

 Mitigating oxidative drug metabolism is an important component of lead optimization. This review focuses on scaffold-hopping strategies used in the recent medicinal chemistry literature to address metabolic liabilities of aromatic compounds.
Mitigating oxidative drug metabolism is an important component of lead optimization. This review focuses on scaffold-hopping strategies used in the recent medicinal chemistry literature to address metabolic liabilities of aromatic compounds.
Abstract
Understanding and minimizing oxidative metabolism of aromatic compounds is a key hurdle in lead optimization. Metabolic processes not only clear compounds from the body, but they can also transform parent compounds into reactive metabolites. One particularly useful strategy when addressing metabolically labile or oxidation-prone structures is scaffold-hopping. Replacement of an aromatic system with a more electron-deficient ring system can often increase robustness towards cytochrome P450-mediated oxidation while conserving the structural requirements of the pharmacophore. The most common example of this substitution strategy, replacement of a phenyl ring with a pyridyl substituent, is prevalent throughout the literature; however scaffold-hopping encompasses a much wider scope of heterocycle replacement. This review will showcase recent examples where different scaffold-hopping approaches were used to reduce metabolic clearance or block the formation of reactive metabolites. Additionally, we will highlight considerations that should be made to garner the most benefit from a scaffold-hopping strategy for lead optimization.
Introduction
Consideration of drug metabolism is central to any multi-parameter lead-optimization campaign. Two major issues relevant to drug metabolism that may arise during hit-to-lead optimization include 1) rapid degradation of the pharmacologically active compound into inactive metabolites and 2) metabolism of the active compound to toxic and/or reactive metabolites.1 Fortunately for medicinal chemists, principles of physical organic chemistry often explain sites of drug metabolism, and this knowledge can be used to mitigate metabolic liabilities.2–6 In this review we will discuss the implication of scaffold choice on drug metabolism in terms of both mitigating rapid metabolism and avoiding the formation of reactive metabolites.
Drug metabolism principles and tools
Several excellent reviews and texts have been written on the fundamentals of drug metabolism,7–9 but for the purposes of this review, a brief introduction is sufficient. Xenobiotics, including drugs, often are cleared from the body through metabolic modification, which tends to increase hydrophilicity so that metabolites can be cleared by the kidneys. The most common routes of metabolism are oxidation, reduction, hydrolysis, and conjugation. Mitigating oxidative drug metabolism is the focus of this review.
Oxidation tends to be focused on electron-rich sites on molecules. Cytochrome P450s are most often responsible for oxidative metabolism of electron-rich molecules. A common strategy, covered in more detail below, is to replace electron-rich aromatic rings with electron-poor heterocycles as a way to mitigate oxidative metabolism.10–12 Tuning the electronic structure in such a way may decrease the rate of metabolism at the off-target (P450 enzymes), but it will hopefully not have a deleterious effect of compound binding to the on-target.13,14 Although this electron-rich to electron-poor switch often holds for compounds that are oxidized by P450 enzymes, it is now widely appreciated that aldehyde oxidase and/or xanthine oxidase may oxidize nitrogen-rich aromatic heterocycles.15–17 While this review will mainly focus on strategies to overcome cytochrome P450-mediated oxidation, it is important to keep in mind the potential for metabolism of electron-poor ring systems by aldehyde and/or xanthine oxidase.
Throughout this review, we will focus on some commonly used tools and techniques of drug metabolism studies. Because the liver is the major site of drug metabolism, many in vitro methods for studying drug metabolism use subcellular liver fractions, hepatocytes, or liver slices.18 Two liver fractions that commonly are used in drug metabolism studies are liver S9 fractions and liver microsomes. S9 fractions are derived from liver homogenates that are moderately centrifuged.19,20 More robust centrifugation is used to prepare liver microsomes, which are self-assembling vesicles that are assembled from pieces of the endoplasmic reticulum.21 S9 fractions contain both P450s and many of the conjugative enzymes, whereas liver microsomes contain P450s but only a few conjugative enzymes.19,20 Both S9 fractions and liver microsomes can be pooled to cover different genotypes and can be obtained from both model organisms (e.g., mouse, rat, dog, cynomolgus monkey) and humans.22 Cultured hepatocytes and liver slices can also be used for drug metabolism studies. Hepatocytes can be cultured as a monolayer, or they can be cultured in three-dimensions.23,24
In vitro drug metabolism stability studies typically are completed using liquid chromatography-mass spectrometry methods. Data may be presented as “percent remaining” at a pre-selected timepoint, or, if concentrations are measured at multiple time-points, a t1/2 may be calculated. Metabolite identification studies are typically carried out using LC-MS/MS and/or NMR methods.25,26 Synthesis of authentic standards is sometimes used to unequivocally assign a structure and may be necessary for establishing mechanism-of-action or for toxicology studies.26
The rate at which a drug is metabolized and eliminated from the body is typically presented as a clearance (CL) value. The clearance of a drug represents the volume of blood from which a drug can be completely removed over a period of time, often given in units of mL min–1 kg–1 or μL min–1 mg–1. Hepatic clearance (Clh), is the clearance of a compound by the liver, and is limited by the blood flow into the liver. In vitro intrinsic clearance (CLint) values can be calculated from the t1/2 of drug in hepatic fractions. CLint values represent the maximum possible hepatic clearance if all external factors, such as blood flow limitations through the liver and plasma protein binding, are not considered.27,28 Plasma clearance (CLp) is a measure of the rate at which a drug is removed from the plasma through all routes of metabolism and excretion. The terms plasma clearance and total clearance can be used synonymously.29 Drug interactions with plasma proteins can effectively restrict metabolic access to a drug, and it can be useful to determine the rate of clearance for the unbound portion of the drug. An unbound in vitro clearance (CLint,u or CL(Free)) can be obtained by dividing the experimental apparent intrinsic clearance value (CLint) by the unbound fraction of drug.30 Addressing metabolic liabilities of compounds early can be thought of as a quality by design (QbD) approach which limits the need to adjust compound metabolism retroactively.31
In vivo drug metabolism studies are often carried out using a radiolabeled compound.32 These studies are inherently low-throughput, and, given the focus of this review on scaffold-hopping in hit-to-lead optimization, they are not considered here.
Electronic properties of commonly encountered heterocycles
Metabolic stability of common heterocycles often can be correlated with electronic structure. In general, for any two given congeners, the molecule with the higher-energy highest occupied molecular orbital (HOMO) will undergo oxidation more easily than the molecule with the lower-energy HOMO.33 This principle is used in medicinal chemistry by substituting electron-rich aromatic systems with electron-deficient heterocycles, which tend to be less prone to oxidative metabolism; of course, steric effects and substrate recognition also play a role in determining the ease of attack of a metabolic enzyme on a particular compound, but this review will focus on the electronic structure of the ring itself rather than the steric implications of its substituents on metabolism.
Three types of aromatic heterocycles are encountered most commonly in drug discovery: five-membered heterocycles, six-membered heterocycles, and benzannulated five- or six-membered rings. Typically, five-membered heterocycles, which contain at least one heteroatom (e.g., pyrrole, furan, thiophene) are considered to be electron-rich, while six-membered heterocycles with one or more heteroatoms (e.g., pyridine, pyrimidine, pyrazine) are considered electron-deficient.34–36 These trends can be seen when examining HOMO energies of various heterocycles (Table 1 (ref. 34–36)). For instance, the HOMO energy of pyrrole (–8.66 eV) is higher than benzene (–9.65 eV), which is higher than pyridine (–9.93 eV; see Table 1). Incorporation of relatively electronegative nitrogen atoms in an aromatic heterocycle will typically decrease its overall electron density and decrease the energy of the HOMO, making the heterocycle less prone to P450-mediated oxidative metabolism.33 Benzannulation of heterocycles tends to increase the energy of the HOMO compared to the standalone five- or six-membered ring systems. For instance, the HOMO energies of quinoline and isoquinoline (–9.18, –9.03 eV, respectively) are higher than the HOMO energy of pyridine (–9.93 eV; see Table 1). When two or more heteroatoms are present in a ring system, the precise placement of heteroatoms in an aromatic system alters both the HOMO and the electron density which the carbon atoms in the ring bear; this can be an important factor in predicting potential sites of metabolic transformations.
Table 1. AM1 energies of the highest occupied molecular orbitals (HOMOs) of some common heterocycles a .
| Ring type | Molecule | HOMO energy (eV) |
| 5-Membered | Pyrrole | –8.66 |
| Furan | –9.32 | |
| Thiophene | –9.22 | |
| Imidazole | –9.16 | |
| Pyrazole | –9.71 | |
| 1H-1,2,3-triazole | –10.18 | |
| 2H-1,2,3-triazole | –10.33 | |
| 1H-1,2,4-triazole | –10.27 | |
| 4H-1,2,4-triazole | –10.03 | |
| 1H-Tetrazole | –11.41 | |
| 2H-Tetrazole | –11.16 | |
| Isoxazole | –10.47 | |
| Oxazole | –9.89 | |
| Isothiazole | –9.54 | |
| Thiazole | –9.70 | |
| 6-Membered | Benzene | –9.65 |
| Pyridine | –9.93 | |
| Pyridazine | –10.67 | |
| Pyrimidine | –10.58 | |
| Pyrazine | –10.25 | |
| 1,2,3-Triazine | –11.31 | |
| 1,2,4-Triazine | –10.71 | |
| 5,6-Fused | Indole | –8.40 |
| Isoindole | –7.80 | |
| Benzo[b]furan | –9.01 | |
| Benzimidazole | –9.00 | |
| 6,6-Fused | Quinoline | –9.18 |
| Isoquinoline | –9.03 |
These types of replacements lend themselves well to Matched Pairwise Analyses, where physicochemical or ADME properties are compared between pairs that differ by replacing atoms in a carbocycle/heterocycle with one or more heteroatoms.13 Useful pairwise analyses have been reported by Dossetter et al., as well as Ritchie and Macdonald, detailing expected ADME changes when substituting phenyl rings with various heteroaromatic replacements.14,37 Likewise, Chang et al.38 reported a comprehensive pairwise analysis between six-membered rings. These pairwise analyses provide a good guide for some heteroaromatic substitutions. In this review, we will highlight additional recent examples of scaffold-hopping to address metabolic liabilities and incorporate other learnings that may not be encompassed by pairwise analyses.
Scaffold-hopping
Scaffold-hopping, a term coined in 1999 by Gisbert Schneider, covers a wide array of structural modifications with the general goal of producing “isofunctional molecular structures with significantly different backbones.”39 Since the term's emergence, it has been used to describe small hops, such as replacements of pendant aromatic structures, and larger hops, which only conserve the structural requirements of the pharmacophore.40–44 While several properties could be addressed by scaffold-hopping, this review focuses on recent applications of scaffold-hopping as a way to address metabolic liabilities.
There are several different strategies used in scaffold-hopping, including ring closing/opening, peptidomimetics, and topology-based scaffold-hopping.41 The most common form of scaffold-hopping, heterocycle replacement, is a particular focus of this review. Modification of a specific heterocyclic core typically involves a rearrangement or an increase in the number of heteroatoms within the core, which modifies the electronic structure of the core and can make it more resistant to oxidative metabolism.11 To aid in substitution-based approaches, computational methods can be used to predict likely sites of metabolism.45 Topology-based scaffold-hopping, which involves larger structural variations, can be streamlined via computational methods to discern similarity between structurally distinct scaffolds. For example, rapid overlay of chemical structures (ROCS) can compare and calculate the 3-dimentional structural and electronic similarities between various scaffolds (for a recent overview of various computational methods for scaffold hopping see the following reference).39
The goal of this review is to provide instructive, rather than comprehensive, examples of scaffolding-hopping in drug metabolism studies. In the sections below, we will outline several recent examples of scaffold-hopping to address metabolic liabilities in six-membered, five-membered, and fused heterocycles. Given the ubiquity of phenyl rings in lead optimization, we begin with six-membered heterocycles, followed by five-membered heterocycles, and, finally, fused heterocycles.
Six-membered heterocycles
One of the simplest examples of the use of scaffold-hopping to impart metabolic stability is the replacement of a phenyl substituent with a pyridyl or pyrimidyl substituent. The incorporation of nitrogen atoms into the aromatic system tends to increase the metabolic stability though decreasing the potential for oxidative metabolism (see above). This is well-illustrated by the pursuit of a hepatitis C virus NS5B replicase palm site allosteric inhibitor by Yeung et al.46 The authors identified a potent compound (1) that suffered from poor metabolic stability. Metabolite identification studies identified the unsubstituted phenyl ring as a metabolic soft spot. To address this issue, the authors hypothesized that adding a nitrogen atom into the ring would increase metabolic stability. It was found that a 2-pyridyl group (2) dramatically increased the half-life of these compounds while maintaining good potency. Furthermore, to tune the physicochemical properties of the molecule, the authors introduced a second nitrogen atom into the ring. This pyrimidyl analog (3) exhibited an increased half-life, and this molecule was selected for further development (Fig. 1).
Fig. 1. Nitrogen incorporation increases metabolic stability. This example from Yeung et al.46 demonstrated how scaffold-hopping from benzene to pyridine to pyrimidine may increase metabolic stability. HLM: human liver microsomes.

In multi-parameter optimization, it is important to remember that substitutions made to address metabolic stability or another property should also benefit on-target binding affinity, when possible. In a campaign to discover disrupters of glucokinase–glucokinase regulatory protein (GK–GKRP) binding, Pennington et al. sought to optimize the potency and high clearance rate of screening hit 4.47 When compared to pyridyl compounds 6a and 6b, pyrimidine 6c and 3-fluoropyridine 6d showed enhanced metabolic stability. Both strategies of CH-to-N and CH-to-CF had similar effects of blocking metabolic sites and reducing the electron density of the aromatic system; however, introduction of an additional nitrogen into the ring served to increase hydrophilicty, while fluorine substitution served to increase hydrophobicity. In this case, the fluoro analog engaged additional hydrophobic interactions with GKRP, making compound 6d a more potent and desirable molecule (Table 2).
Table 2. Structural requirements of the binding pocket can guide scaffold-hopping. Pennington et al.47 showed that binding to a hydrophobic pocket called for the incorporation of a fluoropyridine rather than a similarly stable pyrimidine. RLM: rat liver microsomes.
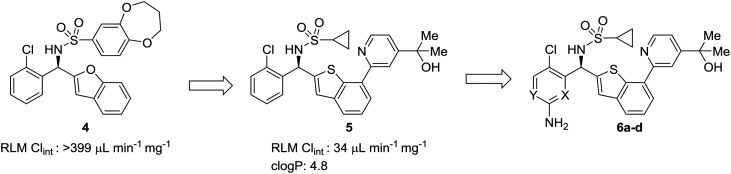
| |||||
| Entry | Compound | X | Y | RLM Clint (μL min–1 mg–1) | clog P |
| 1 | 6a | N | CH | 130 | 3.1 |
| 2 | 6b | CH | N | 67 | 3.1 |
| 3 | 6c | N | N | 28 | 2.3 |
| 4 | 6d | CF | N | 26 | 3.2 |
Metabolite identification can indicate toxic liabilities that may arise through the formation of a reactive metabolite, and scaffold-hopping can be used to address these potentially toxic species. For instance, scaffold-hopping was used to avoid the formation of toxic metabolites arising from metabolism of COX-2-selective inhibitor nimesulide (7).48 Metabolite identification studies indicated that the nitro group was reduced to the aniline, which was then oxidized to a diiminoquinone (8), a reactive functionality, which caused hepatotoxicity.12,49 Replacement of the nitrobenzene with a pyridine (9) removed this metabolic liability. When optimizing the metabolic profile of a compound, addressing the metabolism of one portion of the molecule often leads to the creation of a new metabolic soft spot. In this example, upon replacing the nitrophenyl with a pyridine, further optimization was required to avoid rapid metabolism of the other phenyl ring (Fig. 2).
Fig. 2. Scaffold-hopping can eliminate the formation of reactive metabolites. In this example from Renard et al.,48 replacement of a nitrobenzene with a pyridyl core removed the potential for toxic metabolite formation.

The complexities of addressing cross-species in vivo clearance are well-captured by the pursuit of a microtubule-affinity regulating kinase (MARK) inhibitor. Haidle et al. set out to address significant metabolic liabilities identified in screening hit 10.50 The hydroxyindane of 10 was identified as the target of oxidation and glucuronidation. Replacement with N-acyl piperidine was found to be more metabolically stable, yet conferred poor binding affinity. The N-acyl piperidine was used as a placeholder while other metabolic liabilities were addressed. Specifically, the unsubstituted aniline of 11 was a cause for concern since it can lead to the formation of reactive metabolites.12 Replacement of the phenyl ring with a pyridyl group (12) greatly increased the metabolic stability, likely through reducing the lipophilicity and increasing resistance to oxidative metabolism; however, when returning to optimize the potency of these compounds, all modifications (13) that led to enhanced MARK inhibition led to high in vivo clearance in rats. Despite the fact that good PK was observed in dogs, the practical need for testing in rodents led to deprioritization of this structural class (Fig. 3).
Fig. 3. Metabolic stability across species is crucial for lead compound advancement. This example from Haidle et al.50 highlighted that addressing rapid metabolism in vitro does not always guarantee in vivo stability or metabolic stability in other species. HLM: human liver microsomes; RLM: rat liver microsomes; CLp: total plasma clearance.

Five-membered heterocycles
An attractive aspect of scaffold-hopping is that the new scaffold gives a display of substituents that is similar to the parent scaffold. Computational modeling—and structural overlay, in particular—can be a powerful tool for finding these alternative scaffolds. A good example of this comes from Wavhale et al.51 In this report, the authors searched for a replacement for the pyrrole core of BM212 (14) to identify new leads against drug-resistant Mycobacterium tuberculosis. BM212 and other analogs were found to be cytotoxic and/or metabolically unstable. The pyrrole core—which can generate toxic metabolites52,53—was suspected as the source of the cytotoxicity of these compounds. Wavhale, et al. used the rapid overlay of chemical structures (ROCS) program to search for analogous 3D scaffolds to BM212. Several scaffolds, including benzimidazole, imidazole, and imidazopyridine, were identified as potential replacements. Of the modeled and tested compounds, benzimidazole overlaid best and showed heightened activity against M. tuberculosis. Screening aryl substituents on each of the attached aryl rings yielded 15, which showed similar activity to BM212 but was found to be 25-fold less cytotoxic and more metabolically stable (Fig. 4).
Fig. 4. Computational methods can be used to identify new scaffolds. Wavhale et al.51 used the rapid overlay of chemical structures (ROCS) program to scaffold-hop from pyrrole to benzimidazole, which conserved important aspects of the three-dimensional structure.

Fragment-based approaches to drug design often start with small fragments that are grown into more selective and drug-like molecules. The process of fragment-growing can be thought of as a scaffold-hopping approach, as various functionalities are judiciously investigated to develop a structure–activity relationship, usually with the aid of crystallographic data. Concerns about metabolic stability tend not to arise until a fragment has been grown into a sufficiently potent molecule. In the development of mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) inhibitors by Wang et al., a pyrrolotriazine (16) was identified as a starting fragment.54 A crystal structure led the authors to explore the substitution off of the pyrrole ring to extend into a relatively flat hydrophobic pocket. Various substituents were investigated for their potency and metabolic stability. Phenyl, tetrazole, furan, and pyrazole 18a–d were prepared, but each of these compounds had their own issues for further development: furan had known metabolic liabilities,12 the tetrazole had relatively poor activity, and the phenyl and pyrazole structures were poorly soluble. Another important lesson from this work is that some ring systems readily lend themselves to functionalization. In this work, phenyl and pyrazole structures were investigated further due to their ease of substitution. The solubility of the phenyl compounds could not be rectified satisfactorily, but appending a substituted hydroxyethyl group to the pyrazole yielded alcohol 18e, which showed enhanced solubility and good metabolic stability (Fig. 5).
Fig. 5. Scaffold-hopping paired with fragment-based design. Wang et al.54 showed that a scaffold-hop from a benzene to various five-membered rings imparted enhanced metabolic stability. HLM: human liver microsomes; RLM: rat liver microsomes; CLh: hepatic clearance.

Most examples in this review have showcased replacement of one aromatic system for another, but scaffold hopping also can be used to replace metabolically labile groups with heterocyclic isosteres. In their investigation of benzyl amide mineralocorticoid receptor antagonist 19, Cox et al. showed that replacement of the amide by heterocycles affected physicochemical, ADME, and lipophilic ligand efficiency.55 With respect to microsomal stability, the starting benzyl amide 19 was completely metabolized by liver microsomes after 30 minutes. Cyclization to the oxazole (20a) increased the microsomal stability, which was even further increased by scaffold-hopping to the 4H-1,2,4-triazole and 1,3,4-oxadiazole analogs 20c and 20d. While compounds 20c and 20d have similar metabolic stability, the 1,3,4-oxadiazole core was chosen for further development due to the increased lipophilic ligand efficiency of these compounds versus the 4H-1,2,4-triazole analogs (Table 3).
Table 3. Metabolically labile groups can be stabilized as heteroaromatic isosteres. Cox et al.55 used scaffold-hopping to replace a metabolically labile amide by incorporating a metabolically stable 1,3,4-oxadiazole. H/R LM: human/rat liver microsomes.
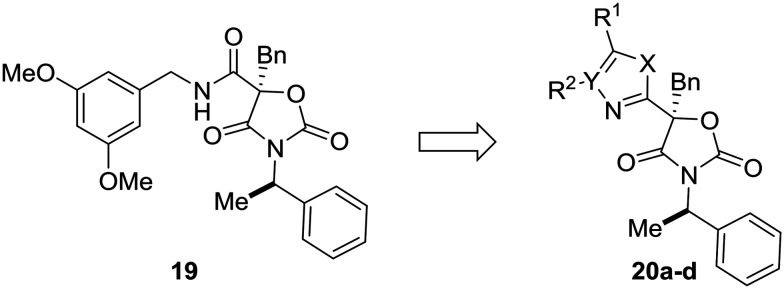
| |||||
| Compound | X | Y | R1 | R2 | LM (H, R) % remaining after 30 min |
| 19 | — | — | — | — | 0, 0 |
| 20a | O | CH | Ph | H | 73, 29 |
| 20b | NH | CH | Ph | H | 62, 4 |
| 20c | NH | N | Ph | — | 78, 29 |
| 20d | O | N | Ph | — | 82, 0 |
Fused heterocycles
When experimental analysis leads to conflicting results, computational methods can be used to elucidate potential metabolic soft spots that may not be apparent through metabolite identification studies. Robarge et al. used MetaSite™, a computational program that predicts potential sites of metabolism, in the development of mitogen-activated protein kinase kinase (MEK) inhibitors.56,57 The authors identified the imidazo[1,5-a] pyridine core as a suitable core for further development of MEK inhibitors, as this substitution would increase the structural diversity of known bicyclic MEK inhibitor cores and would offer enhanced physicochemical properties compared to known scaffolds. 22 was selected for further investigation, due to its lower cross-species clearance values; however, there was a disparity between the in vitro and in vivo metabolic stability values. In vivo metabolite identification of 22 indicated that the major metabolic site was at the hydroxamate, with hydrolysis to the acid as the most prominent metabolite; however, MetaSite™ indicated that the C8 position of the imidazo[1,5-a] pyridine core was a hot spot for metabolism. Initially, the authors tried fluorination at this position, but this led to unacceptably high CYP2C9 inhibition. A second MetaSite™ analysis showed that incorporating a nitrogen atom at the C7 position should block metabolism both at the C7 and C8 position. This replacement shifted the major metabolic site to the five-membered imidazole ring of the core and enhanced the overall metabolic stability of the inhibitor (23) (Fig. 6).
Fig. 6. Computational methods can be used to address discrepancies observed between in vitro and in vivo metabolism. Using the MetaSite™ program,56 Robarge et al.57 identified additional metabolic soft spots that were not obvious through metabolite ID analysis. The authors established a good correlation between in vitro and in vivo clearance. H/R/M CLh: human/rat/mouse hepatic clearance (in mL min–1 kg–1); Clp: total plasma clearance; Clu: unbound clearance. Blue circle indicates most probable location of metabolism; red circles indicate other likely sites of metabolism.

Another prominent effect which nitrogen incorporation yields is a decrease in log P. Lowering log P is often correlated with a better metabolic and safety profile. Cid et al. set out to discover positive allosteric modulators (PAMs) of the mGlu2 receptor;58 however, their compounds suffered from poor metabolic stability, and only specific substituents imparted the desired metabolic stability when using the imidazopyridine core. To improve overall metabolic stability and expand the substituents available for optimization, the authors reduced the lipophilicity of the imidazopyridine core by converting it to the 1,2,4-triazolopyridine core, which greatly enhanced stability in human liver microsomes. It is difficult to pinpoint the exact role the nitrogen plays in this case (i.e., blocking metabolism or reducing lipophilicity), because metabolites of 24 were not identified; thus, the enhanced metabolic stability may arise from decreasing log P, blocking a metabolic soft spot, or a combination of both effects (Fig. 7).59
Fig. 7. Nitrogen incorporation lowers clog P and decreases lipophilicity. Cid et al.58 showed that scaffold-hopping from imidazopyridine to 1,2,4-triazolopyridine increased metabolic stability with a decrease in clog P. HLM: human liver microsomes.

The scaffold-hopping progression presented by Doherty et al. illustrates well how scaffold-hopping can be used in various ways—from nitrogen incorporation to ring contraction—to address oxidative metabolism.60 To reduce intrinsic clearance in human liver microsomes, the investigators performed an in-depth structure–activity relationship analysis of the bicyclic region of lead quinoline 26, an antagonist of the vanilloid receptor. Metabolite identification studies conducted on 26 indicated that oxidation on the quinoline ring was a prevalent route of metabolism. Moving the nitrogen and oxo-linkage around the bicyclic system provided compound 27, which exhibited excellent binding affinity and an encouraging increase in metabolic stability; however, metabolite analysis of this compound indicated that approximately 70% of the metabolites were still generated from hydroxylation and sulfation of the quinoline ring. Converting the quinoline to the quinoxaline was hypothesized to reduce this oxidative metabolism, and, although this appeared to reduce clearance in vitro, the in vivo clearance remained unacceptably high. The major metabolite of quinoxaline 28 was identified as the 3-oxo derivative. To block metabolism at this position, the authors found that eliminating the oxidation-prone carbon atom by ring contraction to the benzothiazole (29) reduced in vivo clearance from 4.3 L h–1 kg–1 to 0.22 L h–1 kg–1 (Fig. 8).
Fig. 8. Carbon atoms prone to oxidation can be excised to improve metabolic stability. Doherty et al.60 showed that nitrogen incorporation was insufficient to completely block oxidative metabolism of quinoline 27; thus, ring contraction from quinoxaline 28 to benzothiazole 29 was used to eliminate the most prominent site of metabolism. HLM CLint: intrinsic clearance in human liver microsomes (in μL min–1 mg–1).

The formation of mutagenic metabolites is also a concern with regards to metabolic stability. Richardson et al. showed that the 1,4-diaminonaphthalene of a known KEAP1-NRF2 protein–protein interaction inhibitor (30) was metabolized to give mutagenic metabolite(s) in a mini-Ames assay.61 The authors hypothesized that this metabolite likely possessed a 1,4-diiminoquinone type structure and that replacement of the naphthalene with a more electron-poor scaffold would reduce the propensity toward oxidative activation of the scaffold.12 After surveying several different heterocyclic cores, only isoquinoline 31 conserved the activity of the initial lead compound. Formation of mutagenic metabolites in the presence of rat S9 fractions was analyzed using a mini-Ames assay. The isoquinoline core showed a reduced mutagenic profile, and this core was used as a lead structure for further optimization (Fig. 9).
Fig. 9. Mutagenic metabolites may arise through oxidative metabolism. Richardson et al.61 used scaffold-hopping to diminish the formation of mutagenic metabolites by replacing a naphthalene core with an isoquinoline ring.

Many of the examples we have seen thus far have used scaffold-hopping to replace just one problematic scaffold, but the strategies of scaffold-hopping can be combined to address numerous liabilities within a compound. In a particularly striking example by Raheem et al., three different heterocycles were replaced with more stable scaffolds in the development of phosphodiesterase 10A inhibitor PyP1 (34).62 Starting from a hit identified by fragment-based screening, several metabolic liabilities and off-target effects were identified in compound 32. Scaffold-hopping approaches were used to mitigate the oxidative metabolism of both the quinoline ring and the thiazole. Replacement of these ring systems with more electron-deficient heterocycles pyridine and pyrazole (33) significantly lowered the clearance of the initial hit compound. To address other off-target effects, the pyrimidine ring was replaced by a pyrazolopyrimidine, which had the additional effect of further increasing the metabolic stability (Fig. 10).
Fig. 10. Scaffold-hopping can be carried out on multiple sites in a molecule to impart desirable metabolic properties. By replacing three separate heterocycles, Raheem et al.62 used scaffold-hopping to enhance metabolic stability and reduce off-target effects.

Lastly, this review would not be complete without addressing the importance of other enzymes which impact drug metabolism, such as aldehyde oxidase. Although nitrogen incorporation into aromatic systems can reduce oxidative metabolism by cytochrome P450 enzymes,10,11 electron-deficient arenes can be suitable substrates for aldehyde oxidase metabolism.15,16 There has been a great deal of recent work to predict and mitigate aldehyde oxidase metabolism; however, the mixture of steric and electronic effects governing aldehyde oxidase metabolism complicate accurate predictive modeling.64–70 Furthermore, AO metabolism varies widely across species and can lead to poor cross-species metabolic stability.15,16 In an example from Linton et al.,63 scaffold hopping was used to mitigate aldehyde oxidase metabolism of an androgen receptor antagonist 35. Modifications on the pyridine ring showed no effect on AO-based metabolism, which pointed to the imidazo[1,2-a]pyrimidine as the site of AO metabolism. In one example, conversion of the imidazo[1,2-a]pyrimidine to an imidazo[1,5-a]pyridine (36) caused the compound to no longer be a substrate for AO, indicating that the carbon adjacent to the removed nitrogen was a likely site of metabolism (Fig. 11).
Fig. 11. Electron-deficient arenes may be susceptible to aldehyde oxidase (AO) metabolism. This example describes scaffold hopping from a imidazo[1,2-a] pyrimidine to a imidazo[1,5-a] pyridine to avoid metabolism by AO.63.

Conclusions
We have presented here several recent examples that highlight the power of using scaffold-hopping as a strategy to address metabolic liabilities during multi-parameter optimization. Several lessons can be learned from the examples here. First, based upon principles from physical organic chemistry, electron-rich aromatic systems can be replaced with electron-poor heterocycles to maneuver around P450-mediated oxidative metabolism. It is important, however, to realize that other enzymes, including aldehyde and xanthine oxidase, may oxidize these newly formed aromatic systems. Replacing a metabolically soft carbon atom with a nitrogen atom can often be used, but it is sometimes sufficient to replace another carbon atom in the ring system with a nitrogen atom, as this can change the overall electronic properties of the heterocycle. In addition to replacing a carbon atom, it may also be possible to excise the metabolically soft carbon atom to give a ring-contracted heterocycle. In lead optimization, it is also important to consider other properties, such as lipophilicity or H-bond acceptors, which scaffold-hopping may affect. Lastly, medicinal chemists should also view scaffold-hopping as a route that opens up further functionalization, through nitrogen-based chemistry (e.g., imidazole alkylation or SNAr reactions on electron-poor heterocycles). Overall, scaffold-hopping has become an important component of the medicinal chemist's toolbox, and employing it to address metabolic liabilities is a particularly beneficial use.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
The authors would like to thank the National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS) for funding this work through a grant to the University of Illinois at Chicago (Principal Investigator: T. W. M. Grant number: 1R01 AR069541-01A1).
References
- Claesson A., Minidis A. Chem. Res. Toxicol. 2018;31:389–411. doi: 10.1021/acs.chemrestox.8b00046. [DOI] [PubMed] [Google Scholar]
- de Groot M. J., Ackland M. J., Horne V. A., Alex A. A., Jones B. C. J. Med. Chem. 1999;42:4062–4070. doi: 10.1021/jm991058v. [DOI] [PubMed] [Google Scholar]
- Singh S. B., Shen L. Q., Walker M. J., Sheridan R. P. J. Med. Chem. 2003;46:1330–1336. doi: 10.1021/jm020400s. [DOI] [PubMed] [Google Scholar]
- Kirchmair J., Goller A. H., Lang D., Kunze J., Testa B., Wilson I. D., Glen R. C., Schneider G. Nat. Rev. Drug Discovery. 2015;14:387–404. doi: 10.1038/nrd4581. [DOI] [PubMed] [Google Scholar]
- Kirchmair J., Williamson M. J., Tyzack J. D., Tan L., Bond P. J., Bender A., Glen R. C. J. Chem. Inf. Model. 2012;52:617–648. doi: 10.1021/ci200542m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzack J. D., Kirchmair J. Chem. Biol. Drug Des. 2019;93:377–386. doi: 10.1111/cbdd.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P. Chem. Res. Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- Croom E. Prog. Mol. Biol. Transl. Sci. 2012;112:31–88. doi: 10.1016/B978-0-12-415813-9.00003-9. [DOI] [PubMed] [Google Scholar]
- Caldwell J., Gardner I., Swales N. Toxicol. Pathol. 1995;23:102–114. doi: 10.1177/019262339502300202. [DOI] [PubMed] [Google Scholar]
- St Jean, Jr. D. J., Fotsch C. J. Med. Chem. 2012;55:6002–6020. doi: 10.1021/jm300343m. [DOI] [PubMed] [Google Scholar]
- Pennington L. D., Moustakas D. T. J. Med. Chem. 2017;60:3552–3579. doi: 10.1021/acs.jmedchem.6b01807. [DOI] [PubMed] [Google Scholar]
- Walsh J. S., Miwa G. T. Annu. Rev. Pharmacol. Toxicol. 2011;51:145–167. doi: 10.1146/annurev-pharmtox-010510-100514. [DOI] [PubMed] [Google Scholar]
- Gleeson P., Bravi G., Modi S., Lowe D. Bioorg. Med. Chem. 2009;17:5906–5919. doi: 10.1016/j.bmc.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J., Macdonald S. J. F. Eur. J. Med. Chem. 2016;124:1057–1068. doi: 10.1016/j.ejmech.2016.10.029. [DOI] [PubMed] [Google Scholar]
- Pryde D. C., Dalvie D., Hu Q., Jones P., Obach R. S., Tran T. D. J. Med. Chem. 2010;53:8441–8460. doi: 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- Sanoh S., Tayama Y., Sugihara K., Kitamura S., Ohta S. Drug Metab. Pharmacokinet. 2015;30:52–63. doi: 10.1016/j.dmpk.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Giulia Battelli M., Polito L., Bortolotti M., Bolognesi A. Curr. Med. Chem. 2016;23:4027–4036. doi: 10.2174/0929867323666160725091915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt R., Hengstler J. G., Muller D., Glockner R., Buenning P., Laube B., Schmelzer E., Ullrich M., Utesch D., Hewitt N., Ringel M., Hilz B. R., Bader A., Langsch A., Koose T., Burger H. J., Maas J., Oesch F. Drug Metab. Rev. 2003;35:145–213. doi: 10.1081/dmr-120023684. [DOI] [PubMed] [Google Scholar]
- Brandon E. F. A., Raap C. D., Meijerman I., Beijnen J. H., Schellens J. H. M. Toxicol. Appl. Pharmacol. 2003;189:233–246. doi: 10.1016/s0041-008x(03)00128-5. [DOI] [PubMed] [Google Scholar]
- De Graaf I. A., Van Meijeren C. E., Pektas F., Koster H. J. Drug Metab. Dispos. 2002;30:1129–1136. doi: 10.1124/dmd.30.10.1129. [DOI] [PubMed] [Google Scholar]
- Pelkonen O., Kaltiala E. H., Larmi T. K. I., Kärki N. T. Chem.-Biol. Interact. 1974;9:205–216. doi: 10.1016/s0009-2797(74)80005-0. [DOI] [PubMed] [Google Scholar]
- Araya Z., Wikvall K. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 1999;1438:47–54. doi: 10.1016/s1388-1981(99)00031-1. [DOI] [PubMed] [Google Scholar]
- Chenery R.R. J.J., Ayrton A., Oldham H. G., Standring P., Norman S. J., Seddon T., Kirby R. Drug Metab. Dispos. 1987;15:312–317. [PubMed] [Google Scholar]
- Puviani A. C., Ottolenghi C., Tassinari B., Pazzi P., Morsiani E. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 1998;121:99–109. doi: 10.1016/s1095-6433(98)10109-5. [DOI] [PubMed] [Google Scholar]
- Saurina J., Sentellas S. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2017;1044–1045:103–111. doi: 10.1016/j.jchromb.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Li L., Ren W., Kong H., Zhao C., Zhao X., Lin X., Lu X., Xu G. Anal. Chim. Acta. 2017;990:96–102. doi: 10.1016/j.aca.2017.07.058. [DOI] [PubMed] [Google Scholar]
- Houston J. B. Biochem. Pharmacol. 1994;47:1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- Wilkinson G. R., Shand D. G. Clin. Pharmacol. Ther. 1975;18:377–390. doi: 10.1002/cpt1975184377. [DOI] [PubMed] [Google Scholar]
- Toutain P. L., Bousquet-Melou A. J. Vet. Pharmacol. Ther. 2004;27:415–425. doi: 10.1111/j.1365-2885.2004.00605.x. [DOI] [PubMed] [Google Scholar]
- Austin R. P., Barton P., Cockroft S. L., Wenlock M. C., Riley R. J. Drug Metab. Dispos. 2002;30:1497–1503. doi: 10.1124/dmd.30.12.1497. [DOI] [PubMed] [Google Scholar]
- Wang J. and Faller B., Progress in Bioanalytics and Automation Robotics for Absorption, Distribution, Metabolism, and Excretion Screening, in Comprehensive Medicinal Chemistry II, ed. B. Testa, 2nd edn, 2007, vol. 5, pp. 341–356. [Google Scholar]
- Atzrodt J., Derdau V., Kerr W. J., Reid M. Angew. Chem., Int. Ed. 2018;57:1758–1784. doi: 10.1002/anie.201704146. [DOI] [PubMed] [Google Scholar]
- Conradie J. J. Phys.: Conf. Ser. 2015;633:012045. [Google Scholar]
- Katritzky A. R., Ramsden C. A., Joule J. A. and Zhdankin V. V., in Handbook of Heterocyclic Chemistry (Third Edition), ed. A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Elsevier, Amsterdam, 2010, pp. 87–138, 10.1016/B978-0-08-095843-9.00004-5. [DOI] [Google Scholar]
- Katritzky A. R., Ramsden C. A., Joule J. A. and Zhdankin V. V., in Handbook of Heterocyclic Chemistry (Third Edition), ed. A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Elsevier, Amsterdam, 2010, pp. 37–86, 10.1016/B978-0-08-095843-9.00003-3. [DOI] [Google Scholar]
- Katritzky A. R., Ramsden C. A., Joule J. A. and Zhdankin V. V., in Handbook of Heterocyclic Chemistry (Third Edition), ed. A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Elsevier, Amsterdam, 2010, pp. 139–209, 10.1016/B978-0-08-095843-9.00005-7. [DOI] [Google Scholar]
- Dossetter A. G., Douglas A., O'Donnell C. MedChemComm. 2012;3:1164. [Google Scholar]
- Chang G., Huard K., Kauffman G. W., Stepan A. F., Keefer C. E. Bioorg. Med. Chem. 2017;25:381–388. doi: 10.1016/j.bmc.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Schuffenhauer A. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012;2:842–867. [Google Scholar]
- Bohm H. J., Flohr A., Stahl M. Drug Discovery Today: Technol. 2004;1:217–224. doi: 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Sun H., Tawa G., Wallqvist A. Drug Discovery Today. 2012;17:310–324. doi: 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G., Schneider P., Renner S. QSAR Comb. Sci. 2006;25:1162–1171. [Google Scholar]
- Kumar A., Zhang K. Y. J. Front. Chem. 2018;6:315. doi: 10.3389/fchem.2018.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Stumpfe D., Bajorath J. J. Med. Chem. 2017;60:1238–1246. doi: 10.1021/acs.jmedchem.6b01437. [DOI] [PubMed] [Google Scholar]
- Peach M. L., Zakharov A. V., Liu R., Pugliese A., Tawa G., Wallqvist A., Nicklaus M. C. Future Med. Chem. 2012;4:1907–1932. doi: 10.4155/fmc.12.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung K. S., Beno B. R., Parcella K., Bender J. A., Grant-Young K. A., Nickel A., Gunaga P., Anjanappa P., Bora R. O., Selvakumar K., Rigat K., Wang Y. K., Liu M., Lemm J., Mosure K., Sheriff S., Wan C., Witmer M., Kish K., Hanumegowda U., Zhuo X., Shu Y. Z., Parker D., Haskell R., Ng A., Gao Q., Colston E., Raybon J., Grasela D. M., Santone K., Gao M., Meanwell N. A., Sinz M., Soars M. G., Knipe J. O., Roberts S. B., Kadow J. F. J. Med. Chem. 2017;60:4369–4385. doi: 10.1021/acs.jmedchem.7b00328. [DOI] [PubMed] [Google Scholar]
- Pennington L. D., Bartberger M. D., Croghan M. D., Andrews K. L., Ashton K. S., Bourbeau M. P., Chen J., Chmait S., Cupples R., Fotsch C., Helmering J., Hong F. T., Hungate R. W., Jordan S. R., Kong K., Liu L., Michelsen K., Moyer C., Nishimura N., Norman M. H., Reichelt A., Siegmund A. C., Sivits G., Tadesse S., Tegley C. M., Van G., Yang K. C., Yao G., Zhang J., Lloyd D. J., Hale C., St Jean, Jr. D. J. J. Med. Chem. 2015;58:9663–9679. doi: 10.1021/acs.jmedchem.5b01367. [DOI] [PubMed] [Google Scholar]
- Renard J. F., Lecomte F., Hubert P., de Leval X., Pirotte B. Eur. J. Med. Chem. 2014;74:12–22. doi: 10.1016/j.ejmech.2013.12.033. [DOI] [PubMed] [Google Scholar]
- Carini M., Aldini G., Stefani R., Marinello C., Maffei Facino R. J. Pharm. Biomed. Anal. 1998;18:201–211. doi: 10.1016/s0731-7085(98)00172-1. [DOI] [PubMed] [Google Scholar]
- Haidle A. M., Childers K. K., Zabierek A. A., Katz J. D., Jewell J. P., Hou Y., Altman M. D., Szewczak A., Chen D., Harsch A., Hayashi M., Warren L., Hutton M., Nuthall H., Stanton M. G., Davies I. W., Munoz B., Northrup A. Bioorg. Med. Chem. Lett. 2017;27:109–113. doi: 10.1016/j.bmcl.2016.08.066. [DOI] [PubMed] [Google Scholar]
- Wavhale R. D., Martis E. A. F., Ambre P. K., Wan B., Franzblau S. G., Iyer K. R., Raikuvar K., Macegoniuk K., Berlicki L., Nandan S. R., Coutinho E. C. Bioorg. Med. Chem. 2017;25:4835–4844. doi: 10.1016/j.bmc.2017.07.034. [DOI] [PubMed] [Google Scholar]
- Assandri A., Tarzia G., Bellasio E., Ciabatti R., Tuan G., Ferrari P., Zerilli L., Lanfranchi M., Pelizzi G. Xenobiotica. 1987;17:559–573. doi: 10.3109/00498258709043963. [DOI] [PubMed] [Google Scholar]
- Murphy P. J., Williams T. L. J. Med. Chem. 1972;15:137–139. doi: 10.1021/jm00272a005. [DOI] [PubMed] [Google Scholar]
- Wang L., Stanley M., Boggs J. W., Crawford T. D., Bravo B. J., Giannetti A. M., Harris S. F., Magnuson S. R., Nonomiya J., Schmidt S., Wu P., Ye W., Gould S. E., Murray L. J., Ndubaku C. O., Chen H. Bioorg. Med. Chem. Lett. 2014;24:4546–4552. doi: 10.1016/j.bmcl.2014.07.071. [DOI] [PubMed] [Google Scholar]
- Cox J. M., Chu H. D., Yang C., Shen H. C., Wu Z., Balsells J., Crespo A., Brown P., Zamlynny B., Wiltsie J., Clemas J., Gibson J., Contino L., Lisnock J., Zhou G., Garcia-Calvo M., Bateman T., Xu L., Tong X., Crook M., Sinclair P. Bioorg. Med. Chem. Lett. 2014;24:1681–1684. doi: 10.1016/j.bmcl.2014.02.057. [DOI] [PubMed] [Google Scholar]
- Cruciani G., Carosati E., De Boeck B., Ethirajulu K., Mackie C., Howe T., Vianello R. J. Med. Chem. 2005;48:6970–6979. doi: 10.1021/jm050529c. [DOI] [PubMed] [Google Scholar]
- Robarge K. D., Lee W., Eigenbrot C., Ultsch M., Wiesmann C., Heald R., Price S., Hewitt J., Jackson P., Savy P., Burton B., Choo E. F., Pang J., Boggs J., Yang A., Yang X., Baumgardner M. Bioorg. Med. Chem. Lett. 2014;24:4714–4723. doi: 10.1016/j.bmcl.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Cid J. M., Tresadern G., Vega J. A., de Lucas A. I., Matesanz E., Iturrino L., Linares M. L., Garcia A., Andres J. I., Macdonald G. J., Oehlrich D., Lavreysen H., Megens A., Ahnaou A., Drinkenburg W., Mackie C., Pype S., Gallacher D., Trabanco A. A. J. Med. Chem. 2012;55:8770–8789. doi: 10.1021/jm3010724. [DOI] [PubMed] [Google Scholar]
- Broccatelli F., Aliagas I., Zheng H. ACS Med. Chem. Lett. 2018;9:522–527. doi: 10.1021/acsmedchemlett.8b00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty E. M., Fotsch C., Bannon A. W., Bo Y., Chen N., Dominguez C., Falsey J., Gavva N. R., Katon J., Nixey T., Ognyanov V. I., Pettus L., Rzasa R. M., Stec M., Surapaneni S., Tamir R., Zhu J., Treanor J. J., Norman M. H. J. Med. Chem. 2007;50:3515–3527. doi: 10.1021/jm070190p. [DOI] [PubMed] [Google Scholar]
- Richardson B. G., Jain A. D., Potteti H. R., Lazzara P. R., David B. P., Tamatam C. R., Choma E., Skowron K., Dye K., Siddiqui Z., Wang Y. T., Krunic A., Reddy S. P., Moore T. W. J. Med. Chem. 2018;61:8029–8047. doi: 10.1021/acs.jmedchem.8b01133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raheem I. T., Schreier J. D., Fuerst J., Gantert L., Hostetler E. D., Huszar S., Joshi A., Kandebo M., Kim S. H., Li J., Ma B., McGaughey G., Sharma S., Shipe W. D., Uslaner J., Vandeveer G. H., Yan Y., Renger J. J., Smith S. M., Coleman P. J., Cox C. D. Bioorg. Med. Chem. Lett. 2016;26:126–132. doi: 10.1016/j.bmcl.2015.11.013. [DOI] [PubMed] [Google Scholar]
- Linton A., Kang P., Ornelas M., Kephart S., Hu Q., Pairish M., Jiang Y., Guo C. J. Med. Chem. 2011;54:7705–7712. doi: 10.1021/jm2010942. [DOI] [PubMed] [Google Scholar]
- Jones J. P., Korzekwa K. R. Mol. Pharmaceutics. 2013;10:1262–1268. doi: 10.1021/mp300568r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori M., Jorgensen F. S., Olsen L. ACS Omega. 2017;2:4237–4244. doi: 10.1021/acsomega.7b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori M., Lyngholm-Kjaerby C., Long A., Olsen L., Jorgensen F. S. Comput. Struct. Biotechnol. J. 2019;17:345–351. doi: 10.1016/j.csbj.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara F., Burns A. C., Collins M. R., Dalvie D., Ornelas M. A., Vaz A. D., Fujiwara Y., Baran P. S. J. Med. Chem. 2014;57:1616–1620. doi: 10.1021/jm4017976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R. A., Korzekwa K. R., McMasters D. R., Fandozzi C. M., Jones J. P. J. Med. Chem. 2007;50:4642–4647. doi: 10.1021/jm0703690. [DOI] [PubMed] [Google Scholar]
- Xu Y., Li L., Wang Y., Xing J., Zhou L., Zhong D., Luo X., Jiang H., Chen K., Zheng M., Deng P., Chen X. J. Med. Chem. 2017;60:2973–2982. doi: 10.1021/acs.jmedchem.7b00019. [DOI] [PubMed] [Google Scholar]
- Cruciani G., Milani N., Benedetti P., Lepri S., Cesarini L., Baroni M., Spyrakis F., Tortorella S., Mosconi E., Goracci L. J. Med. Chem. 2018;61:360–371. doi: 10.1021/acs.jmedchem.7b01552. [DOI] [PubMed] [Google Scholar]