in 2005 that “most published research is false” [2]. Chalmers and Glasziou [3] later quantified the false positive rate of published science at 85%; the false positive rates in translational medicine may be even higher than this estimate. Up to 89% of studies demonstrating significant preclinical effects of novel molecules are nonreplicable [4], and the translation failure rate of novel interventions demonstrating significant effects preclinically that are never approved for clinical use reaches up to 95% [5]. These ranges may themselves be underestimates, since they are based on molecules assessed by pharmaceutical companies and in studies published in the highest-impact journals. The translation failure rate of less promising molecules is likely higher still.
In recent years, the emergence of multidimensional “big data” has endowed clinician investigators with more plentiful research substrate than ever before. However, issues with translation have persisted: despite innumerable statistically significant biomarkers identified in the preclinical setting, few of these generalize effectively. For example, 0% of proposed biomarkers for rheumatoid arthritis have demonstrated generalizability [6]. In addition, since enormous samples contribute sufficient statistical power capable of offsetting minute effect sizes, increasingly voluminous data may cause translation failure to become more rather than less of an endemic problem. Indeed, recent studies have noted a 36% deterioration of clinical effectiveness for molecules in Phase II trials [5].
We do not believe that the “depth” of samples (ie, cohort size) is responsible for the observed patterns in translation failure associated with big data. Rather, we believe that the problem is insufficient “breadth”; that is, the datasets used for discovery and validation fail to represent the diversity observed in distinctive real-world patient populations. In other words, by failing to represent the extent of real-world population diversity, we can define these datasets as inadequately heterogeneous.
There is already evidence for the effectiveness of translational bioinformatics premised on heterogeneity for conditions previously plagued by generalization failures, such as in the derivation of host response–based gene panels to predict sepsis and tuberculosis. These panels have outperformed all precedents developed without accounting for heterogeneity (including those using the most sophisticated machine-learning techniques); have been validated across time points, disease severity cohorts, and comorbidities; and have been generalizable across multiple continents [7-9].
In this paper, we highlight the tendency toward homogeneity in translational discovery and illuminate its negative implications. In contrast, we present heterogeneity as an ally rather than an enemy of meaningful translation. Finally, we describe the potential impact of incorporating heterogeneity into the process of translational bioinformatics for addressing the Institute for Healthcare Improvement’s Triple Aim: facilitating personalized medicine, alleviating a health care cost crisis, and resolving health disparities [10,11].
Homogeneity Inherent to “Big” Translational Datasets
The core benefits of big data can be summarized in terms of volume (how much data are available), velocity (how quickly data are accumulated), and variety (how heterogenous the data are) [12]. Although the former two benefits have been harnessed extensively in translational research, the latter has not.
Datasets used for translational research may lack variety owing to three mechanisms: it may be absent, unevenly distributed, or inaccessible. The absence of variety results from constricted sourcing of data, leading to the funneling of homogenous features. One example is the exclusive use of healthy subjects for benchmarking, such as in immunocellular profiling for autoimmune disease [13]. The uneven distribution of variety within a dataset can lead to unintentional clustering of homogeneity, thereby filtering out heterogeneous characteristics. This is a digitized form of sampling bias: since heritability and penetrance both vary within populations, the findings in genome-wide association studies (GWAS) depend markedly upon the sampled cohorts [14]. Finally, variety may be present in the raw data but difficult to access, sequestering the heterogeneity due to technical hurdles. As dataset complexity increases, the risk of sequestration is amplified [15].
This becomes problematic in translational genomics, such as by producing “missing heritability” that is unexplainable from the processed dataset. It has been theorized that much of this “dark matter” (ie, the factors invisible in the processed dataset) relates to environmental influences. These environmental influences produce endophenotypes (expression profiles remaining latent until specific triggering exposures), which are epigenetic traits that can have strong contributions to phenotypic variation [16].
Homogenous datasets account poorly for differential environmental exposures and thus tend to be unreflective of transcriptomic diversity in broader populations. In turn, findings derived from such datasets may not extrapolate routinely beyond the experimental setting, thus precipitating translation failure.
Homogeneity Rendered From “Big” Translational Datasets
Alternatively, homogeneity may be intentionally selected for within the dataset. The contemporary system of science is lubricated by two forms of currency—financial and academic—both of which present disincentives to embracing heterogeneity. On the one hand, budgetary constraints make inclusive, comprehensive methodologies (for instance, preclinical validation studies on multiple animal cohorts) either impractical or unaffordable [13]. On the other hand, the relentless pursuit of academic currency (reputation, garnered through publication) is more easily facilitated by exclusive, narrow methodologies. The inflation of effect sizes is readily conjured in well-controlled experimental populations subjected to investigator-dependent research methods [17].
This investigator-dependent variability—which produces what has been deemed the “vibration of effects”—fosters significant interstudy dissimilarity [17]. Investigator choices can fragment broad baseline populations into discrete clusters subjected to inconsistent exposures to create unbalanced terminal populations [7]. As Kaptchuk [18] pointed out:
Facts do not accumulate on the blank slates of researchers' minds and data simply do not speak for themselves…[the] evaluative process is never totally objective or completely independent of scientists’ convictions or theoretical apparatus.
Accordingly, one way to reframe the reproducibility crisis is as an exclusivity crisis. Intrinsic homogeneity (native to datasets) compounded by extrinsic homogeneity (rendered to datasets) yields a sort of “private epidemiology,” in which discrete study clusters are nonrepresentative of clinical diversity. This has been observed both in vitro and in vivo, where physiologic models poorly recapitulate real-world biology; up to 100% of findings based on observational data (such as vast catalogs of genetic signals) are not replicable [2,5,19]. Poor reproducibility has also been observed in silico, as predictive models premised on these limited feature sets have low external validity [12].
In short, the forces molding experimental homogeneity sculpt what become N-of-none studies. These are reflective of realities contained neatly within digital cells in spreadsheets rather than organic realities in patients.
Heterogeneity in Translational Big Data: Today
More vivid depictions of organic (rather than spreadsheet) realities can be drawn from the introduction of heterogeneity to translational bioinformatics. Heterogeneity expands the analytical spectrum beyond the monochromatic shades of homogenous datasets to better represent real-world phenomena.
Just as meta-analyses mediate between-study biases in evaluation of treatment effects, the introduction of heterogeneity similarly allows for mediation of between-sample biases. Crucially, heterogeneity does not eliminate differences but rather synthesizes similarities [15]. The utility of heterogeneity comes from deriving commonality across diverse subgroups by including rather than excluding distinctive features.
This adheres to theories of systems biology (beyond Oslerian pathophysiology), which contextualize biological interactions in dynamic settings. Robust evidence has documented the inconsistent behavior of unique biological entities (ie, genomic, proteomic, and transcriptomic) “longitudinally” across time points and “latitudinally” across milieu [20]. Accordingly, cross-sectional studies in well-controlled samples seem to be ill-suited for explaining—much less, solving—polygenic diseases and polymechanistic syndromes.
Heterogeneity may be imputed experimentally by casting a wide net of investigators or of data samples. For the former, crowd-sourced collaboration has improved translational efforts compared with independent analyses across multiple indications (Table 1).
Table 1.
Illustrative applications of crowd-sourced heterogeneity.
| Title | Author | Year | Indication |
| Crowdsourced assessment of common genetic contribution to predicting anti-TNF treatment response in rheumatoid arthritis | Sieberts et al [22] | 2016 | Rheumatoid arthritis |
| Crowdsourced estimation of cognitive decline and resilience in Alzheimer’s disease | Allen et al [23] | 2016 | Alzheimer disease |
| Prediction of overall survival for patients with metastatic castration-resistant prostate cancer: development of a prognostic model through a crowdsourced challenge with open clinical trial data | Guinney et al [24] | 2017 | Prostate cancer |
| A community approach to mortality prediction in sepsis via gene expression analysis | Sweeney et al [25] | 2018 | Sepsis |
For the latter, construction of diverse datasets has yielded durable findings relevant for translation across numerous disorders previously plagued by false positives (Table 2). Protocols for introduction of heterogeneity by the use of multiple datasets are publicly available [21].
Table 2.
Illustrative applications of user-constructed heterogeneity.
| Title | Author | Year | Indication |
| Leveraging heterogeneity across multiple datasets increases cell-mixture deconvolution accuracy and reduces biological and technical biases | Vallania et al [26] | 2018 | Autoimmune disease (systemic lupus erythematosus) |
| Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. | Barth et al [27] | 2006 | Cardiomyopathy |
| A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. | Khatri et al [28] | 2013 | Organ transplantation |
| Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. | Sweeney et al [29] | 2016 | Upper respiratory infection |
| A community approach to mortality prediction in sepsis via gene expression analysis | Sweeney et al [24] | 2018 | Sepsis |
| Integrated, multi-cohort analysis identifies conserved transcriptional signatures across multiple respiratory viruses. | Andres-Terre et al [30] | 2015 | Influenza |
| Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases | Li et al [31] | 2014 | Neurodegenerative disease |
| Integrated, multicohort analysis of systemic sclerosis identifies robust transcriptional signature of disease severity. | Lofgren et al [32] | 2016 | Systemic sclerosis |
| Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis | Sweeney et al [8] | 2016 | (Pulmonary) tuberculosis |
| Meta-analysis of continuous phenotypes identifies a gene signature that correlates with COPD disease status. | Scott et al [33] | 2017 | Chronic obstructive pulmonary disease (COPD) |
| A comprehensive time-course–based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set | Sweeney et al [34] | 2016 | Sepsis |
Benefits to these strategies are exemplified by the studies mentioned in the Background section addressing tuberculosis and sepsis, respectively. The imputation of heterogeneity allowed for a 3-gene tuberculosis panel to be generalizable across 10 African countries [8,35] and an 11-gene panel capable of forming distinctive sepsis patient clusters to be validated in multiple nations [36]. Both of these panels, with their ability to accurately guide care for diverse patient groups (within and between populations), symbolize truly personalized medicine [7].
Heterogeneity in Translational Big Data: Tomorrow
General Prospects
Looking toward the future, the use of heterogeneity may play a prominent role in the advancement of translational bioinformatics by cultivating generalizability as a byproduct of representativeness.
This bears substantial potential at the discovery stage, during which statistical significance is useful but not sufficient for predicting clinical effectiveness [2,19]. A myriad of diagnostic/prognostic and therapeutic modalities are being actively investigated for translation of personalized medicine, and validation will be crucial to distinguish the wheat from the chaff (Figure 1). Validation of novel diagnostics/prognostics (such as biomarkers) stands to benefit from heterogeneity given the aforementioned patient diversity across longitudinal and latitudinal scenarios [20]. Validation of novel therapeutics benefits from heterogeneity by enrichment of preclinical and clinical trials [37].
Figure 1.
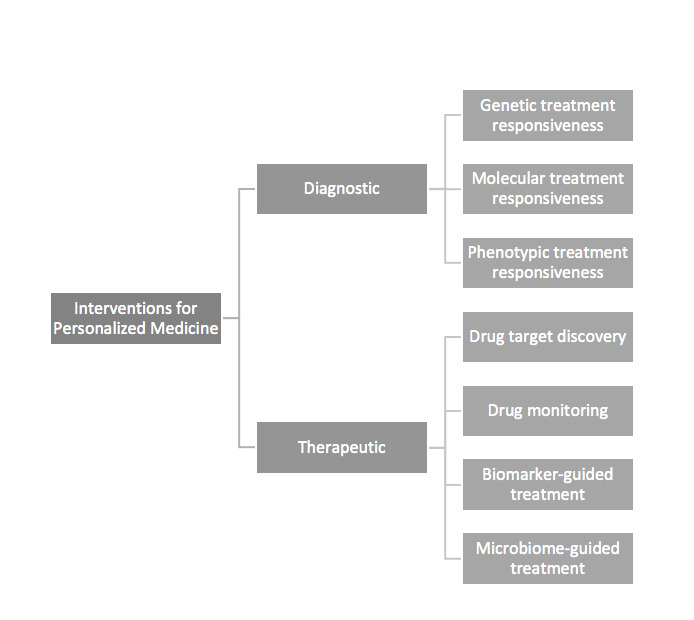
Modalities currently under investigation using translational bioinformatics to promote personalized medicine.
Establishment of data inclusiveness standards to supplement existing research guidelines (such as ARRIVE for preclinical studies and STROBE for observational studies) can accelerate the uptake of heterogeneity into best practices. The assimilation of heterogeneity into research practice in turn bears implications on personalized medicine, health care costs, and health disparities.
Personalized Medicine
Leveraging heterogeneity in translational medicine may offer the quickest path to personalized medicine. It has been noted that increasing the number of datasets included in GWAS samples, controlling for sample size, markedly improves the predictive power of the obtained gene panels to a much greater extent than expanding the sample size alone [15].
This model also incorporates “dark matter” contributing to “missing heritability,” permitting the parsimonious identification of key biological pathways in spite of environmental differences between patient cohorts [16,38]. Moreover, observed differences may be informative rather than confounding: outliers bilaterally (such as weak or strong responders to interventions) are instructive and fertile sources for future investigation rather than “negligible.” N-of-one study becomes feasible within this paradigm.
Finally, while heterogeneity is not necessarily a panacea for discovery—studies utilizing heterogeneity to address acute respiratory distress syndrome have failed to find robust biomarkers—the utility of negative findings is bolstered by the methodology [39]. Evidence-of-absence investigations benefit greatly from additional rigor that more conclusively redirects researchers toward clinically meaningful prospects [13].
Health Care Costs
Health care costs may be targeted from the sides of supply and demand alike. On the supply side, from the perspective of pharmaceutical companies, improved replicability of novel molecules reduces research and development costs devoted toward validation studies, which are currently estimated in the millions of dollars per agent tested [5]. Theoretically, this can allow for reduction in prices with preservation of profit margins. On the demand side, from the payor perspective, improved generalizability first enhances the cost-effectiveness of covered interventions, as clinical effects approach experimental effects [14]. Additionally, more reliable evidence-of-absence studies empower decision making for minimization of overutilized, misutilized, and ineffective interventions [13]. Finally, better understanding of “outlier” pathophysiology can promote the optimal management of “hot spotters”; that is, the oft-cited 1% of the population accounting for 33% of expenditures [40].
Health Disparities
Reductions in payor costs, if passed on to consumers, improve the accessibility of health care. For example, the demonstration of predictive power for tuberculosis diagnosis using 3-gene rather than 71-gene panels implies marked reductions in testing costs (presuming proportional and consistent marginal costs). Furthermore, to the extent that technological barriers for 3-gene sequencing are lower, these diagnostics become available to populations outside of high-resource settings alone [8]. As long as more parsimonious models are adequately representative and maintain predictive power across population groups (as was the case in [8]), accuracy would be preserved in an equitable way while access is simultaneously enhanced.
Heterogeneity may also support the resolution of health disparities by virtue of inclusiveness. As previously discussed, multiplicity of sample sets benefits all populations, with disproportionately greater benefits for traditionally excluded populations [15]. In this way, channeling the “wisdom of crowds” refers not only to wisdom pulled by collaboration between investigators but also to the wisdom pushed by the comprehensiveness of study populations.
Conclusion
In summary, we believe that research practices premised on sample homogeneity are important drivers of shortcomings in contemporary bench-to-bedside informatics. We assert that introduction of heterogeneity can favorably bend this trajectory. Uptake promoted by informal research culture change and formal inclusiveness criteria can lead to meaningful, sustainable, and equitable patient care in the future. In other words, the heterogeneity ethos echoes Osler’s original invocation for personalized medicine: “Just listen to the patient. He is telling you the diagnosis!”
Abbreviations
- GWAS
genome-wide association study
Footnotes
Authors' Contributions: Both authors (EC and PK) equally contributed to conceptualization, editing, and finalizing the manuscript. EC drafted the manuscript and created the figure. Both authors meet the following criteria: (1) substantial contributions to the conception or design of the work or the acquisition, analysis or interpretation of the data; (2) drafting the work or revising it critically for important intellectual content; (3) final approval of the completed version; and (4) accountability for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflicts of Interest: None declared.
References
- 1.Popper K. The logic of scientific discovery. New York: Basic Books; 1959. [Google Scholar]
- 2.Ioannidis J. Why most published research findings are false. PLoS Med. 2005 Aug;2(8):e124. doi: 10.1371/journal.pmed.0020124. http://dx.plos.org/10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chalmers I, Glasziou P. Avoidable waste in the production and reporting of research evidence. Lancet. 2009 Jul 04;374(9683):86–89. doi: 10.1016/S0140-6736(09)60329-9. [DOI] [PubMed] [Google Scholar]
- 4.Begley C, Ellis L. Drug development: Raise standards for preclinical cancer research. Nature. 2012 Mar 28;483(7391):531–533. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- 5.Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011 Aug 31;10(9):712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Rodriguez R, Perez-Pampin E, Marquez A, Blanco F, Joven B, Carreira P, Ferrer MA, Caliz R, Valor L, Narvaez J, Cañete JD, Ordoñez MDC, Manrique-Arija S, Vasilopoulos Y, Balsa A, Pascual-Salcedo D, Moreno-Ramos MJ, Alegre-Sancho JJ, Navarro-Sarabia F, Moreira V, Garcia-Portales R, Raya E, Magro-Checa C, Martin J, Gomez-Reino JJ, Gonzalez A. Validation study of genetic biomarkers of response to TNF inhibitors in rheumatoid arthritis. PLoS One. 2018;13(5):e0196793. doi: 10.1371/journal.pone.0196793. http://dx.plos.org/10.1371/journal.pone.0196793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sweeney TE, Azad TD, Donato M, Haynes WA, Perumal TM, Henao R, Bermejo-Martin JF, Almansa R, Tamayo E, Howrylak JA, Choi A, Parnell GP, Tang B, Nichols M, Woods CW, Ginsburg GS, Kingsmore SF, Omberg L, Mangravite LM, Wong HR, Tsalik EL, Langley RJ, Khatri P. Unsupervised Analysis of Transcriptomics in Bacterial Sepsis Across Multiple Datasets Reveals Three Robust Clusters. Crit Care Med. 2018 Jun;46(6):915–925. doi: 10.1097/CCM.0000000000003084. http://europepmc.org/abstract/MED/29537985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sweeney T, Braviak L, Tato C, Khatri P. Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. Lancet Respir Med. 2016 Mar;4(3):213–224. doi: 10.1016/S2213-2600(16)00048-5. http://europepmc.org/abstract/MED/26907218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warsinske H, Vashisht R, Khatri P. Host-response-based gene signatures for tuberculosis diagnosis: A systematic comparison of 16 signatures. PLoS Med. 2019 Apr;16(4):e1002786. doi: 10.1371/journal.pmed.1002786. http://dx.plos.org/10.1371/journal.pmed.1002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berwick DM, Nolan TW, Whittington J. The triple aim: care, health, and cost. Health Aff (Millwood) 2008;27(3):759–769. doi: 10.1377/hlthaff.27.3.759. [DOI] [PubMed] [Google Scholar]
- 11.IHI Triple Aim Initiative. Institute for Healthcare Improvement. [2020-07-15]. http://www.ihi.org/Engage/Initiatives/TripleAim/Pages/default.aspx.
- 12.Cahan E, Hernandez-Boussard T, Thadaney-Israni S, Rubin D. Putting the data before the algorithm in big data addressing personalized healthcare. NPJ Digit Med. 2019;2:78. doi: 10.1038/s41746-019-0157-2. http://europepmc.org/abstract/MED/31453373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweeney T, Khatri P. Generalizable Biomarkers in Critical Care: Toward Precision Medicine. Crit Care Med. 2017 Jun;45(6):934–939. doi: 10.1097/CCM.0000000000002402. http://europepmc.org/abstract/MED/28509729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vineis P, Schulte P, McMichael A. Misconceptions about the use of genetic tests in populations. Lancet. 2001 Mar 03;357(9257):709–712. doi: 10.1016/S0140-6736(00)04136-2. [DOI] [PubMed] [Google Scholar]
- 15.Sweeney T, Haynes W, Vallania F, Ioannidis J, Khatri P. Methods to increase reproducibility in differential gene expression via meta-analysis. Nucleic Acids Res. 2017 Jan 09;45(1):e1. doi: 10.1093/nar/gkw797. http://europepmc.org/abstract/MED/27634930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reed L, Williams S, Springston M, Brown J, Freeman K, DesRoches C, Sokolowski MB, Gibson G. Genotype-by-diet interactions drive metabolic phenotype variation in Drosophila melanogaster. Genetics. 2010 Jul;185(3):1009–1019. doi: 10.1534/genetics.109.113571. http://www.genetics.org/cgi/pmidlookup?view=long&pmid=20385784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel C, Burford B, Ioannidis J. Assessment of vibration of effects due to model specification can demonstrate the instability of observational associations. J Clin Epidemiol. 2015 Sep;68(9):1046–1058. doi: 10.1016/j.jclinepi.2015.05.029. http://europepmc.org/abstract/MED/26279400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaptchuk T. Effect of interpretive bias on research evidence. BMJ. 2003 Jun 28;326(7404):1453–1455. doi: 10.1136/bmj.326.7404.1453. http://europepmc.org/abstract/MED/12829562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ioannidis J. How to make more published research true. PLoS Med. 2014 Oct;11(10):e1001747. doi: 10.1371/journal.pmed.1001747. http://dx.plos.org/10.1371/journal.pmed.1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwan A, Hubank M, Rashid A, Klein N, Peters M. Transcriptional instability during evolving sepsis may limit biomarker based risk stratification. PLoS One. 2013;8(3):e60501. doi: 10.1371/journal.pone.0060501. http://dx.plos.org/10.1371/journal.pone.0060501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khatri Lab: Tools 2019. Khatri P. [2020-07-14]. https://khatrilab.stanford.edu/tools/
- 22.Sieberts SK, Zhu F, García-García J, Stahl E, Pratap A, Pandey G, Pappas D, Aguilar D, Anton B, Bonet J, Eksi R, Fornés O, Guney E, Li H, Marín MA, Panwar B, Planas-Iglesias J, Poglayen D, Cui J, Falcao AO, Suver C, Hoff B, Balagurusamy VSK, Dillenberger D, Neto EC, Norman T, Aittokallio T, Ammad-Ud-Din M, Azencott CA, Bellón V, Boeva V, Bunte K, Chheda H, Cheng L, Corander J, Dumontier M, Goldenberg A, Gopalacharyulu P, Hajiloo M, Hidru D, Jaiswal A, Kaski S, Khalfaoui B, Khan SA, Kramer ER, Marttinen P, Mezlini AM, Molparia B, Pirinen M, Saarela J, Samwald M, Stoven V, Tang H, Tang J, Torkamani A, Vert JP, Wang B, Wang T, Wennerberg K, Wineinger NE, Xiao G, Xie Y, Yeung R, Zhan X, Zhao C, Members of the Rheumatoid Arthritis Challenge Consortium. Greenberg J, Kremer J, Michaud K, Barton A, Coenen M, Mariette X, Miceli C, Shadick N, Weinblatt M, de Vries N, Tak PP, Gerlag D, Huizinga TWJ, Kurreeman F, Allaart CF, Louis Bridges S, Criswell L, Moreland L, Klareskog L, Saevarsdottir S, Padyukov L, Gregersen PK, Friend S, Plenge R, Stolovitzky G, Oliva B, Guan Y, Mangravite LM. Crowdsourced assessment of common genetic contribution to predicting anti-TNF treatment response in rheumatoid arthritis. Nat Commun. 2016 Aug 23;7:12460. doi: 10.1038/ncomms12460. http://europepmc.org/abstract/MED/27549343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen GI, Amoroso N, Anghel C, Balagurusamy V, Bare CJ, Beaton D, Bellotti R, Bennett DA, Boehme KL, Boutros PC, Caberlotto L, Caloian C, Campbell F, Chaibub Neto E, Chang YC, Chen B, Chen CY, Chien TY, Clark T, Das S, Davatzikos C, Deng J, Dillenberger D, Dobson RJB, Dong Q, Doshi J, Duma D, Errico R, Erus G, Everett E, Fardo DW, Friend SH, Fröhlich H, Gan J, St George-Hyslop P, Ghosh SS, Glaab E, Green RC, Guan Y, Hong MY, Huang C, Hwang J, Ibrahim J, Inglese P, Iyappan A, Jiang Q, Katsumata Y, Kauwe JSK, Klein A, Kong D, Krause R, Lalonde E, Lauria M, Lee E, Lin X, Liu Z, Livingstone J, Logsdon BA, Lovestone S, Ma TW, Malhotra A, Mangravite LM, Maxwell TJ, Merrill E, Nagorski J, Namasivayam A, Narayan M, Naz M, Newhouse SJ, Norman TC, Nurtdinov RN, Oyang YJ, Pawitan Y, Peng S, Peters MA, Piccolo SR, Praveen P, Priami C, Sabelnykova VY, Senger P, Shen X, Simmons A, Sotiras A, Stolovitzky G, Tangaro S, Tateo A, Tung YA, Tustison NJ, Varol E, Vradenburg G, Weiner MW, Xiao G, Xie L, Xie Y, Xu J, Yang H, Zhan X, Zhou Y, Zhu F, Zhu H, Zhu S, Alzheimer's Disease Neuroimaging Initiative Crowdsourced estimation of cognitive decline and resilience in Alzheimer's disease. Alzheimers Dement. 2016 Jun;12(6):645–653. doi: 10.1016/j.jalz.2016.02.006. https://linkinghub.elsevier.com/retrieve/pii/S1552-5260(16)00081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guinney J, Wang T, Laajala TD, Winner KK, Bare JC, Neto EC, Khan SA, Peddinti G, Airola A, Pahikkala T, Mirtti T, Yu T, Bot BM, Shen L, Abdallah K, Norman T, Friend S, Stolovitzky G, Soule H, Sweeney CJ, Ryan CJ, Scher HI, Sartor O, Xie Y, Aittokallio T, Zhou FL, Costello JC, Prostate Cancer Challenge DREAM Community Prediction of overall survival for patients with metastatic castration-resistant prostate cancer: development of a prognostic model through a crowdsourced challenge with open clinical trial data. Lancet Oncol. 2017 Jan;18(1):132–142. doi: 10.1016/S1470-2045(16)30560-5. http://europepmc.org/abstract/MED/27864015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sweeney TE, Perumal TM, Henao R, Nichols M, Howrylak JA, Choi AM, Bermejo-Martin JF, Almansa R, Tamayo E, Davenport EE, Burnham KL, Hinds CJ, Knight JC, Woods CW, Kingsmore SF, Ginsburg GS, Wong HR, Parnell GP, Tang B, Moldawer LL, Moore FE, Omberg L, Khatri P, Tsalik EL, Mangravite LM, Langley RJ. A community approach to mortality prediction in sepsis via gene expression analysis. Nat Commun. 2018 Feb 15;9(1):694. doi: 10.1038/s41467-018-03078-2. http://europepmc.org/abstract/MED/29449546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vallania F, Tam A, Lofgren S, Schaffert S, Azad TD, Bongen E, Haynes W, Alsup M, Alonso M, Davis M, Engleman E, Khatri P. Leveraging heterogeneity across multiple datasets increases cell-mixture deconvolution accuracy and reduces biological and technical biases. Nat Commun. 2018 Nov 09;9(1):4735. doi: 10.1038/s41467-018-07242-6. http://europepmc.org/abstract/MED/30413720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barth AS, Kuner R, Buness A, Ruschhaupt M, Merk S, Zwermann L, Kääb S, Kreuzer E, Steinbeck G, Mansmann U, Poustka AM, Nabauer M, Sültmann H. Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. J Am Coll Cardiol. 2006 Oct 17;48(8):1610–1617. doi: 10.1016/j.jacc.2006.07.026. https://linkinghub.elsevier.com/retrieve/pii/S0735-1097(06)01914-0. [DOI] [PubMed] [Google Scholar]
- 28.Khatri P, Roedder S, Kimura N, De Vusser K, Morgan AA, Gong Y, Fischbein MP, Robbins RC, Naesens M, Butte AJ, Sarwal MM. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J Exp Med. 2013 Oct 21;210(11):2205–2221. doi: 10.1084/jem.20122709. https://rupress.org/jem/article-lookup/doi/10.1084/jem.20122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sweeney TE, Wong HR, Khatri P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci Transl Med. 2016 Jul 06;8(346):346ra91. doi: 10.1126/scitranslmed.aaf7165. http://europepmc.org/abstract/MED/27384347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andres-Terre M, McGuire HM, Pouliot Y, Bongen E, Sweeney TE, Tato CM, Khatri P. Integrated, Multi-cohort Analysis Identifies Conserved Transcriptional Signatures across Multiple Respiratory Viruses. Immunity. 2015 Dec 15;43(6):1199–1211. doi: 10.1016/j.immuni.2015.11.003. https://linkinghub.elsevier.com/retrieve/pii/S1074-7613(15)00455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li MD, Burns TC, Morgan AA, Khatri P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol Commun. 2014 Sep 04;2:93. doi: 10.1186/s40478-014-0093-y. https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-014-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lofgren S, Hinchcliff M, Carns M, Wood T, Aren K, Arroyo E, Cheung P, Kuo A, Valenzuela A, Haemel A, Wolters PJ, Gordon J, Spiera R, Assassi S, Boin F, Chung L, Fiorentino D, Utz PJ, Whitfield ML, Khatri P. Integrated, multicohort analysis of systemic sclerosis identifies robust transcriptional signature of disease severity. JCI Insight. 2016 Dec 22;1(21):e89073. doi: 10.1172/jci.insight.89073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott M, Vallania F, Khatri P. META-ANALYSIS OF CONTINUOUS PHENOTYPES IDENTIFIES A GENE SIGNATURE THAT CORRELATES WITH COPD DISEASE STATUS. Pac Symp Biocomput. 2017;22:266–275. doi: 10.1142/9789813207813_0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sweeney TE, Shidham A, Wong HR, Khatri P. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci Transl Med. 2015 May 13;7(287):287ra71. doi: 10.1126/scitranslmed.aaa5993. http://europepmc.org/abstract/MED/25972003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Francisco N, Fang Y, Ding L, Feng S, Yang Y, Wu M, Jacobs M, Ryffel B, Huang X. Diagnostic accuracy of a selected signature gene set that discriminates active pulmonary tuberculosis and other pulmonary diseases. J Infect. 2017 Dec;75(6):499–510. doi: 10.1016/j.jinf.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 36.Maslove D, Shapira T, Tyryshkin K, Veldhoen R, Marshall J, Muscedere J. Validation of diagnostic gene sets to identify critically ill patients with sepsis. J Crit Care. 2019 Feb;49:92–98. doi: 10.1016/j.jcrc.2018.10.028. [DOI] [PubMed] [Google Scholar]
- 37.Temple R. Enrichment of clinical study populations. Clin Pharmacol Ther. 2010 Dec;88(6):774–778. doi: 10.1038/clpt.2010.233. [DOI] [PubMed] [Google Scholar]
- 38.Raymond S, López MC, Baker H, Larson S, Efron P, Sweeney T, Khatri P, Moldawer LL, Wynn JL. Unique transcriptomic response to sepsis is observed among patients of different age groups. PLoS One. 2017;12(9):e0184159. doi: 10.1371/journal.pone.0184159. http://dx.plos.org/10.1371/journal.pone.0184159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sweeney T, Thomas N, Howrylak J, Wong H, Rogers A, Khatri P. Multicohort Analysis of Whole-Blood Gene Expression Data Does Not Form a Robust Diagnostic for Acute Respiratory Distress Syndrome. Crit Care Med. 2018 Feb;46(2):244–251. doi: 10.1097/CCM.0000000000002839. http://europepmc.org/abstract/MED/29337789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gawande A. The hot spotters: can we lower medical costs by giving the neediest patients better care? New Yorker. The New Yorker. 2011. Jan 24, [2020-07-14]. https://www.newyorker.com/magazine/2011/01/24/the-hot-spotters. [PubMed]