

Abstract
The effectiveness of targeted α-therapy (TAT) can be explained by the properties of α-particles. Alpha particles are helium nuclei and are ~8,000 times larger than β−-particles (electrons). When emitted from radionuclides that decay via an α-decay pathway, they release enormous amounts of energy over a very short distance. Typically, the range of α-particles in tissue is 50–100 μm and they have high linear energy transfer (LET) with a mean energy deposition of 100 keV/μm, providing a more specific tumor cell killing ability without damage to the surrounding normal tissues than β−-emitters. Due to these properties, the majority of pre-clinical and clinical trials have demonstrated that α-emitters such as 225Ac, 211At, 212Bi, 213Bi, 212Pb, 223Ra, and 227Th are ideal for the treatment of smaller tumor burdens, micrometastatic disease, and disseminated disease. Even though these α-emitters have favorable properties, the development of TAT has been limited by high costs, unresolved chemistry, and limited availability of the radionuclides. To overcome these limitations, more potent isotopes, additional sources, and more efficient isotope production methods should be addressed. Furthermore, better chelation and labeling methods with the improvements of isotope delivery, targeting vehicles, molecular targets, and identification of appropriate clinical applications are still required.
Keywords: Radioimmunotherapy, Alpha particles, Antibody, Actinium, Astatine, Bismuth, Lead, Radium, Thorium, Alpha targeted therapy, Alpha emitters
Introduction
Alpha and β−-particles, two kinds of particle radiation, were discovered by Rutherford in 1898 and were characterized as helium nuclei and electrons, respectively. Due to their ability to destroy tissue, these radiation particles were quickly applied to therapeutic applications such as radiation cancer therapy. One proposed approach was to deliver the killing power of these highly toxic radionuclides directly to tumor tissue via the appropriate targeting vectors, for instance, tumor antigen binding monoclonal antibodies (mAbs) and cell surface receptor binding peptides. This approach has continued to be a popular strategy and has been actively pursued for several years. Finally, as a result of these efforts, two radiolabeled antibodies targeting CD20 were approved by the FDA for treatment of non-Hodgkin’s lymphoma (NHL). One was 90Y-labeled ibritumomab tiuxeran (Zevalin) in 2002 and the other is 131I-labeled tositumomab (Bexxar) in 2003.
Even though the clinical utility of α-particle has been traditionally perceived as being limited to clinical applications that permit rapid targeting and cellular uptake to address half-life limitations, interest in α-particle has continued to be a burgeoning area of interest. Treatment of accessible disease, intratumoral administration strategies, and direct targeting of tumor vasculature have all been promoted as means to counter both half-life limitations as well as to better match physical half-life with biological half-life. As a consequence, new radioimmunotherapy (RIT) approaches with α-particle emitters have been considered and developed by numerous researchers. While pre-clinical studies date back in time for decades, targeted α-therapy (TAT) first appeared in the clinical research literature in 1999 with the first patient being treated in 1995 [1]. The effectiveness of TAT can be explained by the properties of α-particles. Alpha particles are helium atoms that are about 8,000 times larger than β−-particles (electrons). When emitted from radionuclides that decay via an α-decay pathway, they release enormous amounts of energy over a very short distance. Typically, the range of α-particles in tissue is 50–100 μm. They have high linear energy transfer (LET) with a mean energy deposition of 100 keV/μm, resulting in a lower number of particle emissions required for cell kill linked, in part, to an inability of DNA double-strand break repair potential and lack of oxygen effects on cytotoxicity [2, 3]. Thus, the cytotoxicity of α-particles may be extremely effective and may also be more dose independent than β−-emissions with cell death occurring from a single or a few α-particle traversals of the cell nucleus [4, 5].
The growing interest of TAT has led to the development of new chelating agents because the stable sequestration of the radionuclide in vivo is a critical component of targeted radiation therapy. In vivo stability of radioimmunoconjugates makes therapy possible by the achievement of maximum delivery of radiation to tumor while minimizing toxicity. Radioimmunoconjugates can also be subject to the direct effects of daughter radionuclides and to catabolism in targeted cells after internalization. There are various chelating agents available dependent on the chemical nature and the coordination chemistry of the radionuclides (Figs. 1 and 2).
Fig. 1.
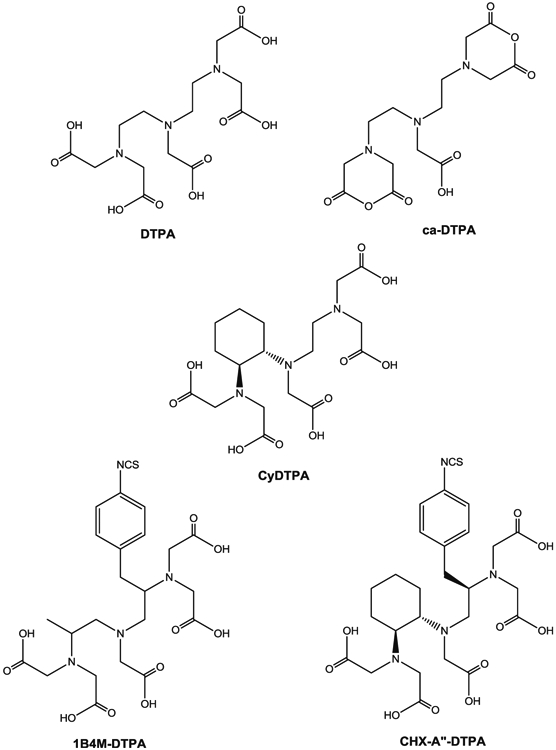
Structures of DTPA derivatives
Fig. 2.
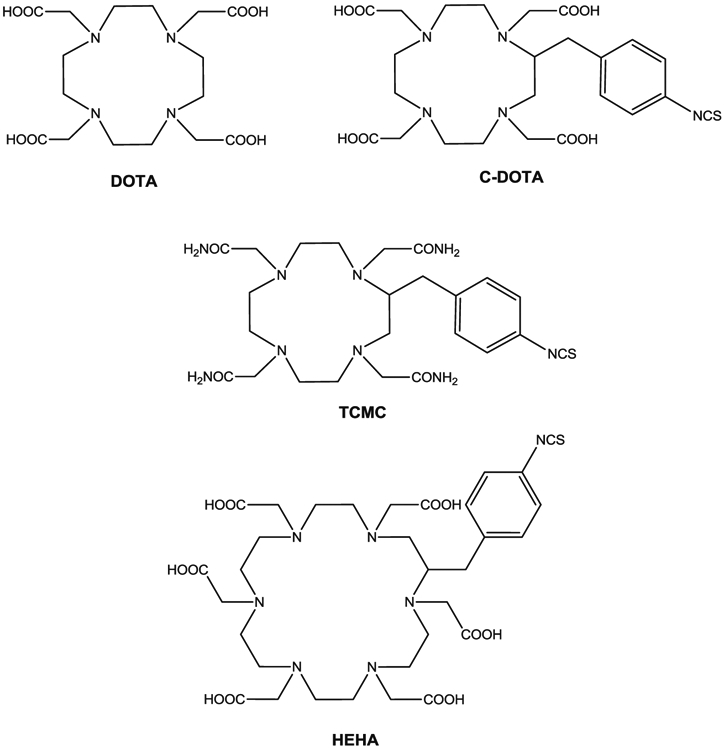
Structures of DOTA, TCMC, and HEHA
The clinical evaluation of RIT with α-emitters generally has been limited due to high costs of the radionuclide, unresolved chemistry, and limited availability of the radionuclide. With the elimination or reduction of many of these obstructions combined with a better understanding and new generation of monoclonal antibodies, numerous research groups have successfully evaluated many radiolabeled monoclonal antibodies for TAT.
There have been a number of α-emitting radionuclides considered for targeted therapy application. However, only a very limited number of α-emitting radionuclides on the list are realistically available for such applications due to either a too short or too long half-life, a lack of realistically viable chemistry, complicated decay pathways, or actual production/availability issues. For instance, 213Bi, an α-emitting nuclide with a half-life of 46 min, has been introduced into the therapeutic field and has been evaluated in the clinic. However, the short half-life of 213Bi remains a major concern regarding its widespread use, limiting its delivery ability to only the readily accessible cancer cells, e.g., to leukemias, to actual tumor vasculature, or through intratumoral administrations. One resolution to this constraint is to deliver the α-emitter in the form of a generator to the target cell, allowing production of radionuclides in situ, such as 225Ac (parent of 213Bi) and 212Pb (parent of 212Bi), which will generate in vivo effective α-particles to eradicate tumor cells. This concept, “in vivo generators”, was later termed, “nanogenerators”. The major advantage of this approach is that the longer half-life parental isotopes may facilitate targeting to and extravasation into tumor tissue. The longer half-life of the parental radionuclides also permit all operations to be executed more efficiently than with the short half-life isotopes as well as than the potential delivery of the accumulated range of emissions associated with the decay chain at the tumor site for greater efficacy.
This review will focus on those α-emitters suitable for pre-clinical and clinical uses such as actinium-225 (225Ac), astatine-211 (211At), bismuth-212 (212Bi), bismuth-213 (213Bi), lead-212 (212Pb), radium-223 (223Ra), and thorium-227 (227Th) (Table 1). New concepts, approaches, and techniques will be described as well. As yet, numerous review articles related to the development of TAT have been published and are readily available [6-21], and this discussion will again be principally limited to providing an update those reviews.
Table 1.
Candidates for targeted α-therapy
| Radionuclide | Daughters | Half-life | Emission | Energy | Production | Chelating agents |
|---|---|---|---|---|---|---|
| 225Ac | 221Fr, 217At, 213Bi, 213Po, 209Tl, 209Pb | 10 days | 5 α, 3 β− | 5.8 MeV | 233U natural decay | HEHA, DOTA |
| Cyclotron | ||||||
| 211At | 211Po, 207Bi | 7.2 h | 2 α, 2 EC | 5.9 MeV | Cyclotron | |
| 212Bi | 212Po, 208Tl | 60.6 min | 2 α, 2 β− | 6.05 MeV | 228Th natural decay | CHX-A″-DTPA |
| 224Ra generator | ||||||
| 213Bi | 213Po, 209Tl, 209Pb | 45.6 min | 2 α, 3 β− | 5.8 MeV | 225Ac generator | CHX-A″-DTPA |
| 212Pb | 212Bi, 212Po, 208Tl | 10.6 h | 2 α, 3 β− | 6.05 MeV | 224Ra generator | DOTA, TCMC |
| 223Ra | 219Rn, 215Po, 211Pb, 211Bi, 211Po, 207Tl | 11.4 days | 5 α, 3 β− | 5.7 MeV | 227Ac generator | |
| 227Th | 223Ra, 219Rn, 215Po, 211Pb, 211Bi, 211Po, 207Tl | 18.7 days | 6 α, 3 β− | 5.9 MeV | 227Ac generator | DOTA |
225Ac
Properties, chemistry, and radiolabeling
Actinium was discovered by Andre Louis Debierne in 1899 and Friedrich Oskar Giesel in 1902 [22]. Actinium occurs naturally in association with uranium radionuclides and can be obtained from either the decay of 233U or by the neutron transmutation of 226Ra (Fig. 3) [23]. Actinium-225 is a radiometal with a half-life of 10 days and there are six radionuclide daughters in the decay path to a final stable state 209Bi. For each decay event of 225Ac, there are four α and two β−-emissions with high energy (8.38 MeV for α-decay). Given the 10-day half-life, the high energy α-particle emission, and the favorable rapid decay chain to stable 209Bi, 225Ac has been considered a potential candidate for use in cancer therapy, particularly in regards for use as an in vivo generator. However, the coordination chemistry of Ac (III) is not well defined primarily due to there being no stable isotopes to promote routine chemical studies. Therefore, use of 225Ac as a therapeutic has been limited by the availability of suitable chelating agents stably binding the radionuclide as well as the daughter radionuclides generated by its decay [24, 25]. In addition, the chelating agent will be challenged by the recoil energy of α-particle emission (100–200 keV), which is higher than the binding energy than that of the environment defined by the coordination complex.
Fig. 3.
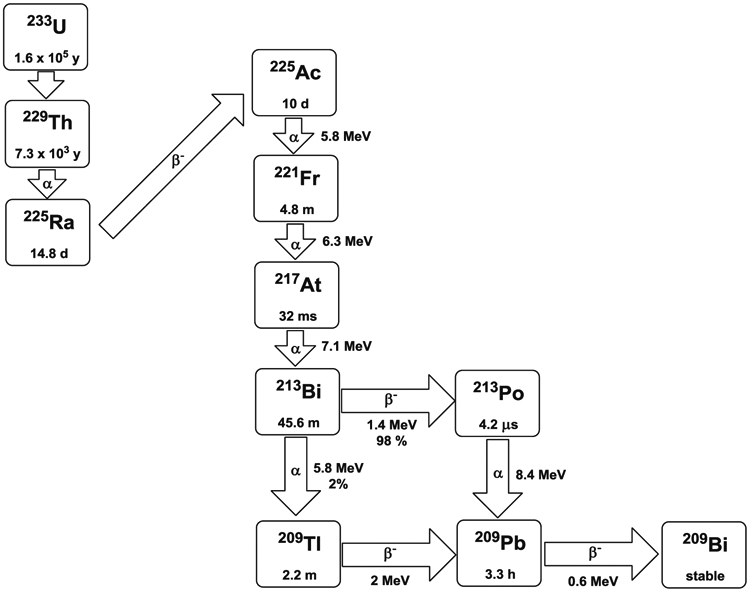
Decay scheme of 225Ac and 213Bi
Chappell et al. developed a chelating agent, 2-(4-isothiocyanatobenzyl)-1,4,7,10,13,16-hexaazacyclohexadecane-1,4,7,10,13,16-hexaacetic acid (HEHA-NCS, Fig. 2), and radiolabeled three mAbs with 225Ac [26]. HEHA is a macrocyclic chelating agent with six nitrogens and six carboxylic acids available as donors to its coordination sphere as well as a large macrocyclic cavity to match the large ionic radius of Ac(III). Due to its properties, HEHA was proposed as potentially useful for forming a complex with 225Ac, but the study concluded that RIT with 225Ac-HEHA was compromised by unstable coordination chemistry and release of the daughter radionuclides in vivo. Another strategy was proposed by the group from the Sloan-Kettering Cancer Center, which was the use of 225Ac envisioned as a “nanogenerator” approach [27]. In this study, McDevitt et al. used 2-(4-isothiocyanatobenzyl)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (C-DOTA, Fig. 2) to form an 225Ac complex which, after transport into the target cells, decayed and daughter isotopes were retained. This system was efficacious in vitro and in vivo for the targeted cells and limited the toxicity to non-target cells.
Pre-clinical studies
Neovascular endothelium is an interesting target for TAT. Tumor vasculature targeting is the rapid, efficient, and selective method of endoradiotherapy delivery to tumor endothelium by peptides, antibodies, antibody fragments, and nanoparticles with α-emitters [28, 29]. This endoradiotherapy directly at tumor vasculature may prove extremely proficient since the endothelium is immediately adjacent to blood which carries the radiolabeled compound so that radiolabeled compounds have no need to be highly diffusible through the tumor interstitium. E4G10, a mAb that targets vascular endothelial cadherin, was radiolabeled with 225Ac and evaluated in mice bearing LNCaP prostate tumor cell xenografts [30]. Treatment with 225Ac-E4G10 resulted in tumor growth inhibition, decreased serum prostate-specific antigen levels, and prolonged survival. This therapeutic benefit was, in turn, enhanced by administration of paclitaxel. Immunohisto-chemistry revealed decreased vessel density and enhanced apoptosis of the tumor. In addition, residual tumor vasculature appeared normalized without toxicity being observed in vascularized normal organs. E4G10 was also evaluated as a single wall carbon nanotube (SWCNT) conjugate constructs with 225Ac-DOTA in a murine xenograft model of human colon adenocarcinoma (LS174T). Therapy using this “nano” assembly resulted in reduced tumor volumes and improved animal survival rates as compared to the experimental controls [31].
Retention of the daughter radionuclides generated by the decay of 225Ac at the desired tumor site would be expected to increase efficacy, while conversely the release and redistribution of those same daughter radionuclides would be predicted to increase toxicity. Sofou et al. [32] proposed liposomal encapsulation of 225Ac to retain the daughters at the target site, but only 14% of daughter radionuclides were retained at the target. In their following study involving multivesicular liposomes (MUVELs) evaluated to increase daughter retention [33], these researchers found that PEGy-lated MUVELs stably retained 98% of the encapsulated 225Ac, as well as 31% of the 213Bi daughter. MUVELs were also conjugated to an anti-HER2/neu antibody and evaluated in vitro with SKOV3-NMP2 ovarian cancer cells, showing significant cellular internalization (83%).
Sgouros’ group compared the efficacy of 225Ac with 213Bi and 90Y in a mouse model bearing NT2.5 xenografts, a rat HER-2/neu-expressing mouse mammary tumor cell line [34]. In this study, anti-rat HER-2/neu monoclonal antibody (7.16.4) was conjugated to SCN-CHX-A″-DTPA (for 213Bi and 90Y) and p-SCN-Bn-DOTA (for 225Ac). Treatment with 225Ac-7.16.4 was found to eradicate the breast tumor, leading to long-term survival (1 year) of the mice. Use of 225Ac was much more effective than either 213Bi-7.16.4 (61 days) or 90Y-7.16.4 (50 days). Dosimetry calculations indicated that the 225Ac-treated tumor (9.6 Gy) received significantly higher dose than either 213Bi (2.0 Gy) or 90Y (2.4 Gy). Biodistribution studies suggested that only a small portion of the 225Ac daughter radionuclides, 221Fr and 213Bi, were released from the target site.
Recently, there has been increasing interest in nanomaterials for use in the diagnostic and therapeutic fields. Woodward et al. [35] synthesized La(225Ac)PO4 nanoparticles conjugated to monoclonal antibody mAb 201B, which targets normal mouse lung endothelium. Based on biodistribution and imaging studies, 225Ac and the associated daughter radionuclides rapidly accumulated in mouse lung after i.v. injection with 30% ID/lung (>200% ID/g) of La(225Ac)PO4 nanoparticles at 1 h p.i. in the lung. In addition, an in vivo release study of 213Bi was performed and the data indicated a deficiency of 213Bi activity relative to the equilibrium 213Bi/225Ac ratio in the lungs. At 24 h p.i., 80% of the 213Bi, one daughter of 225Ac decay, was retained in the target area, and the retention of 213Bi increased to 87% at 120 h p.i. In vitro analysis demonstrated that 50% of the daughter radionuclides were sequestered in the La(225Ac)PO4 nanoparticles. One should also note that with this example of a targeted nanoparticle that access to and penetration into tumor was traversed by the targeting vehicle and target and that extravasation of nanoparticles into tumor remains a challenge to their use regardless of any attractions associated with nanotechnology. If the size of intact an IgG is considered an obstacle to delivery and penetration of solid tumors, then the use of nanoparticles, generally larger than antibodies, remains challenged by these same size limitations.
Clinical studies
There has been only one clinical trial using 225Ac to date. HuM195, a humanized antibody, targets CD33 on acute myeloid leukemia cells and has been extensively investigated in clinical trials with β−-emitters and α-emitters [91, 92]. Studies with HuM195 were extended to 225Ac due to its favorable properties for RIT, and based on pharmacokinetics, dosimetry, and toxicity studies of 225Ac-HuM195 conducted with cynomolgus monkeys [36]. These studies resulted in a phase I clinical trial was started by the Scheinberg group at Memorial Sloan-Kettering Cancer Center and which is still recruiting patients [37]. The main goal of this trial was to define both safety and the maximum tolerated dose of 225Ac-HuM195 for patients with advanced myeloid leukemia. The starting dose was 0.5 μCi/kg of 225Ac-HuM195 with three to six patients being treated at each dose level, with dose escalation proceeding if less than 33% of patients in a cohort experienced dose limiting toxicity. Thereafter, six patients were planned to be treated at the maximum tolerated dose. Blood, urine, and bone marrow were to be measured after treatment to determine the toxicity, pharmacokinetics, immunogenicity, and anti-cancer effects.
211At
Properties, chemistry, and radiolabeling
Astatine-211 is one of the most promising radionuclides for α-particle therapy, and notation of its potential medical utility dates back as far as 50 years [38]. Astatine is the heaviest halogen without a stable isotope and 211At has a half-life of 7.2 h which provides enough time for distillation and radiolabeling. Also, the half-life is reasonably well matched to the pharmacokinetics of a variety of molecular entities such as peptides, mAbs (intact and fragments), and small molecules. Astatine-211 emits two α-particles through a split decay pathway with energies of 5.87 and 7.45 MeV, a range in soft tissue of 55–80 μm, and an LET of 99 KeV/μm. One decay pathway is by direct α-particle emission to 207Bi (42% of decay) followed by electron capture to the stable 207Pb (Fig. 4). The other pathway is by electron capture to 211Po (58% of decay) followed by α-particle emission to the ground state 207Pb. In addition, the 211Po daughter emits 77–92 KeV polonium K X-rays that enable external imaging, including SPECT, and γ counting for biodistribution studies, all additional advantages. Even though 211At has very attractive properties for RIT, its widespread use as a clinical therapeutic has been seriously limited by availability and radiochemical purity. Astatine-211 is cyclotron produced by bombarding a bismuth target with 22–28.5 MeV α-particles via the 209Bi(α, 2n)211At nuclear reaction followed by the dry distillation to isolate 211At in solution, e.g., chloroform or methanol [39, 40]. In order to minimize the production of 210At, which decays to 210Po with a half-life of 138 days and could lead to safety and toxicity issues, the beam energy window should be kept below 28.5 MeV. However, only a few institutions in the USA have cyclotrons available with an appropriate α-particle beam for production of 211At and possess facilities for target processing.
Fig. 4.
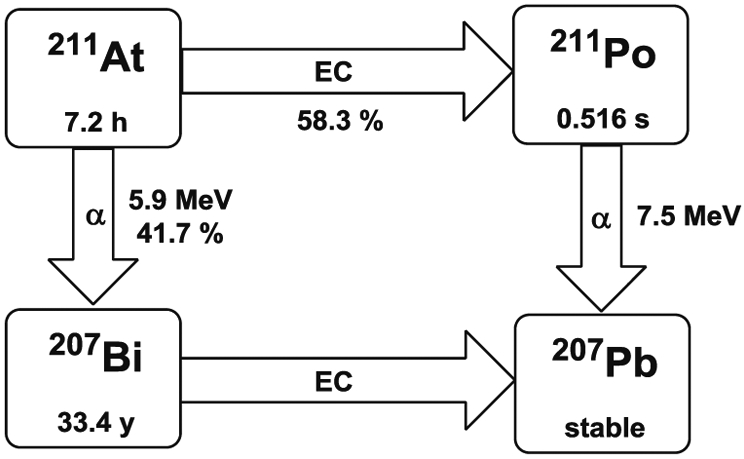
Decay scheme of 211At
Although astatine is generally treated as a halogen, it also has significant metallic characteristics in certain environments [41, 42]. The aryl carbon–halogen bond strength for astatine is significantly lower than that of iodine [8] which precludes using standard direct radio-iodination methods for labeling mAbs with 211At. Such methods lead to labile products resulting in rapid in vivo loss of 211At. To resolve this problem, approaches have been developed in several research groups based on small linker molecules that create an aryl carbon–astatine bond involving an astatodemetallation reaction using tin, silicon, or mercury precursors. Zalutsky et al. [39, 40] developed the two-step method for radiolabeling intact and mAb fragments. In the first step, N-succinimidyl 3-(trimethylstannyl)benzoate and the oxidant tert-butyl hydroperoxide were added into 211At/chloroform followed by 15-min incubation and HPLC purification. The second step was addition of the protein to the purified product followed by 15-min incubation in an ice bath. The whole process takes 1.5 h making this approach practical for providing 211At-labeled mAb with good specific activity, high immunoreactivity as well as with reasonable in vivo stability.
While generally dry distillation is used for 211At purification, wet extraction technique has also been developed due to the demand for a high yield of 211At [43]. In this technique, the irradiated bismuth target was dissolved in nitric acid, and the 211At extracted by di-isopropylether (DIPE) instead of being distilled into a solution. The N-hydroxysuccinimidyl-meta-trimethylstannylbenzoate ester (MeSTB) has been used for radiolabeling with 211At obtained by wet extraction methods [43, 59].
Pre-clinical studies
One leading center for TAT using 211At has been Duke University. The Zalutsky group there has extensively studied production, radiolabeling chemistry and conducted pre-clinical studies with 211At [2, 8, 40, 44, 45]. Recently, Zalutsky and co-workers evaluated the therapeutic effect of intrathecal administration of 211At-labeled trastuzumab in rats bearing MCF-7/HER2-18 breast carcinoma xenograft as a model for breast carcinoma carcinomatous meningitis (CM) [46]. CM is a form of metastatic cancer spreading to the subarachnoid space of the brain and spinal cord; most therapies are ineffective against CM. In this study, they found that median survival increased significantly versus the control group. For instance, the animals that received 33 and 66 μCi of 211At-trastuzumab the median survival was 45 and 48 days, respectively, while for the saline and cold trastuzumab groups for the median survival was 21 days.
Investigators at the Göteborg University at Sweden are also actively involved in TAT using 211At. Elgqvist et al. investigated the efficacy of targeted α-therapy of ovarian cancer in mice using 211At-labeled mAb MX35 and MX35 F (ab′)2 [47-49]. Animals inoculated intraperitoneally (i.p.) with OVCAR-3 cells received TAT treatment by i.p. injection. Two months after treatment, the mice were euthanized and the presence of macroscopic/microscopic tumors and ascites was determined. When tumor radius was ≤30 μm, the tumor-free fraction (TFF) of animals treated with 211At-labeled-MX35 F(ab′)2 was 95%. Compared to those treated with the non-specific antibody [Rituximab F(ab′)2], the specific antibody, 211At-labeled-MX35 F(ab′)2 gave a significantly higher TFF when the tumor radius exceeded the range of the α-particles. This result was explained by a high mean absorbed dose from the activity bound to the tumor surface combined with the ability of the F(ab′)2 to penetrate farther into the tumor as compared to the intact IgG [49]. Another efficacy study of an 211At-labeled antibody was performed with a HER-2 positive ovarian carcinoma by Palm et al. [50], and the responses to single dose and fractionated treatment with unlabeled and 211At-labeled trastuzumab were evaluated. Tumor-bearing mice were injected with 211At-trastuzumab (400 kBq) followed by 5, 50, or 500 μg of trastuzumab 7 days later. Compared to 5 μg group, tumors were reduced by 87% with 50 μg of trastuzumab and completely eradicated in mice given 500 μg of trastuzumab.
The Wilbur group has investigated the application of α-particle emitting radionuclides as a replacement of total body irradiation (TBI) for hematopoietic cell transplantation (HCT) [51, 52]. Allogeneic HCT is an effective method for treating patients with hematopoietic disease; mAb labeled with β−-emitting radionuclides had been previously studied for increasing specificity and efficacy with less toxicity from TBI. However, due to the long path length of their emissions and low dose rates, β−-emitting radionuclides were not suitable for targeting hematopoietic cells as compared to α-emitting radionuclides. Biodistribution and toxicity studies were performed with radioimmunoconjugates of the antimurine CD45 antibody 30F11 labeled with 211At and 213Bi. The results indicated that lower doses of 211At-30F11 were sufficient to achieve myelosuppression and myeloablation with less toxicities compared to 213Bi-30F11 [52].
There are other research groups who have studied the efficacy of TAT with 211At. Almqvist and co-workers targeted the A33 antigen, expressed on 95% of colorectal carcinomas, using 211At-labeled humanized monoclonal antibody A33 (huA33) [53, 54]. At 8 h p.i., tumor had the highest uptake (~15% ID/g) than any other organ except blood. The tumor/blood ratio of 211At-huA33 increased with time and reached 2.5 at 21 h p.i. In a dosimetry study, blood had the highest absorbed dose (8.9 Gy/MBq), and tumor received 6.2 Gy/MBq, which is higher than the normal organs besides the thyroid [54]. Another group from Sweden has focused on targeted α-therapy with 211At using chimeric mAb U36 in head and neck squamous cell carcinoma [55, 56]. Tumor uptake (10% ID/g at 21 h p.i.) increased with time while 211At-U36 activity in other organs decreased excluding thyroid. In the therapy study, 18 of 20 mice responded to therapy, indicated by stabilized or decreased tumor volume [56].
The therapeutic efficacy of 211At-labeled anti-CD25 [interleukin-2 receptor α (IL-2Rα)] monoclonal antibody, 7G7/B6 has also been evaluated as a potential therapeutic agent for CD25 expressing leukemias and lymphomas by the Waldmann group [57]. Their study demonstrated that the combining 211At-7G7/B6 with daclizumab (unmodified anti-Tac) significantly improved therapeutic efficacy [58]. Mice were treated with 0.44 MBq of 211At-7G7/B6 and 100 μg of daclizumab given weekly (0, 7, 14, and 21 days). The combination treatment resulted in 91% of mice surviving through 94 days, whereas only 36% and 50% of mice from the 211At-7G7/B6 and daclizumab groups, respectively, were alive at 94 days.
Clinical studies
A limited number of clinical trials have evaluated the therapeutic potential of 211At. The study with 211At-ch81C6, a chimeric antibody targeting tenascin, a glycoprotein over-expressed in gliomas, was extended to clinical trials. Zalutsky et al. investigated the feasibility and safety of this therapy in patients with recurrent malignant brain tumors [60, 61]. Eighteen patients were treated with 211At-ch81C6 administered into a surgically created resection cavity (SCRC). No patient presented with dose-limiting toxicity and 96.7% of 211At decays occurred inside the SCRC. No toxicities of grade 3 or higher were derivable from the treatment and no patient required repeat surgery for radionecrosis. Eight of 14 patients with recurrent glioblastoma multiforme survived for a year and two patients survived for 3 years after treatment with 144 MBq of 211At-ch81C6. This study clearly demonstrated that the regional administration of 211At-ch81C6 was attainable, safe, and associated with a possible therapeutic benefit for patients with recurrent central nerve system tumors.
Another clinical trial has been initiated at the University of Gothenburg, Sweden. Andersson and co-workers investigated the pharmacokinetics and dosimetry of 211At-MX35 F(ab′)2 [62]. Women with recurrent ovarian carcinoma were enrolled in this phase I study. Nine patients were infused with 211At-MX35 F(ab′)2 via peritoneal catheter, and the result has demonstrated that 211At-MX35 F(ab′)2 by intraperitoneal administration is feasible to achieve therapeutic doses in microscopic tumor clusters without significant toxicity.
212Bi
Properties, chemistry, and labeling
Bismuth-212 can be obtained from the decay chain of 228Th. It has a half-life of 60.6 min and decays to stable 208Pb via either 208Tl (36%) or 212Po (64%) (Fig. 5). Both pathways emit β−-particles and α-particles with an average energy of 7.8 MeV and path length in tissues of 40–100 μm, localizing cellular toxicity in a small region. The combination of α- and β−-particles may help to increase the therapeutic efficacy for tumor heterogeneity. However, the short half-life of 212Bi has in part been a major limitation for radiolabeling processes and RIT applications. This problem was partly resolved by the availability of a 224Ra generator which makes local production of 212Bi feasible [63, 64]. Still, applications including i.v. injection of mAb remain challenging with the short physical half-life of 212Bi. Another serious perceived challenge to the clinical evaluation of 212Bi has been the 2.6 MeV γ-ray originating from the 208Tl daughter produced in 212Bi decay chain, which requires significant shielding to minimize radiation exposure to health care workers, making it unclear what level of shielding is needed for a clinical environment due to the combination of actual dosing logistics, short half-life, and safety concerns.
Fig. 5.
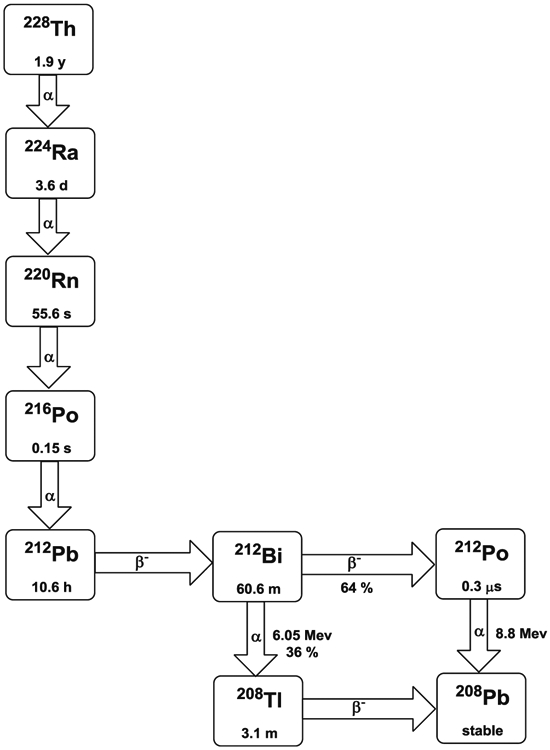
Decay scheme of 212Bi and 212Pb
Bismuth-212 can be reasonably bound by bifunctional metal chelating agents such as derivatives of functionalized diethylenetriamine pentaacetic acid (DTPA, Fig. 1) conjugated to mAbs, their fragments, and peptides including the cyclic DTPA anhydride derivative, 2-(p-isothiocyanatobenzyl)-DTPA (SCN-Bz-DTPA), isothiocyanatobenzyl derivative of C-functionalized trans-cyclohexyl DTPA (CHX-A″-DTPA), and 2-(p-isothiocyanatobenzyl)-6-methyl-DTPA (Mx-DTPA, 1B4M-DTPA). However, only the CHX-A″-DTPA forms suitably stable complexes with bismuth radionuclides in vivo [65, 66]. DOTA has also been successfully used for labeling mAbs with 212Bi and demonstrated in vivo stability in an animal model [67, 68]. However, the slow formation kinetics of radiolabeling DOTA with 212Bi seriously compromises the use of this chelation technology. As such, there have been no recent pre-clinical studies reported in the literature using 212Bi with this Bi(III) radionuclide effectively being supplanted by 213Bi.
213Bi
Properties, chemistry, and labeling
Bismuth-213 is a decay product of 225Ac making it generator available and 213Bi decays via a branched pathway by α-particle emission with an energy of 8.37 MeV to 209Bi (Fig. 3). Bismuth-213 has a half-life of 45.6 min, shorter yet than 212Bi; however, 213Bi additionally emits a 440-keV γ-ray (26.5%) allowing imaging, biodistribution, pharmacokinetics, and dosimetry studies to be performed and, more importantly, also lacks the high energy γ-ray of 212Bi. Bismuth-213 for clinical use requires a generator due to the short half-life, and currently, the Oak Ridge National Laboratory produces the only 225Ac/213Bi generator in the USA. Primarily, methods to separate 213Bi from 225Ac were introduced by Boll et al. [69, 70]. The 225Ac/213Bi generator has since been further developed by Memorial Sloan-Kettering Cancer Center in order to supply carrier-free 213Bi [71-74]; however, the initial generator design dates to Pippin et al. [75]. After elution from the generator, 213Bi is readily used for radiolabeling chelate conjugated mAbs, peptides, or other vectors without significant safety measures or shielding required other than gloves and monitoring devices. Similarly to 212Bi, the bifunctional chelating agent, CHX-A ″-DTPA (Fig. 1), can readily attach covalently to protein molecules and has been proven to be the agent of choice for sequestering radio-bismuth stably in vivo [65, 66, 76].
Pre-clinical studies
The Allen group in Australia has actively studied the efficacy of 213Bi-labeled mAb as TAT agents [77-81]. The mAb 9.2.27 has a high specificity for human melanoma cell surfaces. The biodistribution of 213Bi-labeled mAb 9.2.27 showed no evidence of retention of the α-radioimmunoconjugate in non-targeted organs. Tumor growth was completely inhibited at 2 days post-inoculation. In conclusion, the targeted α-therapy was effective for the treatment of micrometastatic melanoma when the tumor was in a pre-angiogenic stage. In addition, a multiple dose treatment regimen was more effective than a single dose [77].
The toxicity and efficacy of 213Bi-labeled plasminogen activator inhibitor type 2 (PAI2) in a prostate cancer model has been examined [79]. In this particular study, tumor growth was inhibited following the administration of single doses of either 947 or 1,421 MBq/kg at 3 days post-tumor inoculation. Complete tumor growth inhibition was also observed when a total dose of 947 MBq/kg was given on five successive days. Treatments given at 6, 12, and 18 days post-tumor inoculation showed significantly slower tumor growth than the controls. Additionally, all treatments were tolerated in mice and rabbits as monitored by biochemical and hematological studies; multiple doses were no more toxic than the single dose regime [79].
This group also evaluated 213Bi-labeled C595 anti-MUC1 mAb for control of ascites in an ovarian cancer ascites mouse model; MUC1 is over-expressed in ovarian cancer tissues [80]. A single dose of 355 MBq/kg of 213Bi-labeled C595 increased survival by 25 days when administered 9 days after tumor implantation. The maximum tolerated dose was more than 1,180 MBq/kg with monitoring up to 21 weeks. This study demonstrated that 213Bi-C595 can effectively target and kill ovarian cancer cells with acceptable levels of toxicity.
The Sgouros group at Johns Hopkins has focused on the targeting of metastatic breast cancer [82, 83]. They have investigated the efficacy of 213Bi-labeled anti-rat HER-2/neu mAb 7.16.4 in a rat/neu transgenic mouse model for metastatic mammary carcinoma. The maximum tolerated dose was 120 μCi; treatment at this dose prolonged median survival to 41 days as compared to 28 days for treatment with the control antibody. Overall, 213Bi-7.16.4 was effective for treating early stage HER-2/neu metastases [82].
Liposomes have been studied for several decades as vehicles for drug delivery, and incorporating polyethylene glycol (PEG) chains has made it possible to effectively increase their circulatory half-life. Lingappa and co-workers radiolabeled a liposome-CHX-A″-DTPA conjugate with 213Bi and compared the efficacy of that product to a liposome-CHX-A″-DTPA-213Bi-7.16.4 and 213Bi-7.16.4 in rat HER2/neu transgenic mice. Based on the survival times, the 213Bi-labeled liposome was as effective as antibody targeted 213Bi in this animal model [83].
Park et al. were interested in evaluating a pre-targeting system using antibody–streptavidin (Ab-SA) constructs and radiolabeled biotin [84]. In this study, mice with Ramos lymphoma cells were given anti-CD20 1F5(scFv)4SA fusion protein. A dendrimeric clearing agent and 213Bi-DOTA-biotin were then injected as per protocol. Tumor uptake was 16.5±7.0% ID/g 90 min p.i. compared to 2.3±0.9% ID/g for the control. Tumor growth was significantly delayed by treatment with 1F5(scFv)4SA and 213Bi-DOTA-biotin (600 μCi). The median survival time was 90 days compared to 23 days for the control. In addition, the treatment was well tolerated with no significant toxicity reported.
Locoregional RIT of disseminated gastric cancer in mice using 213Bi showed good therapeutic results with a single dose depending on the time interval between tumor inoculation and therapy. Bloechl et al. compared single versus double i.p. injections of mAb d9MAb labeled with 213Bi for efficacy and toxicity [85]. Single and double applications of 0.37, 0.74, or 1.48 MBq of 213Bi-d9MAb were injected into mice at 1 and 8 days after tumor inoculation. Double doses of 0.37 MBq significantly prolonged the median survival as compared with a single dose. In an advanced stage of the disease, double injection of 0.74 MBq was much more effective than a single dose of 1.48 MBq without any toxicity.
Milenic et al. has investigated the dual targeting efficacy of targeted α-therapy using trastuzumab, which binds to HER-2 receptor and humanized CC49 mAb (HuCC49ΔCH2), which binds to tumor-associated glycoprotein 72 (TAG-72) [86]. A therapy study was performed in which 500 μCi of 213Bi-trastuzumab and 213Bi-HuCC49ΔCH2 was injected (i.p.) alone and concurrently to mice with 3-day tumor burden. Sequential administration, 213Bi-trastuzumab followed by 213Bi-HuCC49ΔCH2 or vice versa, resulted in greater therapeutic efficacy than single dose and produced a median survival of 36 days and 40 days, respectively. The highest therapeutic efficacy was achieved with the concurrent administration of 213Bi-trastuzumab and 213Bi-HuCC49ΔCH2. The median survival was 147 days, which translates to a therapeutic index of 9.8.
The short half-life of 213Bi (45.6 min) is well matched with the peptide-receptor radionuclide α-therapy. Wild and co-workers recently prepared two 213Bi-labeled peptides, DOTA-PEG4-bombesin (DOTA-PESIN) and DO3A-CH2CO-8-aminooctanoyl-QWAVGHLM-NH2 (AMBA), and compared them with 177Lu-DOTA-PESIN in a human prostate carcinoma xenograft model (PC-3 tumor) [87]. The maximum tolerated dose of 213Bi-DOTA-PESIN and AMBA was 25 MBq (5×5 MBq) while 177Lu-DOTA-PESIN was 112 MBq (4×28 MBq). With this dose level, 213Bi-labeled peptides were clearly more efficient than 177Lu-DOTA-PESIN. For example, an additional injection of 213Bi-labeled peptides reduced the risk of recurrent disease by 57%, whereas 177Lu-DOTA-PESIN treatment showed no effect on abatement of recurrent disease. Moreover, the median survival time with 213Bi-labeled peptides was extended by 15 weeks, five times greater than that obtained from 177Lu-DOTA-PESIN treatment.
Clinical studies
With the replacement of 212Bi with 213Bi, a number of clinical trials have been initiated and executed. The therapeutic efficacy of α-emitting 213Bi on B-cell-lineage lymphoma has been investigated with various antibodies. In pre-clinical experiments, 213Bi-labeled anti-CD20-CHX-A″-DTPA showed a strong cytotoxic potency for targeting lymphoma cells. This radioimmunoconjugate also demonstrated a favorable in vivo stability and biodistribution pattern. No other toxic effects occurred beside the induced myelosuppression [88]. Consequently, Heeger and co-worker at German Cancer Research Center started a phase I dose escalation trial to determine toxicity and feasibility as well as dosimetry and pharmacokinetics [89]. Nine patients with B-cell malignancy were enrolled and received doses of 213Bi-anti-CD20 from 385 to 1,640 MBq. No acute or extramedullary toxicity were observed except a mild leukopenia (two patients). Two patients responded to radioimmunotherapy. Currently, this trial has been continued at the University Hospital Düsseldorf, Germany.
An open-label phase I dose escalation study of 213Bi-labeled mAb 9.2.27 for metastatic melanoma was initiated by the Allen group [90]. Twenty-two patients with stage IV/in-transit metastasis were treated with 55–947 MBq of 213Bi-9.2.27. Overall, 6% demonstrated almost complete response, 14% showed partial response, 50% stable disease and 30% progressive disease. Most of the patients showed reductions of disease in 8 weeks based on the tumor marker melanoma inhibitory activity protein. No toxicity was detected during the dose escalation study.
A humanized anti-CD33 mAb, HuM195, targeting myeloid leukemia cells, radiolabeled with 213Bi was translated to a phase I dose-escalation trial at Memorial Sloan-Kettering Cancer Center [91]. Eighteen patients with advanced myeloid leukemia were treated with 10.36 to 37.0 MBq/kg of 213Bi-HuM195. No significant toxicity was detected. Uptake of 213Bi, determined by γ-camera imaging, was found to be mainly in the bone marrow, liver, and spleen without significant uptake in other organs. Absorbed dose ratios between bone marrow, liver, and spleen and the whole body were 1,000 times higher with 213Bi-HuM195 than with β−-emitters labeled HuM195. Fourteen of 18 patients had reductions in the percentage of bone marrow blasts, and 14 patients had reductions in circulating blasts after therapy. This study is the first proof-of-concept for systemic targeted α-particle immunotherapy in humans.
A phase I/II trial followed to determine the maximum tolerated dose and effects of 213Bi-lintuzumab (HuM195) by Jurcic et al. at Memorial Sloan-Kettering Cancer Center [92]. Thirteen patients newly diagnosed and 18 patients with acute myeloid leukemia were enrolled and treated with cytarabine continuous infusion for 5 days. Doses of 213Bi-lintuzumab (18.5–46.25 MBq/kg) were then injected over 1 to 2 days. Myelosuppression was the primary toxicity, and the maximum tolerated dose was 37 MBq/kg. Two of 21 patients treated with the maximum tolerated dose died. At all dose levels, marrow blasts reductions were observed. Based on biodistribution and pharmacokinetic studies, CD33 sites were saturated by 213Bi-lintuzumab after partial cytoreduction by cytarabine.
Substance P is tachykinin peptide neurotransmitter which targets the neurokinin type-1 receptor (NK-1), and NK-1 is consistently over-expressed in World Health Organization (WHO) grade II–IV gliomas. The Merlo group conjugated 1,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid (DOTAGA) to substance P and was then performed a pilot trial of TAT using 213Bi [93, 94]. In this pilot study, five patients with WHO grades II–IV were enrolled and the treatments were given via an implanted catheter system (intratumoural injection). Four patients received 1.07–2.0 GBq of 213Bi-DOTA-substance P within one therapeutic cycle, and one patient received 7.36 GBq with four therapeutic cycles. Lengthy retention of 213Bi-DOTA-substance P was observed at the target site, and no local or systemic toxicity was detected according to the NCI Common Toxicity Criteria (CTC) scale. The pre-therapeutic functional score (the Barthel Index) was 75 and 80 with the two glioblastoma multiforme (GBM) which means mild restrictions in activities of daily living. After treatment, both patients improved to 90 out of 100. MR imaging demonstrated that there were radiation-induced necrosis and demarcation of the tumors [94].
212Pb
Properties, chemistry, and labeling
Lead-212 is a product of the 228Th decay chain and also can be generated by the previously mentioned 224Ra generator that again facilitates on-site production [63]. Pb-212 (10.6 h half-life) is a β−-emitter and is the parent of 212Bi (Fig. 5). One strategy which has been proposed and investigated has been to label a mAb with 212Pb and use this conjugate again as an in vivo generator for 212Bi to effectively traverse the problem of short half-life 212Bi [95]. Several lead and bismuth complexes have been studied for use in this in vivo generator system [96-98].
There are distinct advantages of targeting tumor with 212Pb over 212Bi and 213Bi. Pb-212 can deliver more than 10 times the dose per unit of administered activity as compared to either 212Bi or 213Bi. In addition, the 10.6 h of half-life is very reasonable for dose preparation and administration. However, there is major complication associated with radiolabeling a mAb with 212Pb due to the electron capture and Auger electrons which may compromise the chelating moiety. Many bifunctional DOTA derivatives have been used for labeling antibodies with 212Pb, but the Pb(II)–DOTA complex has been shown to permit dissociation of Pb(II) at a pH comparable to that in lysozymes post-internalization into cells as compared with the tetra amide derivative, 1,4,7,10-tetraaza-1,4,7,10-tetra-(2-carbamoyl methyl)-cyclododecane (TCMC, Fig. 2) [99]. The subsequently free 212Pb has been reported as a source of bone marrow toxicity [100]. Therefore, TCMC has become the chelation chemistry of choice for sequestrating 212Pb.
Pre-clinical studies
Milenic and co-workers have proposed and demonstrated the effective use of 212Pb-labeled trastuzumab for the treatment of disseminated peritoneal disease [101]. In studies using mice bearing LS-174T i.p. xenografts, the maximum tolerated dose was 0.74–1.48 MBq, and the median survival of animals with 0.37 MBq was prolonged from 19 to 56 days. A multi-dose regime of 212Pb–TCMC–trastuzumab administered monthly (up to 3 months) increased the median survival of mice bearing 3-day LS-174T i.p. xenografts to 110 days.
This group also performed a combination study of 212Pb–TCMC–trastuzumab and gemcitabine (GEM) for treating disseminated peritoneal disease [102]. Treatment with GEM followed 24–30 h later by 0.19 or 0.37 MBq showed an improvement of median survival from 31 (without GEM) to 51 (with GEM) days at the 0.19 MBq, and from 45 (without GEM) to 70 (with GEM) days at the 0.37 MBq compared with 16 days for the untreated group. Three weekly treatments of GEM with 212Pb prolonged the median survival to 90 days as compared with 21 days for the untreated mice. Treatment with two cycles of 0.37 MBq of 212Pb–TCMC–trastuzumab with two doses of GEM achieved the greatest therapeutic efficacy with a median survival of 196.5 days.
Milenic et al. have also evaluated the efficacy of the combination treatment of paclitaxel with 212Pb–TCMC–trastuzumab [103]. Paclitaxel was given 24 h prior to, concurrent with, or 24 h post-administration of 212Pb–TCMC–trastuzumab to study the optimal order for administration. Enhanced therapeutic efficacy was found in the group receiving 600 μg of paclitaxel administered 24 h before the RIT. The median survival increased from 44 to 171 days compared with the group that received 212Pb–TCMC–trastuzumab alone.
The strategy of antibody pre-targeting has been developed to address the limitations of antibodies in the delivery of radiation to tumor [104, 105]. Su et al. evaluated the potential of antibody pretargeting therapy with 212Pb as an in vivo generator of 212Bi for α-particle radiotherapy [106]. In this study, NR-LU-10 antibody–streptavidin conjugate was administrated first followed 20–24 h later by the N-acetyl-galatos-amine-biotin clearing agent. Thereafter, 212Pb/212Bi-DOTA-biotin was injected 6 h after. Their results demonstrated that the tumor uptake of DOTA-biotin labeled with 212Pb and 212Bi was higher than 25% ID/g and remained at that level for up to 24 h p.i while the non-targeted radioactivity was rapidly cleared via the renal excretion route. Consequently, biodistribution data showed the high kidney uptake at an early time point (14.5% ID/g at 2 h p.i.).
Radiolabeled peptides have also been evaluated as potential therapeutic agents for TAT during the last decade. The advantages of using radiolabeled peptides over antibodies are simple chemical synthesis, easy labeling, fast uptake, rapid clearance, uniform distribution into tissues, and a lower potential of immunogenicity; however, that same rapid clearance can also be a source of renal toxicity as well as an economic drain of an expensive radionuclide. Due to their properties, some peptides, RGD, octreotide, α-melanocyte-stimulating hormone analogues, and bombesin derivatives, are feasible for targeted α-therapy [107]. Miao et al. investigated the therapeutic efficacy of the melanoma targeting rhenium-cyclized peptide [Re(Arg11)CCMSH] radiolabeled with the 212Pb/212Bi in vivo generator on B16/F1 murine melanoma bearing C57 mice [108]. In this study, treatment with 50, 100, and 200 μCi of 212Pb[DOTA]-Re(Arg11)CCMSH significantly decreased tumor growth rates and extended the mean survival times to 22, 28, and 49.8 days, respectively, as compared with 14.6 days of the untreated control group. 212Pb[DOTA]-Re (Arg11)CCMSH exhibited a rapid tumor uptake and prolonged retention with rapid clearance. Consequently, only minimal amounts of measurable 212Bi were released.
Clinical studies
Due to the Brechbiel group’s pre-clinical data of 212Pb–TCMC–trastuzumab [101-103], the first phase I clinical trial employing 212Pb has been sponsored by Areva Med LLC. This phase I trial, opened at the University of Alabama, Birmingham in 2011, is designed to determine the toxicity profile of 212Pb–TCMC–trastuzumab, its dose-limiting toxicities, and its anti-tumor ability in patients with HER-2 positive intraperitoneal cancers, specifically in the area of ovarian cancer. The number of participants with adverse events after treatment of 212Pb–TCMC–trastuzumab will be measured as an indicator of dose-limiting toxicity. The human immune response against 212Pb–TCMC–trastuzumab via i.p. infusion will also be characterized. Anti-tumor effects will be monitored by physical examination, radiographic imaging, and tumor marker studies. Furthermore, the plasma pharmacokinetics and excretion mechanism of radioactivity from the peritoneal cavity will be determined by γ-camera imaging.
223Ra and 227Th
Properties, chemistry, and labeling
Radium-223 can be produced from a 227Ac generator [109] and is also available from uranium mill tailings in large quantities [110]. Radium-223 has a half-life of 11.4 days and emits four α-particles and two β−-particles in its decay pathway to stable 207Pb (Fig. 6). There are advantages and disadvantages with these four α-particles. From a therapeutic point of view, having four α-particles available to provide a therapy dose is an advantage. On the other hand, they may be a disadvantage from the radiolabeling and toxicity point of view For example, 219Rn, the gaseous decay product daughter of 223Ra, can redistribute in the body and damage the non-target tissues, particularly from the subsequent decays of that radionuclide. Thus, development of a suitable ligand for in vivo sequestration of radium remains an area of opportunity since those ligands currently in use have simply been adopted from other applications or for chelating other radionuclides. Even though various chelating agents have been evaluated, development of Ra(II) chelation chemistry for in vivo therapy applications has been held back considerably due to the lack of stable isotopes with which the chemistry can be evaluated.
Fig. 6.
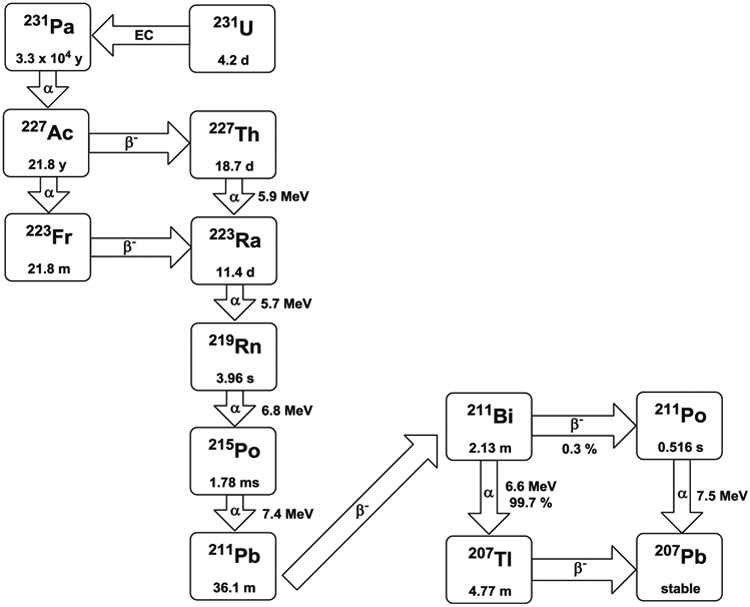
Decay scheme of 223Ra and 227Th
223Ra has been known for a potential candidate for delivery of radiation to cancer cell on bone surface due to its propensity for bone and has been extensively investigated in pre-clinical studies. As a consequence, Alpharadin (223Ra chloride) was developed by the Norwegian company Algeta ASA partnered with Bayer Schering Pharma AG and has a natural affinity for bone metastases with the potent and localized tumor cell killing ability of α-particle emission, which leads to minimal damage of non-targeted tissues. Currently, Alpharadin is in several phase II and III clinical trials as a potential α-radiation treatment for patients with bone metastases associated with cancers such as prostate and breast [111-113].
Thorium-227 (Fig. 6) has a half-life of 18.7 days and can be obtained continuously from 227Ac, which in turn can be generated by thermal neutron irradiation of 226Ra [114]. Due to a long half-life, 227Th may be used to radiolabel, administrate, and target the tumor before a significant amount of 223Ra is generated. Even though a considerable amount of 223Ra is accumulated in the bone, this may not cause bone marrow toxicity due to the short range of the α-particle [115]. Therefore, there may be an opportunity for use of this radionuclide as a therapeutic with reasonable toxicity incurred. Furthermore, the low γ-components greatly reduce the necessity for patient shielding.
The therapeutic candidate, 227Th, was successfully conjugated to two mAbs, rituximab and trastuzumab using the bifunctional chelator, p-SCN-Bn-DOTA by Dahle and co-workers [116, 117]. The immunoreactivity, serum and in vivo stability, antigen-binding ability, and the effect on in vitro cell growth as well as in vivo biodistribution were then explored. Overall, 227Th-labeled radioimmunoconjugates demonstrated a relevant stability in serum and in vivo, with a significant antigen-dependent inhibition of cell growth. The overall radiolabeling yield was 17% and the immunoreactive fraction of 227Th–DOTA–rituximab was 56–65%.
Pre-clinical studies
Norwegian Radium Hospital in Norway is the leading center for targeted therapy using 227Th. As mentioned above, the Dahle group has developed the novel low-dose rate α-emitting radionuclide 227Th on rituximab conjugated to DOTA, and tested the efficacy in mice bearing Raji lymphoma xenografts [118]. Treatment with 200 to 1,000 kBq/kg demonstrated delay in tumor growth and prolonged mean survival as compared with the control without any serious toxicity. A significant increase in growth delay with a dose of 1,000 kBq/kg was achieved compared to 200 kBq/kg (from 40 days to 17 days), whereas the median survival showed an inverse result (from 75 days to 119 days). Overall, the low-dose rate strategy using 227Th seems to be feasible against macroscopic tumor and single tumor cells.
The relative biologic effects (RBE) of α-radiation from 227Th–rituximab have been also investigated by Dahle et al. [119]. In this study, Zevalin and external beam radiation were used for comparison purposes. Tumor growth was measured two to three times a week after injection of 227Th–rixuximab, Zevalin, or external beam radiation. For the first week, the absorbed radiation dose rate in tumor reached 0.1 Gy/day and then gradually decreased to 0.03 Gy/day at 21 days. With Zevalin, the maximum dose rate (0.2 Gy/day) was obtained 6 h p.i. which then decreased to 0.01 Gy/day after 7 days. In terms of RBE, treatment with 227Th–rituximab was 2.5 to 7.2 times more effective in inhibiting tumor growth than external beam radiation while the Zevalin treatment was 1 to 1.3 times more effective external beam radiation.
227Th–rituximab has been investigated for long-term radiotoxicity to evaluate possible side effects [120]. Various doses, saline, cold rituximab, or 50, 200, and 1,000 kBq/kg 227Th–rituximab, were injected into tumor-bearing animals and observed for 1 year. Mice with Raji lymphoma xenografts were included in this study as well. Mice treated with 1,000 kBq/kg experienced weight loss as well as a decrease in WBC and platelet count. Therefore, the no-observed-adverse-effect level (NOAEL) was 200 kBq/kg. The maximum tolerated activity was 600 to 1,000 kBq/kg, and the maximum tolerated dose for bone marrow was 2.1 to 3.5 Gy. Based on these results, the dose-limiting toxicity was determined to be bone marrow suppression.
Heyerdahl et al. evaluated the cytotoxicity of low-dose rate 227Th–DOTA–trastuzumab for treating HER-2 positive breast and ovarian cancer [121]. In this study, three HER-2 positive cell lines were investigated, the breast cancer cell lines BT-474 and SKBR-3 and the ovarian cancer cell line SKOV-3. MCF-7 (HER-2 negative breast cancer cell line) was included as a control. SKBR-3 cells received 50% of the mean absorbed radiation dose from internalized activity and 50% from cell surface bound activity, whereas BT-474 and SKOV-3 cells got 75% of the radiation dose from internalized activity and 25% from cell surface bound activity. BT-474 cells treated with 2.5 kBq/ml 227Th–trastuzumab showed a mean absorbed radiation dose of 2 to 2.5 Gy. Cell growth inhibition and apoptosis were induced in all cell lines at the clinically relevant activity concentration. The cytotoxic effect was higher than that of external beam radiation (relative biological effectiveness=1.2).
Clinical studies
As noted above, Alpharadin (223Ra chloride) is currently in several phase II and III clinical trials as a potential α-radiation treatment agent and has also been evaluated in two open-label phase I trials and three double-blind phase II trials for castration-resistant prostate cancer (CRPC) and bone metastases. Based on trial data (37 patients for phase I and 255 patients for phase II), Parker et al. released the hematologic and safety profile [122]. Out of 292 patients treated with 233Ra, less than 1% experienced common toxicity criteria (CTC) grade 4 hematologic toxicity, 4% showed grade 3 anemia, and less than 3% had grade 3 toxicity for platelets, neutrophils, or WBC. Mild reversible neutropenia was found after repeated 223Ra treatments (25, 50, 80 kBq/kg given every 4 or 6 weeks). There was no sign of renal or hepatic toxicity. More adverse events and serious adverse events were observed in the placebo group than in the 223Ra-treated group in phase II trials. In the phase I trials, there was no dose-limiting toxicity. Consistent improvement in disease-related biomarkers and pain, and a highly favorable safety profile was observed. In one trial, the median overall survival increased by 4.5 months with 223Ra treatment as compared with placebo group.
Prospects and conclusions
Compared with β−-emitters, α-particle emitters have a shorter range and higher LET. Due to these properties, the majority of pre-clinical and clinical trials have demonstrated that α-emitters are ideal for the treatment of smaller tumor burdens, micrometastatic disease, and disseminated disease where in fact the α-emitters could be efficiently delivered. As such, the rational matching of disease presentation with realistic accessibility and delivery is a key criterion to their successful use in a clinical setting. Even though α-particle emitters have favorable properties, the development of TAT has been limited by high costs, unresolved chemistry, and limited availability of the radionuclides. To overcome these limitations, new sources and better production methods of medical grade radionuclides are issues that continue to remain unresolved. Current supplies for some α-emitters, especially for those radionuclides associated with the 225Ac and 224Ra decay pathways, remain limited by naturally isolated by-products from weapons development within the USA with the parental feedstock being slated for disposal. Currently, only two sources are available for “clinical grade” 225Ac, Oak Ridge National Laboratory in Oak Ridge, TN, USA, and the Institute for Transuranium Elements in Karlsruhe, Germany. Production of 211At is also hampered by production limits both in amounts and feasible locations due to cyclotron energy constraints. Besides the immediate production limits, assembly and transport of generators of sufficient activity amounts to support multiple clinical trials really have yet to be produced at adequate levels. Furthermore, the costs of these radionuclides have continued to increase over the past decade, which poses a serious obstacle that challenges both those researchers who currently work on or are entering this area of research.
There are still remaining issues associated with “nanogenerators” even though the strategy of the long half-life parental radionuclides serving as in vivo generators for the delivery and production of desired therapeutic daughter radionuclides effectively extends their respective short half-life radionuclides. Attempts to sequester daughter radionuclides have led to the creation of larger and larger constructs. As a consequence of such strategies, this comes at a cost associated with limits or compromises to the use of efficient targeting systems combined with then limitations imposed on both penetration and extravasation into tumor. In addition, the in vivo stability and metabolism of these constructs is not altogether well defined, and radiologic side effects are observed which are likely to be mediated by release of daughter radionuclides from the chelate and construct.
From a chemistry point of view, there are still challenges remaining regarding the coordination and conjugation chemistry for the multiple decay pathway radionuclides. More efficient conjugation and radiolabeling methods need to be developed that lead to stable products with higher specific activities to maximize therapeutic efficacy. Better pharmacokinetics and dosimetry studies are also required that actually address the cellular micro-dosimetry aspect of targeted α-emitters since their targets exist at the individual cell level for therapy.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We also thank Diane Milenic for assistance in assembling the manuscript.
Footnotes
Conflicts of interest None
References
- 1.Sgouros G, Ballangrud AM, Jurcic JG, McDevitt MR, Humm JL, Erdi YE, Mehta BM, Finn RD, Larson SM, Scheinberg DA. Pharmacokinetics and dosimetry of an α-particle emitter antibody: 213Bi-HuM195 (anti-CD33) in patients with leukemia. J Nucl Med. 1999;40:1935–46. [PubMed] [Google Scholar]
- 2.Zalutsky MR, Bigner DD. Radioimmunotherpy with α-particle emitting radioimmunoconjugates. Acta Oncol. 1996;35:373–9. [DOI] [PubMed] [Google Scholar]
- 3.Zalutsky MR, Schuster JM, Garg PK, Archer GE, DeWhirst MW, Binger DD. Two approaches for enhancing radioimmunotherapy: alpha emitters and hyperthermia. Recent Results Cancer Res. 1996;141:101–22. [DOI] [PubMed] [Google Scholar]
- 4.Nikula TK, McDevitt MR, Finn RD, Wu CC, Kozak RW, Garmestani K, Brechbiel MW, Curcio ML, Pippin CG, Tiffany-Jones L, Geerlings MW, Apostolidis C, Molinet R, Gansow OA, Sheinberg DA. Alpha-emitting bismuth cyclohexylbenzyl DTPA constructs of recombinant humanized anti-CD33 antibodies: pharmacokinetics, bioactivity, toxicity and chemistry. J Nucl Med. 1999;40:166–76. [PubMed] [Google Scholar]
- 5.Raju MR, Eisen Y, Carpenter S, Inkret WC. Radiobiology of alpha-particles. 3. Cell inactivation by alpha-particle traversals of the cell-nucleus. Radiat Res. 1991;128:204–9. [PubMed] [Google Scholar]
- 6.Vaidyanathan G, Zalutsky MR. Targeted therapy using alpha emitters. Phys Med Biol. 1996;41:1915–31. [DOI] [PubMed] [Google Scholar]
- 7.McDevitt MR, Sgouros G, Finn RD, Humm JL, Jurcic JG, Larson SM, Scheinberg DA. Raioimmunotherapy with alpha-emitting nuclides. Eur J Nucl Med. 1998;25:1341–51. [DOI] [PubMed] [Google Scholar]
- 8.Zalutsky MR, Vaidyanathan G. Astatine-211-labeled radiotherapeutics: an emerging approach to targeted alpha-particle radiotherapy. Curr Pharm Des. 2000;6:1433–55. [DOI] [PubMed] [Google Scholar]
- 9.Imam SK. Advancements in cancer therapy with alpha-emitters: a review. Int J Radiat Oncol Biol Phys. 2001;51:271–8. [DOI] [PubMed] [Google Scholar]
- 10.Bolch WE. α-particle emitters in radioimmunotherapy: new and welcome challenges to medical internal dosimetry. J Nucl Med. 2001;42:1222–4. [PubMed] [Google Scholar]
- 11.Zalutsky MR, Pozzi OR. Radioimmunotherapy with α-particle emitting radionuclides. Q J Nucl Mol Imaging. 2004;48:289–96. [PubMed] [Google Scholar]
- 12.Couturier O, Supiot S, Degraef-Mougin M, Faivre-Chauvet A, Carlier T, Chatel J-F, Davodeau F, Cherel M. Cancer radioimmunotherapy with alpha-emitting nuclides. Eur J Nucl Med Mol Imaging. 2005;32:601–14. [DOI] [PubMed] [Google Scholar]
- 13.Mulford DA, Scheinberg DA, Jurcic JG. The promise of targeted α-particle therapy. J Nucl Med. 2005;46:199S–204S. [PubMed] [Google Scholar]
- 14.Welch MJ. Potential and pitfalls of therapy with α-particles. J Nucl Med. 2005;46:1254–5. [PubMed] [Google Scholar]
- 15.Cherel M, Davodeau F, Kraeber-Bodere F, Chatal JF. Current status and perspectives in alpha radioimmunotherapy. Q J Nucl Med Mol Imaging. 2006;50:322–9. [PubMed] [Google Scholar]
- 16.Brechbiel MW. Targeted α-therapy: past, present, future? Dalton Trans. 2007;21:4918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sgouros G Alpha-particles for targeted therapy. Adv Drug Deliv Rev. 2008;60:1402–6. [DOI] [PubMed] [Google Scholar]
- 18.Miederer M, Scheinberg DA, McDevitt MR. Realizing the potential of the Actinium-225 radionuclide generator in targeted alpha particle therapy applications. Adv Drug Deliv Rev. 2008;60:1371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbet J, Cherel M, Chatal J-F. Alpha particles more promising than toxins? Eur J Nucl Med Mol Imaging. 2010;37:849–50. [DOI] [PubMed] [Google Scholar]
- 20.Barbet J, Chatal J-F. The best radionuclide for radioimmunotherapy of small tumors: beta- or alpha-emitter? Eur J Nucl Med Mol Imaging. 2011;38:271–3. [DOI] [PubMed] [Google Scholar]
- 21.Yong K, Brechbiel MW. Towards translation of 212Pb as a clinical therapeutic: getting the lead in! Dalton Trans. 2011;40:6068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adloff JP. The centenary of a controversial discovery: actinium. Radiochim Acta. 2000;88:123–7. [Google Scholar]
- 23.Mirzadeh S Generator-produced alpha-emitters. Appl Radiat Isot. 2000;49:383–95. [Google Scholar]
- 24.Lambrecht RM, Tomiyoshi K, Sekine T. Radionuclide generators. Radiochim Acta. 1997;77:103–23. [Google Scholar]
- 25.Davis IA, Glowienka KA, Boll RA, Deal KA, Brechbiel MW, Stabin M, Bochsler PN, Mirzadeh S, Kennel SJ. Comparison of 225Actinium chelates: tissue distribution and radiotoxicity. Nucl Med Biol. 1999;26:581–9. [DOI] [PubMed] [Google Scholar]
- 26.Chappell LL, Deal KA, Dadachova E, Brechbiel MW. Synthesis, conjugation and radiolabeling of a novel bifunctional chelating agent for Ac-225 radioimmunotherapy application. Bioconjug Chem. 2000;11:510–9. [DOI] [PubMed] [Google Scholar]
- 27.McDevitt MR, Ma DS, Lai LT, Simon J, Borchardt P, Frank RK, Wu K, Pellegrini V, Crucio MJ, Miederer M, Bander NH, Scheinberg DA. Tumor therapy with targeted atomic nanogenerators. Science. 2001;294:1537–40. [DOI] [PubMed] [Google Scholar]
- 28.Akabani G, McLendon RE, Bigner DD, Zalutsky MR. Vascular targeted endoradiotherapy of tumors using alpha-particle-emitting compounds: theoretical analysis. Int J Radiat Oncol Biol Phys. 2002;54:1259–75. [DOI] [PubMed] [Google Scholar]
- 29.Kennel SJ, Mirzadeh S, Eckelman WC, Waldmann TA, Garmestani K, Yordanov AT, Stabin MG, Brechbiel MW. Vascular-targeted radioimmunotherapy with the alpha-particle emitter 211At. Radiat Res. 2002;157:633–41. [DOI] [PubMed] [Google Scholar]
- 30.Jaggi JS, Henke E, Seshan SV, Kappel BJ, Chattopadhyay D, May C, McDevitt MR, Nolan D, Mittal V, Benezra R, Scheinberg DA. Selective alpha-particle mediated depletion of tumor vasculature with vascular normalization. PLoS One. 2007;3:e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruggiero A, Villa CH, Holland JP, Sprinkle SR, May C, Lewis JS, Scheinberg DA, McDevitt MR. Imaging and treating tumor vasculature with targeted radiolabeled carbon nanotubes. Int J Nanomed. 2010;5:783–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sofou S, Thomas JL, Lin HY, McDevitt MR, Scheinberg DA, Sgouros G. Engineered liposomes for potential α-particle therapy of metastatic cancer. J Nucl Med. 2004;45:253–60. [PubMed] [Google Scholar]
- 33.Sofou S, Kappel BJ, Jaggi JS, McDevitt MR, Scheinberg DA, Sgouros G. Enhanced retention of the α-particle-emitting daughters of Actinium-225 by liposome carriers. Bioconjug Chem. 2007;18:2061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song H, Hobbs RF, Vajravelu R, Huso DL, Esaias C, Apostolidis C, Morgenstern A, Sgouros G. Radioimmunotherapy of breast cancer metastases with α-particle emitter 225Ac: comparing efficacy with 213Bi and 90Y. Cancer Res. 2009;69:8941–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woodward J, Kennel SJ, Stuckey A, Osborne D, Wall J, Rondinone AJ, Standaert RF, Mirzadeh S. LaPO4 nanoparticles doped with Actinium-225 that partially sequester daughter radionuclides. Bioconjug Chem. 2011;22:766–76. [DOI] [PubMed] [Google Scholar]
- 36.Miederer M, McDevitt MR, Sgouros G, Kramer K, Cheung NKV, Scheinberg DA. Pharmacokinetics, dosimetry, and toxicity of the targetable atomic generator, 225Ac-HuM195, in nonhuman primates. J Nucl Med. 2004;45:129–37. [PubMed] [Google Scholar]
- 37.ClinicalTrials.gov. Targeted atomic nano-generators (Actinium-225-labeled humanized anti-CD33 monoclonal antibody HuM195) in patients with advanced myeloid malignancies. 2011. http://clinicaltrials.gov/show/NCT00672165. June 9 2011.
- 38.Durbin PW, Asling CW, Johnston ME, Parrott MW, Jeung N, Williams MH, Hamilton JG. The induction of tumors in the rat by astatine-211. Radiat Res. 1958;9:378–97. [PubMed] [Google Scholar]
- 39.Zalutsky MR, Narula AS. Astatination of proteins using an N-succimidyl tri-n-butylstannyl benzoate intermediate. Appl Radiat Isot. 1988;39:227–32. [DOI] [PubMed] [Google Scholar]
- 40.Zalutsky MR, Garg PK, Friedman HS, Bigner DD. Labeling monoclonal antibodies and F(ab′)2 fragments with the alpha particle emitting nuclide atstatine-211: preservation of immunoreactivity with in vivo localizing capacity. Proc Natl Acad Sci U S A 1989;86:7149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown I Astatine—its organonuclear chemistry and biomedical applications. Adv Inorg Chem. 1987;31:43–88. [Google Scholar]
- 42.Visser GWM. Inorganic astatine chemistry 2. The chameleon behavior and electrophilicity of At-species. Radiochim Acta. 1989;47:97–103. [Google Scholar]
- 43.Yordanov AT, Pozzi O, Carlin S, Akabani G, Wieland B, Zalutsky MR. Wet harvesting of no-carrier-added At-211 from an irradiated Bi-209 target for radiopharmaceutical applications. J Radioanal Nucl Chem. 2004;262:593–9. [Google Scholar]
- 44.Zalutsky MR, Zhao XG, Alston KL, Bigner D. High-level production of alpha-particle emitting 211At and preparation of 211At-labeled antibodies for clinical use. J Nucl Med. 2001;42:1508–15. [PubMed] [Google Scholar]
- 45.Zalutsky MR, Reardon DA, Pozzi OR, Vaidyanathan G, Bigner DD. Targeted α-particle radiotherapy with 211At-labeled monoclonal antibodies. Nucl Med Biol. 2007;34:779–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boskovitz A, McLendon RE, Okamura T, Sampson JH, Bigner DD, Zalutsky MR. Treatment of HER2-positive breast carcinomatous meningitis with intrathecal administration of α-emitting 211At-labeled trastuzumab. Nucl Med Biol. 2009;36:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elgqvist J, Andersson H, Bäck T, Hultborn R, Jensen H, Karlsson B Lindegren S, Palm S, Warnhammar E, Jacobsson L. Therapeutic efficacy and tumor dose estimations in radioimmunotherapy of intraperitoneally growing OVCAR-3 cells in nude mice with 211At-labeled monoclonal antibody MX35. J Nucl Med. 2005;46:1907–15. [PubMed] [Google Scholar]
- 48.Elgqvist J, Andersson H, Bernhardt P, Bäck T, Claesson I, Hultborn R, Jensen H, Johansson BR, Lindergren S, Olsson M, Palm S, Warnhammar E, Jacobsson L. Administered activity and metastatic cure probability during radioimmunotherapy of ovarian cancer in nude mice with 211At-MX35 F(ab′)2. Int J Radiat Oncol Biol Phys. 2006;66:1228–37. [DOI] [PubMed] [Google Scholar]
- 49.Elgqvist J, Andersson H, Bäck T, Claesson I, Hultborn R, Jensen H, Johansson BR, Lindergren S, Olsson M, Palm S, Warnhammar E, Jacobsson L. α-radioimmunotherapy of intraperitoneally growing OVCAR-3 tumors of variable dimensions: outcome related to measured tumor size and mean absorbed dose. J Nucl Med. 2006;47:1342–50. [PubMed] [Google Scholar]
- 50.Palm S, Back T, Claesson I, Danielsson A, Elgqvist J, Frost S, Hultborn R, Jensen H, Lindegren S, Jacobsson L. Therapeutic efficacy of astatine-211-labeled trastuzumab on radioresistant SKOV-3 tumors in nude mice. Int J Radiat Oncol Biol Phys. 2007;69:572–9. [DOI] [PubMed] [Google Scholar]
- 51.Wilbur DS, Thakar MS, Hamlin DK, Santos EB, Chyan MK, Nakamae H, Pagel JM, Press OW, Sanmaier BM. Reagent for astatination of biomolecules. 4. Comparison of maleimido-closo-decarborate(2-) and meta-[211At]Astatobenzoate conjugates for labeling anti-CD45 antibodies with [211At]Astatine. Bioconjug Chem. 2009;20:1983–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakamae H, Wilbur DS, Hamlin DK, Thakar MS, Santos EB, Fisher DR, Kenoyer AL, Pagel JM, Press OW, Storb R, Sandmaier BM. Biodistribution, myelosuppression, and toxicities in mice treated with an anti-CD45 antibody labeled with α-emitting radionuclides Bismuth-213 or Astatine-211. Cancer Res. 2009;69:2408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Almqvist Y, Orlova A, Sjostrom A, Jensen HJ, Lundqvist H, Sundin A, Tolmachev V. In vitro characterization of 211At-labeled antibody A33—a potential therapeutic agent against metastatic colorectal carcinoma. Cancer Biother Radiopharm. 2005;20:514–23. [DOI] [PubMed] [Google Scholar]
- 54.Almqvist Y, Steffen AC, Lundqvist H, Jensen H, Tolmachev V, Sundin A. Biodistribution of 211At-labeled humanized monoclonal antibody A33. Cancer Biother Radiopharm. 2007;22:480–7. [DOI] [PubMed] [Google Scholar]
- 55.Nestor M, Persson M, van Dongen GA, Jensen HJ, Lundqvist H, Anniko M, Tolmachev V. In vitro evaluation of the astatinated chimeric monoclonal antibody U36, a potential candidate for treatment of head and neck squamous cell carcinoma. Eur J Nucl Med Mol Imaging. 2005;32:1296–304. [DOI] [PubMed] [Google Scholar]
- 56.Cheng J, Ekberg T, Engstrom M, Nestor M, Jensen HJ, Tolmachev V, Anniko M. Radioimmunotherapy with astatine-211 using chimeric monoclonal antibody U36 in head and neck squamous cell carcinoma. Laryngoscope. 2007;117:1013–8. [DOI] [PubMed] [Google Scholar]
- 57.Zhang M, Yao Z, Zhang Z, Garmestani K, Talanov VS, Plascjak PS, Yu S, Kim HS, Goldman CK, Paik CH, Brechbiel MW, Carrasquillo JA, Waldmann TA. The anti-CD25 monoclonal antibody 7G7/B6, armed with the α-emitter 211At, provided effective radioimmunotherapy for murine model of leukemia. Cancer Res. 2006;66:8227–32. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Z, Zhang M, Garmestani K, Talanov VS, Plascjak PS, Beck B, Goldman C, Brechbiel MW, Waldmann TA. Effective treatment of a murine model of adult T-cell leukemia using 211At-7G7/B6 and its combination with unmodified anti-Tac (daclizumab) directed toward CD25. Blood. 2006;108:1007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bourgeois M, Guerard F, Alliot C, Mougin-Degraef M, Rajerison H, Remaud-Le Saec P, Gestin JF, Davodeau F, Cherel M, Barbet J, Faivre-Chauvet A. Feasibility of the radioastatination of a monoclonal antibody with astatine-211 purified by wet extraction. J Label Compd Radiopharm. 2008;51:379–83. [DOI] [PubMed] [Google Scholar]
- 60.Zalutsky MR, McLendon RE, Garg PK, Archer GE, Schuster JM, Bigner DD. Radioimmunotherapy of neoplastic meningitis in rats using an α-particle emitting immunoconjugate. Cancer Res. 1994;54:4719–25. [PubMed] [Google Scholar]
- 61.Zalutsky MR, Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, McLendon RE, Wong TZ, Bigner DD. Clinical experience with α-particle emitting 211At: treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med. 2008;49:30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andersson H, Cederkrantz E, Back T, Divgi C, Elgqvist J, Himmelman J, Horvath G, Jacbsson L, Jensen H, Lindegren S, Palm S, Hultborn R. Intraperitoneal α-particle radioimmunotherapy of ovarian cancer patients: pharmacokinetics and dosimetry of 211At-MX35 F(ab′)2—a phase I study. J Nucl Med. 2009;50:1153–60. [DOI] [PubMed] [Google Scholar]
- 63.Atcher RW, Friedman AM, Hines JJ. An improved generator for the production of 212Pb and 212Bi from 224Ra. Int J Radiat Appl Instrum A. 1988;39:283–6. [DOI] [PubMed] [Google Scholar]
- 64.Gansow OA, Atcher RW, Link DC, Friedman AM, Seevers RH, Anderson W, Scheinberg DA, Strand M. Generator-produced Bi-212: chelated to chemically modified monoclonal antibody for use in radiotherapy. Am Chem Soc Symp Ser. 1984;241:215–27. [Google Scholar]
- 65.Brechbiel MW, Gansow OA. Synthesis of C-functionalized trans-cyclohexyldiethylenetriaminepenta-acetic acids for labeling of monoclonal antibodies with the bismuth-212 α-particle emitter. J Chem Soc Perkin Trans. 1992;1:1173–8. [Google Scholar]
- 66.Wu C, Kobayashi H, Sun B, Yoo TM, Paik CH, Gansow OA, Carrasquillo JA, Pastan I, Brechbiel MW. Stereochemical influence on the stability of radio-metal complexes in vivo. Synthesis and evaluation of the four stereoisomers of 2-(p-nitrobenzyl)-trans-CyDTPA. Bioorg Med Chem. 1997;5:1925–34. [DOI] [PubMed] [Google Scholar]
- 67.Ruegg CL, Anderson-Berg WT, Brechbiel MW, Mirzadeh S, Gansow OA, Strand M. Improved in vivo stability and tumor targeting of bismuth-labeled antibody. Cancer Res. 1990;50:4221–6. [PubMed] [Google Scholar]
- 68.Junghans RP, Dobbs D, Brechbiel MW, Mirzadeh S, Raubitschek AA, Gansow OA, Waldmann TA. Pharmacokinetics and bioactivity of 1, 4, 7, 10-tetraazacyclododecane N, N’N”, N’”-tetraacetic acid (DOTA)-bismuth conjugated anti-Tac antibody for alpha-emitter (212Bi) therapy. Cancer Res. 1993;53:5683–9. [PubMed] [Google Scholar]
- 69.Boll RA, Malkemus D, Mirzadeh S. Production of actinium-225 for alpha particle mediated radioimmunotherapy. Appl Radiat Isot. 2005;62:667–79. [DOI] [PubMed] [Google Scholar]
- 70.Boll RA, Mirzadeh S, Kennel SJ. Optimizations of radiolabeling of immunoprotein with Bi-213. Radiochim Acta. 1997;79:145–9. [Google Scholar]
- 71.McDevitt MR, Nikula TN, Finn RD, Curcio MJ, Gansow OA, Geerlings MW, Larson SM, Scheinberg DA. Bismuth labeled antibodies for therapy of leukemias, lymphomas, and carcinomas: preclinical studies. Tumor Target. 1996;2:182. [Google Scholar]
- 72.Finn RD, McDevitt MR, Scheinberg DA. Refinements and improvements for Bi-213 production and use as a targeted therapeutic radiopharmaceutical. J Label Compd Radiopharm. 1997;40:293. [Google Scholar]
- 73.McDevitt MR, Finn RD, Sgouros G, Sheinberg DA. An Ac-225/Bi-213 generator system for therapeutic clinical applications: construction and operation. Appl Radiat Isot. 1999;50:895–904. [DOI] [PubMed] [Google Scholar]
- 74.Ma DH, McDevitt MR, Finn RD, Scheinberg DA. Breakthrough of Ac-225 and its radionuclide daughters from an 225Ac/213Bi generator: development of new methods, quantitative characterization, and implications for clinical use. Appl Radiat Isot. 2001;55:667–78. [DOI] [PubMed] [Google Scholar]
- 75.Pippin CG, Gansow OA, Brechbiel MW, Koch L, Molinet R, van Geel J, Apostolidis C, Geerlings MW, Scheinberg DA. Recovery of Bi-213 from an Ac-225 cow: application to the radiolabeling of antibodies with Bi-213 In: Emran AM, editor. Chemists’ views of imaging centers. New York: Plenum; 1995. p. 318–22. [Google Scholar]
- 76.Ma DH, McDevitt MR, Finn RD, Scheinberg DA. Rapid preparation of short-lived alpha particle emitting radioimmunopharmaceuticals. Appl Radiat Isot. 2001;55:463–70. [DOI] [PubMed] [Google Scholar]
- 77.Rizvi SMA, Qu CF, Song YJ, Raja C, Allen BJ. In vivo studies of pharmacokinetics and efficacy of bismuth-213 labeled antimelanoma monoclonal antibody 9.2.27. Cancer Biol Ther. 2005;4:763–8. [DOI] [PubMed] [Google Scholar]
- 78.Song YJ, Qu CF, Rizvi SMA, Li Y, Robertson G, Raja C, Morgenstern A, Apostolidis C, Perkins AC, Allen BJ. Cytotoxicity of PAI2, C595 and herceptin vectors labeled with the alpha-emitting radioisotope bismuth-213 for ovarian cancer cell monolayers and clusters. Cancer Lett. 2006;234:176–83. [DOI] [PubMed] [Google Scholar]
- 79.Rizvi SMA, Li Y, Song EYJ, Qu CF, Raja C, Morgenstern A, Apostolidis C, Allen BJ. Preclinical studies of bismuth-213 labeled plasminogen activator inhibitor type 2 (PAI2) in a prostate cancer nude mouse xenograft model. Cancer Biol Ther. 2006;5:386–93. [DOI] [PubMed] [Google Scholar]
- 80.Song EY, Qu CF, Rizvi SMA, Raja C, Beretov J, Morgenstern A, Apostolidis C, Bruchertseifer F, Perkins A, Allen BJ. Bismuth-213 radioimmunotherapy with C595 anti-MUC1 monoclonal antibody in an ovarian cancer ascites model. Cancer Biol Ther. 2008;7:76–86. [DOI] [PubMed] [Google Scholar]
- 81.Rizvi SMA, Song EY, Raja C, Beretov J, Morgenstern A, Apostolidis C, Russel PJ, Kearsley JH, Allen BJ. Preparation and testing of bevacizumab radioimmunoconjugates bismuth-213 and bismuth205/bismuth-206. Cancer Biol Ther. 2008;7:1547–54. [DOI] [PubMed] [Google Scholar]
- 82.Song H, Shahverdi K, Huso DL, Esaias CK, Fox J, Liedy A, Zhang Z, Reilly RT, Apostolidis C, Morgenstern A, Sgouros G. 213Bi (α-emitter)-antibody targeting of breast cancer metastases in the neu-N transgenic mouse model. Cancer Res. 2008;68:3873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lingappa M, Song H, Thompson S, Bruchertseifer F, Morgenstern A, Sgouros G. Immunoliposomal delivery of 213Bi for α-emitter targeting of metastatic breast cancer. Cancer Res. 2010;70:6815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park SI, Shenoi J, Pagel JM, Hamlin DK, Wilbur DS, Orgun N, Kenoyer AL, Frayo S, Axtman A, Back R, Lin Y, Fisher DR, Gopal AK, Green DJ, Press OW. Conventional and pretargeted radioimmunotherapy using bismuth-213 to target and treat non-Hodgkin lymphomas expressing CD20: a preclinical model toward optimal consolidation therapy to eradicated minimal residual disease. Blood. 2011;116:4231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bloechl S, Beck R, Seidl C, Morgenstern A, Schwaiger M, Senekowitsch-Schmidtke R. Fractionated locoregional low-dose radioimmunotherapy improves survival in a mouse model of diffuse-type gastric cancer using a 213Bi-conjugated monoclonal antibody. Clin Cancer Res. 2005;11:7070s–4s. [DOI] [PubMed] [Google Scholar]
- 86.Milenic DM, Brady ED, Garmestani K, Albert PS, Abdulla A, Brechbiel MW. Improved efficacy of α-particle-targeted radiation therapy. Cancer. 2010;116:1059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wild D, Frischknecht M, Zhang H, Morgenstern A, Bruchertseifer F, Boisclair J, Provencher-Bolliger A, Reubi JC, Maecke HR. Alpha- versus beta-particle radiopeptide therapy in a human prostate cancer model (213Bi-DOTA-PESIN and 213Bi-AMBA versus 177Lu-DOTA-PESIN). Cancer Res. 2011;71:1009–18. [DOI] [PubMed] [Google Scholar]
- 88.Heeger SW, Moldenhauer G, Egerer G, Wesch H, Eisenmenger A, Nikula T, Apostolidis C, Janssens W, Brechbiel MW, Haberkorn U, Ho AD, Haas R. Treatment of B-lineage non-Hodgkin’s lymphoma using 213Bi-labeled anti-CD20-CHX-A″-DTPA anti body conjugate. J Nucl Med. 2002;43:116P. [Google Scholar]
- 89.Heeger S, Moldenhauer G, Egerer G, Wesch H, Martin S, Nikula T, Aposstolidis C, Brechbiel MW, Ho AD, Haas R. Alpha-radioimmunotherapy of B-lineage non-Hodgkin’s lymphoma using 213Bi-labeled anti-CD19 and anti-CD20-CHX-A″-DTPA conjugates. ACS Meet Abstr. 2003;225:U261. [Google Scholar]
- 90.Raja C, Graham P, Rizvi SMA, Song E, Goldsmith H, Thompson J, Bosserhoff A, Morgenstern A, Apostolidis C, Kearsley J, Reisfeld R, Allen BJ. Interim analysis of toxicity and response in phase I trial of systemic targeted alpha therapy for metastatic melanoma. Cancer Biol Ther. 2007;6:846–52. [DOI] [PubMed] [Google Scholar]
- 91.Jurcic JG, Larson SM, Sgouros G, McDevitt MR, Finn RD, Divgi CR, Ballangrud AM, Hamacher KA, Ma D, Humm JL, Brechbiel MW, Molinet R, Scheinberg DA. Targeted α particle immunotherapy for myeloid leukemia. Blood. 2002;100:1233–9. [PubMed] [Google Scholar]
- 92.Rosenblat TL, McDevitt MR, Mulford DA, Pandit-Taskar N, Divgi CR, Panageas KS, Heaney ML, Chanel S, Morgenstern A, Sgouros G, Larson SM, Scheinberg DA, Jurcic JG. Sequential cytarbine and α-particle immunotherapy with bismuth-213-lintuzumab (HuM195) for acute myeloid leukemia. Clin Cancer Res. 2010;16:5303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kneifel S, Cordier D, Good S, Ionescu MCS, Ghaffari A, Hofer S, Kretzschmar M, Tolnay M, Apostolidis C, Waser B, Arnold M, Mueller-Brand J, Maecke HR, Reubi JC, Merlo A. Local targeting of malignant gliomas by the diffusible peptidic vector 1,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance P. Clin Cancer Res. 2006;12:3843–50. [DOI] [PubMed] [Google Scholar]
- 94.Cordier D, Forrer F, Bruchertseifer F, Morgenstern A, Apostolidis C, Good S, Muller-Brand J, Macke H, Reubi JC, Merlo A. Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8, Met(O2)11]-substance P: a pilot trial. Eur J Nucl Med Mol Imaging. 2010;37:1335–44. [DOI] [PubMed] [Google Scholar]
- 95.Mausner LF, Straub RE, Srivastava SC. The “in vivo” generator for radioimmunotherapy. J Label Compd Radiopharm. 1989;26:177–8. [Google Scholar]
- 96.Kumar K, Magerstadt M, Gansow OA. Lead (II) and bismuth (III) complexes of the polyazacyclo-alkane-N-acetic acids NOTA, DOTA, and TETA. J Chem Soc Chem Commun. 1989:145–6. [Google Scholar]
- 97.Gansow OA, Brechbiel MW, Pippin CG, McMurry TJ, Lambrecht R, Colcher D, Schlom J, Roselli M, Strand M, Huneke RB, Ruegg CL. Lead and bismuth complexes of functionalized DTPA ligands and of the polyazacycloalkane-N-acetic acids DOTA. Utility for radioimmunoimaging and radioimmunotherapy. Antibody Immunoconj Radiopharm. 1991;4:413–25. [Google Scholar]
- 98.Pippin CG, McMurry TJ, Brechbiel MW, McDonald R, Lambrecht R, Milenic D, Roselli M, Colcher D, Gansow OA. Lead (II) complexes of 1,4,7,10-tetraazacyclododecane-N, N’, N”, N’”-tetraacetate: solution chemistry and application to tumor localization with 203Pb labeled monoclonal antibodies. Inorg Chim Acta. 1995;239:43–51. [Google Scholar]
- 99.Chappell LL, Dadachova E, Milenic DE, Garmestani K, Wu C, Brechbiel MW. Synthesis, characterization and evaluation of a novel bifunctional chelating agent for the lead isotopes 203Pb and 212Pb. Nucl Med Biol. 2000;27:93–100. [DOI] [PubMed] [Google Scholar]
- 100.Rubel G, Wu C, Squire RA, Gansow OA, Strand M. The use of 212Pb-labeled monoclonal antibody in the treatment of murine erythroleukemia. Int J Radiat Oncol Biol Phys. 1996;34:609–16. [DOI] [PubMed] [Google Scholar]
- 101.Milenic DE, Garmestani K, Brady ED, Albert PS, Ma D, Abdulla A, Breachbiel MW. α-particle radioimmunotherapy of disseminated peritoneal disease using a 212Pb-labeled radioimmunoconjugates targeting HER2. Cancer Biother Radiopharm. 2005;20:557–68. [DOI] [PubMed] [Google Scholar]
- 102.Milenic DE, Garmestani K, Brady ED, Abdulla A, Flynn J, Brechbiel MW. Potentiation of high-LET radiation by gemcitabine: targeting HER-2 with trastuzumab to treat disseminated peritoneal disease. Clin Cancer Res. 2007;13:1926–35. [DOI] [PubMed] [Google Scholar]
- 103.Milenic DE, Garmestani K, Brady ED, Baidoo KE, Albert PS, Wong KJ, Flynn J, Brechbiel MW. Multimodality therapy: potentiation of high linear energy transfer radiation with paclitaxel for the treatment of disseminated peritoneal disease. Clin Cancer Res. 2008;14:5108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roseborough SF. Two-step immunological approaches for imaging and therapy. Q J Nucl Med. 1996;40:234–51. [PubMed] [Google Scholar]
- 105.Goodwin DA. Tumor pretargeting: almost the bottom line. J Nucl Med. 1995;36:876–9. [PubMed] [Google Scholar]
- 106.Su FM, Beaumier P, Axworthy D, Atcher R, Fritzberg A. Pretargeted radioimmunotherapy in tumored mice using an in vivo 212Pb/212Bi generator. Nucl Med Biol. 2005;32:741–7. [DOI] [PubMed] [Google Scholar]
- 107.Dijkgraaf I, Boerman OC, Oyen WJG, Corstens FHM, Gotthardt M. Development and application of peptide-based radiopharmaceuticals. Anti Cancer Agents Med Chem. 2007;7:543–51. [DOI] [PubMed] [Google Scholar]
- 108.Miao Y, Hylarides M, Fisher DR, Shelton T, Moore H, Wester DW, Fritzberg AR, Winkelmann CT, Hoffman T, Quinn TP. Melanoma therapy via peptide-targeted α-radiation. Clin Cancer Res. 2005;11:5616–21. [DOI] [PubMed] [Google Scholar]
- 109.Atcher RW, Friedman AM, Huizenga JR, Spencer RP. A radionuclide generator for the production of Pb-211 and its daughters. J Radioanal Nucl Chem. 1989;135:215–21. [Google Scholar]
- 110.Wilbur DS. Potential use of alpha emitting radionuclides in the treatment of cancer. Antibody Immunoconj Radiopharm. 1991;4:85–97. [Google Scholar]
- 111.ClinicalTrials.gov. A phase III study of Alpharadin (Radium-223) in patients with symptomatic hormone refractory prostate cancer with skeletal metastases (ALSYMPCA). 2011. http://clinicaltrials.gov/ct2/show/NCT00699751. 6 January 2011.
- 112.ClincalTrials.gov. A study of Alpharadin with docetaxel in patients with bone metastasis from castration-resistant prostate cancer (CRPC). 2010. http://clinicaltrials.gov/ct2/show/NCT01106352. 10 December 2010.
- 113.ClinicalTrials.gov. A study of Alpharadin in breast cancer patients with bone dominant disease no longer considered suitable for hormone therapy. 2011. http://clinicaltrials.gov/ct2/show/NCT01070485. 7 March 2011.
- 114.Henriksen G, Bruland OS, Larsen RH. Thorium and actinium polyphosphonate compounds as bone-seeking alpha particle emitting agents. Anticancer Res. 2004;24:101–5. [PubMed] [Google Scholar]
- 115.Larsen RH, Saxtorph H, Skydsgaard M. Radiotoxicity of the alpha-emitting bone-seeker Ra-223 injected intravenously into mice: histology, clinical chemistry and hematology. In Vivo. 2006;20:325–31. [PubMed] [Google Scholar]
- 116.Dahle J, Borrebæk MKB, Bruland ØS, Salberg G, Olsen DR, Larsen RH. Initial evaluation of 227Th-p-benzyl-DOTA-rituximab for low-dose rate α-particle radioimmunotherapy. Nucl Med Biol. 2006;33:271–9. [DOI] [PubMed] [Google Scholar]
- 117.Larsen RH, Borrebæk J, Dahle J, Melhus KB, Krogh C, Valan MH, Bruland ØS. Preparation of 227Th-labeled radioimmunoconjugates, assessment of serum stability and antigen binding ability. Cancer Biother Radiopharm. 2007;22:431–7. [DOI] [PubMed] [Google Scholar]
- 118.Dahle J, Borrebæk J, Jonasdottir TJ, Hjelmerud AK, Melhus KB, Bruland ØS, Press OW, Larsen RH. Targeted cancer therapy with a novel low-dose rate α-emitting radioimmunoconjugate. Blood. 2007;110:2049–56. [DOI] [PubMed] [Google Scholar]
- 119.Dahle J, Bruland ØS, Larsen RH. Relative biologic effects of low-dose-rate α-emitting 227Th-rituximab and β-emitting 90Y-tiuexetan-ibritumomab versus external beam X-radiation. Int J Radiat Oncol Biol Phys. 2008;72:186–92. [DOI] [PubMed] [Google Scholar]
- 120.Dahle J, Jonasdottir TJ, Heyerdahl H, Nesland JM, Borrebæk J, Hjelmerud AK, Larsen RH. Assessment of long-term radiotoxicity after treatment with the low-dose alpha-particle-emitting radioimmunoconjugate 227Th-rituximab. Eur J Nucl Med Mol Imaging. 2010;37:93–102. [DOI] [PubMed] [Google Scholar]
- 121.Heyerdahl H, Krogh C, Borrebæk J, Larsen A, Dahle J. Treatment of HER2-expressing breast cancer and ovarian cancer cells with alpha particle-emitting 227Th-trastuzumab. Int J Radiat Oncol Biol Phys. 2011;79:563–70. [DOI] [PubMed] [Google Scholar]
- 122.Parker C, Aksnes A, Haugen I, Bolstad B, Nilsson S. Radium-223 chloride, a novel, highly targeted alpha-pharmaceutical for treatment of bone metastases from castration-resistant prostate cancer (CRPC): hematologic and safety profile with repeated dosing. Ann Oncol. 2010;21 Suppl 8:viii277–8. [Google Scholar]