

The human pathogen Mycobacterium tuberculosis produces a toxin that can stall bacterial growth by blocking the activity of tRNAs.
Abstract
Toxin-antitoxin systems are widespread stress-responsive elements, many of whose functions remain largely unknown. Here, we characterize the four DUF1814-family nucleotidyltransferase-like toxins (MenT1–4) encoded by the human pathogen Mycobacterium tuberculosis. Toxin MenT3 inhibited growth of M. tuberculosis when not antagonized by its cognate antitoxin, MenA3. We solved the structures of toxins MenT3 and MenT4 to 1.6 and 1.2 Å resolution, respectively, and identified the biochemical activity and target of MenT3. MenT3 blocked in vitro protein expression and prevented tRNA charging in vivo. MenT3 added pyrimidines (C or U) to the 3′-CCA acceptor stems of uncharged tRNAs and exhibited strong substrate specificity in vitro, preferentially targeting tRNASer from among the 45 M. tuberculosis tRNAs. Our study identifies a previously unknown mechanism that expands the range of enzymatic activities used by bacterial toxins, uncovering a new way to block protein synthesis and potentially treat tuberculosis and other infections.
INTRODUCTION
Toxin-antitoxin (TA) systems are widely distributed throughout prokaryotic genomes and have been shown to help bacteria to survive predation by bacteriophages, immune responses, and antibiotic treatments (1–5). In many cases, however, the roles of chromosomal TA systems remain largely unknown, primarily due to the lack of a phenotype associated with deletion mutants under in vitro laboratory conditions (6–9). TA systems are also widespread among mobile genetic elements, including plasmids, superintegrons, cryptic prophages, and conjugative transposons, where they contribute to their stability (10, 11).
TA systems encode two components, a toxic protein that targets an essential cellular process and an antagonistic antitoxin, which blocks toxin activity when cells are growing under favorable conditions. Although the processes that lead to toxin activation remain under debate, it has been proposed that under certain stress conditions, increased toxin transcription and synthesis may lead to activation (8, 12). This, in turn, reduces growth rate, which can provide a means to survive with minimal metabolic burden until favorable conditions return (13).
TA systems are divided into six types according to the nature of the toxin and antitoxin (whether they are RNA or protein) and the mechanism of toxin antagonism (3). Type II systems, in which a protein toxin is sequestered by a protein antitoxin, have been most extensively studied. They are also remarkably abundant in Mycobacterium tuberculosis, which potentially encodes more than 80 type II TA systems, and are thought to have contributed to the success of M. tuberculosis as a human pathogen (14–16). Many of the putative M. tuberculosis toxins tested thus far were shown to inhibit bacterial growth, suggesting that these TA systems are functionally active and could modulate M. tuberculosis growth under certain conditions, thereby contributing to survival in the human host (15, 17). Accordingly, many M. tuberculosis TA operons were shown to be induced in response to relevant stressors, including hypoxia, the presence of antimicrobial drugs, or macrophage engulfment (14, 17). As M. tuberculosis encodes, among others, more than 50 VapBC, 10 MazEF, 3 HigBA, and 3 RelBE TA systems, it might be expected that there is redundancy between them, alongside condition-specific applications for each system. Furthermore, the highly toxic nature of some of these toxins suggests that their antibacterial mechanisms could be developed into antimicrobials (18).
This study focuses on a family of four putative toxins from M. tuberculosis, namely, Rv0078A, Rv0836c, Rv1045, and Rv2826c, which share a conserved nucleotidyltransferase (NTase)–like domain annotated as domain of unknown function (DUF) 1814 (Fig. 1A). The most well-characterized example of this DUF1814 family is AbiEii from Streptococcus agalactiae, which shares 18.3% sequence identity with Rv1045, and was identified within the AbiE abortive infection bacteriophage-defense systems (19). AbiEii was shown to constitute a new type of TA system, type IV, based on the observation that no interaction could be detected between the toxin and the antitoxin proteins (20). The DUF1814 family of proteins is widespread in bacterial, archaeal, and fungal genomes (20), though not all examples are genetically linked to putative antitoxins. As putative NTases, DUF1814 proteins contain four conserved motifs. The N-terminal motifs I and II are found in DNA polymerase β and are proposed to coordinate a metal ion for nucleotide binding and transfer (20). The C-terminal motif III is similar to that of tRNA NTases that add the 3′-CCA motif to immature tRNAs and may be important for base stacking with substrates (21). The C-terminal motif IV is unique to DUF1814 proteins and is proposed to form a catalytic site with motif III (20).
Fig. 1. Analysis of the four TA systems with NTase-like toxins encoded by the M. tuberculosis genome.
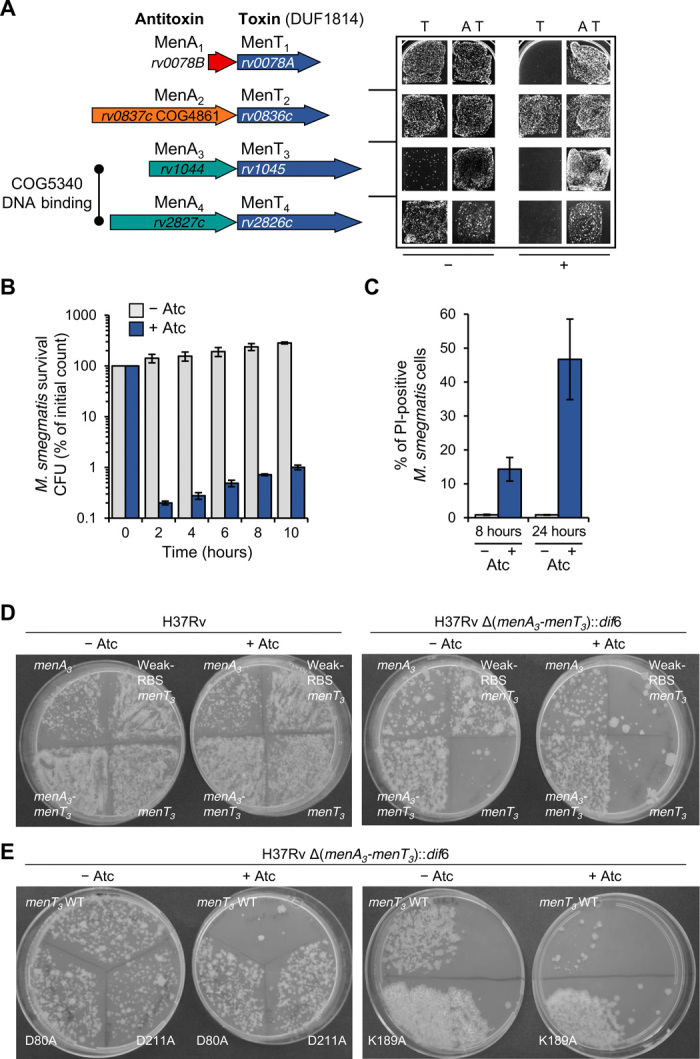
(A) Scaled representation of the four M. tuberculosis TA systems containing NTase-like toxin genes with original and revised nomenclature (left), and corresponding toxicity and antitoxicity assays in M. smegmatis (right). For toxicity and antitoxicity assays, cotransformants of M. smegmatis mc2 155 containing pGMC-vector, -MenT1, -MenT2, -MenT3, or -MenT4 (toxins) and pLAM-vector, -MenA1, -MenA2, -MenA3, or -MenA4 (antitoxins) were plated on LB-agar in the presence or absence of anhydrotetracycline (Atc; 100 ng ml−1) and acetamide (Ace; 0.2%) inducers for toxin and antitoxin expression, respectively. Plates were incubated for 3 days at 37°C. “T” and “A” denote toxin and antitoxin, respectively. “−” and “+” represent absence or presence of inducer, respectively. (B) M. smegmatis strain mc2 155 transformed with plasmid pGMCS-TetR-P1-RBS1-MenT3 was grown in complete 7H9 medium with Sm. At time 0, the culture was divided into two. Half was kept in the same medium (pale blue bars) and half was additionally treated with Atc (200 ng ml−1) (dark blue bars). Samples were harvested at the indicated times, washed, diluted, and plated on LB-agar with Sm but without Atc. Colonies were counted after 3 days at 37°C. Shown values are the average of three biological replicates with SD. CFU, colony-forming unit. (C) Samples of the same cultures as in (B) were harvested after 8 or 24 hours, labeled with the LIVE/DEAD BacLite dyes [Syto 9; propidium iodide (PI)], and analyzed by fluorescence-activated cell sorting. The percentage of PI-positive cells is shown for each sample (pale blue bars, no Atc; dark blue bars, 200 ng ml−1 Atc). Shown values are the average of three biological replicates with SD. (D) M. tuberculosis wild-type (WT) H37Rv or mutant strain H37Rv Δ(menA3-menT3)::dif6 were transformed with 100 ng of plasmids expressing either menA3, menT3, or menA3-menT3. These plasmids encode a consensus Shine-Dalgarno sequence (RBS1), except for “Weak-RBS-menT3,” which encodes a near-consensus sequence (RBS4) to weaken expression. After phenotypic expression, half of the transformation mix was plated on 7H11 oleic acid–albumin-dextrose-catalase (OADC) plates with Sm, and the other half was plated on 7H11 OADC Sm plates supplemented with Atc (200 ng ml−1). Plates were imaged after 20 days at 37°C; data are representative of three independent experiments. (E) Mutant strain H37Rv Δ(menA3-menT3)::dif6 was transformed with 100 ng of plasmids expressing either menT3 WT or mutant alleles introducing the D80A, K189A, or D211A substitutions. After phenotypic expression, half of the transformation mix was plated on 7H11 OADC plates with Sm, and the other half was plated on 7H11 OADC Sm plates supplemented with Atc (200 ng ml−1). Pictures were taken after 20 days at 37°C; data are representative of three independent experiments.
In M. tuberculosis, the DUF1814 toxins are encoded downstream of a variety of putative antitoxins (Fig. 1A). The toxin gene rv0078A is paired with a short upstream open reading frame encoding a 68–amino acid antitoxin, Rv0078B, related to MazE antitoxins, which is predicted to be disordered and lacking a DNA-binding domain (16). Toxin gene rv0836c lies downstream of a COG4861 gene, encoding a much larger putative antitoxin than the cognate toxin (Fig. 1A). Rv1045 and Rv2826c toxins are downstream of their cognate putative antitoxins Rv1044 and Rv2827c, respectively, both of which are COG5340 transcriptional regulator family proteins (Fig. 1A). COG5340 proteins include the S. agalactiae AbiEi antitoxin partner of AbiEii, which has previously been shown to bind to and repress the abiE promoter, similar to autoregulation observed in type II TA loci (22). An earlier transposon site hybridization study identified both the Rv1044 and Rv2827c antitoxins as essential for growth (23). Saturating transposon mutagenesis has additionally demonstrated that Rv1044 is essential, while transposon insertions in Rv2827c impart a growth defect (24). The fact that both antitoxins are important for M. tuberculosis growth strongly suggests that their putative cognate Rv1045 and Rv2826c toxins inhibit growth in M. tuberculosis.
Here, we undertook a series of microbiological, structural, genetic, and biochemical studies to investigate the DUF1814 toxins of M. tuberculosis and reveal their mode of action. We show that the Rv1045 toxin is a tRNA NTase that is active in M. tuberculosis and blocks translation through a previously undescribed mechanism involving inactivation of serine tRNAs.
RESULTS
Three DUF1814 proteins are part of bona fide TA systems
We first investigated the activity of the putative TA systems containing NTase-like DUF1814 toxins in Mycobacterium smegmatis, which is closely related to M. tuberculosis and does not encode similar antitoxins (15). On the basis of the findings presented below, we renamed these putative systems as “mycobacterial AbiE-like NTase toxins” (MenT) and antitoxins (MenA), numbered according to their order in the M. tuberculosis genome (Fig. 1A). Toxins and antitoxins were expressed in trans, with the toxins cloned into the pGMC-integrative plasmid under the control of an anhydrotetracycline (Atc)–inducible promoter and the antitoxins into the compatible pLAM plasmid under the control of an acetamide (Ace)–inducible promoter (Fig. 1A). Among the four putative toxins, only MenT1 has been tested so far and was shown to be toxic in M. smegmatis when expressed without the upstream open reading frame encoding MenA1, suggesting that MenA1-MenT1 form a functional TA system (16). Accordingly, the data presented in Fig. 1A show that MenT1 toxicity was efficiently counteracted by MenA1 expressed in trans. Both MenA3-MenT3 and MenA4-MenT4 also acted as TA pairs, while MenT2 expression was not toxic (Fig. 1A). Inhibition of MenT4 toxicity could only be achieved when the putative antitoxin was expressed in the context of the menA4-menT4 operon (Fig. 1A). Expression of MenA4 alone from pLAM was toxic (fig. S1A), indicating that MenA4-MenT4 might not function as a typical TA pair under these conditions. Similar experiments performed in Escherichia coli confirmed the phenotypes observed in M. smegmatis for MenA2-MenT2, MenA3-MenT3, and MenA4-MenT4 (including MenA4 toxicity), but not for MenT1, which exhibited no detectable toxicity in E. coli (fig. S1B). Last, coexpression of the active toxins with noncognate antitoxins did not reveal any detectable cross-talk between the different TA pairs (fig. S1, A and C). Note that cross-talk assays with MenA4 antitoxin expressed from pLAM in M. smegmatis could not be performed because of its toxicity.
Ectopic expression of MenT3 in the presence of inducer showed the most robust toxicity in both M. smegmatis and E. coli when compared to the other toxins (Fig. 1A and fig. S1). In M. smegmatis, only a few MenT3 transformants were obtained, even in the absence of inducer. Ectopic expression of MenT3 in M. smegmatis induced a rapid drop of about 3-log10 in colony-forming units only 2 hours after induction with Atc (Fig. 1B). LIVE/DEAD BacLight stains have previously been used to study the effects of toxin expression on cell viability in M. tuberculosis (18). Flow cytometry analysis of M. smegmatis expressing MenT3 revealed that the proportion of propidium iodide–permeable cells was substantially higher in MenT3-induced versus noninduced cells 8 or 24 hours after induction with Atc (Fig. 1C), indicating that MenT3 strongly affects cell viability.
MenA3-MenT3 is a functional TA system in M. tuberculosis
To investigate the impact of MenA3 and MenT3 on M. tuberculosis growth, plasmids encoding the toxin, the antitoxin, or both, were introduced into H37Rv wild-type (WT) strain. The resulting transformants were not sensitive to ectopic expression of MenT3 (Fig. 1D), presumably because endogenous MenA3 was sufficient to neutralize the sum of endogenous and ectopic MenT3. To confirm this hypothesis, we attempted to construct a strain deleted for the menA3-menT3 operon. Previous work showed that menA3 cannot be disrupted by transposon insertion (24). Accordingly, we found that deletion of the menA3-menT3 operon in M. tuberculosis H37Rv strain could not be achieved, most likely because simultaneous disruption of both genes resulted in a toxic effect from residual MenT3. To circumvent this problem, we constructed the deletion in a derivative of H37Rv carrying a second copy of menA3 constitutively expressed from a pGMC integrative plasmid. Once the menA3-menT3 operon was deleted, it was then possible to remove the ectopic copy of menA3 by pGMC plasmid replacement (fig. S2). The ∆menA3-menT3 mutant became highly sensitive to the MenT3 toxin, even in the absence of inducer (Fig. 1D). Therefore, to finally obtain transformants, menT3 was cloned downstream of a weaker Shine-Dalgarno sequence. Using this construct, we observed inducible MenT3 toxicity, which was fully abolished by the presence of the antitoxin (Fig. 1D). Together, these data demonstrate that the MenT3 toxin inhibits growth and that MenA3-MenT3 functions as a bona fide TA pair in M. tuberculosis.
A previous amino acid sequence alignment of DUF1814 putative NTases highlighted conserved residues, a number of which were confirmed as essential for AbiEii toxicity in S. agalactiae (20). To investigate whether some of these residues were important for MenT3 toxicity, we selected and engineered three conserved residues for substitution: D80A, localized in the DNA polβ superfamily motif, and K189A and D211A, both toxin-specific residues. We then tested the impact of these substitutions on M. tuberculosis growth (Fig. 1E). All three substitutions abolished MenT3 toxicity in both M. tuberculosis (Fig. 1E) and E. coli (fig. S3A).
Next, we investigated whether the MenT3 toxin and MenA3 antitoxin could interact in vivo. Since this TA pair is functional in E. coli, we performed affinity-tagged in vivo copurification experiments in E. coli using His-tagged variants of MenT3 and MenA3, which were first confirmed to be active as toxin and antitoxin, respectively (fig. S4A). In strains coexpressing both the toxin and the antitoxin (with either the toxin or the antitoxin tagged), and with tagged toxin and tagged antitoxin alone as controls, the in vivo copurification revealed that a small but significant fraction of the MenT3 toxin and the MenA3 antitoxin copurified, whether the toxin or the antitoxin was used as bait (fig. S4, C and D). Similar results were obtained with the MenA1-MenT1 pair, which encodes a much shorter, unrelated antitoxin (fig. S4, B, E, and F). Together, these data show that both TA partners can interact, but it remains to be determined whether a direct interaction between an NTase toxin and its cognate antitoxin is required for toxin inhibition.
MenT3 and MenT4 are structural homologs
To begin investigations into the mechanism of toxicity of MenT3, we solved its structure to 1.6 Å resolution by x-ray crystallography (Fig. 2A and Table 1). MenT3 is a monomeric bi-lobed globular protein, with two hemispheres connected by a short linker (Fig. 2A). This monomeric assembly matches the expected size observed by size exclusion chromatography. Surface electrostatics show a distinct electropositive surface leading to a deeper recess (Fig. 2B, left), which contains residues D80, K189, and D211 that were needed for toxicity in vivo (Fig. 1E). This potentially indicates the position of the active site, and the electropositive surface may facilitate interaction with electronegative substrates such as nucleic acids. To further characterize the DUF1814 family, we also solved the MenT4 toxin structure to 1.2 Å resolution (Fig. 2C and Table 1). MenT4 is also monomeric (also observed by size exclusion chromatography) and the overall architecture is similar to, but not exactly the same as, MenT3. MenT4 has a bi-lobed globular structure and distinct electropositive patches close to a similarly positioned active site region (Fig. 2, C and D). Aligning MenT3 and MenT4 by sequence gave a poor root mean square deviation (RMSD) of 13.4 Å; however, this can be improved to 4.7 Å using sequence-independent superposition, which demonstrates similarity in overall fold (Fig. 2E). A close-up of MenT3 residues D80, K189, and D211 show them clustered at the putative active site, and when overlaid, the homologous MenT4 residues, D69, K171, and D186, respectively, take up similar positions (Fig. 2F). There was also density for a phosphoserine at MenT3 S78, but the corresponding residue in MenT4, S67, was not phosphorylated (Fig. 2F). Searches for structural homologs of MenT3 and MenT4 were performed using the DALI server (25). Among multiple hits for NTases, the best match was for JHP933 from Helicobacter pylori, a predicted NTase encoded by the jhp0933 gene (26). JHP933 aligned to MenT3 with an RMSD of 2.4 Å, though multiple additional helices were resolved in the MenT3 structure (Fig. 2G). An analysis of the H. pylori genome revealed that the jhp0932 gene lies just upstream of jhp0933 and partially overlaps its coding sequence. The presence of these genes in what appears to be a classic TA configuration suggests that JHP933 may belong to the MenT3/MenT4 family of NTase-like toxins.
Fig. 2. Crystal structure of the MenT3 and MenT4 toxins.
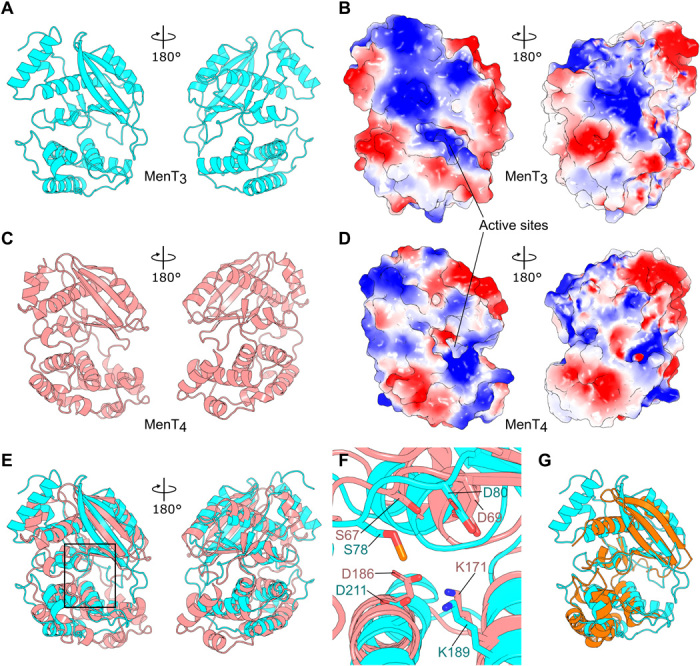
(A) Structure of monomeric MenT3 toxin, with views from front and back, shown as cyan cartoon representations. (B) Surface electrostatics of MenT3, viewed as in (A), with red for electronegative and blue for electropositive potential. (C) Structure of monomeric MenT4, with views from front and back, shown as salmon cartoon representations. (D) Surface electrostatics of MenT4, viewed as in (C), colored as per (B). (E) Superposition of MenT4 onto MenT3, viewed and colored as per (A) and (C). (F) Tilted close-up view of the toxin active sites, as indicated by the boxed region of (E). MenT3 residues S78 (phosphorylated), D80, K189, and D211 are indicated, along with the homologous MenT4 residues S67, D69, K171, and D186. (G) Alignment of JHP933 (PDB: 4O8S) as orange cartoon representation, against MenT3 viewed and colored as per (A, left).
Table 1. Crystallographic data collection and refinement statistics.
| MenT3 native | MenT3 Se-peak | MenT3 Se-high remote | MenT3 Se-inflection | MenT4 native | |
| Data collection | |||||
| PDB ID code | 6Y5U | - | - | - | 6Y56 |
| Beamline | Diamond I04 | Diamond I03 | Diamond I03 | Diamond I03 | Diamond I24 |
| Wavelength (Å) | 0.9795 | 0.9793 | 0.9641 | 0.9795 | 0.9781 |
| Resolution range (Å) | 47.70–1.59 (1.65–1.59)* | 47.78–2.19 (2.26–2.19) | 47.83–2.05 (2.11–2.05) | 53.13–2.04 (2.11–2.04) | 42.23–1.23 (1.27–1.23) |
| Space group | P3221 | P3221 | P3221 | P3221 | P21 |
| Unit cell, a b c (Å), α β γ (°) | 95.4 95.4 69.0, 90.0 90.0 120.0 | 95.6 95.6 69.2, 90.0 90.0 120.0 | 95.7 95.7 69.3, 90.0 90.0 120.0 | 95.6 95.6 69.3, 90.0 90.0 120.0 | 42.3 57.8 54.7, 90.0 92.3 90.0 |
| Total reflections | 98,016 (9668) | 36,407 (3179) | 44,514 (3476) | 47,255 (4637) | 149,653 |
| Unique reflections | 49,008 (4834) | 19,130 (1646) | 23,313 (1788) | 23,628 (2319) | 75,996 (7206) |
| Multiplicity | 2.0 | 1.9 | 1.9 | 2.0 | 2.0 |
| Completeness (%) | 99.95 (99.83) | 100.00 (100.00) | 100.00 (99.80) | 100.00 (99.70) | 98.80 (88.97) |
| Mean I/σ(I) | 16.7 | 6.5 | 7.6 | 8.9 | 7.0 |
| Rmerge | 0.016 (0.486) | 0.055 (0.373) | 0.055 (0.522) | 0.048 (0.463) | 0.060 (0.926) |
| Rmeas | 0.022 (0.687) | 0.077 (0.528) | 0.078 (0.739) | 0.068 (0.654) | 0.085 (1.310) |
| CC1/2 | 1.0 (0.672) | 0.995 (0.803) | 0.997 (0.544) | 0.997 (0.641) | 0.996 (0.294) |
| Refinement | |||||
| Rwork | 0.2024 (0.2924) | - | - | - | 0.1840 (0.3174) |
| Rfree | 0.2242 (0.3108) | - | - | - | 0.1950 (0.3352) |
| No. of non-hydrogen atoms | 2494 | - | - | - | 2649 |
| Macromolecules | 2213 | - | - | - | 2322 |
| Solvent | 281 | - | - | - | 327 |
| Protein residues | 288 | - | - | - | 292 |
| RMSD (bonds, Å) | 0.006 | - | - | - | 0.005 |
| RMSD (angles, °) | 0.940 | - | - | - | 0.830 |
| Ramachandran favored (%) | 97.53 | - | - | - | 98.28 |
| Ramachandran allowed (%) | 2.47 | - | - | - | 1.72 |
| Ramachandran outliers (%) | 0.00 | - | - | - | 0.00 |
| Average B factor | 34.1 | - | - | - | 20.6 |
| Macromolecules | 33.4 | - | - | - | 19.3 |
| Solvent | 39.4 | - | - | - | 29.9 |
*Statistics for the highest-resolution shell are shown in parentheses.
RNase PH overexpression confers resistance to MenT3
MenT3 is the most toxic of the four M. tuberculosis NTase-like toxins tested, both in mycobacteria and in E. coli (Fig. 1 and fig. S1). We therefore took advantage of this robust toxicity to search for E. coli genes that were able to suppress MenT3-mediated growth inhibition when overexpressed. We reasoned that identification of such suppressors might potentially shed light on the cellular processes affected by the toxin. Details of the genetic selection used are described in Materials and Methods. Among the approximately 60,000 clones of the E. coli genomic plasmid library tested in this work, we identified 18 plasmids that passed two rounds of selection and appeared to encode bona fide suppressors of MenT3 toxicity. We observed that the toxin-resistant colonies were noticeably smaller and translucent compared to noninduced cells, indicating that, although notably reduced, MenT3 toxicity is not fully suppressed. Sequencing of the genomic regions encoded by the 18 suppressor plasmids revealed that several of these candidate plasmids harbored the same genomic fragments. Six different suppressor clones encompassing two different regions of the E. coli chromosome were identified. Two of the six suppressor plasmids harbored the ydeA gene, encoding an l-arabinose (l-ara) exporter protein known to decrease l-ara levels in E. coli (27). These suppressors were discarded as YdeA overexpression would presumably decrease toxicity of many toxic proteins expressed from the araBAD promoter. The four other suppressor plasmids harbored the rph gene, encoding the phosphorolytic ribonuclease (RNase PH), involved in the 3′ processing of RNAs (Fig. 3A). RNase PH removes nucleotides downstream of the 3′-CCA sequence, required for aminoacylation of tRNAs, from tRNA precursors with 3′ extensions. It is also involved in other RNA maturation and quality control processes, including the maturation of rRNA (28).
Fig. 3. RNase PH suppresses MenT3 toxicity and inhibition of translation.

(A) The E. coli K-12 genomic region containing the rph gene is shown. Suppressor plasmids that counteract MenT3 toxicity encoded rph, as depicted by small arrows under the adjacent genes pyrE, yicC, and dinD. The positions in base pair of the ends of each suppressor fragment, in relation to the E. coli K-12 chromosome, are indicated between brackets. (B) Overexpression of E. coli RNase PH partially suppresses MenT3 toxicity. E. coli DLT1900 strains containing either pK6-vector (−) or pK6-MenT3 (+) were cotransformed with p29SEN-vector (−) or p29SEN-Rph (RNase PH) (+). The resulting cotransformants were serially diluted, spotted onto LB-agar plates in the presence or absence of l-ara (0.1%) and IPTG (200 μM) inducers, and incubated at 37°C. (C) Deletion of rph further increases MenT3 toxicity. Transformants of E. coli DLT1900 WT and ∆rph mutant strains containing plasmid pK6-MenT3 were serially diluted, spotted onto LB-agar plates with or without l-ara (0.01%), and incubated at 37°C. (D) In vitro transcription/translation reactions assessing levels of DHFR control protein produced in the absence or presence of increasing concentrations of MenT3 toxin. Samples were separated by SDS–polyacrylamide gel electrophoresis and stained with InstantBlue. (E) For in vivo assays, transformants of E. coli BL21 (λDE3) containing plasmid pET-MenT3 or the empty vector were grown in M9M at 37°C. Following overexpression of MenT3, tRNAs were extracted, separated, and visualized by Northern blot using specific radiolabeled probes against tRNATrp. For in vitro assays, purified MenT3 (10 μM) was added to transcription/translation assays producing GatZ protein. After 2 hours at 37°C, tRNAs were extracted, separated, and visualized by Northern blot as performed for the in vivo samples. All images are representative of triplicate data.
Suppression of MenT3 toxicity by RNase PH overexpression was confirmed by cloning rph alone in a low–copy number plasmid under the control of an isopropyl-β-d-thiogalactopyranoside (IPTG)–inducible promoter and assaying for growth in the presence of MenT3 in E. coli (Fig. 3B). We also showed that the toxicity of MenT3 was enhanced when expressed in E. coli carrying a deletion of the rph gene, even with a 10-fold decrease in inducer levels (Fig. 3C), further reinforcing the genetic link between menT3 and rph. The primary role of RNase PH in processing tRNAs suggests that DUF1814 NTase-like toxins could act directly at the site of aminoacylation at the 3′-end of tRNA, thus inhibiting translation. Whether endogenous RNase PH would be sufficiently induced in response to toxin expression to help restore the functional tRNA pool in recovering M. tuberculosis cells remains to be determined.
MenT3 inhibits tRNA charging
MenT3 WT and the MenT3(D80A) and MenT3(K189A) substitutions were overexpressed and purified for biochemical characterization. When tested in an in vitro transcription/translation reaction that uses recombinant E. coli components, purified MenT3 WT reduced production of the E. coli dihydrofolate reductase (DHFR) control protein in a concentration-dependent manner (Fig. 3D). Compared to MenT3 WT, MenT3(D80A) and MenT3(K189A) had a markedly reduced impact on the production of DHFR (fig. S5A). The same trend was observed when MenT3 WT, MenT3(D80A), and MenT3(K189A) were used in in vitro reactions producing WaaF and GatZ as test proteins (fig. S5, B and C). We also expressed and purified MenT4 WT and demonstrated that this, too, prevented the production of DHFR in a concentration-dependent manner in in vitro transcription/translation assays (fig. S5D).
The fact that MenT3 inhibited protein synthesis, and that RNase PH is involved in the removal of nucleotides following the 3′-CCA sequence required for tRNA aminoacylation, suggested that tRNA charging might be affected by MenT3 expression in vivo. To address this hypothesis, we first used a method developed for E. coli, which separates charged from uncharged tRNAs and allows their detection by Northern blot after extraction in vivo (29). We chose tRNATrp as a model tRNA because (i) the tryptophanyl-tRNA can be well separated from uncharged tRNATrp and (ii) there is only one tRNATrp in E. coli (29). No charged tryptophanyl-tRNATrp could be detected following overexpression of MenT3 when compared to the empty vector control (Fig. 3E and fig. S5E). tRNATrp charging levels were also investigated in vitro by adding purified MenT3 to the transcription/translation assay described above (Fig. 3E). In this case, MenT3 also affected tRNATrp charging in vitro, thus supporting the hypothesis that the toxin inhibits protein synthesis by preventing aminoacylation of tRNA.
MenT3 transfers pyrimidines to the 3′ acceptor stem of specific tRNAs
The observation that MenT3 is related to NTases (Fig. 2G) suggests that its mode of action is to directly transfer nucleotides to tRNAs, thereby preventing aminoacylation. We performed assays using radiolabeled tRNAs to track the addition of nucleotides by MenT3 WT, MenT3(D80A), and MenT3(K189A) (Fig. 4).
Fig. 4. Toxin MenT3 adds pyrimidines to the 3′-CCA acceptor stem of tRNA.
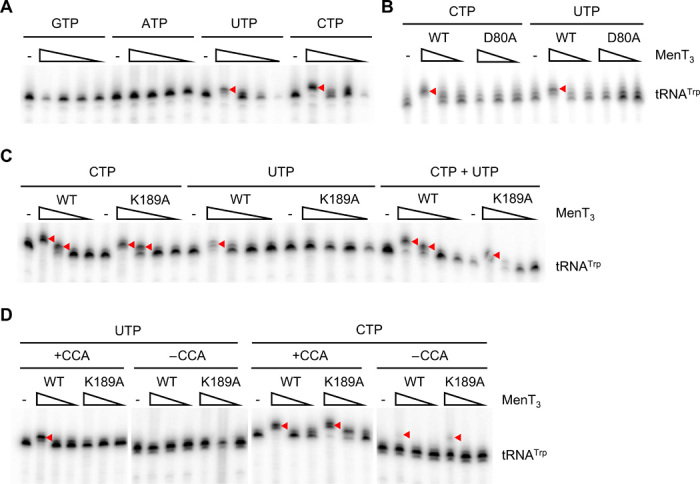
(A) Radiolabeled E. coli tRNATrp was incubated with 1, 0.1, 0.01, or 0.001 μg of MenT3 WT or no toxin (−) for 20 min at 37°C in the presence of unlabeled GTP, ATP, UTP, or CTP. Extended products are indicated with arrowheads throughout all panels. (B) Radiolabeled E. coli tRNATrp was incubated with 1, 0.1, or 0.01 μg of MenT3 WT or MenT3(D80A) with CTP or UTP, as per conditions in (A). (C) Incubation of radiolabeled E. coli tRNATrp with 1, 0.1, 0.01, or 0.001 μg of MenT3 WT or MenT3(K189A), with CTP, UTP, or a mixture of both, as per conditions in (A). (D) Radiolabeled E. coli tRNATrp preparations, made with or without a 3′-CCA motif, were incubated with 1, 0.1, or 0.01 μg of either MenT3 WT, MenT3(K189A), or no toxin (−), for 20 min at 37°C in the presence of unlabeled UTP or CTP. Note that the (−) CCA lanes have been overexposed to equalize intensity to the (+) CCA lanes of the same gel. Assays of the individual WT and MenT3 substitution proteins and tRNATrp ± CCA substrates shown in (A) to (D) were performed between two and four times.
MenT3 WT was incubated with tRNATrp from E. coli, as a model recipient tRNA, in the presence of guanosine 5′-triphosphate (GTP), adenosine 5′-triphosphate (ATP), uridine 5′-triphosphate (UTP), or cytidine 5′-triphosphate (CTP), and nucleotide transfer was monitored as an increase in tRNA size by high-resolution polyacrylamide gel electrophoresis (PAGE; Fig. 4A). At high concentrations of the enzyme, we found that MenT3 can add two to three extra nucleotides to tRNATrp in the presence of CTP or UTP, with a slight preference for CTP, suggesting that MenT3 is a pyrimidine-specific NTase (Fig. 4A). No transfer was observed with purines ATP or GTP as substrates (Fig. 4A). MenT3(D80A), which was unable to inhibit in vitro protein synthesis (fig. S5, A to C), had no NTase activity with either UTP or CTP (Fig. 4B). MenT3(K189A), which was also inactive in the in vitro transcription/translation assay (fig. S5, A to C), only lost its NTase activity in the presence of UTP, but retained some activity (albeit less than WT) in the presence of CTP, or both nucleotides (Fig. 4C). This could imply that K189A is important for substrate nucleotide selectivity. No synergistic effect was seen when MenT3 WT was incubated with a mixture of CTP and UTP, as the pattern with both nucleotides together resembled that of CTP alone (Fig. 4C).
Canonical tRNA NTases typically add the 3′-CCA motif to tRNAs lacking an encoded 3′-CCA that are processed at the level of the discriminator nucleotide (nucleotide 73). They also repair this motif when 3′-exoribonucleases, such as RNase PH, fail to stop at the 3′-CCA motif when processing tRNA precursors containing an encoded 3′-CCA, typically removing the terminal A residue. Since M. tuberculosis contains a mixture of tRNA genes encoding or lacking a 3′-CCA motif, we wondered whether MenT3 had a preference for one class (or another class) of substrate. While faint NTase activity was observed when MenT3 WT and MenT3(K189A) were incubated with CTP and tRNATrp lacking a 3′-CCA, the data show that MenT3 had a clear preference for tRNAs that already possessed a 3′-CCA motif (Fig. 4D). This is in contrast to the normal function of tRNA NTases, which prefer tRNAs lacking an intact 3′-CCA. Again, MenT3 WT modified tRNATrp using both CTP and UTP as substrate, while MenT3(K189A) could only use CTP (Fig. 4D). Addition of nucleotides to mature tRNAs by MenT3 would completely abolish the ability of these tRNAs to be charged with their cognate amino acid and take part in translation, accounting for their cellular toxicity.
Our in vivo data show that toxins MenT3, as well as MenT1 and MenT4, are significantly less toxic in E. coli than in mycobacteria (Fig. 1 and fig. S1), which suggests that these toxins may have a tRNA target preference. We therefore asked whether MenT3 would exhibit some specificity toward the different tRNAs of M. tuberculosis. We made polymerase chain reaction (PCR) templates allowing us to in vitro transcribe the 45 different tRNAs of M. tuberculosis, each with a 3′-CCA motif (fig. S6). As before, each radiolabeled tRNA was incubated with MenT3 and nonradiolabeled CTP (Fig. 5A). To our surprise, MenT3 appeared to be highly specific, preferentially modifying the four M. tuberculosis tRNASer isoacceptors, along with weak modification of tRNALeu5 (Fig. 5A). Although we cannot exclude that MenT3 can modify other tRNAs in vivo, the data show that the toxin presents a high degree of specificity toward different tRNAs in vitro, which may explain the variable toxicity observed in different bacteria.
Fig. 5. Screening for MenT3 M. tuberculosis tRNA targets.
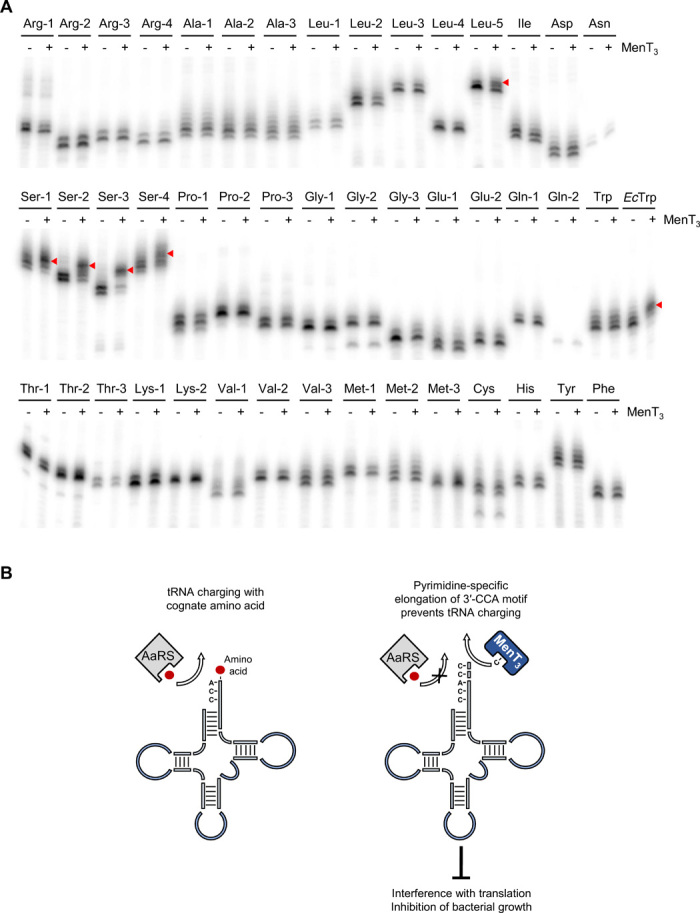
(A) Radiolabeled M. tuberculosis tRNAs were incubated with 0.1 μg of MenT3 WT (+) or no toxin (−) for 20 min at 37°C in the presence of unlabeled CTP. E. coli tRNATrp (EcTrp) was used as a positive control. The global screen of all M. tuberculosis tRNA was performed once and the effect of MenT3 tRNASer2 was confirmed twice independently. (B) Schematic diagram of the MenT3 toxin mechanism of action. MenT3 elongates the 3′-CCA motif of specific tRNAs, preventing their charging by aminoacyl-tRNA synthetases (AaRS), thereby interfering with translation and inhibiting bacterial growth.
Last, we asked whether the antitoxin MenA3 inhibited the NTase activity of MenT3 directly, or whether it could simply reverse its action by removing the added nucleotides in a manner similar to the RNase PH multicopy suppressor. Addition of MenA3 strongly inhibited the NTase activity of MenT3 on the natural substrate M. tuberculosis tRNASer2 when coincubated with the toxin at a molar ratio > 2.5. However, MenA3 failed to remove the added nucleotides from tRNASer2 when added after a preincubation of tRNASer2 with MenT3, even at high concentrations (fig. S7). This suggests that the antitoxin is likely to inhibit the toxin rather than reverse the reaction on the substrate.
DISCUSSION
This study has characterized a family of TA systems from M. tuberculosis containing NTase-like DUF1814 toxins, establishing MenT3 as a potent toxin in this problematic pathogen. We have solved the structures of the homologous toxins MenT3 and MenT4 by x-ray crystallography, revealing fold similarity and conserved residues within the proposed active sites, and have observed a similar mode of toxin activity, targeting protein synthesis. We have further elucidated the mechanism of toxicity for MenT3, showing that it functions as a pyrimidine-specific NTase preferentially targeting M. tuberculosis tRNASer in vitro (Fig. 5B).
The observation that the three NTase toxins identified in this work show different levels of toxicity when expressed in the same host, and that such toxic signatures can vary when expressed in different bacterial hosts (i.e., E. coli versus mycobacteria), is intriguing (Fig. 1 and fig. S1). The most marked example is MenT1, which shows robust toxicity in M. smegmatis but no toxicity in E. coli (Fig. 1A and fig. S1B). Although we cannot exclude this being a result of improper folding or expression of the toxin in E. coli, it is also reasonable to assume that the toxin may not be able to recognize its tRNA targets due, for example, to tRNA modification, or the absence of its preferred tRNA target (30). Another possibility is that tRNA targets are expressed at higher levels in E. coli and are thus sufficiently abundant to overcome the noxious effect of the toxin in vivo. The fact that M. tuberculosis and M. smegmatis only have 45 and 46 tRNA genes, respectively, while E. coli has 86, is in line with this hypothesis (30, 31).
The apparent in vitro specificity of MenT3 for certain M. tuberculosis tRNAs, especially tRNASer, is remarkable (Fig. 5A). We did not test other tRNAs in E. coli besides tRNATrp; it may well have been fortuitous that the only tRNA we tested in this organism was detectably modified by the toxin in vitro (Fig. 4A) and in vivo, inferred from the reduced charging levels following toxin expression (Fig. 3E). We checked whether the M. tuberculosis tRNAs that were substrates of MenT3 had any distinguishing features and were struck by the fact that all serine tRNAs and several leucine tRNAs were unique among M. tuberculosis tRNAs in that they had long variable arms (fig. S8A) (32). While this is intriguing and may contribute to substrate specificity, it cannot be the only recognition element because (i) two leucine tRNAs besides tRNALeu5 have variable loops but are not MenT3 substrates in vitro and (ii) E. coli tRNATrp does not have a variable loop (fig. S8B), but can be extended by the NTase activity of the toxin. It is also intriguing in this regard that M. tuberculosis tRNATrp is not a MenT3 substrate in vitro. E. coli and M. tuberculosis tRNATrp are highly homologous but do show differences in their variable- and T-arm sequences (fig. S8C). Substrate specificity therefore appears to come from a combination of multiple sequence and structure motifs. Having identified these tRNA targets in vitro, further work is now needed to confirm targeting in vivo in M. tuberculosis.
Our observed TA interactions raise questions regarding the molecular mechanisms of antitoxicity for DUF1814 toxins (fig. S4, C to F). Typically, in type II TA systems, antitoxin function is in part driven by its strong and direct interaction with the cognate toxin (3). While we have shown interactions between cognate toxins and antitoxins (fig. S4, C to F), the antitoxin interaction in vivo appears weak. We additionally demonstrated that coincubation of the MenA3 antitoxin with MenT3 is able to neutralize the NTase activity (fig. S7). This suggests that any interaction-based antitoxicity might be a transient and labile mechanism and, due to the difference in size and sequence between antitoxins MenA1 and MenA3 (Fig. 1A), may well differ between these systems.
The DUF4433 DarT toxin from M. tuberculosis was recently identified as a single-stranded DNA NTase that specifically and reversibly adenosine 5′-diphosphate (ADP)–ribosylates thymidines (33). Our study identifies MenT3 as an NTase toxin from the unrelated DUF1814 protein family. In comparison to DarT, MenT3 acts via a distinct and novel mode of toxicity where the MenT3 toxin preferentially targets M. tuberculosis tRNAs in vitro, preventing their charging with cognate amino acids by adding nucleotides to the 3′-CCA acceptor stem (Fig. 5B). Accordingly, antitoxin function also appears to differ between these systems. Whereas DarT is counteracted enzymatically by the cognate antitoxin DarG via target de-ADP-ribosylation (33), we found that MenA3 was unable to reverse MenT3 toxicity by removing nucleotides, suggesting that MenA3 likely inhibits the toxin activity.
Increasing numbers of toxins have been identified that target tRNAs by various mechanisms (13). The M. tuberculosis type II VapC toxins function as endoribonucleases cleaving tRNAs (34), whereas TacT from Salmonella Typhimurium and AtaT from E. coli are tRNA acetyltransferases, modifying charged tRNAs to block translation (35, 36). That MenT3 provides yet another way to inhibit tRNA activity is perhaps not unusual, given the essential nature of translation to cellular growth and survival. This likely reflects the value of possessing multiple TA systems to promote adaptability to different stressful environments via tRNA metabolism, with downstream effects ranging from stalling cell growth to potentially altering translation output (13). It remains to be seen whether this mechanism is conserved among DUF1814-toxins; while MenT4 shares structural similarities to MenT3 and inhibits protein synthesis in vitro (Fig. 2 and fig. S5D), we have not yet explored the molecular mechanism behind its toxicity. Given the continued significance of M. tuberculosis worldwide, the mechanism used by the MenA3-MenT3 TA system highlights a new way to block protein synthesis. We propose that further exploring the molecular mechanisms of both toxicity and antitoxicity will provide useful insights into the regulation of bacterial growth.
MATERIALS AND METHODS
Bacterial strains and culture conditions
E. coli DH5α (Invitrogen), DH10B (Thermo Fisher Scientific), BL21 (λDE3) (Novagen), ER2566 (New England Biolabs), W3110 [strain American Type Culture Collection (ATCC) 27325], DLT1900 (37), and M. smegmatis mc2 155 (strain ATCC 700084) are as previously described. To construct BL21 (λDE3) ∆slyD, the ∆slyD::KmR allele from JW3311 (Keio collection) was moved into BL21 (λDE3) using bacteriophage P1-mediated transduction. To construct the unmarked DLT1900 ∆rph mutant, the ∆rph::KmR allele from JW3618 (Keio collection) was first moved into DLT1900 using bacteriophage P1-mediated transduction and by subsequent removing of the kanamycin (Km) resistance cassette using plasmid pCP20, as previously described (38). E. coli were routinely grown at 37°C in LB medium or M9 minimal (M9M) medium supplemented when necessary with Km (50 μg ml−1), ampicillin (Ap; 50 μg ml−1), chloramphenicol (Cm; 34 μg ml−1), streptomycin (Sm; 25 μg ml−1), spectinomycin (Sp; 50 μg ml−1), IPTG (1 mM), l-ara (0.1% w/v), or d-glucose (glu; 0.2% w/v). M. smegmatis mc2 155 strains were routinely grown at 37°C in either LB or 7H9 medium (Difco). M. tuberculosis H37Rv (WT; ATCC 27294) and mutant strains were routinely grown at 37°C in complete 7H9 medium (Middlebrook 7H9 medium, Difco) supplemented with 10% albumin-dextrose-catalase (ADC; Difco) and 0.05% Tween 80 (Sigma-Aldrich), or on complete 7H11 solid medium (Middlebrook 7H11 agar medium, Difco) supplemented with 10% oleic acid–ADC (OADC; Difco). When required, mycobacterial growth media were supplemented with Km (50 μg ml−1), hygromycin (Hm; 50 μg ml−1), Sm (25 μg ml−1), zeocin (Zc; 25 μg ml−1), Ace (0.2% w/v), or Atc (100 or 200 ng ml−1).
Plasmid constructs
Plasmids pMPMK6 (39), p29SEN (40), pGMCS (41), pGMCZ (42), pLAM12 (43), pETDuet-1, pET15b and pRARE (Novagen), pBAD30 (44), and pTA100 (4) have been described. Primers used for plasmid construction are described in table S1. All the plasmids constructed in this work have been verified by sequencing. The pMPMK6 derivatives expressing the toxins, namely, pK6-MenT1, pK6-MenT2, pK6-MenT3, and pK6-MenT4, were constructed as follows: menT1, menT2, menT3, and menT4 were PCR-amplified from the M. tuberculosis H37Rv genome and cloned as Eco RI/Hind III fragments (menT1 and menT2) and Mfe I/Hind III fragments (menT3 and menT4) into Eco RI/Hind III–digested pMPMK6.
The p29SEN plasmid derivatives encoding the antitoxins, namely, p29SEN-MenA1, p29SEN-MenA2, p29SEN-MenA3, and p29SEN-MenA4, were constructed as follows: menA1, menA2, menA3, and menA4 were PCR-amplified from the M. tuberculosis H37Rv genome and cloned either as Eco RI/Hind III fragments (menA1, menA2, and menA3) or as Mfe I/Hind III fragments (menA4) into Eco RI/Hind III–digested p29SEN. For p29SEN-Rph, the rph gene was PCR-amplified from the E. coli DLT1900 genome and cloned as an Eco RI/Hind III fragment into Eco RI/Hind III–digested p29SEN.
To construct pGMC-MenT2, pGMC-MenT3, and pGMC-MenT4, menT2, menT3, and menT4 were PCR-amplified using pK6-MenT2, pK6-MenT3, and pK6-MenT4 templates, respectively, and cloned into pGMCS using In-Fusion HD Cloning Kits (Takara Bio). Plasmid pGMC-MenT1 and pGMC-MenT1-His were obtained following PCR amplification of menT1 and menT1-His using pK6-MenT1 as a template and homologous recombination in linearized pGMCS plasmid by In-Fusion HD Cloning Kits (Takara Bio). For pGMC-*MenA4-MenT4, the menA4-menT4 operon was PCR-amplified from the H37Rv genome and cloned into linearized pGMCS plasmid by In-Fusion HD Cloning Kits (Takara Bio).
To construct plasmids pLAM-MenA2, pLAM-MenA3, and pLAM-MenA4, menA2, menA3, and menA4 were PCR-amplified using p29SEN-MenA2, p29SEN-MenA3, and p29SEN-MenA4 as templates, respectively. These were cloned as Nde I/Eco RI fragments (menA2 and menA3) and Nde I/Mfe I fragments (menA4) into Nde I/Eco RI–digested pLAM12. Plasmid p29SEN-MenA1 was used to amplify menA1 and menA1-His, which were then cloned as Nde I/Eco RI fragments into Nde I/Eco RI–digested pLAM12 to produce pLAM-MenA1 and pLAM-MenA1-His, respectively.
The pET vector derivatives used in this work were constructed as follows. To construct plasmid pET-MenT3-His, menT3-His (with an added fragment encoding a Ser-Ser-Gly-His6 C-terminal tag) was PCR-amplified from pK6-MenT3 template and cloned as an Nde I/Mfe I fragment into Nde I/Mfe I–digested pETDuet-1. Plasmid pET-MenT3-His was used as a template to construct pET-MenT3-His(D80A) and pET-MenT3-His(K189A) by QuikChange site-directed mutagenesis (Agilent) using appropriate primers. Plasmid pET-MenA3-His, encoding an N-terminal His6-tagged MenA3 antitoxin, was constructed by PCR amplification of menA3-His using p29SEN-MenA3 as a template, Nde I/Hind III digestion, and cloning into Nde I/Hind III–digested pET15b plasmid. To construct plasmid pET-MenT3/MenA3-His, menA3-His was first PCR-amplified from p29SEN-MenA3 template and cloned as an Nco I/Hind III fragment into Nco I/Hind III–digested pETDuet-1. menT3 was then PCR-amplified from pK6-MenT3, digested with Nde I/Mfe I, and cloned into Nde I/Mfe I–digested pET-MenA3-His. To construct pET-MenT3-His/MenA3, menA3 was first PCR-amplified using p29SEN-MenA3 as a template and cloned as an Nco I/Hind III fragment into Nco I/Hind III–digested pET-MenT3-His. To generate pET-MenT1-His (expressing MenT1 with an N-terminal His6-Ser-Ser-Gly-tag), menT1-His was PCR-amplified from pK6-MenT1 and cloned as an Nde I/Mfe I fragment into Nde I/Mfe I–digested pETDuet-1. For pET-MenA1-His (expressing MenA1 with an N-terminal His6-Ser-Ser-Gly-tag), menA1-His was PCR-amplified from p29SEN-MenA1 template and cloned as an Nco I/Bam HI fragment into Nco I/Bam HI–digested pETDuet-1. For pET-MenT1/MenA1-His, menT1 was PCR-amplified from pK6-MenA1 and cloned as an Nde I/Mfe I fragment into Nde I/Mfe I–digested pET-MenA1-His. For pET-MenT1-His/MenA1, menA1 was PCR-amplified from p29SEN-MenA1 and cloned as an Nco I/Bam HI fragment into Nco I/Bam HI–digested pET-MenT1His.
To generate MenT3 and MenT4 expression constructs for crystallization and biochemistry, overlap PCRs were performed to fuse a sentrin protease (SENP)–cleavable N-terminal His6-SUMO tag, amplified from the pBAT4 derivative (45), pSAT1-LIC (this study), to either menT3 or menT4, amplified from H37Rv genomic DNA. The resulting PCR products were cloned as either Kpn I/Hind III fragments into Kpn I/Hind III–digested pBAD30 (menT3), producing pTRB517, or as Xma I/Hind III fragments into Xma I/Hind III–digested pBAD30 (menT4) to generate pTRB544.
Plasmids pPF656 and pPF657 were constructed by amplifying menA3 and menT3 from H37Rv genomic DNA and cloning as Mfe I/Xma I fragments into Eco RI/Xma I–digested pTA100 and pBAD30, respectively. To express His6-SUMO-tagged MenT3(D80A), site-directed mutagenesis was carried out using pTRB517 as a template. Briefly, nonoverlapping inverse primers were used to amplify menT3(D80A), followed by incubation with a mix of T4 DNA ligase, T4 polynucleotide kinase, and DpnI at 37°C to remove template and circularize amplified DNA. This reaction was then used to transform E. coli DH5α, resulting in pTRB593. Similarly, this method was used to generate MenT3(D80A), MenT3(K189A), and MenT3(D211A) for functional testing, using pPF657 as a template, resulting in pTRB591, pTRB562, and pTRB592, respectively.
Plasmid pTRB491 was generated by amplifying menA3 from H37Rv genomic DNA and cloning into pSAT1-LIC via ligation-independent cloning (LIC). The pSAT1-LIC plasmid features a LIC site that fuses an N-terminal His6-SUMO tag to the target protein. To produce MenT3(K189A) protein, the mutated gene was amplified from pTRB562 and similarly cloned into pTRB550 via LIC, resulting in pTRB577. The pTRB550 plasmid features a His6-SUMO LIC site, originally amplified from pSAT1-LIC and cloned as an Eco RI/Hind III fragment into Eco RI/Hind III–digested pBAD30.
To produce plasmids for use in M. tuberculosis, menA3, menT3, or both genes were amplified by PCR using PrimeSTAR GXL DNA polymerase, with M. tuberculosis H37Rv genomic DNA as template and primer pairs clo-RBS1-MenA3-attB2/clo-MenA3-attB3, clo-RBS1-MenT3-attB2/clo-MenT3-attB3, clo-RBS4-MenT3-attB2/clo-MenT3-attB3, or clo-RBS1-MenA3-attB2/clo-MenT3-attB3, respectively (tables S1 and S2). RBS1 (AGGAAGACAGGCTGCCC) and RBS4 (ACGAAGACAGGCTGCCC), corresponding to a strong or weak Shine-Dalgarno sequence, respectively, were placed upstream from the ATG translation start of MenA3 or the GTG translation start of MenT3. Plasmids pGMCS-TetR-P1-RBS1-MenA3, pGMCS-TetR-P1-RBS1-MenA3-MenT3, pGMCS-TetR-P1-RBS1-MenT3, or pGMCS-TetR-P1-RBS4-MenT3 were constructed by multisite gateway recombination (18), using plasmid pDE43-MCS as the destination vector. These plasmids are integrative vectors (insertion at the attL5 mycobacteriophage insertion site in the glyV tRNA gene) and express MenA3, MenT3, or MenA3-MenT3 under the control of P1 (Pmyc1 tetO), a tetracycline-inducible promoter (table S2) (46).
Construction of MenT3 D80A, D211A, and K189A substitutions for use in M. tuberculosis was performed as follows: Plasmid pGMCS-TetR-P1-RBS4-MenT3 was amplified by PCR with PrimeSTAR GXL DNA polymerase and the oligonucleotides pairs InFus-MenT3D80A-right/InFus-MenT3D80A-left, InFus-MenT3D211A-right/InFus-MenT3D211A-left, or InFus-MenT3K189A-right/InFus-MenT3K189A-left (table S1). The amplified linear fragments were purified on agarose gels and circularized using the In-Fusion HD Cloning Kit (Takara), as recommended by the manufacturer. Plasmids used to transform Stellar recipient cells were verified by sequencing and introduced by electroporation into M. tuberculosis ∆(menA3-menT3)::dif6/pGMCZ (see the next paragraph).
Construction of M. tuberculosis mutants
Mutant strains of M. tuberculosis H37Rv were constructed by allelic exchange using recombineering (43), as previously described (fig. S2) (47). Two ~0.5-kb DNA fragments flanking the menA3-menT3 operon were amplified by PCR using PrimeSTAR GXL DNA polymerase (Takara), M. tuberculosis H37Rv genomic DNA, and the primer pairs MenA3Am-For/MenA3Zc-Am-Rev or MenT3Zc-Av-For/MenT3Av-Rev, respectively (table S1). A three-fragment PCR fused these two fragments to a Zc-resistance cassette flanked by two dif6 variants of the M. tuberculosis dif site and the recombination substrate was recovered by agarose gel purifications. The recipient strain for recombineering was a derivative of M. tuberculosis H37Rv carrying two plasmids: pJV53H, an Hm-resistant pJV53-derived plasmid expressing recombineering enzymes (43), and the integrative plasmid pGMCS-P1-MenA3, constitutively expressing menA3 (table S2). This strain was grown in complete 7H9 medium supplemented with Hm until mid-log phase and expression of recombineering enzymes was induced by Ace (0.2%) overnight at 37°C. After induction, electrotransformation was performed with 100 ng of the linear DNA fragment for allelic exchange. After a 48-hour incubation at 37°C, mycobacteria were plated onto agar supplemented with Zc. Zc-resistant clones were restreaked on the same medium, grown in complete 7H9 without antibiotic, and verified to be carrying the expected allele replacement by PCR amplification of chromosomal DNA and subsequent DNA sequencing, using primers MenA3Am-For/MenT3Av-Rev (fig. S1C and table S1). Spontaneous loss of the Zc-resistance cassette by XerCD-dependent recombination and of the pJV53H plasmid was obtained by serial rounds of culture without antibiotics and phenotypic tests for ZcS and HmS. Plasmid pGMCS-P1-MenA3 was then removed by transformation with pGMCZ, a similar integrative vector but carrying resistance to Zc, resulting in the deleted strain M. tuberculosis ∆(menA3-menT3)::dif6/pGMCZ.
E. coli multicopy plasmid library
E. coli MC4100 ∆dnaKdnaJ::KmR ∆tig:CmR double mutant (40) was partially digested with Sau3 AI restriction enzyme and DNA fragments of about 1.5 to 4 kb in size were purified, then ligated into linearized and dephosphorylated Bam HI–digested pMPM2 (ColE1 origin) plasmid (39), and used to transform E. coli DH10B. About 25,000 independent transformants were pooled to constitute the multicopy library. This library has previously been used as a tool to identify multicopy suppressors of chaperone mutants (48).
Bacterial growth assays
In vivo toxicity and antitoxicity assays by cognate or noncognate antitoxins in E. coli were performed as follows. E. coli DLT1900 were cotransformed with pMPMK6-vector, pK6-MenT1, -MenT2, -MenT3, or -MenT4 (toxins), and p29SEN-vector, p29SEN-MenA1, -MenA2, -MenA3, or -MenA4 (antitoxins). Transformants were re-seeded from overnight cultures and grown at 37°C to mid-log phase in LB supplemented with Km and Ap, and then serially diluted and spotted on LB-agar plates supplemented with Km and Ap, with or without l-ara (0.1%) and/or IPTG (200 μM). Plates were incubated at 37°C overnight and then imaged and counted. MenT3 substitutions were tested for toxicity in E. coli DH5α carrying pBAD30-vector, -MenT3 WT (pPF657), -MenT3(D80A) (pTRB591), -MenT3(K189A) (pTRB562), or -MenT3(D211A) (pTRB592). Strains were grown to mid-log phase, then serially diluted, and spotted onto M9M-agar plates supplemented with Ap, with or without l-ara (0.1%). After a 2-day incubation at 37°C, plates were imaged and counted.
In vivo toxicity and rescue assays by cognate or noncognate antitoxins in M. smegmatis were performed as follows. Cultures of mc2 155 strain grown in LB at 37°C were cotransformed with the integrative pGMC-vector, -MenT1, -MenT2, -MenT3, or -MenT4 (toxins), and with pLAM12-vector, pLAM-MenA1, -MenA2, -MenA3, or -MenA4 (antitoxins). Samples were selected on LB-agar plates supplemented with Km and Sm for 3 days at 37°C, in the presence or absence of Atc (100 ng ml−1) and Ace (0.2%) for toxin and antitoxin expression, respectively. A similar procedure was applied for pGMC-*MenA4-MenT4 carrying the menA4-menT4 operon, with the exception that no cotransformation with pLAM12 derivatives or selection on Km was needed.
Viability staining and flow cytometry
Exponentially growing cultures [OD600 (optical density at 600 nm) between 0.05 and 0.2] of M. smegmatis strain mc2 155 containing plasmid pGMCS-TetR-P1-RBS1-MenT3 were divided in two: Half was left in complete 7H9 growth medium with Sm (uninduced cultures), while the other half was additionally treated with Atc (200 ng ml−1) to induce expression from the P1 promoter. For labeling with LIVE/DEAD BacLight (Molecular Probes) dyes, cells were harvested 8 hours after Atc induction. Cells were centrifuged, resuspended in phosphate-buffered saline buffer, and stained as recommended by the manufacturer. Labeled cells were analyzed by fluorescence-activated cell sorting using a BD LSRFortessa X20 flow cytometer. Flow cytometry data analysis was performed using FlowJo software.
Toxicity assays in M. tuberculosis
M. tuberculosis strains H37Rv or H37Rv ∆(menA3-menT3)::dif6/pGMCZ were transformed by electroporation with 100 ng of plasmids pGMCS-TetR-P1-RBS1-MenA3, pGMCS-TetR-P1-RBS1-MenA3-MenT3, pGMCS-TetR-P1-RBS1-MenT3, pGMCS-TetR-P1-RBS4-MenT3, pGMCS-TetR-P1-RBS4-MenT3(D80A), pGMCS-TetR-P1-RBS4-MenT3(K189A), or pGMCS-TetR-P1-RBS4-MenT3(D211A). After 3 days of phenotypic expression in 7H9 ADC Tween at 37°C, the transformation mix was divided into two halves. One half was plated on 7H11 OADC with Sm; the other half was plated on 7H11 OADC Sm supplemented with Atc (200 ng ml−1). Plates were imaged after 20 days of incubation at 37°C.
In vivo coaffinity purification assays
To perform in vivo copurification assays, E. coli BL21 ∆slyD was transformed with (i) pET-MenT3-His, pET-MenA3-His, pET-MenT3/MenA3-His, or pET-MenT3-His/MenA3, or with (ii) pET-MenT1-His, pET-MenA1-His, pET-MenT1/MenA1-His, or pET-MenT1-His/MenA1, and selected on LB-agar plates supplemented with Ap and glu (20%). Transformants were grown at 37°C to an OD600 of approximately 0.4 and then protein expression was induced overnight at 20°C with 1 mM IPTG. Cell lysis and affinity purification of the protein complexes were performed as described below for MenT3-His purification. Elution fractions were separated on SDS-PAGE and proteins revealed using InstantBlue Protein Stain (Expedeon, catalog no. ISB1L).
Recombinant protein production
To purify MenT3 for biochemistry, BL21 (λDE3) ∆slyD transformed with pET-MenT3-His, pET-MenT3-His(D80A), or pET-MenT3-His(K189A) was grown to an OD600 of approximately 0.4 at 37°C. IPTG (1 mM) was then added, and the culture was incubated overnight at 20°C. Under such conditions, MenT3 expression in E. coli was better tolerated and led to a reasonable amount of soluble MenT3 that could be collected for purification. Cultures were centrifuged at 5000g for 10 min at 4°C, pellets were resuspended in Lysis buffer [300 mM NaCl, 50 mM tris (pH 7.5), and protease inhibitor tablet (Roche); 20 ml of buffer per 1 liter of cell culture] and incubated for 30 min on ice. Lysis was performed using the One Shot cell disrupter at 1.5 kbar (One Shot model, Constant Systems Ltd.). Lysates were centrifuged for 30 min at 30,000g in 4°C, and the resulting supernatants were gently mixed at 4°C for 30 min with Ni–nitrilotriacetic acid agarose beads (Qiagen, catalog no. 30230) preequilibrated with buffer PD [300 mM NaCl and 50 mM tris (pH 7.5)], using a 10-ml poly-prep column (Bio-Rad, catalog no. 7311550). Columns were stabilized for 10 min at 4°C and washed three times with 10 ml of buffer PD plus 25 mM imidazole, and proteins were then eluted with buffer PD containing 250 mM imidazole. Elutions (500 μl) were collected and PD MiniTrap G-25 columns (GE Healthcare, catalog no. 16924748) were used to exchange buffer with buffer PD supplemented with 10% glycerol. Proteins were concentrated using Vivaspin 6 columns with a 5000-Da cutoff (Sartorius, catalog no. 184501257). Proteins were stored at −80°C until further use.
For additional MenT3 and MenT3(K189A) expression, either for crystallization or biochemistry, E. coli ER2566 pRARE pPF656 was transformed with either pTRB517 or pTRB577, respectively. For MenT3(D80A) expression, E. coli ER2566 pRARE was transformed with pTRB593. MenT4 was expressed in E. coli BL21 (λDE3) transformed with pTRB544. MenA3 was expressed in E. coli ER2566 transformed with pTRB491. For these expressions, the same procedure was followed: Overnight cultures were re-seeded 1:100 into 2-liter flasks containing 1-liter 2× YT. Cells were grown at 175 rpm in 37°C until an OD600 of 0.3 was reached and then at 22°C until OD600 0.5, whereupon expression was induced by the addition of l-ara (0.1%) for toxins and IPTG (1 mM) for antitoxins. Cells were left to grow overnight at 16°C, shaking at 175 rpm.
For selenomethionine incorporation, starter cultures of ER2566 pRARE pPF656 pTRB517 were grown overnight in LB at 37°C with 200 rpm shaking. Cells were pelleted, washed, and resuspended in M9M, and then sub-cultured into 500 ml of M9M in 2-liter baffled flasks to a starting OD600 of 0.075. Cells were grown at 37°C with 175 rpm shaking until an OD600 of 0.6, whereupon cells were centrifuged at 4200g and resuspended in fresh M9M. This sample was divided between separate 2-liter baffled flasks containing new M9M and shaken at 175 rpm for a further 1 hour at 37°C. Once an OD600 of 0.7 was reached, 12 ml of nutrient mix [l-lysine hydrate (4 mg ml−1), l-threonine (4 mg ml−1), l-phenylalanine (4 mg ml−1), l-leucine (2 mg ml−1), l-isoleucine (2 mg ml−1), l-valine (2 mg ml−1), and 4 mM CaCl2] was added to each flask to promote feedback inhibition of methionine synthesis, followed by 250× SelenoMethionine Solution (Molecular Dimensions) to a final concentration of 40 μg ml−1, and cells were left to incubate for 1 hour at 20°C. Last, toxin and antitoxin expression were induced by the addition of l-ara (0.1%) and IPTG (1 mM), and samples were left to grow overnight at 175 rpm in 16°C.
All five proteins were purified in the same manner. Bacteria were harvested by centrifugation at 4200g, and the pellets were resuspended in buffer A500 [20 mM tris-HCl (pH 7.9), 500 mM NaCl, 5 mM imidazole, and 10% glycerol]. Cells were lysed by sonication at 40 kpsi and then centrifuged (45,000g, 4°C). The clarified lysate was next passed over a HisTrap HP column (GE Healthcare), washed for 10 column volumes with A500, followed by 10 column volumes of buffer A100 [20 mM tris-HCl (pH 7.9), 100 mM NaCl, 5 mM imidazole, and 10% glycerol], and then eluted directly onto a HiTrap Q HP column (GE Healthcare) with buffer B100 [20 mM tris-HCl (pH 7.9), 100 mM NaCl, 250 mM imidazole, and 10% glycerol]. The Q HP column was transferred to an Äkta Pure (GE Healthcare), washed with 3 column volumes of A100, and then proteins were eluted using a gradient from 100% A100 to 100% buffer C1000 [20 mM tris-HCl (pH 7.9), 1000 mM NaCl, and 10% glycerol]. Fractions containing the protein peak were analyzed by SDS-PAGE, pooled, and incubated overnight at 4°C with hSENP2 SUMO protease to cleave the His6-SUMO tag from the target protein. The following day, the samples were passed through a second HisTrap HP column and the flow-through fractions containing untagged target protein were collected. These samples were concentrated and run over a HiPrep 16/60 Sephacryl S-200 size exclusion column (GE Healthcare) in buffer S [50 mM tris-HCl (pH 7.9), 500 mM KCl, and 10% glycerol]. Peak fractions were analyzed by SDS-PAGE, pooled, and concentrated. Optimal fractions were separated and either flash-frozen in liquid N2 for storage at −80°C or dialyzed overnight at 4°C into buffer X [20 mM tris-HCl (pH 7.9), 150 mM NaCl, and 2.5 mM dithiothreitol (DTT)] for crystallographic studies. Crystallization samples were quantified and stored on ice and then either used immediately or flash-frozen in liquid N2 for storage at −80°C. Frozen crystallization samples still formed usable crystals 15 months after storage.
Protein crystallization
Native and selenomethionine-derivatized MenT3 were concentrated to 12 mg ml−1 and MenT4 was concentrated to 6 mg ml−1, all in buffer X (see above). Initial crystallization screens were performed using a Mosquito Xtal3 robot (TTP Labtech) to set 200:100 nl and 100:100 nl protein:condition sitting drops. After initial screening and optimization, both MenT3 protein samples formed thick, six-sided needles in condition G5 [0.2 M calcium acetate hydrate, 0.1 M tris (pH 8.5), and 25% w/v polyethylene glycol 2000 monomethyl ether] of Clear Strategy II HT-96 (Molecular Dimensions). MenT4 formed thin, six-sided needles in the same condition as MenT3. To harvest, 20 μl of condition reservoir was added to 20 μl of cryo buffer [25 mM tris-HCl (pH 7.9), 187.5 mM NaCl, 3.125 mM DTT, and 80% glycerol] and mixed quickly by vortexing; an equal volume of this mixture was then added to the drop. After addition of cryo buffer, crystals were immediately extracted using a nylon loop and flash-frozen in liquid N2.
Data collection and structure determination
Diffraction data were collected at Diamond Light Source on beamlines I04 (MenT3 native), I03 (MenT3 selenomethionine-derivatized), and I24 (MenT4 native) (Table 1). Single 360° datasets were collected for native MenT3 and MenT4. Two 360° datasets from MenT3 selenomethionine-derivatized crystals measured at the selenium peak (0.9793 Å) were merged using iSpyB (Diamond Light Source). Additional MenT3 selenomethionine-derivatized datasets were collected at selenium high remote (0.9641 Å) and inflection (0.9795 Å) wavelengths. Diffraction data were processed with XDS (49), and then AIMLESS from CCP4 (50) was used to corroborate the space groups (Table 1). The crystal structure of MenT3 was solved by MAD by providing the SHELX suite in CCP4 with the native and three anomalous MenT3 datasets. The solved starting model for MenT3 was built in REFMAC within CCP4. The crystal structure of MenT4 was solved ab initio using ARCIMBOLDO (51). Both models were then iteratively refined and built using PHENIX (52) and COOT (53), respectively. The quality of the final model was assessed using COOT and the wwPDB validation server. Structural figures were generated using PyMOL (Schrödinger). Comparison against models within the Protein Data Bank (PDB) was performed using DALI (25).
Genetic screen for suppressors of toxicity
The following genetic procedure was developed and applied to select for E. coli genes that confer resistance to the MenT3 toxin. E. coli strain DLT1900 was first transformed with pK6-MenT3 (KmR) plasmid and transformants were selected at 37°C on LB-agar plates supplemented with Km and glu (0.2%) to repress toxin expression from the araBAD promoter of pK6-MenT3. DLT1900 containing pK6-MenT3 was then grown in LB supplemented with Km and glu, transformed with the pMPMA2-based multicopy library of E. coli genes, and plated on selective LB-agar supplemented with Km, Ap, and l-ara (0.1%) to induce toxin expression. Plates were incubated for 24 hours at 37°C. A control aliquot of transformants plated on nonselective plates (no l-ara) indicated that the number of transformants tested during the selection procedure was approximately 60,000. Note that under such conditions, E. coli DLT1900 pK6-MenT3 transformed with pMPMA2 empty vector did not produce any colonies on selective plates. We identified 72 toxin-resistant colonies that grew on selective plates after 24 hours, although they were smaller and translucent, indicating that growth inhibition by the toxin is not fully blocked by the suppressors identified. Of the 72 toxin-resistant colonies identified, only 41 were able to grow in culture. Plasmids were extracted from the 41 cultures, used to re-transform DLT1900 pK6-MenT3, and plated as above, to validate growth rescue in the presence of MenT3. Of 41 clones, 18 suppressors passed the second round of selection and were sequenced using the pMPMA2-For and -Rev primers (table S1).
In vitro transcription/translation assays
Assays were performed as previously described (54). Briefly, template DNAs of DHFR (P0ABQ4), WaaF-Strep (P37692), and GatZ-Strep (P0C8J8) were used for in vitro transcription/translation coupled assays (PURExpress, New England Biolabs). These were performed according to the manufacturer’s instructions, in the presence or absence of the toxin. Following protein synthesis reactions of 2 hours at 37°C, samples were separated on SDS-PAGE and visualized by InstantBlue staining (DHFR) or Western blots using anti-Strep tag antibodies (WaaF-Strep and GatZ-Strep).
Identification of uncharged tRNAs in vivo and in vitro
Prevention of E. coli tRNATrp aminoacylation by MenT3 was monitored using a combination of two previously published methods (29, 55). E. coli BL21 (λDE3) transformed with pETDuet or pET-MenT3 was grown at 37°C to OD600 0.1 in M9M, whereupon expression of MenT3 was induced with 1 mM IPTG until an OD600 of about 0.4. The bacterial culture (25 ml) was then kept on ice and centrifuged for 10 min at 5000g in 4°C. The pellet was resuspended in 0.5 ml of cold 0.3 M sodium acetate (pH 4.5) and 10 mM EDTA and transferred to a precooled 1.5-ml microcentrifuge tube, and 0.5 ml of phenol (equilibrated with the same buffer) was then added. After gentle pipetting, the sample was transferred into phase-lock tubes with an additional 400 μl of cold chloroform. After 30 seconds shaking, the sample was first incubated on ice for 15 min and then centrifuged for 20 min at 20,000g in 4°C. The aqueous phase was then transferred to a new cold 1.5-ml tube. Five hundred microliters of cold isopropanol was added and immediately mixed. RNA was precipitated for 1 hour at −20°C, before the sample was centrifuged for 30 min at 20,000g in 4°C (55). The supernatant was discarded and 1 ml of cold 75% ethanol was carefully added without disturbing the RNA pellet. After further centrifugation for 10 min at 20,000g in 4°C, the supernatant was removed and the pellet was air-dried until no ethanol remained. The pellet was then resuspended by vigorously mixing in 20 μl of cold 10 mM sodium acetate (pH 4.5) and 1 mM EDTA. Samples were stored at −80°C. Samples were separated on a denaturing urea acrylamide gel for 3 hours at 100 V in 4°C, as previously described (29). Northern blot and visualization with a radiolabeled DNA probe against tRNATrp was performed as previously described (56). Note that to distinguish the band of aminoacylated tRNA from its deacylated counterpart on the Northern blot, a chemically deacylated aliquot of RNA sample prepared from strain containing the empty vector was subjected to alkaline treatment. In this case, 46 μl of tris-HCl (pH 9.0) was added to a 4-μl aliquot of the RNA sample and incubated for 2 hours at 37°C. Fifteen microliters of 0.3 M sodium acetate at pH 4.5 was added and followed by 125 μl of 96% ethanol. RNA was precipitated at −20°C for 1 hour, resuspended, and separated as described above.
For in vitro tRNA charging, in vitro transcription/translation assays were performed as above, using gatZ as DNA template. After a 2-hour reaction at 37°C with or without MenT3 toxin (10 μM), tRNA extraction, separation, and visualization were performed as described for the in vivo samples.
In vitro transcription of tRNAs
Labeled tRNAs were prepared by in vitro transcription of PCR templates containing an integrated T7 RNA polymerase promoter sequence. The template for E. coli tRNATrp was made by PCR amplification of chromosomal DNA from strain MG1655 with the primers CC2556 and CC2557 (CC2591 for tRNATrp without CCA) (table S1). The oligos for M. tuberculosis tRNAs are given in table S1. The T7 RNA polymerase in vitro transcription reactions were performed in 25-μl total volume, with a 5-μl nucleotide mix of 2.5 mM ATP, 2.5 mM CTP, 2.5 mM GTP, and 60 μM UTP and 2 to 4 μl of 10 mCi ml−1 of radiolabeled UTP [α-P32]. Template (0.1 to 0.2 μg) was used per reaction with 1.5 μl of rRNasin (40 U ml−1) (Promega), 5 μl of 5× optimized transcription buffer (Promega), 2 μl of T7 RNA polymerase (20 U ml−1), and 2.5 μl of 100 mM DTT. Template DNA was removed by the addition of 2 μl of RQ DNase (1 U ml−1) (Promega). Unincorporated nucleotides were removed by G50 spin columns (GE Healthcare) according to the manufacturer’s instructions, in a final volume of 30 μl. For E. coli tRNATrp, the transcript reaction was gel-purified on a denaturing 5% acrylamide gel and eluted in 0.3 M sodium acetate for 4 hours overnight at 4°C. The supernatant was removed, ethanol-precipitated, and resuspended in 20 to 30 μl of nuclease-free H2O.
Nucleotide transfer assays
MenT3 NTase activity was assayed in 10-μl reaction volumes containing 50 mM tris-HCl (pH 9.5), 10 mM MgCl2, and 2.5 mM rNTPs and incubated for 20 min at 37°C. Fresh, uniformly labeled tRNA (0.5 μl) was used per assay, with different dilutions of the protein (1, 0.1, 0.01, and 0.001 mg ml−1) in 50 mM tris-HCl (pH 7.8), 300 mM NaCl, and 10% glycerol. The 10-μl reactions were mixed directly with 10 μl of RNA loading dye (95% formamide, 1 mM EDTA, 0.025% SDS, xylene cyanol, and bromophenol blue), denatured at 90°C, and applied to 5% polyacrylamide-urea gels. The gel was vacuum-dried at 80°C and exposed to a PhosphorImager screen.
In vitro antitoxicity assays
The effect of MenA3 antitoxin was assayed using in vitro-transcribed tRNASer2 as a substrate. For the coincubation assay, MenT3 (5 μM) and increasing molar ratios of MenA3 were incubated with tRNASer2 and 2.5 mM CTP in 10-μl reaction volumes containing 50 mM tris-HCl (pH 9.5) and 10 mM MgCl2 for 20 min at 37°C. For the postincubation assay, the reactions were first incubated for 20 min at 37°C with MenT3 alone in 7-μl reaction volumes, then 3 μl containing different concentrations of MenA3 were added, and the reactions were incubated for a further 20 min at 37°C.
MenT3 tRNA screening
The tRNA screening was performed using 0.5 μl of uniformly labeled M. tuberculosis tRNAs, all containing the CCA motif. The activity was tested in 50 mM tris-HCl (pH 9.5), 10 mM MgCl2, and 2.5 mM rCTP in 10-μl reaction volumes and incubated for 20 min at 37°C. The transcripts were incubated with 1 μl of MenT3 (0.1 mg ml−1), or with nuclease-free water as a control. The reaction was stopped with 10 μl of RNA loading dye (95% formamide, 1 mM EDTA, 0.025% SDS, xylene cyanol, and bromophenol blue), denatured at 90°C, and applied to 5% polyacrylamide-urea gels. The gel was vacuum-dried at 80°C and exposed to a PhosphorImager screen.
Supplementary Material
Acknowledgments
We thank D.-J. Bigot for plasmid constructs, and P. Bordes, M.-P. Castanié-Cornet, L. Falquet, L. Poljak, L. Hadjeras, and H. Akarsu for valuable advice. We also thank K. Semeijn and R. Dy for initial plasmid construction and testing, and E. Naser (Genotoul TRI-IPBS imaging facility) for help with flow cytometry analysis. Funding: This work was supported by a scholarship from the China Scholarship Council (CSC) as part of a joint international PhD program with Toulouse University Paul Sabatier (Y.C.); Springboard Award (SBF002\1104) from the Academy of Medical Sciences (B.U. and T.R.B.); University of Otago Research Grant (P.C.F.), the School of Biomedical Sciences Bequest Fund, and University of Otago (P.C.F.); CNRS (UPR 9073), Université Paris VII-Denis Diderot, the Agence Nationale de la Recherche (ARNr-QC), and the Labex (Dynamo) program (A.T. and C.C.); European Commission (contracts NEWTBVAC n°241745 and TBVAC2020 n°643381), Centre National de la Recherche Scientifique, Université Paul Sabatier, Agence Nationale de la Recherche (ANR-13-BSV8-0010-01), and Fondation pour la Recherche Médicale (DEQ20160334902) (C.G. and O.N.); and grant SNF CRSII3_160703 (P.G.). Author contributions: Conceptualization, all authors. Investigation, Y.C., B.U., C.G., A.T., and M. M. Writing, all authors. Funding acquisition, P.C.F., C.C., O.N., P.G., and T.R.B. Supervision, C.C., O.N., P.G., and T.R.B. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The crystal structures of MenT3 and MenT4 have been deposited in the Protein Data Bank under accession numbers 6Y5U and 6Y56, respectively. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eabb6651/DC1
REFERENCES AND NOTES
- 1.Norton J. P., Mulvey M. A., Toxin-antitoxin systems are important for niche-specific colonization and stress resistance of uropathogenic Escherichia coli. PLOS Pathog. 8, e1002954 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helaine S., Cheverton A. M., Watson K. G., Faure L. M., Matthews S. A., Holden D. W., Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Page R., Peti W., Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Fineran P. C., Blower T. R., Foulds I. J., Humphreys D. P., Lilley K. S., Salmond G. P., The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl. Acad. Sci. U.S.A. 106, 894–899 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pecota D. C., Wood T. K., Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J. Bacteriol. 178, 2044–2050 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goormaghtigh F., Fraikin N., Putrinš M., Hallaert T., Hauryliuk V., Garcia-Pino A., Sjödin A., Kasvandik S., Udekwu K., Tenson T., Kaldalu N., Van Melderen L., Reassessing the role of Type II toxin-antitoxin systems in formation of Escherichia coli Type II persister cells. MBio 9, e00640–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pontes M. H., Groisman E. A., Slow growth determines nonheritable antibiotic resistance in Salmonella enterica. Sci. Signal. 12, eaax3938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeRoux M., Culviner P. H., Liu Y. J., Littlehale M. L., Laub M. T., Stress induces the transcription of toxin-antitoxin systems but does not activate the toxin. bioRxiv, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronneau S., Helaine S., Clarifying the link between toxin–antitoxin modules and bacterial persistence. J. Mol. Biol. 431, 3462–3471 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Fraikin N., Goormaghtigh F., Van Melderen L., Type II toxin-antitoxin systems: Evolution and revolutions. J. Bacteriol. 202, e00763-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szekeres S., Dauti M., Wilde C., Mazel D., Rowe-Magnus D. A., Chromosomal toxin-antitoxin loci can diminish large-scale genome reductions in the absence of selection. Mol. Microbiol. 63, 1588–1605 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Song S., Wood T. K., Toxin/antitoxin system paradigms: Toxins bound to antitoxins are not likely activated by preferential antitoxin degradation. Adv. Biosyst. 4, 1900290 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Hall A. M., Gollan B., Helaine S., Toxin–antitoxin systems: Reversible toxicity. Curr. Opin. Microbiol. 36, 102–110 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Keren I., Minami S., Rubin E., Lewis K., Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio 2, e00100–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sala A., Bordes P., Genevaux P., Multiple toxin-antitoxin systems in Mycobacterium tuberculosis. Toxins 6, 1002–1020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akarsu H., Bordes P., Mansour M., Bigot D.-J., Genevaux P., Falquet L., TASmania: A bacterial Toxin-Antitoxin Systems database. PLoS Comput. Biol. 15, e1006946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramage H. R., Connolly L. E., Cox J. S., Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLOS Genet. 5, e1000767 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freire D. M., Gutierrez C., Garza-Garcia A., Grabowska A. D., Sala A. J., Ariyachaokun K., Panikova T., Beckham K. S. H., Colom A., Pogenberg V., Cianci M., Tuukkanen A., Boudehen Y.-M., Peixoto A., Botella L., Svergun D. I., Schnappinger D., Schneider T. R., Genevaux P., de Carvalho L. P. S., Wilmanns M., Parret A. H. A., Neyrolles O., An NAD+ phosphorylase toxin triggers Mycobacterium tuberculosis cell death. Mol. Cell 73, 1282–1291.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garvey P., Fitzgerald G. F., Hill C., Cloning and DNA sequence analysis of two abortive infection phage resistance determinants from the lactococcal plasmid pNP40. Appl. Environ. Microbiol. 61, 4321–4328 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dy R. L., Przybilski R., Semeijn K., Salmond G. P. C., Fineran P. C., A widespread bacteriophage abortive infection system functions through a Type IV toxin-antitoxin mechanism. Nucleic Acids Res. 42, 4590–4605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li F., Xiong Y., Wang J., Cho H. D., Tomita K., Weiner A. M., Steitz T. A., Crystal structures of the Bacillus stearothermophilus CCA-adding enzyme and its complexes with ATP or CTP. Cell 111, 815–824 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Hampton H. G., Jackson S. A., Fagerlund R. D., Vogel A. I. M., Dy R. L., Blower T. R., Fineran P. C., AbiEi binds cooperatively to the Type IV abiE toxin–antitoxin operator via a positively-charged surface and causes DNA bending and negative autoregulation. J. Mol. Biol. 430, 1141–1156 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Sassetti C. M., Boyd D. H., Rubin E. J., Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Dejesus M. A., Gerrick E. R., Xu W., Park S. W., Long J. E., Boutte C. C., Rubin E. J., Schnappinger D., Ehrt S., Fortune S. M., Sassetti C. M., Ioerger T. R., Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio 8, e02133–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holm L., Sander C., Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 (1993). [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y., Ye X., Su Y., Sun L., She F., Wu Y., Crystal structure confirmation of JHP933 as a nucleotidyltransferase superfamily protein from Helicobacter pylori strain J99. PLOS ONE 9, e104609 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bost S., Silva F., Belin D., Transcriptional activation of ydeA, which encodes a member of the major facilitator superfamily, interferes with arabinose accumulation and induction of the Escherichia coli arabinose PBAD promoter. J. Bacteriol. 181, 2185–2191 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bechhofer D. H., Deutscher M. P., Bacterial ribonucleases and their roles in RNA metabolism. Crit. Rev. Biochem. Mol. Biol. 54, 242–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.B. D. Janssen, E. J. Diner, C. S. Hayes, in Bacterial Regulatory RNA (Humana Press, 2012; www.ncbi.nlm.nih.gov/pubmed/22736012), vol. 905, pp. 291–309.
- 30.Chan P. P., Lowe T. M., GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samhita L., Nanjundiah V., Varshney U., How many initiator tRNA genes does Escherichia coli need? J. Bacteriol. 196, 2607–2615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenhard B., Orellana O., Ibba M., Weygand-Durasević I., tRNA recognition and evolution of determinants in seryl-tRNA synthesis. Nucleic Acids Res. 27, 721–729 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jankevicius G., Ariza A., Ahel M., Ahel I., The toxin-antitoxin system DarTG catalyzes reversible ADP-ribosylation of DNA. Mol. Cell 64, 1109–1116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winther K. S., Brodersen D. E., Brown A. K., Gerdes K., VapC20 of Mycobacterium tuberculosis cleaves the Sarcin–Ricin loop of 23S rRNA. Nat. Commun. 4, 2796 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Cheverton A. M., Gollan B., Przydacz M., Wong C. T., Mylona A., Hare S. A., Helaine S., A Salmonella toxin promotes persister formation through acetylation of tRNA. Mol. Cell 63, 86–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jurėnas D., Chatterjee S., Konijnenberg A., Sobott F., Droogmans L., Garcia-Pino A., Van Melderen L., AtaT blocks translation initiation by N-acetylation of the initiator tRNAfMet. Nat. Chem. Biol. 13, 640–646 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Bouet J.-Y., Bouvier M., Lane D., Concerted action of plasmid maintenance functions: Partition complexes create a requirement for dimer resolution. Mol. Microbiol. 62, 1447–1459 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Datsenko K. A., Wanner B. L., One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayer M. P., A new set of useful cloning and expression vectors derived from pBlueScript. Gene 163, 41–46 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Genevaux P., Keppel F., Schwager F., Langendijk-Genevaux P. S., Hartl F. U., Georgopoulos C., In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 5, 195–200 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blumenthal A., Trujillo C., Ehrt S., Schnappinger D., Simultaneous analysis of multiple Mycobacterium tuberculosis knockdown mutants in vitro and in vivo. PLOS ONE 5, e15667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Botella L., Vaubourgeix J., Livny J., Schnappinger D., Depleting Mycobacterium tuberculosis of the transcription termination factor Rho causes pervasive transcription and rapid death. Nat. Commun. 8, 14731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Kessel J. C., Hatfull G. F., Recombineering in Mycobacterium tuberculosis. Nat. Methods 4, 147–152 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Guzman L. M., Belin D., Carson M. J., Beckwith J., Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peränen J., Rikkonen M., Hyvönen M., Kääriäinen L., T7 vectors with a modified T7lac promoter for expression of proteins in Escherichia coli. Anal. Biochem. 236, 371–373 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Ehrt S., Guo X. V., Hickey C. M., Ryou M., Monteleone M., Riley L. W., Schnappinger D., Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 33, e21 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boudehen Y.-M., Wallat M., Rousseau P., Neyrolles O., Gutierrez C., An improved Xer-cise technology for the generation of multiple unmarked mutants in mycobacteria. Biotechniques 68, 106–110 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Anglès F., Castanié-Cornet M.-P., Slama N., Dinclaux M., Cirinesi A.-M., Portais J.-C., Létisse F., Genevaux P., Multilevel interaction of the DnaK/DnaJ(HSP70/HSP40) stress-responsive chaperone machine with the central metabolism. Sci. Rep. 7, 41341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodríguez D. D., Grosse C., Himmel S., González C., de Ilarduya I. M., Becker S., Sheldrick G. M., Usón I., Crystallographic ab initio protein structure solution below atomic resolution. Nat. Methods 6, 651–653 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Bordes P., Sala A. J., Ayala S., Texier P., Slama N., Cirinesi A. M., Guillet V., Mourey L., Genevaux P., Chaperone addiction of toxin-antitoxin systems. Nat. Commun. 7, 13339 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stenum T. S., Sørensen M. A., Lo Svenningsen S., Quantification of the abundance and charging levels of transfer RNAs in Escherichia coli. J. Vis. Exp. 22, 56212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khemici V., Prados J., Linder P., Redder P., Decay-initiating endoribonucleolytic cleavage by RNase Y is kept under tight control via sequence preference and sub-cellular localisation. PLOS Genet. 11, e1005577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eabb6651/DC1