

Abstract
Thorough characterization of protein assemblies is required for the control of structure and robust performance in any given application, especially for the safety and stability of protein therapeutics. Here, we report the use of multiple, orthogonal characterization techniques to enable control over the structure of a multivalent antibody carrier for future use in drug delivery applications. The carrier, known as Hex, contains six antibody binding domains that bind the Fc region of antibodies. Using size exclusion chromatography, analytical ultracentrifugation, and dynamic light scattering, we identified the stoichiometry of assembled Hex−antibody complexes and observed changes in the stoichiometry of nanocarriers when incubated at higher temperatures over time. The characterization data informed the modification of Hex to achieve tighter control over the protein assembly structure for future therapeutic applications. This work demonstrates the importance of using orthogonal characterization techniques and observing protein assembly in different conditions over time to fully understand and control structure and dynamics.
Graphical Abstract
INTRODUCTION
Many naturally existing proteins have self-assembly capabilities that have been exploited to create new nanostructures for various applications in biosensing, biocatalysis, and therapeutics.1 As the understanding of protein self-assembly has progressed, de novo design of proteins has also yielded new self-assembling protein and peptide building blocks.2 Rational protein engineering can be used to develop functional protein materials by combining self-assembling domains with functional domains through recombinant fusion, covalent cross-linking, or affinity interactions.1,3 Such protein assemblies have been used in a variety of applications such as biosensing, enzyme immobilization, and drug delivery.4–7
When developing protein assemblies for these applications, extensive characterization of the structure and stability is required for robust and predictable performance, including the effective and safe translation of therapeutics.8 First and foremost, the complexity of protein assemblies requires characterization to determine their size, polydispersity, and in the case of multicomponent assemblies, their stoichiometry.9 This is critical for knowing exactly how many functional or therapeutic protein domains are in the assembly to accurately determine the dose of the therapy or expected response of a diagnostic or enzymatic assembly.8 Furthermore, as protein assemblies are formed by non-covalent, reversible interactions, any dynamics or loss in stability that occur over time or upon exposure to different conditions must be identified. Dynamics in protein assemblies have previously been identified and sometimes even been used to tune the structure. In one such case of an allosteric protein amphiphile, the protein assembled into long nanofibers but transitioned into a rectangular nanosheet structure when a small molecule ligand was added.10 In another case, self-assembled multicomponent protein hydrogels have been engineered to be shear-thinning and self-healing due to weak, transient cross-links between monomers.11 Finally, a peptide nanofiber assembly that was initially made up of α-helical coiled coils underwent a transition to a β-sheet structure that could be accelerated by higher temperatures.12 In all of these cases, protein assemblies exhibited dynamics when exposed to different conditions such as ligand concentrations, temperatures, and mechanical shear stresses. In the peptide nanofiber example, the morphology remained the same when viewed with transmission electron microscopy, but the protein secondary structure changed and was only observed through solid-state nuclear magnetic resonance spectroscopy.12 These examples show how characterization of the protein assemblies, often with multiple techniques, was needed to identify the material dynamics.
Given the importance of characterizing protein assemblies, many techniques have already been adopted widely in the field. Due to the limitations present with any individual technique, the use of multiple, orthogonal techniques is critical to fully characterize a system. In many examples of protein self-assembly, only one or two techniques are combined to understand the structure. For example, dynamic light scattering (DLS) or size exclusion chromatography (SEC) are used to provide information about an assembly’s size, and atomic force microscopy or transmission electron microscopy are used to visualize the morphology of protein assemblies.13–16 The microscopy techniques require rigorous sample preparation that may alter the native structure of the proteins during analysis. Dynamic light scattering estimates the particle sizes of samples in their native state, but if a sample is polydisperse, DLS cannot distinguish between the size differences of less than 3 orders of magnitude.17 SEC improves upon DLS by providing size-based separation of different molecular species and quantification of their relative abundance. However, the analysis can be affected by an assembly’s interactions with column matrices or the inability of large aggregates or assemblies to enter the column and be detected.18 Therefore, using only DLS or SEC for estimating the size of protein assemblies may not be sufficient, and even using both may require a third technique for validation. Analytical ultracentrifugation (AUC) is an orthogonal method to SEC that can provide similar information about protein assemblies in their solution state without potential artifacts from column interactions.18–22 Both AUC and SEC can also provide estimates of molecular weights of different species through analysis of AUC data or connecting SEC columns to downstream light scattering detectors, as in the case of the Malvern OmniSEC instrument.23 Regardless of the type of protein assembly, using multiple techniques of characterization can overcome the limitations of any one technique. When characterizing pharmaceutical products, in particular, the FDA recommends using orthogonal methods to characterize complex biomolecules and linking quality attributes to safety and clinical performance.8,24 Therefore, characterization of therapeutic protein assemblies in development should follow these guidelines to improve the likelihood of translation.
In this work, we demonstrate the use of multiple, orthogonal techniques to characterize and control the structure of a self-assembled protein drug complex. This complex consists of two components, a highly stable coiled coil peptide assembly, called the Hex carrier, and multiple therapeutic antibodies bound to the carrier (Figure 1a). The Hex carrier was previously developed and reported to deliver antibodies intracellularly to cytosolic targets.25 The carrier is comprised of a self-assembled hexameric coiled coil core, where each coil is fused by a flexible glycine-serine linker to an antibody binding domain. The coiled coil at the center is made up of six identical alpha helices designed de novo to self-assemble via hydrophobic and electrostatic interactions.26 The antibody binding domain, Staphyloccocus aureus Protein A domain B (SPAB), noncovalently binds the Fc (constant) region of antibodies with nanomolar affinity, enabling the formation of a protein complex for intracellular delivery of antibodies to cytosolic targets.27 The drug carrier contains six SPAB domains, and antibodies contain two SPAB binding sites, yielding a theoretical stoichiometric loading of three antibodies to the Hex carrier and a maximum possible loading of six antibodies to the carrier. Because antibodies have twice the molecular weight of the Hex carrier, the binding of antibodies constitutes self-assembly of the overall Hex−antibody protein complex. Previous work with the Hex carrier assessed the ability of Hex to deliver antibodies inside of cells.25 The work presented here focuses on the structural characterization of the Hex−antibody complexes, both to understand Hex for future use and to provide more general knowledge applicable to other multivalent, affinity-based assemblies.
Figure 1.
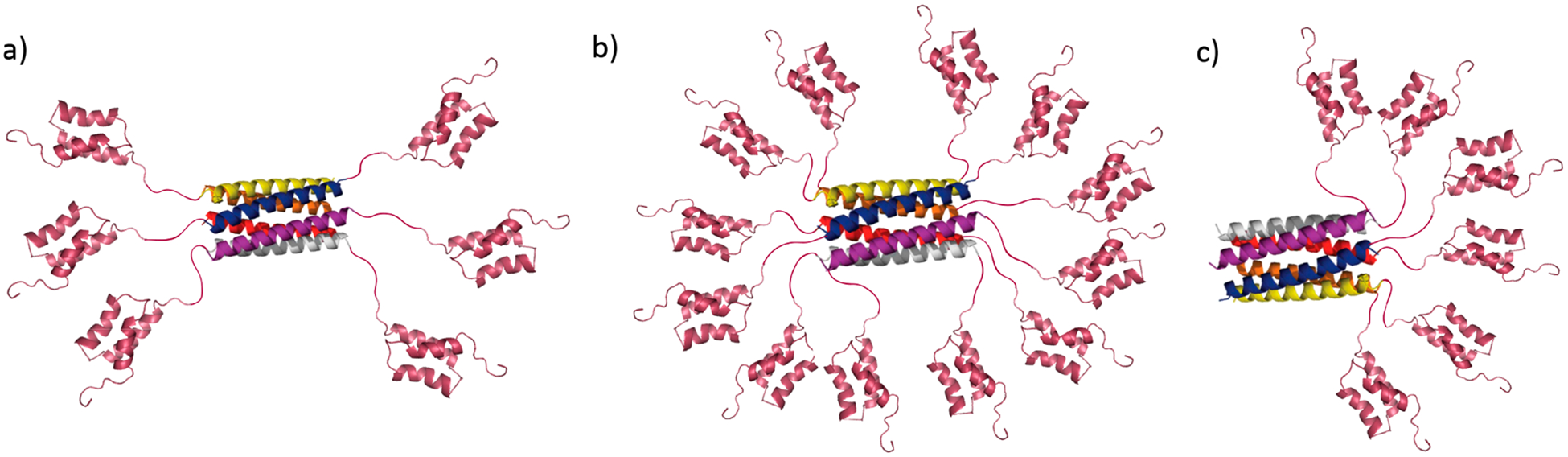
(a) Representative secondary structure of the Hex carrier comprising a Hex coiled coil core (PDB ID: 3R47) and six SPAB domains flanking the core (PDB ID: 1BDC) connected by glycine-serine linkers. (b) Structure of SPAB–Hex–SPAB protein, which contains 12 SPAB domains. (c) Structure of Hex–SPAB protein with six SPAB domains on one side of the Hex coiled coil. Images are not drawn to scale.
To determine the particle size, molecular weight, and stoichiometry of the Hex carrier loaded with antibodies, we used DLS, SEC, and AUC. While DLS has previously been used to report the size of the loaded Hex carriers,25 OmniSEC and AUC were utilized to determine the optimal binding ratio and resulting stoichiometry of the drug carriers. Characterizing the loaded Hex carriers after exposure to different temperatures revealed a rearrangement of the complexes over different time scales. Furthermore, similar binding ratios and dynamic assembly behavior were observed with a modified version of Hex, SPAB–Hex–SPAB (SHS) with twice the SPAB domains (Figure 1b), and a one-sided version of Hex, Hex–SPAB (HS) (Figure 1c). By using multiple techniques to characterize the self-assembled Hex–antibody complexes, we were able to control the structure of these proposed therapeutic proteins. This work highlights the importance of using orthogonal techniques for characterizing therapeutic protein assemblies and the value of this information in designing new assemblies. Furthermore, changes in structure at different temperatures and times highlighted the potential for protein self-assemblies to exhibit dynamic rearrangement, which should also be characterized.
MATERIALS AND METHODS
Protein Expression and Purification.
The Hex carrier was produced by the separate production of two proteins, Hex–SPAB (HS) and SPAB–Hex (SH), each with the SPAB domain on different termini of the coiled coil α helix. The genes for HS and SH were previously cloned into pQE80 vectors and transformed into Top10 Escherichia coli.25 The gene for SPAB–Hex–SPAB (SHS) was synthesized and cloned into a pQE-60 vector by Genscript and transformed into a BL21-AFIQ strain of E. coli. Each protein was expressed in a Lysogeny Broth media at 37 °C in 1 L cultures supplemented with 0.2 mg/mL ampicillin for HS/SH or 0.2 mg/mL ampicillin and 0.034 mg/mL chloramphenicol for SHS. Protein expression was induced by 1 mM Isopropyl-β-thiogalactoside at an optical density of 0.6, and the cells were grown for an additional 4 h after induction. After collecting the cells by centrifugation, the proteins were purified using Ni-NTA (Qiagen) affinity chromatography under native conditions according to manufacturer’s instructions. A concentration of 80 mM imidazole was used in the wash buffer for all three protein purifications. The purity of the proteins was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) following boiling in a gel loading buffer containing a reducing agent. The purified protein samples were stored in the elution buffer (from the purification protocol) containing 250 mM imidazole for long-term 4 °C storage, no longer than 2 months. Proteins were filtered (0.2 μm syringe filter) and characterized by DLS to ensure aggregates were not present.
Sample Preparation.
To produce Hex carriers with a balanced distribution of SPAB domains on either side of the coiled coil, SH and HS proteins needed to be “reassembled”, as previously reported.25 Volumes corresponding to 0.85 mg of each HS and SH were combined, 33 μL of a 20% w/v solution of sodium dodecyl sulfate (SDS) was added, and phosphate buffered saline (PBS) was added to a final volume of 2.5 mL. SDS is added to the reassembling buffer to break the coiled coils in HS and SH into monomers. The solution was passed through a PD-10 column (GE Healthcare) five times to remove the SDS. After removing the SDS from the HS and SH monomer mixture, the monomeric alpha helices reassemble into coiled coils, with a mixture of HS and SH in each hexamer, resulting in a more balanced distribution of SPAB domains on either side. The resulting, diluted Hex solution was concentrated using centrifugal filters (MWCO 3 kDa, Millipore Sigma) to a final volume of 0.5 to 1 mL in PBS. The concentrated Hex carrier solution was syringe filtered with a 0.2 μm membrane filter, and the protein concentration was measured using the Protein A280 settings on a NanoDrop 2000 (Thermo Fisher).
For SHS and HS, the purified protein solutions were buffer exchanged into PBS using centrifugal filters, exchanging 15 mL of PBS for 1 mL of SHS in elution buffer. The resulting SHS solution in PBS was also syringe filtered and concentration quantified as above.
To prepare samples of Hex, SHS, or HS mixed with rabbit immunoglobulin G (IgG, Sigma-Aldrich), the desired amount of IgG (depending on the carrier to IgG ratio) was diluted into PBS. Hex, SHS, or HS solutions were added to a final concentration of 1.667 μM Hex and HS or 0.833 μM SHS. The mixture was placed on a shaker for 10 min prior to use in experiments. Samples that were aged at different temperatures were placed in either a 37 °C incubator, left on the bench at room temperature (25 °C), or placed in a 4 °C refrigerator for indicated time periods.
Circular Dichroism.
Circular dichroism (CD) spectra were acquired on the Applied Photophysics Chirascan-plus spectrometer with the sample chamber maintained at 25 °C. Measurements were made using a 0.1 mm path length quartz cell cuvette. For each sample, the average spectra of three measurements were obtained from a wavelength range of 200–280 nm, with 1 nm increments. The spectra for PBS without protein were obtained as a blank. Spectra for Hex, SHS, HS, and IgG proteins were measured at 1 uM protein concentration. Spectra for all Hex–IgG, HS–IgG, and SHS–IgG were taken at concentrations of 0.75 uM IgG, 0.25 uM Hex or HS, and 0.125 uM SHS, after incubation at 37 °C for 18 h.
Spectra were acquired in millidegrees and converted to mean residue ellipticity using this equation: [θ] = ([θ]obs*MW)/(10*l*C*n). [θ]obs is the observed sequence in millidegrees, MW is the molecular weight in g/mol, l is path length in cm, C is protein concentration in g/L, and n is the total number of amino acids per protein. For Hex–IgG, HS–IgG, and SHS–IgG, the molecular weights and amino acids were determined by adding each individual proteins MW or n and multiplying by its respective molar ratio.
Dynamic Light Scattering.
The particle size of the samples was analyzed by dynamic light scattering using a Malvern Zetasizer Nano ZS equipped with a 4 mW He–Ne laser light source (633 nm). The following solvent settings were used to acquire size and polydispersity index for each sample reported: medium PBS, refractive index of 1.33, viscosity of 0.8872 cP, measurement temperature of 25 °C, cuvette type of ZEN 0040, laser wavelength of 633 nm, and scattering angle of 173 degrees. Three separate readings were taken for each sample, and the resulting intensity plots and particle sizes were averaged before reporting.
SEC.
SEC analysis of samples (IgG, Hex–IgG 1:2, 1:3, 1:4, 1:5, 1:6, 1:12) was conducted using a HiPrep Sephacryl S-300 size exclusion column (GE Life Sciences) attached to an AKTAxpress FPLC system (GE Life Sciences). A volume of 5 mL of protein in PBS was loaded on the column with an automatic sample pump, and the absorbance at 280 nm was monitored for 1.5 column volumes of PBS flowing at 0.5 mL/min through the column. Each sample was analyzed at least twice, using different batches of Hex proteins.
Analytical Ultracentrifugation.
Hex–IgG samples were prepared at a Hex concentration of 1.13 μM such that the total absorbance of the sample would be between 0.1 and 1 for both the 1:3 and 1:4 Hex:IgG ratios. Samples were loaded into an AUC cell with 12 mm double-sector Epon centerpieces and quartz windows. The centerpieces were then incubated at 20 °C in an AN50 Ti rotor for 1 h. Data were collected using a Beckman Optima AUC analytical ultracentrifuge, with a rotor speed of 40000 rpm at 20 °C. The data were obtained by monitoring the sedimentation of absorbance at 280 nm for the protein, using a radial step size of 0.001 cm. The resulting data were analyzed by SEDFIT, and the baseline, meniscus, frictional coefficient, systematic time-invariant, and radial invariant noise were fit.
OmniSEC Analysis.
Hex–IgG and SHS–IgG samples were analyzed by a Malvern OmniSEC integrated system (Malvern Panalytical) with a SRT SEC-300 analytical SEC column (Sepax). Samples were loaded from an autoinjector sample tray, kept at 20 °C. Calibration was performed using a bovine serum albumin standard. Data from a refractive index, right angle light scattering (RALS), low angle light scattering (LALS), and a UV polydiode array detector were collected. The resulting chromatograms were analyzed using triple detection (RI, RALS, and viscometer) and the dn/dc from sample concentrations were used to calculate the MW of the peaks. The UV signal chromatogram at 254 nm was used to quantify peak areas. Molecular weights were calculated with Malvern OmniSEC software version 10.41. Each sample was analyzed at least twice by OmniSEC, and multiple batches of Hex and SHS proteins were used throughout all experiments.
Data Processing.
Raw data were extracted from all instruments and related software, processed in Microsoft Excel, and then plotted using GraphPad Prism. For SEC chromatograms where the baseline drifted below the x-axis of a chromatogram, the minimum value of the entire chromatogram was added to all data points in the chromatogram to shift the baseline upward.
RESULTS AND DISCUSSION
Determination of Maximum Antibody Loading in Hex Carriers.
Knowing the optimal ratio of Hex carriers to antibodies is required to produce a uniform protein assembly and to know how many therapeutic antibodies can be loaded on the carrier for future applications. To determine the loading ratio, Hex carriers were mixed with rabbit serum IgG in various ratios for analysis by SEC. The SPAB domain has a high binding affinity to the Fc region of rabbit IgG, similar to the IgG2a isotype of mouse serum IgG and multiple isotypes of human IgG,28 both of which would be used for translation of the Hex carrier. Hex carriers were produced by expression in E. coli and purified by affinity chromatography. Protein purity was analyzed by SDS-PAGE (Figure S1). The secondary structure of Hex proteins was analyzed by CD (Figure S2a) and showed two peaks at 208 and 222 nm, indicative of α helical content.
The stoichiometry of the Hex–IgG assembly was initially undetermined because antibodies contain a binding site for SPAB in the Fc domain of each heavy chain, yielding two binding sites per IgG molecule.29 Upon mixing with Hex carriers, which contain six SPAB domains, the maximum possible loading is six antibodies if a single SPAB domain binds each antibody, and the stoichiometric loading is three if two SPAB domains bind each antibody. When mixing Hex with IgG in different ratios, the concentration of Hex was kept at 1.66 μM so that loadings of three and six antibodies would represent possible doses for future therapeutic in vivo studies. To determine the actual loading, Hex carriers were mixed with IgG in various ratios and analyzed by SEC (Figure 2). Mixing ratios of 1:3, 1:4, and 1:5 (Hex:IgG) showed peaks at 45 and 55 mL elution volumes. Ratios of 1:4 and 1:5 exhibited an additional peak at 68 mL elution volume, which was assigned to be unbound IgG since it was identical to the elution volume of an IgG standard (Figure S3a). The analysis of 1:6 and 1:12 mixing ratios (Figure S3c and d) shows the same peaks as 1:4 and 1:5 but with larger 68 mL free IgG peaks corresponding to the increased amount of unbound IgG. The two peaks at 45 and 55 mL most likely represent different binding ratios of Hex–IgG complexes. Analysis of a 1:2 Hex–IgG (Figure S3b) showed peaks that eluted around 60 mL, suggesting the formation of a smaller Hex–IgG complex without enough IgG to form the 45 and 55 mL peaks in samples of higher IgG ratios. Taken together, these results indicate that three antibodies represent maximum loading of the Hex carrier since this is the largest ratio that did not have an unbound IgG peak. However, the presence of two peaks at 45 and 55 mL reveals that different forms of the Hex–IgG assemblies exist. SEC by itself cannot provide more detailed information, such as absolute molecular weight estimates, so further characterization was required to identify these two complexes.
Figure 2.
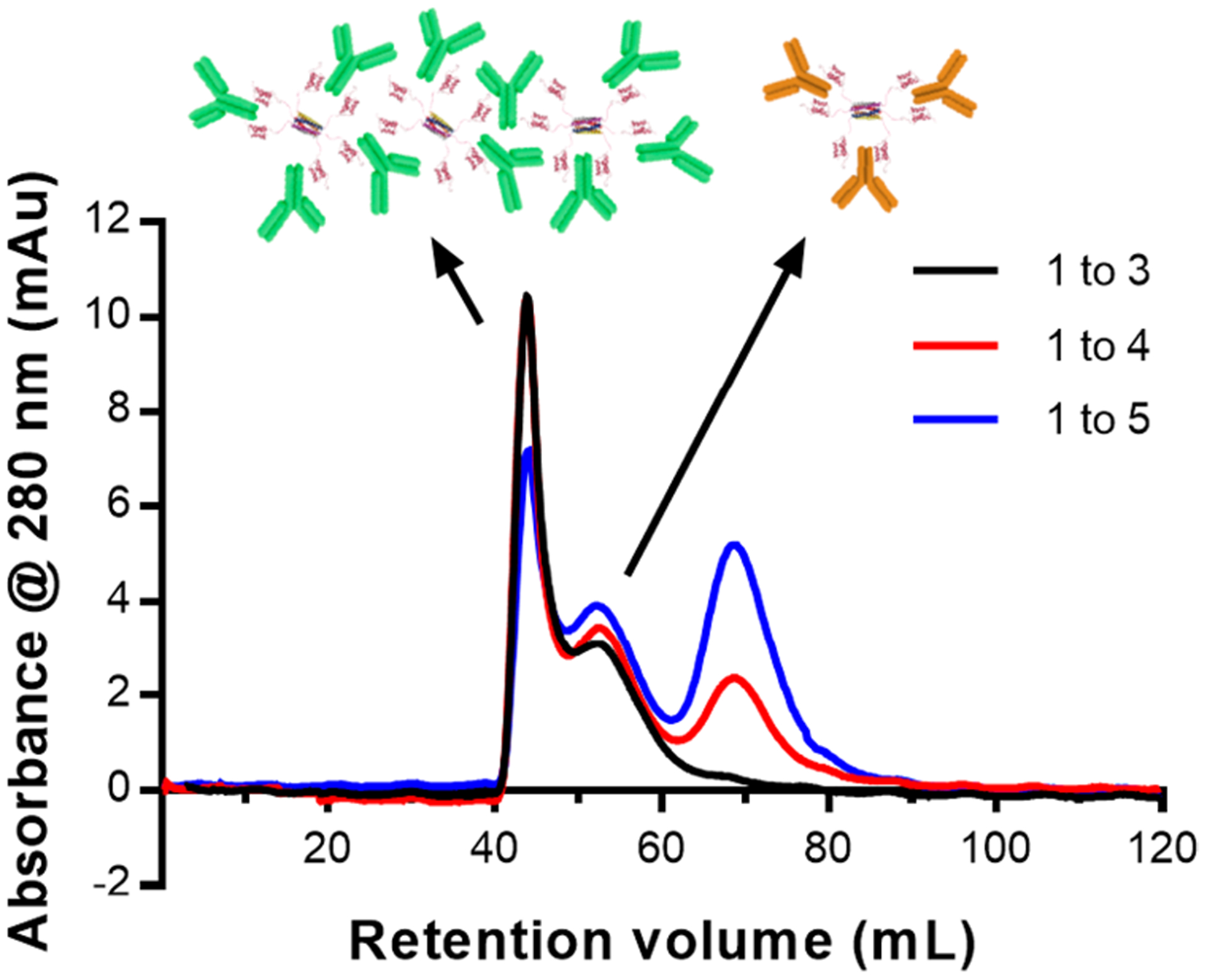
SEC chromatogram of Hex carriers and IgG mixed in ratios of 1 Hex to 3, 4, and 5 IgG. The peak at 68 mL is indicative of unbound IgG. The cartoon above the chromatogram shows the possible form of complexes in each peak.
Estimation of Species Molecular Weights.
AUC and OmniSEC analysis were used to estimate the molecular weights of the two different species in the mixture and determine their composition. Because AUC characterizes samples in their native solution conditions and concentration without a column, it can also confirm the SEC results. Species of different molecular weights, shapes, or densities sediment differently during ultracentrifugation.30 Analysis of the sedimentation gradient from a UV detector provides relative abundance and molecular weight. Hex carriers and IgG were mixed in ratios of 1:3 and 1:4 for analysis by AUC. The resulting sedimentation coefficient distributions and molecular weight estimations were obtained by analysis in SEDFIT.31,32 Sedimentation coefficient is proportional to molecular weight of the species, so smaller species appear first on the distribution curve of sedimentation coefficients (Figure 3a and b).
Figure 3.
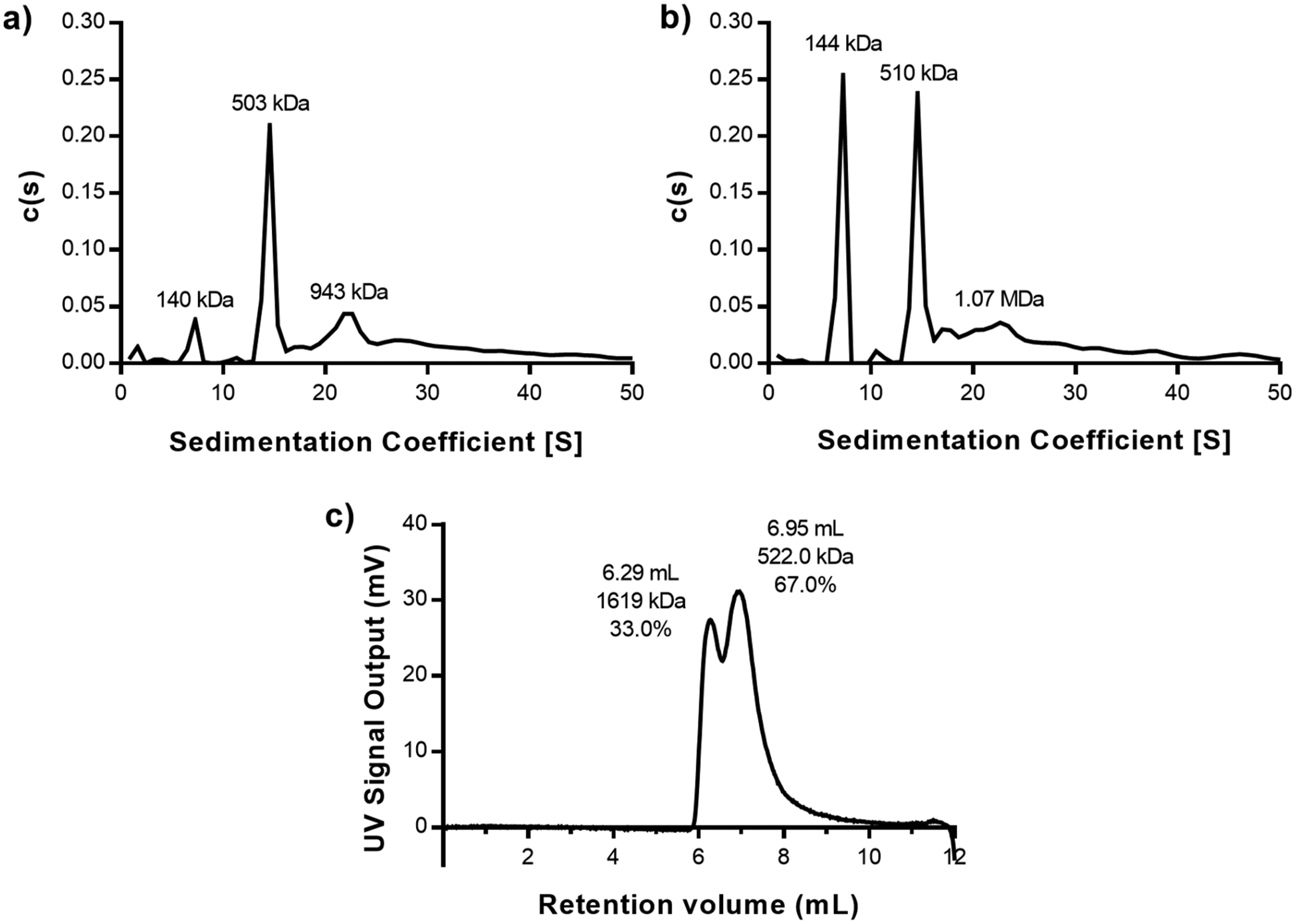
(a and b) Sedimentation coefficient distributions obtained from AUC for a Hex–IgG sample in a (a) 1:3 ratio or a (b) 1:4 ratio. Molecular weights above peaks represent estimates of each species in the mixture. (c) OmniSEC chromatogram of Hex–IgG sample in a 1:3 ratio analyzed immediately after mixing. Retention volume, molecular weight estimate, and peak area % of sample are listed above each identified peak.
AUC results confirmed the maximum loading of 3 antibodies on the carrier and identified the potential stoichiometry of the different species in the mixture. In the 1:4 Hex–IgG sample, a peak with an estimated molecular weight (MW) of 144 kDa was present, indicating the presence of excess IgG (MW = 150 kDa) (Figure 3b). This IgG peak was present at a much lower intensity in the 1:3 sample, matching the SEC results for maximum antibody loading (Figure 3a). Importantly, the AUC result confirmed that the stoichiometry of Hex–IgG complexes is not affected by dilution of the sample on a large (120 mL) SEC column or by potential interactions with the column matrix. In both samples, a major peak was observed with estimated molecular weights of 503 kDa (1:3) and 510 kDa (1:4). Out of the multiple stoichiometries possible for a Hex–IgG mixture, this most closely matches the theoretical molecular weight of a 1:3 complex at 521 kDa (Figure 4a). The Hex coiled coil at the core of the assembly maintains its hexamer structure during OmniSEC analysis, so it cannot contribute to different oligomeric states of the assembly (Figure S4a). In both samples, a third peak was also observed with an estimated molecular weight of ∼1 MDa, possibly corresponding to a larger oligomer seen as the 45 mL peak in the SEC results. The structure of this species likely involves the binding of an IgG by two different Hex carriers in order to form a larger oligomer where IgG molecules serve as “crosslinkers” between the Hex carriers (Figure 4b).
Figure 4.
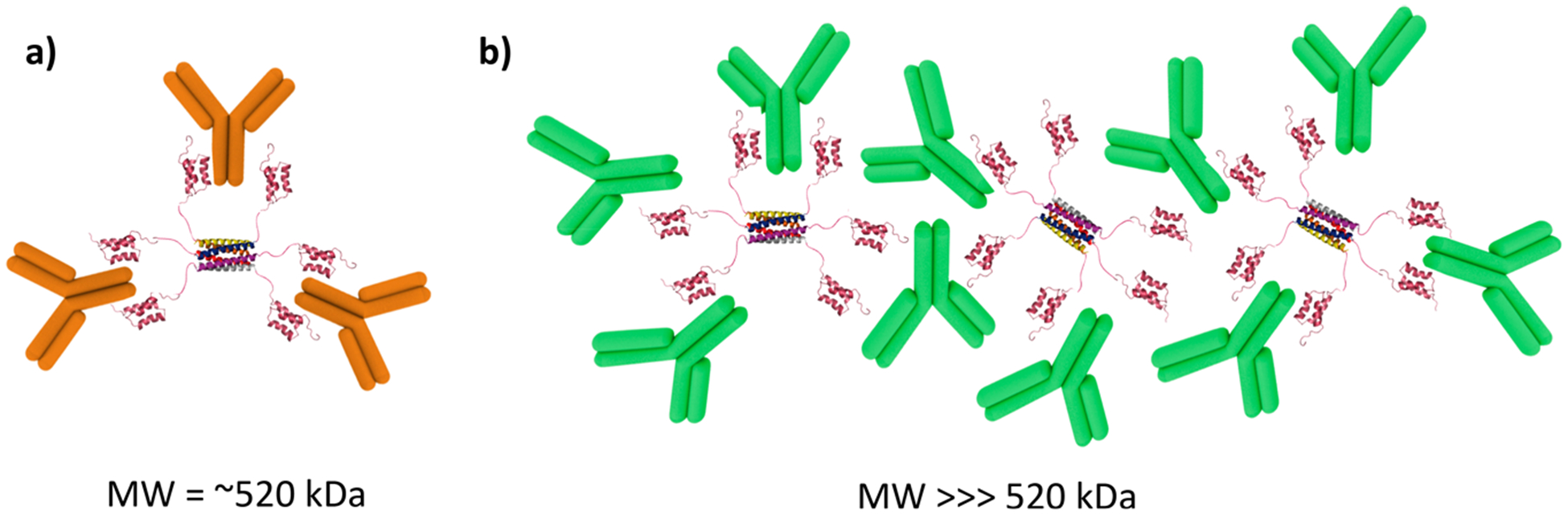
(a) Schematic of a Hex–antibody complex in a 1:3 binding ratio. The theoretical molecular weight of this complex is 520 kDa. (b) Schematic of a theoretical Hex–antibody oligomer where some antibodies are bound by multiple carriers, causing formation of a network of proteins. The molecular weight of a large oligomer is much larger than that of the 1:3 Hex–antibody complex. Not drawn to scale.
Samples of Hex–IgG 1:3 were also analyzed by the OmniSEC instrument to confirm the results of the AUC analysis (Figure 3c). The OmniSEC instrument consists of an analytical SEC column (12 mL) followed by an integrated detector module with a refractive index (RI) detector, low-angle and right-angle light scattering (LALS/RALS), and a viscometer. Species separated by the SEC column are analyzed by the detector module, and the combination of the signals when compared with a BSA standard provides molecular weight estimates for each distinct peak. The UV chromatogram of a 1:3 Hex–IgG sample showed two peaks, analogous to the SEC data obtained with the same concentration of Hex and IgG. The elution volumes for the populations are different than for SEC data because the column used for separation on OmniSEC is smaller. The relative intensity of these peaks is also different in the OmniSEC data, potentially because the sample is run at 20 °C and SEC is run at 4 °C. AUC is run at 24 °C. The next section describes in more detail how temperature affects the populations of different Hex–IgG complexes.
Upon analyzing the OmniSEC Data, the molecular weight of the first peak was estimated to be 1.6 MDa, again indicating the formation of a large oligomer with different carriers bound to the same IgG. The discrepancy in this larger species observed with AUC and OmniSEC may be due to the time scale of analysis relative to the dynamics of the complexes, as discussed in the next section. The OmniSEC experiment is performed in 20 min, whereas the AUC data is collected during a 20 to 30 h centrifugation period. The OmniSEC results confirm the presence of a 1:3 Hex–IgG complex in the second peak. The estimated molecular weight of 522 kDa aligns with the theoretical molecular weight of 521 kDa for a 1:3 complex. Thus, both the AUC and OmniSEC results confirm that a 1:3 Hex–IgG mixing ratio saturates the Hex carriers with IgG. However, mixing in this ratio led to the formation of two separate species, one with 1:3 stoichiometry as expected and another larger oligomer of more complex stoichiometry. For the case of therapeutic applications or pharmaceuticals, the presence of multiple species without tight control over which is more likely to be formed is not acceptable. Given the wide differences in size of the two identified species, they may behave differently as well, complicating the assessment of the Hex carrier’s drug delivery capabilities. For any protein assembly, even those without therapeutic applications, the existence of multiple populations obscures the assembly properties and makes it difficult to determine the structure(s) and structure–function relationships. Further characterization was therefore required to determine the source of the polydispersity of Hex–IgG complexes and if it can be reduced.
Dynamic Rearrangement of Hex–IgG Complexes.
The formation of Hex–IgG complexes is driven by the affinity binding interaction between one SPAB domain and one Fc binding site. Due to the reversible nature of affinity interactions as well as different molecular weight estimates of the larger Hex–IgG oligomer from AUC and OmniSEC, we hypothesized that the formation of Hex–IgG complexes was dynamic, and the stoichiometry could change over time. Since binding kinetics are affected by temperature,33 we used dynamic light scattering to measure the average size of Hex–IgG complexes stored at different temperatures over the course of 2 weeks. Hex and IgG were mixed in a 1:3 ratio at the same concentrations used for SEC and OmniSEC and were then incubated at 4 °C, room temperature (25 °C), and 37 °C. During the course of incubation, we observed that precipitates formed in samples stored at 4 °C after a few hours of mixing Hex and IgG together, making DLS analysis infeasible. Some samples stored at 25 °C also showed precipitation following a few days of incubation. The 25 °C data presented here are from samples that did not precipitate. DLS analysis shows that the Hex–IgG complexes stored at both 25 and 37 °C initially had a particle size of 40–45 nm and a polydispersity index (PDI) of 0.203 (Figure 5). For reference, Hex has a DLS size of ∼11 nm and IgG ∼15 nm.25 Over time, the size increased rapidly, then decreased to 25 nm and a PDI of 0.189 (25 °C) and 0.225 (37 °C), and stabilized by the end of the 2-week study. However, samples stored at 37 °C reached 25 nm with a PDI of 0.287 after just 48 h as opposed to 1 week for the 25 °C samples. Secondary structure analysis of a 1:3 Hex–IgG mixture after incubation at 37 °C showed that the formation of protein complexes did not alter the secondary structure of IgG (Figure S2b). The CD spectra of IgG alone shows a peak at 218 nm, characteristic of β sheet content signature. Upon mixing with Hex, the spectra widened to resemble the possible sum of Hex and IgG structural contributions, including the α helical content of Hex at 208 and 222 nm. The CD results confirm the structural integrity of both proteins upon formation of a higher order assembly. These results confirm that the Hex–IgG complexes are dynamic and stable upon binding and that temperature affects the rate of rearrangement.
Figure 5.
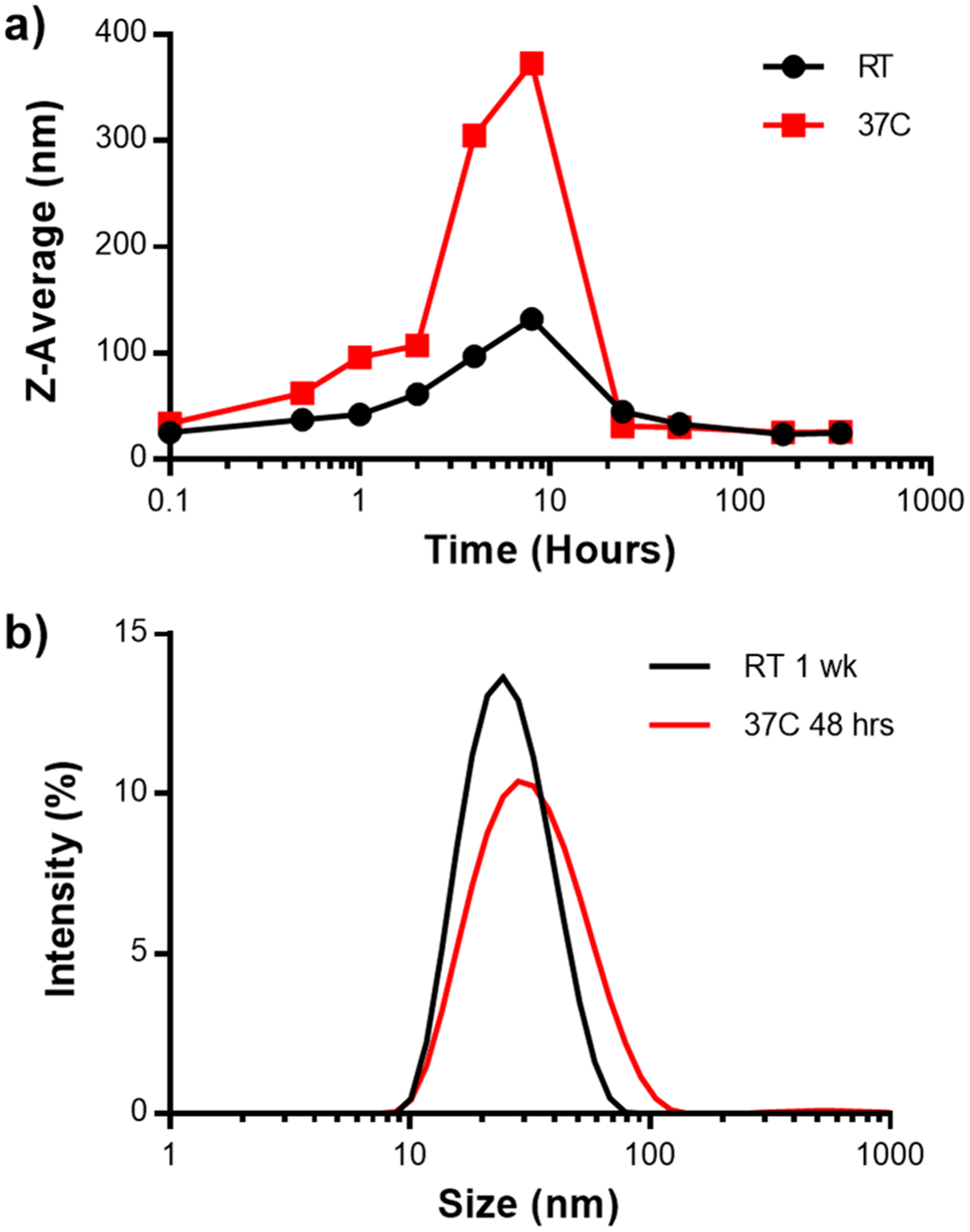
(a) Average particle size of Hex–IgG complexes as measured by DLS over a two week time period, incubated at 25 or 37 °C. (b) DLS intensity plot of Hex–IgG samples after incubating for 1 week at room temperature (RT) or 48 h at 37 °C.
Since DLS can only provide average particle size estimates for the samples tested, OmniSEC was used to estimate the molecular weight and identify the stoichiometry of the 25 nm average diameter complexes formed at the two storage conditions. Analysis of the Hex–IgG samples stored at 37 °C for 48 h sample showed a primary peak on the chromatogram, with a slight shoulder to the left (Figure 6a). The OmniSEC software estimated a molecular weight of 532 kDa (37 °C) and 612 kDa (25 °C, Figure 6b) for the primary peak, both of which most closely correspond to a 1:3 Hex–IgG stoichiometry. Using analytical software for the OmniSEC instrument, peak areas were integrated to give the relative composition of each sample. The shoulders of the peaks were treated as individual peaks only if the RALS/LALS chromatograms exhibited distinct differences in those peaks, as light scattering signals are more sensitive to different-sized assemblies (Figure S5a and b). Using this methodology, we found that the larger oligomer peak area was reduced from 33% initially (Figure 3c) to less than 16% of the total 25 °C sample at 1 week and less than 14% of the total 37 °C sample at 48 h. This result suggested that the larger complex rearranges into the 1:3 orientation over longer time periods and that higher incubation temperatures accelerates this rearrangement. Next, multiple time periods of Hex–IgG incubation at 37 °C were tested to determine how much time was required to reach equilibrium between populations. (Figure S6a and b) Interestingly, a dramatic reduction in the proportion of the larger Hex–IgG oligomer was seen at 10 h, which was the earliest time point tested. Over time, the proportion of the oligomer fluctuated between 15 and 21%. The retention volume of the shoulder for the oligomer peak also fluctuated over time, suggesting that the larger oligomer exhibits more dynamics than the 1:3 complex and is likely not as stable over time.
Figure 6.
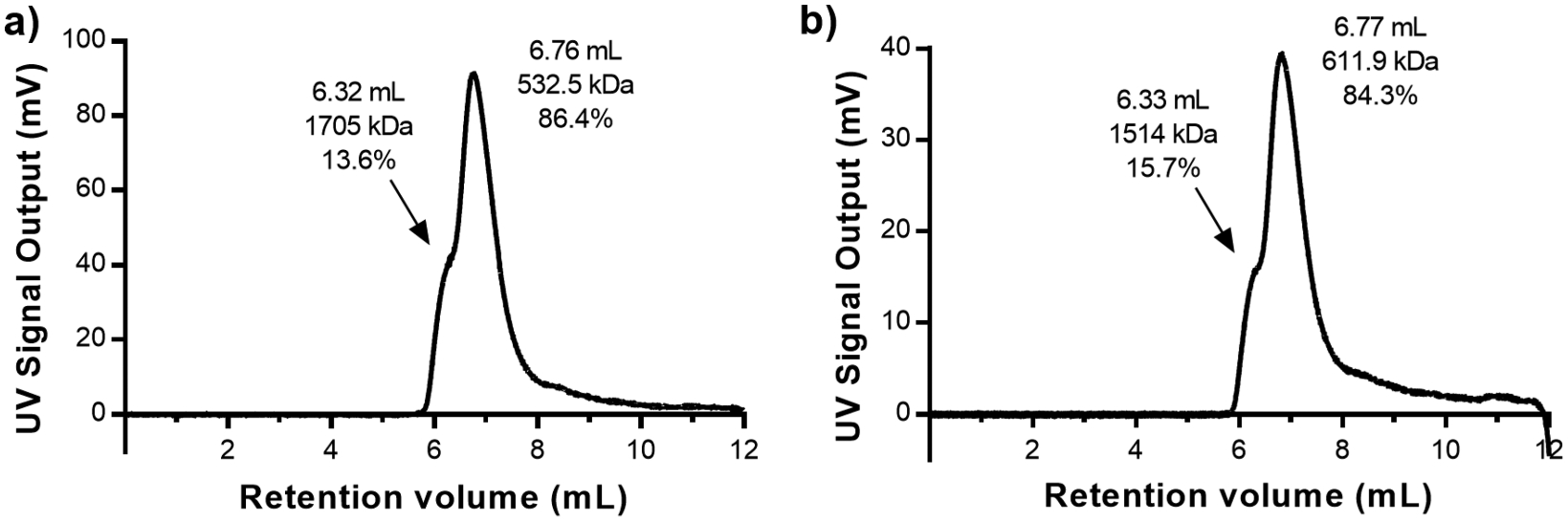
OmniSEC chromatogram and peak analysis for Hex–IgG samples incubated at 37 °C for 48 h (a) and room temperature for 1 week (b).
The transition that Hex–IgG complexes undergo over time at different incubation temperatures can be explained by the path they travel across the energy landscape. In an analogous process of protein folding, proteins sample various intermediate states before reaching the most stable state with the lowest energy. Some of the states sampled along this path can be local minima, and the protein can be kinetically trapped in those states.34 A similar process occurs with intermolecular interactions between proteins and can be used to explain how Hex and IgG rearrange into different stoichiometric orientations.35 The kinetically favorable orientation of Hex–IgG complexes is one where rapid binding of SPAB domains to IgG causes oligomerization into a large complex. At low incubation temperatures such as 4 °C and sometimes at 25 °C, this rapid binding may cause formation of large precipitates. Due to the low temperatures, both the dissociation and local diffusion of the SPAB domains and IgG molecules is reduced and may be trapping the proteins in the precipitated state, a local energy minimum. At higher temperatures (37 °C), the increased association and dissociation of SPAB domains and IgG molecules and faster diffusion of the proteins allows the intermolecular assemblies to sample more conformations. Finally, when two SPAB domains are bound to one IgG molecule, as in the case of 1:3 Hex–IgG complexes, a thermodynamically stable state is reached, where the energy barrier to sample an alternate orientation is much higher, as it requires the simultaneous dissociation of two SPAB domains as well as the diffusion of IgG molecules to and from the SPAB domains. This represents a global energy minimum, which is critical for controlling the loading of antibodies onto the Hex carrier and maintaining this orientation during drug delivery.
Characterization of SPAB–Hex–SPAB complexes.
The larger Hex–IgG oligomer most likely exists because of the ability of SPAB domains from different Hex carriers to bind to the same antibody, since both the Hex and IgG are multivalent. We hypothesized that increasing the number of SPAB domains on one Hex carrier would reduce the presence of large oligomers by increasing the chances that an antibody is bound by two SPAB domains from the same carrier. To address this question, we designed and produced SPAB–Hex–SPAB (SHS), a variant of the Hex carrier with one SPAB domain on each terminus of a single Hex monomer, for a total of 12 SPAB domains on the self-assembled SHS hexameric assembly (Figure 1b). We characterized SHS–IgG complexes in a similar manner to Hex–IgG to identify if the same trends of antibody loading and temperature-dependent dynamics occurred. First, multiple mixing ratios of SHS to IgG were evaluated using OmniSEC to identify the preferred IgG loading stoichiometry (Figure 7a). The SHS concentration in all samples was half of the Hex sample, as SHS contains twice the number of binding domains. OmniSEC results showed that ratios of 1:8 and 1:12 SHS:IgG exhibited a free IgG peak at an elution volume of 8.3 mL (Figure S4b). The 1:6 ratio did not show an unbound IgG peak, suggesting that 6 antibodies is the maximum loading of SHS carriers containing 12 SPAB domains. This stoichiometry is consistent with the observed Hex:IgG stoichiometry of 3 antibodies for 6 SPAB domains.
Figure 7.
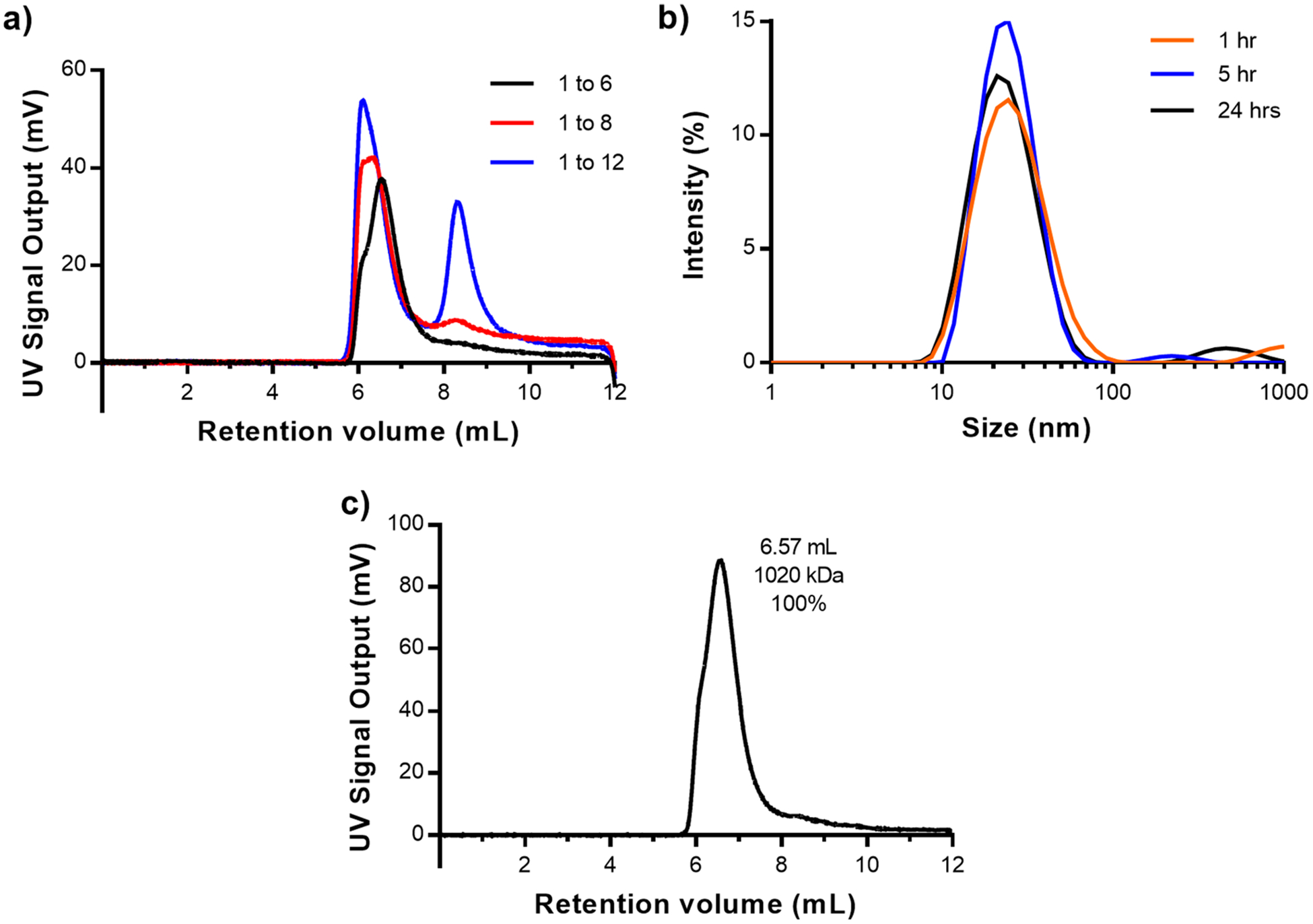
(a) OmniSEC chromatogram of SHS and IgG mixed in ratios of 1 SHS to 6, 8, and 12 IgG. Peaks at 8.5 mL are indicative of unbound IgG. (b) DLS intensity plot of SHS–IgG in a 1:6 ratio incubated at 37 °C over 24 h. (c) OmniSEC chromatograms and peak analysis for SHS–IgG samples incubated at 37 °C for 10 h.
Next, the dynamics of SHS and IgG at different temperatures were evaluated using DLS. Precipitation of proteins was observed for samples incubated at 4 °C within a few hours of sample preparation and for samples incubated at 25 °C within 24 h of preparation. Unlike the Hex samples, where precipitates at 25 °C were observed in 2 of the 4 cases tested, SHS–IgG samples showed precipitation in all 25 °C cases tested. Only the samples incubated at 37 °C did not show precipitation. These visual observations suggest that the increase in SPAB domains on a single carrier can promote the formation of a kinetically trapped precipitated state. In this state, the SHS oligomer may require even more energy than the Hex oligomers to overcome the energy barrier and move to a different configuration. Due to this precipitation, only samples of SHS–IgG in a 1:6 ratio at 37 °C were tested by DLS. Results showed that the particle size of SHS–IgG complexes stayed constant around 28 nm with a PDI of 0.256 to 0.293 over 24 h of storage at 37 °C (Figure 7b). Compared with Hex–IgG complexes, the size of SHS–IgG complexes did not fluctuate as much and reached a stable particle size sooner. Additionally, the secondary structure of SHS–IgG complexes resembled that of Hex–IgG, with a broader CD spectra than IgG alone to account for α helical contributions of SHS (Figure S2a and b).
OmniSEC analysis was then used to characterize the distribution of different species of SHS–IgG proteins when stored at 37 °C over time. Compared to Hex–IgG, the chromatograms of SHS–IgG suggested a tighter distribution of species with a much smaller shoulder on the left of the primary peak (Figure 7c). At a time point of 10 h after incubation, the molecular weight estimate of 1020 kDa almost exactly matched the theoretical molecular weight of a 1:6 SHS to IgG complex. Given that the retention volume of this peak did not change drastically over 48 h (Figure S6c and d), we conclude that a stable 1:6 SHS–IgG complex was also formed within 10 h of incubation and that the proportion of a larger oligomer is much smaller compared to that of the Hex–IgG mixtures. Using the criteria of a distinct peak in the light scattering chromatogram, the subtle shoulder on the primary SHS peak corresponding to a large oligomer was not treated as a separate peak. Combined with the reduced fluctuation in particle sizes, the OmniSEC results show that increasing the number of SPAB domains was able to reduce the amount of large oligomers and promote formation of a monodisperse, well-defined 1:6 complex of SHS and IgG.
Stabilization of HS–IgG Complexes.
After conditions to produce a monodisperse population of SHS–IgG complexes were identified, we applied the same conditions to a previously unstable mixture of Hex–SPAB (HS) (Figure 1c) and IgG. As previously reported, the mixing of HS and IgG, without a reassembly step to produce balanced Hex carriers, led to precipitation within 1 h at 25 °C.25 Since we observed similar precipitation with SHS–IgG complexes, we aimed to improve the stability of HS–IgG complexes using the same 37 °C incubation temperature as SHS–IgG. Similar to SHS, the HS protein has six SPAB domains in close proximity on a single side of the coiled coil. To test if HS could form stable complexes with IgG at higher temperatures, HS and IgG were mixed in a 1:3 ratio and incubated at either 25 or 37 °C. As expected, the HS–IgG samples stored at 25 °C showed visible precipitation after 1 h. The 37 °C samples did not show precipitation and were analyzed by DLS at 1 and 18 h after mixing (Figure 8). The results showed that HS–IgG complexes had reached a size of 25 nm with a PDI of 0.161 in just 1 h after mixing. After 18 h, the size was 28 nm with a PDI of 0.224. This result matched the stable particle sizes seen with SHS–IgG samples at early time points, demonstrating an improvement over Hex–IgG complexes. As with both Hex–IgG and SHS–IgG complexes, the CD spectra for HS–IgG was wider than that of IgG alone, including the IgG contribution and α helical contribution from HS (Figure S2a and b). Again, the close proximity of multiple SPAB domains on one side of the coiled coil may have promoted the formation of a monodisperse, well-defined HS–IgG complex.
Figure 8.
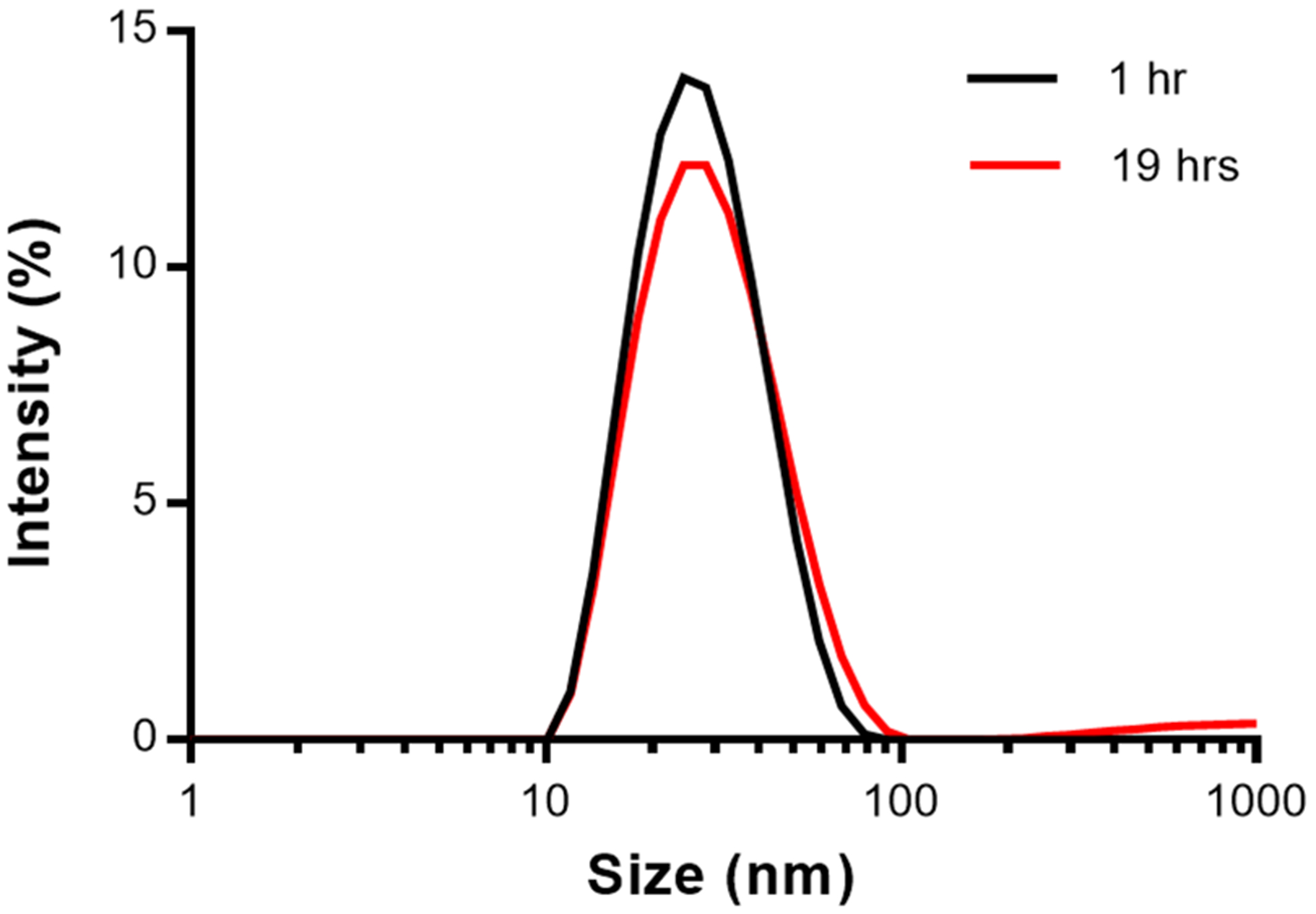
DLS intensity plot of HS–IgG in a 1:3 ratio, incubated at 37 °C for 1 and 19 h.
Post-Equilibrium Storage Analysis.
Since the use of the Hex proteins is for antibody drug delivery, the stability of Hex–IgG complexes after equilibrium at different storage temperatures was analyzed. Although incubation of IgG mixtures with Hex, HS, and SHS at 37 °C was needed for improving the monodispersity of the complexes and preventing precipitation, long-term storage of 37 °C is not feasible for a potential therapeutic. We hypothesized that monodisperse complexes that had reached an equilibrium would maintain their size when stored at lower temperatures. To test this, we prepared IgG mixtures with Hex, SHS, and HS proteins, incubated them at 37 °C for 18 h to reach equilibrium, and then stored them at either 25 or 4 °C for 2 days. Samples were analyzed by DLS to observe changes in particle size during this storage period. All three complexes showed particle sizes between 27 to 35 nm with a PDI between 0.110 and 0.350 immediately after the 37 °C incubation period. As we hypothesized, Hex–IgG samples maintained their size when stored at 25 and 4 °C over the course of at least 2 days (Figure S6a and b). However, HS–IgG and SHS–IgG showed reduced stability compared to Hex. HS–IgG maintained its size at 25 °C over 2 days, but the DLS analysis at 1 and 2 day time points at 4 °C indicated an aggregation of complexes (Figure S6c and d). The particle sizes of SHS–IgG slightly increased at 25 °C storage but also aggregated at 4 °C (Figure S6d and f). For all room temperature readings, the PDI remained between 0.121 and 0.315, indicating little fluctuation in monodispersity. These results suggest that, at storage temperatures of 25 °C, the equilibrium of complexes was maintained, and long-term storage at 25 °C could be explored if these proteins were used as therapeutics. The 4 °C results for HS and SHS, however, suggested that the equilibrium may be disrupted and complexes could escape the energy minimum and transition back to a kinetically trapped state of a larger oligomer. While unexpected, the results indicate that strategies to covalently link monodisperse protein assemblies may be needed to prevent further dynamics and improve long-term stability at lower temperatures.
CONCLUSION
In this work, we used multiple, orthogonal characterization techniques to characterize and subsequently control the structure of a self-assembled protein complex with an eye toward future therapeutic applications of the assembly. The Hex and SHS carriers bind antibodies through multivalent affinity interactions between SPAB domains and the Fc region of antibodies. SEC analysis demonstrated that the most stable loading of both Hex and SHS was in a 2:1 SPAB to IgG stoichiometry, which results in the loading of 3 antibodies for the Hex carrier and 6 antibodies for SHS. When Hex–IgG complexes were characterized over a two-week time frame at different incubation temperatures, a transition between larger Hex–IgG oligomers to the 1:3 complex was seen. The larger oligomer most likely represents a kinetically favorable assembly, driven by nanomolar binding of the SPAB domains to antibodies. Increasing the time or temperature of incubation promoted the formation of a thermodynamically stable 1:3 Hex:IgG complex; however, a small proportion of the Hex–IgG complexes were still present as an oligomer. The uniformity of assemblies was improved in the case of SHS–IgG, where doubling the SPAB domains via fusion protein design increased the likelihood of reaching thermodynamic equilibrium earlier. SHS–IgG complexes showed improved size stability compared to Hex–IgG, and most of the SHS–IgG complexes were in a 1:6 orientation, with little to no detectable oligomer. Furthermore, a version of the Hex carrier, HS, which was previously shown to be unstable in mixtures with IgG, improved its stability and monodispersity when incubated at higher temperatures. For these particular assemblies, Hex–IgG and SHS–IgG, a lower proportion of oligomers results in a more monodisperse population of antibody carriers with known loading, which improves their potential for therapeutic intracellular antibody delivery applications.
More generally, this work highlights the need to perform characterization with multiple orthogonal techniques to fully understand multivalent, affinity-based protein assemblies. While DLS gives quick readings of average particle size over time/temperature, SEC, AUC, and OmniSEC together provided a much more detailed breakdown of the proportion of different species in the sample and the stoichiometry and molecular weight of the assemblies. It is important to compare a technique such as AUC, which assesses protein complexes in their native state (buffer and concentration), free from artifacts due to a column matrix or increased sample concentration needed for detection. The combination of DLS, SEC, OmniSEC, and AUC make up a comprehensive and orthogonal suite of characterization techniques that can be applied to other nanoscale protein assemblies, including those with therapeutic applications. This type of analysis could also prove useful for other types of drug nanocarriers such as those made from polymers, polysaccharides, or lipids to complex with different drug cargos, especially given the importance of understanding drug loading, aggregation propensity, and stability as well as the use of FDA-recommended orthogonal techniques. With this characterization in place, Hex, SHS, and other assemblies can be better designed and evaluated for therapeutic, diagnostic, and biocatalytic applications.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the support of the Biopolymer Characterization Core in the Parker H. Petit Institute for Bioengineering and Bioscience at the Georgia Institute of Technology, specifically Dr. Bettina Bommarius for help with OmniSEC experiments and Dr. John Robbins for help with AUC experiments.
Funding
This research was financially supported by the National Institutes of Health, Award No. 5R21EB022794-02, from the National Institute of Biomedical Imaging and Bioengineering, and by The Shurl and Kay Curci Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.9b01712.
SDS-PAGE analysis of protein purity, CD analysis of carriers with and without IgG, SEC chromatograms of multiple Hex–IgG binding ratios, OmniSEC analysis of Hex and IgG controls, OmniSEC analysis of RALS/ LALS data showing the methodology for peak detection, OmniSEC analysis of multiple incubation time points for Hex–IgG and SHS-IgG samples, and DLS analysis of Hex-IgG, SHS-IgG, and HS-IgG incubated at lower temperatures after reaching equilibrium (PDF)
The authors declare no competing financial interest.
Contributor Information
Anshul Dhankher, School of Chemical & Biomolecular Engineering and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
Manuel E. Hernandez, School of Chemical & Biomolecular Engineering and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States
Hannah C. Howard, School of Chemical & Biomolecular Engineering and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States
Julie A. Champion, School of Chemical & Biomolecular Engineering and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States;.
REFERENCES
- 1.Luo Q; Hou C; Bai Y; Wang R; Liu J Protein Assembly: Versatile Approaches to Construct Highly Ordered Nanostructures. Chem. Rev 2016, 116 (22), 13571–13632. [DOI] [PubMed] [Google Scholar]
- 2.Dawson WM; Rhys GG; Woolfson DN Towards Functional de Novo Designed Proteins. Curr. Opin. Chem. Biol 2019, 52, 102–111. [DOI] [PubMed] [Google Scholar]
- 3.Herrera Estrada LP; Champion JA Protein Nanoparticles for Therapeutic Protein Delivery. Biomater. Sci 2015, 3 (6), 787–799. [DOI] [PubMed] [Google Scholar]
- 4.Leng Y; Wei HP; Zhang ZP; Zhou YF; Deng JY; Cui ZQ; Men D; You XY; Yu ZN; Luo M; Zhang XE Integration of a Fluorescent Molecular Biosensor into Self-Assembled Protein Nanowires: A Large Sensitivity Enhancement. Angew. Chem., Int. Ed 2010, 49 (40), 7243–7246. [DOI] [PubMed] [Google Scholar]
- 5.Porter JR; Stains CI; Jester BW; Ghosh I A General and Rapid Cell-Free Approach for the Interrogation of Protein-Protein, Protein-DNA, and Protein-RNA Interactions and Their Antagonists Utilizing Split-Protein Reporters. J. Am. Chem. Soc 2008, 130 (20), 6488–6497. [DOI] [PubMed] [Google Scholar]
- 6.Liu J; Hou C; Luo Q; Liu J; Miao L; Zhang C; Gao Y; Zhang X; Xu J; Dong Z Construction of GPx Active Centers on Natural Protein Nanodisk/Nanotube: A New Way to Develop Artificial Nanoenzyme. ACS Nano 2012, 6 (10), 8692–8701. [DOI] [PubMed] [Google Scholar]
- 7.Williams EM; Jung SM; Coffman JL; Lutz S Pore Engineering for Enhanced Mass Transport in Encapsulin Nano-compartments. ACS Synth. Biol 2018, 7 (11), 2514–2517. [DOI] [PubMed] [Google Scholar]
- 8.Fisher AC; Lee SL; Harris DP; Buhse L; Kozlowski S; Yu L; Kopcha M; Woodcock J Advancing Pharmaceutical Quality: An Overview of Science and Research in the U.S. FDA’s Office of Pharmaceutical Quality. Int. J. Int. J. Pharm 2016, 515 (1–2), 390–402. [DOI] [PubMed] [Google Scholar]
- 9.Roberts CJ Therapeutic Protein Aggregation: Mechanisms, Design, and Control. Trends Biotechnol. 2014, 32 (7), 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu M; Zeng R; Xiang J; Yan Q Shaping Protein Amphiphilic Assemblies via Allosteric Effect: From 1D Nanofilament to 2D Rectangular Nanosheet. J. Am. Chem. Soc 2019, 141 (35), 13724–13728. [DOI] [PubMed] [Google Scholar]
- 11.Wong Po Foo CTS; Lee JS; Mulyasasmita W; Parisi-Amon A; Heilshorn SC Two-Component Protein-Engineered Physical Hydrogels for Cell Encapsulation. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (52), 22067–22072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts EK; Wong KM; Lee EJ; Le MM; Patel DM; Paravastu AK Post-Assembly α-Helix to β-Sheet Structural Transformation within SAF-P1/P2a Peptide Nanofibers. Soft Matter 2018, 14 (44), 8986–8996. [DOI] [PubMed] [Google Scholar]
- 13.Sutter M; McGuire S; Ferlez B; Kerfeld CA Structural Characterization of a Synthetic Tandem-Domain Bacterial Micro-compartment Shell Protein Capable of Forming Icosahedral Shell Assemblies. ACS Synth. Biol 2019, 8 (4), 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun H; Miao L; Li J; Fu S; An G; Si C; Dong Z; Luo Q; Yu S; Xu J; Liu J Self-Assembly of Cricoid Proteins Induced by “Soft Nanoparticles”: An Approach to Design Multienzyme-Cooperative Antioxidative Systems. ACS Nano 2015, 9 (5), 5461–5469. [DOI] [PubMed] [Google Scholar]
- 15.Kitagishi H; Kakikura Y; Yamaguchi H; Oohora K; Harada A; Hayashi T Self-Assembly of One-And Two-Dimensional Hemoprotein Systems by Polymerization through Heme-Heme Pocket Interactions. Angew. Chemie -Int. Angew. Chem., Int. Ed 2009, 48 (7), 1271–1274. [DOI] [PubMed] [Google Scholar]
- 16.Hattori M; Hibbs RE; Gouaux E A Fluorescence-Detection Size-Exclusion Chromatography-Based Thermostability Assay for Membrane Protein Precrystallization Screening. Structure 2012, 20 (8), 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharjee S DLS and Zeta Potential -What They Are and What They Are Not? J. Controlled Release 2016, 235, 337–351. [DOI] [PubMed] [Google Scholar]
- 18.Carpenter JF; Randolph TW; Jiskoot W; Crommelin DJA; Middaugh CR; Winter G Potential Inaccurate Quantitation and Sizing of Protein Aggregates by Size Exclusion Chromatography: Essential Need to Use Orthogonal Methods to Assure the Quality of Therapeutic Protein Products. J. Pharm. Sci 2010, 99 (5), 2200–2208. [DOI] [PubMed] [Google Scholar]
- 19.Balbo A; Minor KH; Velikovsky CA; Mariuzza RA; Peterson CB; Schuck P Studying Multiprotein Complexes by Multisignal Sedimentation Velocity Analytical Ultracentrifugation. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (1), 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demeule B; Shire SJ; Liu J A Therapeutic Antibody and Its Antigen Form Different Complexes in Serum than in Phosphate-Buffered Saline: A Study by Analytical Ultracentrifugation. Anal. Biochem 2009, 388 (2), 279–287. [DOI] [PubMed] [Google Scholar]
- 21.Liu J; Yadav S; Andya J; Demeule B; Shire SJ Analytical Ultracentrifugation and Its Role in Development and Research of Therapeutical Proteins. Methods Enzymol. 2015, 562, 441–476. [DOI] [PubMed] [Google Scholar]
- 22.van Dieck J; Fernandez-Fernandez MR; Veprintsev DB; Fersht AR Modulation of the Oligomerization State of P53 by Differential Binding of Proteins of the S100 Family to P53 Monomers and Tetramers. J. Biol. Chem 2009, 284 (20), 13804–13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malvern Panalytical Worldwide. Characterization of IgG Monomers and Their Aggregates; 2018. [Google Scholar]
- 24.U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research; Center for Veterinary Medicine. Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Bioanalytical Method Validation Guidance for Industry Biopharmaceutics; 2018. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidancefor-Industry.pdf.
- 25.Lim SI; Lukianov CI; Champion JA Self-Assembled Protein Nanocarrier for Intracellular Delivery of Antibody. J. Controlled Release 2017, 249, 1–10. [DOI] [PubMed] [Google Scholar]
- 26.Zaccai NR; Chi B; Thomson AR; Boyle AL; Bartlett GJ; Bruning M; Linden N; Sessions RB; Booth PJ; Brady RL; Woolfson DN A de Novo Peptide Hexamer with a Mutable Channel. Nat. Chem. Biol 2011, 7 (12), 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhde-Holzem K; McBurney M; Tiu BD; Advincula RC; Fischer R; Commandeur U; Steinmetz NF Production of Immunoabsorbent Nanoparticles by Displaying Single-Domain Protein A on Potato Virus X. Macromol. Biosci 2016, 16 (2), 231–241. [DOI] [PubMed] [Google Scholar]
- 28.Grodzki AC; Berenstein E Affinity Chromatography Vol. 1: Antibodies; GE Healthcare: 2015; Vol. 1 https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/General_Information/1/ge-affinity-chromatography.pdf. [Google Scholar]
- 29.Hutt M; Färber-Schwarz A; Unverdorben F; Richter F; Kontermann RE Plasma Half-Life Extension of Small Recombinant Antibodies by Fusion to Immunoglobulin-Binding Domains. J. Biol. Chem 2012, 287 (7), 4462–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howlett GJ; Minton AP; Rivas G Analytical Ultracentrifugation for the Study of Protein Association and Assembly. Curr. Opin. Chem. Biol 2006, 10 (5), 430–436. [DOI] [PubMed] [Google Scholar]
- 31.Robbins JM; Bommarius AS; Gadda G Mechanistic Studies of Formate Oxidase from Aspergillus Oryzae: A Novel Member of the Glucose-Methanol-Choline Oxidoreductase Enzyme Superfamily That Oxidizes Carbon Acids. Arch. Biochem. Biophys 2018, 643, 24–31. [DOI] [PubMed] [Google Scholar]
- 32.Krayukhina E; Uchiyama S; Fukui K Effects of Rotational Speed on the Hydrodynamic Properties of Pharmaceutical Antibodies Measured by Analytical Ultracentrifugation Sedimentation Velocity. Eur. J. Pharm. Sci 2012, 47 (2), 367–374. [DOI] [PubMed] [Google Scholar]
- 33.Scott Fogler H Elements of Chemical Reaction Engineering, 5th ed.; Prentice Hall: 2016; Vol. 42. . [Google Scholar]
- 34.Eichner T; Radford SE A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Mol. Cell 2011, 43 (1), 8–18. [DOI] [PubMed] [Google Scholar]
- 35.Korevaar PA; George SJ; Markvoort AJ; Smulders MMJ; Hilbers PAJ; Schenning APHJ; De Greef TFA; Meijer EW Pathway Complexity in Supramolecular Polymerization. Nature 2012, 481 (7382), 492–496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.