

Abstract
Over millennia, natural evolution has allowed for the emergence of countless biomolecules with highly specific roles within natural systems. As seen with peptides and proteins, often evolution produces molecules with a similar function but with variable amino acid composition and structure but diverging from a common ancestor, which can limit sequence diversity. Using antimicrobial peptides as a model biomolecule, we train a generative deep learning algorithm on a database of known antimicrobial peptides to generate novel peptide sequences with antimicrobial activity. Using a variational autoencoder, we are able to generate a latent space plot that can be surveyed for peptides with known properties and interpolated across a predictive vector between two defined points to identify novel peptides that show dose-responsive antimicrobial activity. These proof-of-concept studies demonstrate the potential for artificial intelligence-directed methods to generate new antimicrobial peptides and motivate their potential application toward peptide and protein design without the need for exhaustive screening of sequence libraries.
1. Introduction
The rising specter of broad antibiotic resistance has created a critical need for the rapid development of new antimicrobials. At the current rate, the annual global death toll due to antibiotic resistance is expected to exceed 10 million by 2050 at a cost of 100 trillion USD.1 One avenue to combat this problem is the use of antimicrobial peptides (AMPs). As integral components of the innate immune system of humans and other organisms, naturally occurring AMPs have remained effective antimicrobials despite their ancient origins and widespread continual contact with pathogens. For this reason, among others, peptide antibiotics including colistin have been deemed “drugs of last resort” for their ability to kill multidrug-resistant bacteria.2
AMPs generally act through mechanisms associated with membrane disruption, as well as other routes, including binding to DNA and essential cytoplasmic proteins, inhibiting their normal function.3 The relative immutability of bacterial membranes and other AMP targets makes the development of resistance to AMPs rare; however, the need for the development of new AMPs remains essential for combating multidrug-resistant bacteria.
Many attempts at both generating new AMPs and improving their activity have been made, resulting in some success.4,5 These peptides have largely been generated via expert knowledge, low-throughput design methods, including rational design and specific amino acid substitution, or random sequence mutation and template-based methods.
Various computational tools have been developed for performing AMP design in a more sophisticated manner than those previously used that require expert knowledge, including statistical modeling, quantitative structure–activity relationship studies, genetic algorithms,6 machine learning,7 and deep learning.8−10 Using these computational methods, key biophysical attributes can be extracted from sequence information, which can be used to provide model creation information to predict or further enhance the antimicrobial activity of AMPs. In general, these tools require large databases of peptides with known antimicrobial activity. There are currently several thousand AMP sequences in various databases, including Antimicrobial Peptide Database 3 (APD3)11 and A Database of Anti-Microbial (ADAM)12 peptides that contain AMPs with experimentally determined activity against Gram-negative and Gram-positive bacteria, fungi, HIV, and cancer cells. An extended overview of AMP databases and data mining was recently described by Porto et al.13 and new databases of various forms are regularly being released.14
Since peptides and proteins can be represented as sequential amino acid residues in the form of a string of characters, several groups have made use of previously developed methods for extracting sequence information in an order-dependent manner, which have long been used in learning language, including the recursive neural network (RNN) called long short-term memory (LSTM) networks.15 In the few applications of LSTMs for peptide design recently reported, the promising ability to generate sequences of active AMPs, with experimental verification, has demonstrated the usefulness of these established deep learning techniques for new AMP generation.8,10
In another recently developed approach, a number of reports have demonstrated the utility of generative deep learning, including generative adversarial networks (GANs) and variational autoencoders (VAEs),16 for the generation of new objects from existing data. Among a broad array of attempted applications, these have been successfully applied to the generation of new images17 and new chemical compounds via generation of simplified molecular-input line-entry system (SMILES) strings.18,19 A noteworthy attribute of VAEs that distinguishes it from other generative deep learning techniques is their ability to create a continuous latent space that can be used to smoothly interpolate between objects. This capability was extensively exhibited by Gómez-Bombarelli et al., whereby interpolating between two selected FDA-approved drugs, they generated novel chemical structures with a smooth transition between structures, enabling the potential for the optimization of known drugs, among other applications.18
In this study, the latent space generated by a VAE trained using an AMP database was utilized for the discovery of new AMPs. The continuous representation of peptides allows for the generation of novel AMPs in an automated fashion, enables the smooth interpolation between AMP sequences, and shows the potential for optimization of peptide characteristics. Furthermore, we experimentally examine AMPs generated via interpolation and explore the potential of the method for AMP design.
2. Results
2.1. Model Creation and Generation of Active Peptides
We implemented and trained a VAE for modeling peptide sequences with antimicrobial activity. We then used the model for generative peptide de novo design. The general design of the standard VAE architecture is shown in Figure 1. Five-fold cross-validation on different network architectures was tested for best performance as measured by loss, where networks with 512 or 1024 intermediate dimensions performed best, with no observable advantage with larger networks. Training was stopped after 250 epochs as the loss decreased at a sufficiently low rate (less than 0.1 per 10 epochs; Figure S1) and results did not noticeably improve with longer periods of training. The final state of the model was saved and used for sampling novel sequences (Figure 2A).
Figure 1.

Machine learning for antimicrobial design. (A) Flow diagram of VAE design. During training, both known and scrambled peptides are fed into the encoder, generating their latent codes (z). The latent codes are then decoded into the peptide sequence by the decoder. The two terms of the loss function (KL and reconstruction loss) and model are updated using stochastic gradient descent. (B) Here, we train the VAE using an AMP dataset, which allows for experimental validation of results using common microbiological assays. From starting input AMP sequences, the encoder network converts each peptide into a vector in the latent space, which can be viewed as a continuous AMP representation. Provided a point in the latent space, the decoder network will output a corresponding AMP sequence. The output sequences can then be experimentally tested for activity.
Figure 2.

Antimicrobial activity assays. (A) Three peptides, P1, P2, and P3, were selected from point locations near short active peptides described in the literature, NA-CATH, KR-12, and Tet110, generating three peptides sequences designated P1, P2, and P3, respectively, and three corresponding scrambled control peptides (SP1, SP2, and SP3). (B) Experimental assessment of active (P1–P3) AMPs and scrambled (SP1–SP3) pairs on E. coli, A. baumannii, and S. aureus. Peptide concentrations were 0, 0.64, 3.2, 16, 80, and 400 μg/mL. Peptide sequences are listed in Table 1. Inhibition of colony formation is indicative of AMP activity.
Since the model was trained using two sets of sequences composed of “active” and “scrambled” peptides, the implicit starting assumption was that the sequence order, or “peptide grammar”,20 and characteristics dependent on that sequence were the components that would be learned by the model. Characteristics related to amino acid frequency, such as net charge, would not directly contribute to model training as these are also present in the scrambled peptides.
Using the generated latent space, new peptides were decoded using locations near short active peptides described in the literature. Latent space positions for the peptides NA-CATH (AP00897; its 11-mer N-terminal variant, ATRA-1, has been extensively studied21), KR-12 (AP00608),22 and Tet110 (AP02874)23 were identified and used to generate three peptides sequences designated P1, P2, and P3, respectively. From these sequences, three scrambled control peptides (SP1, SP2, and SP3) were produced (Figure 2A), only altering the sequence order while maintaining amino acid frequency and length (see Table 1). Each of these peptides was then assessed for antimicrobial activity against Escherichia coli, Acinetobacter baumannii, and Staphylococcus aureus (Figure 2B).
Table 1. Peptides Used in the Study and Associated Characteristics.
| name/series | increment | sequence | length | molecular weight | net charge | hydrophobicity | hydrophobic moment |
|---|---|---|---|---|---|---|---|
| P1 | RKLKKLWRKFR | 11 | 1558.983 | 6.997 | –1.782 | 0.969 | |
| P2 | RRFVKKVRKLVK | 12 | 1557.008 | 6.997 | –0.825 | 0.976 | |
| P3 | FRWLRKWFRR | 10 | 1550.878 | 4.998 | –1.430 | 1.045 | |
| SP1 | KKRRFRWLKLK | 11 | 1558.983 | 6.997 | –1.782 | 0.161 | |
| SP2 | RKLRKKVFVKRV | 12 | 1557.008 | 6.997 | –0.825 | 0.253 | |
| SP3 | WKRLRWRRFF | 10 | 1550.878 | 4.998 | –1.430 | 0.173 | |
| series 1 | 0.00 | VIREHKYVLLL | 11 | 1382.712 | 1.090 | 0.718 | 0.366 |
| series 1 | 0.33 | LPKIKKTVSTR | 11 | 1270.582 | 3.997 | –0.682 | 0.345 |
| series 1 | 0.66 | LLKSGRLLMKI | 11 | 1271.671 | 2.997 | 0.736 | 0.637 |
| series 1 | 0.99 | KKIKRFLRKIG | 11 | 1386.793 | 5.997 | –0.855 | 1.015 |
| series 2 | 0.00 | GLGIIPHRRYGK | 12 | 1366.632 | 3.088 | –0.617 | 0.325 |
| series 2 | 0.33 | GIMSLFKGVLKT | 12 | 1293.631 | 1.997 | 0.908 | 0.589 |
| series 2 | 0.66 | GLFKIIKNIFSG | 12 | 1336.640 | 1.997 | 0.833 | 0.676 |
| series 2 | 0.99 | KLFRIIKRIFKG | 12 | 1518.955 | 4.997 | 0.150 | 1.075 |
By spot-plating the bacteria following incubation with a range of AMP concentrations, half-maximal effective concentration (EC50) values were calculated from triplicate experiments. For E. coli, the EC50 values of P1, P2, and P3 were determined to be 2.9, 3.1, and 2.9 μg/mL, respectively, while only SP3 had a determinable EC50 of 3.1 μg/mL; the others, SP1 and SP2, did not display significant killing at the concentrations of peptide tested (<400 μg/mL). The EC50 values of P1, P2, and P3 against A. baumannii were found to be 3.1, 5.9, and 1.8 μg/mL, respectively. Both SP1 and SP3 obtained calculable EC50’s of 10.6 and 19 μg/mL, respectively, while SP2 did not display antimicrobial activity at the concentrations used. For the Gram-positive S. aureus, the EC50 values of P1, P2, and P3 were determined to be 0.4, 6.6, and 2.2 μg/mL, respectively. Both SP1 and SP3 obtained calculable EC50’s of 10.6 and 16.5 μg/mL, respectively, while SP2 was found to be inactive. EC50 values are shown in Table S3. Overall, by mean activity, the scrambled peptides were assessed as significantly less active (p < 0.05; Welch’s t-test) than those generated by sampling near active peptides in the latent space. This result is visible in representative plating images shown in Figure 2B. This result suggested that the model learned a degree of sequence-dependent information considering that the scrambled peptides maintained identical net charge, hydrophobicity, and other sequence order-independent attributes (listed in Table 1). Of note was the significant difference in hydrophobic moment between P1, P2, and P3 peptides when compared to SP1, SP2, and SP3: mean hydrophobic moment values of 1.0 and 0.2, respectively. None of the other biophysical characteristics examined were found to be significantly different between the groups.
2.2. Interpolation and Dataset Characterization
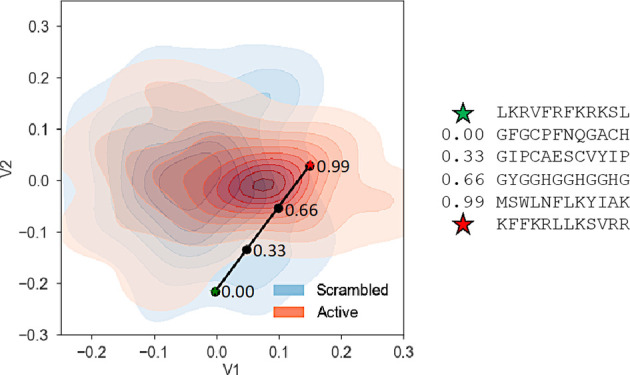
To test the effect of interpolation between active and scrambled peptides, the overall characteristics of the decoded peptides were examined to determine the effect of different sampling points in the latent space. A representation of the latent space generated from 2971 active (obtained from APD311) and 2971 corresponding scrambled peptide sequences is shown in Figure 3A,B. Here, two series of peptides were generated, between SP1 and P1 and between SP2 and P2. Linear interpolation was performed, and four peptides were generated along what can be called an antimicrobial concept vector (black line) in stepwise sampling (Figure 3). These four peptide sequences were generated at increments of 0.33: 0.00, 0.33, 0.66, and 0.99. The sequence at 0.00 was generated close to scrambled peptide (SP) and the sequence at 0.99 was generated close to the active peptide (P). For each series, peptides were chosen at relative distances of 0.33 and 0.66 along the prediction line or antimicrobial concept vector (Table 1). A series was not generated for SP3–P3 since both were found to be active.
Figure 3.

Latent space plots. Latent space maps were generated from the APD3 database (plus scrambled sequences). Antimicrobial concept vector (black line) with stepwise sampling shown, extending from the scrambled peptide (A) SP1 (green star) to P1 (red star) and (B) SP2 (green star) to P2 (red star). V1 and V2 are latent variables 1 and 2, respectively. Antimicrobial activity (black arrow) is expected to increase or decrease proportional to the distance from the scrambled/inactive (SP) or active (P) position. Peptide sequences for each point in the space can be found in Table 1.
To determine the different distributions of characteristics of the peptides generated using interpolation, as shown in Figure 3, we performed this for every active and scrambled pair in the dataset (2971 pairings, plus the P1–SP1 and P2–SP2 pairs), resulting in a list of 17,838 sequences each with a location label (scrambled, 0.00, 0.33, 0.66, 0.99, and active; see Table S2). Attributes that are considered to be important for antimicrobial activity of AMPs, such as hydrophobic moment,24 and others that are likely uncorrelated with activity, such as length, were calculated as a function of step (location of sampling). Although the AMP length was not restricted solely to 12 amino acids, but was trimmed to ≤12, the majority of AMPs were at that length (96%), with the remainder consisting of shorter peptides (Figure 4A). Lengths were similar, regardless of sampling location, with each group containing between 95 and 98% peptides of length 12.
Figure 4.

Comparison of peptide characteristics for the entire generated dataset. (A) Length, (B) amino acid frequency, (C) net charge, (D) hydrophobicity, and (E) hydrophobic moment. For (D) and (E), the middle line is displayed at the median. The sequence list for the generated dataset is provided in Table S2.
Like length, the amino acid frequency was similar regardless of sampling location while not being perfectly uniform (Figure 4B). This intergroup similarity was also the case for net charge (Figure 4C), amino acid type (Figure S2A), isoelectric point (pI; Figure S2B), and molecular weight (Figure S2C). With the exception of net charge, each of these attributes is generally considered to be largely uncorrelated to antimicrobial activity in AMPs. The net charge within the group was positive (1.24 ± 0.01 (mean ± SEM)) ranging between +1.19 and +1.31. While a net positive charge is common among AMPs, between 12 and 15% of naturally occurring AMPs are thought to be anionic (net negative charge).25 Beyond this, as net charge is an attribute resulting from amino acid frequency and the active and scrambled group AMPs are identical in this regard, the sameness of the groups in net charge was expected.
Peptide hydrophobicity and protein hydrophobicity are dependent on amino acid frequency and structural elements. Within this study and dataset, all groups have a hydrophobicity value between 0.08 and 0.2 (overall, 0.13 ± 0.01 (mean ± SEM)) as measured by the Kyte–Doolittle scale.26 Interestingly, there was an observable trend upward between scrambled and active groups (Figure 4D). When examining the larger dataset of active and scrambled peptides, at steps 0.00, 0.33, 0.66, and 0.99, the hydrophobicity values were determined to be 0.08 ± 0.02, 0.11 ± 0.02, 0.14 ± 0.02, and 0.19 ± 0.02, respectively. Both scrambled and active groups were 0.11 ± 0.02. Since overall hydrophobicity is known to be positively correlated with antimicrobial activity,24 this result is not surprising; however, interestingly, the trend is not continued in scrambled and active groups.
To estimate the proportion of the secondary structure of the groups, the GOR IV method was used, which provided predicted propensities for helix, beta sheet, and random coil. As expected, the average peptide in the active group is predicted to be primarily helical, while their scrambled counterparts have a median predicted helicity of ∼15%, with the steps in between increasing in helicity when sampled nearer to active (Figure S3). A related, significant trend was observed in comparing the predicted hydrophobic moments between the sampling step locations. The hydrophobic moments were calculated using a default angle of 100°, which assumes a helical secondary structure. Hydrophobic moments for each group were found to be 0.26 ± 0.002, 0.28 ± 0.002, 0.30 ± 0.003, 0.32 ± 0.003, 0.35 ± 0.003, and 0.38 ± 0.003 for scrambled, 0.00, 0.33, 0.66, 0.99, and active, respectively. This means that the spatial orientations of hydrophobic and polar amino acids differ between the groups along the interpolation path, leading to significantly increased hydrophobic moments (p < 0.01, two-sided Welch’s t-test) compared to those of the scrambled peptide set (Figure 4E). Also, importantly, the mean value of each group is significantly different from each adjacent set, suggesting a gradual shift in spatial orientations along the route from scrambled to active.
Next, to quantify the potential antimicrobial activity of the generated peptides, we made use of the CAMPR3 AMP prediction tool. We decided to use the CAMPR3 classifier as it has been empirically determined to be superior to other classifiers in a recent benchmark report,27 best reflecting real antimicrobial activity as determined in experiments. The probability of antimicrobial activity (P(AMP)) of the AMPs was obtained and plotted in Figure 5A. In a trend similar to the hydrophobic moment described above, the P(AMP) increased as a function of the sampling step locations between scrambled and active. The P(AMP) for each group was, as output by CAMPR3, 0.40 ± 0.006, 0.41 ± 0.006, 0.41 ± 0.006, 0.44 ± 0.006, 0.49 ± 0.007, and 0.50 ± 0.007 for scrambled, 0.00, 0.33, 0.66, 0.99, and active, respectively. Meanwhile, the overall P(AMP) was 0.44 ± 0.002 (mean ± SEM), suggesting that, on average, the peptides generated in this study are predicted to be inactive (where a P(AMP) of ≥0.5 is classified as active). However, several of these are clearly active, as seen in Figure 5B and discussed below. Critically, Welch’s t-test rejected the hypothesis that the scrambled and active datasets have equal means of activity predictions (p < 0.05), as well as 0.66 and 0.99 datasets when compared to the scrambled dataset (p < 0.05). Interestingly, at the 0.33 point, the peptides were not of higher P(AMP).
Figure 5.

(A) Active AMP prediction using CAMPR3. (B) Peptide series 1 and 2 tested against E. coli, A. baumannii, and S. aureus. Peptide concentrations were 0, 0.64, 3.2, 16, 80, and 400 μg/mL. Row labels are interpolation step for each peptide series. The absence or inhibition of colony formation is indicative of AMP activity.
The relatively low P(AMP) prediction for those sequences in the active peptide group may result from the following: (1) the sequences in the training set for CAMPR3 are substantially longer on average than those used in this study (≤12), (2) short peptides may not contain sufficient information for most models to properly assess likelihood of activity, and (3) it is possible that the truncation of peptides in the APD3-based dataset did impact real activity. Overall, according to the CAMPR3 predictions, the VAE model can be used to smoothly interpolate between AMPs predicted to be active (or borderline active) and those that are predicted to be inactive.
2.3. Evaluation of Antimicrobial Activity
As described above (Section 2.2), following their testing, the peptides P1, P2, SP1, and SP2 were used as markers to test the capability of interpolating between the two groups, scrambled and active. Two series of interpolating peptides were generated at four increments between each of the scrambled–active pairs of P1–SP1 and P2–SP2 at increments of 0.00, 0.33, 0.66, and 0.99, as shown in Figure 3. The resulting peptides were next experimentally investigated by assessing their antimicrobial activities on E. coli, A. baumannii, and S. aureus (Figure 5B). By spot-plating the bacteria following incubation with a range of AMP concentrations, EC50’s were calculated from triplicate experiments. EC50’s for both series 1 and 2 peptides against each of the three species tested are found in Table S4 and corresponding EC50 curves are shown in Figure S4.
For E. coli, the EC50 values of peptides 0.66 and 0.99 for series 1 were found to be 308 and 11 μg/mL, respectively, while neither preceding peptides in the series displayed activity. Similarly, for series 2, peptides 0.66 and 0.99 were found to have EC50’s of 84 and 0.2 μg/mL, respectively, while both peptides 0.00 and 0.33 were inactive at the concentrations tested. When tested against A. baumannii, the EC50 values of peptides 0.66 and 0.99 for series 1 were found to be 400 and 36 μg/mL, respectively, while neither of the preceding peptides 0.00 or 0.33 display activity. For series 2, the EC50’s were observed to be 65, 13, and 0.8 μg/mL for 0.33, 0.66, and 0.99, respectively; peptide 0.00 was inactive. For S. aureus, the EC50 values of peptides 0.33, 0.66, and 0.99 for series 1 were determined as 69, 11, and 0.4 μg/mL, respectively, while peptide 0.00 did not show activity. For series 2, all of the peptides were found inactive at the concentrations examined. Overall, these results appear to confirm the P(AMP) predictions by CAMPR3 in Figure 5A, suggesting that the peptides sampled from regions closer to the active space in latent space are more active than those decoded in close proximity to scrambled. This trend is further supported by comparing the overall mean activities, which show that 0.99 group AMPs have a significantly lower EC50 than those from 0.66 (p < 0.05; Welch’s t-test). Although the EC50 values for many of those in group 0.33 and all of those in group 0.00 are not available due to insufficient activity for calculation, Figure 5B suggests the overall trend of increasing activity along the path between scrambled and active groups.
3. Discussion
This study demonstrates the use of a VAE for de novo peptide sequence design using AMPs as an example. The VAE described has the potential for sequence design automation provided that a large database of AMP sequences is available, a requirement similar to other computational techniques. The increasing number of both AMP databases and entries per database makes AMPs a viable proof of concept for assessing the ability of VAEs to generate new peptides. This approach is distinct from many previously reported design techniques, such as random mutagenesis, template-based peptide design strategies, and other random sequence generation methods, where amino acids are drawn from a predefined distribution and then concatenated.5 The encoder and decoder of the VAE rely on LSTM RNNs that have high-dimensional sequence representations, meaning that it does not simply reproduce sequence templates but learns important features present in the training dataset.
The results achieved suggest that the peptide VAE model was capable of generating an internal representation of AMPs from the APD3 database, in that the model did not simply reproduce the training data, supported by the absence of any previously known sequences in those experimentally examined. However, when sampled from similar locations in the latent space, the decoded peptides do have statistically similar distributions to those found in the training data (either active or scrambled). Critically, the features generally reported to be important for the antimicrobial activity of AMPs, whether for bacterial membrane targeting or otherwise, such as high hydrophobic moment in alpha helical peptides,24 are retained in the peptides decoded near active peptides, while properties less correlated with antimicrobial activity, such as length, molecular weight, and pI, are roughly constant regardless of location in the latent space. In fact, of all of the peptide attributes examined in the study, the main difference between the active model-generated sequences and scrambled random sequences is their amphipathicity, which is illustrated by a higher hydrophobic moment for the model-generated sets close to active peptides in the latent space, consistent with the results reported by Müller et al. utilizing a LSTM RNN model.8
Since the hydrophobic moment translates back to a regular pattern of charged and hydrophobic residues at specific locations in the sequence, this demonstrates that this has been learned by the model. It is very likely that the increasing hydrophobic moment yielded higher probabilities in the CAMPR3 AMP prediction model,28 suggesting that simply alternating groups of positively ionizable and hydrophobic amino acids will score highly by the CAMPR3 predictor, a hypothesis that has been manipulated by Müller et al.,8 highlighting the importance of experimentally verifying the antimicrobial activity of generated AMPs. This, along with preserving the simplicity of the method presented, is a possible advantage of avoiding pairing generative models with predictive models, which has been done elsewhere:29 we can avoid any possibility of forcing the generative model to “learn” to tailor its output to what scores highly in a predictive model.
To verify antimicrobial activity, the interpolation feature provided by VAEs was exploited. By interpolating between two known peptides, an active peptide found in APD3 and an assumed inactive scrambled partner, increasing gradations of antimicrobial activity can be sampled at regular steps along the line (Figure 2). This study demonstrated that two series of peptides, between P1–SP1 and P2–SP2, sampled at four steps each could yield an increasing spectrum of activity between two points. The results indicate that not only did the VAE actually learn to recognize the grammar of amphipathicity in peptides but also it may have learned the underlying grammar of what features contribute to antimicrobial activity. The critical finding is that the VAE learned the grammar of amphipathicity and antimicrobial activity in peptides, which can then be tuned in a controllable manner.
As observed in these studies, the intermediates of P3–SP3 displayed high antimicrobial activity as both the predicted and scrambled peptides. With the P1, P2, SP1, and SP2 constructs, the reduced activity of the scrambled peptides can likely be attributed to a change in the hydrophobic moment and, therefore, the ability for the peptide to readily insert into the bacterial membrane. For the P3–SP3 pair, however, the significant change in hydrophobic moment does not appear to affect the antimicrobial activity. The P3–SP3 peptides are composed of multiple tryptophan and arginine residues. In a publication by Wessolowski et al., a similar phenotype was observed with short peptides of similar composition, suggesting antimicrobial activity through noncanonical mechanisms.30 While their consistent activity as native and scrambled peptides is interesting, neighboring peptides were not examined in these studies as similar activity was expected. This observation, however, illustrates the potential need for pairing the VAE with a filtering algorithm to remove certain classes of AMPs depending on the application.
As confirmation of the active AMP prediction model,28 the two interpolated series of AMPs were tested against E. coli, A. baumannii, and S. aureus and, overall, yielded the same trend identified by the AMP prediction, where peptides sampled displayed more activity as a function of their proximity to known active peptides. Interestingly, the peptide series may show a Gram bias, in that the series 2 peptides did not display any measurable activity against S. aureus but were active against Gram-negative bacteria E. coli and A. baumannii. More investigation is required to determine if this result was predictable prior to synthesis. Although, for simplicity, no predictive model was paired with VAE in the current study, various other groups using generative models for chemical or peptide generation have output shuttled through predictive models prior to use.18 Such a pairing could be developed for future applications to potentially wield some control over Gram bias in antimicrobial activity and other attributes of peptides prior to experiment assessment.
4. Conclusions
The results of the study demonstrate the use of a VAE for the automated generation of novel AMPs. The model was trained on thousands of known and scrambled AMP sequences from APD3 and created a readily usable tool, the form of a continuous representation of peptides. The latent space allowed us to generate novel AMPs in a controllable fashion, which we experimentally verified to be active. Despite the shortcomings of this proof-of-concept study, including the focus on truncated peptides, this favorable result will drive future work in the application of VAEs toward various peptide and protein-designing projects, including full-length AMPs. This work suggests that the VAE could operate with greater utility when paired with a predictive model, or other characteristic classifier, in a manner described by Gómez-Bombarelli et al. for molecule generation to better predict AMP activity and for optimization purposes. Beyond this, it is unclear whether the VAE technique is specifically well suited for peptide and protein applications or could other recently developed and improved generative deep learning techniques such as GANs result in superior performance. This idea, use of a conditional VAE or otherwise pairing with a characteristic classifier, and assessment of VAE-generated AMPs on other bacteria will be the subject of future research.
5. Experimental Section
5.1. Dataset and Preprocessing
AMP sequences and other information, including activity (Gram-negative bacteria, Gram-positive bacteria, cancer cells, etc.), were downloaded from APD3,11 yielding 2971 peptide sequences (accessed in mid-2018). Despite the flaws of APD3, such as high homogeneity and incomplete annotation of post-translationally modified sequences, it remains to be one of largest AMP databases available. Since synthesis of long peptides can be prohibitively expensive, and to reduce the computational load downstream, the sequences were truncated such that all peptides were ≤12 amino acids in length, retaining the N-terminal sequence. The length of ≤12 amino acids was chosen based on several reports that have shown that short (10–12 amino acids), truncated AMPs are capable of maintaining high levels of antimicrobial activity.4,31,32 The list of trimmed peptide sequences was then duplicated and randomly scrambled with the stringi R package21 function stri_rand_shuffle to create a scrambled peptide pair for each peptide found in APD3, resulting in a list of 5942 sequences. Duplicate active peptides were retained as all scrambled pairings were unique. The sequences were then tokenized (each residue separated; see Table S1) and represented by a one-hot encoding scheme using binary vectors with length equal to the size of the amino acid vocabulary: “<end>”, “a”, “c”, “d”, “e”, “f’”, “g”, “h”, “i”, “k”, “l”, “m”, “n”, “p”, “q”, “r”, “s”, “t”, “v”, “w”, “x”, and “y”, where “x” is an unknown amino acid present in some of the APD3 sequences. This resulted in a 3D data matrix of dimensions 5942, 24, and 12 for the number of sequences, length of the vocabulary, and feature vector length, respectively.
5.2. Training and Sequence Generation
The architecture of the VAE was implemented as described by Bowman et al.,33 including use of character dropout, which replaces a random fraction of characters (listed in Section 5.1) with an <unk> character and utilization of Kullback–Leibler (KL) annealing, comprising an additional variable weight added to the cost function KL term while training. The loss function, as described by Bowman et al., was composed of reconstruction loss and KL loss to penalize poor reconstruction of the data by the decoder and encoder output representations of z (latent space variables) that are different from a standard normal distribution, respectively (Figure 1A). Training stoppage criteria were met when loss values did not decrease for 10 iterations through the entire dataset (epochs). The preprocessed data were encoded into vectors using LSTMs. The encoder LSTM was paired with a decoder LSTM to do sequence-to-sequence learning. The decoder results were converted from binary one-hot encoded vectors to peptide sequences (Figure 1B). The VAE was trained using the Keras34 library with a TensorFlow35 backend via the Adam optimizer. The number of neurons for the LSTM layers found in both encoder and decoder was both set to 1024. All models were trained on an Ubuntu workstation with a Nvidia Geforce GTX1070 GPU. The LSTM RNNs used in the decoder (and encoder) are stochastic, meaning that decoding from the same point in the latent space may result in a different peptide being generated and is dependent on the random seed set prior to running. Sequence sampling was performed using linear interpolation between active peptides and their scrambled pairs, which is expected to be inactive or less functional as an AMP against target bacteria. For each pairing, four points were generated at equally spaced increments of 0.33: 0.00, 0.33, 0.66, and 0.99, where the scrambled pair is encoded at point 0 and the active pair is encoded at 1 such that points 0.00 and 0.99 are adjacent to AMPs in the dataset, while points decoded at 0.33 and 0.66 are intermediates. Here, it was theorized that antimicrobial activity would correspond to the latent space position or distance from either the active (position 1.0) or inactive/scrambled peptide (position 0.0).
5.3. Examination of Sequence Characteristics
The characteristics of sequences generated, including peptide length, amino acid composition, net charge, hydrophobicity, and hydrophobic moment, were assessed using the Peptides R package36 and Python library modlAMP.37 Prediction of the three-state protein secondary structure (helix, beta, and coil) was carried out using the Garnier–Osguthorpe–Robson (GOR) IV method38 implemented in the Decipher R package.39 The probability of AMP activity (P(AMP)) was predicted using CAMPR327 by uploading sequences to its server. Exported results from CAMPR3, as well as characteristic values output from the Peptides package and modlAMP, were visualized using ggplot2 in the tidyverse R package.40
5.4. Antimicrobial Assays
Peptides synthesized for use in this study are listed in Table 1. Peptides were synthesized by Genscript, Inc. (Piscataway, NJ), and each was confirmed to have greater than 90% purity. Lyophilized peptides were solubilized in water, aliquoted, and stored at −20 °C. Overnight cultures of E. coli BL21, A. baumannii ATCC 17978, and S. aureus ATCC 12600 were grown in Lysogeny Broth (LB). EC50 assays were performed as previously described.41 Overnight cultures of bacteria were enumerated by a standard curve using optical density vs CFU/mL. Cultures were resuspended in 10 mM sodium phosphate buffer (pH 7) to a final concentration of ∼2 × 106 CFU/mL. For determination of EC50 values, peptides were assessed at a range of concentrations (0.64–400 μg/mL) with bacteria for 1 h at 37 °C. Following incubation, bacteria were spotted for colony counting as previously described;42 full spread plating was performed for confirmation of colony counting.
Acknowledgments
S.N.D. acknowledges the funding support through the National Research Council Research Associateship Program and additional funding through the Jerome and Isabella Karle Distinguished Scholar Fellowship provided by the Naval Research Laboratory. S.A.W. was supported by Core funds provided to the Naval Research Laboratory through Office of Naval Research (WU# MA041–06–41).
Glossary
Abbreviations
- AMPs
antimicrobial peptides
- VAE
variational autoencoder
- RNN
recursive neural network
- LSTM
long short-term memory
- EC50
half-maximal effective concentration
- SMILES
simplified molecular-input line-entry system
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00442.
EC50 values for P1, P2, and P3 and their scrambled pairs (Table S3); EC50 values corresponding to the survival curves found in Figure S3 (Table S4); representative loss curve from the training of the VAE (Figure S1); comparison of peptide characteristics (Figure S2); secondary structure prediction of generated peptides across each group (Figure S3); and EC50 curves for the peptide series examined (Figure S4) (PDF)
Input dataset for training (Table S1) (XLSX)
Generated peptides (Table S2) (XLSX)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- O’Neill J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. 2014, 1. [Google Scholar]
- Lewies A.; Du Plessis L. H.; Wentzel J. F. Antimicrobial Peptides: the Achilles’ Heel of Antibiotic Resistance?. Probiotics Antimicrob. Proteins 2019, 370–381. 10.1007/s12602-018-9465-0. [DOI] [PubMed] [Google Scholar]
- Chung E. M. C.; Dean S. N.; Propst C. N.; Bishop B. M.; van Hoek M. L. Komodo dragon-inspired synthetic peptide DRGN-1 promotes wound-healing of a mixed-biofilm infected wound. npj Biofilms Microbiomes 2017, 3, 9. 10.1038/s41522-017-0017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean S. N.; Bishop B. M.; van Hoek M. L. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 2011, 11, 114. 10.1186/1471-2180-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M. D. T.; Sothiselvam S.; Lu T. K.; de la Fuente-Nunez C. Peptide Design Principles for Antimicrobial Applications. J. Mol. Biol. 2019, 3547. 10.1016/j.jmb.2018.12.015. [DOI] [PubMed] [Google Scholar]
- Cardoso M. H.; Oshiro K. G. N.; Rezende S. B.; Cândido E. S.; Franco O. L. The Structure/Function Relationship in Antimicrobial Peptides: What Can we Obtain From Structural Data?. Adv. Protein Chem. Struct. Biol. 2018, 112, 359–384. 10.1016/bs.apcsb.2018.01.008. [DOI] [PubMed] [Google Scholar]
- Lima A. N.; Philot E. A.; Trossini G. H. G.; Scott L. P. B.; Maltarollo V. G.; Honorio K. M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discovery 2016, 11, 225–239. 10.1517/17460441.2016.1146250. [DOI] [PubMed] [Google Scholar]
- Müller A. T.; Hiss J. A.; Schneider G. Recurrent Neural Network Model for Constructive Peptide Design. J. Chem. Inf. Model. 2018, 58, 472–479. 10.1021/acs.jcim.7b00414. [DOI] [PubMed] [Google Scholar]
- Nagarajan D.; Nagarajan T.; Roy N.; Kulkarni O.; Ravichandran S.; Mishra M.; Chakravortty D.; Chandra N. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 2018, 293, 3492–3509. 10.1074/jbc.M117.805499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P.; Müller A. T.; Gabernet G.; Button A. L.; Posselt G.; Wessler S.; Hiss J. A.; Schneider G. Hybrid Network Model for ″Deep Learning″ of Chemical Data: Application to Antimicrobial Peptides. Mol. Inf. 2017, 36, 1600011. 10.1002/minf.201600011. [DOI] [PubMed] [Google Scholar]
- Wang G.; Li X.; Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-T.; Lee C.-C.; Yang J.-R.; Lai J. Z. C.; Chang K. Y. A large-scale structural classification of antimicrobial peptides. BioMed Res. Int. 2015, 2015, 1–6. 10.1155/2015/475062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto W. F.; Pires A. S.; Franco O. L. Computational tools for exploring sequence databases as a resource for antimicrobial peptides. Biotechnol. Adv. 2017, 35, 337–349. 10.1016/j.biotechadv.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Nagarajan D.; Nagarajan T.; Nanajkar N.; Chandra N. A uniform in vitro efficacy dataset to guide antimicrobial peptide design. Data 2019, 4, 27. 10.3390/data4010027. [DOI] [Google Scholar]
- Gers F. A.; Schmidhuber E. LSTM recurrent networks learn simple context-free and context-sensitive languages. IEEE Trans. Neural Networks 2001, 12, 1333–1340. 10.1109/72.963769. [DOI] [PubMed] [Google Scholar]
- Kingma D. P.; Welling M.. Auto-encoding variational bayes. 2013, arXiv:1312.6114. arXiv.org e-Print archive. https://arxiv.org/abs/1312.6114. [Google Scholar]
- White T.Sampling generative networks. 2016, arXiv:1609.04468. arXiv.org e-Print archive. https://arxiv.org/abs/1609.04468. [Google Scholar]
- Gömez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusner M. J.; Paige B.; Hernández-Lobato J. M.. Grammar variational autoencoder . In Proceedings of the 34th International Conference on Machine Learning-Volume 70, JMLR.org: 2017; pp 1945–1954.
- Loose C.; Jensen K.; Rigoutsos I.; Stephanopoulos G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867. 10.1038/nature05233. [DOI] [PubMed] [Google Scholar]
- Dean S. N.; Bishop B. M.; van Hoek M. L. Susceptibility of Pseudomonas aeruginosa Biofilm to Alpha-Helical Peptides: D-enantiomer of LL-37. Front. Microbiol. 2011, 2, 128. 10.3389/fmicb.2011.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- Hilpert K.; Elliott M.; Jenssen H.; Kindrachuk J.; Fjell C. D.; Körner J.; Winkler D. F. H.; Weaver L. L.; Henklein P.; Ulrich A. S.; Chiang S. H. Y.; Farmer S. W.; Pante N.; Volkmer R.; Hancock R. E. W. Screening and characterization of surface-tethered cationic peptides for antimicrobial activity. Chem. Biol. 2009, 16, 58–69. 10.1016/j.chembiol.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Pathak N.; Salas-Auvert R.; Ruche G.; Janna M. h.; McCarthy D.; Harrison R. G. Comparison of the effects of hydrophobicity, amphiphilicity, and α-helicity on the activities of antimicrobial peptides. Proteins: Struct., Funct., Bioinf. 1995, 22, 182–186. 10.1002/prot.340220210. [DOI] [PubMed] [Google Scholar]
- Harris F.; Dennison S. R.; Phoenix D. A. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. 10.2174/138920309789630589. [DOI] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Gabere M. N.; Noble W. S. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017, 33, 1921–1929. 10.1093/bioinformatics/btx081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghu F. H.; Barai R. S.; Gurung P.; Idicula-Thomas S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. 10.1093/nar/gkv1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Lin K.; Wang S.; Wang L.; Cai C.; Song C.; Lai L.; Pei J. Deep learning for molecular generation. Future Med. Chem. 2019, 567. 10.4155/fmc-2018-0358. [DOI] [PubMed] [Google Scholar]
- Wessolowski A.; Bienert M.; Dathe M. Antimicrobial activity of arginine- and tryptophan-rich hexapeptides: the effects of aromatic clusters, D-amino acid substitution and cyclization. J. Pept. Res. 2004, 64, 159–169. 10.1111/j.1399-3011.2004.00182.x. [DOI] [PubMed] [Google Scholar]
- Bruni N.; Capucchio M.; Biasibetti E.; Pessione E.; Cirrincione S.; Giraudo L.; Corona A.; Dosio F. Antimicrobial activity of lactoferrin-related peptides and applications in human and veterinary medicine. Molecules 2016, 21, 752. 10.3390/molecules21060752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasetty G.; Papareddy P.; Kalle M.; Rydengård V.; Mörgelin M.; Albiger B.; Malmsten M.; Schmidtchen A. Structure-activity studies and therapeutic potential of host defense peptides of human thrombin. Antimicrob. Agents Chemother. 2011, 55, 2880–2890. 10.1128/AAC.01515-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman S. R.; Vilnis L.; Vinyals O.; Dai A. M.; Jozefowicz R.; Bengio S.. Generating sentences from a continuous space. 2015, arXiv:1511.06349. arXiv.org e-Print archive. https://arxiv.org/abs/1511.06349. [Google Scholar]
- Chollet F.Keras. https://github.com/fchollet/keras.
- Abadi M.; Barham P.; Chen J.; Chen Z.; Davis A.; Dean J.; Devin M.; Ghemawat S.; Irving G.; Isard M.. Tensorflow: A system for large-scale machine learning . In 12th {USENIX} Symposium on Operating Systems Design and Implementation ({OSDI} 16); USENIX Association: 2016; pp 265–283.
- Osorio D.; Rondón-Villarrea P.; Torres R. Peptides: a package for data mining of antimicrobial peptides. R Journal 2015, 7, 4–14. 10.32614/rj-2015-001. [DOI] [Google Scholar]
- Müller A. T.; Gabernet G.; Hiss J. A.; Schneider G. modlAMP: Python for antimicrobial peptides. Bioinformatics 2017, 33, 2753–2755. 10.1093/bioinformatics/btx285. [DOI] [PubMed] [Google Scholar]
- Garnier J.; Gibrat J.-F.; Robson B.. GOR method for predicting protein secondary structure from amino acid sequence. In Methods in enzymology, Elsevier: 1996; Vol. 266, pp 540–553, 10.1016/S0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- Wright E. S. DECIPHER: harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinf. 2015, 16, 322. 10.1186/s12859-015-0749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H.Tidyverse: Easily install and load’tidyverse’packages. R package version; R Core Team: 2017,1 ( (1), ).
- Chung M. C.; Dean S. N.; van Hoek M. L. Acyl carrier protein is a bacterial cytoplasmic target of cationic antimicrobial peptide LL-37. Biochem. J. 2015, 470, 243–253. 10.1042/BJ20150432. [DOI] [PubMed] [Google Scholar]
- Sieuwerts S.; de Bok F. A. M.; Mols E.; de vos W. M.; van Hylckama Vlieg J. E. T. A simple and fast method for determining colony forming units. Lett. Appl. Microbiol. 2008, 47, 275–278. 10.1111/j.1472-765X.2008.02417.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.