

Abstract
Symptomatic plasma cell dyscrasias can arise in patients following renal transplantation. These patients are likely to have higher serum creatinine and proteinuria as well as higher levels of M-protein, free light chain ratio, and percentage of plasma cells in bone marrow. There is a risk of progression to multiple myeloma, and these patients should have close follow-up and monitoring.
Introduction:
Plasma cell disorders (PCDs) are clonal plasma cell disorders that include conditions such as monoclonal gammopathy of undetermined significance (MGUS), monoclonal gammopathy of renal significance (MGRS), multiple myeloma (MM), smoldering MM (SMM), solitary plasmacytoma, and light-chain (AL) amyloidosis. The risk factors associated with and the clinical course of PCDs after renal transplantation is not well established although immunosuppressive protocols may impact the incidence and natural history of PCDs posttransplant.
Patients and Methods:
This single-center retrospective study evaluated patients with a history of renal transplant who developed a PCD between January 1, 2014-December 31, 2018.
Result:
A total of 41 patients met the inclusion criteria including 29 with MGUS and 12 with symptomatic PCD (4 with MM, 2 with SMM, 4 with MGRS, 1 with AL amyloidosis, and 1 with solitary plasmacytoma). The median follow-up of survivors was 41.6 months. Three patients (1 with MGUS and 2 with MGRS) progressed to MM during the follow-up period. There was a male preponderance in both groups. There was no correlation between the donor and immunosuppressive regimen and the development of a PCD. Patients with symptomatic PCD had higher serum creatinine and M-protein levels at diagnosis and higher free light chain ratio and plasma cell burden. There was also a higher percentage of allograft failure noted in the symptomatic PCD subset 50% (n = 6), whereas only 23% (n = 7) of patients had allograft failure in the MGUS group.
Conclusion:
This study shows the importance of considering monoclonal gammopathy in the differential of renal dysfunction after kidney transplant and the need to follow these patients closely to monitor for progression to symptomatic PCD.
Keywords: Amyloidosis, Monoclonal gammopathy, Multiple Myeloma, Plasma Cell Disorders, Renal transplant
Introduction
Plasma cell disorders (PCDs) are characterized by a clonal proliferation of plasma cells in the bone marrow. These cells produce monoclonal intact immunoglobulins and/or light chains (M protein), which are detectable as a paraprotein in the patient’s blood or urine. PCDs encompass a spectrum of hematologic diseases including monoclonal gammopathy of unknown significance (MGUS), monoclonal gammopathy of renal significance (MGRS), light-chain (AL) amyloidosis, smoldering multiple myeloma (SMM), solitary plasmacytoma, and MM. These entities are defined by distinct hematologic and clinical manifestations. MGUS is characterized by < 10% plasma cells in bone marrow and paraprotein (M-protein) levels < 3 g/dL. MGRS shares hematologic features with MGUS (or a small B-cell clone) and includes all renal disorders attributable to monoclonal immunoglobulin, which is distinctly nephrotoxic. MM is defined on the basis of a higher clonal burden defined as ≥ 10% plasma cells in bone marrow and M-protein levels ≥ 3 g/dL with evidence of myeloma-defining events including renal failure, anemia, hypercalcemia, and bone lytic lesions, whereas SMM shares hematologic features with MM in the absence of any myeloma-defining events.1–3 Systemic AL amyloidosis is associated with insoluble fibrillar deposits from clonal immunoglobulins causing multiorgan dysfunction with high morbidity and mortality.4
Monoclonal gammopathy is an important cause of kidney disease. It is estimated that 50% of patients with MM will develop renal impairment, with 20% to 50% presenting with renal dysfunction at the time of diagnosis.5 Renal insufficiency can worsen over time and lead to end-stage renal disease6 with progression of the underlying gammopathy. In a study by Nasr et al,7 renal survival was reported to be 20 months in patients with MM, whereas in AL amyloidosis this was only marginally better at 30 months.
Renal transplantation is a well-established alternative to dialysis for patients with end-stage renal disease and relies on the use of long-term immunosuppression to promote allograft tolerance and survival.8 It is known that long-standing immunosuppression pre-disposes these patients to an increased risk of posttransplant lymphoproliferative disorders as well as other malignancies (eg, skin cancers), but it is unclear if these patients are also at higher risk for PCD.9 Additionally, the outcomes of these diseases in the posterenal transplant setting are unclear. In the last 2 decades, several successful plasma celledirected therapies have emerged, including proteasome inhibitors (bortezomib, carfilzomib, and ixazomib), immunomodulatory drugs (thalidomide, lenalidomide, and pomalidomide), and monoclonal antibodies (daratumumab and elotuzumab), in addition to autologous stem cell transplantation, which have resulted in improved renal outcomes and overall survival of patients with symptomatic PCD.5,10–14
We undertook this single-center study to evaluate and describe the spectrum of PCD after renal transplantation. We also assessed the risk factors, impact of treatment, and clinical outcomes of different PCDs in the posttransplant period.
Methods
This was a single-center, retrospective study involving patients with a history of renal transplant in whom a first monoclonal gammopathy was first identified between January 1, 2014, and December 31, 2018. The monoclonal gammopathy was defined by the presence of a monoclonal disorder on serum or urine electrophoresis, immunofixation studies and/or abnormal free light chain levels, or bone marrow examination. Chart review was conducted to confirm the monoclonal gammopathy, which was established on 2 separate laboratory tests. The algorithm that was followed for screening and inclusion of patients is depicted in Figure 1. Subjects who developed a monoclonal gammopathy outside the study time frame and those who had other solid organ transplants (liver, lung, or heart) were excluded from the study. The study was approved by the Institutional Review Board of the Medical College of Wisconsin. Descriptive statistical analysis was performed. Continuous variables were analyzed using the t test, whereas the χ2 and Fisher exact tests were used for categoric variables.
Figure 1.
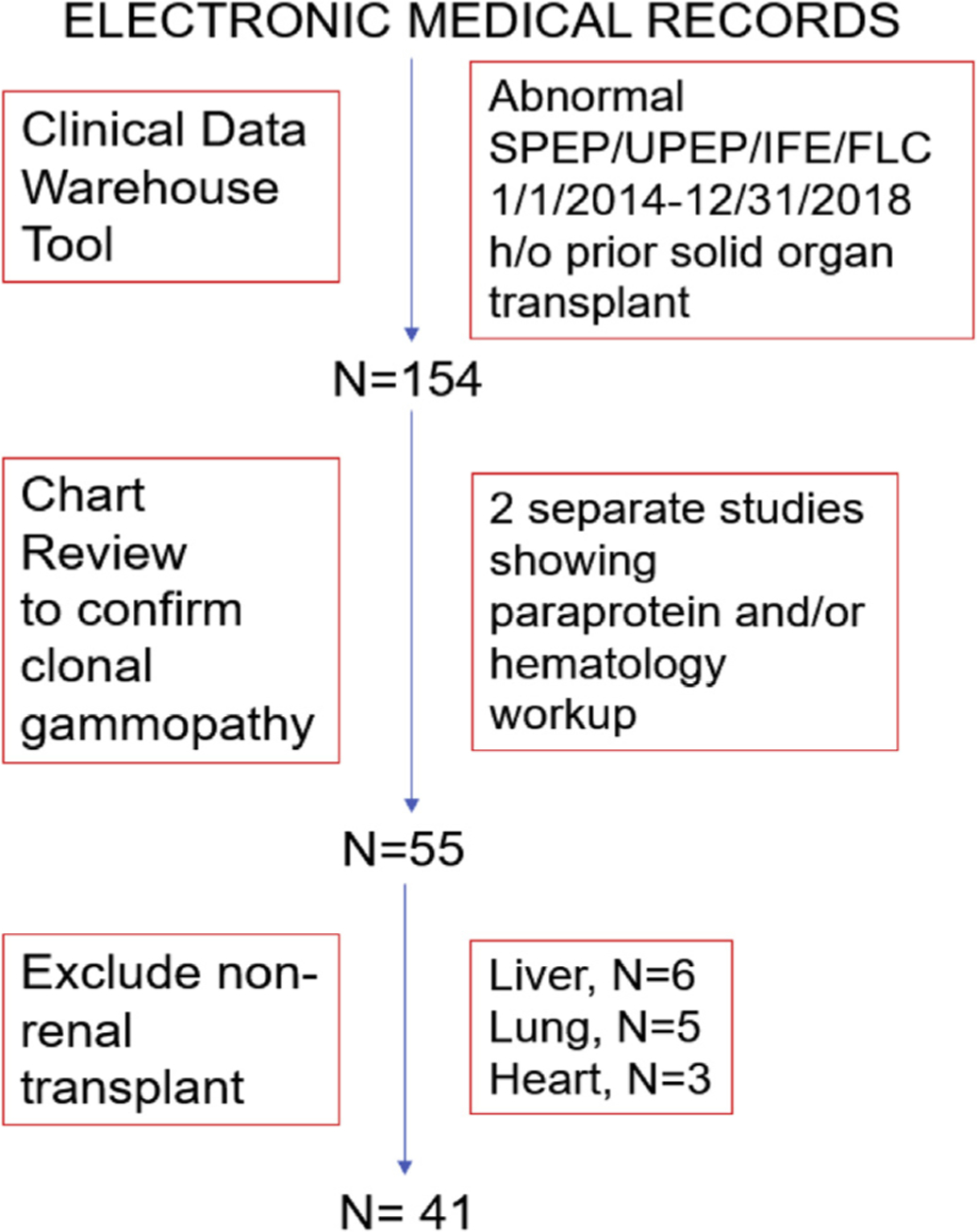
The Algorithm for the Screening and Inclusion of Patients
Results
A total of 41 patients with PCDs met the inclusion criteria; 29 patients had MGUS, whereas the remaining 12 (29%) had a symptomatic PCD, including MM in 4, smoldering myeloma in 2, MGRS in 4, AL amyloidosis in 1, and solitary extramedullary plasmacytoma in 1. The clinical characteristics of the cohort are shown in Table 1. The median age at transplant for the study population was 51 years (range, 17–81 years), whereas the median age at PCD diagnosis was 62 years (range, 37–87 years). The subject population was predominantly male (63%) and white (66%). The median free light chain ratio was 2.3 mg/L (range, 0.4–538 mg/L), whereas the median M-protein level was 0.4 g/dL (range, 0–3.8 g/ dL). A lambda clone was noted in 66% of the subjects. The primary indication for workup for PCD was abnormal creatinine or proteinuria in 41% patients followed by hematologic indication (23%). The median hemoglobin level was 11.1 g/dL (range, 6.5–15.8 g/dL), whereas calcium and albumin levels were within normal limits across the group. The median serum creatinine for subjects was similar at diagnosis (1.7 mg/dL; range, 0.7–9.8 mg/dL) and at last follow-up (1.8 mg/dL; range, 0.6–9.9 mg/dL). The median level of 24-hour proteinuria was 0.3 g (range, 0–4.7 g) based on spot urine protein/urine creatinine ratios.
Table 1.
Clinical Characteristics of Subjects With Plasma Cell Disorder (PCD) After Renal Transplant
| Total (n = 41) | MGUS (n = 29) | Symptomatic PCD (n = 12) | P Value | |
|---|---|---|---|---|
| Age at transplant, y | 51 (17–81) | 46 (17–67) | 57 (31–81) | .08 |
| Etiology of end-stage renal disease | .1 | |||
| Diabetes mellitus | 8(19) | 6(21) | 2(17) | |
| HTN | 6 (15) | 3(10) | 3(25) | |
| Glomerular diseasea | 15 (36) | 12 (41) | 3(25) | |
| Polycystic kidney disease | 6 (15) | 3(10) | 3(25) | |
| Other | 4(10) | 3(10) | 1 (8) | |
| Unknown | 2(5) | 2(7) | 0 | |
| Age at PCD diagnosis, y | 62 (37–87) | 62 (37–80) | 67 (47–87) | .2 |
| Time from transplant to PCD, y | 9.2 (0.07–31.6) | 12 (0.1–31.6) | 6.6 (1.3–23.4) | .2 |
| Sex, male (%) | 26 (63) | 16 (55) | 10 (83) | .2 |
| Race, black (%) | 14 (34) | 10 (35) | 4 (33) | 1.0 |
| Reason for testing, n = 40 | .01 | |||
| Renal, n (%) | 16 (40) | 9 (30) | 7(70) | |
| Other, n (%) | 24 (60) | 21 (70) | 3 (30) | |
| Clone type, lambda (%) | 27 (66) | 16 (55) | 11 (92) | .03 |
| Bone marrow plasma cells, %, n = 28 | 6.6 (0–90) | 2.4 (0–7.5) | 13.1 (0–90) | .006 |
| M protein, g/dL, n = 40 | 0.4 (0–3.8) | 0.3 (0–1.6) | 0.8 (0–3.8) | .04 |
| Free light chain ratio | 2.3 (0.4–538) | 1.9 (0.4–18.4) | 12.7 (0.5–538) | .006 |
| Creatinine, mg/dL | 1.7 (0.7–9.8) | 1.5 (0.7–3.3) | 2.1 (1.4–9.8) | .02 |
| Hemoglobin, g/dL | 11.2 (6.5–15.8) | 11.3 (6.5–15.8) | 10.6 (8.8–13.6) | .4 |
| Calcium, mg/dL | 9.3 (7.6–12.8) | 9.4 (8.2–10.6) | 8.7 (7.6–12.8) | .2 |
| Albumin, g/L | 3.9 (2.2–4.5) | 4 (2.2–4.5) | 3.9 (2.4–4.4) | .5 |
| Urine protein/creatinine ratio, n = 33 | 0.3 (0–4.7) | 0.3 (0–3.3) | 0.3 (0–4.7) | .5 |
| Allograft rejection after diagnosis of PCD (%) | 13 (31) | 7(23) | 6 (50) | .1 |
| Maintenance immunosuppression, n = 40 | .2 | |||
| CNI + MMF + prednisone | 22 (54) | 13 (45) | 9(75) | |
| Other | 18 (46) | 16 (55) | 2(25) | |
| Creatinine at last follow-up, mg/dL | 1.8 (0.6–9.9) | 1.6 (0.6–9.9) | 1.9 (1.1–8.8) | .1 |
Abbreviations: CNI = calcineurin inhibitor (tacrolimus or cyclosporine); HTN = hypertension; MGUS = monoclonal gammopathy of undetermined significance; MMF = mycophenolate mofetil.
Glomerular disease includes the diagnosis of membranous nephropathy, focal segmental glomerulosclerosis, systemic lupus erythematosus, Alport disease, and membranoproliferative glomerulonephritis.
The majority (61%) of subjects had undergone a deceased donor transplant, whereas the remainder had living donor transplants. End-stage renal disease was attributed to glomerular disease in 15 patients, and the other reported etiologies included diabetes mellitus (n = 8), hypertension (n = 6), and polycystic kidney disease (n = 6). Data on induction therapy pretransplant were available for only 12 subjects, and thymoglobulin was most frequently used for induction. A variety of maintenance regimens were used across the group; the most common ones included tacrolimus, mycophenolate mofetil, and prednisone. Allograft rejection was noted in 31% of the patients after PCD diagnosis during the follow-up period. The median follow-up of survivors was 41.6 months (range, 5.7–157.8 months).
When comparing the MGUS and symptomatic PCD groups (Table 1), the age at transplant and at PCD diagnosis was higher in the symptomatic PCD group. The median time from transplant to diagnosis was 6.6 years in the symptomatic PCD group compared with 12 years in the MGUS group. As expected, a higher percentage of bone marrow plasma cells, a higher median M-protein level, and a higher free light chain ratio were noted in the symptomatic PCD group compared with the MGUS group. A renal indication for testing was the most common reason leading to the diagnosis of symptomatic PCD, whereas nonrenal indications (fatigue, neuropathy, cardiomyopathy, and osteopenia) for testing for paraproteinemia predominated in the MGUS group. The median serum creatinine at diagnosis was higher in the symptomatic PCD subset compared with the MGUS subset (2.1 vs. 1.5, P = .02). The difference in the serum creatinine between the 2 groups at the last follow-up was also higher in the symptomatic PCD group but did not reach statistical significance (1.9 vs. 1.6, P = .1). There were no differences in the median hemoglobin, serum albumin, serum calcium, or amount of proteinuria.
The management of symptomatic PCD is shown in Table 2. The treatment consisted of a combination of bortezomib, cyclophosphamide, and dexamethasone in patients with MGRS and AL amyloidosis and 2 patients with MM. One patient with MM was treated with bortezomib and dexamethasone, whereas another received thalidomide and dexamethasone. One patient with MM also underwent an autologous stem cell transplant. One patient with a solitary plasmacytoma was given radiation therapy. The renal allograft rejection rate during the follow-up period was higher in the symptomatic PCD group at 50% (n = 6) versus 23% (n = 7) in the MGUS group.
Table 2.
Clinical Characteristics of the Symptomatic Plasma Cell Disorder (PCD) Subset
| PCD | Sex | Race | Age at Renal Transplant | Age at PCD Diagnosis | Clone Subtype | Initial Treatment | PCD at Last Follow-up | Renal Allograft Status at Last Follow-up | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | SMM | Male | Black | 61 | 62 | Lambda | None | SMM | No rejection |
| 2 | SMM | Male | Black | 52 | 67 | Lambda | None | SMM | Rejection |
| 3 | MM | Male | White | 67 | 68 | Lambda | VD-AutoHCT | MM | Rejection |
| 4 | MM | Male | White | 49 | 66 | Kappa | Hospice | MM | No rejection |
| 5 | MM | Male | Black | 63 | 67 | Lambda | VCD | MM | No rejection |
| 6 | MM | Male | White | 46 | 46 | Lambda | VCD-AutoHCT | MM | No rejection |
| 7 | MGRS | Male | White | 81 | 86 | Lambda | VCD | MM | Rejection |
| 8 | MGRS | Male | White | 70 | 75 | Lambda | VCD | MGRS | No rejection |
| 9 | MGRS | Male | White | 41 | 47 | Lambda | VCD | MM | Rejection |
| 10 | MGRS | Male | White | 31 | 54 | Lambda | VCD | MGRS | No rejection |
| 11 | AL | Female | White | 62 | 69 | Lambda | VCD | AL | Rejection |
| 12 | SEP | Female | White | 53 | 62 | Lambda | Radiation | SEP | Rejection |
Abbreviations: AL = light-chain amyloidosis; MGRS = monoclonal gammopathy of renal significance; MM = multiple myeloma; SEP = solitary extramedullary plasmacytoma; SMM = smoldering multiple myeloma; VD = velcade dexamethasone; VCD = velcade cyclophosphamide dexamethasone; VCD-AutoHCT = velcade cyclophosphamide dexamethasone-autologous hematopoietic stem cell transplant.
Discussion
In our study of posterenal transplant patients with PCD, we make the following observations: (1) PCD can occur after renal allograft and range across the entire spectrum of diseases; (2) patients with symptomatic PCD were more likely to have a lambda clone; (3) symptomatic PCD was more likely to present with renal indication for testing (elevated creatinine and proteinuria); and (4) during a relatively short follow-up of under 4 years, 3 patients with low clonal burden (MGUS or MGRS) progressed to MM.
The spectrum of symptomatic PCD in renal transplant recipients includes MM, SMM, solitary plasmacytoma, AL amyloidosis, and MGRS. Three patients (1 with MGUS and 2 with MGRS) progressed to MM during the follow-up period. There was a male preponderance in the MGUS and symptomatic PCD groups similar to PCD in the general population.15 Although not statistically significant, the symptomatic PCD patients were older at the time of renal transplant and at the time of PCD diagnosis. This is consistent with our current knowledge that PCD occurs more frequently in the older population.16 There was also a trend toward earlier posttransplant diagnosis for the symptomatic PCD subset when compared with the MGUS subset. However, there was no difference between the type of transplant (deceased donor vs. living donor) and maintenance immunosuppressive regimen and the occurrence of symptomatic PCD. Half of the patients with symptomatic PCD showed evidence of allograft rejection during follow-up and before the diagnosis of PCD, which could have impacted the occurrence of symptomatic PCD, the timing of symptomatic PCD diagnosis, the clinical course, and renal outcomes in this group.
A higher level of M protein, free light chains ratio, and percentage of plasma cells in bone marrow distinguished the symptomatic PCD group from the MGUS subset. This is consistent with the higher burden of the abnormal proliferating clone in the former group and expected per disease definitions. In previous studies, excess lambda light chain has been associated with higher rates of renal involvement in AL amyloid patients.17 In this study, in the symptomatic PCD patient group, the predominant clone was lambda light chain restricted. The indication for paraprotein testing was primarily renal in the symptomatic PCD group in contrast to the MGUS group where other nonrenal etiologies (fatigue, neuropathy, cardiomyopathy, and osteopenia) prompted the workup in MGUS. There was also a trend toward a higher creatinine level at diagnosis and the last follow-up in the symptomatic PCD group. This is not surprising because a serum creatinine level of 2.0 mg/dL or higher is a defining criteria for renal involvement in MM, and many patients with MM have renal impairment at the time of diagnosis.18 Patients with symptomatic PCD tended to have a lower hemoglobin level than patients with MGUS, which could be attributed to plasma cell expansion in the bone marrow.
All patients with MGRS, AL amyloid, and MM (with the exception of 1 patient) were treated with a bortezomib-based chemotherapy regimen, which is a standard of care treatment in these patients. Additionally, because bortezomib is safe in patients with renal failure, it constitutes the backbone of treatment in patients with renal dysfunction when diagnosed with symptomatic PCD.5,10,11 Despite treatment, we identified a high rate of allograft failure of 50% in the symptomatic PCD group in this cohort. Thus, our study affirms that renal impairment secondary to symptomatic PCD portends a poorer prognosis, even in the post–renal transplant setting. This finding is similar to the prognostic value of renal involvement in MM in the general population.5
The risk of MGUS has been reported to be significantly higher after kidney transplantation in contrast to the general population.19,20 These patients may progress to MM, which may complicate their posttransplant course. The expected risk of progression of MGUS to MM is about 1% per year, whereas SMM carries a higher risk of 10% per year.21,22 In our study, 1 patient with MGUS and 2 with MGRS progressed to MM during the relatively short follow-up period of our study. This may be reflective of a higher risk of progression in the renal transplant population than appreciated in previously published studies, but our numbers are small.15,19,22 This could be similar to the risk of posttransplant lymphoproliferative disorders in the posteorgan transplant period driven by the selection and expansion of unique plasma cell clones in the setting of therapeutic immune suppression causing impaired cancer immune surveillance, especially of B-cell lineages.15 Thus, these patients may need closer follow-up than recommended for asymptomatic PCD in order to assess the progression or transformation to MM.
We are limited by the small sample size from a single center giving us limited power for statistical tests and the retrospective nature of the study. Because our study only captured newly diagnosed monoclonal gammopathy in renal transplant patients during a set time period instead of longitudinally following patients who had kidney transplant within a specific time interval, it does not allow for estimation of the incidence or prevalence of symptomatic PCD after renal transplantation. Our study is a snapshot of renal transplant recipients who may have had transplant at different time points before or during the study interval and those who may have been lost to follow-up during the study interval. Although this allowed us to capture all patients with PCD and prior renal transplant, our study does not allow us to determine the incidence or prevalence of new PCD. The data on induction immunosuppression were only available in a subset of patients, and, in addition, there was considerable heterogeneity in the maintenance immunosuppressive regimens used. Thus, a causal relationship between immunosuppressive agents and the development of symptomatic PCD after transplant could not be ascertained. None of the patients included in this study had been screened for monoclonal gammopathy before renal transplant. Therefore, it is unclear if any of these patients had a preexisting monoclonal gammopathy before kidney transplant and if this could have impacted the occurrence and clinical outcomes of symptomatic PCD after transplant. Additionally, Epstein-Barr virus testing was not available in the majority of patients at the time of PCD diagnosis. Thus, the role of immune modulation after Epstein-Barr virus infection and its contribution to the development of PCD could not be assessed in our study. Our next steps would be to conduct a multi-institutional longitudinal cohort study of a larger number of kidney transplant patients who are followed from the time of transplant in order to determine the incidence of new PCD after renal transplant and establish the risk factors for the development of posttransplant PCD comparing between centers that do or do not check for monoclonal gammopathy before renal transplant. Future studies should also evaluate and compare patients developing MM after renal transplant versus those who have preexisting MM and undergo renal transplantation because these may be potentially 2 different cohorts with a distinct clinical course and clinical outcomes. In conclusion, our study shows that patients can develop PCD in the post–renal transplant period. A lambda clone appears to be more commonly involved in pathogenesis. Patients with a new PCD have a high incidence of renal allograft rejection, particularly among the symptomatic PCD group. It is also important to consider monoclonal gammopathy in the differential diagnosis and workup of renal dysfunction in the posttransplant period in addition to the other etiologies for acute kidney injury in patients with renal allografts. Testing for monoclonal gammopathy as part of the pretransplant evaluation may allow for better risk stratification of patients and timely referral to the hematology team for closer monitoring and follow-up to assess for the progression to symptomatic PCD after kidney transplantation.
Clinical Practice Points.
Plasma cell disorder (PCD) can occur after renal transplantation.
Symptomatic PCD (sPCD) can often be detected because of renal issues (elevated serum creatinine and proteinuria).
sPCD is associated with higher M protein, a higher free light chain ratio, and a higher percentage of bone marrow plasma cells.
There is a risk of progression of sPCD to multiple myeloma.
Monoclonal protein studies should be done in renal allograft patients who develop renal failure or have other suggestive symptoms.
Screening patients for monoclonal protein studies before renal allograft should be done to perform appropriate risk stratification.
Disclosures
Dr. D’Souza was funded by K23HL141445. Its contents are solely the responsibility of the authors and do not necessarily represent the official vies of the National Institutes of Health. Dr. Hari received grant funding and honoraria from Takeda, Celgene, BMS, Janssen, Kite, Juno, Pharmacyclics, and Karyopharm. Dr. Dhakal received grant funding and honoraria from Takeda, Celgene, Janssen, Amgen, and Sanofi. Dr. D’Souza received grant funding and honoraria from Prothena, Sanofi, EDO Mundipharma, TeneoBio, Pfizer, Imbrium, and Akcea. The remaining authors have stated that they have conflicts of interest.
Footnotes
Presented in part as a poster presentation at the 4th International Kidney Monoclonal Gammopathy Meeting, May 23–24, 2019, Montreal, Canada
References
- 1.Leung N, Bridoux F, Hutchison CA, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 2012; 120:4292–5. [DOI] [PubMed] [Google Scholar]
- 2.Safadi S, Dispenzieri A, Amer H, et al. Multiple myeloma after kidney transplantation. Clin Transplant 2015; 29:76–84. [DOI] [PubMed] [Google Scholar]
- 3.Sethi S, Fervenza FC, Rajkumar SV. Spectrum of manifestations of monoclonal gammopathy-associated renal lesions. Curr Opin Nephrol Hypertens 2016; 25: 127–37. [DOI] [PubMed] [Google Scholar]
- 4.Merlini GAL. amyloidosis: from molecular mechanisms to targeted therapies. Hematology Am Soc Hematol Educ Program 2017; 2017:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lum EL, Bunnapradist S. Current opinions in nephrology and hypertension: kidney transplantation in patients with plasma cell dyscrasias. Curr Opin Nephrol Hypertens 2019; 28:573–80. [DOI] [PubMed] [Google Scholar]
- 6.Leung N, Drosou ME, Nasr SH. Dysproteinemias and glomerular disease. Clin J Am Soc Nephrol 2018; 13:128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasr SH, Valeri AM, Sethi S, et al. Clinicopathologic correlations in multiple myeloma: a case series of 190 patients with kidney biopsies. Am J Kidney Dis 2012; 59:786–94. [DOI] [PubMed] [Google Scholar]
- 8.Bancu I, Canas L, Juega FJ, et al. Outcomes of monoclonal gammopathy of undetermined significance in patients who underwent kidney transplantation. Transplant Proc 2015; 47:2344–5. [DOI] [PubMed] [Google Scholar]
- 9.Goebel TE, Schiltz NK, Woodside KJ, et al. Neoplastic and non-neoplastic complications of solid organ transplantation in patients with preexisting monoclonal gammopathy of undetermined significance. Clin Transplant 2015; 29:851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fermand JP, Bridoux F, Kyle RA, et al. How I treat monoclonal gammopathy of renal significance (MGRS). Blood 2013; 122:3583–90. [DOI] [PubMed] [Google Scholar]
- 11.Heher EC, Rennke HG, Laubach JP, Richardson PG. Kidney disease and multiple myeloma. Clin J Am Soc Nephrol 2013; 8:2007–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan HSH, Chen CI, Reece DE. Current review on high-risk multiple myeloma. Curr Hematol Malig Rep 2017; 12:96–108. [DOI] [PubMed] [Google Scholar]
- 13.Chhabra S Novel proteasome inhibitors and histone deacetylase inhibitors: progress in myeloma therapeutics. Pharmaceuticals (Basel) 2017; 10:E40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhakal B, Szabo A, Chhabra S, et al. Autologous transplantation for newly diagnosed multiple myeloma in the era of novel agent induction: a systematic review and meta-analysis. JAMA Oncol 2018; 4:343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naina HV, Harris S, Dispenzieri A, et al. Long-term follow-up of patients with monoclonal gammopathy of undetermined significance after kidney transplantation. Am J Nephrol 2012; 35:365–71. [DOI] [PubMed] [Google Scholar]
- 16.Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol 2014; 32:2531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sidiqi MH, Aljama MA, Muchtar E, et al. Light chain type predicts organ involvement and survival in AL amyloidosis patients receiving stem cell transplantation. Blood Adv 2018; 2:769–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121:749–57. [PubMed] [Google Scholar]
- 19.Cuellar-Garcia C, Sevillano Ruiz-Mateos C, Mazuecos Blanca MA, et al. Follow-up monoclonal gammopathy of undetermined significance in kidney transplant. Transplant Proc 2015; 47:78–80. [DOI] [PubMed] [Google Scholar]
- 20.Rostaing L, Modesto A, Abbal M, Durand D. Long-term follow-up of monoclonal gammopathy of undetermined significance in transplant patients. Am J Nephrol 1994; 14:187–91. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Calle V, Mateos MV. Monoclonal gammopathies of unknown significance and smoldering myeloma: assessment and management of the elderly patients. Eur J Intern Med 2018; 58:57–63. [DOI] [PubMed] [Google Scholar]
- 22.Alfano G, Fontana F, Colaci E, et al. Monoclonal gammopathy of undetermined significance after kidney transplantation: single-center experience. Transplantation 2017; 101:e337–42. [DOI] [PubMed] [Google Scholar]