

Abstract
Methylation of organohalides represents a valuable transformation, but typically requires harsh reaction conditions or reagents. We report a radical approach for the methylation of (hetero)aryl chlorides using nickel/photoredox catalysis wherein trimethyl orthoformate, a common laboratory solvent, serves as a methyl source. This method permits methylation of (hetero)aryl chlorides and acyl chlorides at an early and late-stage with broad functional group compatibility. Mechanistic investigations indicate that trimethyl orthoformate serves as a source of methyl radical via β-scission from a tertiary radical generated upon chlorine-mediated hydrogen atom transfer.
Graphical Abstract
INTRODUCTION
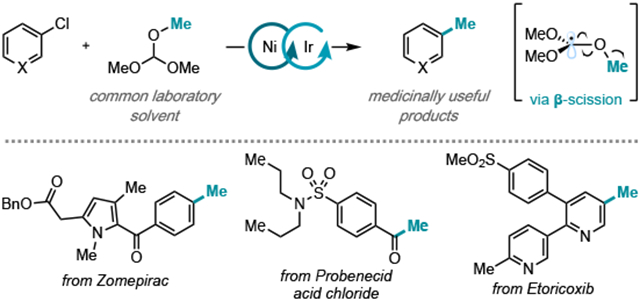
The methyl group is one of the most commonly occurring structural motifs in medicinal compounds, appearing in 80% of top-selling small-molecule pharmaceuticals in 2018.1 In drug development, installation of a methyl substituent - on an aromatic ring, for example - is a common strategy for rendering compounds with improved binding affinity, bioavailability, and metabolic stability (Figure 1A).2 The impact of methylation on the biological and physical properties of a molecule has been so pervasive that it has been named the “magic methyl effect”.3 As such, synthetic reactions that enable the installation of methyl groups site-selectively and under mild conditions are of broad value. Transition metal-catalyzed cross-coupling is one of the most robust and modular methods for site-selective carbon–carbon bond formation.4 However, most traditional cross-coupling methods for methylation rely upon acutely toxic alkylating reagents or highly reactive organometallic reagents such that compatibility with common functional groups found in bioactive small molecules is problematic.5 Accordingly, there remains a great demand for cross-coupling methods that enable methylation at a late-stage for medicinal and/or process chemistry applications.3
Figure 1.

A Examples of aryl methylation that have resulted in enhanced bioactivity. B Established radical-based methylation strategies in cross-coupling. C This work.
Recently, researchers have turned to radical-based methods to overcome challenging C(sp3)–C cross coupling. For example, Minisci-type methylation reactions of heteroarenes have been reported with a variety of methyl radical sources, including acetic acid,6a tert-butylperacetate,6b dicumyl peroxide,6c methane,6d and methanol.6e-h Nevertheless, these methods are only effective for electron-deficient heteroarenes and their site selectivity can limit broad applicability. To address these challenges, a few radical-based methylation reactions of aryl halides have been described (Figure 1B). The Weix group demonstrated that N-hydroxy-phthalimide esters can serve as competent sources of methyl, primary, and secondary alkyl radicals for the alkylation of aryl iodides with nickel catalysis.7 Additionally, using nickel/photoredox catalysis in combination with a supersilane reagent, the MacMillan group achieved cross-coupling between aryl and alkyl bromides to furnish new C(sp3)–C(sp2) bonds, including one example of methylation using methyl tosylate.8 Methyl tosylate has also recently found application in nickel-catalyzed methylation of aryl bromides and tosylates, alkyl halides, and acid chlorides, as reported by Gong and coworkers.9 While highly enabling, these methods still require preparation of the methyl radical source or employment of electrophilic methylating reagents. Furthermore, a radical-based method that permits methylation of aryl chlorides, the most abundant and inexpensive aryl halide coupling partner, has yet to be reported. In addition to the benefits that use of aryl chlorides would permit for early-stage methylation, late-stage conversion of aryl chlorides to toluenes has demonstrable value in medicinal chemistry as well: for example, methylation of the aryl chloride in a Celecoxib precursor shortened its half-life such that it could be administered as the first selective COX-2 non-steroidal anti-inflammatory drag.10
Our group has recently reported an approach to the cross-coupling of chloride-containing electrophiles with C(sp3)–H bonds via nickel and photoredox catalysis.11 The chloride-containing electrophile serves as both the coupling partner and the source of chlorine radical for C(sp3)–H bond activation via hydrogen atom transfer (HAT). We questioned whether this reaction platform could be adapted to enable methylation of (hetero)aryl chlorides. While methane is the most analogous methyl radical source, initial reactions employing methane under our previously optimized conditions proved unfruitful. Instead, we sought an alternative methyl radical source that could be accessed via HAT and that would have similar attributes to methane, including its abundance, cost, and functional group compatibility.
In this context, we considered that trimethyl orthoformate, a common laboratory solvent, could serve as a methyl radical source (Figure 1C). Owing to its weak tertiary C(sp3)–H bond (88.7 kcal/mol), preferential HAT at the methine over the methyl positions was computed to be favorable (ΔBDFE = −1.2 kcal/mol) (Figure 2A). If addition of the resultant tertiary radical were slow to rebound into a nickel catalyst, we posited that unimolecular β-scission could occur to generate methyl radical and dimethyl carbonate (ΔG = −25.1 kcal/mol and ΔG‡ = 11.4 kcal/mol).12 Coupling the generation of high energy alkyl radicals with the formation of a stable carbonyl byproduct via β-scission has been well-studied,13 but only recently emerged as a strategy in transition metal-catalyzed cross coupling. Reported examples of β-scission from carbon-centered radicals in cross coupling are limited to xanthate esters14 that must be pre-synthesized and afford only stabilized radical species, thus precluding access to methyl radical.15 In contrast, trimethyl orthoformate would permit access to methyl radical from a commercial and abundant reagent.
Figure 2.

A Computed reaction coordinate (CBS-QB3). B Initial result. a [Ir[dF(CF3)ppy]2(dtbbpy)]PF6 (1 mol%), Ni(cod)2 (10 mol%), dtbbpy (15 mol%), K3PO4 (2 equiv), (MeO)3CH:PhH (1:1) (0.05 M).
RESULTS AND DISCUSSION
Reaction Optimization.
To evaluate the feasibility of using trimethyl orthoformate as a source of methyl radical, we investigated the coupling of 4′-chloroacetophenone with trimethyl orthoformate (Figure 2B).11a Using Ni(cod)2 (10 mol%), 4,4'-di-tert-butylbipyridine (dtbbpy) (15 mol%), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%), and K3PO4 (2 equiv) in a 1:1 mixture of trimethyl orthoformate and benzene afforded the desired toluene 3 in 38% yield. Two other products were formed alongside the methylated product: ester product 2 (8% yield), arising from coupling at the tertiary position of trimethyl orthoformate (B in Figure 2), and benzylic ether product 1 (27% yield), arising from coupling at the primary C–H bonds (A in Figure 2).
To improve the yield and selectivity of the reaction, we undertook an optimization of the reaction conditions. Use of NiCl2•glyme as the Ni source resulted in a slightly improved yield of 43% (Table 1, Entry 1). Both yield and selectivity between 3 and 1 could be further improved by employing tert-butylbenzene as the reaction cosolvent, likely a result of greater stabilization of chlorine radical by the more electron-rich solvent (Table 1, Entry 2). Previous studies have shown that chlorine radical, an electrophilic radical species, can be stabilized by arenes to deliver more selective C(sp3)–H abstraction according to the Hammond postulate.16 Reducing the loading of trimethyl orthoformate to ten equivalents in benzene led to 26% yield of the desired toluene in 24 hours (Table 1, Entry 3). Conversely, performing reactions in trimethyl orthoformate without cosolvent led to the largest increase in yield and selectivity, providing the desired toluene in 61% yield (Table 1, Entry 4). Selectivity between 3 and 1 reached a ratio of 3.9:1, while ester formation was minimized (< 5% yield) relative to reactions run with aromatic cosolvents. Upon omission of the base, 3 was obtained in 9% yield, suggesting that HCl formation without sequestration may be deleterious to reactivity (Table 1, Entry 5). All other components of the reaction were required for productive methylation, as individual omission of NiCl2•glyme, dtbbpy, photocatalyst, and light source resulted in no product formation and complete recovery of the aryl chloride (Table 1, Entries 6-9).
Table 1.
Reaction evaluation for aryl methylation.
 | ||||
|---|---|---|---|---|
| Entrya | Deviation | % Yield 1 | % Yield 2 | % Yield 3 |
| 1 | C6H6 : (MeO)3CH (1 : 1) | 32 | 9 | 43 |
| 2 | tert-butylbenzene : (MeO)3CH (1 : 1) | 24 | 3 | 52 |
| 3 | 10 equiv. (MeO)3CH in C6H6b | 17 | 5 | 26 |
| 4 | none | 15 | 3 | 61 |
| 5 | without base | n.d.c | n.d.c | 9 |
| 6 | without NiCl2·glyme | 0 | 0 | 0 |
| 7 | without dtbbpy | 0 | 0 | 0 |
| 8 | without photocatalyst | 0 | 0 | 0 |
| 9 | without light | 0 | 0 | 0 |
Reactions performed on 0.1 mmol scale with 1,3,5-trimethoxybenzene added as an external standard (GC yield). 0.05 M trimethyl orthoformate = 182 equiv.
24 h.
n.d. = not determined.
Substrate Scope.
With optimized conditions, we examined the reaction scope (Figure 3A). Generally, electron-deficient aryl chlorides underwent methylation in higher yields than electron-rich aryl substrates, consistent with their relative reactivity to Ni(0) oxidative addition. Unlike methods that employ reactive nucleophilic or electrophilic methylating reagents, a variety of sensitive functionality was well tolerated, including ketones 3 and 4, nitriles 5 and 6, aldehyde 7, and ester 8. Ortho-substituted aryl chlorides (5, 11) also delivered methylated product in moderate to high yield. Methylation of substrates containing heteroaryl functionality distal to the site of cross-coupling, including pyridines 13, 16, and 17, furan 14, and pyrrole 15, could also be achieved. Biologically relevant aryl chlorides loratadine 19, fenofibrate 20, and zomepirac 21 provided the corresponding methylated product in high yields, indicating that this method is amenable to late-stage functionalization of bioactive compounds.10 In the methylation of perphenazine (22), exclusive methylation of the aryl chloride was observed, in contrast to methods employing electrophilic methylating reagents that would be expected to methylate the primary alcohol. Finally, procymidone, which contains two chemically equivalent aryl chlorides, underwent selective mono-methylation to produce 23 in 55% yield, likely a result of the sensitivity of the catalytic system to electronic effects.
Figure 3.

Methylation of (hetero)aryl chlorides (0.5 mmol scale). a GC yield. b Yield after hydrogenolysis of reaction mixture (5 mol% Pd/C, H2 (balloon), MeOH (0.05 M), 10 h). c Isolated with 10% homocoupled product; d 19F NMR yield. e 0.2 mmol scale reaction. f1H NMR yield. g0.25 mmol scale reaction (48 h) in trimethyl orthoformate:benzene (1:1).
Employment of electron-rich aryl chlorides in the nickel/photoredox cross-coupling reaction delivered low yields of methylated products, likely due to sluggish oxidative addition (Table 2, Entries 1 and 4). To overcome this challenge, we turned to aryl bromides as substrates, but productive methylation was not observed from these substrates either, presumably because the weak H–Br bond (BDE = 88 kcal/mol) renders HAT from bromine radical less favorable. However, reactivity could be restored by using aryl bromides in conjunction with an exogenous chloride additive for halide exchange (Table 2, Entries 3 and 6), delivering 24 and 25 in 56% and 52% yield, respectively.
Table 2.
Methylation of aryl bromides.
 | ||||
|---|---|---|---|---|
| Entrya | R | X | Additive | % Yield |
| 1 | OMe | Cl | none | <2 |
| 2 | OMe | Br | none | <2 |
| 3 | OMe | Br | TBACI | 56 |
| 4 | Me | Cl | none | 8 |
| 5 | Me | Br | none | <2 |
| 6 | Me | Br | TBACI | 52 |
Reactions performed on 0.25 mmol scale with 1,3,5-trimethoxybenzene added as an external standard (GC yield). TBAC1 = tetrabutylammonium chloride.
Next, we sought to explore the scope of heteroaryl chloride coupling partners (Figure 3B). A variety of nitrogen, oxygen, and sulfur-containing heteroaryl chlorides underwent methylation in moderate to high yields, including pyridines 26–29, quinolines 30–34 and 42, quinoxaline 35, quinazoline 41, pyrimidines 36–37, thiophenes 38 and 39, and thiazole 40. Importantly, this method for radical methylation enables functionalization at sites that are not accessible via Minisci-type reactivity; for example, the 3-, 6-, and 7-positions of quinolines (32, 33, and 34, respectively) and positions meta to nitrogen atoms in pyridines (27–29) underwent site-selective methylation. Biologically-relevant heteroaryl chlorides, such as etoricoxib (43), also underwent methylation in good yield.
The primary byproduct in this methodology is derived from alkoxymethylation of the aryl chloride. Since this byproduct is a benzylic ether, a solution that we pursued was subjecting the reaction to Pd/C hydrogenolysis to convert the byproduct into methylated product. Select examples of the improvement in yields afforded by this workup protocol, including for high-value targets 19 and 20, are shown in Figure 3.
Furthermore, our group has previously demonstrated that acid chlorides can be used as coupling partners in Ni/photo-redox-catalyzed C(sp3)–H functionalization of alkanes.11c We recognized that application of the methylation conditions to acid chloride coupling partners could potentially deliver a mild synthesis of aliphatic and aromatic methyl ketones9b as compared to traditional protocols which rely on harsh organometallic reagents or strong Lewis acids.17 Gratifyingly, we found that application of the optimized methylation conditions to this substrate class afforded access to methyl ketones from primary (44), secondary (45), tertiary (46), and aryl (47) acid chlorides (Figure 3C). The method was also applicable to acid chlorides prepared from biologically relevant ketoprofen (45) and probenecid (47). Additionally, ester 48 and tertiary amide 49 were prepared from the corresponding chloroformate and carbamoyl chloride.18
Mechanistic Investigations.
Having explored the scope of this transformation, we next sought to evaluate the mechanism of methyl incorporation from trimethyl orthoformate. According to our prior mechanistic work,11a we propose that photoelimination from Ni(III) intermediate 52 affords a chlorine radical which mediates HAT with trimethyl orthoformate (Figure 4A).19 Observation of methylated product 3, ester product 2, and benzylic ether product 1 lends initial support to a HAT-initiated process in the Ni-catalyzed cross-coupling. As further evidence for the intermediacy of organic radical intermediates, when the methylation of 4′-chloroacetophenone was conducted under standard conditions with one equivalent of TEMPO, none of these three products were observed.20
Figure 4.

A Proposed catalytic cycle. B Reaction progress by ReactIR (normalized single wavelength kinetics at 762 cm−1). C Investigation of hydrodealkoxylation pathway. D Alternative mechanistic possibility for methylation. E Methyl radical trapping experiment in the absence of Ni. a Optimized Ni/photoredox methylation conditions in Figure 3.
To further evaluate the mechanism, we monitored the reaction course via ReactIR. The reaction progress showed that as 4′-chloroacetophenone is consumed, dimethyl carbonate and 4′-methylacetophenone (3) are generated in a 1:1 ratio (Figure 4B, right). The formation of dimethyl carbonate and 3 could also be traced in a 1:1 ratio via quantitative 13C NMR experiments and both experiments suggest overall non-zeroth order kinetics (see SI, Figures S19-S20). While this observation is consistent with a β-scission mechanism which requires stoichiometric formation of dimethyl carbonate at a comparable rate to product formation, we also considered that methylated product 3 and dimethyl carbonate could arise from Ni-catalyzed hydrodealkoxylation of byproduct 1. However, a control reaction subjecting 1 to the photocatalytic Ni conditions did not generate methylated product 3 (Figure 4C).
Another pathway that is consistent with these results is the one shown in Figure 4D, wherein Ni(II)(Ar) species 53 mediates oxidation of the tertiary radical of trimethyl orthoformate (B), delivering Ni(I)–X that undergoes addition to the oxocarbenium intermediate (56).21 Such a process would produce Ni(III) intermediate 54 and dimethyl carbonate, which are both invoked in the proposed catalytic cycle (Figure 4A). A key difference between these proposals is that, according to the β-scission mechanism, methylation with trimethyl orthoformate should be possible in the absence of nickel. Indeed, when diethyl vinylphosphonate was reacted with benzoyl peroxide at 80 °C in trimethyl orthoformate, the methylated Giese product 57 was observed in 22% yield (Figure 4E). Taken together, these experiments provide support for the proposed β-scission mechanism for methyl radical generation from trimethyl orthoformate.
Expansion of the Methodology.
More generally, the results gathered in interrogating the β-scission mechanism suggest that trialkyl orthoformates can serve as broadly useful and practical aliphatic radical sources for the design of new synthetic methods. For example, in this Ni/photoredox cross-coupling, simple modification of the solvent to triethyl or triisopropyl orthoformate allowed access to the respective alkylated product 58 or 59 (Figure 5A).22
Figure 5.

A Alkylation with trialkyl orthoformates. B Alkylation with acetals. Reaction conditions indicated in Figure 3 with the following modifications: a Reaction performed in triethyl orthoformate. b Reaction performed 1:1 with orthoformate or acetal in benzene. c Reaction performed with 12 equiv acetal in benzene.
This finding prompted us to consider using acetals as sources of aliphatic radicals in this cross-coupling reaction. Dialkyl acetals have been shown to undergo β-scission with liberation of aliphatic radicals;13d-e however, the reactions are initiated under harsh conditions (homolysis from peroxides or high temperatures) and, to the best of our knowledge, acetals have not been used as a source of radicals in metallaphotoredox cross-coupling. For the transfer of more complex aliphatic radicals, we envisioned that these reagents could be attractive alternatives to orthoformates given the facile synthesis of acetals from aldehydes and a diverse array of alcohols and because acetals feature only two equivalents of the alkyl cross-coupling partner per molecule rather than three in the related orthoformate derivative. In a preliminary study, we were delighted to find that replacing the orthoformate cosolvent with a 1:1 mixture of benzaldehyde dimethyl acetal:benzene provided methylated product 4 in 50% yield (Figure 5B). Furthermore, reaction of 4-chlorobenzophenone with 12 equivalents of benzaldehyde dineopentyl acetal afforded the resulting alkylated product 60 in 41% yield. These preliminary results offer promise for utilizing β-scission of acetals for installing aliphatic groups selectively and under mild reaction conditions.
CONCLUSION
In conclusion, we have developed a Ni/photoredox approach to the site-selective methylation of chloride-containing electrophiles using trimethyl orthoformate as an abundant, nontoxic, and functional group-compatible methylating reagent. Methylation of feedstock, as well as chemically complex, (hetero)aryl and acyl chlorides is possible such that we anticipate that this method could find application in the pharmaceutical industry. Mechanistic investigations indicate that trimethyl orthoformate serves as a source for methyl radical via β-scission from a tertiary radical generated upon chlorine-mediated hydrogen atom transfer. As such, this approach offers an opportunity to circumvent traditional protocols for accessing low molecular weight aliphatic radicals from toxic or high-energy reagents.
Supplementary Material
ACKNOWLEDGMENT
This material is based on work supported by the National Science Foundation Graduate Research Fellowship Program under Grant Number DGE-1656466 (to S.K.K.). M.A.T.-S. wishes to thank Princeton’s Presidential Postdoctoral Fellowship for funding. A.G.D. gratefully acknowledges Celgene, Princeton Innovation Fund, and NIGMS (R35 GM126986) for financial support. We thank Prof. David MacMillan and Dr. Laura K. G. Ackerman for helpful suggestions.
Funding Sources
The authors declare no competing financial interest.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, spectroscopic data, and details of the computational study
REFERENCES
- (1).McGrath NA; Brichacek M; Njardarson JT A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ 2010, 87, 1348–1349. [Google Scholar]
- (2).(a) Barreiro EJ; Kümmerle AE; Fraga CAM The Methylation Effect in Medicinal Chemistry. Chem. Rev 2011, 111, 5215–5246. [DOI] [PubMed] [Google Scholar]; (b) Leung CS; Leung SSF; Tirado-Rives J; Jorgensen WL Methyl Effects on Protein-Ligand Binding. J. Med. Chem 2012, 55, 4489–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Schönherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C-H Methylation Reactions. Angew. Chem. Int. Ed 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- (4).(a) Yan G; Borah AJ; Wang L; Yang M Recent Advances in Transition Metal-Catalyzed Methylation Reactions. Adv. Synth. Catal 2015, 557, 1333–1350. [Google Scholar]; (b) Hu L; Liu YA; Liao X Recent Progress in Methylation of (Hetero)Arenes by Cross-Coupling or C-H Activation. Synlett 2018, 29, 375–382. [Google Scholar]
- (5).For a recent example that overcomes many existing limitations through the use of a new methylating agent, see: He Z-T; Li H; Haydl AM; Whiteker GT; Hartwig JF Trimethylphosphate as a Methylating Agent for Cross Coupling: A Slow-Release Mechanism for the Methylation of Arylboronic Esters. J. Am. Chem. Soc 2018, 140, 17197–17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Minisci F; Bernardi R; Bertini F; Galli R; Perchinummo M Nucleophilic character of alkyl radicals-VI. A new convenient selective alkylation of heteroaromatic bases. Tetrahedron 1971, 27, 3575–3579. [Google Scholar]; (b) DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem. Int. Ed 2014, 55, 4802–4806. [DOI] [PubMed] [Google Scholar]; (c) Kubo T; Chatani N Dicumyl Peroxide as a Methylating Reagent in the Ni-Catalyzed Methylation of Ortho C–H Bonds in Aromatic Amides. Org. Lett 2016, 18, 1698–1701. [DOI] [PubMed] [Google Scholar]; (d) Hu A; Guo J-J; Pan H; Zuo Z Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 2018, 561, 668–672. [DOI] [PubMed] [Google Scholar]; (e) Ochiai M; Morita K A Novel Photo-Induced Methylation of Pyrimidines and Condensed Pyrimidine Compounds. Tetrahedron Lett. 1967, 8, 2349–2351. [Google Scholar]; (f) Akira S; Tetsuo Y; Hiroaki I; Masayuki N; Keiko T; Noriko O Radiation-Induced Alkylation of Quinoline Derivatives with Alcohol. Bull. Chem. Soc. Jpn 1986, 59, 3905–3909. [Google Scholar]; (g) Jin J; MacMillan DWC Alcohols as alkylating agents in heteroarene C-H functionalization. Nature 2015, 525, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liu W; Yang X; Zhou Z-Z; Li C–J Simple and Clean Photo-induced Methylation of Heteroarenes with MeOH. Chem 2017, 2, 688–702. [Google Scholar]
- (7).Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 158, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang P; Le C. “Chip”; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 158, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Wang J; Zhao J; Gong H Nickel-Catalyzed Methylation of Aryl Halides/Tosylates with Methyl Tosylate. Chem. Commun 2017, 55, 10180–10183. [DOI] [PubMed] [Google Scholar]; (b) Liang Z; Xue W; Lin K; Gong H Nickel-Catalyzed Reductive Methylation of Alkyl Halides and Acid Chlorides with Methyl p-Tosylate. Org. Lett 2014, 16, 5620–5623. [DOI] [PubMed] [Google Scholar]
- (10).Sun S; Fu J Methyl-containing pharmaceuticals: Methylation in drug design. Bioorg. & Med. Chem. Lett 2018, 28, 3283–3289. [DOI] [PubMed] [Google Scholar]
- (11).(a) Shields BJ; Doyle AG Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 158, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nielsen MK; Shields BJ; Liu J; Williams MJ; Zacuto MJ; Doyle AG Mild, Redox-Neutral Formylation of Aryl Chlorides through the Photocatalytic Generation of Chlorine Radicals. Angew. Chem. Int. Ed 2017, 129, 7297–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C–C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 14059–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).CBS–QB3. See SI (Section II) for details.
- (13).(a) Kochi JK Chemistry of Alkoxy Radicals: Cleavage Reactions. J. Am. Chem. Soc 1962, 84, 1193–1197. [Google Scholar]; (b) Bacha JD; Kochi JK Polar and Solvent Effects in the Cleavage of t-Alkoxy Radicals. J. Org. Chem 1965, 50, 3272–3278. [Google Scholar]; (c) Walling C Some Aspects of the Chemistry of Alkoxy Radicals. Pure Appl. Chem 1967, 15, 69–80. [Google Scholar]; (d) Kuhn LP; Wellman C Reaction of t-Butyl Peroxide with Acetals. J. Org. Chem 1957, 22, 774–776. [Google Scholar]; (e) Hartzell GE; Huyser ES Generation of Methyl Radicals by Decomposition of Bibenzyl Compounds Containing α-Methoxy Substituents. J. Org. Chem 1964, 29, 3341–3344. [Google Scholar]
- (14).(a) Vara BA; Patel NR; Molander GA O-Benzyl Xanthate Esters under Ni/Photoredox Dual Catalysis: Selective Radical Generation and Csp3 –Csp2 Cross-Coupling. ACS Catal. 2017, 7, 3955–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu J; Bär RM; Guo L; Noble A; Aggarwal VK Photoinduced Deoxygenative Borylations of Aliphatic Alcohols. Angew. Chem. Int. Ed 2019, 58, 18830–18834. [DOI] [PubMed] [Google Scholar]
- (15).Huang Q; Zard SZ Inexpensive Radical Methylation and Related Alkylations of Heteroarenes. Org. Lett 2018, 20, 1413–1416. [DOI] [PubMed] [Google Scholar]
- (16).(a) Poutsma ML Methods in Free-Radical Chemistry, 1st ed.; Huyser ES, Ed.; Marcel Dekker: New York, 1969. [Google Scholar]; (b) Skell PS; Baxter HN; Taylor CK π Complexing of Chlorine Atoms: Is That All There Is? J. Am. Chem. Soc 1983, 105, 120–121. [Google Scholar]; (c) Skell PS; Baxter HN; Tanko JM; Chebolu V Chlorine atom/benzene system. 1. The role of the 6-chlorocyclohexadienyl radical. J. Am. Chem. Soc 1986, 108, 6300–6311. [Google Scholar]; (d) Breslow R; Brandl M; Hunger J; Turro N; Cassidy K; Krogh-Jespersen K; Westbrook JD Pyridine complexes of chlorine atoms. J. Am. Chem. Soc 1987, 109, 7204–7206. [Google Scholar]
- (17).(a) Tegner C On the Reaction between Methyllithium and Carboxylic Acids. Acta. Chem. Scand 1952, 6, 782–790. [Google Scholar]; (b) Jorgenson MJ Preparation of Ketones from the Reaction of Organolithium Reagents with Carboxylic Acids. Org. React 1970, 18, 1–97. [Google Scholar]
- (18).Preliminary efforts to use alkyl halides as coupling partners under the methylation conditions have been met with limited success.
- (19).(a) Hwang SJ; Anderson BL; Powers DC; Maher AG; Hadt RG; Nocera DG Halogen Photoelimination from Monomeric Nickel(III) Complexes Enabled by the Secondary Coordination Sphere. Organometallics 2015, 34, 4766–4774. [Google Scholar]; (b) Hwang SJ; Powers DC; Maher AG; Anderson BL; Hadt RG; Zheng S-L; Chen Y-S; Nocera DG Trap-Free Halogen Photoelimination from Mononuclear Ni(III) Complexes. J. Am. Chem. Soc 2015, 137, 6472–6475. [DOI] [PubMed] [Google Scholar]
- (20).TEMPO can interfere with transition metal-catalyzed processes independent of the intermediacy of organic radicals. See: Albéniz AC; Espinet P; López-Fernández R; Sen A A Warning on the Use of Radical Traps as a Test for Radical Mechanisms: They React with Palladium Hydrido Complexes. J. Am. Chem. Soc 2002, 124, 11278–11279. [DOI] [PubMed] [Google Scholar]
- (21).Chan KS; Li XZ; Dzik WI; de Bruin B Carbon-Carbon Bond Activation of 2,2,6,6-Tetramethyl-piperidine-1-oxyl by a RhII Metalloradical: A Combined Experimental and Theoretical Study. J. Am. Chem. Soc 2008, 150, 2051–2061. [DOI] [PubMed] [Google Scholar]
- (22).See SI for reaction details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.