

Abstract
Dilemmaones A-C are naturally occurring tricyclic indole alkaloids possessing a unique hydroxymethylene or methoxymethylene substituent at the C2 position of the indole core and a C6-C7 fused cyclopentanone. Dilemmaone B has been prepared in 5 steps from 5-methylindan-1-one, and dilemmaone A has been prepared in 3 steps from a common precursor, 6-bromo-5-methyl-7-nitroindan-1-one. In both syntheses, key steps include a Kosugi-Migita-Stille cross coupling and a reductive cyclization using hydrogen gas and a transition metal catalyst.
Graphical Abstract
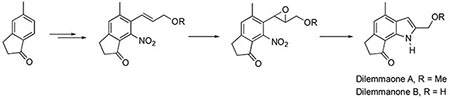
Introduction
In 1997, a variety of sponge specimens were collected off the coast of Cape Town, South Africa in order to explore the biomedical potential of South African marine invertebrates. During preliminary cytotoxicity screening, an extract of a mixed collection of sponges was selected for investigation. A bioassay directed fractionation of the crude extract led to the isolation of a sphingolipid as the active metabolite. Subsequent 1H NMR investigation of the inactive fractions led to the structural elucidation of dilemmaones A (1), B (2) and C (3) (Figure 1.).1 These indole alkaloids were originally named based upon the dilemma surrounding their sponge source. However, a process of elimination suggested that one of the sponges in the mixed collection, Ectyonanchora flabellata, is the source of the dilemmaones.
Figure 1.

Structures of the dilemmaones.
The functional group dense structures of dilemmaones A-C are unusual. To the best of our knowledge, no other 2-hydroxymethylene- or 2-methoxymethylene-substituted indoles have been isolated and fully characterized to date. In addition to the three dilemmaones, eighteen related 6,8-dimethyl-cyclopenta[g]indole natural products have been reported to date. These include the trikentrins (4-8, Figure 2), a family of compounds displaying antibacterial activity that were first isolated by Capon et al from the marine sponge Trikentrion flabelliforme.2 The structurally similar herbindoles were isolated from the Australian sponge Axinella sp. by Scheuer et al (9-11)3 and very recently from Trikentrion flabelliforme by Salib and Molinski (12, Figure 2).4 Herbindoles possess both cytotoxic and antifeedant properties.
Figure 2.

Structures of trikentrins (4-8) and herbindoles (9-12).
Subsequently, the related trikentramides have all been isolated from sponges belonging to the genus Trikentrion (Figure 3).5
Figure 3.

Structures of trikentramides (13-21).
Two related compounds are monomargine (22) and monomarginine (23), cytotoxic lactams isolated from Monocarpia marginalis (Figure 4).6 These 6,7-annulated compounds possess the indanone-like framework also seen in the dilemmaones.
Figure 4.

Structures of monomargine and monomarginine.
The interesting biological profiles of the trikentrins, herbindoles, and trikentramides combined with their uncommon structural motifs have made them attractive targets for total synthesis. The significant challenges associated with the construction of these complex systems are reflected in the many distinct approaches that have been reported for the racemic7, 8 and enantioselective total syntheses.9, 10 To date, there has been no reported synthesis of any member of the dilemmaone family, despite their skeletal resemblance to the trikentrins and herbindoles. Hence, a route to the dilemmaones was envisioned which involves a palladium catalyzed, carbon monoxide mediated, reductive cyclization as the key step.11 Herein we report the first total syntheses of dilemmaones A and B.
Results and Discussion
Retrosynthetically, dilemmaone B (2) can be constructed from the palladium-catalyzed reductive cyclization of the 2-nitrostyrene 24. Compound 24 may arise from a Kosugi-Migita-Stille cross coupling between an aryl halide (26) and vinyl stannane 25. Arylindanone 26 could be fashioned through the manipulation of commercially available 2,3-dihydro-5-methylinden-1-one (27).
For the synthesis of dilemmaone B, 6-amino-5-methylindan-1-one (28) was prepared in two steps from commercially available 5-methylindan-1-one (27) according to Pinna et al (Scheme 2).12 In order to install the nitro group present in the reductive cyclization precursor 24 (Scheme 1), a direct nitration of 28 to intermediate 31 was attempted. Lemaire et al have reported a one-step nitration of several anilines using 2,3,5,6-tetrabromo-4-methyl-4-nitrocyclohexa-2,5-dienone (34)13 as the nitronium ion source.14 Utilizing Lemaire’s protocol, aniline 28 was seemingly converted to nitroaniline 31 in good yield. An electrophilic aromatic substitution was confirmed by the presence of only one aromatic proton by 1H NMR. However, no molecular ion or protonated molecular ion was observed by HRMS of the presumed nitration product, 31. Instead, the HRMS spectrum surprisingly displayed two equally intense peaks at m/z 240 and 242, indicating the presence of a bromine atom. Thus, it was determined that aniline 28 was brominated by 33 to give bromoindanone 34 in 59% isolated yield (Scheme 2). The dual reactivity of 33, nitration or bromination, has previously been demonstrated.15 Iranpoor and Firouzabadi have also reported a nitration of free aromatic amines using a triphenylphosphine-bromine-silver nitrate reagent system.16 Interestingly, treatment of aniline 28 with PPh3-Br2-AgNO3 also produced the undesired bromoindanone 34 as the sole reaction product in comparable yield (Scheme 2).
Scheme 2.

Unexpected bromination of aniline 28.
Scheme 1.

Retrosynthetic route to dilemmaone B.
After the failure of a direct nitration of aniline 28, a more traditional route to nitroaniline 31 was envisioned via a protection-nitration-deprotection sequence. Aniline 28 was first converted to acetanilide 29 in good yield using acetyl chloride in the presence of triethyl amine (Scheme 3). Nitration of 29 initially proved to be problematic. However, after some optimization of the reaction conditions, the most effective and concise procedure was the use of fuming HNO3 and concentrated H2SO4 at a reaction temperature of −20 °C for 90 min.
Scheme 3.

Initial synthetic approach to dilemmaone B.
With nitroindanone 30 in hand, deprotection to 2-nitroaniline 31 was attempted using a procedure reported by Pouli et al.17 These conditions afforded nitroaniline 31, however, the reactions were highly inconsistent, with yields ranging from 5-84%. Conversion of 31 to the corresponding iodide or bromide also proved challenging. Sequential diazotization-iodination of aromatic amine 31 furnished iodoindanone 32, but we were unable to prepare the corresponding bromoindanone from 31 using a similar methodology. With iodoindanone 32 in hand, Kosugi-Migita-Stille cross-coupling with (E)-3-(tributylstannyl)-2-propen-1-ol (25)18 was attempted. Under all reaction conditions screened, only starting material 32 was isolated. Since iodoindanone 32 proved to be an unsuitable candidate for the preparation of cyclization precursor 24, a different approach was envisioned.
The second iteration toward dilemmaone B sought to install an aromatic halogen prior to nitration, thus avoiding the more circuitous five-step route from 27 involving a protection-deprotection protocol. To that end, various conditions were screened in an attempt to access bromoindanone 35. Diazotization of aniline 28 followed by addition of copper bromide proceeded smoothly to afford desired bromoindanone 35 (Scheme 4). The overall three-step yield of 35 from 5-methylindan-1-one (27) was, in our hands, 37%. In an effort to directly convert commercially available 27 to 35, bromination with Br2-AlCl3 was attempted, however, only the undesired isomer 36 was observed under the reaction conditions (Scheme 5, Conditions A). However, when 27 was treated with N-bromosuccinimide (NBS) in sulfuric acid, conditions previously employed in our laboratory,19 both the desired bromoindanone 35 and its regioisomer 36 were obtained in 32% and 41% isolated yields, respectively. The isomers were readily separated by chromatography. Although the yields of the desired product 35 were similar either by a bromination of 5-methylindanone (27) (Scheme 5) or in three steps from 27 via aniline 28 (Schemes 2 and 4), the former approach was more direct.
Scheme 4.

Synthesis of intermediate 35 via diazotation- bromination of 28.
Scheme 5.

Synthesis of intermediate 35 by direct bromination of 27.
Nitration of 35 using fuming HNO3 and concentrated H2SO4 gave, as expected, two isomeric products (Scheme 6). The desired nitroindanone 38 was isolated as the major product (64%) and the isomer 37 as the minor product (17%). Nitroindanone 38 was then subjected to a Kosugi-Migita-Stille cross coupling with (E)-tributyl(3-hydroxy-1-propenyl)stannane (25).20 Upon optimization, we were gratified to find that cyclization precursor 24 could be obtained cleanly in 55% yield. Compound 24 was then subjected to reductive cyclization conditions previously developed in our laboratories.21 In the event, compound 24 was treated with carbon monoxide in the presence of a catalytic amount of bis(dibenzylidene)acetone palladium, 1,10-phenanthroline, and 1,2-bis(diphenylphophino)propane. A number of related palladium-ligand combinations were examined including palladium diacetate - triphenylphosphine.22 However, all attempts to form dilemmaone B were unsuccessful only resulting in recovery of starting material (24).
Scheme 6.

Second synthetic approach to dilemmaone B.
Theorizing that the free alcohol or the electron withdrawing effect of the ketone could affect the cyclization, compound 38 was first converted to 2-nitrostyrene derivative 40 in 80% yield via a Kosugi-Migita-Stille cross coupling with (E]-tributyl(3-methoxy-1-propenyl)stannane (39)23 (Scheme 7). However, cyclization of 40 using a palladium diacetate - triphenyl phosphine catalyst system in the presence of carbon monoxide did again fail. Finally, compound 40 was treated with trimethyl orthoformate under acidic conditions in an attempt to isolate the corresponding dimethylacetal. Surprisingly the corresponding methoxyindene (41) was isolated in place of the acetal in low yield. Surmising that this unexpected product was sufficiently altered in its electronics from compound 24, derivative 41 was subjected to reductive cyclization conditions. Upon examination of the crude 1H NMR, no indole product (42) was observed. Rather, chemical shifts indicated conversion back to indanone 40. Based upon these results, the free alcohol or electronics of the system were likely not the reason for the lack of reactivity under palladium catalyzed, carbon monoxide mediated conditions.
Scheme 7.

Synthesis and attempted cyclization of derivative 41.
In an effort to effect cyclization by alternative methods, both 2-nitrostyrene derivatives, 24 and 40, were subjected to an array of other reductive cyclizations conditions. Thus, in addition to cyclization conditions described above, TiCl3-promoted reductive cyclization,24 diborane-mediated deoxygenation,25 and a modified Cadogan-Sundberg cyclization26 were attempted. In all cases, cyclization to the desired indole was never observed, but rather starting material 24 or 40 was recovered in various amounts. It is plausible that the substrates are too sterically encumbered to undergo reductive cyclization. This is supported by the lack of examples of reductive cyclizations of 3,6-disubstituted-2-nitrostyrenes to give 4,7-disubstituted indoles, using a variety of reducing agents. The only previously described sterically encumbered starting material successfully cyclized to form the corresponding indole is methyl 3-(3,6-dibromo-4,5-dimethoxy-2-nitrophenyl)propanoate. This transformation was achieved under modified Cadogan-Sundberg conditions using triphenyphosphine and a catalytic amount of MoO2Cl2(dmf)2 under microwave heating.27
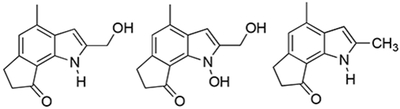
Due to the steric encumbrance of the dilemmaone core being unavoidable, and our proposed late-stage cyclization of 2-nitrostyrene 24 having thus been ruled out as a possible mean of access, an alternate pathway was envisioned. Compound 24 was transformed into epoxide 43 using 4 equivalents of 3-chloroperbenzoic acid (3-CPBA, Scheme 8). It was anticipated that treatment of 43 with Pd/C and H2 would not only result in a reduction of the nitro group to the corresponding amine or hydroxylamine, but also a subsequent epoxide ring-opening and elimination to afford the desired product, dilemmaone B (2). To the best of our knowledge, this type of transformation has only been reported once in the literature.28 Our first attempt did afford an indole from 43, however, the 1H NMR chemical shifts were similar but not in complete agreement with the shifts reported by Faulkner et al for dilemmaone B. The structure of the isolated indole product 44 was determined to be the N-hydroxy analog of dilemmaone B (Scheme 8), the product derived from incomplete reduction of the nitro group to a hydroxylamine. Additionally, 46% of unreacted starting material 43 was also isolated.
Scheme 8.

Synthesis of derivative 44 via sequential epoxidation-reductive cyclization.
Encouraged by this result, we examined this reductive cyclization for its viability toward the synthesis of dilemmaone B. As our first attempt had yielded 27% of the N-hydroxy derivative (44) and had not run to completion, the reaction was performed at a slightly elevated temperature (50 °C, Table 1, entry 2). In this case, the reaction proceeded to completion, but the only product isolated was N-hydroxyindole 44, in a somewhat higher yield. N-Hydroxyindoles have been shown to be readily deoxygenated using Pd/C-H2 (pCO = 1 atm) at ambient temperature.29 Thus, N-hydroxyindole (44) was treated with H2 (2.7 atm) in the presence of Pd/C and the reaction was carefully monitored by TLC. It was observed that starting material had been completely converted to dilemmaone B (2) in 27% yield after 2 h (Table 1, entry 3). The successful formation of dilemmaone B from N-hydroxyindole 44 suggests that a one pot reductive cyclization to give 44 followed by deoxygenation to afford dilemmaone B (2) should be possible using the conditions described in entry 3. However, after complete consumption of the starting material, it was found that over-reduction had occurred, resulting in the formation of 2,4-dimethylindole derivative 45 (Table 1, entry 4). Reductive transformations of 2-hydroxymethylindoles to give 2-methylindoles have been described using platinum black under 2.7 atm of H2 in methanol.30
Table 1.
Reductive cyclizations to give dilemmaone B (2), 44, or 45.
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Catalyst | Time | 2 | 44 | 45 | |
| 1b | 43 | Pd/C (0.02 equiv Pd) | 16 h | - | 27% (50%)c | - |
| 2 | 43 | Pd/C (0.02 equiv Pd) | 16 h | - | 37% | - |
| 3 | 44 | Pd/C (0.05 equiv Pd) | 2 h | 27% | - | - |
| 4 | 43 | Pd/C (0.05 equiv Pd) | 16 h | - | - | 16% |
| 5 | 43 | PtO2 (0.02 eauiv) | 3 h | 48% | - | - |
All reactions were performed in EtOH under 2.7 atm of H2 using Pd/C (10 mol% Pd) at 50 °C unless otherwise noted. The starting material was completely consumed. Yields reported are isolated yields after chromatography. See experimental section for details.
Reaction performed at ambient temperature.
Based on recovered starting material.
In an effort to obtain dilemmaone B (2) directly from the epoxide precursor, a different reducing agent was selected. Upon subjecting 43 to PtO2 and H2 (pCO = 2.7 atm) at 50 °C, we were gratified to find that dilemmaone B was obtained in 48% isolated yield after 3 h. Neither N-hydroxyindole 44 nor 2,5-dimethylindole 45 were observed (Table 1, entry 5).
It is worth noting that, while the identities of N-hydroxyindole 44 and dilemmaone B (2) can be confirmed and differentiated via 1H NMR analysis in CDCl3, their solubilities are poor in CDCl3 and thus splitting patterns are unclear. Conversely, both compounds are readily soluble in DMSO-d6, however, their 1H NMR chemical shifts are strikingly similar in this solvent, necessitating structural confirmation by 1H-15N gHMBCAD experiments (See supplemental materials. Dilemmaone B (2) exhibits a 1H-15N correlation between the 1H resonance at 11.30 ppm and the 15N peak at −246.6 ppm. No correlation is seen between the 1H peak of N-hydroxyindole 44 at 11.30 ppm and its 15N resonance at −199.0 ppm. Full characterization data for both compounds can be found in the experimental section.
Synthesis of Dilemmaone A
With the synthesis of dilemmaone B successfully completed, we turned our attention to dilemmaone A (1), which differs in structure only at the C2 position with the presence of a 2-methoxymethyl rather than a 2-hydroxymethyl substituent. Epoxidation of 40 proceeded smoothly to furnish compound 46 in 51% yield (Scheme 9). Cyclization precursor 46 was then subjected to reductive conditions using both Pd/C and PtO2.
Scheme 9.

Synthesis of dilemmaone A.
a) Pd/C (0.02 equiv Pd), 2.7 atm H2, EtOH, 16 h, 50 °C. 30%.
b) PtO2 (0.02 equiv), 2.7 atm H2, EtOH, 4 h, 50 °C. 25%.
Interestingly, both conditions led to clean conversion of epoxide 45 to dilemmaone A (1) in comparable yields. While Pd/C (0.02 equiv) consistently led to the production of N-hydroxyindole 44 while attempting to form dilemmaone B, the same conditions fully reduced precursor 45 to dilemmaone A, and the corresponding N-hydroxyindole was not observed. While the use of PtO2 worked cleanly toward the production of both dilemmaones A and B, it seems that the use of Pd/C is marginally more effective for the reductive cyclization of epoxide 45.
Conclusions
Dilemmaones A and B can be efficiently prepared from 2,3-dihydro-5-methylinden-1-one (27), with both syntheses featuring a Kosugi-Migita-Stille coupling and an interesting one-pot reductive cyclization using H2 and either a Pd/C or PtO2 catalyst. Dilemmaone B (2) was synthesized in 5 steps with an overall yield of 4.0 %. From a common intermediate 6-bromo-5-methyl-7-nitroindan-1-one (38), dilemmaone A (1) was synthesized in 3 additional steps with an overall yield of 12.2 %. The !H NMR data in CDCl3 for synthetic dilemmaone A and B and the compounds isolated by Faulkner et al from the sponge Ectyonanchora flabellata are almost identical. The range in difference in 1H NMR chemical shifts between isolated and synthetic material was 0.07 ppm (−0.01 to 0.06 ppm) and 0.13 ppm (−0.12 to 0.01) for dilemmaone A (1) and dilemmaone B (2), respectively. Due to their low solubility in CDCl3, the synthetically prepared alkaloids were fully characterized by 1H and 13C NMR in DMSO-d6. Probably due to the very limited amount of dilemmaone 2 isolated, Faulkner et al did not observe any of the seven quaternary 13C NMR resonances. It should be noted that while the melting point for synthetic 1 was only slightly higher compared to the isolated compound, the melting point of synthetic 2 was twenty degrees higher compared to isolated 2.
As the scope of reductive cyclization of 2-nitrostyrene derived epoxides using either Pd/C or PtO2 has not yet been explored, studies are ongoing in our laboratory to determine its versatility.
Experimental Section
General Procedures.
NMR spectra were determined in CDCl3 or DMSO-d6 at 400 or 600 MHz (1H NMR) and at 100 or 151 MHz (13C NMR). 15N NMR chemical shifts were determined in DMSO-d6 at 60.6 MHz via 1H-15N gHSQCAD experiments. The chemical shifts are expressed in δ values relative to SiMe4 (0.0 ppm, 1H and 13C) or CDCl3 (77.0 ppm, 13C) internal standards. The multiplicity of each resonance observed in the 1H NMR spectra are reported as, br = broad; s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet. Hexanes and ethyl acetate (EtOAc) were distilled prior to use. Tetrahydrofuran (THF) was purified/dried via two consecutive columns composed of activated alumina and Q5 catalyst on a Glass Contours solvent purification system. Toluene and 1,4-dioxane were purified/dried via two consecutive columns composed of activated alumina on a Glass Contours solvent purification system. N-bromosuccinimide (NBS) was recrystallized prior to use. Chemicals prepared according to literature procedures have been footnoted the first time discussed in the Results and Discussion section; all other reagents were obtained from commercial sources and used as received. Solvents were removed from reaction mixtures and products on a rotary evaporator at water aspirator pressure. Melting points are uncorrected.
N-(5-Methyl-1-oxo-1H-indan-6-yl)acetamide (29).
To a solution of 28 (4.62 g, 28.7 mmol) in dichloromethane (DCM, 100 mL) was added triethylamine (3.19 g, 31.5 mmol) at ambient temperature. After 15 min, ethanoyl chloride (2.70 g, 34.4 mmol) was added drop wise and the reaction mixture was stirred for 22 h at ambient temperature. The crude reaction mixture was washed with water (2 × 100 mL) and brine (100 mL), dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by chromatography (EtOAc) to give 29 (4.86 g, 23.9 mmol, 84%) as a tan solid. mp = 221-223 °C; 1H NMR (600 MHz, CDCl3) δ 7.99 (s, 1H), 7.33 (s, 1H), 6.98 (br s, 1H), 3.07 (t, J = 4.8 Hz, 2H), 2.68 (t, J = 5.4 2H), 2.35 (s, 3H), 2.23 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 206.1, 168.6, 152.4, 138.9, 135.9, 135.2, 128.4, 119.2, 36.6, 25.3, 24.0, 18.9; IR (ATR) 3310, 1691, 1656, 1527, 864 cm−1; HRMS (ESI) calculated for C12H14NO2 (M+H+) 204.1019, found 204.1019.
N-(5-Methyl-7-nitro-1-oxo-1H-indan-6-yl)acetamide (30).
Fuming nitric acid (10 mL) was stirred at −20 °C for 5 min, 25 drops of H2SO4 (conc.) was added and the resulting mixture was stirred at −20 °C for 20 min. This mixture was added drop wise to 29 (1.00 g, 4.92 mmol), cooled to −20 °C and stirred at the same temperature for 1.5 h. Water (20 mL) and saturated NH4G (aqueous, 20 mL) was sequentially added and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with H2O (3 × 50 mL), dried (MgSO4), filtered, and concentrated in vacuo. The resulting residue was purified by chromatography (EtOAc) to afford 30 (860 mg, 3.46 mmol, 70%) as a white solid. mp = 225-227 °C; 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H), 7.17 (br s, 1H), 3.14 (t, J = 6.0 Hz, 2H), 2.79 (t, J = 6.3 Hz, 2H), 2.39 (s, 3H), 2.18 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 200.4, 168.9, 154.6, 146.3, 142.4, 130.7, 126.7, 126.1, 36.8, 25.3, 23.2, 19.5; IR (ATR) 3347, 1718, 1673, 1536, 782 cm−1; HRMS (ESI) calculated for C12H13N2O4 (M+H+) 249.0870, found 249.0871.
6-Amino-5-methyl-7-nitroindan-1-one (31).
A solution of 30 (140 mg, 0.56 mmol) and 5 M HCl (aqueous, 10 mL) in absolute ethanol (15 mL) was heated at reflux for 18 h. The ethanol was removed in vacuo and the resulting aqueous solution was made alkaline (pH 8-9) with a saturated NaOH solution (aqueous). The solution was extracted with DCM (3 × 20 mL), dried (MgSO4), filtered, and concentrated in vacuo. The resulting residue was purified by chromatography (hexanes/EtOAc, 1:1) to afford 31 (98 mg, 0.48 mmol, 84%) as a bright yellow solid. mp = 229-231 °C; 1H NMR (600 MHz, CDCl3 + DMSO-d6) δ 7.31 (s, 1H), 5.10 (br s, 2H), 2.98 (t, J = 5.6 Hz, 2H), 2.73 (t, J = 5.7 Hz, 2H), 2.32 (s, 3H); 13C NMR (151 MHz, CDCl3-DMSO-d6) δ 199.6, 143.9, 139.2, 133.5, 129.8, 127.5, 127.0, 35.8, 23.3, 17.8; IR (ATR) 3460, 3362, 1689, 1634, 1518, 1313, 887 cm−1; HRMS (ESI) calculated for C10H11N2O3 (M+H+) 207.0764, found 207.0764.
6-Iodo-5-methyl-7-nitroindan-1-one (32).
To an ice cold solution of 31 (21 mg, 0.10 mmol) in HCl (conc., 1 mL) was added drop wise a solution of sodium nitrite (8 mg, 0.11 mmol) in H2O (1 mL). After 30 min of at 0 °C, the resulting mixture was added drop wise to a solution of potassium iodide (50 mg, 0.30 mmol) in H2O (1 mL) at ambient temperature. After 16 h at ambient temperature, the mixture was extracted with DCM (3 × 20 mL). The combined organic phases were washed in sequence with 10% NaOH (aqueous, 20 mL), 5% NaHCO3 (aqueous, 20 mL), and H2O (20 mL) before being dried (MgSO4), filtered, and concentrated in vacuo. The resulting dark yellow residue was purified by chromatography (hexanes/EtOAc, 9:1 then 8:2) to afford 32 (20 mg, 0.063 mmol, 63%) as an off-white solid. mp = 173-175 °C; *H NMR (400 MHz, CDCl3) δ 7.29 (s, 1H), 3.00 (t, J = 6.0 Hz 2H), 2.75 (t, J = 6.6 Hz, 2H), 2.61 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 201.8, 156.8, 150.4, 136.1, 131.6, 125.8, 107.3, 36.9, 31.1, 24.5; IR (ATR) 2923, 1694, 1432, 1149, 871 cm−1; HRMS (ESI, negative mode) calculated for C10H7INO3 (M-H−) 315.9471, found 315.9479.
6-Amino-7-bromo-5-methylindan-1-one (34).
2,3,5,6-Tetrabromo-4-methyl-4-nitrocyclohexa-2,5-dienone (33)13 (2.13 g, 4.55 mmol) was added to a solution of 28 (733 mg, 4.55 mmol) in ethanoic acid (5 mL). The resulting solution was stirred for 4 h at ambient temperature and then poured into a 10% NaHCO3 solution (aqueous, 15 mL). The resulting suspension was extracted with DCM (3 × 30 mL) and the combined organic phases were washed with H2O (2 × 30 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. The resulting residue was purified by chromatography (hexanes/EtOAc, 8:2) to afford 34 (640 mg, 2.66 mmol, 59%) as a brown solid. mp=177-179 °C; 1H NMR (400 MHz, CDCl3) δ 7.11 (s, 1H), 4.26 (br s, 2H), 2.95 (t, J = 5.7 Hz, 2H), 2.69 (t, J = 6.0 Hz, 2H), 2.30 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 204.2, 147.4, 142.4, 132.5, 131.2, 126.7, 103.6, 37.4, 23.8, 19.2; IR (ATR) 3378, 3352, 1688, 1621, 1469, 1318, 871 cm−1; HRMS (ESI) calculated for C10H11BrNO (M+H+) 240.0024, found 240.0018.
6-Bromo-5-methylindan-1-one (35).
A solution of 28 (100 mg, 0.62 mmol) in 48% HBr (aqueous, 500 μL), H2O (1 mL), and 1,4-dioxane (500 μL) was heated at reflux for 20 min. The resulting solution was cooled to ambient temperature and placed in an ice bath. A solution of NaNO2 (40 mg, 0.62 mmol) in H2O (1 mL) was added dropwise, and the resulting orange solution was stirred at 0 °C for 20 min. This solution was added drop wise to a solution of CuBr (100 mg, 0.68 mmol) in 48% HBr (aqueous, 500 μl) and H2O (1 mL) at 0 °C, during which time the evolution of gas was observed. The resulting dark red-brown solution was warmed to ambient temperature and stirred for 16 h. Saturated NaHCO3 (aqueous) was slowly added and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with H2O (10 mL), dried (MgSO4), filtered, and concentrated in vacuo. The resulting residue was purified by chromatography (hexanes/EtOAc 8:2) to afford 35 (104 mg, 0.46 mmol, 75%) as an orange solid.
6-Bromo-5-methylindan-1-one (35) and 4-bromo-5-methylindan-1-one (36).
Compound 27 (2.00 g, 13.7 mmol) was added to H2SO4 (conc., 15 mL) at ambient temperature. N-bromosuccinimide (NBS, 3.05 g, 17.1 mmol) was added in portions, and the resulting orange solution was stirred at ambient temperature for 3 h. The reaction was quenched by addition of ice and the solution was extracted with EtOAc (3 × 15 mL). The combined organic phases were washed with saturated NaHCO3 (aqueous, 15 mL) and brine (15 mL), dried (MgSO4), filtered, and concentrated in vacuo. The resulting residue was purified by chromatography (hexanes/EtOAc, 9:1 then 7:3) to afford, in order of elution, 35 (973 mg, 4.33 mmol, 32%) as an orange solid and 36 (1.26 g, 5.61 mmol, 41%) as a pale orange solid.
Analytical data for 35: mp = 114-116 °C; 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.36 (s, 1H), 3.05 (t, J = 5.8 Hz, 2H), 2.70 (t, J = 6.2 Hz, 2H), 2.48 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 205.1, 154.0, 144.9, 136.7, 128.6, 127.4, 124.3, 36.5, 25.3, 23.9; IR (ATR) 1702, 1597, 1432, 1379, 1171, 884 cm−1; HRMS (ESI) calculated for C10H10BrO (M+H+) 224.9915, found 224.9918.
Analytical data for 36: mp = 65-68 °C; 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 7.7 Hz, 1H), 7.27 (d, J = 7.2 Hz, 1H), 3.07 (t, J = 5.8 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.51 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 205.8, 155.4, 145.1, 136.8, 130.2, 124.1, 122.0, 36.4, 27.5, 23.1; IR (ATR) 1702, 1595, 1258, 1116, 1038, 808 cm−1; HRMS (ESI) calculated for C10H10BrO (M+H+) 224.9915, found 224.9917.
4-Bromo-5-methylindan-1-one (36).
A solution of 27 (500 mg, 3.42 mmol) in DCM (4 mL) was added drop wise over 30 min to a suspension of aluminum trichloride (910 mg, 6.84 mmol) in DCM (4 mL). To the resulting solution was added dropwise a solution of Br2 (190 μL, 3.76 mmol) in DCM (4 mL). The dark red-orange solution was heated at 50 °C for 22 h. The solution was cooled to ambient temperature and water (5 mL) was slowly added. The organic phase was washed with water (10 mL) and brine (10 mL), dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by chromatography (hexanes/EtOAc 9:1) to afford 36 (92 mg, 0.41 mmol, 12%) as a pale orange solid.
6-Bromo-5-methyl-4-nitroindan-1-one (37) and 6-Bromo-5-methyl-7-nitroindan-1-one (38).
Compound 35 (1.52 g, 6.77 mmol) was treated with fuming HNO3 (15 mL) and conc. H2SO4 (40 drops) as described for 30 (−20 °C, 90 min). Purification by chromatography (hexanes/EtOAc, 7:3 then EtOAc) gave, in order of elution, 37 (317 mg, 1.18 mmol, 17%) as an off-white solid and 38 (1.15 g, 4.30 mmol, 64%) as an orange solid. Analytical data for 37: mp = 145 °C (sublimation); 1H NMR (600 MHz, CDCl3) δ 8.11 (s, 1H), 3.18 (t, J = 5.8 Hz, 2H), 2.78 (t, J = 6.1 Hz, 2H), 2.56 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.6, 149.3, 145.7, 137.8, 137.3, 130.1, 126.2, 35.9, 23.8, 19.6; IR (ATR) 3069, 1712, 1527, 1356, 1241, 1199, 1161 cm−1; HRMS (ESI) calculated for C10H9BrNO3 (M+H+) 271.9766, found 271.9748.
Analytical data for 38: mp = 137-138 °C; 1H NMR (600 MHz, CDCl3) δ 7.51 (s, 1H), 3.12 (t, J = 5.8 Hz, 2H), 2.79 (t, J = 6.1 Hz, 2H), 2.57 (s, 3H); 13C NMR (151 MHz, CDCl3-DMSO-d6) δ 199.7, 154.7, 147.0, 145.4, 129.7, 126.3, 114.1, 36.3, 25.0, 23.9; IR (ATR) 2928, 1704, 1599, 1538, 1431, 1375, 1309, 1165, 887, 801 cm−1; HRMS (ESI) calculated for C10H9BrNO3 (M+H+) 271.9766, found 271.9745.
6-(3-Hydroxy-1-propenyl)-5-methyl-7-nitroindan-1-one (24).
To an oven-dried flask equipped with a condenser was added 38 (688 mg, 2.56 mmol), PdCl2(PPh3)2 (36 mg, 0.05 mmol) and PPh3 (27 mg, 0.10 mmol), and the system was purged with N2. 1,4-dioxane (anhydrous, 15 mL) was added via syringe, and the solution was stirred at ambient temperature. (E)-3-(Tributylstannyl)-2-propen-1-ol (25)18 (1.15 g, 3.33 mmol) dissolved in 1,4-dioxane (5 mL) was added via syringe and the solution was heated at 110 °C for 120 h. After allowing the mixture to cool to ambient temperature, water (20 mL) was added and the solution was extracted with EtOAc (3 × 30 mL). The combined organic phases were washed with brine (3 × 30 mL), dried (MgSO4), filtered, and concentrated in vacuo. The resulting residue was purified by chromatography (hexanes/EtOAc, 2:8) to afford 24 (345 mg, 1.40 mmol, 55%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3, HO not observed) δ 7.42 (s, 1H), 6.51 (d, J = 16.2 Hz, 1H), 6.06 (dt, J = 16.2, 4.6 Hz, 1H), 4.31 (br s, 2H), 3.13 (t, J = 6.0 Hz, 2H), 2.76 (t, J = 5.8 Hz, 2H), 2.43 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 201.2, 154.8, 146.3, 144.0, 137.2, 129.3, 128.1, 125.1, 120.5, 62.7, 36.5, 25.2, 21.3; IR (ATR) 3409, 2924, 1710, 1614, 1536, 1381, 1314, 1124, 1022 cm−1; HRMS (ESI) calculated for C13H14NO4 (M+H+) 248.0923, found 248.0928.
6-(3-Methoxy-1-propenyl)-5-methyl-7-nitroindan-1-one (40).
To an oven-dried flask equipped with a cold water condenser were added 38 (601 mg, 2.23 mmol), PdCl2(PPh3)2 (32 mg, 0.05 mmol), and PPh3 (24 mg, 0.09 mmol), and the system was purged with N2. 1,4-dioxane (anhydrous, 15 mL) was added via syringe, and the solution was stirred at ambient temperature. (E)-Tributyl(3-methoxy-1-propenyl)stannane (39)23 (1.05 g, 2.90 mmol) in 1,4-dioxane (anhydrous, 7 mL) was added via syringe and the solution was heated to 110 °C for 120 h. The resulting reaction mixture was cooled to ambient temperature and H2O (20 mL) was added. The solution was extracted with EtOAc (3 × 30 mL) and the combined organic phases were washed with brine (3 × 30 mL), dried (MgSO4), filtered, and concentrated in vacuo. The resulting residue was purified by chromatography (hexanes/EtOAc 1:1) to afford an approximately 10:1 ratio of E- to Z-40 (467 mg, 1.79 mmol, 80%) as an orange solid. Analytical data of the major isomer from the mixture. mp = 109-113 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (s, 1H), 6.48 (d, J = 16.3 Hz, 1H), 5.99 (dt, J =16.3, 5.0 Hz, 1H), 4.05 (dd, J = 5.0, 1.8 Hz, 2H), 3.39 (s, 3H), 3.13 (t, J = 5.8 Hz, 2H), 2.74 (t, J = 6.0 Hz, 2H), 2.43 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 200.9, 154.8, 146.2, 134.9, 131.9, 129.3, 128.2, 125.2, 122.0, 72.2, 58.3, 36.5, 25.2, 21.4; IR (ATR) 2929, 1716, 1615, 1539, 1441, 1382, 1315, 1186, 1120 cm−1; HRMS (ESI) calculated for C14H16NO4 (M+H+) 262.1074, found 262.1084. Partial analytical data of non-overlapping signals of the minor isomer from the mixture. 1H NMR (400 MHz, CDCl3) δ 6.26 (d, J = 14.4 Hz, 1H), 5.75 (m, 1H), 3.33 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 72.6, 57.9.
3-Methoxy-5-(3-methoxy-1-propen-1-yl)-6-methyl-4-nitroind-1-ene (41).
To a solution of 40 (101 mg, 0.39 mmol) in MeOH (3 mL) was added p-toluenesulfonic acid (3 mg, 0.02 mmol) followed by trimethyl orthoformate (84 μL, 0.77 mmol). The resulting solution was stirred at ambient temperature for 16 h before being diluted with EtOAc (5 mL) and NaOH (aqueous, 1M, 5 mL). The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic phases were dried (MgSO4), filtered, and concentrated in vacuo. Purification by chromatography (hexanes/EtOAc 7:3 then 1:1) gave, in order of elution, 41 (13 mg, 0.05 mmol, 12%) as a viscous yellow oil and 40 (11 mg, 0.04 mmol, 11%). 1H NMR (600 MHz, CDCl3) δ 7.30 (s, 1H), 6.53 (d, J = 16.3 Hz, 1H), 5.94 (dt, J = 16.2, 5.4 Hz, 1H), 5.35 (t, J = 2.4 Hz, 1H), 4.07-4.05 (m, 2H), 3.79 (s, 3H), 3.38 (s, 3H), 3.30 (d, J = 2.3 Hz, 2H), 2.36 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 156.2, 143.8, 135.0, 133.3, 128.0, 126.9, 126.8, 124.2, 110.0, 100.0, 72.6, 58.0, 57.3, 33.6, 20.6; IR (ATR) 2927, 1712, 1613, 1533, 1379, 1116 cm−1; HRMS (ESI) calculated for C15H18NO4 (M+H+) 276.1236, found 276.1232.
6-(1,2-Epoxy-3-hydroxypropyl)-5-methyl-7-nitroindan-1-one (43).
Compound 24 (340 mg, 1.40 mmol) was dissolved in DCM (10 mL), and the solution was cooled to 0 °C. 3-Chloroperoxybenzoic acid (3-CPBA, 70-75%/wt, 1.38 g, 5.60 mmol) was added in portions. The resulting suspension was warmed to ambient temperature and stirred for 16 h. The reaction mixture was quenched by slow addition of saturated Na2SO3 (aqueous, 10 mL) and diluted with DCM (5 mL), then washed with saturated NaHCO3 (aqueous, 10 mL) and brine (10 mL). The organic phases were dried (Na2SO4), filtered, and concentrated in vacuo. The resulting residue was purified by chromatography (EtOAc) to afford 43 (268 mg, 1.02 mmol, 74%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.45 (s, 1H), 4.09 (s, 1H), 4.02 (d, J = 13.4 Hz, 1H), 4.02 (d, J = 13.2, 1H), 3.85 (t, J = 14.9 Hz, 1H), 3.19 (s, 1H), 3.13 (t, J = 5.9 , 3H), 2.76 (t, J = 5.8 Hz, 2H), 2.57 (s, 3H), 1.63 (t, J = 6.8 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 200.8, 155.9, 147.0, 144.0, 130.0, 126.1, 125.3, 60.5, 59.2, 51.5, 36.5, 25.2, 20.1; IR (ATR) 3419, 2927, 1713, 1618, 1541, 1372, 1309, 1121, 903 cm−1; HRMS (ESI) calculated for C13H14NO5 (M+H+) 264.0866, found 264.0876.
6,7-Dihydro-2-(hydroxymethyl)-4-methylcyclopent[g]-(N-hydroxy)indol-8(1H)-one (44).
To a thick-walled pressure tube was added 43 (84 mg, 0.32 mmol) and Pd/C (10%-Pd, 7 mg, 0.006 mmol). Absolute EtOH (3 mL) was added and the resulting suspension was charged with hydrogen gas (2.7 atm). The reaction mixture was heated at 50 °C for 12 h. The solution was cooled to ambient temperature and filtered through Celite. The Celite was washed with hot absolute ethanol and the filtrate was concentrated in vacuo. The residue was purified by chromatography (hexanes/EtOAc, 1:1) affording 44 (27 mg, 0.12 mmol, 37%) as a yellow solid. mp = 164-166 °C; 1H NMR (400 MHz, DMSO-d6/CDCl3) δ 11.70 (br s, 1H), 6.87 (s, 1H), 6.39 (s, 1H), 4.98 (t, J = 5.8 Hz, 1H), 4.73 (d, J = 5.6 Hz, 2H), 3.26-3.22 (m, 2H), 2.77-2.73 (m, 2H), 2.55 (s, 3H); 1H NMR (400 MHz, CDCl3) δ 12.17 (s, 1H), 6.88 (s, 1H), 6.39 (s, 1H), 4.86 (d, J = 6.6 Hz, 2H), 3.39 (t, J = 5.1 Hz, 2H), 2.84-2.79 (m, 2H), 2.57 (s, 3H), 2.23 (t, J = 6.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6/CDCl3) δ 206.8, 153.1, 140.4, 135.3, 125.6, 121.1, 118.4, 116.6, 95.2, 54.3, 35.5, 27.0, 18.7; 15N NMR (60.8 MHz, DMSO-d6) δ - 199.0; IR (ATR) 3350, 2925, 1614, 1367, 1299, 1158, 1076, 1006 cm−1; HRMS (ESI) calculated for C13H14NO3 (M+H+) 232.0968, found 232.0978.
6,7-Dihydro-2,4-dimethylcyclopent[g]indol-8(1H)-one (45).
Treatment of 43 (58 mg, 0.22 mmol) with Pd/C (10% Pd, 12 mg, 0.011 mmol) in absolute ethanol (3 mL) under 2.7 atm of H2, as described for 44 (50 °C, 16 h), gave after chromatography (hexanes/EtOAc, 1:1) 45 as a pale-orange viscous oil. 1H NMR (600 MHz, CDCl3) δ 9.30 (br s, 1H), 6.93 (s, 1H), 6.27 (s, 1H), 3.19 (t, J = 5.4 Hz, 2H), 2.71 (t, J = 5.5 Hz, 2H), 2.58 (s, 3H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.7, 151.4, 138.7, 135.0, 130.3, 128.1, 119.2, 117.7, 99.0, 36.5, 26.6, 19.7, 13.6; IR (ATR) 3327, 2921, 1678, 1633, 1554, 1298 cm−1; HRMS (ESI) calculated for C13H14NO (M+H+) 200.1075, found 200.1070.
Dilemmaone B (2).
Treatment of 44 (17 mg, 0.074 mmol) with Pd/C (10%-Pd, 4 mg, 0.004 mmol) in EtOH (3 mL) under 2.7 atm of H2, as described for 45 (50 °C, 2 h), gave after chromatography (hexanes/EtOAc, 2:8) 2 (4 mg, 0.02 mmol, 27%) as an off-white solid.
Alternative method: Treatment of 43 (126 mg, 0.48 mmol) with PtO2 (3 mg, 0.010 mmol) in EtOH (3 mL) under 2.7 atm H2, as described for 45 (50 °C, 3 h), gave after chromatography (hexanes/EtOAc, 2:8) 2 (49 mg, 0.23 mmol, 48%) as an off-white solid. mp = 188-190 °C; 1H NMR (400 MHz, CDCl3, broad signals observed due to low solubility in CDCl3) δ 9.70 (1H), 6.97 (1H), 6.49 (1H), 4.87 (2H), 3.21 (2H), 2.73 (2H), 2.60 (3H), 2.04 (1H); 1H NMR (600 MHz, DMSO-d6) δ 11.33 (br s, 1H), 6.94 (s, 1H), 6.43 (s, 1H), 5.09 (t, J = 6.0 Hz, 1H), 4.61 (d, J = 6.1 Hz, 2H), 3.13 (t, J = 5.4 Hz, 2H), 2.62 (t, J = 5.5 Hz, 2H), 2.53 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 204.7, 151.4, 140.8, 138.1, 129.4, 127.3, 119.3, 117.2, 98.1, 56.6, 35.9, 26.0, 19.2; 15N NMR (60.8 MHz, DMSO-d6) δ −246.6; IR (ATR) 3376, 2851, 1634., 1556, 1439, 1299, 1233, 1125, 1025 cm−1; HRMS (ESI) calculated for C13H14NO2 (M+H+) 216.1019, found 216.1028.
6-(1,2-Epoxy-3-methoxypropyl)-2,3-dihydro-5-methyl-7-nitroinden-1-one (46).
Treatment of a solution of 40 (220 mg, 0.84 mmol) in DCM (5 mL) with m-CPBA (70-75%/wt, 830 mg, 3.36 mmol), as described for 43, gave after chromatography (hexanes/EtOAc 2:8) 46 (119 mg, 0.43 mmol, 51%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 3.95 (d, J = 1.8 Hz, 1H), 3.77(dd, J = 11.7, 2.8 Hz, 1H), 3.54 (dd, J = 11.7, 4.8 Hz, 1H), 3.42 (s, 3H), 3.18-3.10 (m, 3H), 2.77-2.70 (m, 2H), 2.56 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 216.0, 200.5, 186.8, 155.9, 147.2, 130.0, 126.0, 125.2, 71.2, 59.4, 57.7, 51.4, 36.5, 25.2, 20.2; IR (ATR) 2929, 1714, 1618, 1541, 1449, 1371, 1309, 1110, 907 cm−1; HRMS (ESI) calculated for C14H16NO5 (M+H+) 278.1028, found 278.1025.
Dilemmaone A (1).
Method A (Scheme 8): 46 (88 mg, 0.32 mmol) was treated with Pd/C (10% Pd, 7 mg, 0.006 mmol) and EtOH (3mL) under 2.7 atm H2 as described for 2 (50 °C, 16 h) to afford after purification (hexanes/EtOAc, 1:1) 1 (22 mg, 0.10 mmol, 30%) as an off-white solid.
Method B (Scheme 8): 46 (119 mg, 0.43 mmol) was treated with PtO2 (2 mg, 0.009 mmol) and EtOH (3 mL) under 2.7 atm of H2 as described for 2 (50 °C, 4 h) to afford after chromatography (hexanes/EtOAc, 1:1) 1 (25 mg, 0.11 mmol, 25%) as an off-white solid. mp = 151-154 °C; 1H NMR (400 MHz, CDCl3) δ 9.52 (br s, 1H), 6.95 (s, 1H), 6.50 (s, 1H), 4.61 (s, 2H), 3.37 (s, 3H), 3.21 (t, J = 5.6 Hz, 2H), 2.72 (t, J = 5.5 Hz, 2H), 2.59 (s, 3H); 1H NMR (600 MHz, DMSO-d6) δ 11.59 (s, 1H), 6.95 (s, 1H), 6.52 (s, 1H), 4.54 (s, 2H), 3.28 (s, 3H), 3.12 (t, J = 5.6 Hz, 2H), 2.62 (t, J = 5.6 Hz, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 204.6, 152.1, 138.4, 136.3, 129.5, 127.3, 119.5, 117.3, 100.4, 66.4, 57.2, 35.9, 26.1, 19.3; 15N NMR (60.8 MHz, DMSO-d6) δ −241.2 ppm; IR (ATR) 3339, 1658, 1560, 1294, 1129, 971, 793, 708 cm−1; HRMS (ESI) calculated for C14H16NO2 (M+H+) 230.1181, found 230.1174.
Supplementary Material
Highlights.
Total syntheses and structure corroboration of dilemmaones A-B
Acknowledgements
We gratefully acknowledge the C. Eugene Bennett Department of Chemistry and funding from the National Institutes of Health (1 R15 GM122002-01) for support. The National Science Foundation-MRI program is also gratefully acknowledged for the funding of a 400 MHz NMR system (CHE-1228366). The authors would like to thank the WVU BioNano Research Facilities for HRMS analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data
Supplementary data associated with this article, including 1H NMR and 13C NMR spectra of all novel compounds, 1H-15N gHSQCAD spectra for compounds 1, 2 and 44 and Tables comparing the NMR data for isolated and synthetic dilemmaones (1-2) can be found, in the online version.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References and Footnotes
- 1.Beukes DR; Davies-Coleman MT; Kelly-Borges M; Harper MK; Faulkner DJ Dilemmaones A-C, unusual indole alkaloids from a mixed collection of South African sponges, J. Nat. Prod 1998, 61, 699–701. [DOI] [PubMed] [Google Scholar]
- 2.Capon RJ; Macleod JK; Scammells PJ The trikentrins: novel indoles from the sponge Trikentrion flabelliforme, Tetrahedron 1986, 42, 6545–6550. [Google Scholar]
- 3.Herb R; Carroll AR; Yoshida WY; Scheuer PJ; Paul VJ Polyalkylated cyclopentindoles: cytotoxic fish antifeedants from a sponge, Axinella sp, Tetrahedron. 1990, 46, 3089–3092. [Google Scholar]
- 4.Salib MN; Molinski TF Six trikentrin-like cyclopentanoindoles from trikentrin flabelliforme. Absolute structural assignment by NMR and ECD, J. Org. Chem 2018, 83, 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Khokhar S; Feng Y; Campitelli MR; Quinn RJ; Hooper JNA; Ekins MG; Davis RA Trikentramides A-D, indole alkaloids from the Australian sponge Trikentrion flabelliforme, J. Nat. Prod 2013, 76, 2100–2105. [DOI] [PubMed] [Google Scholar]; b) Salib MN; Molinski TF Six trikentrin-like cyclopentanoindoles from Trikentrin flabelliforme. Absolute structural assignment by NMR and ECD, J. Org. Chem 2018, 83, 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim S-H; Mahmood K; Komiyama K; Kam T-S A Cycloartane incorporating a fused tetrahydrofuran ring and a cytotoxic lactam from Monocarpia marginalis, J. Nat. Prod 2008, 71, 1104–1106. [DOI] [PubMed] [Google Scholar]
- 7.(a) Herbindoles: Wiedenau P; Monse B; Blechert S Total synthesis of (±)-cis-trikentrin A, Tetrahedron 1995, 51, 1167–1176. [Google Scholar]; (b) Jackson SK; Banfield SC; Kerr MA Total Synthesis of (±)-Herbindole B and (±)-cis-Trikentrin B, Org. Lett 2005, 7, 1215–1218. [DOI] [PubMed] [Google Scholar]; (c) Chandrasoma N; Pathmanathan S; Buszek KR A practical, multi-gram synthesis of (±)-herbindole A, (±)-herbindole B, and (±)-herbindole C from a common intermediate via 6,7-indole aryne cycloaddition and Pd(0)-catalyzed cross-coupling reactions, Tetrahedron Lett. 2015, 56, 3507–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Trikentrins: Huntley RJ; Funk RL Total syntheses of (±)-cis-trikentrin A and (±)-cis-trikentrin B via electrocyclic ring closures of 2,3-divinylpyrrolines, Org. Lett 2006, 8, 3403–3406. [DOI] [PubMed] [Google Scholar]; b) Leal RA; Bischof C; Lee YV; Sawano S; McAtee CC; Latimer LN; Russ ZN; Dueber JE; Yu J-Q; Sarpong R Application of a palladium-catalyzed C-H functionalization/indolization method to syntheses of cis-trikentrin A and herbindole B, Angew. Chem., Int. Ed 2016, 55, 11824–11828. [DOI] [PubMed] [Google Scholar]; c) Jackson SK; Kerr MA Total synthesis of (±)-herbindole A, (±)-herbindole B, and (±)-cis-trikentrin A, J. Org. Chem 2007, 72, 1405–1411. [DOI] [PubMed] [Google Scholar]; d) Macleod J; Monahan L The total synthesis of (±)-cis- and trans-trikentrin A, Aust. J. Chem 1990, 43, 329–337. [Google Scholar]; e) Boger DL; Zhang M Total synthesis of (±)-cis and (±)-trans-Trikentrin A: Diels-Alder reactions of heteroaromatic azadienes, J. Am. Chem. Soc 1991, 113, 4230–4234. [Google Scholar]
- 9.Ent-Herbindole: Muratake H; Mikawa A; Natsume M Total synthesis of (+)-herbindole A, (+)-herbindole B, and (+)-herbindole C. Deterination of the absolute configuration of the natural herbindoles, Tetrahedron Lett. 1992, 33, 4595–4598. [Google Scholar]
- 10.a) Trikentrines: Muratake H; Natsume M Determination of the absolute structures of cis-trikentrin A and trans-trikentrin A by synthesis of their enantiomers, Tetrahedron Lett. 1989, 30, 5771–5772. [Google Scholar]; b) Muratake H; Watanabe M; Goto K; Natsume M Syntheses of (±)- and (−)-cis-trikentrin A, (±)- and (−)-trans-trikentrin A, (±)-cis-trikentrin B, (±)-trans-trikentrin B, and (±)-iso-trans-trikentrin B, Tetrahedron 1990, 46, 4179–4192. [Google Scholar]; c) Muratake H; Seino T; Natsume M Total synthesis of (6R,8S)-cis-trikentrin B, (6R,8R)-trans-trikentrin B, and (6R,8R)-iso-trans-trikentrin B. Determination of the absolute structures of the natural trikentrins B, Tetrahedron Lett. 1993, 34, 4815–4818. [Google Scholar]; d) Lee M; Ikeda I; Kawabe T; Mori S; Kanematsu K Enantioselective total synthesis of cis-trikentrin B, J. Org. Chem 1996, 61, 3406–3416. [Google Scholar]; e) Silva LF; Craveiro MV A Diastereoselective total synthesis of trans-trikentrin A: A ring contraction approach, Org. Lett 2008, 10, 5417–5420. [DOI] [PubMed] [Google Scholar]; f) Tebeka IRM; Longato GB; Craveiro MV; de Carvalho JE; Ruiz ALTG; Silva LF Total synthesis of (+)-trans-trikentrin A, Chem. Eur. J 2012, 18, 16890–16901. [DOI] [PubMed] [Google Scholar]; h) Liu W; Lim HJ; RajanBabu TV Asymmetric hydrovinylation of vinylindoles. A facile route to cyclopenta[g]indole natural products (+)-cis-Trikentrin A and (+)-cis-Trikentrin B, J. Am. Chem. Soc 2012, 134, 5496–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) For recent examples of this type of cyclization in total synthesis of indole alkaloids, see: Ansari NH; Taylor MC; Söderberg BCG Syntheses of three naturally occurring polybrominated 3,3 '-bi-1H-indoles. Tetrahedron Lett. 2017, 58, 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang YL; McArdle IW; Hubbard JW; Akhmedov NG; Söderberg BCG Total synthesis of the tetracyclic indole alkaloid ht-13-A. Tetrahedron Lett. 2016, 57, 2865–2867. [DOI] [PubMed] [Google Scholar]; c) Ghimire G; Söderberg BCG Short syntheses of the tricyclic indole alkaloids cimitrypazepine and fargesine. Tetrahedron Lett. 2016, 57, 3873–3875. [Google Scholar]; d) Ansari NH; Söderberg BCG Short syntheses of the indole alkaloids alocasin A, scalaridine A, and hyrtinadine A-B. Tetrahedron 2016, 72, 4214–4221. [Google Scholar]; e) Zhang YL; Hubbard JW; Akhmedov NG; Petersen JL; Söderberg BCG Total Synthesis of the Tetracyclic lndole Alkaloid Ht-13-B. J. Org. Chem 2015, 80, 4783–4790. [DOI] [PubMed] [Google Scholar]
- 12.Murineddu G; Lazzari P; Ruiu S; Sanna A; Loriga G; Manca I; Falzoi M; Dessi C; Curzu M; Chelucci G; Pani L; Pinna G Tricyclic pyrazoles. 4. Synthesis and biological evaluation of analogues of the robust and selective CB2 cannabinoid ligand 1-(2’,4’-dichlorophenyl)-6-methyl-N-piperidin-1-yl-dihydroindeno[1,2-c]pyrazole-3-carboxamide, J. Med. Chem 2006, 49, 7502–7512. [DOI] [PubMed] [Google Scholar]
- 13.Lemaire M; Guy A; Roussel J; Guette J Nitrocyclohexadienones. A new class of nitrating agents, Tetrahedron. 1987, 43, 835–844. [Google Scholar]
- 14.Lemaire M; Guy A; Boutin P; Guette J Direct nitration of anilines using nitrocyclohexadienones, Synthesis. 1989, 10, 761–763. [Google Scholar]
- 15.Coombes RG Dual reactivity if 2,3,5,6-tetrabromo-4-methyl-4-nitrocyclohexa-2,5-dienone., J. Chem. Soc. Perkin Trans 1, 1992, 1007–1008. [Google Scholar]
- 16.Iranpoor N; Firouzbadi H; Nowrouzi N; Firouzabadi D Highly chemoselective nitration of aromatic amines using the PPh3/Br2/AgNO3 system. Tetrahedron Lett. 2006, 47, 6879–6881. [Google Scholar]
- 17.Panteleon V; Marakos P; Pouli N; Mikros E; Andreadou I Synthesis, conformational analysis and free radical scavenging activity of some new spiropyranoquinolinones, Chem. Pharm. Bull 2003, 51, 522–529. [DOI] [PubMed] [Google Scholar]
- 18.Prepared according to: Oda H; Kobayashi T; Kosugi M; Migita T Synthesis of 5,12-DiHETE derivative by palladium-catalyzed ternary coupling between vinylic halide, a vinylic tin compound, and norbornadiene, Tetrahedron 1995, 51, 695–702. [Google Scholar]
- 19.Cummings MM; Söderberg BCG Reexamination of the bromination of 2-nitrobenzaldehyde with NBS or NaBr-NaIO4 in sulfuric acid, Synth. Comm 2014, 44, 954–958. [Google Scholar]
- 20.Prepared according to: Oda H; Kobayashi T; Kosugi M; Migita T Synthesis of 5,12-DiHETE derivative by palladium-catalyzed ternary coupling between vinylic halide, a vinylic tin compound, and norbornadiene, Tetrahedron 1995, 51, 695–702. [Google Scholar]
- 21.Scott TL; Söderberg BCG Palladium-catalyzed synthesis of 1,2-dihydro-4(3H)-carbazolones. Formal total synthesis of murrayaquinone A. Tetrahedron 2003, 59, 6323–6332. [Google Scholar]
- 22.Söderberg BCG; Shriver JA Palladium-catalyzed synthesis of indoles by reductive N-heteroannulation of 2-nitrostyrenes. J. Org. Chem 1997, 62, 5838–5845. [Google Scholar]
- 23.Prepared according to: Brown JM; Leppard SJ; Oakes J; Thornthwaite D Diastereoselectivity in scalemic tartrate/titanium epoxidations, Chirality 2000, 12, 496–504. [DOI] [PubMed] [Google Scholar]
- 24.Tong S; Xu Z; Mamboury M; Wang Q; Zhu J Aqueous titanium trichloride-promoted reductive cyclization of o-nitrostyrenes to indoles: Development and application to the synthesis of rizatriptan and aspidosperidine, Angew. Chem., Int. Ed 2015, 54, 11809–11812. [DOI] [PubMed] [Google Scholar]
- 25.Yang K; Zhou F; Kuang Z; Gao G; Driver TG Diborane-mediated deoxygenation of o-nitrostyrenes to form indoles, Org. Lett 2016, 18, 4088–4091. [DOI] [PubMed] [Google Scholar]
- 26.Li B; Williams JD; Peet NP Two concise total syntheses of the wasabi phytoalexin methyl 1-methoxyindole-3-carboxylate, Tetrahedron Lett. 2013, 54, 3124–3126. [Google Scholar]
- 27.a) Selvaraju S; Sachinthani KAN; Hopson RA; McFarland FM; Guo S; Rheingold AL; Nelson TL Eumelanin-inspired core derived from vanillin: a new building block for organic semiconductors. Chem. Commun 2015, 51, 2957–2959. [DOI] [PubMed] [Google Scholar]; b) Huleatt PB; Lau J; Chua S; Tan YL; Duong HA; Chai CLL Concise, efficient and practical assembly of bromo-5,6-dimethoxyindole building blocks. Tetrahedron Lett. 2011, 52, 1339–1342. [Google Scholar]
- 28.Watanabe T; Hamaguchi F; Ohki S Indoles. III. New synthesis of 4-indolecarboxylic acid, Chem. Pharm. Bull 1972, 20, 2123–2127. [Google Scholar]
- 29.Penoni A; Volkmann J; Nicholas KM Regioselective synthesis of indoles via reductive annulation of nitrosoaromatics with alkynes. Org. Lett 2002, 4, 699–701. [DOI] [PubMed] [Google Scholar]
- 30.For examples, see: Smart BP; Oslund RC; Walsh LA; Gelb MH The first potent inhibitor of mammalian group X secreted phospholipase A2: elucidation of sites for enhanced binding. J. Med. Chem 2006, 49, 2858–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.