

Abstract
Worldwide, about half a million people are diagnosed with pancreatic cancer every year, with mortality rates of more than 90%. T cells within pancreatic tumors are generally infrequent and incapable of eliciting anti-tumor immunity. Thus, pancreatic cancer is considered an “immunologically cold” tumor. However, recent studies clearly show that when T-cell immunity in pancreatic cancer is sufficiently induced, T cells become effective weapons. This fact suggests that in order to improve pancreatic cancer patients’ clinical outcomes, we need to unveil the complex immune biology of this disease. In this review, we discuss the elements of tumor immunogenicity in the specific context of pancreatic malignancy.
Keywords: pancreatic cancer, PDAC, immunotherapy, immunogenicity, T cells
PANCREATIC CANCER
Several types of cancer can arise from the pancreas. Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic malignancy (75–95%). Other types of pancreatic cancer include, but are not limited to, neuroendocrine tumors (15–20%), colloid carcinoma (2%), solid-pseudopapillary tumors (2%), acinar cell carcinoma (1%), and pancreatoblastoma (0.5%).1 In this review, PDAC is referred to as pancreatic cancer.
Worldwide, nearly half a million people were diagnosed with pancreatic cancer in 2018. This equates to more than 1200 individuals daily. The highest incidence is in North America and Europe.2 In the United States, pancreatic cancer became the third leading cause of cancer death in 2016.3 In 2019 alone, 56,770 patients were expected to be diagnosed with pancreatic cancer, and 45,750 people died from this disease.4
From 1975 to 2014, the 5-year survival rate for pancreatic cancer increased from ~3% to ~9%.5 However, this figure does not provide a full picture of pancreatic cancer survival. Currently, the five-year survival rate for people with early stage local pancreatic cancer is more than 30%, but it is only 3% for individuals diagnosed when the cancer has already metastasized. Early diagnosis is still a challenge, and the vast majority of patients are diagnosed with advanced or metastatic disease.4 Even among individuals with early stage, resectable pancreatic tumors, the majority will experience local and systemic relapse after curative-attempt surgery.6–8 Given the significant progress that has been made for other cancer types over the past four decades, pancreatic cancer is projected to become the second leading cause of cancer-related mortality in the United States by 2025, surpassing colorectal cancer.9
CANCER ANTIGENICITY AND IMMUNOGENICITY
Cancer antigenicity and immunogenicity are terms with distinct meanings. Cancer antigenicity refers to the extent to which tumors present antigens that can be specifically recognized by the adaptive immune system. However, this antigen recognition does not always lead to an effective immune response. In contrast, cancer immunogenicity refers to the extent to which tumors present antigens that are recognizable to the immune system and stimulate efficient tumor host immunity, which is largely mediated by T cells. Hence, while all tumor immunogens are antigens, not all tumor antigens are immunogens.10,11
Tumor immunogens are antigens that can initiate and propagate the cancer immunity cycle (Fig. 1). In the course of cancer development, some neoplastic cells die. During the continuous sampling of their microenvironment, antigen presenting cells (APCs) within the tumors take up dead cancer cells and/or their corresponding particles. These engulfed materials then will be degraded, processed into smaller peptides and presented on the surface of APCs carried by major histocompatibility complex (MHC) molecules. In order for these intra-tumoral APCs to assess the foreignness of presented peptides, they migrate to T cell-rich areas of secondary lymphoid tissues, such as lymph nodes. Since cancer cells contain protein sequences that differ either in form or in level of expression than the proteins of their normal counterparts, peptide-MHC molecules on APCs can be recognized specifically by T-cell receptors (TCRs) on naïve T cells, resulting in T-cell priming and activation. Through a coordinated cascade of cytokines and chemokines, active T cells migrate through the vasculature to infiltrate and reside in the tumor. Like all nucleated cells in the body, cancer cells process and present their own materials on MHC class I (MHC-I). Consequently, tumor-infiltrating CD8+ T cells recognize the same peptide that they previously encountered in the secondary lymphoid organs but is now presented by cancer cells. This T-cell-antigen interaction stimulates CD8+ T-cell cytotoxic responses toward cancer cells. Eventually, this can lead to the release of more immunogenic peptides, creating a continuous feedback loop.12–14 It is worth mentioning, however, that some cancer cells are also capable of displaying their peptides carried on MHC class II (MHC-II) to CD4+ T cells, mimicking professional APCs. Considering the diversity of CD4+ T-cell subsets, CD4+ T-cell-antigen recognition can result in a wide variety of immune responses ranging from anti-tumor immunity via enhancing CD8+ T-cell activity, to immune suppression, as in the case of Treg cells.15
FIGURE 1.
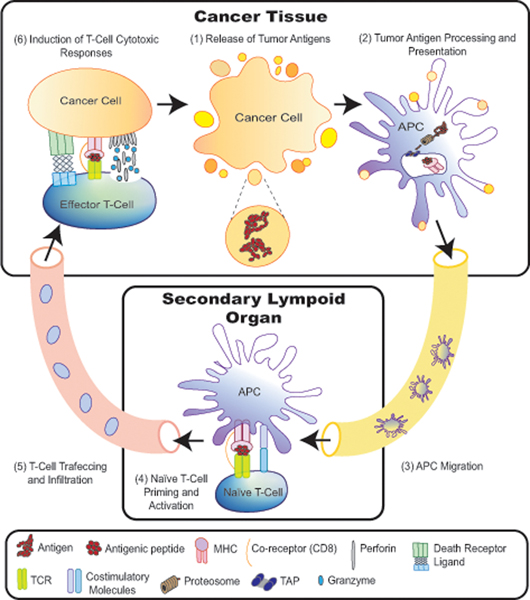
Cancer–immunity cycle. During cancer development, cancer cells die. Antigen presenting cells (APCs) within the tumors engulf dead cancer cells and/or their corresponding particles. APCs process engulfed materials into smaller peptides to be presented on their surface carried on major histocompatibility complex (MHC) molecules. Those APCs migrate through the lymphatics to secondary lymphoid organs and activate antigen-specific- naive T cells. Then, active T cells travel through blood vessels to infiltrate into the tumor. Intratumoral effector T cells can recognize tumor antigens on the surface of cancer cells and kill them, resulting in the release of more antigens. Cytotoxic T cells kill their cellular targets via either the death receptor pathway or the granule exocytosis pathway. Endoplasmic reticulum (ER); T-cell receptor (TCR); transporter associated with antigen processing (TAP).
Nonetheless, this picture of the cancer immunity cycle does not fully represent the complexity of tumor immunogenicity. In fact, many factors can tip the balance between cancer antigenicity and immunogenicity. These include the quantity and quality of tumor antigens, the kinetics of antigen appearance and discontinuity,16 and the complex interplay between cancer cells and the surrounding stroma.17,18 Unsurprisingly, though the concept of tumor immunogenicity was proposed more than a century ago, our understanding of this sophisticated biological process is still evolving.10
PANCREATIC CANCER CHARACTERISTICS
Before we discuss the immunogenicity of pancreatic cancer, it is important to first describe its complex biology. Pancreatic ductal adenocarcinoma is characterized by atypical neoplastic glands surrounded by dense collagenous stroma (desmoplasia). Pancreatic cancer stroma accounts for more than 50% of the total tumor mass,1,19 and exhibits a wide range of immunosuppressive properties,17,18,20 which will be discussed later in this review.
The tumor microenvironment serves as a well-organized physical and biochemical system (Fig. 2). It is made up of diverse cellular types, including immune cells, fibroblasts, adipocytes, neuronal cells and cells of the surrounding blood vessels and the lymphatics. Focusing on immune cells, a wide range of cellular populations commonly infiltrate pancreatic cancers. These include tumor-associated macrophages (TAMs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), NK cells, T cells, B cells, innate lymphoid cells (ILCs) and mast cells. Moreover, many of these immune cell types involve several specialized subtypes that exhibit distinct molecular and functional activity profiles, including T cells.1,17,18,20,21
FIGURE 2.
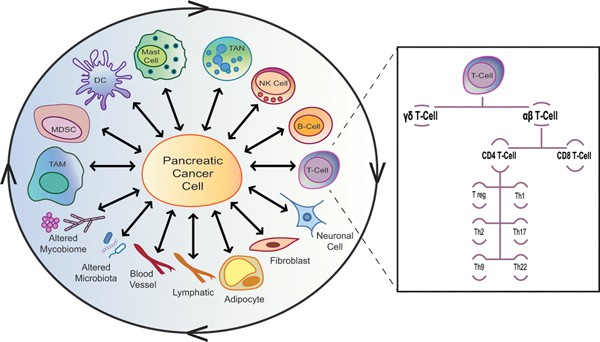
Pancreatic cancer stroma is a well-organized, dynamic and complex system.
Pancreatic cancer stroma is made up of diverse cellular types, including immune cells, fibroblasts, adipocytes, neuronal cells, blood vessels and lymphatics. Also, a wide range of immune cellular populations commonly infiltrate pancreatic cancers. These include tumor-associated macrophages (TAMs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), natural killer (NK) cells, T cells, B cells, innate lymphoid cells (ILCs) and mast cells. Many of these stromal cell types involve several specialized types and subtypes that exhibit distinct molecular and functional activity profiles, including T cells. Conventional T lymphocytes express TCRs on their surface that are composed of α and β chains (αβ T cells). αβ T cells are generally either CD4+ (T helper cells; Th cells) or CD8+ (T cytotoxic lymphocytes; CTL). When naïve CD4+ T cells get activated, they differentiate into very distinct Th subsets, including Th1, Th2, Th17, Treg, Th9 and Th22. In addition to αβ T cells, some T lymphocytes express γ and δ TCR chains (γδ T cells). All these stroma components continuously interact with each other and with cancer cells, creating a very dynamic and complex stromal-cancer crosstalk network.
Conventional T lymphocytes express TCRs on their surface that are composed of α and β chains (αβ T cells). αβ T cells are generally either CD4+ (T helper cells; Th cells) or CD8+ (T cytotoxic lymphocytes; CTL). When naïve CD4+ T cells get activated, they differentiate into very distinct Th subsets, including Th1, Th2, Th17, Treg, Th9 and Th22.22–25 Besides conventional αβ T cells, some T lymphocytes express γ and δ TCR chains (γδ T cells).26,27 Each of these T-cell populations secretes distinct cytokines and chemokines, and thus regulate the immune response in synergistic and opposing manners. Moreover, the continuous epigenetic modifications of T-cell transcription factors can ultimately result in a dynamic switch between some of these various T-cell lineages. Even within the same sub-population, T cells of different clones may distinctly alter tumor properties.28–30 Notably, although immune cell complexity is described here as an example, other stromal cells, such as fibroblasts, exhibit similar degrees of heterogeneity and plasticity.31–34
In addition, the pancreatic tumor microenvironment is comprised of many non-cellular extracellular matrix (ECM) components. These include collagen, fibronectin, laminin, hyaluronic acid, growth factors, cytokines and chemokines. Also, the pancreatic cancer stroma is associated with abnormal conditions of hypoxia and acidic extracellular pH.1,20 Interestingly, the deposition of some of ECM molecules results in increased pancreatic tumor stiffness and hydrostatic pressures. While this observation had first led to the assumption that the pancreatic cancer stroma acts as a physical barrier, recent studies refute this hypothesis. Indeed, it is now evident that some stromal elements can restrain pancreatic tumor growth.18 In line with that observation, PDAC desmoplasia does not correlate with paucity in intra-tumoral T-cell accumulation.21
Furthermore, during PDAC tumorigenesis, some specific classes of gut microorganisms (i.e. bacteria and fungi) can migrate to the pancreas. Later, in the pancreas, these microorganisms can modulate the immune system, and, ultimately, affect both tumor growth and response to therapy.35–38 These studies clearly support the notion that there is a complex interplay between various components of the immune system, cancer cells, and other parts of stroma. Though they point to the possibility of new therapeutic approaches for pancreatic cancer, many questions remain to be answered. For example, it is still unclear how fungal and bacterial communities that coexist in many areas of the body can influence each other.39
Similar to stromal cell diversity, malignant epithelial pancreatic cells are often phenotypically and functionally heterogeneous. Cancer cells tend to continuously manipulate their genetic, epigenetic, proteomic and/or metabolic profiles, leading to a diverse population of cancer cells (i.e., tumor heterogeneity). These cellular differences at the molecular level contribute to distinctive behaviors of cancer cells with respect to their motility, proliferation, and metastatic potentials, as well as their differentiation and growth patterns.40–44
There are multiple levels of tumor heterogeneity. Although this is not exclusive to PDAC, it is a common finding in this disease. It is exemplified by significant variations between tumors from different patients (i.e. interpatient heterogeneity).41 Also, tumor heterogeneity can be detected within a single patient. Indeed, metastatic tumors at different locations as well as multiple primary tumor cell clusters in the same organ are likely to exhibit distinct biological features (i.e. multi-focal or inter-tumor, intra-patient heterogeneity). Tumor heterogeneity can also be observed between different cancer clones in different parts of the same tumor, as well as between cancer cells that are immediately adjacent to each other (ie, intra-tumor heterogeneity).40,45 More intriguingly, it is not uncommon to identify varying mutational and epigenetic patterns that co-exist within one neoplastic cell (ie, mutational and epigenetic heterogeneity).46
The KRAS oncogene serves as a good example of how mutational heterogeneity adds to the complexity of pancreatic cancer. Indeed, KRAS is mutated in more than 90% of pancreatic cancer cases. While the type of mutation that is commonly detected in KRAS is a missense point mutation, gene amplification has been reported in about 4% of PDAC patients. Also, KRAS may contain missense mutations at different residues. However, the most common mutational hotspot is the glycine in codon 12 (G12). Even this specific glycine residue can be substituted by various amino acids, resulting in distinct KRAS mutant variants (G12D, G12A, G12R, G12C, G12S, and G12V). Interestingly, many KRAS mutant variants can be simultaneously detected within a single tumor. Moreover, an individual pancreatic cancer cell can harbor more than one mutant KRAS allele (ie, bi-allelic KRAS mutations). Despite this mutational diversity, most of these KRAS mutations selectively modify the gene’s function to favor tumor initiation, progression, metastasis and/or therapeutic resistance.45–47
This significant molecular heterogeneity in pancreatic cancer is clearly reinforced by the morphological heterogeneity of the disease. Indeed, based on the grade of cellular differentiation, growth pattern and desmoplastic stroma, several morphological subtypes have been defined. These include pancreatobiliary, intestinal, and clear cell patterns. Also, there are additional variants that have not yet been formally described in the World Health Organization (WHO) classification. Similar to molecular heterogeneity, these distinct morphological variants can be seen between different patients as well as within different parts of the same tumor.44
Taken together, pancreatic cancers are typically stroma-rich tumors, and are highly immunosuppressive. Both the stroma and epithelial pancreatic cancer cells are very diverse and heterogeneous. All tumor components continuously interact with each other, creating a very dynamic and complex stroma-cancer crosstalk network.
PANCREATIC CANCER ANTIGENICITY AND IMMUNOGENICITY
The essential building blocks of tumor immune responses are (i) the generation of tumor antigens; (ii) the efficiency of tumor antigen processing and presentation; (iii) the infiltration of tumor-specific cytotoxic T cells; and (iv) the induction of T-cell effector anti-tumor responses.12 In order to understand the relevance of immunogenicity to pancreatic malignancy, we will discuss each of these four T-cell immunity elements in turn by raising several fundamental questions.
Question 1: Is There Sufficient Tumor Antigen Expression to Elicit an Immune Response?
The significance of studying tumor mutational load to evaluate its immunogenicity relies on the idea that some of these mutations can generate tumor antigens, which may induce T-cell immunity.48 That being noted, the vast majority of pancreatic cancers (97%) have gene alterations, including amplifications, deletions, translocations, inversions, frameshifts and substitutions (Table 1).49 Alexandrov et al found that the median number of mutations per Mega base in pancreatic cancer is 1 (ranging from 0.1 to 10). Assuming that an average genome has 2.8 gigabases, this study suggests that each pancreatic cancer patient has 280 to 28,000 mutations.50 However, a major challenge in genomic sequencing is the low malignant epithelial cell content, which can adversely impact the sensitivity of mutation detection. As mentioned above, pancreatic tumors are associated with extensive desmoplastic stroma and low tumor cellularity. Thus, estimates based on early studies had to be revisited.17
TABLE 1.
A Summary of Human Pancreatic Cancer Sequencing Studies
| Study | Frequency of Mutations | Sample Size | Sequencing Technique | Evaluation of Tumor Cellularity |
|---|---|---|---|---|
| Alexandrov et al50 | The median number of mutations per Mega base is 1, ranging from 0.1 to 10 mutations per Mega base (ie, 280 to 28,000 mutations per patient) | ~120 | Either whole genome sequencing (15 samples) and whole exome sequencing (the remaining samples). | |
| Jones et al51 | The average number of mutations detected per patient is 48 | 24 | Microarrays-based exome sequencing: protein-coding exons from more than 20,000 genes were identified. Then, using microarrays containing probes for ~106 single-nucleotide polymorphisms, homozygous deletions and amplifications in the tumor samples were detected. | To remove contaminating non-neoplastic cells, tumor samples were passaged in vitro as cell lines or in nude mice. Then, to validate somatic mutations, exons containing variant sequences were reamplified and resequenced from both tumor and normal tissues |
| Biankin et al19 | The average number of mutations detected per patient is 26, ranging from 1 to 116. | 99 | Whole exome sequencing: using exome capturing and sequencing of different mixtures of cancer cell line and matched germline DNA as a standard, tumor samples with greater than 20% epithelial cellularity and/or ≥10 validated somatic mutations were included in the study. The average sequence depth was 26,608-fold, ranging from 609 to 213,544. | The cellularity of each primary sample was estimated through pathological review, deep amplicon-based sequencing of exons 2 and 3 of KRAS and single nucleotide polymorphism (SNP) array-based cellularity estimates. |
| Balachandran et al, the MSKCC cohort52 | The median number of mutations detected per patient is 171 The median number of neoantigen-related mutations detected per patient is 38 |
58 | Whole exome sequencing: whole exome sequencing was performed at 150× coverage for tumor samples and 70× for matched normal | Only tumor islands of more than 70% cellularity were included in the study based on expert PDAC pathologic review. |
| Balachandran et al, the ICGC cohort52 | The median number of mutations detected per is 135 The median number of neoantigen-related mutations detected per patient is 32 |
166 | Either whole genome sequencing or whole exome sequencing: primary tumors and patient-derived cell lines with more than 40% cellularity underwent whole genome sequencing at 75× mean coverage. Samples with 12–40% cellularity underwent deep-exome sequencing at 400× mean coverage. | Tumor cellularity was estimated for each sample using a combination of qPure analysis and KRAS amplicon sequencing. |
| Waddell et al54 | The average number of mutations per Mega base is 2.64, ranging from 0.65 to 28.2 mutations per Mega base (ie, 1820 to 78,960 mutations per patient) | 100 | Whole-genome sequencing: Tumor samples with more than 40% cellularity and patient-derived cell lines whole genome sequencing at 65× mean coverage and compared to the germline at an average coverage of 38× |
Tumor cellularity was estimated for each sample using qPure analysis |
| Bailey et al41 | The median number of coding mutations detected per patient is 62 The number of neoantigen-related mutations detected per sample ranges from 4 to 4000 |
456 | Either whole genome sequencing or whole exome sequencing: primary tumors and patient-derived cell lines with more than 40% cellularity underwent whole genome sequencing at 75× mean coverage. Samples with 12–40% cellularity underwent deep-exome sequencing at 400× mean coverage. | Tumor cellularity was estimated for each sample using a combination of qPure analysis and KRAS amplicon sequencing. |
| Bailey et al65 |
To overcome this challenge, several subsequent human pancreatic cancer sequencing studies took into account tumor cellularity. Using microarray-based exome sequencing, Jones and colleagues reported that human PDAC tumors contain an average of 63 genetic alterations, 48 of which were validated to be somatic mutations. The majority of these mutations were point mutations (ie, among all detected genetic alterations, ~84% were missense mutations and ~5% were nonsense mutations).51 In a different study, using whole exome sequencing, Biankin and colleagues identified an average of 26 mutations per patient, with a total of 2627 mutations (2016 of which were non-silent mutations) and 1628 copy-number variations in 99 evaluated pancreatic tumors.19 In another recent study, Balachandran et al evaluated two cohorts: the MSKCC (Memorial Sloan Kettering Cancer Center) and ICGC (International Cancer Genome Consortium) collections, and determined that the median numbers of mutations detected per patient are 171 and 135, respectively.52
Besides focal mutations, Notta et al showed that chromothripsis and polyploidization, phenomena of massive chromosomal rearrangements, were also very common. They found that among the 107 PDAC tumors evaluated in the study, 48 (45%) displayed changes in DNA copy number consistent with polyploidization, and 70 (65.4%) harbored at least one chromothripsis event.53 In line with those observations, Waddell et al reported that the average number of mutations per Mega base in 100 PDAC samples was 2.64, ranging from 0.65 to 28.2 mutations per Mega base (ie, 1820 to 78,960 mutations per patient). Among these PDAC evaluated tumors, the genome of only 20% of the samples contained ≤ 50 structural variation events. On the other hand, the remaining samples had either local rearrangements in one or two chromosomes (30%), 50–200 structural variation events (36%) or more than 200 structural variation events (14%). Interestingly, this paper suggested a direct link between large chromosomal rearrangements and the disruption of particular genes known to be important in pancreatic cancer.54
In addition, intra-tumoral heterogeneity of both cancer cells and T lymphocytes can determine T-cell efficacy, and thus plays an important role in tumor immunogenicity.55–58 The core of this theory embraces two concepts; T-cell clonality and T-cell immunological adaptation. Once a T-cell recognizes an immunogenic peptide, it undergoes explosive proliferation, generating a clone of active T cells that express identical TCRs specifically against that particular antigen (ie, clonal expansion). Accordingly, T-cell clonality is a metric of T-cell expansion and reactivity. Additionally, the dynamic changes in the spectrum of tumor antigens is concordant with changes in the repertoire of antigen-specific T cells over time (ie, immunological adaptation). Hence, the enrichment of many unique low abundant mutations within different regions of the same tumor could be associated with low antigen dosage, and thus induce T cells unresponsiveness. Even if some subclonal mutations (i.e. mutations that are not present in the entire proportion of neoplastic cells) were highly immunogenic, responsive T cells would be unable to target every tumor cell but rather only a small fraction, limiting overall T-cell anti-tumor efficacy. This hypothesis has been supported by studies in melanoma55 and lung cancer.57 Nonetheless, high degrees of T-cell infiltration may overcome the disadvantages of intra-tumor heterogeneity. Concomitant high levels of cytotoxic T-cell infiltration with T-cell diversity may indicate that many T-cell clones in the tumor bed are reactive with a broader variety of antigens, leading to broader and more efficient anti-tumor T-cell immunity. In line with this theory, Balachandran et al showed that tumors of long-term pancreatic cancer survivors (median survival 6 years) exhibited both greater densities of cytolytic CD8+ T cells (ie, 12-fold) and greater TCR repertoire diversity compared to short-term survivors.52 However, this may be a consequence rather than a cause of improved survival following therapy. Hence, mechanistic studies are needed to deeply understand how TCR intra-tumor heterogeneity can impact on cancer prognosis.
In addition to antigen quantity and intra-tumor heterogeneity, antigen quality can highly influence its immunogenicity. Important measures of antigen quality include the degree of molecular differences between mutant variants and wild-type peptides, and the affinity of the antigenic peptide to both MHC and TCR molecules. In fact, by integrating these antigen quality factors with mutational burden, TCR diversity and intra-tumoral cytotoxic T-cell frequency, Balachandran et al were able to develop a “neoantigen quality fitness” model that can predict PDAC patients’ clinical outcomes. Notably, this study also showed that tumor antigen quantity alone does not correlate with PDAC patients’ survival.52
Finally, it should be remembered that tumor antigens are not limited to protein mutant sequences. The vast majority of proteins in eukaryotic cells are subjected to post-translational modifications (PTMs), which refer to the enzymatic alterations of proteins following their synthesis in the endoplasmic reticulum (ER) and Golgi. These modifications include phosphorylation, glycosylation, nitrosylation, ADP-ribosylation, ubiquitination, and acetylation.59 Since PTMs affect the solubility, folding, localization, and half-life of proteins, this phenomenon is highly relevant to different aspects of cellular biology.60 A detailed discussion of this area is beyond the scope of this review. However, since PTM proteins have distinct tertiary structures and biochemistry, PTMs can alter how corresponding peptides are processed and presented to T cells. Also, PTMs can change the affinity between these peptides and MHC molecules as well as between peptide-MHC complexes and TCRs.60,61 While these data may suggest that PTMs can manipulate tumor immunogenicity, data from human PDAC tumors are still lacking. The significance of this information relies on two facts. First, aberrant PTMs, including glycosylation, are very common in PDAC. Second, aberrant PTMs are associated with pancreatic cancer disease progression and poor prognosis.62–64
Together, recent sequencing studies demonstrate that the genetic landscape of malignant epithelial pancreatic cells is associated with the presence of both focal mutations and large structural rearrangements. It is noteworthy, however, that pancreatic cancers still have lower mutational loads compared with several other cancers such as melanoma,50,65 a disease that is typically associated with increased T-cell infiltration and immunotherapy responsiveness. However, this does not preclude the possibility that the frequency of genetic alterations in pancreatic cancer is sufficient to stimulate effective immune responses. Besides the frequency of tumor antigens, antigen quality, intra-tumoral cancer heterogeneity and TCR diversity are all important determinants of pancreatic cancer immunogenicity. Finally, although pure amino acid sequence-based protein epitopes are considered the primary targets for T cells, it is possible that post-translationally modified proteins play a role in pancreatic cancer immunogenicity.
Question 2: Are Pancreatic Cancer Antigens Processed and Presented on the Surface of Cancer Cells?
To address the concept that in order for antigens to be immunogens, they must be carried on the surface of MHC molecules and presented to T cells, neoantigen prediction computational algorithms have been employed. These algorithms translate all mutations detected by whole exome sequencing to short peptides, and then evaluate the putative MHC-I binding of these mutant peptides compared to wild type sequences. By employing this in silico approach, Balachandran reported that the median numbers of neoantigen-related mutations detected per pancreatic cancer patient are 38 in the MSKCC cohort and 32 in the ICGC cohort.52 Also, using in silico predictions and genomic profiling of 221 PDAC cases extracted from large publicly available datasets, Bailey et al demonstrated that nearly all evaluated pancreatic cancer samples express mutations that are predicted to be immunogenic, ranging from 4 to 4000 neoantigens per sample.65 Nonetheless, it should be noted that only a few computationally predicted immunogenic antigens have been successful in inducing effective tumor-specific T-cell responses, and that these algorithms only consider MHC-antigen interactions but not TCR binding.
Furthermore, although in silico strategies predict that mutations in PDAC tumors can bind to MHC-I, molecules important for antigen processing and presentation are frequently reduced or lost in the context of pancreatic cancer. These include human leukocyte antigen (HLA) class I (ie, human MHC-I), and transporter for antigen presentation (TAP). The loss or downregulation of peptide-MHC complexes on the surface of pancreatic cancer cells may render T cells blind to their targets, and thus it could represent an immune escape mechanism.66,67
Question 3: Do T Cells Infiltrate Pancreatic Cancer Tissue?
Based on immunohistochemistry (IHC) staining and multi-epitope imaging, the majority of human pancreatic cancer tissues are infiltrated with T cells. Nonetheless, while there is substantial inter-patient heterogeneity in intra-tumoral T-cell densities, most studies suggest a relative paucity of intra-tumoral T cells in the majority of pancreatic cancer samples. Hence, pancreatic cancers have been considered immunologically “cold” tumors.21,68–73
Genomic profiling of human PDAC samples, using the Cancer Genome Atlas (TCGA) and the Australian Pancreatic Cancer Genome Initiative, shows high expression of Lymphocyte Cell-Specific Protein-Tyrosine Kinase (LCK), a T-cell marker, implying robust T-cell infiltration. More strikingly, this study also showed that the level of LCK expression in PDAC is not statistically significantly different from skin cutaneous melanoma. However, it is necessary to take into consideration that sample preparations for sequencing and imaging techniques are very different.65 The preparation of sequencing samples may result in the enrichment of immune cells, creating a bias toward immune signatures. On the other hand, IHC quality depends on both optimal sample fixation, and antibody specificity and sensitivity. Also, while most PDAC patients present with inoperable disease, the majority of human pancreatic cancer specimens included in both imaging and sequencing studies were collected from resectable tumors during surgeries. Therefore, all these estimates could be partially biased toward the minority of patients with potential curable malignancies, and thus require careful interpretation.
Furthermore, pancreatic cancer is typically infiltrated by heterogeneous T-cell subpopulations. These subpopulations are skewed toward CD4+ T cells, whereas cytotoxic CD8+ T cells seem to be rare. Also, CD4+ T cells are skewed toward Th2, which is associated with tumor immune tolerance, more than Th1, which enhances CD8+ T-cell tumor-killing responses.70,74 Other CD4+ T-cell subtypes that are commonly detected in PDAC tumors are Treg cells and Th17 cells. The number of Treg cells typically increases during pancreatic cancer development both within tumor tissues70,75,76 and in the peripheral blood,77 and they are associated with worse outcomes.78 Interestingly, through various immunosuppressive mechanisms, Treg cells play an important role in pancreatic cancer immune evasion.70,75,76 However, the role of Th17 cells in pancreatic cancer tumorigenesis is highly controversial. Notably, Th17 cells have the ability to shift into Treg cells, resulting in intermediate stage Th17/reg cells. Thus, this plasticity between Treg and Th17 cells may, at least partially, explain the inconsistency among published reports.30,79 In addition to conventional αβ T cells, non-canonical γδ T cells, which are typically abundant only in the gut mucosa, account for up to 40% of tumor-infiltrating T cells in human pancreatic cancers. These γδ T cells have been shown to support PDAC oncogenesis by restraining the activity of conventional αβ T cells.26,27
Collectively, pancreatic tumors are typically infiltrated with T cells. However, a larger proportion of intra-tumoral T cells promote tumorigenesis, whereas intra-tumoral cytotoxic T cells are generally infrequent in pancreatic cancer.
Question 4: Are Cytotoxic T Cells Within Pancreatic Tumors Capable of Eliciting Anti-tumor Responses?
Even when cytotoxic T lymphocytes infiltrate PDAC tumors, pancreatic cancer cells find ways to escape T-cell anti-tumor immunity. In order for T cells to elicit their cytotoxic responses, they first need to exit the adjacent blood vessels and enter into the tumor tissue. Then, they need to navigate within the complex tumor microenvironment until they reach their target antigens expressed on cancer cells.12 Interestingly, cytotoxic T cells are commonly excluded from the vicinity of pancreatic malignant cells.80–82 Using a novel computational imaging analysis, Carstens et al have demonstrated that the spatial distribution of cytotoxic CD8+ T cells, but not CD4+ T cells or total T cells, in proximity to pancreatic cancer cells correlates with increased overall patient survival.21 This observation implies that pancreatic malignant cells are being protected from cytotoxic T-cell recognition and destruction. Also, using pancreatic cancer mouse models, it has been shown that pancreatic neoplastic cells, through the secretion of the chemokine (C-X-C motif) ligand 1 (CXCL1), and extra-tumoral macrophages can divert T cells away from their target cancer cells, allowing tumors to escape T-cell recognition and attack.80
Furthermore, substantial evidence, using both human and murine samples, suggests that even when effector CD8+ T cells directly interact with pancreatic cancer cells, their cytotoxic activity could be suppressed (Fig. 3). Tumor-related-immune evasion in PDAC is created and mediated by neoplastic cells, stromal cells, altered microorganisms and non-cellular stromal components. Many stromal cells in pancreatic cancer, including cancer-associated fibroblasts (CAFs), MDSCs, TAMs, γδ T cells and Treg cells, directly suppress T-cell anti-tumor immunity via the secretion of a wide variety of immunosuppressive molecules, including interleukin (IL)-6, IL-10, IL-11, CXCL12, transforming growth factor beta (TGFβ), vascular endothelial growth factor (VEGF), arginase 1, nitric oxide synthase (iNOS), indoleamine 2,3-dioxygenase (IDO) and matrix metalloproteinases (MMPs). Immunosuppressive stromal cells can also directly inhibit T-cell immunity by expressing immune checkpoints (such as PD-1, PD-L1, CTLA4, and VISTA), resulting in T-cell exhaustion and/or inactivation. Moreover, stromal cells can indirectly induce T-cell suppression by stimulating the immunosuppressive properties of cancer cells. On the other hand, cancer cells can mediate T-cell immune evasion by acquiring immunosuppressive mutations (such as mutations in KRAS), upregulating immune checkpoints (such as PD-L1 and CTLA4), secreting immunosuppressive molecules (such as TGFβ), downregulating their immunogenicity, and stimulating the recruitment of immunosuppressive stromal cells. These T-cell immune evasion mechanisms in pancreatic cancer have been extensively reviewed elsewhere.17,20,83
FIGURE 3.
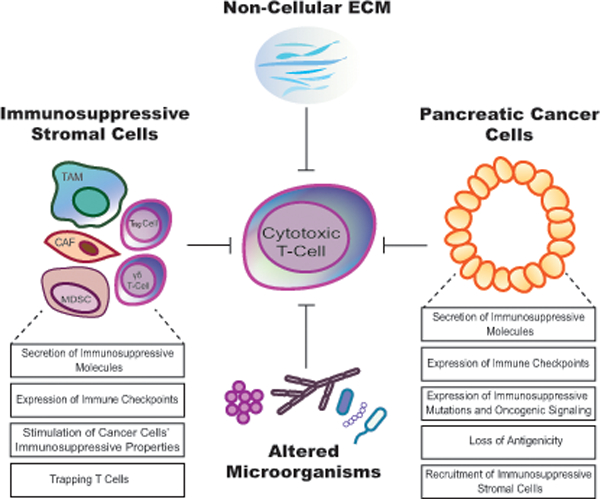
The pancreatic tumor microenvironment exhibits immunosuppressive properties.
Pancreatic cancer cells, several stromal cell types, some altered microorganisms and some non-cellular stromal components can suppress anti-tumor cytotoxic T-cell responses. Tumor-associated macrophage (TAM); regulatory T-cell (Treg); myeloid-derived suppressor cell (MDSC); cancer-associated fibroblast (CAF); T lymphocyte expresses γ and δ TCR chain (γδ T-cell); extracellular matrix (ECM).
To summarize, pancreatic cancers are likely to express immunogenic mutations and they are typically infiltrated by T cells, but generally not at high frequency. Also, a large proportion of intra-tumoral T cells have pro-tumor effects rather than anti-tumor properties. Even when cytotoxic T cells successfully infiltrate the tumors, various pancreatic cancer components tend to prevent anti-tumor T-cell immunity by either physically trap T cells within the stroma or suppress their anti-tumor activity.
FUTURE PROSPECTS
Several therapeutic interventions have been developed to target immune escape routes in pancreatic cancer. These strategies include (i) eliciting tumor antigenicity via a wide range of vaccine strategies, chemotherapy, radiation therapy and targeted therapy,17,84 (ii) augmenting T-cell responses by either boosting T-cell co-stimulatory signals,85 or by inhibiting immune checkpoint molecules, (iii) CAR-T cells and86 (iv) targeting the immunosuppressive properties of the stroma or the neoplastic cells.17 The question now, with a more than 90% mortality rate, and the failure of the vast majority of PDAC clinical trials, is whether there is hope to win the war against pancreatic cancer by harnessing the power of the patient’s immune system?
To answer this question, it is important to remember that checkpoint blockade has demonstrated efficacy in pancreatic cancer patients with high microsatellite instability (MSI-high), who represent less than 2% of all PDAC cases.87 Also, tumors of long-term pancreatic cancer survivors harbor immunogenic mutations and exhibit potent cytotoxic T -cell responses.40,52 These facts indicate that T-cell immunity is relevant to pancreatic cancer and exclude the possibility that the pancreas has organ site-specific resistance to immunotherapy. Also, these findings suggest that in order to improve the efficacy of pancreatic cancer immunotherapy, further resolution of the complex biology of pancreatic cancer immunity is needed.
Furthermore, several lines of evidence point to the idea that combined therapeutic strategies are likely to be required to adequately overcome pancreatic cancer immunotherapy resistance. For instance, pancreatic cancer vaccines alter the immune microenvironment and stimulate tumor-specific T-cell infiltration. However, pancreatic cancers eventually find a way to suppress T-cell immunity.88–90 Similarly, when CAR-T cells that are specifically generated against pancreatic cancer antigens infiltrate pancreatic tumors, they demonstrate poor persistence and induce the expression of checkpoint inhibitory molecules, rendering them ineffective.91 Also, the fact that checkpoint blockade can be effective only in MSI-high PDAC cases,87 typically with higher mutation burden, suggests that augmenting pancreatic cancer antigenicity and simultaneously targeting relevant immune suppressive mechanisms is a potential effective therapeutic strategy. Not surprisingly, several clinical trials combining either vaccines, radiation therapy and/or chemotherapy with immune checkpoint inhibitors and/or myeloid inhibitors are undergoing. These clinical trials are nicely summarized in another review.17
Among the few clinical trials that reported promising therapeutic responses is NCT02482168. It is a small phase Ib study of the CD40 agonist monoclonal antibody APX005M. CD40 is a co-stimulator molecule that is predominantly expressed by APCs, and it is required for their function. Hence, the major mechanism of CD40 therapeutic agonists is to stimulate APC proliferation and activity. However, CD40 can also be expressed by some malignant cells, and thus CD40 agonists can induce targeted cancer cell death.92 Previous pre-clinical studies determined that combining CD40 agonists with chemotherapy promotes the activation of myeloid cells, enhances T-cell priming and stimulates T-cell-dependent anti-tumor immunity.93–96 In this clinical trial, 30 previously untreated PDAC patients received therapy and 24 were dose-limiting toxicity-evaluable. The 24 patients were divided into 4 cohorts: (a) Gemcitabine/nab-paclitaxel/APX005M at 0.1 mg/kg, (b) Gemcitabine /nab-paclitaxel/APX005M at 0.3 mg/kg, (c) Gem/nab-paclitaxel/nivolumab/APX005M at 0.1 mg/kg, and (d) Gem/nab-paclitaxel/ nivolumab/APX005M at 0.3 mg/kg. Those 24 treated patients demonstrated manageable safety profiles and promising therapeutic responses: 14 (58%) patients had partial response, 8 (33%) patients had stable disease, 1 patient (4%) had progressive disease, and 1 patient (4%) had no treatment evaluation. Based on these encouraging results, a randomized phase II clinical trial evaluating chemotherapy and APX005M, with or without nivolumab is currently ongoing.85
CONCLUSION
T cells within PDAC tumors are generally infrequent and/or incapable of eliciting anti-tumor responses. Hence, pancreatic cancer is considered an immunologically cold tumor. However, when T-cell immunity is sufficiently induced in pancreatic cancer, T cells become effective weapons. Thus, the old dogmas that T-cell immunity is not relevant to pancreatic cancer, and that pancreatic cancer is unlikely to respond to immunotherapy are clearly beginning to evolve.
Acknowledgments
Funding:
This research was supported by NCI R01 CA50633 (LMW) and National Institutes of Health (NIH)/National Cancer Institute (NCI) Grant P30-CA051008. RA was supported by King Saud bin Abdulaziz University for Health science (KSAU-HS) and the Saudi Arabian Cultural Mission (SACM).
REFERENCES
- 1.Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Prim. 2016;2:16022 [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69=:7–34. [DOI] [PubMed] [Google Scholar]
- 5.American Cancer Society. Cancer Facts & Figures 2019. Atlanta, GA: American Cancer Society;2019. [Google Scholar]
- 6.Tesfaye AA, Philip PA. Adjuvant treatment of surgically resectable pancreatic ductal adenocarcinoma. Clin Adv Hematol Oncol. 2019;17:54–63. [PubMed] [Google Scholar]
- 7.Lee JC, Ahn S, Cho IK,, et al. Management of recurrent pancreatic cancer after surgical resection: A protocol for systematic review, evidence mapping and meta-analysis. BMJ Open. 2018;106:898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA. 2013;310:1473–1481. [DOI] [PubMed] [Google Scholar]
- 9.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014;74:2913–21. [DOI] [PubMed] [Google Scholar]
- 10.Blankenstein T, Coulie PG, Gilboa E, et al. The determinants of tumour immunogenicity. Nat Rev Cancer. 2012;12:307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arruebo M, Vilaboa N, Sáez-Gutierrez B, et al. Assessment of the evolution of cancer treatment therapies. Cancers (Basel). 2011;3:3279–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity. 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 13.Ajina R, Zamalin D, Weiner LM. Functional genomics: Paving the way for more successful cancer immunotherapy. Brief Funct Genomics. 2019;18:86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seo YD, Pillarisetty VG. T-cell programming in pancreatic adenocarcinoma: A review. Cancer Gene Ther. 2017;24:106–113. [DOI] [PubMed] [Google Scholar]
- 15.Thibodeau J, Bourgeois-Daigneault MC, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology. 2012;1:908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pradeu T, Carosella ED. On the definition of a criterion of immunogenicity. Proc Natl Acad Sci U S A. 2006;103:17858–17861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balachandran VP, Beatty GL, Dougan SK. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology. 2019;156:2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neesse A, Bauer CA, Öhlund D, et al. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut. 2019;68:159–171. [DOI] [PubMed] [Google Scholar]
- 19.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wörmann SM, Diakopoulos KN, Lesina M, et al. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33:2956–2967. [DOI] [PubMed] [Google Scholar]
- 21.Carstens JL, De Sampaio PC, Yang D, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 2017; 8:15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niccolai E, Taddei A, Ricci F, et al. Intra-tumoral IFN-γ-producing Th22 cells correlate with TNM staging and the worst outcomes in pancreatic cancer. Clin Sci. 2016;130:247–258. [DOI] [PubMed] [Google Scholar]
- 23.Piro G, Simionato F, Carbone C, et al. A circulating TH2 cytokines profile predicts survival in patients with resectable pancreatic adenocarcinoma. Oncoimmunology. 2017; 6:e1322242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang XW, Jiang HX, Lei R, et al. Recruitment and significance of Th22 cells and Th17 cells in malignant ascites. Oncol Lett. 2018;16:5389–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Y, Hong S, Li H, et al. Th9 cells promote antitumor immune responses in vivo. J Clin Invest. 2012;122:4160–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lafont V, Sanchez F, Laprevotte E, et al. Plasticity of γδ T cells: Impact on the anti-tumor response. Front Immunol. 2014;5:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daley D, Zambirinis CP, Seifert L, et al. γδ T Cells Support Pancreatic Oncogenesis by Restraining αβ T Cell Activation. Cell. 2016;166:1485–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu J T helper cell differentiation, heterogeneity, and plasticity. Cold Spring Harb Perspect Biol. 2018;10:a030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thommen DS, Schumacher TN. T Cell Dysfunction in Cancer. Cancer Cell. 2018;33:547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Wang L, Mo Q, et al. Changes of Th17/Treg cell and related cytokines in pancreatic cancer patients. Int J Clin Exp Pathol. 2015; 8:5702–5708. [PMC free article] [PubMed] [Google Scholar]
- 31.Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belle JI, Denardo DG. A single-cell window into pancreas cancer fibroblast heterogeneity. Cancer Discov. 2019;9:1001–1002. [DOI] [PubMed] [Google Scholar]
- 33.Neuzillet C, Tijeras-Raballand A, Ragulan C, et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol. 2019;248:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aykut B, Pushalkar S, Chen R, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pushalkar S, Hundeyin M, Daley D, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riquelme E, Zhang Y, Zhang L, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geller LT, Barzily-Rokni M, Danino T, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dambuza IM, Brown GD. Fungi accelerate pancreatic cancer. Nature. 2019;574:184–185. [DOI] [PubMed] [Google Scholar]
- 40.Peng J, Sun BF, Chen CY, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 42.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verbeke C Morphological heterogeneity in ductal adenocarcinoma of the pancreas - Does it matter? Pancreatology. 2016;16:295–301. [DOI] [PubMed] [Google Scholar]
- 45.Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raphael BJ, Hruban RH, Aguirre AJ, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pedersen K, Bilal F, Bernadó Morales C, et al. Pancreatic cancer heterogeneity and response to Mek inhibition. Oncogene. 2017;36:5639–5647. [DOI] [PubMed] [Google Scholar]
- 48.Gubin MM, Artyomov MN, Mardis ER, et al. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cicenas J, Kvederaviciute K, Meskinyte I, et al. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers (Basel). 2017;9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Notta F, Chan-Seng-Yue M, Lemire M, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 2016;538:378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verdegaal EME, De Miranda NFCC, Visser M, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536:91–95. [DOI] [PubMed] [Google Scholar]
- 56.McGranahan N, Furness AJS, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reuben A, Gittelman R, Gao J, et al. TCR repertoire intratumor heterogeneity in localized lung adenocarcinomas: an association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discov. 2017;7:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kristensen VN. The Antigenicity of the tumor cell - Context matters. N Engl J Med. 2017;376:491–493. [DOI] [PubMed] [Google Scholar]
- 59.Raposo B, Merky P, Lundqvist C, et al. T cells specific for post-translational modifications escape intrathymic tolerance induction. Nat Commun. 2018;9:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santos AL, Lindner AB. Protein posttranslational modifications: roles in aging and age-related disease. Oxid Med Cell Longev. 2017;2017:5716409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alpízar A, Marino F, Ramos-Fernández A, et al. A molecular basis for the presentation of phosphorylated peptides by HLA-B antigens. Mol Cell Proteomics. 2017;16:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pan S, Chen R, Tamura Y, et al. Quantitative glycoproteomics analysis reveals changes in N-glycosylation level associated with pancreatic ductal adenocarcinoma. J Proteome Res. 2014;13:1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holst S, Belo AI, Giovannetti E, et al. Profiling of different pancreatic cancer cells used as models for metastatic behaviour shows large variation in their N-glycosylation. Sci Rep. 2017;7:16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Munkley J The glycosylation landscape of pancreatic cancer (Review). Oncol Lett. 2019;17:2569–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bailey P, Chang DK, Forget MA, et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci Rep. 2016;6:35848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pandha H, Rigg A, John J, et al. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol. 2007;148:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ryschich E, Nötzel T, Hinz U, et al. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res. 2005;11:498–504. [PubMed] [Google Scholar]
- 68.Lohneis P, Sinn M, Bischoff S, et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer. 2017;83:290–301. [DOI] [PubMed] [Google Scholar]
- 69.Emmrich J, Weber I, Nausch M, et al. Immunohistochemical characterization of the pancreatic cellular infiltrate in normal pancreas, chronic pancreatitis and pancreatic carcinoma. Digestion. 1998;59:192–198. [DOI] [PubMed] [Google Scholar]
- 70.Ino Y, Yamazaki-Itoh R, Shimada K, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108:914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsujikawa T, Kumar S, Borkar RN, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 2017;19:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shibuya KC, Goel VK, Xiong W, et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS One. 2014;9:e96565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tang ES, Newell PH, Wolf RF, et al. Association of immunologic markers with survival in upfront resectable pancreatic cancer. JAMA Surg. 2018;153:1055–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Foucher ED, Ghigo C, Chouaib S, et al. Pancreatic ductal adenocarcinoma: A strong imbalance of good and bad immunological cops in the tumor microenvironment. Front Immunol. 2018;9:1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hiraoka N, Onozato K, Kosuge T, et al. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423–5434. [DOI] [PubMed] [Google Scholar]
- 76.Jang JE, Hajdu CH, Liot C, et al. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. 2017;20:558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. [DOI] [PubMed] [Google Scholar]
- 78.Liu C, Cheng H, Luo G, et al. Circulating regulatory T cell subsets predict overall survival of patients with unresectable pancreatic cancer. Int J Oncol. 2017;51:686–694. [DOI] [PubMed] [Google Scholar]
- 79.He S, Fei M, Wu Y, et al. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int J Mol Sci. 2011;12:7424–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li J, Byrne KT, Yan F, et al. Tumor Cell-Intrinsic Factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. 2018;49:178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beatty GL, Winograd R, Evans RA, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6Clow F4/80+ extratumoral macrophages. Gastroenterology. 2015;149:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci U S A. 2018; 115:E4041-E4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Looi CK, Chung FFL, Leong CO, et al. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J Exp Clin Cancer Res. 2019;38:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Hara MH, O’Reilly EM, Rosemarie M, et al. Abstract CT004: A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PDAC) patients. Cancer Res. 2019;79(13 Suppl):CT004.abstract. [Google Scholar]
- 86.Akce M, Zaidi MY, Waller EK, et al. The potential of CAR T cell therapy in pancreatic cancer. Front Immunol. 2018;9:2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Le DT, Lutz E, Uram JN, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36:382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Soares KC, Rucki AA, Wu AA, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stromnes IM, Schmitt TM, Hulbert A, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell. 2015;28:638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. 2018;17:509–527. [DOI] [PubMed] [Google Scholar]
- 93.Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 2016;15:2719–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beatty GL, Torigian DA, Gabriela Chiorean E, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;9:6286–6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Long KB, Gladney WL, Tooker GM, et al. IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 2016;6:400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]