

Abstract
Gyrase and topoisomerase IV are the targets of fluoroquinolone antibacterials. However, the rise in antimicrobial resistance has undermined the clinical use of this important drug class. Therefore, it is critical to identify new agents that maintain activity against fluoroquinolone-resistant strains. One approach is to develop non-fluoroquinolone drugs that also target gyrase and topoisomerase IV, but interact differently with the enzymes. This has led to the development of the “novel bacterial topoisomerase inhibitor” (NBTI) class of antibacterials. Despite the clinical potential of NBTIs, there is a relative paucity of data describing their mechanism of action against bacterial type II topoisomerases. Consequently, we characterized the activity of GSK126, a naphthyridone/aminopiperidine-based NBTI, against a variety of Gram-positive and Gram-negative bacterial type II topoisomerases including gyrase from Mycobacterium tuberculosis, and gyrase and topoisomerase IV from Bacillus anthracis and Escherichia coli. GSK126 enhanced single-stranded DNA cleavage and suppressed double-stranded cleavage mediated by these enzymes. It was also a potent inhibitor of gyrase-catalyzed DNA supercoiling and topoisomerase IV-catalyzed decatenation. Thus, GSK126 displays a similar bimodal mechanism of action across a variety of species. In contrast, GSK126 displayed a variable ability to overcome fluoroquinolone resistance mutations across these same species. Our results suggest that NBTIs elicit their antibacterial effects by two different mechanisms: inhibition of gyrase/topoisomerase IV catalytic activity or enhancement of enzyme-mediated DNA cleavage. Furthermore, the relative importance of these two mechanisms appears to differ from species to species. Therefore, we propose that the mechanistic basis for the antibacterial properties of NBTIs is bimodal in nature.
Graphical Abstract
INTRODUCTION
In addition to their essential cellular functions,1–3 the bacterial type II topoisomerases, gyrase and topoisomerase IV, are the targets for fluoroquinolone antibacterials.4–11 These broad-spectrum drugs are among the most widely prescribed antibacterials worldwide,4,5,7,12 and act by stabilizing covalent protein-cleaved DNA complexes that are generated during the catalytic cycles of the type II enzymes.7,8,11,13,14 By inserting between the bases at the cleaved scissile bonds, fluoroquinolones increase the cellular concentration of gyrase- and topoisomerase IV-mediated DNA breaks and block the essential catalytic functions of these enzymes.4–11,13,14 Clinically relevant fluoroquinolones interact with the bacterial type II topoisomerases through a water-metal ion bridge.7,15–19 The C3/C4 ketoacid of the fluoroquinolone skeleton chelates a divalent metal ion, which is coordinated by four water molecules. Two of these water molecules interact with a highly conserved serine and an acidic residue that is located four amino acids upstream in the GyrA or GrlA/ParC subunits of gyrase or topoisomerase IV (Gram-positive/Gram-negative), respectively.15
Despite the importance of fluoroquinolones in the treatment of bacterial infections, their overuse has contributed to a rise in resistance that spans their entire spectrum of clinical usage.4–7,10 This resistance is most often associated with mutations in the two amino acid residues that anchor the water-metal ion bridge.4–7,10 Given the broad clinical applications of fluoroquinolones, it is critical to discover new drug classes that can supplement their use in the clinic. One approach is to develop novel drugs that act on validated targets, such as gyrase and topoisomerase IV, but interact differently with the enzymes. This approach has led to the development of compounds known as “novel bacterial topoisomerase inhibitors” (NBTIs) (Figure 1).10,11,20,21
Figure 1.
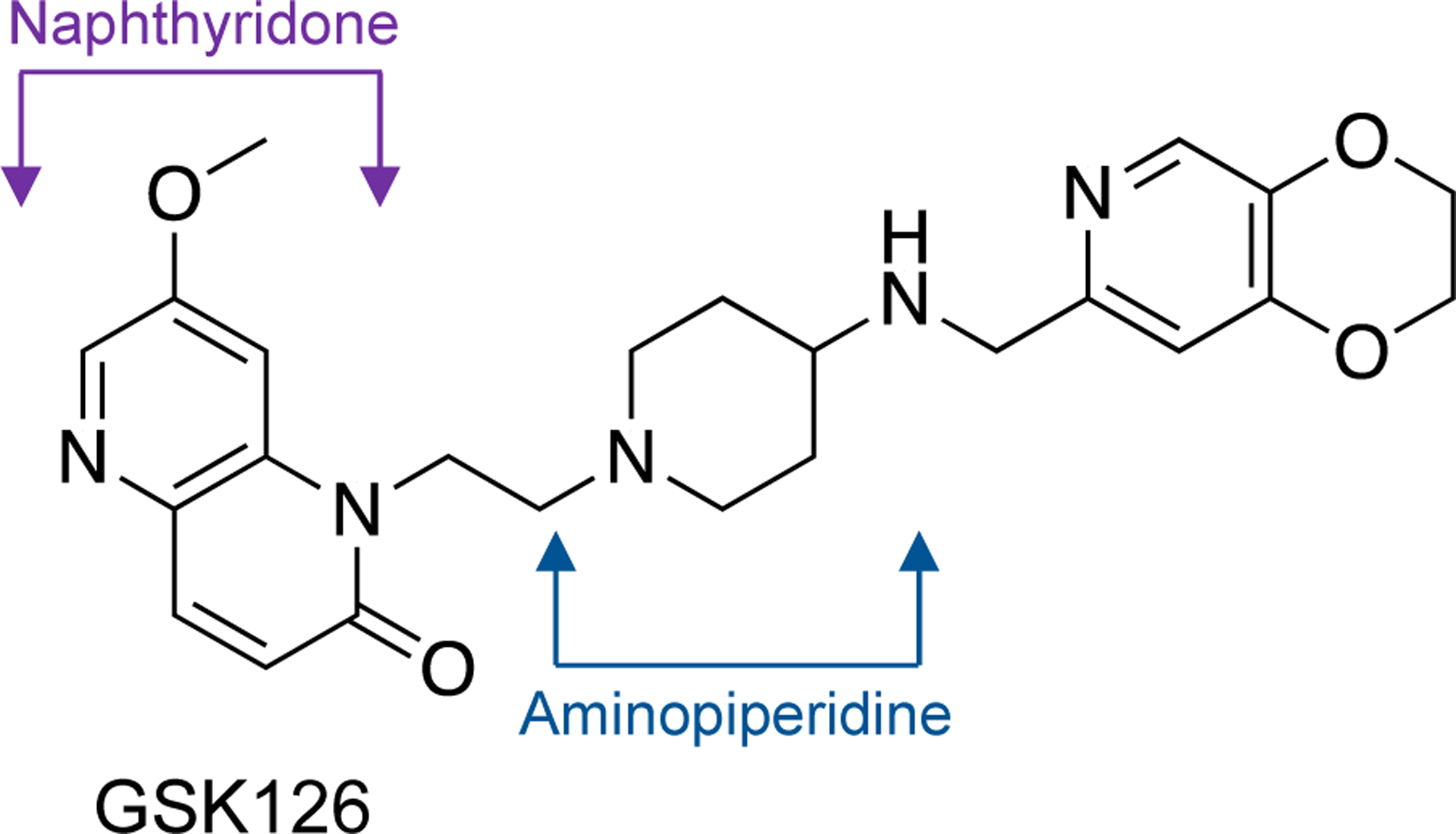
Structure of the NBTI GSK126. The naphthyridone (purple) and aminopiperidine (blue) moieties are indicated.
Several NBTIs display activity against clinically relevant bacterial species.9,10,21–28 However, most show poor activity against Mycobacterium tuberculosis.29 GSK126 (Figure 1), which is a naphthyridone/aminopiperidine-based NBTI, was identified in a screen as having moderate activity against the bacterium.29 This discovery led to a subset of NBTIs that were derived from GSK126 and displayed high activity against M. tuberculosis in culture and in mouse infection models.29 Compounds in this subclass are known as M. tuberculosis gyrase inhibitors (MGIs). Whereas a recent study characterized the mechanistic basis for the actions of MGIs against M. tuberculosis gyrase,30 relatively little is known about the actions of the parent NBTI class against gyrase and topoisomerase IV.9–11
In order to address this important issue, we analyzed the actions of GSK126 against a variety of bacterial type II topoisomerases, including gyrase from M. tuberculosis, and gyrase and topoisomerase IV from Bacillus anthracis and Escherichia coli. This NBTI was chosen for initial studies to afford direct comparisons with the MGIs. Results indicate that GSK126 has a broader spectrum of activity against gyrase and topoisomerase IV than the MGIs, but displays similar mechanistic characteristics. GSK126 acted in a bimodal fashion; it was a potent inhibitor of gyrase-catalyzed DNA supercoiling and topoisomerase IV-catalyzed decatenation and also induced single-stranded DNA cleavage (i.e., acted as a topoisomerase poison) with both enzymes. However, the relative importance of these two modes of NBTI action (inhibiting vs. poisoning) appears to differ among bacterial species. Thus, we propose that the mechanistic basis for the antibacterial properties of NBTIs is bimodal in nature.
EXPERIMENTAL PROCEDURES
Enzymes and Materials.
Full-length wild-type B. anthracis gyrase subunits (GyrA and GyrB) and GyrA mutant (GyrAS85L), as well as wild-type B. anthracis topoisomerase IV subunits (GrlA and GrlB) and GrlA mutant (GrlAS81F), were expressed and purified as described by Dong et al.31 Wild-type E. coli gyrase subunits (GyrA and GyrB) were expressed and purified as described by Maxwell et al.32 Mutant E. coli GyrAS83L was expressed and purified as described by Dong et al.31 Wild-type E. coli topoisomerase IV and ParC mutant (ParCS80L) were expressed and purified as described by Peng and Marians33 or by a minor modification of Corbett et al.34 Wild-type M. tuberculosis gyrase subunits (GyrA and GyrB) and GyrA mutant (GyrAA90V) were expressed and purified as described by Blower et al.35 as modified by Aldred et al.18
Negatively supercoiled pBR322 DNA was prepared from E. coli using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Relaxed pBR322 plasmid DNA was generated by treating negatively supercoiled pBR322 with calf thymus topoisomerase I (Invitrogen) and purified as described previously.17 Kinetoplast DNA (kDNA) was isolated from Crithidia fasciculata as described by Englund.36
The NBTI GSK126 was synthesized as described previously by Blanco et al.29 In the paper by Blanco et al., GSK126 was identified as compound 1. The NBTI was stored at 4 °C as a 1–2 mM stock solution in 10% dimethylsulfoxide.
DNA Cleavage.
DNA cleavage assays were based on the procedure of Aldred et al.16 Reactions were carried out in the absence of GSK126 or presence of increasing concentrations of the NBTI and, unless stated otherwise, contained 500 nM B. anthracis wild-type or mutant (GyrAS85L) gyrase (1:2 GyrA:GyrB ratio), 150 nM B. anthracis wild-type or 125 nM mutant (GrlAS81F) topoisomerase IV (1:2 GrlA:GrlB ratio), 50 nM E. coli wild-type or mutant (GyrAS83L) gyrase (1:1 GyrA:GyrB ratio), 20 nM E. coli wild-type or 10 nM mutant (ParCS80L) topoisomerase IV (1:1 ParC:ParE ratio), or 100 nM M. tuberculosis wild-type or mutant (GyrAA90V) gyrase (1.5:1 or 1:1 GyrA:GyrB ratio, respectively). Stated enzyme concentrations reflect those of the holoenzyme (A2B2) calculated on the basis of the limiting subunit. The subunit ratios that were employed represent the minimal amount of protein required to generate maximal cleavage at 37 °C. Assays contained 10 nM negatively supercoiled pBR322 in a total volume of 20 μL of reaction buffer: B. anthracis gyrase: 50 mM Tris-HCl (pH 7.5), 100 mM KGlu, 5 mM MgCl2, 1 mM dithiothreitol (DTT) and 50 μg/mL BSA, M. tuberculosis gyrase: 10 mM Tris-HCl (pH 7.5), 40 mM KCl, 6 mM MgCl2, 0.1 mg/mL bovine serum albumin, and 10% glycerol, all other enzymes: 40 mM Tris-HCl (pH 7.9), 50 mM NaCl, 10 mM MgCl2 and 12.5% glycerol. In some cases, MgCl2 in reaction buffers was replaced with an equivalent concentration of 5 mM CaCl2 or 1.5 mM ATP was included in reaction mixtures. Unless stated otherwise, reactions were incubated at 37 °C for 30 min with B. anthracis gyrase and 10 min with all other enzymes. Enzyme-DNA cleavage complexes were trapped by adding 2 μL of 5% sodium dodecyl sulfate (SDS) followed by 2 μL of 250 mM Na2EDTA and 2 μL of 0.8 mg/mL Proteinase K (Sigma Aldrich). Reaction mixtures were incubated at 45 °C for 30 min to digest the enzyme. Samples were mixed with 2 μL of loading buffer [60% sucrose, 10 mM Tris-HCl (pH 7.9), 0.5% bromophenol blue, and 0.5% xylene cyanol FF] and incubated at 45 °C for 2 min before loading onto 1% agarose gels. Reaction products were subjected to electrophoresis in 40 mM Tris-acetate (pH 8.3) and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. DNA bands were visualized with medium-range ultraviolet light and quantified by scanning densitometry using a Protein Simple AlphaImager HP digital imaging system. DNA single- or double-stranded cleavage was monitored by the conversion of supercoiled plasmid to nicked or linear molecules, respectively, and quantified in comparison to a control reaction in which an equal amount of DNA was digested by EcoRI (New England BioLabs). EC50 values were determined using Prism and represent the concentration of GSK126 at which 50% maximal single-stranded DNA cleavage was observed.
Gyrase-catalyzed DNA Supercoiling.
DNA supercoiling assays were based on previously published protocols.16,18 Assays contained 200 nM B. anthracis wild-type or mutant (GyrAS85L) gyrase (1:2 GyrA:GyrB ratio), 10 nM E. coli wild-type or mutant (GyrAS83L) gyrase (1:1 GyrA:GyrB ratio), or 75 nM M. tuberculosis wild-type or mutant (GyrAA90V) gyrase (1.5:1 GyrA:GyrB ratio), 5 nM relaxed pBR322, and 1.5 mM ATP in a total volume of 20 μL of 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 175 mM KGlu, and 50 μg/mL BSA for all enzymes except M. tuberculosis gyrase or 10 mM Tris-HCl (pH 7.5), 40 mM KCl, 6 mM MgCl2, 0.1 mg/mL BSA, and 10% glycerol for M. tuberculosis gyrase. Stated enzyme concentrations reflect those of the holoenzyme (A2B2) calculated on the basis of the limiting subunit. In reactions with gyrase from B. anthracis and M. tuberculosis, 5 mM DTT and 2 mM DTT, respectively, were included. Reactions were incubated at 37 °C for 30 min, which represents the minimum time required to completely supercoil the DNA in the absence of drug. Reaction mixtures were stopped by the addition of 3 μL of a mixture of 0.77% SDS and 77.5 mM Na2EDTA. Samples were mixed with 2 μL of loading buffer and incubated at 45 °C for 2 min before loading onto 1% agarose gels in 100 mM Tris-borate (pH 8.3) and 2 mM EDTA. Gels were stained with 1 μg/mL ethidium bromide for 30 min. DNA bands were visualized and quantified as described above. IC50 values were determined using Prism and represent the concentration of GSK126 that decreased activity by 50%.
Topoisomerase IV-catalyzed DNA Decatenation.
DNA decatenation assays were based on previously published protocols.16,37 Assays contained 25 nM B. anthracis wild-type or mutant (GrlAS81F) topoisomerase IV (1:2 GrlA:GrlB ratio) or 10 nM E. coli wild-type or mutant (ParCS80L) topoisomerase IV (1:1 ParC:ParE ratio), 5 nM kDNA, and 1.5 mM ATP in 20 μL of 40 mM HEPES (pH 7.6), 100 mM KGlu, 10 mM Mg(OAc)2, and 25 mM NaCl. Stated enzyme concentrations reflect those of the holoenzyme (A2B2) calculated on the basis of the limiting subunit. Reactions were incubated at 37 °C for 30 min, which represents the minimum time required to completely decatenate the DNA in the absence of drug. Reaction mixtures were stopped by the addition of 3 μL of a mixture of 0.77% SDS and 77.5 mM Na2EDTA. Samples were mixed with 2 μL of loading buffer and incubated at 45 °C for 2 min before loading onto 1% agarose gels in 100 mM Tris-borate (pH 8.3) and 2 mM EDTA. Gels were stained with 1 μg/mL ethidium bromide for 30 min. DNA bands were visualized and quantified as described above. IC50 values were determined using Prism and represent the concentration of GSK126 that decreased activity by 50%.
Results
MGIs are the best-characterized members of the naphthyridone/aminopiperidine-based NBTI drug class.29,30 However, their selection for activity against M. tuberculosis resulted in a group of compounds with narrow specificity.30 This narrow specificity range raises questions regarding the spectrum of action of the parent NBTI series and whether the mechanism of the naphthyridone/aminopiperidine-based compounds are translatable across species.
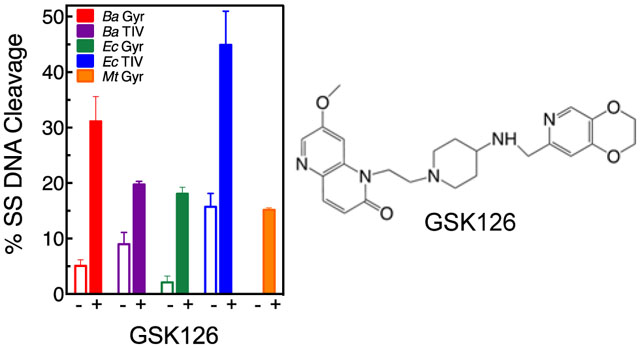
Therefore, the ability of GSK126 (the parent compound of MGIs) to induce enzyme-mediated single-stranded breaks was examined against gyrase and topoisomerase IV from a variety of Gram-negative and Gram-positive bacteria (Figure 2). In contrast to MGIs, the NBTI displayed a much broader spectrum of activity. The compound showed activity against all the enzymes examined, including those from B. anthracis, E. coli, and M. tuberculosis. In order to assess the basis for the actions of GSK126 against bacterial type II topoisomerases, we characterized its activity against all the enzymes shown in Figure 2. This allowed us to characterize the actions of a NBTI against Gram-negative and Gram-positive gyrase and topoisomerase IV as well as M. tuberculosis gyrase (this species does not encode topoisomerase IV38) to afford direct comparisons to MGIs.
Figure 2.
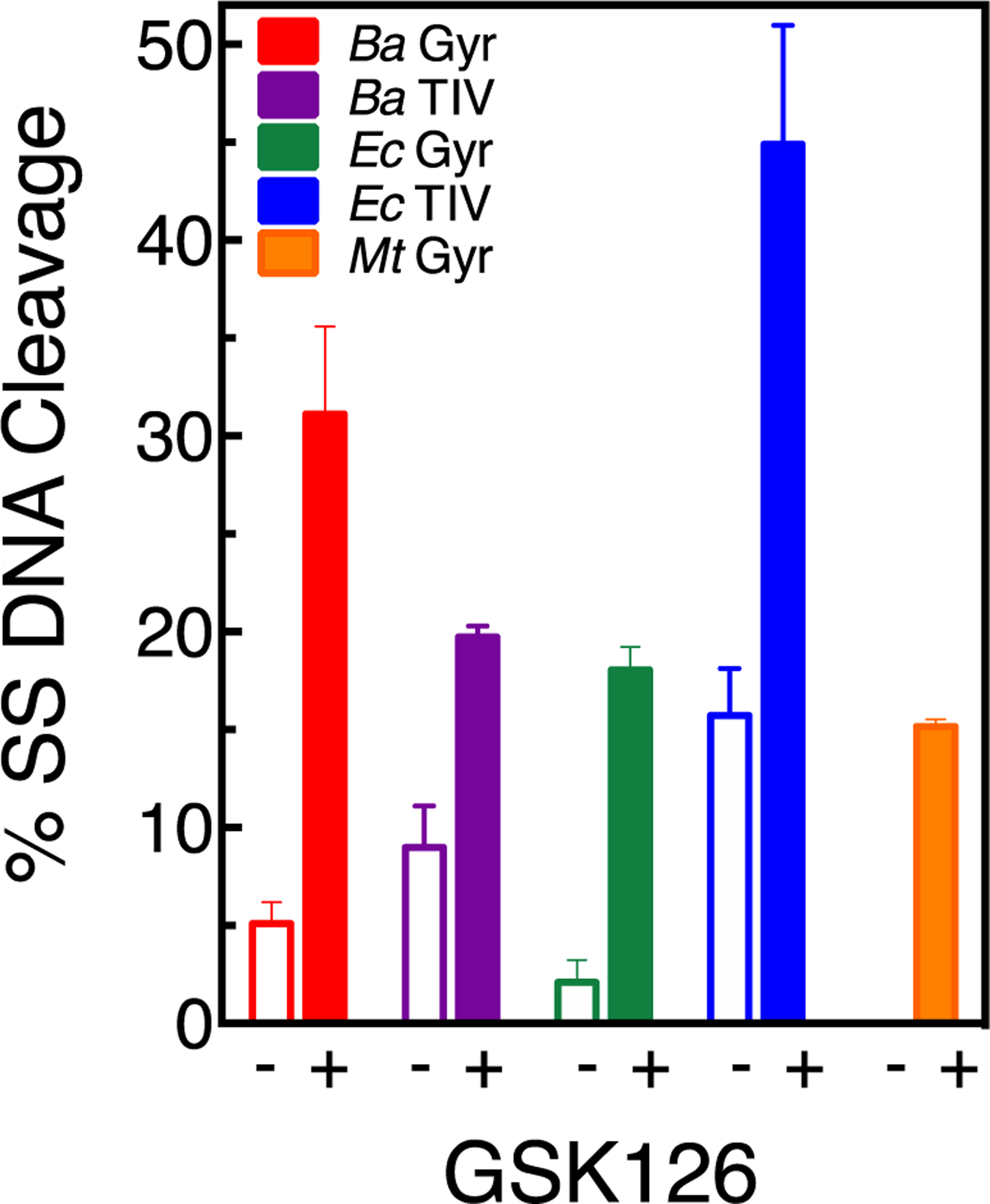
GSK126 displays a broad spectrum of DNA cleavage enhancement against gyrase and topoisomerase IV. The effects of GSK126 (solid bars) on single-stranded DNA cleavage mediated by gyrase and topoisomerase IV are shown for the NBTI concentration that generated the highest levels of single-stranded breaks (see Figures 4 and 9). Data are shown for B. anthracis gyrase (Ba Gyr, red, 0.5 μM GSK126) and topoisomerase IV (Ba TIV, purple, 0.5 μM), E. coli gyrase (Ec Gyr, green, 15 μM) and topoisomerase IV (Ec TIV, blue, 1 μM), and M. tuberculosis gyrase (Mt Gyr, orange, 10 μM) are shown. The corresponding single-stranded DNA cleavage in the absence of GSK126 is shown as empty bars. Error bars represent the SD (standard deviation) of at least three independent experiments.
DNA Gyrase
Effects of GSK126 on the DNA Supercoiling Activity of Wild-type and Fluoroquinolone-resistant Gyrase.
NBTIs have been shown to inhibit the catalytic function of gyrase, but it is not known how enzyme inhibition vs. the ability of NBTIs to enhance DNA cleavage affects drug-induced cell death. Therefore, the effects of GSK126 on DNA supercoiling catalyzed by B. anthracis, E. coli, and M. tuberculosis gyrase were examined (Figure 3). The NBTI was a potent catalytic inhibitor of the B. anthracis and E. coli enzymes, but displayed lower activity against M. tuberculosis gyrase (IC50 ~ 0.4 μM and 2 μM compared to IC50 ~ 75 μM, respectively). In addition, GSK126 was able to decrease the activity of the latter enzyme by only 60%. Furthermore, the compound displayed a variable ability to overcome the effects of the common clinical fluoroquinolone resistance mutations (all are at corresponding serine residues) in B. anthracis (GyrAS85L), E. coli (GyrAS83L), and M. tuberculosis (GyrAA90V) gyrase (Figure 3). Whereas GSK126 maintained activity against all three mutant enzymes, its potency dropped considerably against the B. anthracis and M. tuberculosis gyrase mutants (compared to the wild-type enzyme). In contrast, the compound was 5–10 fold more potent against the E. coli gyrase that harbored the fluoroquinolone resistance mutation.
Figure 3.
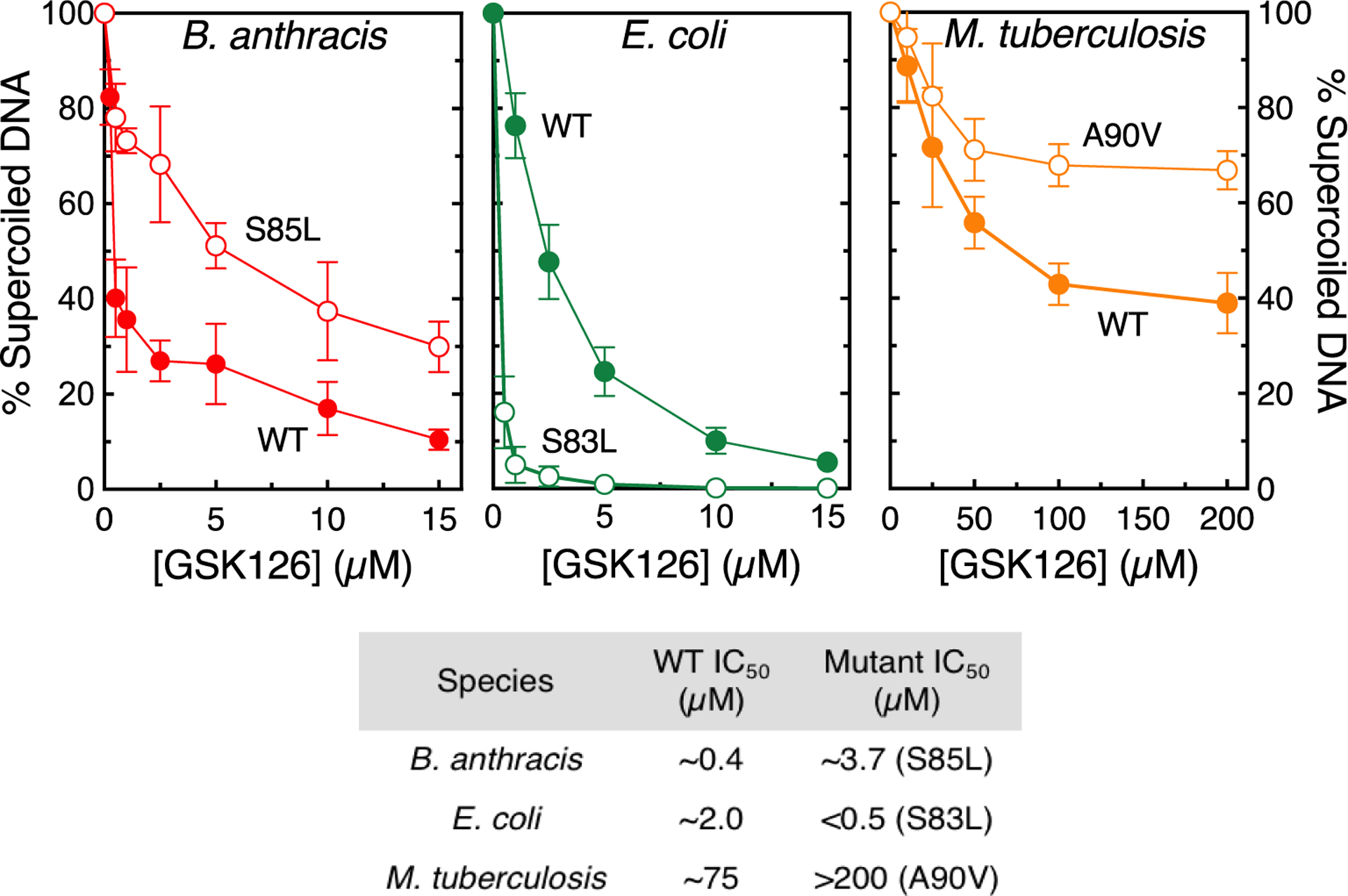
GSK126 inhibits DNA supercoiling catalyzed by wild-type (WT) and fluoroquinolone-resistant gyrase. The effects of GSK126 on the supercoiling of relaxed DNA by WT (filled circles) and fluoroquinolone-resistant (empty circles) B. anthracis WT and GyrAS85L (top left, red), E. coli WT and GyrAS83L (top middle, green), and M. tuberculosis WT and GyrAA90V (top left, orange) gyrase are shown. Error bars represent the SD of at least three independent experiments. The bottom panel shows a table of IC50 values. Representative agarose gels are shown in Figure S1.
Effects of GSK126 on the DNA Cleavage Activity of Wild-type and Fluoroquinolone-resistant Gyrase.
In contrast to studies on the inhibition of gyrase supercoiling,22–25,27,28,39–42 relatively little is known about how NBTIs affect gyrase-mediated DNA cleavage.21,22,25,30,43 Therefore, the effects of GSK126 on DNA cleavage mediated by wild-type gyrase from B. anthracis, E. coli, and M. tuberculosis was examined (Figure 4). Paralleling previous results for MGIs and other classes of NBTIs,21,25,30,43 GSK126 displayed a potent ability to induce single-stranded DNA breaks generated by all three enzymes [EC50 (concentration of GSK126 required to obtain 50% maximal cleavage) values ranged from sub-micromolar to ~1 μM], but displayed no ability to induce double-stranded breaks.
Figure 4.
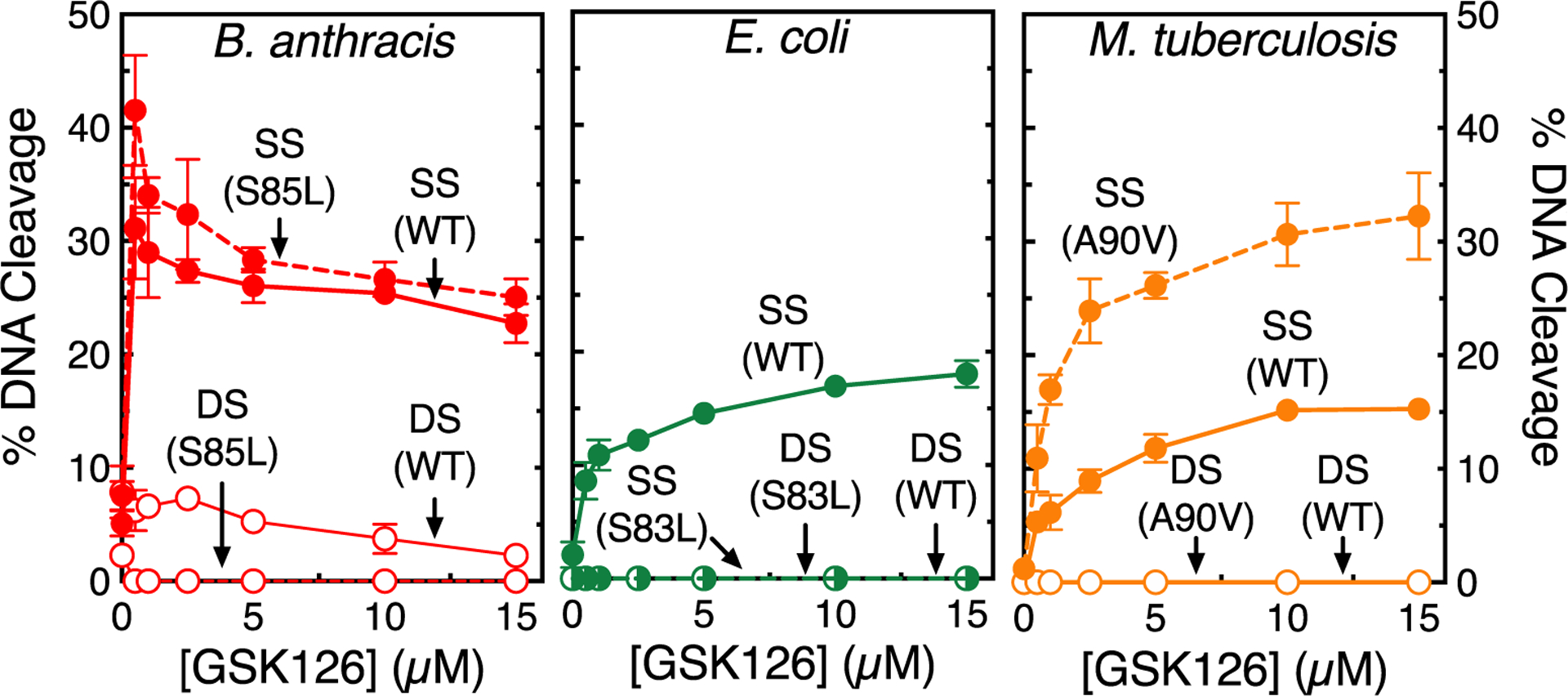
Effects of GSK126 on DNA cleavage mediated by wild-type (WT) and fluoroquinolone-resistant gyrase. The effects of GSK126 on single-stranded (SS, filled circles) and double-stranded (DS, empty circles) DNA cleavage mediated by WT (solid line) and fluoroquinolone-resistant (dashed line) B. anthracis WT and GyrAS85L (left, red), E. coli WT and GyrAS83L (middle, green), and M. tuberculosis WT and GyrAA90V (right, orange) gyrase are shown. Some of the data shown for M. tuberculosis WT and GyrAA90V gyrase are from Gibson et. al.30 Error bars represent the SD of at least three independent experiments. Representative agarose gels are shown in Figure S2.
In contrast to the concentration dependence for the enhancement of single-stranded DNA breaks with E. coli and M. tuberculosis gyrase, GSK126 induced a sharp increase in DNA cleavage mediated by the B. anthracis enzyme (Figure 4). This presumably reflects a strong binding of the NBTI to the B. anthracis gyrase-DNA complex, which is consistent with the sharp decline in supercoiling rates with this enzyme in the presence of GSK126 (Figure 3).
Although NBTIs and fluoroquinolones both bind in the active site of gyrase in the cleavage complex, they do not interact with the same amino acid residues. However, very little is known about how the presence of common fluoroquinolone resistance mutations in gyrase affect the ability of NBTIs to induce enzyme-mediated DNA cleavage.30 As shown in Figure 4, GSK126 was slightly more efficacious against B. anthracis GyrAS85L and induced substantially more single-stranded DNA cleavage against M. tuberculosis GyrAA90V gyrase than their wild-type counterparts. In contrast, GSK126 displayed no ability to enhance DNA cleavage mediated by E. coli GyrAS83L gyrase (control experiments carried out in the presence of Ca2+ demonstrate that GyrAS83L displays wild-type levels of baseline DNA cleavage, not shown). This is the first reported instance in which DNA cleavage enhancement by a NBTI or related compound has failed to overcome resistance induced by a fluoroquinolone-resistant mutation in gyrase.
GSK126 Enhances only Single-stranded DNA Cleavage Mediated by Gyrase and Suppresses Enzyme-generated Double-stranded DNA Breaks.
To further assess the ability of GSK126 to induce gyrase-mediated single- vs. double-stranded breaks with the wild-type enzyme, DNA cleavage reactions were carried out at NBTI concentrations as high as 200 μM and at reaction times up to six times longer than required to achieve DNA cleavage-ligation equilibrium. Under all conditions examined, only the enhancement of single-stranded breaks was observed (Figure 5). Furthermore, no enhancement of double-stranded breaks was seen in the presence of ATP, which is required for overall catalytic activity (Figure 6). However, it is notable that levels of single-stranded DNA cleavage generated by E. coli gyrase in the presence of ATP were considerably lower than those seen in the absence of the high energy cofactor (see Figure 4). This finding opens the possibility that under physiological conditions, GSK126 acts primarily as a catalytic inhibitor (as opposed to a poison) of E. coli gyrase. Conversely, the potent DNA cleavage enhancement with M. tuberculosis gyrase vs. the relatively weak inhibitory properties of GSK126 against this enzyme suggest that the opposite may be the case for GSK126 and M. tuberculosis.
Figure 5.
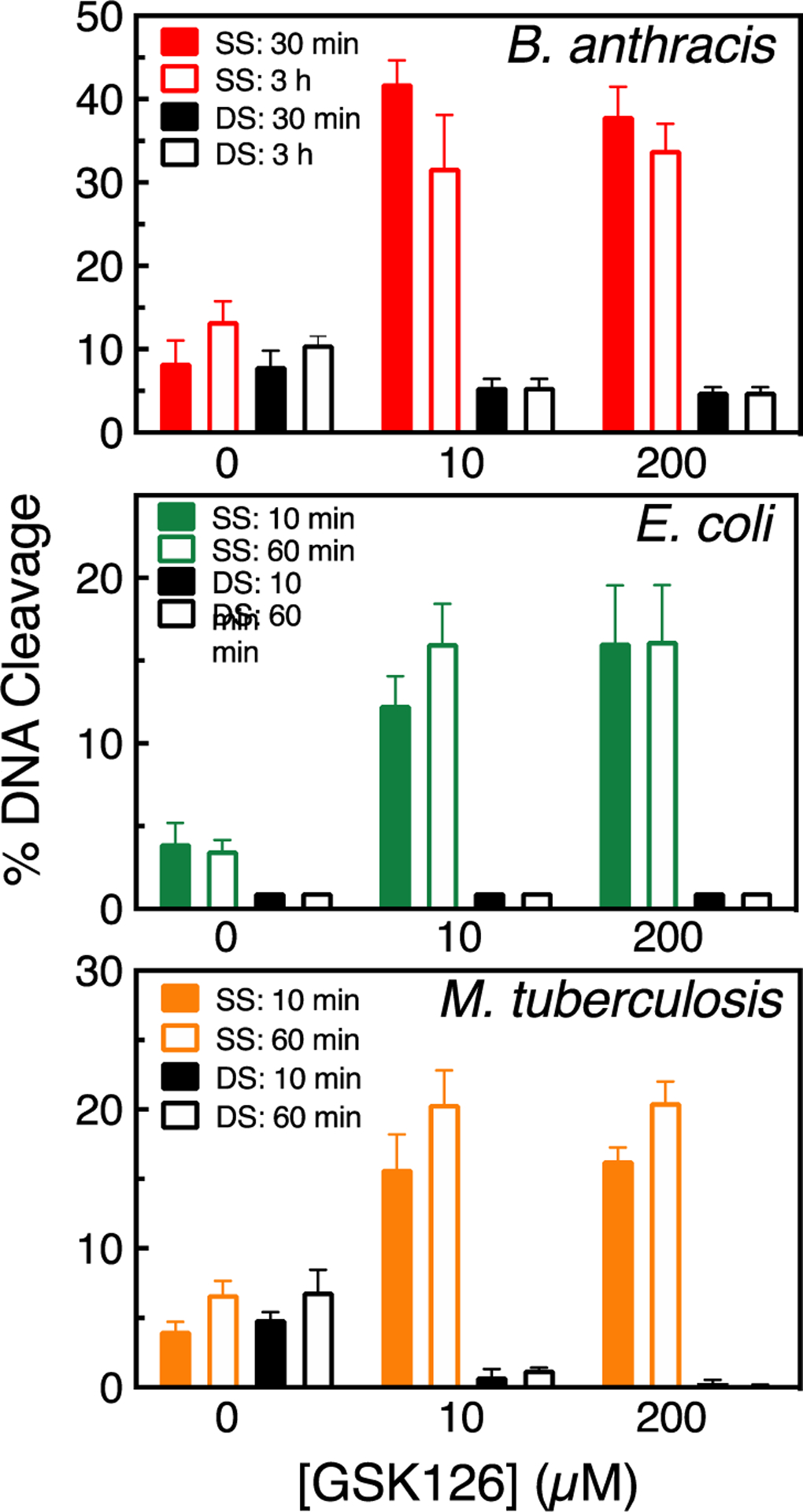
GSK126 enhances only single-stranded DNA breaks mediated by gyrase. The top panel shows the enhancement of B. anthracis gyrase-mediated single- (red) or double-stranded (black) DNA breaks at 30 min (filled bar) or 3 h (empty bar) in the absence or presence of 10 μM or 200 μM GSK126. The middle panel shows the enhancement of E. coli gyrase-mediated single- (green) or double-stranded (black) DNA breaks at 10 min (filled bar) or 60 min (empty bar) in the absence or presence of 10 μM or 200 μM GSK126. The bottom panel shows the enhancement of M. tuberculosis gyrase-mediated single- (orange) or double-stranded (black) DNA breaks at 10 min (filled bar) or 60 min (empty bar) in the absence or presence of 10 μM or 200 μM GSK126. Error bars represent the SD of at least 3 independent experiments. Representative agarose gels are shown in Figure S3.
Figure 6.
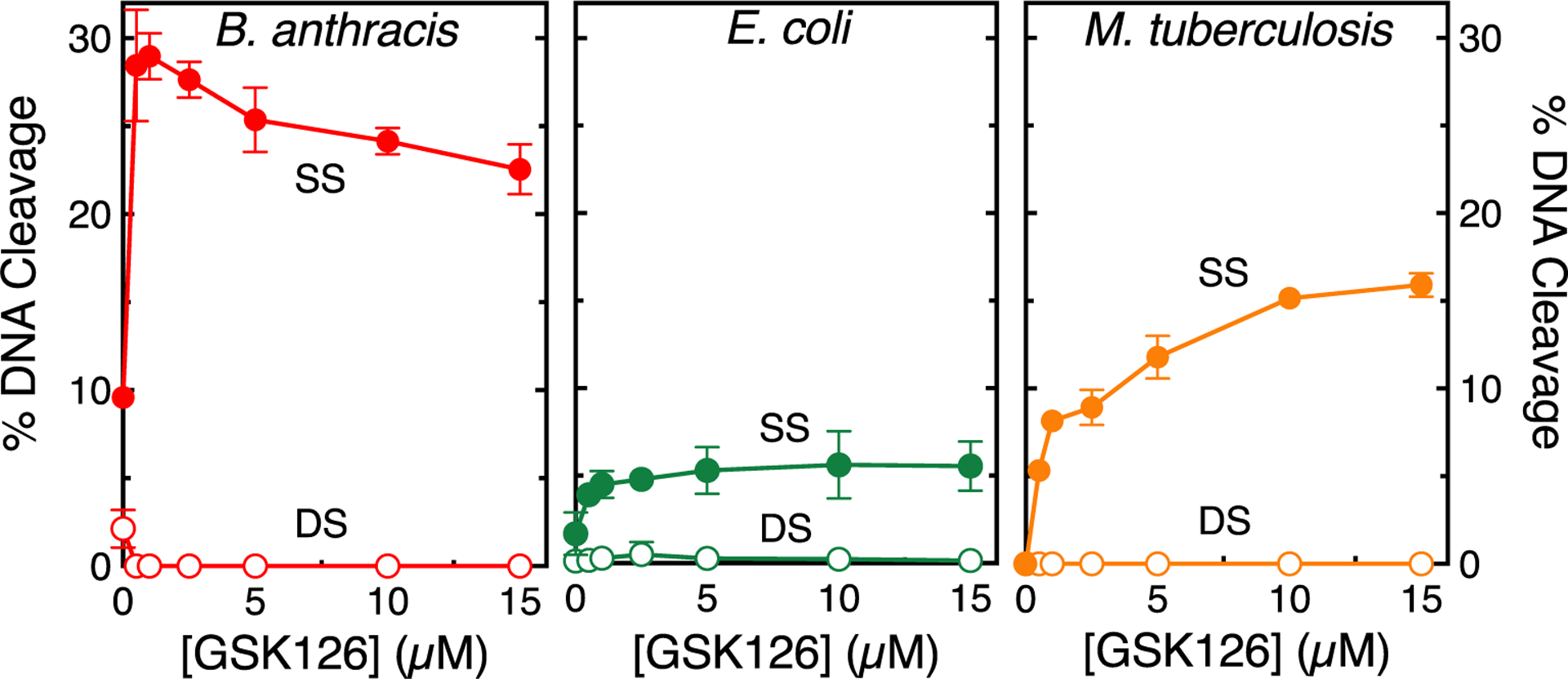
GSK126 enhances only single-stranded DNA breaks mediated by gyrase in the presence of ATP. The effects of GSK126 on single-stranded (SS, filled circles) and double-stranded (DS, empty circles) DNA cleavage mediated by B. anthracis (left, red), E. coli (middle, green), and M. tuberculosis (right, orange) gyrase in the presence of 1.5 mM ATP are shown. Error bars represent the SD of at least three independent experiments. Representative agarose gels are shown in Figure S4.
The double-stranded cleavage of DNA by type II topoisomerases is carried out by two coordinated single-stranded cleavage events.14,44,45 Thus, the generation of single-stranded DNA breaks that are observed in the presence of GSK126 can reflect multiple mechanisms. For example, the compound in any given cleavage complex may stabilize the break only at one of the scissile bonds. Alternatively, after cleavage at one scissile bond, the presence of GSK126 may alter the structure of the enzyme-DNA complex such that the second DNA strand cannot be cut. This latter mechanism is favored by structural studies that indicate distortion in the cleavage complex in the presence of NBTIs.21,43 To help elucidate the mechanisms by which GSK126 induces only single-stranded DNA breaks, the MgCl2 in DNA cleavage assays was replaced with CaCl2. This latter divalent metal ion raises baseline levels of DNA cleavage with most type II topoisomerases, so that double-stranded breaks can be observed more easily even in the absence of drug (compare levels of double-stranded DNA breaks in the absence of GSK126 in Figure 4 with those in Figure 7). As seen in Figure 7 (or more clearly in Figures 4 and 6 with B. anthracis gyrase), the rise in single-stranded cleavage mediated by gyrase at increasing concentrations of GSK126 was accompanied by a coordinate decrease in double-stranded breaks. This finding suggests that cleavage of one scissile bond in the presence of GSK126 suppresses cleavage at the opposite scissile bond.
Figure 7.
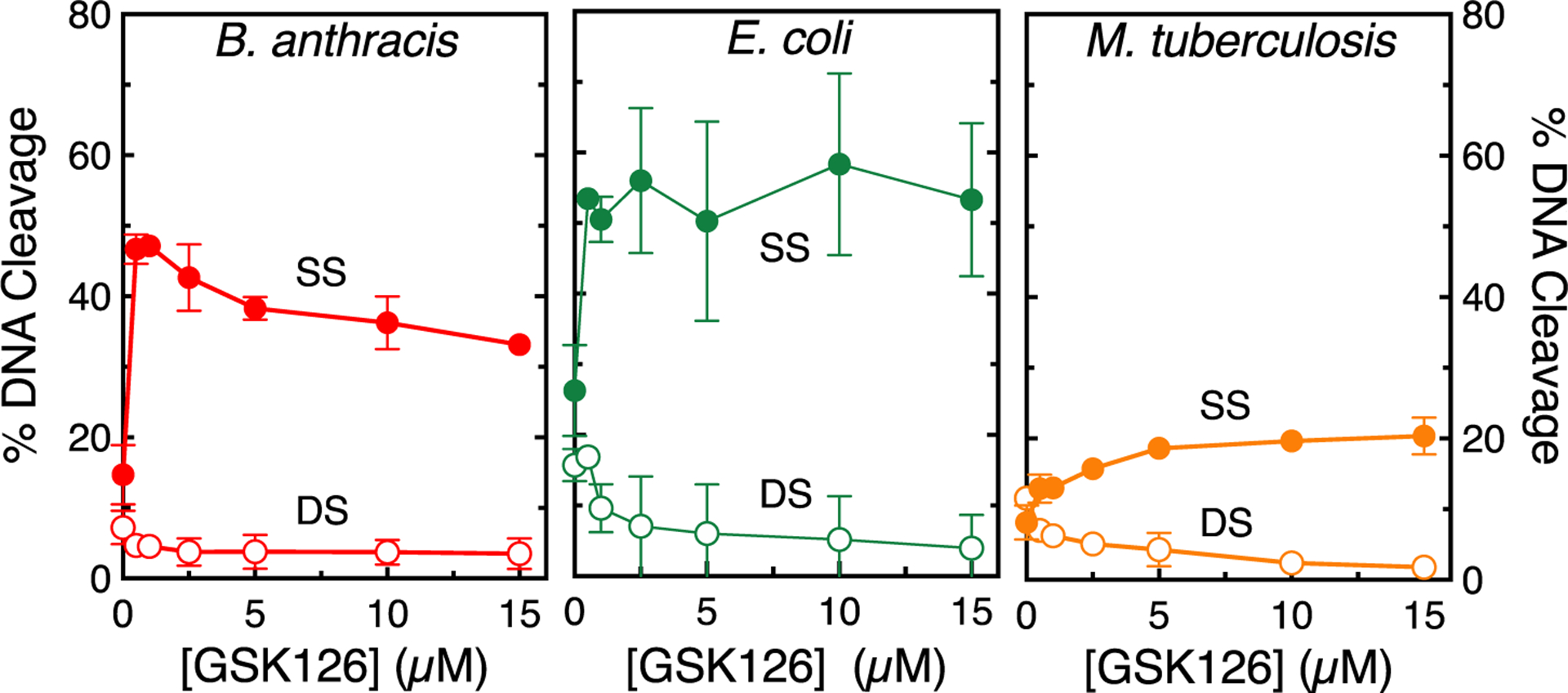
GSK126 suppresses double-stranded DNA breaks generated by gyrase. The effects of GSK126 on single-stranded (SS, filled circles) and double-stranded (DS, empty circles) DNA cleavage mediated by B. anthracis (left, red), E. coli (middle, green), and M. tuberculosis (right, orange) gyrase are shown. Reaction mixtures contained 5 mM CaCl2 in place of MgCl2 to increase baseline levels of DNA cleavage. Error bars represent the SD of at least three independent experiments. Representative agarose gels are shown in Figure S5.
Taken together, the above data provide strong evidence that GSK126 (a naphthyridone/aminopiperidine-based NBTI), like the naphthyridone/aminopiperidine-based MGIs (GSK000 and GSK325)30 and other structural classes of NBTIs (NBTI GSK299423, 5643, and gepotidacin),21,25,43 induces only gyrase-mediated single-stranded DNA breaks across a variety of species.
Topoisomerase IV
Effects of GSK126 on the DNA Decatenation Activity of Wild-type and Fluoroquinolone-resistant Topoisomerase IV.
Depending upon the bacterial species and the specific drug, the primary cellular target of fluoroquinolones may be gyrase, topoisomerase IV, or both enzymes.6,7,10,13 However, less is known about the targeting of NBTIs. Therefore, to be able to make direct comparisons with DNA gyrase, the ability of GSK126 to inhibit decatenation catalyzed by B. anthracis and E. coli topoisomerase IV was characterized.
Consistent with studies with other structural classes of NBTIs21–25,27,28,30,39–43,46 and as seen with gyrase (Figure 3), GSK126 was a potent inhibitor of topoisomerase IV function (IC50 ~ 2.3 μM for B. anthracis and 1.4 μM for E. coli, respectively) (Figure 8). Furthermore, GSK126 maintained activity against the ParCS80L fluoroquinolone-resistant mutant of E. coli topoisomerase IV (IC50 ~ 1.4 μM) and displayed even greater potency against the GrlAS81F mutant of the B. anthracis enzyme (Figure 8). As above, these fluoroquinolone resistance mutations are at the corresponding serine residues.
Figure 8.
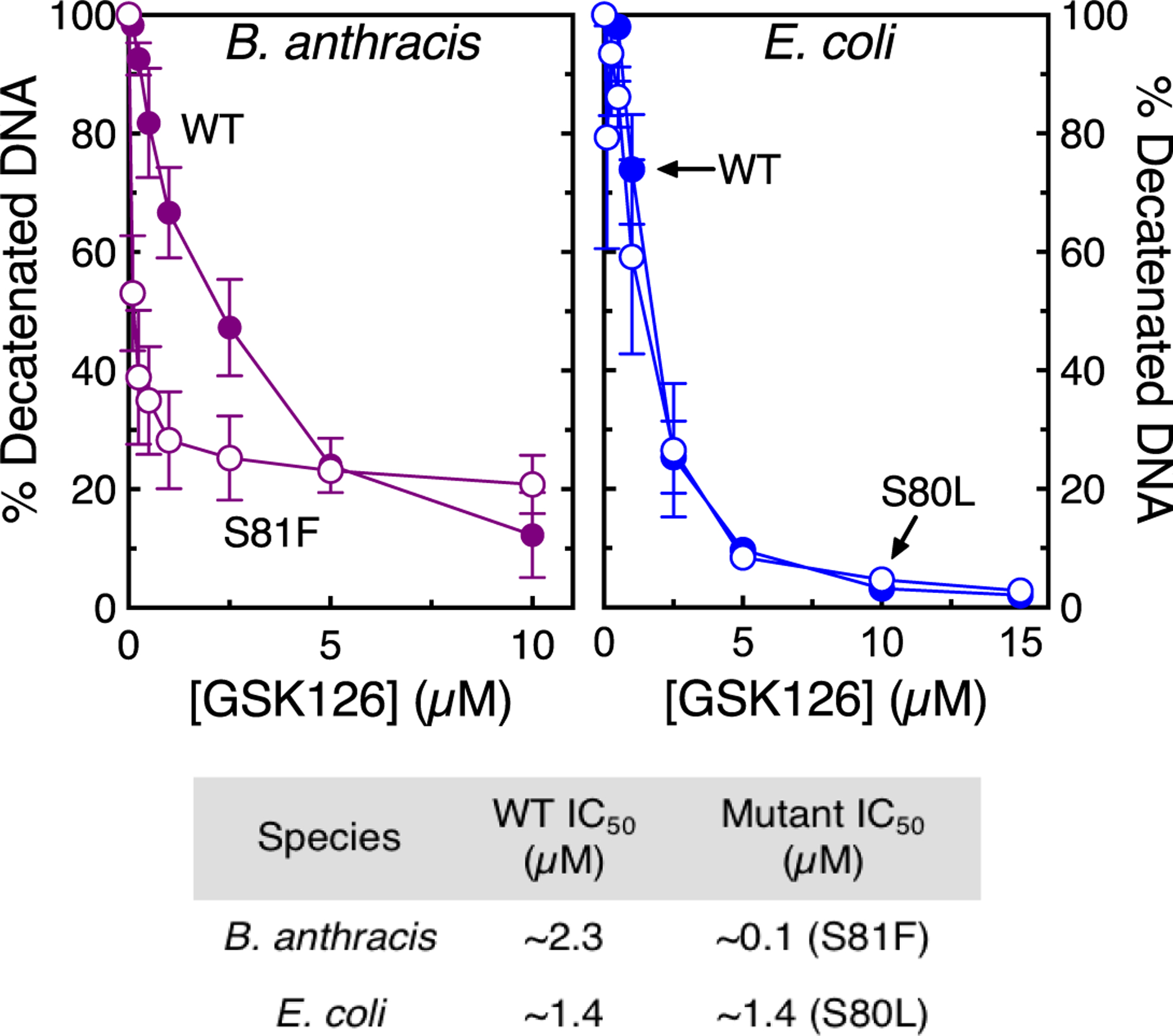
GSK126 inhibits DNA decatenation catalyzed by wild-type (WT) and fluoroquinolone-resistant topoisomerase IV. The effects of GSK126 on DNA decatenation by WT (filled circles) and fluoroquinolone-resistant (empty circles) B. anthracis WT and GrlAS81F (top left, purple) and E. coli WT and ParCS80L (top right, blue) topoisomerase IV are shown. Error bars represent the SD of at least three independent experiments. The bottom panel shows a table of IC50 values. Representative agarose gels are shown in Figure S6.
Effects of GSK126 on the DNA Cleavage Activity of Wild-type and Fluoroquinolone-resistant Topoisomerase IV.
Virtually nothing has been reported regarding the effects of NBTIs on DNA cleavage mediated by topoisomerase IV. Consequently, we examined the ability of GSK126 to enhance DNA cleavage mediated by the enzyme (Figure 9). The NBTI was a potent enhancer of single-stranded DNA cleavage with wild-type B. anthracis and E. coli topoisomerase IV (EC50 values for both enzymes were <0.5 μM). As seen with gyrase in Figure 4, no enhancement of double-stranded breaks was observed. Furthermore, in parallel with results with B. anthracis gyrase, there was a sharp increase in DNA cleavage mediated by B. anthracis topoisomerase IV in the presence of GSK126. Once again, this likely reflects a tight binding between the NBTI and the topoisomerase IV-DNA complex.
Figure 9.
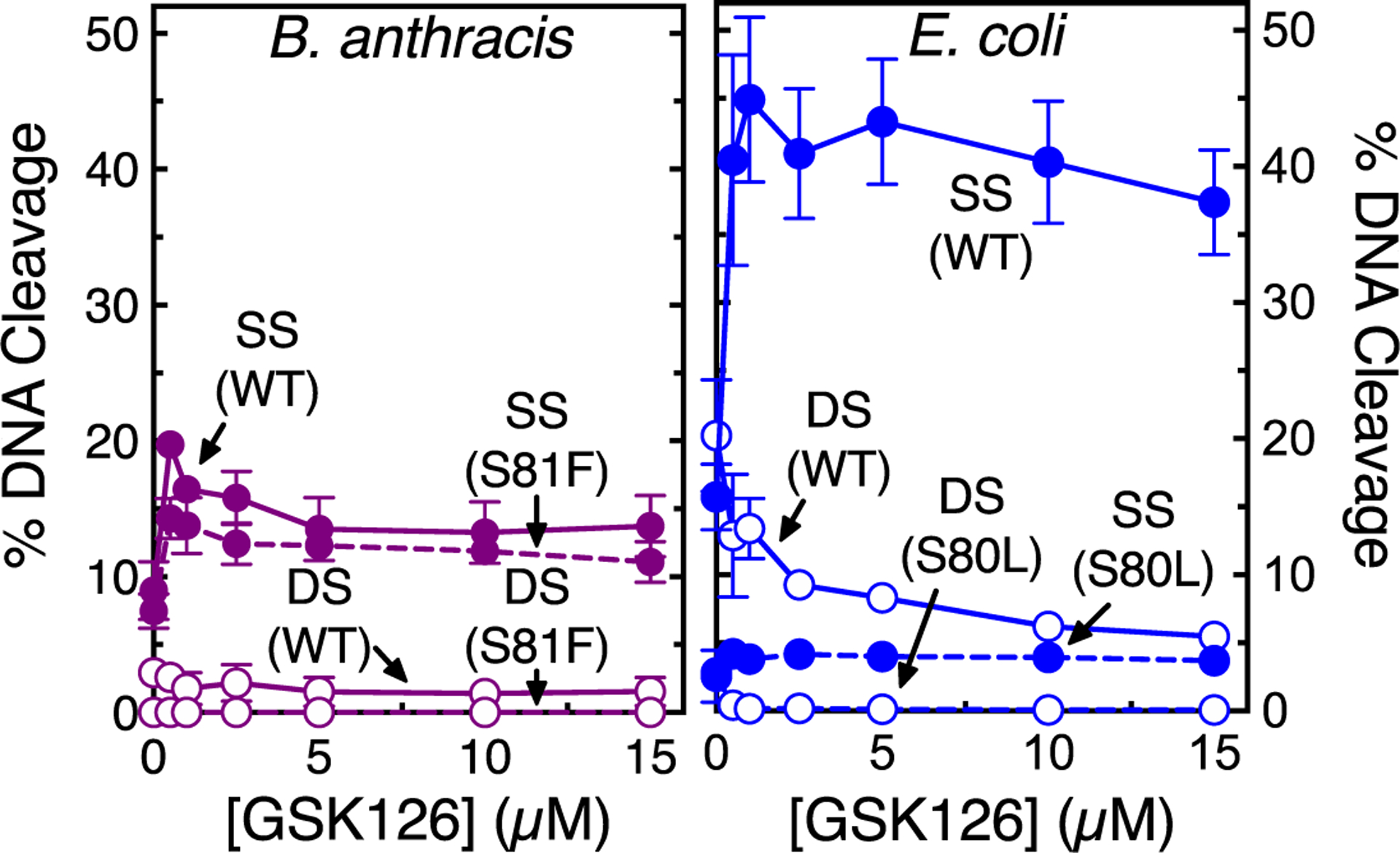
Effects of GSK126 on DNA cleavage mediated by wild-type (WT) and fluoroquinolone-resistant topoisomerase IV. The effects of GSK126 on single-stranded (SS, filled circles) and double-stranded (DS, empty circles) DNA cleavage mediated by WT (solid line) and fluoroquinolone-resistant (dashed line) B. anthracis WT and GrlAS81F (left, purple) and E. coli WT and ParCS80L (right, blue) topoisomerase IV are shown. Error bars represent the SD of at least three independent experiments. Representative agarose gels are shown in Figure S7.
The effects of fluoroquinolone resistance mutations on the induction of topoisomerase IV-mediated DNA cleavage by GSK126 were also examined (Figure 9). Both the potency and efficacy of the NBTI against the B. anthracis GrlAS81F mutant enzyme were similar to those observed for wild-type topoisomerase IV. In contrast, similar to results with E. coli gyrase (see Figure 4), GSK126 was ineffective at inducing DNA cleavage with the E. coli topoisomerase IV fluoroquinolone-resistant ParCS80L mutant. This is despite the fact that the NBTI maintained the ability to inhibit overall catalytic activity.
GSK126 Enhances only Single-stranded DNA Cleavage Mediated by Topoisomerase IV and Suppresses Enzyme-generated Double-stranded DNA Breaks.
As found with gyrase, GSK126 induced only single-stranded DNA breaks mediated by B. anthracis and E. coli topoisomerase IV. Even at high concentrations of GSK126 and longer reaction times, no enhancement of double-stranded breaks was observed (Figure 10). Furthermore, paralleling the results with gyrase, no double-stranded DNA breaks were observed in the presence of ATP (Figure 11). However, the ability of GSK126 to induce DNA cleavage with E. coli topoisomerase IV was marginal in the presence of the high energy cofactor. Thus, as was seen with E. coli gyrase (see Figure 6), this finding suggests that under physiological conditions, GSK126 acts primarily as a catalytic inhibitor of E. coli topoisomerase IV.
Figure 10.
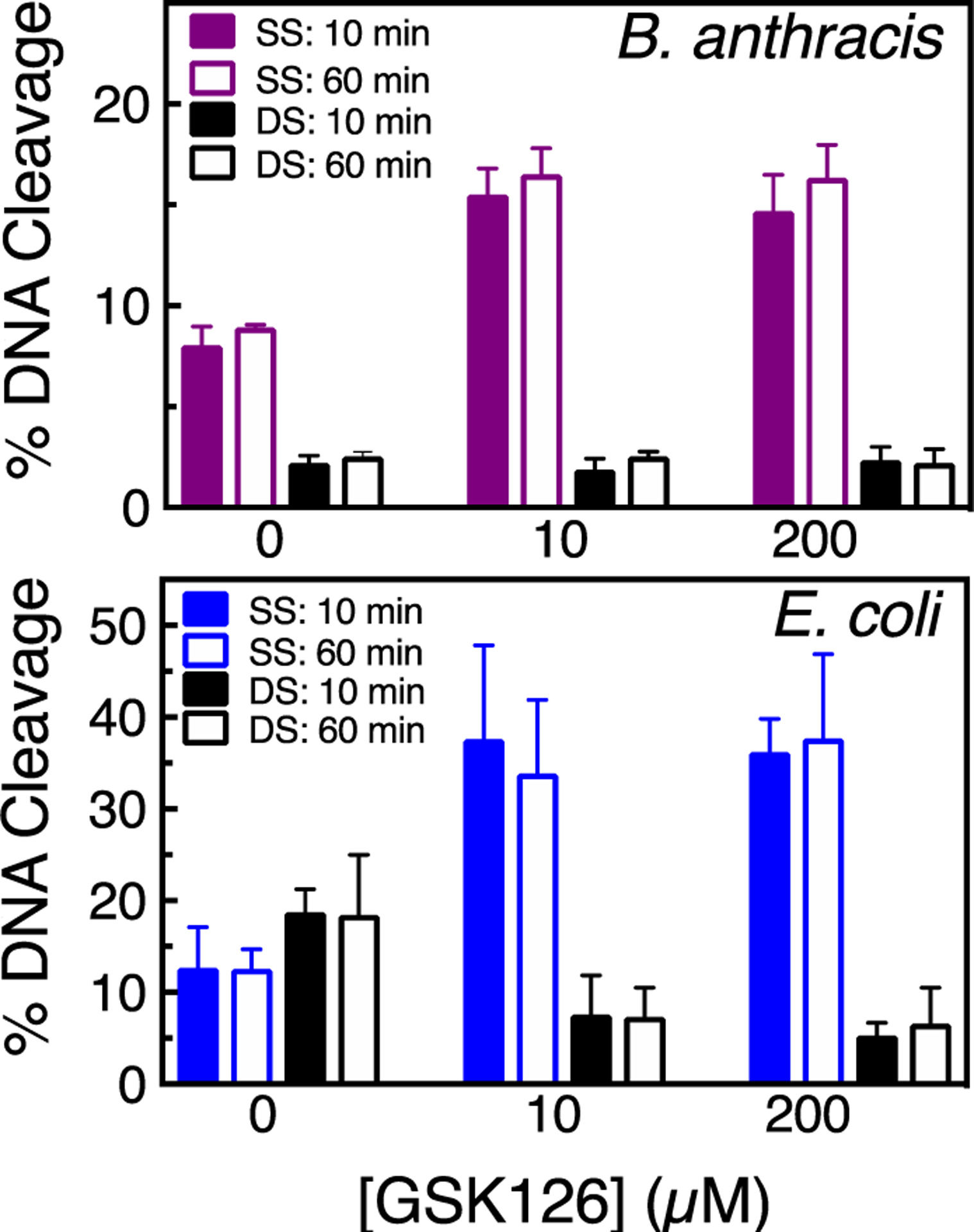
GSK126 enhances only single-stranded DNA breaks mediated by topoisomerase IV. The top panel shows the enhancement of B. anthracis topoisomerase IV-mediated single- (purple) or double-stranded (black) DNA breaks at 10 min (filled bar) or 60 min (empty bar) in the absence or presence of 10 μM or 200 μM GSK126. The bottom panel shows the enhancement of E. coli topoisomerase IV-mediated single- (blue) or double-stranded (black) DNA breaks at 10 min (filled bar) or 60 min (empty bar) in the absence or presence of 10 μM or 200 μM GSK126. Error bars represent the SD of at least 3 independent experiments. Representative agarose gels are shown in Figure S8.
Figure 11.
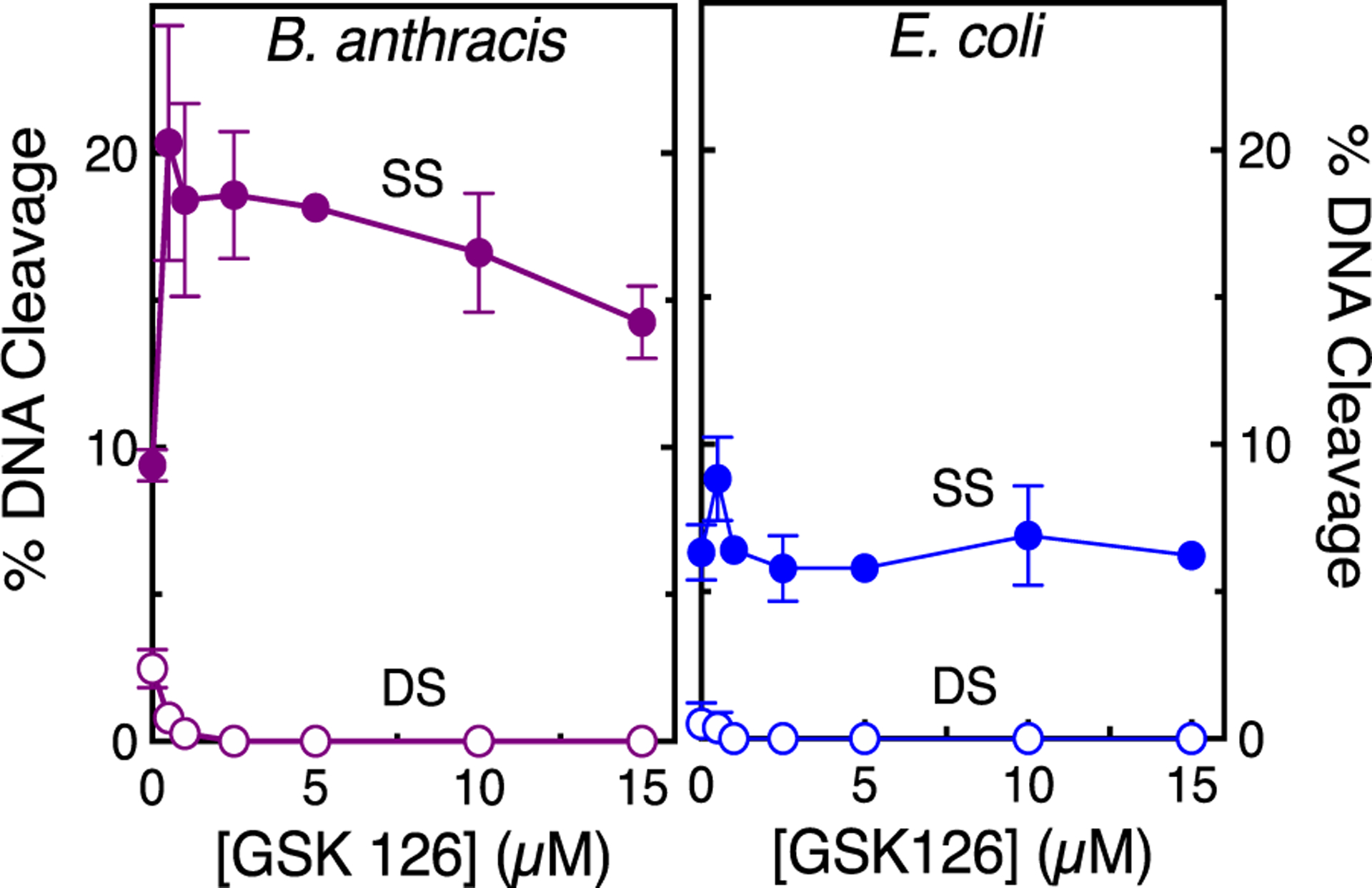
GSK126 enhances only single-stranded DNA breaks mediated by topoisomerase IV in the presence of ATP. The effects of GSK126 on single-stranded (SS, filled circles) and double-stranded (DS, empty circles) DNA cleavage mediated by B. anthracis (left, purple) and E. coli (right, blue) topoisomerase IV in the presence of 1.5 mM ATP are shown. Error bars represent the SD of at least three independent experiments. Representative agarose gels are shown in Figure S9.
Because topoisomerase IV generally maintains higher levels of baseline DNA cleavage than gyrase, double-stranded DNA breaks can be monitored in Mg2+-containing reactions. As seen in Figure 9 with E. coli topoisomerase IV and Figure 11 with B. anthracis topoisomerase IV, the increase in NBTI-induced single-stranded breaks was accompanied by a decrease in double-stranded DNA breaks. This suppression of double-stranded breaks becomes even more apparent for both species of topoisomerase IV in Ca2+-containing reactions (Figure 12). Note that data with E. coli topoisomerase IV are shown using a lower ratio (1:1) of enzyme to DNA than in Mg2+-containing reactions (2:1) because levels of double-stranded breaks are often so high with this enzyme in the presence of Ca2+ that baseline reactions contain multiply cleaved DNA products (not shown). Taken together, the above results indicate that GSK126 enhances single-stranded and suppresses double-stranded DNA breaks generated by bacterial type II topoisomerases.
Figure 12.
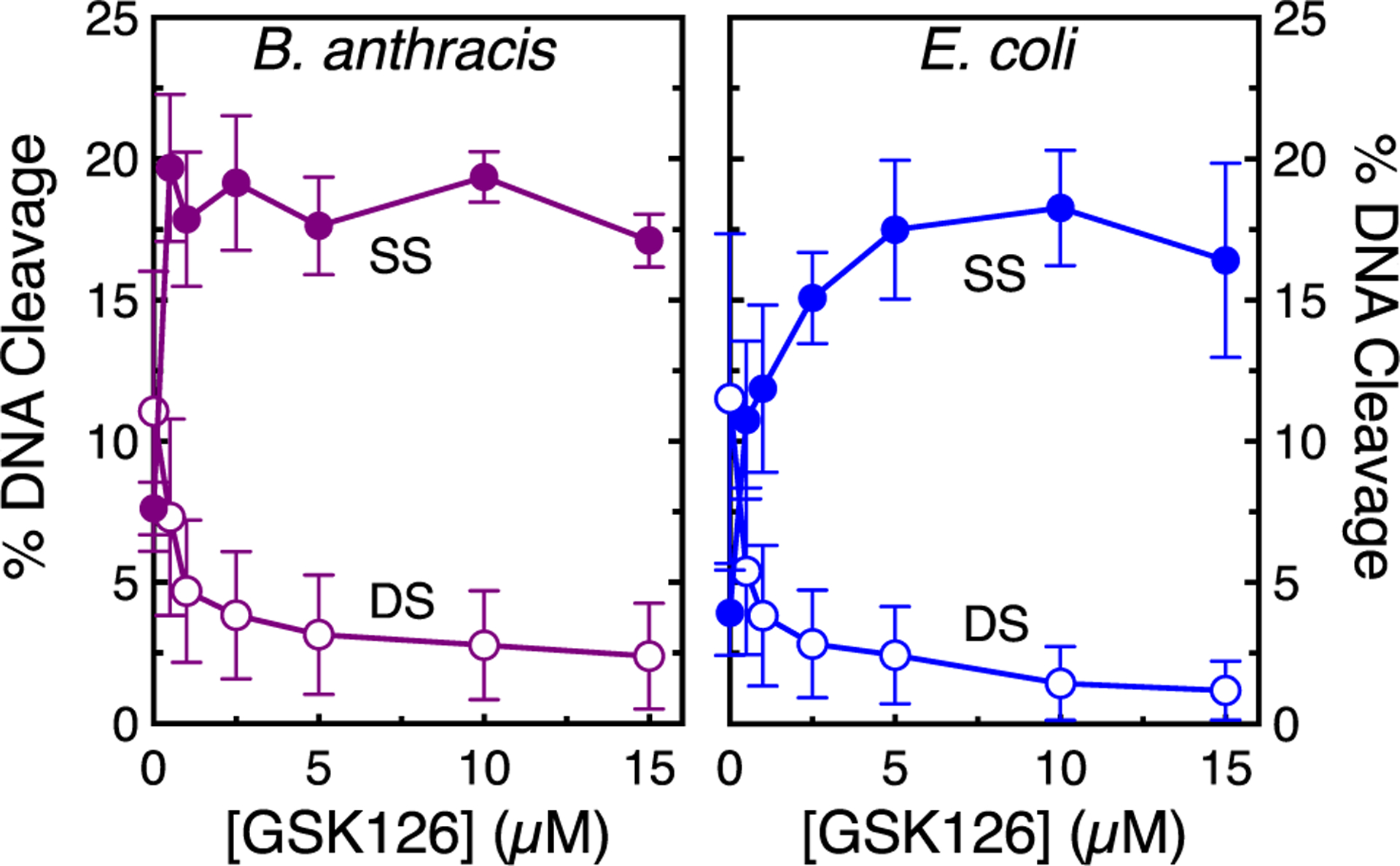
GSK126 suppresses double-stranded DNA breaks generated by topoisomerase IV. The effects of GSK126 on single-stranded (SS, filled circles) and double-stranded (DS, empty circles) DNA cleavage mediated by B. anthracis topoisomerase IV (left, purple) and E. coli (right, blue) topoisomerase IV are shown. Reaction mixtures contained 5 mM CaCl2 in place of MgCl2 to increase baseline levels of DNA cleavage. Note that data shown for E. coli topoisomerase IV were generated at a 1:1 enzyme:plasmid ratio to ensure that baseline reactions carried out in the absence of GSK126 did not include DNA molecules that contained multiple double-stranded breaks. Error bars represent the SD of at least three independent experiments. Representative agarose gels are shown in Figure S10.
Discussion
Given the rise in fluoroquinolone resistance, there is a need to supplement or eventually replace these drugs. The NBTIs display promise to address this unmet medical need. However, there is a paucity of published data describing the mechanism of action of these compounds against their type II topoisomerase targets. Structural work with NBTIs has focused on the interaction of these compounds with gyrase.21,43,47,48 Whereas previous studies have reported the inhibition of DNA supercoiling by gyrase or decatenation or relaxation by topoisomerase IV,21–25,27,28,30,39–43 only a handful of studies have examined the effects of NBTIs on DNA cleavage.21,22,25,30,43 Thus, it was not obvious that previous conclusions could be extended across bacterial species or from gyrase to topoisomerase IV. Consequently, we examined the effects of the NBTI GSK126 on the catalytic and DNA cleavage activities of wild-type and fluoroquinolone-resistant gyrase and topoisomerase IV from B. anthracis and E. coli and gyrase from M. tuberculosis.
NBTIs as bimodal agents that target bacterial type II topoisomerases.
Results of the present work indicate that GSK126 is a potent catalytic inhibitor of gyrase and topoisomerase IV from a variety of gram-positive and gram-negative bacterial species. Furthermore, the NBTI enhances single-stranded DNA cleavage and suppresses double-stranded cleavage mediated by these enzymes. These findings make it likely that NBTIs (at least those that enhance DNA cleavage) display a similar bimodal mechanism of action against gyrase and topoisomerase IV across a spectrum of bacteria. However, the role that “inhibition of catalytic activity” vs. “enhancement of cleavage” plays in the antibacterial properties of NBTIs remains an enigma and may differ from species to species. To this point, results with E. coli gyrase and topoisomerase IV suggest that under physiological conditions (i.e., in the presence of ATP) GSK126 acts primarily as a catalytic inhibitor rather than a poison with these enzymes. This finding opens the possibility that the antibacterial activity of GSK126 against E. coli (Minimal Inhibitory Concentration = 0.26 μM)30 may be more closely linked to its ability to inhibit the critical catalytic activities of gyrase and topoisomerase IV as opposed to its ability to enhance DNA strand breaks. Conversely, the potent DNA cleavage enhancement with M. tuberculosis gyrase vs. the relatively weak inhibitory properties of GSK126 against this enzyme suggest that DNA cleavage enhancement may be the more important mode of action in this species. Alternatively, the more balanced effects of GSK126 on the catalytic and DNA cleavage activities of the B. anthracis type II topoisomerases leave the door open for either mode of action (or a combination of both) to predominate in cells. Taken together, the results of the present study suggest that NBTIs are antibacterial agents that act in a bimodal fashion. Despite the fact that they target the same enzymes across a wide range of bacterial species, the mechanism by which they manifest their antibacterial properties in distinct species may be fundamentally different.
At the present time, it is not possible to predict the actions of any NBTI against any given bacterial type II topoisomerase a priori. Eventually, mechanistic in vitro studies with wild-type and resistant enzymes will have to be coupled with parallel cellular studies to develop a set of “rules” that describe the actions of NBTIs. If this information can be generated, it should hasten the development of this potentially important class of compounds.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Esha D. Dalvie for critical reading of the manuscript.
Funding
This work was supported by the US Veterans Administration (Merit Review Award I01 Bx002198 to N.O.), the National Institutes of Health (R01 GM126363 to N.O.), and the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health (HL001056-07 to K.C.N.). E.G.G. was supported by the Pharmacology Training Grant (T32 GM007628) and pre-doctoral fellowships from the PhRMA Foundation and the American Association of Pharmaceutical Scientists. A.A.O. is a member of the Chemical and Physical Biology Program and was a trainee under NIH grant T32 CA009582.
Footnotes
The authors declare no competing financial interests.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: Gel images of individual experiments (PDF).
REFERENCES
- (1).Levine C, Hiasa H, and Marians KJ (1998) DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta 1400, 29–43. [DOI] [PubMed] [Google Scholar]
- (2).Wang JC (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol 3, 430–440. [DOI] [PubMed] [Google Scholar]
- (3).Chen SH, Chan NL, and Hsieh TS (2013) New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem 82, 139–170. [DOI] [PubMed] [Google Scholar]
- (4).Hooper DC (2001) Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin. Infect. Dis 32 Suppl. 1, S9–S15. [DOI] [PubMed] [Google Scholar]
- (5).Andriole VT (2005) The quinolones: past, present, and future. Clin. Infect. Dis 41 Suppl. 2, S113–119. [DOI] [PubMed] [Google Scholar]
- (6).Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, and Zhao X (2009) Quinolones: action and resistance updated. Curr. Top. Med. Chem 9, 981–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Aldred KJ, Kerns RJ, and Osheroff N (2014) Mechanism of quinolone action and resistance. Biochemistry 53, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bush NG, Evans-Roberts K, and Maxwell A (2015) DNA topoisomerases. EcoSal Plus 6. [DOI] [PubMed] [Google Scholar]
- (9).Hiasa H (2018) DNA topoisomerases as targets for antibacterial agents. Methods Mol. Biol 1703, 47–62. [DOI] [PubMed] [Google Scholar]
- (10).Gibson EG, Ashley RE, Kerns RJ, and Osheroff N (2018) Fluoroquinolone interactions with bacterial type II topoisomerases and target-mediated drug resistance, In Resistance Antimicrobial and Implications for the 21st Century (Drlica K, Shlaes D, and Fong IW, Eds.), pp 507–529, Springer, New York. [Google Scholar]
- (11).Bax BD, Murshudov G, Maxwell A, and Germe T (2019) DNA topoisomerase inhibitors: trapping a DNA-cleaving machine in motion. J. Mol. Biol 431, 3427–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mitscher LA (2005) Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev 105, 559–592. [DOI] [PubMed] [Google Scholar]
- (13).Anderson VE and Osheroff N (2001) Type II topoisomerases as targets for quinolone antibacterials: turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des 7, 337–353. [DOI] [PubMed] [Google Scholar]
- (14).Deweese JE and Osheroff N (2009) The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 37, 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, Leydon VR, Miles TJ, Pearson ND, Perera RL, Shillings AJ, Gwynn MN, and Bax BD (2010) Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol 17, 1152–1153. [DOI] [PubMed] [Google Scholar]
- (16).Aldred KJ, McPherson SA, Wang P, Kerns RJ, Graves DE, Turnbough CL, and Osheroff N (2012) Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry 51, 370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Aldred KJ, Breland EJ, Vlčková V, Strub MP, Neuman KC, Kerns RJ, and Osheroff N (2014) Role of the water-metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry 53, 5558–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Aldred KJ, Blower TR, Kerns RJ, Berger JM, and Osheroff N (2016) Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. U. S. A 113, E839–E846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ashley RE, Lindsey RH Jr., McPherson SA, Turnbough CL Jr., Kerns RJ, and Osheroff N (2017) Interactions between quinolones and Bacillus anthracis gyrase and the basis of drug resistance. Biochemistry 56, 4191–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Coates WJ, Gwynn MN, Hatton IK, Masters PJ, Pearson ND, Rahman SS, Slocombe B, and Warrack JD (1999) Preparation of piperidinylalkylquinolines as antibacterials, In United States Patent W09937635. [Google Scholar]
- (21).Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewis CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, and Gwynn MN (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940. [DOI] [PubMed] [Google Scholar]
- (22).Black MT, Stachyra T, Platel D, Girard AM, Claudon M, Bruneau JM, and Miossec C (2008) Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother 52, 3339–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Surivet JP, Zumbrunn C, Rueedi G, Hubschwerlen C, Bur D, Bruyere T, Locher H, Ritz D, Keck W, Seiler P, Kohl C, Gauvin JC, Mirre A, Kaegi V, Dos Santos M, Gaertner M, Delers J, Enderlin-Paput M, and Boehme M (2013) Design, synthesis, and characterization of novel tetrahydropyran-based bacterial topoisomerase inhibitors with potent anti-gram-positive activity. J. Med. Chem 56, 7396–7415. [DOI] [PubMed] [Google Scholar]
- (24).Singh SB, Kaelin DE, Wu J, Miesel L, Tan CM, Meinke PT, Olsen D, Lagrutta A, Bradley P, Lu J, Patel S, Rickert KW, Smith RF, Soisson S, Wei C, Fukuda H, Kishii R, Takei M, and Fukuda Y (2014) Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med. Chem. Lett 5, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dougherty TJ, Nayar A, Newman JV, Hopkins S, Stone GG, Johnstone M, Shapiro AB, Cronin M, Reck F, and Ehmann DE (2014) NBTI 5463 is a novel bacterial type II topoisomerase inhibitor with activity against gram-negative bacteria and in vivo efficacy. Antimicrob. Agents Chemother 58, 2657–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Biedenbach DJ, Bouchillon SK, Hackel M, Miller LA, Scangarella-Oman NE, Jakielaszek C, and Sahm DF (2016) In vitro activity of gepotidacin, a novel triazaacenaphthylene bacterial topoisomerase inhibitor, against a broad spectrum of bacterial pathogens. Antimicrob. Agents Chemother 60, 1918–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Charrier C, Salisbury AM, Savage VJ, Duffy T, Moyo E, Chaffer-Malam N, Ooi N, Newman R, Cheung J, Metzger R, McGarry D, Pichowicz M, Sigerson R, Cooper IR, Nelson G, Butler HS, Craighead M, Ratcliffe AJ, Best SA, and Stokes NR (2017) Novel bacterial topoisomerase inhibitors with potent broad-spectrum activity against drug-resistant bacteria. Antimicrob. Agents Chemother 61, e02100–02116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mitton-Fry MJ, Brickner SJ, Hamel JC, Barham R, Brennan L, Casavant JM, Ding X, Finegan S, Hardink J, Hoang T, Huband MD, Maloney M, Marfat A, McCurdy SP, McLeod D, Subramanyam C, Plotkin M, Reilly U, Schafer J, Stone GG, Uccello DP, Wisialowski T, Yoon K, Zaniewski R, and Zook C (2017) Novel 3-fluoro-6-methoxyquinoline derivatives as inhibitors of bacterial DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett 27, 3353–3358. [DOI] [PubMed] [Google Scholar]
- (29).Blanco D, Perez-Herran E, Cacho M, Ballell L, Castro J, Gonzalez Del Rio R, Lavandera JL, Remuinan MJ, Richards C, Rullas J, Vazquez-Muniz MJ, Woldu E, Zapatero-Gonzalez MC, Angulo-Barturen I, Mendoza A, and Barros D (2015) Mycobacterium tuberculosis gyrase inhibitors as a new class of antitubercular drugs. Antimicrob. Agents Chemother 59, 1868–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gibson EG, Blower TR, Cacho M, Bax B, Berger JM, and Osheroff N (2018) Mechanism of action of Mycobacterium tuberculosis gyrase inhibitors: a novel class of gyrase poisons. ACS Infect. Dis 4, 1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dong S, McPherson SA, Wang Y, Li M, Wang P, Turnbough CL Jr., and Pritchard DG (2010) Characterization of the enzymes encoded by the anthrose biosynthetic operon of Bacillus anthracis. J. Bacteriol 192, 5053–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Maxwell A and Howells AJ (1999) Overexpression and purification of bacterial DNA gyrase. Methods Mol. Biol 94, 135–144. [DOI] [PubMed] [Google Scholar]
- (33).Peng H and Marians KJ (1993) Escherichia coli topoisomerase IV. Purification, characterization, subunit structure, and subunit interactions. J. Biol. Chem 268, 24481–24490. [PubMed] [Google Scholar]
- (34).Corbett KD, Schoeffler AJ, Thomsen ND, and Berger JM (2005) The structural basis for substrate specificity in DNA topoisomerase IV. J. Mol. Biol 351, 545–561. [DOI] [PubMed] [Google Scholar]
- (35).Blower TR, Williamson BH, Kerns RJ, and Berger JM (2016) Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 113, 1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Englund PT (1978) The replication of kinetoplast DNA networks in Crithidia fasciculata. Cell 14, 157–168. [DOI] [PubMed] [Google Scholar]
- (37).Anderson VE, Gootz TD, and Osheroff N (1998) Topoisomerase IV catalysis and the mechanism of quinolone action. J. Biol. Chem 273, 17879–17885. [DOI] [PubMed] [Google Scholar]
- (38).Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, and Barrell BG (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. [DOI] [PubMed] [Google Scholar]
- (39).Surivet JP, Zumbrunn C, Rueedi G, Bur D, Bruyere T, Locher H, Ritz D, Seiler P, Kohl C, Ertel EA, Hess P, Gauvin JC, Mirre A, Kaegi V, Dos Santos M, Kraemer S, Gaertner M, Delers J, Enderlin-Paput M, Weiss M, Sube R, Hadana H, Keck W, and Hubschwerlen C (2015) Novel tetrahydropyran-based bacterial topoisomerase inhibitors with potent anti-gram positive activity and improved safety profile. J. Med. Chem 58, 927–942. [DOI] [PubMed] [Google Scholar]
- (40).Tan CM, Gill CJ, Wu J, Toussaint N, Yin J, Tsuchiya T, Garlisi CG, Kaelin D, Meinke PT, Miesel L, Olsen DB, Lagrutta A, Fukuda H, Kishii R, Takei M, Oohata K, Takeuchi T, Shibue T, Takano H, Nishimura A, Fukuda Y, and Singh SB (2016) In vitro and in vivo characterization of the novel oxabicyclooctane-linked bacterial topoisomerase inhibitor AM-8722, a selective, potent inhibitor of bacterial DNA gyrase. Antimicrob. Agents Chemother 60, 4830–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Surivet JP, Zumbrunn C, Bruyere T, Bur D, Kohl C, Locher HH, Seiler P, Ertel EA, Hess P, Enderlin-Paput M, Enderlin-Paput S, Gauvin JC, Mirre A, Hubschwerlen C, Ritz D, and Rueedi G (2017) Synthesis and characterization of tetrahydropyran-based bacterial topoisomerase inhibitors with antibacterial activity against gram-negative bacteria. J. Med. Chem 60, 3776–3794. [DOI] [PubMed] [Google Scholar]
- (42).Li L, Okumu AA, Nolan S, English A, Vibhute S, Lu Y, Hervert-Thomas K, Seffernick JT, Azap L, Cole SL, Shinabarger D, Koeth LM, Lindert S, Yalowich JC, Wozniak DJ, and Mitton-Fry MJ (2019) 1,3-dioxane-linked bacterial topoisomerase inhibitors with enhanced antibacterial activity and reduced hERG inhibition. ACS Infect. Dis 5, 1115–1128. [DOI] [PubMed] [Google Scholar]
- (43).Gibson EG, Bax B, Chan PF, and Osheroff N (2019) Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect. Dis 5, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Deweese JE and Osheroff N (2009) Coordinating the two protomer active sites of human topoisomerase IIα: nicks as topoisomerase II poisons. Biochemistry 48, 1439–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pitts SL, Liou GF, Mitchenall LA, Burgin AB, Maxwell A, Neuman KC, and Osheroff N (2011) Use of divalent metal ions in the DNA cleavage reaction of topoisomerase IV. Nucleic Acids Res. 39, 4808–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Magaro G, Prati F, Garofalo B, Corso G, Furlotti G, Apicella C, Mangano G, D’Atanasio N, Robinson D, Di Giorgio FP, and Ombrato R (2019) Virtual screening approach and investigation of structure-activity relationships to discover novel bacterial topoisomerase inhibitors targeting gram-positive and gram-negative pathogens. J. Med. Chem 62, 7445–7472. [DOI] [PubMed] [Google Scholar]
- (47).Miles TJ, Hennessy AJ, Bax B, Brooks G, Brown BS, Brown P, Cailleau N, Chen D, Dabbs S, Davies DT, Esken JM, Giordano I, Hoover JL, Huang J, Jones GE, Sukmar SK, Spitzfaden C, Markwell RE, Minthorn EA, Rittenhouse S, Gwynn MN, and Pearson ND (2013) Novel hydroxyl tricyclics (e.g., GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett 23, 5437–5441. [DOI] [PubMed] [Google Scholar]
- (48).Miles TJ, Hennessy AJ, Bax B, Brooks G, Brown BS, Brown P, Cailleau N, Chen D, Dabbs S, Davies DT, Esken JM, Giordano I, Hoover JL, Jones GE, Kusalakumari Sukmar SK, Markwell RE, Minthorn EA, Rittenhouse S, Gwynn MN, and Pearson ND (2016) Novel tricyclics (e.g., GSK945237) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett 26, 2464–2469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.