

Abstract
Transcriptomic profiling of metastatic cancer can illuminate mechanisms of progression and lead to new therapies, but standard biopsy is invasive and reflects only a single metastatic site. In contrast, circulating tumor cell (CTC) profiling is noninvasive and repeatable, reflecting the dynamic and systemic nature of advanced disease. To date, transcriptomic profiling of CTCs has not delivered on its full potential, because white blood cells (WBCs) vastly outnumber CTCs. Current profiling strategies either lack cancer sensitivity and specificity or require specialized CTC capture protocols that are not readily scalable to large patient cohorts. Here, we describe a new strategy for rapid CTC enrichment and transcriptomic profiling using commercially available WBC depletion, microfluidic enrichment and RNA sequencing. When applied to blood samples from patients with advanced prostate cancer (PC), transcriptomes from enriched samples cluster with cancer positive controls and previously undetectable prostate-specific transcripts become readily measurable. Gene set enrichment analysis reveals multiple significantly enriched signaling pathways associated with PC, as well as novel pathways that merit further study. This accessible and scalable approach yields cancer-specific transcriptomic data and can be applied repeatedly and noninvasively in large cancer patient cohorts to discover new therapeutic targets in advanced disease.
Keywords: circulating tumor cells, expression profiling, oncology, liquid biopsy
Introduction
Treatment of advanced prostate cancer (PC) has improved dramatically in recent years. New chemotherapies, hormonal agents and targeted therapies have successfully delayed progression and extended survival.1–7 At the same time, new questions have emerged about optimal combination and sequencing of these treatments and about the molecular mechanisms that drive resistance. In an effort to address these questions, several landmark studies set out to map the molecular landscape of metastatic PC.8–14 Although these studies produced valuable insights about genomic and transcriptomic alterations, they were dependent on tissue biopsies, which are limited by patient safety and comfort, cost and technical considerations. Due to these constraints, tissue biopsy studies offered a static “snapshot” from a subset of metastatic sites, usually at a single point in time. To overcome these limitations, significant efforts have been directed toward liquid biopsies that can be studied repeatedly and noninvasively as the disease progresses. By enriching and analyzing circulating tumor cells (CTCs) or circulating tumor DNA (ctDNA) from standard blood samples, a new wave of seminal studies has helped to identify candidate biomarkers and molecular disease drivers.15–25
To date, liquid biopsy studies have focused predominantly on CTC enumeration, staining and DNA profiling, as well as on ctDNA profiling, because these phenotypes are readily preserved and reproducibly analyzed with available techniques. For example, somatic DNA mutations are qualitatively different from germline and can be detected by next-generation techniques despite leukocyte background if sequenced at sufficient depth. In contrast, whole transcriptome profiling by next-generation sequencing of CTCs has been more challenging, because CTC-derived transcripts are effectively masked by far more abundant transcripts from the vast numbers of background leukocytes. This signal-to-noise challenge is further complicated by RNA cross-linking in the presence of preservatives or RNA degradation in their absence. Due to these challenges, CTC transcriptomic profiling has lagged behind other liquid biopsy assay development. A handful of studies did undertake whole transcriptome profiling of the cellular component of blood using one of two tacks: One approach employed whole blood fixation and RNA preservation to profile all blood cells rather than just the rare CTCs, yielding expression signatures predictably consisting of genes expressed by leukocytes, which contributed >99.99% of the RNA.26–28 Conversely, the other approach profiled only the CTCs by enriching, recovering and analyzing these rare cells singly or in clusters from small numbers of patient samples29–33 to produce cancer-specific gene signatures. Although this approach has yielded valuable mechanistic insights, it is not readily scalable to large patient cohorts due to its inherently greater cost, technical expertise, specialized equipment and processing time.
In this study, we set out to address the need for efficient, high-throughput cancer-specific gene expression profiling from CTCs. We reasoned that a successful strategy must be robust and user-friendly, enriching live CTCs rapidly and cost-effectively without the need for specialized cell-by-cell micromanipulation, yet also yielding high enough CTC purity levels for detection of cancer-specific genes over background. By testing multiple enrichment workflows, we optimized and analytically validated an approach that combines immunomagnetic leukocyte depletion with microfluidic live CTC enrichment. These new techniques recovered CTCs that yielded high-quality RNA for downstream RNA-seq marked by PC-specific targets and signaling pathways. To our knowledge, this approach is the first to offer cancer-specific gene expression analysis of CTCs in a manner that can be readily applied by the broader cancer research community to profile large numbers of PC patient samples.
Materials and Methods
Cell lines
The human PC cell line 22Rv1 (RRID:CVCL_1045) was purchased from the American Type Culture Collection (Manassas, VA) while LNCaP cells (RRID:CVCL_0395) were a gift from Dr. Jacek Pinski (University of Southern California). Cell lines were authenticated by IDEXX BioAnalytics using short tandem repeat profiling within the last 3 years and confirmed to be mycoplasma free. Cell lines were maintained at 37° C, 5% CO2 in Roswell Park Memorial Institute 1640 supplemented with 10% fetal bovine serum (Omega, Tarzana, CA), penicillin (100 units/ml, Invitrogen, Carlsbad, CA) and streptomycin (100 μg/ml). Cells were harvested once they reached 75% confluency while subsequent spike-in experiments were performed 48 hr later.
Study approval and patient blood samples
Healthy donor and patient blood samples were collected and processed at the USC Norris Comprehensive Cancer Center under protocols HS-11-0054 and HS-17-00639 that were approved by the USC Health Science Institutional Review Board. All blood samples were collected after obtaining informed consent from patients or healthy donors prior to their participation in the study. Blood was drawn into two types of tubes in this study: K2ethylenediaminetetraacetic acid (EDTA) tube (BD, Franklin Lakes, NJ; 10 ml) and CellSave tube (Menarini Silicon Biosystems, Bologna, Italy; 10 ml). Blood drawn into K2EDTA tube was stored at room temperature and processed for gene expression profiling within 4 hr.
CD45 depletion and microfluidic enrichment
Harvested LNCaP cells were labeled using Vybrant CFDA SE cell tracer (Life Technologies, Camarillo, CA) per manufacturer’s instructions. Serial dilutions of determined cell counts using a hemocytometer were performed until a final concentration of 10,000 cells/ml was estimated. Accurate cell concentration was then determined using an INCYTO C-Chip hemocytometer, and volumes equaling 200, 100, 50 or 25 cells were transferred into Pluronic F-68-coated wells of a 96-well plate for accurate cell count determination. Cells were then resuspended and dispensed into healthy donor blood (7.5 ml), ensuring no cells were remaining in the wells and processed immediately. In short, RosetteSep™ CD45 depletion cocktail (Stemcell Technologies, Vancouver, Canada) was added directly to the vacutainer (50 μl per 1 ml blood) and incubated on a nutating mixer for 20 min at room temperature. Layering the blood mixture into a SepMate tube containing Lymphoprep (15 ml; Stemcell Technologies) was then followed by centrifugation at 1200g for 20 min with brake set to off. The upper plasma layer was retained, and further enrichment was performed using a Parsortix system (6.5 μM cassette; Angle PLC, Guildford, UK) that separates by size and deformability. For evaluation of target cell recovery and background leukocyte counts, the Parsortix output was collected directly into a 96-well plate and incubated with Hoechst® 33342 dye for 5 min (5 μg/ml). Images were captured using both fluorescein isothiocyanate (FITC) and 4′,6-diamidino-2-phenylindole (DAPI) filters at ×10 magnification using a Zeiss Axio Observer A1 (Zeiss Microscope, Jena, Germany). Total background leukocyte counts were determined by subtracting the total number of carboxyfluorescein succinimidyl ester (CFSE)-stained LNCaP cells from the total number of Hoechst-stained nucleated cells. For double microfluidic enrichment only, healthy donor blood (7.5 ml) spiked with LNCaP cells (200 cells) was initially processed using a Parsortix system (6.5 μM cassette; Angle PLC), with the harvest collected directly into 1 ml matched plasma. A second separation followed on the Parsortix system (8 μM cassette; Angle PLC), and target cell recovery and background leukocyte counts were evaluated. For microfluidic separation and EasySep CD45 depletion (Stemcell Technologies), healthy donor blood (7.5 ml) spiked with LNCaP cells (200 cells) was initially processed using a Parsortix system (6.5 μM cassette; Angle PLC), with the harvest directly incubated with EasySep CD45 antibody (5 μl; 1:10 dilution) for 10 min. Magnetic particles (10 μl) were then added and incubated for 10 min, before separation on 96-well magnetic plate. Target cell recovery and background leukocyte counts were evaluated.
For gene expression profiling, highly enriched cells were collected directly into 600 μl of RLT buffer (Qiagen, Germantown, MD) with 0.1% β-mercaptoethanol, and RNA extraction was performed using RNeasy Micro Kit (Qiagen) per manufacturer’s instructions (see later) with a final elution volume of 10 μl CTC enumeration from a concurrent CellSave blood draw was performed using the FDA-approved CellSearch system (Menarini Silicon Biosystems) as previously described.34
Multiplexed quantitative polymerase chain reaction
All steps were performed using MyiQ™ single-color real-time polymerase chain reaction (PCR) detection system (Bio-Rad, Hercules, CA). cDNA synthesis was performed with qScript™ cDNA supermix (Quantabio, Beverly, MA) under the following conditions: 22°C for 5 min; 42°C for 30 min; 85°C for 5 min. cDNA multiplex preamplification (14 cycles; 180 nM per primer) was performed with PerfeCTa SYBR Green Supermix for iQ (Quantabio) under the following conditions: 95°C for 10 min (95°C for 15 sec, 60°C for 4 min) × 14 cycles; 99°C for 10 min. PCR product was then diluted 20-fold with nuclease-free water and used as template (1 μl for standard quantitative PCR (qPCR) (40 cycles) as described earlier. All primers were synthesized and purchased from Integrated DNA Technologies (Suppporting Information Table S1).
RNA sequencing
RNA-sequencing libraries were simultaneously constructed from either extracted total RNA (see earlier) or enriched direct cell input using the SMARTer Stranded Total RNA-seq kit v2 according to the manufacturer’s protocol (Cat#634413, Clontech, Mountain View, CA). Quality control of the final sequencing libraries were performed, with size distribution assessed using a BioAnalyzer 2100 (Agilent, Santa Clara, CA) and quantified using a Qubit Fluorometer (Invitrogen). Finally, prepared libraries were quantified using the qPCR-based Kapa Library Quantification Kit (Cat# KK4828, Roche, Basel, Switzerland). Prepared libraries were pooled and sequenced on the NextSeq500 (Illumina, San Diego, CA) at 2 × 75 cycles.
Bioinformatic analysis
The industry standard Illumina’s bcl2fastq was used to convert BCL files to FASTQ files. Sequencing reads are initially aligned to the GRCh37 human genome reference using STAR v2.5.3a, followed by mark duplicates and haplotype caller GATK v3.5.0. Picard Multi/HS/GC Bias Metric and SAMtools v1.235 stats are calculated for overall run summary statistics. STAR-Fusion v0.8.0 was used to identify candidate fusion transcripts and map junction reads based on reference annotation. CuffDiff v2.2.1 was used for isoform quantification and Salmon 0.7.2 for transcript quantification.36
Statistics
All data with error bars represent the mean ± standard error of the mean (SEM). Secondary analysis of RNA-sequencing data was performed without modification using DESeq2 v1.24.0 package37 for differential expression analysis and Ingenuity pathway analysis (IPA v1.14) Qiagen for pathway enrichment analysis. For differential expression analyses, an adjusted p value of less than 0.01 was considered statistically significant. For pathway enrichment analysis, a p value of less than 0.05 was considered statistically significant. Comparison of prostate-specific gene expression profile was performed using log2-transformed Sailfish transcripts per million (TPM) values.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Enrichment of live cancer cells from blood
Live CTC enrichment was first optimized using engineered samples of LNCaP PC cells spiked into 7.5 ml healthy donor blood. Samples were processed on a Parsortix microfluidic device, a size and deformability-based enrichment platform, alone or in combination with immunomagnetic human CD45 depletion. Of the workflows tested, RosetteSep immunomagnetic CD45 depletion followed by Parsortix separation resulted in the greatest capture and enrichment, with 30% cancer cell recovery and a background of only 100 white blood cells (WBCs), a 5-log enrichment (Fig. 1a). This enrichment strategy performed similarly with decreasing numbers of spiked-in cancer cells (Fig. 1b). Hence, the optimized workflow (Fig. 1c) could rapidly (<3 hr) enrich a significant portion of cancer cells in a blood sample while dramatically reducing background peripheral blood mononuclear cell (PBMCs), potentially enabling detection of PC-specific transcripts.
Figure 1.
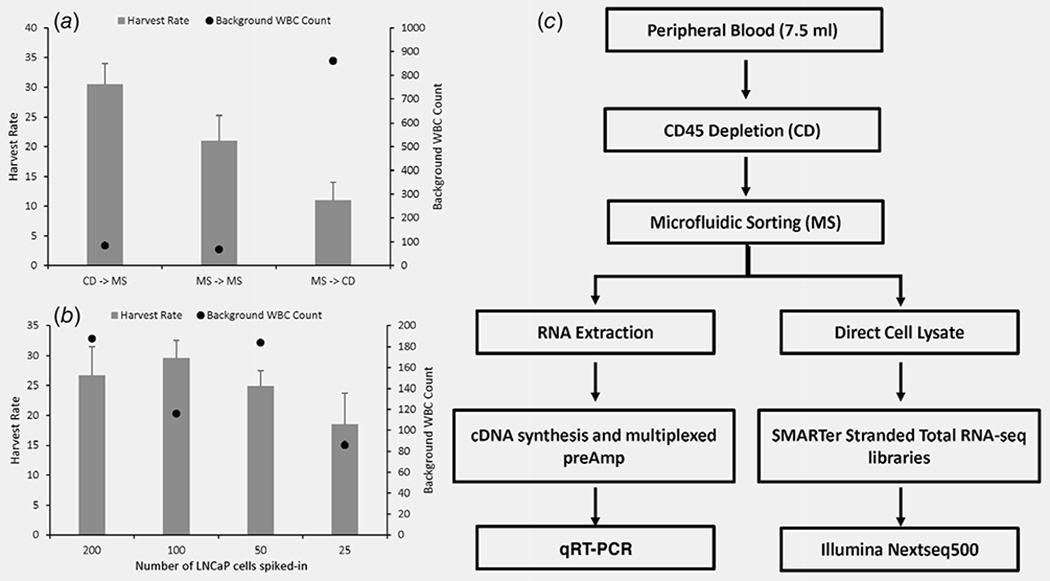
Relative efficacy of CTC enrichment workflows. (a) LNCaP cells (200 cells) were spiked into healthy donor blood prior to assess enrichment using various combinations of microfluidic sorting (MS) and CD45 depletion (CD). Harvest rate = (number of LNCaP cells recovered/number of LNCaP cells spiked in) × 100. (b) Increasing number of LNCaP cells (25, 50, 100 and 200 cells) were spiked into healthy blood and enriched using the CD− >MS method to evaluate the effect on performance. (c) Schema of optimal CTC enrichment and gene expression workflow. Experiments were performed in triplicates and data represents mean ± SEM.
Targeted gene expression analysis of cancer cells enriched from blood
Prior to proceeding to RNA-seq, multiplexed quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed to test whether PC-specific and epithelial-specific gene transcripts could be detected from cancer cells enriched by immunodepletion plus microfluidic capture. First, engineered samples were prepared by spiking LNCaP cells into 7.5 ml of healthy donor blood, followed by CD45 depletion and microfluidic enrichment. Recovered cells were lysed and RNA extracted and subjected to multiplexed preamplification followed by qPCR for the specified genes, which were robustly and linearly detected across input levels (Fig. 2a). Based on these encouraging results, we applied the same enrichment and qPCR workflow to three blood samples from men with metastatic castrate-resistant PC (mCRPC). From each patient, two 7.5-ml EDTA tubes of blood samples were analyzed for live CTC gene expression, and a third 7.5-ml tube (CellSave) of blood sample was analyzed for fixed CTC enumeration using the FDA-cleared CellSearch platform.38 Two of the patients had no detectable CTCs by CellSearch and also no PC-specific or epithelial-specific gene expression detectable by qRT-PCR, whereas the third patient had 88 CTCs by CellSearch and also robust PC-specific and epithelial-specific gene expression detectable by qRT-PCR from enriched CTCs (Fig. 2b). Hence, when CTCs were present, the CD45 depletion plus microfluidic capture protocol was capable of enriching them sufficiently for PC-specific gene detection by qRT-PCR.
Figure 2.
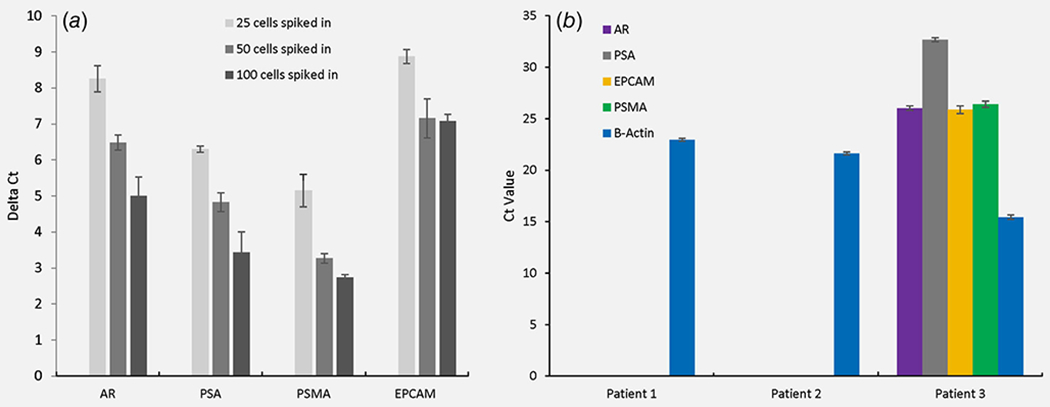
Prostate cancer-relevant gene targets identified by multiplexed qRT-PCR. (a) PC-specific and epithelial-specific gene expression (normalized to −β-actin) from engineered samples of LNCaP prostate cells spiked into 7.5 ml healthy donor blood and enriched using CD45 depletion plus microfluidics. (b) PC-specific and epithelial-specific gene expression from mCRPC patient blood samples enriched using CD45 depletion plus microfluidics. Ct values greater than or equal to 35 were recorded as not detected. CellSearch count for Patients 1 and 2 was 0 CTCs, whereas Patient 3 had 88 CTCs. Results are displayed as the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
RNA-seq of cancer cells enriched from blood
Next, we tested whether the new CTC enrichment workflow could yield cancer transcript levels detectable by RNA-seq, a higher bar as RNA-seq libraries is prepared with universal primers rather than PC-specific primers used in qRT-PCR. Prior to testing actual patient samples, RNA-seq was performed using engineered RNA samples representing the low RNA starting amounts expected from our enrichment method. An amount of 1 ng RNA (~100 cell equivalent) consisting of various ratios of 22Rv1 PC RNA and WBC RNA was subjected to whole transcriptome amplification followed by sequencing and quantitation (TPM) of four PC-specific genes: androgen receptor (AR), Forkhead box A1 (FOXA1), Kallikrein-related peptidase 2 (KLK2) and Grainyhead-like transcription factor 2 (GRHL2). Gene expression was robustly detected in the engineered samples containing PC gene transcripts, decreasing linearly with successively lower CTC equivalents down to as little as five CTC equivalents (Fig. 3a). The engineered samples consisting exclusively of WBC RNA appropriately yielded no detectable PC gene transcripts. Hence, PC-specific transcripts could be detected reliably and linearly from libraries prepared from 1 ng (~100 cell equivalent) starting material consisting of as little as five cancer cells with 95 background PBMCs.
Figure 3.
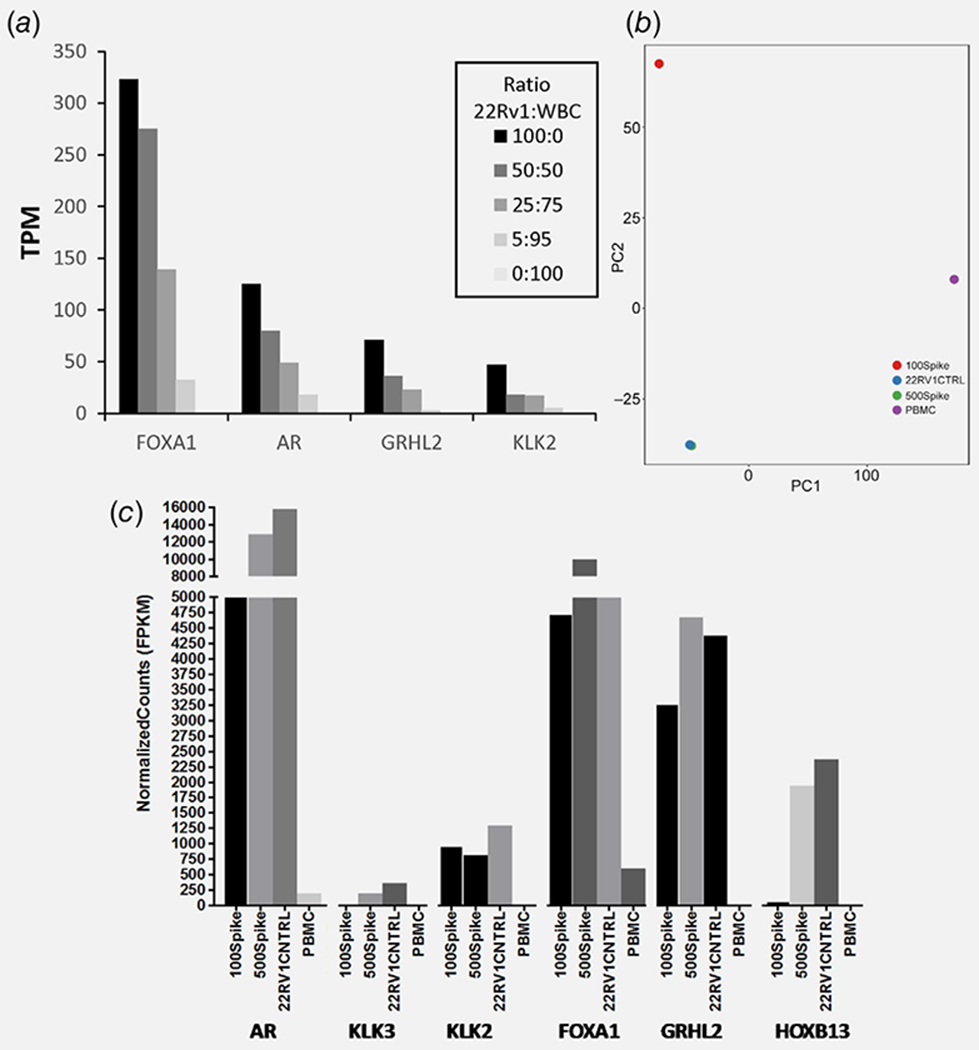
Feasibility of low-input RNA-seq analysis using cell line engineered controls as surrogates for PC patient samples. (a) 22Rv1 RNA and WBC RNA were mixed at the ratios specified, each representing different numbers of CTC equivalents. Each sample equaled 1 ng total RNA (~100 cell) and was used for low-input library prep and RNA-seq, and the PC-specific transcripts were quantified. (b) 22Rv1 cells (100 and 500 cells) were spiked into 7.5 ml healthy donor blood prior to enrichment, low-input library prep and RNA-seq. PCA discriminating 22Rv1 spiked-in samples from bulk PBMCs and cell line control is shown. (c) PC-specific transcript levels quantified from the RNA-seq data for these engineered blood samples. [Color figure can be viewed at wileyonlinelibrary.com]
Given these encouraging results, enrichment and RNA-seq were next applied to engineered blood sample controls consisting of 22Rv1 (500 or 100 cells) spiked into healthy donor blood and enriched by CD45 depletion plus microfluidics. Additional RNA-seq controls consisted of bulk RNA purified from healthy donor buffy coats (negative control) and from 22Rv1 cell line (positive control). Principal component analysis (PCA) clearly discriminated healthy donor buffy coat negative controls from 22Rv1 cell line positive control, which grouped more closely with both the 100 and 500 22Rv1 cell spike-in samples (Fig. 3b). In particular, the 500 22Rv1 cell spike-in sample grouped very closely with the bulk RNA positive control, reflecting a high level of enrichment. As predicted from the PCA, the RNA-seq data yielded progressively higher levels of PC-specific transcripts in the samples enriched from 100 cell spike-in, 500 cell spike-in and bulk 22Rv1 positive control, in contrast to the PBMC controls that had no PC-specific transcripts (Fig. 3c). Hence, CD45-depletion plus microfluidics successfully enriched cancer cells from standard blood volumes in a manner that was reliably and proportionally detected by RNA-seq.
Next, to test the CTC enrichment and RNA-seq workflow on clinical samples, blood was collected from five men with mCRPC into CellSave and EDTA tubes for CellSearch enumeration and RNA-seq, respectively. All five patients had detectable CTCs, with a range of 17–1,192 CTCs/7.5 ml (Supporting Information Table S2). CTC-enriched samples and matched unenriched PBMCs (buffy coat) from the same EDTA tubes were sequenced, yielding a median of 16 million uniquely aligned sequencing reads (range: 5.7–22.0 M; Supporting Information Table S3). Four of the five CTC-enriched patient samples yielded high-quality aligned sequencing data and were used for subsequent analysis. Unsupervised PCA of CTC-enriched patient samples, matched buffy coat specimens and engineered controls resulted in tight grouping of sample groups and discrimination of enriched CTC specimens from either unenriched buffy coat or engineered controls (Fig. 4a). Gratifyingly, engineered control samples were distributed in a manner consistent with their cancer:WBC ratio, with 0:100 closest to patient unenriched PBMCs and 100:0 having farthest distance. Unsupervised hierarchical clustering of the top 1,000 expressed genes for CTC-enriched patient samples, matched buffy coat specimens and engineered controls revealed clustering of patient-enriched CTCs with the 22Rv1 cell line (Fig. 4b). Hence, CD45-depletion plus microfluidic enrichment of blood samples from men with mCRPC yielded RNA-seq data and transcriptional profiles that clustered with PC cell lines, while data from unenriched samples comprised chiefly of leukocytes formed a separate cluster.
Figure 4.
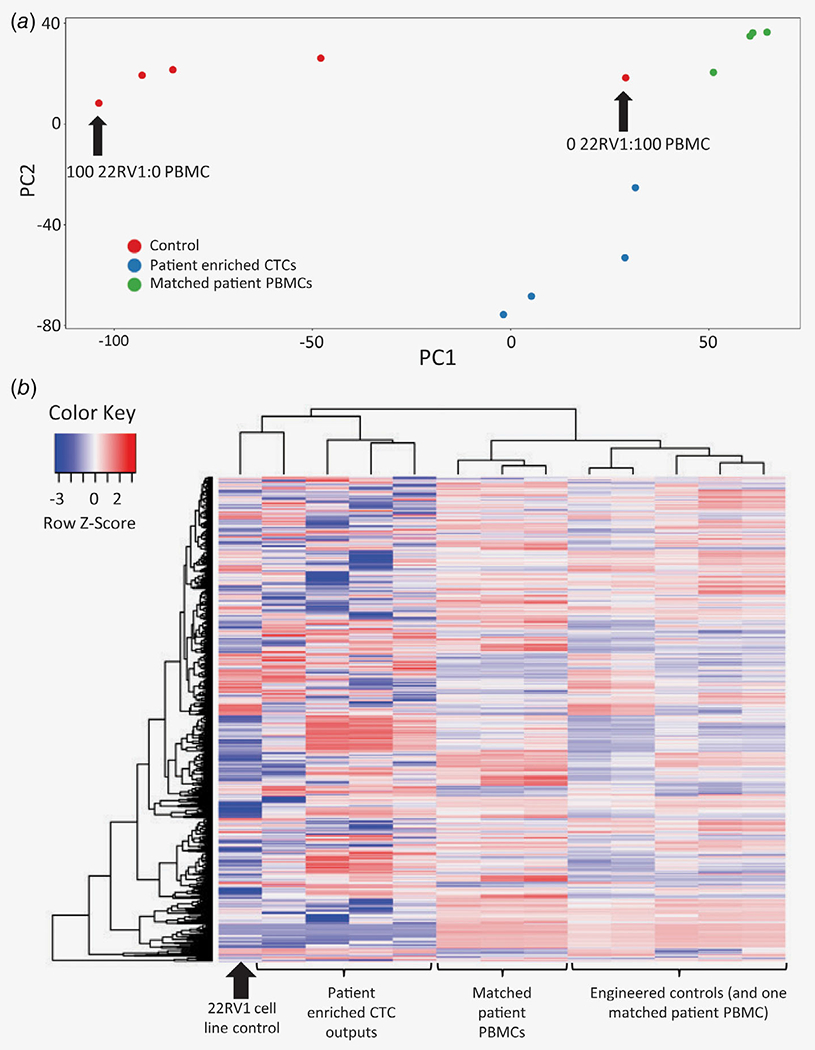
RNA-seq of clinical samples enriched by CD45-depletion plus microfluidics. (a) PCA from enriched cells versus matched unenriched cells from the same samples, as well as from engineered controls comprised of serially diluted PBMCs. (b) Heat map representation of unsupervised hierarchical clustering analysis using the top 1,000 expressed genes. [Color figure can be viewed at wileyonlinelibrary.com]
To determine whether CTC-enriched samples have the potential to yield gene expression data that is clinically and biologically relevant to PC, patient samples were analyzed for expression of PC-relevant genes and pathways. These were identified by curating the PC literature and by querying the expression of candidate genes in whole blood using the Genotype Tissue Expression portal.13,29,39–42 Predictably, in the engineered cancer:WBC control samples, PC-relevant gene expression increased with higher proportions of cancer cells (Fig. 5a). Consistent with this, patient samples enriched for CTCs clustered with engineered control samples comprised high numbers of cancer cells; conversely, matched unenriched patient samples clustered with engineered control samples comprised few or no cancer cells (Fig. 5a). Accordingly, within-patient comparison of CTC-enriched versus matched unenriched samples showed robust detection of PC-specific genes, androgen signaling genes and Wnt signaling genes in the CTC-enriched samples, but little or no detection of these genes in matched unenriched samples (Figs. 5b–5d). Hence, CTC enrichment and RNA-seq of clinical samples generated cancer-relevant expression profiles and effectively “unmasked” PC-specific transcripts that were undetectable without enrichment.
Figure 5.
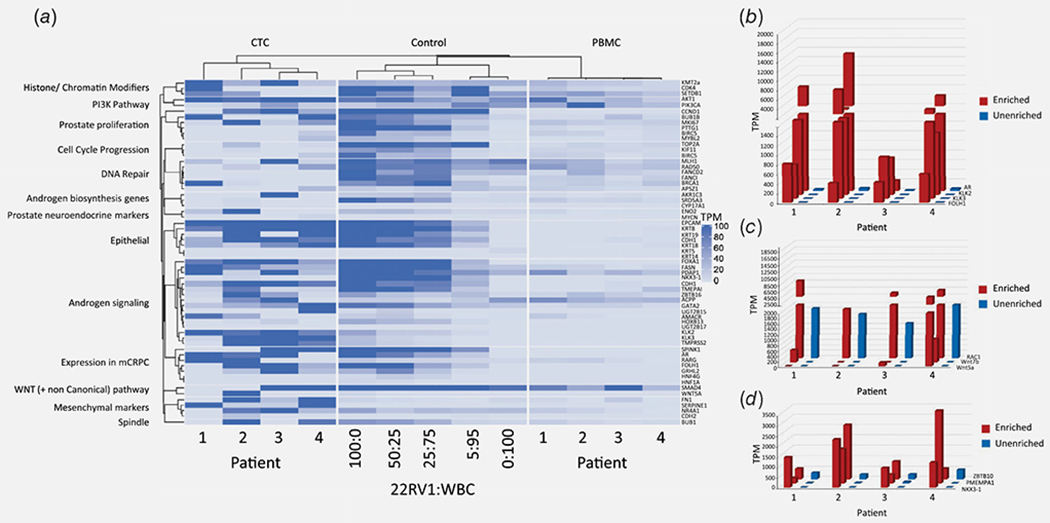
Detection of PC-relevant genes. (a) Heat map representation comparing CTC-enriched, unenriched and engineered spike in controls for expression levels of PC-relevant genes arranged by function and/or pathway. (b–d) PC-relevant gene expression in CTC-enriched versus matched unenriched samples, focusing on examples of PC-specific genes (b), androgen signaling genes (c) and Wnt signaling genes (d). [Color figure can be viewed at wileyonlinelibrary.com]
Finally, we tested whether these techniques could be valuable for unsupervised discovery of gene expression in CTCs, beyond detection of known, well-annotated transcripts. We queried 12,760 genes for statistically significant differential expression between CTC-enriched and matched unenriched PBMCs. A total of 4,956 genes met that criterion, with 976 upregulated genes and 3,980 downregulated genes identified with an adjusted p value of <0.01. Reassuringly, a few highly differentially expressed genes that were upregulated in CTC-enriched samples included the PC-related AR, EGFR and FOXA1 (Fig. 6a). Differentially expressed genes were analyzed using IPA, revealing that many of these are categorized with high significance in cancer and PC-related pathways such as migration, proliferation, invasion and epithelial-mesenchymal transition (EMT) (Fig. 6b). As additional validation, transcripts normally associated with WBC canonical pathways (e.g., CD40) were identified as significantly downregulated in the enriched samples. Furthermore, a long list of as-yet unannotated differentially expressed transcripts (long noncoding RNAs) was also detected. Hence, CTC enrichment and RNA-seq not only detected known PC-specific transcripts but also yielded extensive differential expression profiles attributable to CTCs, providing fertile soil for novel discovery.
Figure 6.
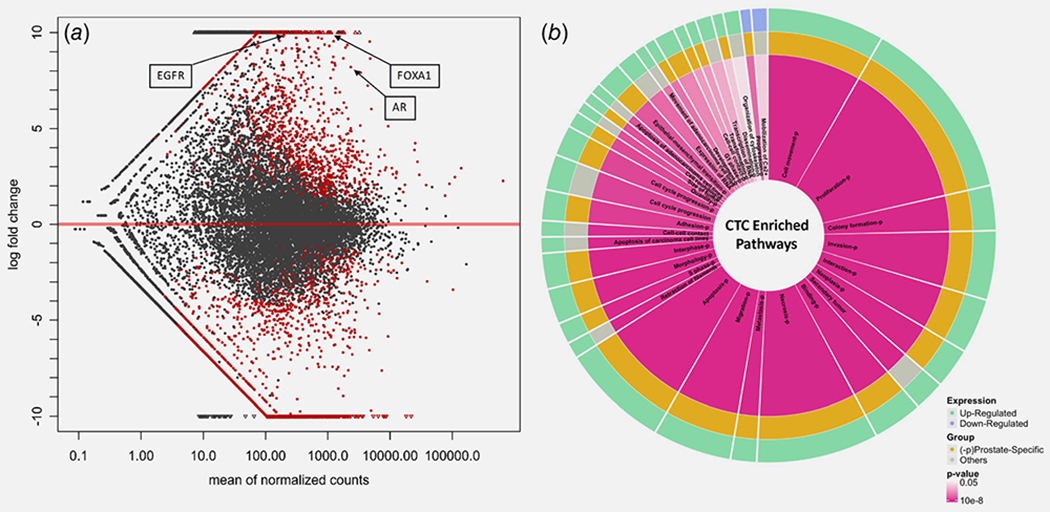
Differential gene expression and enriched signaling pathways identified in CTC-enriched blood samples from mCRPC patients. (a) Differentially expressed genes in CTC-enriched samples compared to matched unenriched patient PBMCs. Genes that are upregulated (>0-fold change) and downregulated (<0-fold change) are shown. Small triangles fall outside of the plotting window. Red dots show significant genes with adjusted p value of <0.01, generated using DESeq2 package.37 Three genes implicated in prostate cancer disease progression are highlighted. (b) Ingenuity pathway analysis of pathways enriched in prostate patient CTCs, derived using differentially regulated significantly expressed genes from (a). Pathways that are upregulated are labeled green and downregulated in blue. Orange and “−P” both indicate that a given pathway is prostate specific. Red intensity indicates significance with range of 0.05 > p < 0.0000001, and the area of each pathway is representative of the number of significant differentially expressed genes contributing to that pathway. Genes associated with each pathway are shown in Supporting Information Data S1. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
Gene expression analysis is a powerful analytic tool that provides valuable insights into the functional pathways driving cancer resistance and progression that can only be implied by genomic phenotypes. In PC, RNA expression signatures have been used to identify predictors of treatment outcome, early dissemination and mechanisms of resistance.13, 29,43 Characterization of CTC transcriptomes provides a means to noninvasively identify unique dissemination phenotypes (e.g., EMT) and to interrogate multiple pathways driving disease progression. Studies of CTC gene expression are increasing; however, each has had to contend with formidable technical challenges, including the rarity of CTCs, limited RNA starting material, amplification bias, a large number of background WBCs and lack of truly cancer-specific gene targets. Gene expression studies on single CTCs have offered the greatest level of resolution and sensitivity. Using this approach, signaling pathways enriched in enzalutamide-resistant mCRPC together with CTC heterogeneity have been identified.29 However, these single cell studies are laborious and expensive and can be confounded by cell surface markers with limited specificity that may lead to false-positive CTC identification. An alternate whole blood lysis method has provided useful prognostic biomarker information in mCRPC patients,26,28 but this approach has inherent sensitivity, specificity and target scalability issues owing to the fact that the RNA is derived almost entirely from leukocytes. A third intermediate approach involves immunomagnetic or microfluidic enrichment of CTCs, which has been applied to develop RNA-based assays of AR-V7 as well as digital CTC signatures associated with response to androgen signaling inhibitors.43–45 However, because this approach is associated with significant background RNA from contaminating WBCs (104–105), it is incompatible with cancer-specific whole transcriptome discovery and can only be used to query a known set of amplified cancer-related genes that are specified a priori.
To address this, we have developed a new workflow that combines CD45 leukocyte depletion with microfluidic CTC enrichment for rapid and efficient CTC recovery with minimal WBC contamination. The strategy has several advantages, including (i) high enrichment levels; (ii) minimal manipulation; (iii) recovered target cells free of antibodies or magnetic beads; (iv) epitope agnostic; (v) rapid, straightforward and robust workflow; (vi) flexible downstream applications. One limitation of this approach is that—in order to achieve simple, rapid, maximal enrichment—the specific numbers of CTCs and leukocytes in an enriched sample are not directly quantified. Thus, if expression of a particular gene is found to increase with progressing or more aggressive disease, it is not immediately apparent whether this reflects an actual biologic increase in per-cell expression or a higher number of CTCs reflecting greater cancer volume and dissemination, but each with the same per-cell gene expression. This can be addressed in three ways: The first is to identify genes whose expression significantly decreases with aggressive or progressing disease and therefore cannot possibly be a simple surrogate for increasing numbers of CTCs in the sample. The second is to perform CTC counts (e.g., CellSearch) in parallel and to normalize gene expression to those counts, an approach that would have to be validated. The third and perhaps simplest approach is to normalize cancer-related genes of interest to the expression of “housekeeping” PC genes that reflect the number of cells rather than a specific cancer function, such as PSA, PSMA or KLK2.
Using this approach first with multiplexed qPCR, we observed that engineered controls and patient samples with identifiable CTCs consistently yielded prostate- and epithelial-specific genes in a sensitive and specific manner. Next for RNA-seq, we sought to take advantage of a modification in the SMARTer Stranded Total RNA-Seq kit that allowed for direct cell input as starting material rather than purified RNA, thus reducing the inherent loss occurred during this isolation step. As expected, in enriched samples from patients with no identifiable CTCs, prostate-specific transcripts were not identified, as these genes have low, if any, expression in background leukocytes. Conversely, prostaterelevant transcripts were robustly detected in engineered controls (as low as five cancer cell equivalents) and in blood enriched from mCRPC patient samples with identifiable CTCs. Enriched patient samples clustered with one another and with the PC cell line control when analyzed by unsupervised hierarchical clustering, and also when analyzed using a PC-specific gene set. Gratifyingly, this proof of concept study identified many of the same targets and signaling pathways implicated in prior mCRPC single cell expression profiles (e.g., Wnt,29). In a prospective agnostic approach to differential gene expression analysis, IPA revealed enrichment of differentially expressed genes (relative to unenriched WBC signatures) in pathways such as proliferation, cell cycle progression, invasion and metastasis. Hence, this CTC enrichment and RNA-seq strategy are capable of detecting numerous cancer-related genes at many-fold levels relative to their detection in unenriched samples, opening potential avenues for new discovery studies using liquid biopsies. When applied to clinically annotated patient cohorts (e.g., good vs. poor outcome), this straightforward and scalable approach can be leveraged to identify novel candidate biomarkers and therapeutic targets in advanced disease.
Supplementary Material
What’s new?
Current strategies for the transcriptomic profiling of circulating tumor cell (CTCs) either lack cancer sensitivity and specificity or are not readily scalable to large patient cohorts. Here, the authors describe a new strategy using commercially-available technologies to rapidly and effectively enrich CTC RNA for whole transcriptome sequencing. This straightforward approach is readily scalable to large numbers of patient blood samples and yields expression profiles marked by numerous cancer-specific genes and pathways. Hence, the new strategy can be used to sample cancer transcriptomes repeatedly and noninvasively in advanced malignancies in order to gain new mechanistic insights and identify novel therapeutic targets.
Acknowledgments
We thank Stephanie Tring and Suyeon Ryu from the USC Molecular Genomics Core of the USC Norris Cancer Center for their help in performing the RNA-seq library generation and sequencing. We thank the patients for their participation in this study. This work has been supported by grant NIH/NCI P30CA014089. Angle PLC provided some of the consumables for the Parsortix platform.
Abbreviations:
- AR
androgen receptor
- CTC
circulating tumor cell
- ctDNA
circulating tumor DNA
- EMT
epithelial–mesenchymal transition
- FOXA1
Forkhead box A1
- GRHL2
Grainyhead-like transcription factor 2
- IPA
Ingenuity pathway analysis
- KLK2
Kallikrein-related peptidase 2
- mCRPC
metastatic castrate-resistant prostate cancer
- PBMC
peripheral blood mononuclear cell
- PC
prostate cancer
- PCA
principal component analysis
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- SEM
standard error of the mean
- TPM
transcripts per million
- WBCs
white blood cells
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflict of Interest
Peggy Robinson was formerly affiliated with Angle PLC which provided some of the consumables for the Parsortix platform used in this work. All other authors have declared that no conflict of interest exists.
References
- 1.Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. New Engl J Med 2015;373:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. New Engl J Med 2017;377:352–60. [DOI] [PubMed] [Google Scholar]
- 3.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and Olaparib in metastatic prostate cancer. New Engl J Med 2015;373:1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010;376:1147–54. [DOI] [PubMed] [Google Scholar]
- 5.Hoskin P, Sartor O, O’Sullivan JM, et al. Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: a prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. Lancet Oncol 2014;15:1397–406. [DOI] [PubMed] [Google Scholar]
- 6.Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 2015;16:152–60. [DOI] [PubMed] [Google Scholar]
- 7.Sternberg CN, de Bono JS, Chi KN, et al. Improved outcomes in elderly patients with metastatic castration-resistant prostate cancer treated with the androgen receptor inhibitor enzalutamide: results from the phase III AFFIRM trial. Ann Oncol 2014;25:429–34. [DOI] [PubMed] [Google Scholar]
- 8.Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;162:454. [DOI] [PubMed] [Google Scholar]
- 9.Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015;520:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper CS, Eeles R, Wedge DC, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet 2015;47:367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boutros PC, Fraser M, Harding NJ, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet 2015;47:736–45. [DOI] [PubMed] [Google Scholar]
- 13.Kumar A, Coleman I, Morrissey C, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 2016;22:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018;174:758–69.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scher HI, Graf RP, Schreiber NA, et al. Nuclear-specific AR-V7 protein localization is necessary to guide treatment selection in metastatic castration-resistant prostate cancer. Eur Urol 2017;71:874–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scher HI, Lu D, Schreiber NA, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol 2016;2:1441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohr JG, Adalsteinsson VA, Cibulskis K, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol 2014;32:479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice Variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol 2017;35:2149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wyatt AW, Azad AA, Volik SV, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol 2016;2:1598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azad AA, Volik SV, Wyatt AW, et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res 2015;21:2315–24. [DOI] [PubMed] [Google Scholar]
- 21.Ulz P, Belic J, Graf R, et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nature communications 2016;7:12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodara E, Morrison G, Cunha A, et al. Multi-parametric liquid biopsy analysis in metastatic prostate cancer. JCI insight 2019;4:e125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldkorn A, Ely B, Tangen CM, et al. Circulating tumor cell telomerase activity as a prognostic marker for overall survival in SWOG 0421: a phase 3 metastatic castration resistant prostate cancer trial. Int J Cancer 2015;136:1856–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodall J, Mateo J, Yuan W, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov 2017;7:1006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandekerkhove G, Struss WJ, Annala M, et al. Circulating tumor DNA abundance and potential utility in de novo metastatic prostate cancer. Eur Urol 2019;75:667–75. [DOI] [PubMed] [Google Scholar]
- 26.Olmos D, Brewer D, Clark J, et al. Prognostic value of blood mRNA expression signatures in castration-resistant prostate cancer: a prospective, two-stage study. Lancet Oncol 2012;13:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ross RW, Galsky MD, Scher HI, et al. A whole-blood RNA transcript-based prognostic model in men with castration-resistant prostate cancer: a prospective study. Lancet Oncol 2012;13:1105–13. [DOI] [PubMed] [Google Scholar]
- 28.Todenhofer T, Azad A, Stewart C, et al. AR-V7 transcripts in whole blood RNA of patients with metastatic castration resistant prostate cancer correlate with response to abiraterone acetate. J Urol 2017;197:135–42. [DOI] [PubMed] [Google Scholar]
- 29.Miyamoto DT, Zheng Y, Wittner BS, et al. RNA-Seq of single prostate CTCs implicates non-canonical Wnt signaling in antiandrogen resistance. Science 2015;349:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cann GM, Gulzar ZG, Cooper S, et al. mRNA-Seq of single prostate cancer circulating tumor cells reveals recapitulation of gene expression and pathways found in prostate cancer. PloS One 2012;7:e49144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gkountela S, Castro-Giner F, Szczerba BM, et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 2019;176:98–112.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szczerba BM, Castro-Giner F, Vetter M, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019;566:553–7. [DOI] [PubMed] [Google Scholar]
- 33.Kulasinghe A, Kapeleris J, Cooper C, et al. Phenotypic characterization of circulating lung cancer cells for clinically actionable targets. Cancers 2019;11:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldkorn A, Ely B, Quinn DI, et al. Circulating tumor cell counts are prognostic of overall survival in SWOG S0421: a phase III trial of docetaxel with or without atrasentan for metastatic castration-resistant prostate cancer. J Clin Oncol 2014;32:1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol 2014;32:462–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res 2008;14:6302–9. [DOI] [PubMed] [Google Scholar]
- 39.Pal SK, Patel J, He M, et al. Identification of mechanisms of resistance to treatment with abiraterone acetate or enzalutamide in patients with castration-resistant prostate cancer (CRPC). Cancer 2018;124:1216–24. [DOI] [PubMed] [Google Scholar]
- 40.Danila DC, Anand A, Schultz N, et al. Analytic and clinical validation of a prostate cancer-enhanced messenger RNA detection assay in whole blood as a prognostic biomarker for survival. Eur Urol 2014;65:1191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer cell 2010;18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuzick J, Swanson GP, Fisher G, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol 2011;12:245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyamoto DT, Lee RJ, Kalinich M, et al. An RNA-based digital circulating tumor cell signature is predictive of drug response and early dissemination in prostate cancer. Cancer Discov 2018;8:288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armstrong AJ, Halabi S, Luo J, et al. Prospective Multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: the PROPHECY study. J Clin Oncol 2019;37:1120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jan YJ, Yoon J, Chen JF, et al. A circulating tumor cell-RNA assay for assessment of androgen receptor signaling inhibitor sensitivity in metastatic castration-resistant prostate cancer. Theranostics 2019;9:2812–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.