

Abstract
Current approaches for generating major histocompatibility complex (MHC) Class-I proteins with desired bound peptides (pMHC-I) for research, diagnostic and therapeutic applications are limited by the inherent instability of empty MHC-I molecules. Using the properties of the chaperone TAP-binding protein related (TAPBPR), we have developed a robust method to produce soluble, peptide-receptive MHC-I molecules in Chinese Hamster Ovary cells at high yield, completely bypassing the requirement for laborious refolding from inclusion bodies expressed in E.coli. Purified MHC-I/TAPBPR complexes can be prepared for multiple human allotypes, and exhibit complex glycan modifications at the conserved Asn 86 residue. As a proof of concept, we demonstrate both HLA allele-specific peptide binding and MHC-restricted antigen recognition by T cells for two relevant tumor-associated antigens. Our system provides a facile, high-throughput approach for generating pMHC-I antigens to probe and expand TCR specificities present in polyclonal T cell repertoires.
Keywords: HLA, TAPBPR, MHC tetramer, molecular diagnostics, TCR repertoire
Introduction
The Class-I proteins of the major histocompatibility complex (MHC-I) play a pivotal role in orchestrating immune responses through their interactions with specialized receptors on T cells and Natural Killer (NK) cells (Germain and Margulies, 1993; Jiang et al., 2019). Immune surveillance by αβ T cell receptors (TCRs) is achieved through the display of short (8–15 residue) peptides derived from viral proteins (or mutated oncogenes) via tight capture within the MHC-I peptide-binding groove as an obligate protein complex (Rossjohn et al., 2015). MHC-I molecules are assembled on the endoplasmic reticulum (ER) from component heavy and β2 microglobulin (β2m) light chains, and loaded with peptides in the context of a multisubunit membrane complex (Cresswell et al., 1999). Interactions of nascent MHC-I with molecular chaperones [tapasin and TAP-binding protein related (TAPBPR)] select for high-affinity peptides to ensure the prolonged stability and immunogenicity of resulting peptide/MHC-I (pMHC-I) complexes (Blum et al., 2013). As an additional quality control step, glucose trimming of the ER-associated Glc3Man9GlcNAc2 moiety found at the conserved Asn 86 (N86) residue ensures that only correctly folded molecules are trafficked further along the antigen processing pathway toward the cell surface (Wearsch et al., 2011). Although tapasin is an integral part of the ER-anchored peptide loading complex, TAPBPR is found throughout the ER and cis-Golgi network and has independent, auxiliary functions in MHC-I quality control (Neerincx et al., 2017), which ultimately affects the displayed peptide repertoire (Boyle et al., 2013; Hermann et al., 2015a, b).
Detecting and quantifying antigen-specific TCRs during the course of disease, treatment or immunization was revolutionized by the use of multivalent, fluorescent, pMHC-I molecules (Altman et al., 1996; Hadrup and Schumacher, 2010). Empty MHC-I molecules are unstable and highly prone to aggregation, so pMHC-I proteins are commonly produced by in vitro refolding of light and heavy chain components, derived from E.coli inclusion bodies, in the presence of large molar excess of a synthetic peptide, which involves a laborious multistep process with typical yields of <5% by weight (Garboczi et al., 1992). There have been considerable efforts to develop peptide-exchange methods, including photolabile peptides (Bakker et al., 2008), dipeptide catalysts (Saini et al., 2015), thermal exchange (Luimstra et al., 2018) or disulfide-linked MHC-I molecules (Moritz et al., 2019). All these methods make use of refolded MHC-I molecules, lacking important posttranslational modifications, which are likely to influence peptide repertoire selection, and T cell and NK cell recognition. Methods for producing pMHC-I molecules in mammalian cells using covalently linked peptides as single-chain (Jurewicz et al., 2019) or fused Antibody-pMHC-I constructs (Schmittnaegel et al., 2016) have been described, however both approaches necessitate cleavage of the bound peptide for exchange to occur, which results in low protein yields.
The use of molecular chaperones for MHC-I peptide exchange applications was first explored in the context of tapasin (Chen and Bouvier, 2007). TAPBPR, known to stabilize the empty MHC-I peptide-binding groove in a widened conformation (Jiang et al., 2017; Thomas and Tampé, 2017) and to promote peptide exchange in vitro (Morozov et al., 2016), offers an attractive alternative to tapasin from a biochemical perspective and has been exploited to load MHC-I molecules with peptides directly on the cell surface, independently of the peptide-loading complex (Ilca et al., 2018). A detailed characterization of the TAPBPR catalytic cycle (McShan et al., 2018) has recently been leveraged to develop a high-throughput exchange methodology for multiple murine and human MHC-I allotypes expressed in E.coli and refolded with an exchangeable peptide (Overall et al., 2020). Here, we exploit the chaperone function of TAPBPR to develop a similar approach for producing soluble pMHC-I molecules, using a mammalian protein expression system. We engineer suitable MHC-I and TAPBPR constructs with a cleavable heterodimeric leucine-zipper, a system which enables the production of pMHC-I molecules of desired peptide specificities at mg quantities. Recombinant MHC-I/TAPBPR complexes produced in mammalian cells bypass the requirements and restrictions of the peptide loading complex, are subject to standard eukaryotic posttranslational modification, and can be readily loaded with peptides toward functional, biochemical and structural characterization of interactions with their cognate immune receptors.
Materials and Methods
Molecular cloning of protein expression constructs
Synthetic, codon-optimized single-chain β2m_HLA-A*02:01, β2m_HLA*A68:02, β2m_HLA*A24:01 and TAPBPR genes were purchased from IDT (Coralville, IA) and ligated into pCDNA3.1 (Thermo Fisher Life Technologies, Carlsbad CA). HLA-A*02:01 S88A (Δ glycan) was produced by site-directed mutagenesis (Quick Change Lightening, Agilent, Santa Clara CA).
Antibodies and peptides
FITC conjugated anti-CD8 was purchased from BD Biosciences (San Jose, CA). Unlabeled peptides at ≥98% purity were prepared by chemical synthesis (Genscript, Piscataway, USA). Sequences are documented in Supplementary Table S1.
Cells and culture conditions for transient production of protein complexes
CHO-S cells obtained from Thermo Fisher, Life Technologies, (Carlsbad, CA) were propagated and transfected as previously described (Byrne et al., 2018) with the exception that we used a 1:1 molar ratio of MHC single chain: TAPBPR expression plasmids.
Purification of native MHC-I/TAPBPR complexes
Culture media was harvested and precleared by centrifugation at 250 xg for 10 min before adjusting to 25 mM Tris pH 8, 1 mM ethylenediaminetetraacetic acid (EDTA) and adding 27 mg/l of avidin. The media was filtered (0.22 μm) and affinity purified on a StrepTrap HP affinity column (GE Healthcare, Chicago IL). Bound protein was washed with 10 column volumes of wash buffer (25 mM Tris pH 8, 100 mM NaCl, 1 mM EDTA) and eluted with 5 mM desthiobiotin/wash buffer. Leucine zippers were removed by a 2-h digestion at room temperature with Tobacco Etch Virus (TEV) protease in 25 mM Tris pH 8, 100 mM NaCl, 1 mM EDTA, 3 mM/0.3 mM glutathione redox buffer. Complex was purified by size exclusion gel filtration (SEC) at room temperature on a Superdex 200 10/300 increase column (GE Healthcare, Chicago IL) at a flow rate of 0.5 ml/min in 50 mM Tris pH 7.5 buffer containing 100 mM NaCl. Protein concentrations were determined using A280 measurements on a NanoDrop spectrophotometer.
Native gel-shift assay of peptide binding to MHC-I/TAPBPR
Peptide-deficient MHC-I/TAPBPR complexes were incubated with a 10-molar excess of high affinity or nonbinding peptide overnight at 4°C, pH 7.2 in phosphate buffer with 150 mM NaCl. Samples were electrophoresed at 90 V on a 12% polyacrylamide gel in 25 mM Tris pH 8.8, 192 mM glycine, at 4°C for 5 h, and developed using InstantBlue (Expedeon San Diego, CA). Gels were imaged using an Innotech FluoChem2 system (Genetic Technologies Grover, MO).
PNGase F digestion assay
Five microgram aliquots of purified HLA-A*02:01 and HLA-A*02:01 S88A (ΔN86 glycan) TAPBPR complex were denatured then either treated with PNGase F (NEB, Ipswich, MA) or incubated in glycosidase buffer alone at 37°C for 1 h, then reduced with DTT and electrophoresed.
Glycan mapping by LC-MS/MS N-Glycan analysis
All materials were purchased from Millipore-Sigma unless otherwise noted. Purified MHC-I (20 μg) was buffer exchanged into 50 mM ammonium bicarbonate (pH 8.0) and incubated at 90°C for 5 min. Following trypsin digestion and reduction, the sample was iodoacetamide treated. Glycopeptides were then enriched using the ProteoExtract Glycopeptide Enrichment Kit according to the manufacturing guidelines, and lyophilized before resuspension in 20 μl of 5% acetonitrile and 0.1% trifluoroacetic acid in ddH2O. Glycopeptides (5 μl) were injected on to a 75 mm × 20 cm column packed with C18 Zorbax resin equipped using a Thermo Scientific EASY-nLC 1200 nanopump. Analytes were eluted with a linear gradient of increasing acetonitrile and injected into a Q Exactive hybrid quadrupole mass spectrometer (Thermo Scientific). MS2 spectra for intense ions were collected with stepped NCE energies of 15, 25 and 35 eV. The 10 most abundant N-glycopeptides, based on spectra counts, were annotated using GlycoWorkbench Version 2.1.
MHC-I tetramer formation
MHC-I molecules were biotinylated using biotin ligase (BirA) (Avidity.com, Co.). Tetramerization of empty-MHC-I/TAPBPR was performed by adding a 4:1:1 molar ratio of biotinylated MHC-I/TAPBPR to streptavidin-PE or streptavidin-APC (Prozyme Hayward, CA) in five additions over 1 h on ice. Peptide-deficient MHC-I/TAPBPR tetramers were then exchanged with peptides of interest by adding a 20-molar excess of peptide and incubating for 1 h. A solution of 8 M biotin (to block any free streptavidin sites) was added and incubated for a further 1 h at room temperature. After exchange, tetramers were transferred to 100 kDa spin columns (Amicon, Millipore, Burlington, Ma) and washed with 1000 volumes of PBS to remove TAPBPR and excess peptide. For T-cell receptor staining comparison, conventionally produced MHC-I molecules were biotinylated and assembled onto streptavidin-PE.
Cells and culture conditions for FACS
We used a Jurkat-MA (DMF5) T cell line which expresses the Melan-A receptor, and the NY-ESO-1 line which expresses the NY-ESO-1 antigen, and standard tetramer staining protocols (Overall et al., 2020). Flow cytometry protocols, including gating strategies, are shown in Supplementary Figure S3. Tetramer analysis of cell lines was carried out by staining 2 × 105 DMF5 cells, with an anti-CD8α FITC conjugated mAb (BD Biosciences) and 1 μg/ml of either conventionally refolded PE-HLA-A*02:01/MART-1 tetramer, CHO-derived PE-HLA-A*02:01 tetramer loaded with MART-1 peptide, CHO-derived PE-HLA-A*02:01 tetramer loaded with the neo-antigen NY-ESO-1, or left peptide-free and incubated for 1 h on ice. Cells were washed twice with 30 volumes of FACS buffer (PBS, 1% BSA, 2 mM EDTA) before analysis, and gated by forward and side scattering properties. All experiments were performed using an LSRII (BD) and data analysis performed using FACSDiva (BD) and FlowJo (Tree Star, Ashland, OR). Cells tested negative for mycoplasma using the universal mycoplasma test kit (ATCC).
Results
Generating properly conformed MHC-I molecules in CHO cells
An overview of our technology and its applications is outlined in Fig. 1, with the full arrangement of the MHC-I and TAPBPR transgenes engineered to co-express a leucine zippered MHC-I/TAPBPR complex shown in Fig. 2A. First, we constructed a cytomegalovirus (CMV) expression cassette, which incorporated the endogenous human secretory β2m, signal-peptide and coding sequences linked via a (GGGS)4 spacer to the ectodomain of human allele HLA-A*02:01 heavy chain (Hansen et al., 2010). The gene was further engineered to express an AVITAG sequence for in vitro biotinylation (Fairhead and Howarth, 2015), and an FOS leucine coil motif (Kouzarides and Ziff, 1988). The TAPBPR expression cassette similarly comprised of a secretory peptide, the TAPBPR ectodomain (with a single C94A mutation to prohibit oligomerization and aggregation at high protein concentration due to free thiol groups), a tag for affinity purification, and a JUN leucine-coil motif. All sequences included a Tobacco Etch Virus (TEV) protease site for leucine zipper removal. Co-expression of the zippered single-chain and TAPBPR proteins permitted folding of the β2m and heavy chain domains. The chaperoned MHC-I complex was secreted and purified directly from conditioned media. As a proof of concept, pilot studies were small scale with no media optimization (100 ml volume resulting in 10–30 mg/l of zippered complex per batch) but the process is highly scalable (Steger et al., 2015). After TEV digestion, the complex remained stable during preparative chromatography (Fig. 2B), and the individual MHC-I and TAPBPR components were resolved using SDS-PAGE as discrete protein bands with apparent molecular mass 51 and 43.5 kDa, respectively (Fig. 2C). While TAPBPR is known to form a 1:1 stoichiometric complex with MHC molecules (Morozov et al., 2016; McShan et al., 2018), it appeared as a more intense band than HLA molecules, due to higher staining efficiency using Coomassie protein stain. The CHO-derived MHC-I was fully glycosylated, as shown by PNGaseF cleavage of all N-linked oligosaccharides from the wildtype molecule resulting in a distinct band shift, but not from the S88A MHC-I mutant, lacking the N-X-S/T glycosylation motif (Yan and Lennarz, 2005) (Fig. 2D).
Fig. 1.
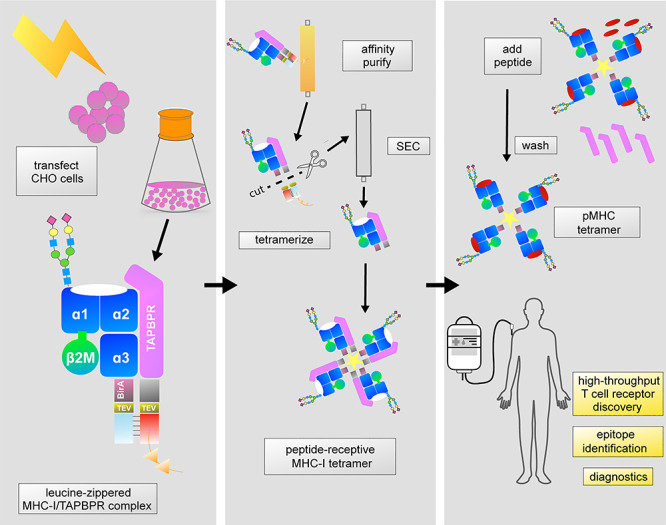
Generation of soluble, peptide-receptive MHC-I molecules in mammalian cells. MHC-I/TAPBPR complexes assembled using leucine zippers are isolated directly from conditioned media via an affinity tag on the TAPBPR protein. After zipper digestion and size exclusion chromatography, MHC-I can be biotinylated and tetramerized while still complexed with TAPBPR. High-affinity peptides displace TAPBPR, releasing functional pMHC-I molecules toward a range of research, diagnostic and therapeutic applications.
Fig. 2.
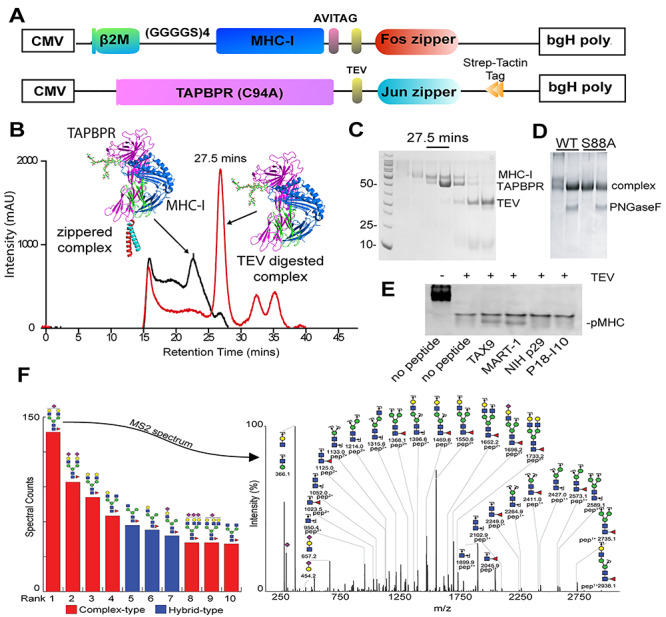
Design and biochemical analysis of native MHC-I/TAPBPR complexes.
(A) CMV expression-cassette organization. (B) Chromatography of zippered (black line) and TEV-digested (red line) HLA-A*02:01/TAPBPR complexes on a S200 16/300 increase column (50 mM Tris, 100 mM, NaCl, running at a flow rate of 0.5 ml/min). (C) Elution of unzippered complex (red line) at 27.5 min was confirmed by SDS PAGE. While the protein peak at 27.5 min is consistent with a 1:1 stoichiometric protein complex, TAPBPR has a significantly higher staining efficiency on Coomassie protein gels. (D) PNGaseF-digested wild-type HLA-A02:01 and HLA-A02:01Δglycan (S88A) TAPBPR complex migration on a 12% SDS PAGE gel. (E) Native gel shift electrophoresis of HLA-A*02:01/TAPBPR complexes, showing pMHC-I formation in the presence of high-affinity peptides TAX9, MART-1 relative to the nonbinding peptides NIH p29 and P18-I10. Gels were stained using InstantBlue (Expedeon). (F) Mass spectroscopy of recombinant MHC-I (HLA-A*02:01) with predominant N-glycan species ranked by spectral counts. MS2 spectrum of the predominant N86 glycopeptide indicating charge, composition and m/z for the individual fragment ions. Individual carbohydrate residues are shown according to the SNFG convention, with red triangles indicating fucose residues.
We further confirmed that the chaperoned MHC-I protein was peptide-receptive using an electrophoretic mobility shift assay on a nondenaturing (native) gel (Morozov et al., 2016). In this assay, incubation of the cleaved complex with 10-fold molar excess of two high-affinity peptides (TAX9 or MART-1) led to the formation of discrete bands corresponding to properly conformed pMHC species of slightly different electrophoretic mobilities (Fig. 2E, lanes 3 and 4). In contrast, incubation with buffer or two nonspecific peptides produced no distinguishable pMHC band (Fig. 2E, lanes 2, 5 and 6), while the leucine-zippered complex barely entered the gel (Fig. 2E, lane 1). Following protein purification using preparative size exclusion chromatography, the cleaved complex was snap frozen and stored at −80°C for up to several months, while remaining peptide-receptive.
Complex-type glycosylation patterns of MHC-I molecules
Glycans play important roles in the immune response by affecting folding, multimerization, trafficking, cell surface stability and half-life of both antigens and their receptors (Baum and Cobb, 2017). Despite the highly polymorphic nature of HLA-A, HLA-B and HLA-C alleles, all class-I molecules share a conserved glycan at Asn 86, and the oligosaccharide structures that predominate appear to be highly processed, biantennary N-linked-oligosaccharides (Parham et al., 1977). Mass-spectroscopy confirmed that proteolysis of CHO-derived recombinant MHC-I resulted in isolation of a single N-glycosylation site at N86 within a unique 15-residue fragment. Peaks corresponding to this peptide revealed high intensities in both the MS1 and MS2 dimensions following analysis by ESI-MS/MS (Fig. 2F). We observed a predominant peak in the MS2 spectra of all N86 glycopeptide species corresponding to peptide GYYNQSEAGSHTVQR from MHC-I, plus a single N-acetylhexosamine residue in MS2 spectra. The first and second most abundant species of glycan detected were highly processed, biantennary di-sialylated N86-glycans containing fucose and one or two terminal N-acetylneuraminic acid residues, respectively. This result is similar to those reported using primary and immortalized human cells (Barber et al., 1996), suggesting that the CHO-expressed MHC-I molecules recapitulate functionally critical glycan modifications.
We modeled a biantennary glycan bearing two nonreducing terminal sialic acid residues on the HLA-A*02:01/MART-1 X-ray structure (Supplementary Figure S1) (Sliz et al., 2001). The glycan moiety occupies a region adjacent to the F-pocket of the peptide binding groove, and overlaps with the Bw4 epitope recognized by the KIR3DL1 NK receptor, consistent with reported effects of glycan modifications on NK cell function (Salzberger et al., 2015). Finally, we considered potential long-range effects of the charged glycan on pMHC-I surface chemistry, by calculating the electrostatic potential using CHARMM (Jo et al., 2008). Display of the potential features on the X-ray structure of HLA-A*02:01 reveals a significant effect on the characteristics of the molecular surface that is displayed to TCRs, suggesting the utility of glycosylated pMHC-I proteins as molecular probes of functionally relevant interactions with T cells (Supplementary Figure S1).
Screening peptide binding on CHO-derived MHC-I/TAPBPR complexes
CHO-derived HLA-A*02:01/TAPBPR complexes can be loaded with high-affinity TAX9 and MART-1 peptides (Fig. 2D). To test if other MHC-I allotypes might be assembled and isolated using our system, we engineered and expressed a single-chain HLA-A*68:02/TAPBPR complex, and assayed it for pMHC-I formation in parallel with HLA-A*02:01 (Fig. 3). The two alleles have considerable sequence homology in the peptide-binding groove formed by the ⍺1/⍺2 domains, resulting in a shared preference for a Val, Leu, Ala or Ile at the peptide anchor position 9, but marked variation at anchor position 2 where HLA-A*02:01 has a preference for Leu, while HLA-A*68:02 prefers Thr or Val (Fig. 3A). Guided by NetMHCpan (Jurtz et al., 2017), we selected a panel of high- to low-affinity peptides for HLA-A*02:01 and HLA-A*68:02, and directly probed pMHC-I formation using our system (Supplementary Table S1). Structure-based modeling (Nerli and Sgourakis, 2020) confirmed A-pocket hydrogen bonding interactions between peptide anchor position L2 with the K66 sidechain, Y7 with Y99 from HLA-A*02:01, and anchor position T2 with N66 and N63 from HLA-A*68:02 (Fig. 3B). According to our electrophoretic mobility shift assay, all predicted high-affinity peptides bound to HLA-A*02:01 and HLA-A*68:02, resulting in discrete pMHC-I protein bands (Fig. 3C). Our results demonstrate that the HLA-A*68:02/TAPBPR complex is stable and peptide-receptive, and that differences in peptide-binding preferences can be recapitulated using our chaperone-based system, suggesting that the isolated MHC-I/TAPBPR complexes exhibit a properly conformed peptide-binding groove. The single, unexpected result was significant binding of peptide GLLGIGILTV on HLA-A*68:02, despite the high μM predicted affinity by NetMHCpan (Jurtz et al., 2017). Structural modeling of the GLLGIGILTV/HLA-A*68:02 complex (Fig. 3B—right panel) suggested the potential for stabilizing interactions at both peptide anchor positions, with leucine (L2) capable of hydrogen-bonding with N63 and N66. We further expanded our results to demonstrate peptide-receptive HLA-A*24:02/TAPBPR complex formation (Supplementary Figure S2), suggesting the utility of our system for producing native pMHC-I molecules for different HLA alleles. Previous work has established that TAPBPR has a restricted specificity range toward murine and human MHC-I allotypes (Morozov et al., 2016), with a preference toward HLA proteins of the A*02, A*24 and A*28 serotypes (Ilca et al., 2019a). The specificity of MHC-I/TAPBPR interactions is fine-tuned by individual amino acid polymorphisms along the MHC-I peptide binding groove, particularly polar interaction patterns at the F-pocket (Ilca et al., 2019a), and at the heavy chain/β2m interface (Overall et al., 2020), which ultimately define a network of correlated dynamic motions connecting the primary interaction surfaces with TAPBPR (McShan et al., 2018, 2019), and mediating transitions between different empty and peptide-bound states (Anjanappa et al., 2020).
Fig. 3.
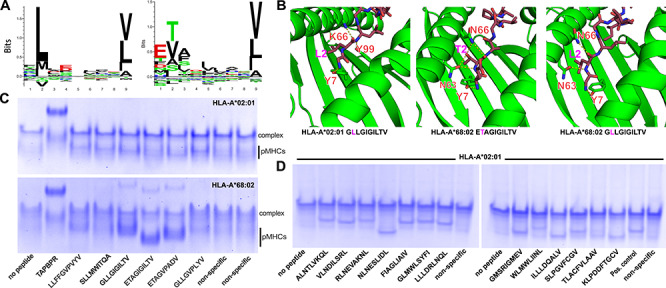
Specificity of peptide loading on CHO-derived HLA-A*02:01 and HLA-A*68:02. (A) Sequence logos of 9 mer epitopic peptides (IEDB database https://www.iedb.org), ranked by frequency (Seq2logo https://doi.org/10.1093/bioinformatics/btp033). (B) Structure models of peptides GLLGIGILTV, ETAGIGILTV and GLLGIGILTV bound to HLA-A*02:01 and HLA-A*68:02. We used a comparative modeling approach in the program Rosetta, with templates obtained from X-ray structures with PDB accession codes 4JFO, 3MRK and 4JFF, respectively (Nerli and Sgourakis, 2020). The dashed yellow lines indicate polar contacts. (C) Native gel shift assay showing pMHC-I formation in the presence of a panel of peptides with predicted affinities for HLA-A*02:01 and HLA-A*68:02 shown in Supplementary Table S1. Nonspecific peptides are YPNVNIHNF and RGPGRAFVTI, respectively (D) Same assay as in (C) showing peptide binding on HLA-A*02:01 for a panel of SARS-CoV-2 epitopic peptides predicted using a structure-based algorithm (Nerli and Sgourakis, 2020) (Supplementary Table S1). As a positive control (pos. control), we used the confirmed epitopic peptide NLVPMVATV from CMV, and as a negative control a nonspecific peptide with sequence YPNVNIHNF (NIH p29). Native gels (12% polyacrylamide) were electrophoresed at 90 V for 5 h at 4°C before visualization with InstantBlue (Expedeon).
We demonstrated peptide/MHC-I complex assembly for a panel of HLA-A*02:01-restricted epitopic peptides predicted from the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome using a structure-guided method developed in our group (Nerli and Sgourakis, 2020) (Supplementary Table S2). Peptides were incubated with MHC-I/TAPBPR complex then analyzed for binding using our electrophoretic mobility shift assay. Of 13 predicted peptides, 11 produced strong pMHC-I bands, while peptides TLACFVLAAV and WLMWLIINL had little or no binding. The specificity of peptide binding was reflected by the observed electrophoretic mobilities of the resulting pMHC-I species, which generally correlate with the overall charges and hydrodynamic radii of the resulting protein complexes (Fig. 3D). According to our native gel analysis (Figs 2E and 3C, D), the efficiency of peptide loading on purified MHC-I/TAPBPR complexes was ~50%, however, this estimate is likely impacted by differences in Coomassie staining efficiency between the TAPBPR and MHC-I components of the complex. Maximal peptide loading on all HLA/TAPBPR complexes and reconstitution of functional pHLA-I molecules was achieved using 10-fold molar excess by overnight incubation at 4°C.
Antigen-specific T cell recognition of native peptide/HLA-A*02:01 antigens
CHO-derived MHC-I/TAPBPR complexes may be readily multimerized via a streptavidin fluorophore conjugate (Altman et al., 1996). TAPBPR-promoted peptide loading can be then utilized to generate pMHC-I tetramers of desired peptide specificities. We investigated the efficiency of antigen-specific staining of a human lymphocyte line (DMF5) transduced with a TCR (Melan-A) specific for the melanoma-associated MART-1 peptide (Johnson et al., 2006). We incubated DMF5 cells with phycoerythrin (PE) labeled MART-1/MHC-I tetramers prepared using either (i) in vitro refolded pMHC-I (Garboczi et al., 1992) (used as a positive control) vs (ii) empty MHC/TAPBPR complexes or (iii) TAPBPR-exchanged pMHC-I molecules loaded with the heteroclitic MART-1 peptide or (iv) a different antigenic peptide, NY-ESO-1, corresponding to the cancer-testis antigen 1B (Gnjatic et al., 2006). To demonstrate antigen/receptor-specific tetramer staining, a complementary set of flow cytometry experiments was performed using a NY-ESO-1 specific T cell line (Bethune et al., 2018). Flow cytometry (Fig. 4 and Supplementary Figure S3) showed that CHO-derived pMHC-I tetramers behaved in a manner comparable to tetramers generated from refolded proteins, specifically staining cells expressing the cognate but not the irrelevant TCRs, for both peptides.
Fig. 4.
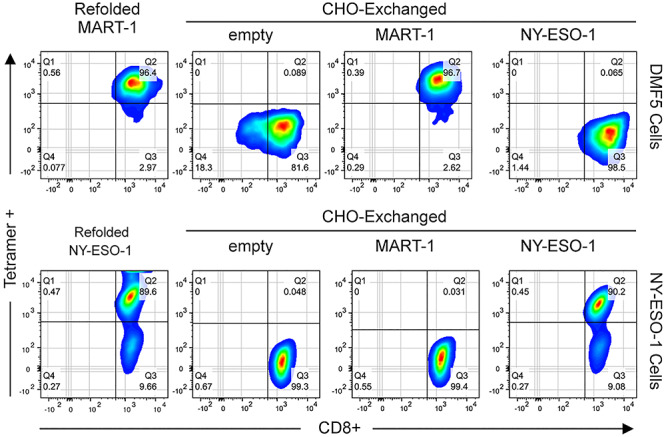
Exchanged HLA-A*02:01 tumor antigens are recognized by their cognate TCRs. Top panel: DMF5+ cells were examined by flow cytometry following staining with 1 μg/ml HLA-A*02:01 tetramers plus anti-CD8 FITC antibody (BD). Tetramers were prepared using (i) in vitro refolded HLA-A*02:01/MART-1, (ii) empty HLA-A*02:01/TAPBPR complexes and (iii) TAPBPR-exchanged HLA-A*02:01 with the specific MART-1 or (iv) nonspecific NY-ESO-1 peptide. Bottom panel: the complementary experiment using T cells expressing a NY-ESO-1 specific receptor. The ~9% tetramer-negative population of NY-ESO-1 cells, seen using both refolded and exchanged tetramers, corresponds to cells that have reduced expression levels of the αβ TCR during culture. All flow cytometric analysis was performed using a BD LSR II instrument equipped with FACSDiva software (BD Biosciences) and FlowJo software (Ashland Oregon). Gating strategy is shown in Supplementary Figure S3.
Discussion
Murine MHC-I molecules engineered as single-chain constructs with a covalently linked peptide were first expressed in mammalian cells (Mage et al., 1992) and are reported to stimulate both antigen-specific B and T cells (Yu et al., 2002). Mammalian expression systems for human HLA antigens with the potential for immune stimulation include both single-chain constructs (Jurewicz et al., 2019), and pMHC-IgG fusions (Wooster et al., 2019), but significant challenges remain with respect to loading such molecules with high-affinity epitopic peptides of choice. Here, we describe systems and methods to produce soluble, peptide-receptive human MHC-I proteins in the biopharmaceutical standard Chinse Hamster Ovary line, suitable for generating natively folded and glycosylated pMHC-I with controlled peptide specificities. Our results demonstrate reconstitution of three commonly occurring human HLA allotypes (HLA-A*02:01, HLA-A*68:02 and HLA-A*24:02) as fully functional, properly conformed pMHC-I molecules. Our system is highly modular, and can be readily adapted to incorporate different HLA allotypes, and affinity purification tags, including the SpyTag-SpyCatcher technology (Reddington and Howarth, 2015). We confirm that antigen-specific staining of T cells with CHO-derived pMHC-I tetramers encompassing known A*02-restricted tumor antigens is comparable to that of refolded pMHC-I molecules, while allowing for a significantly more convenient process which includes functionally important post translational modifications. Finally, we demonstrate an application of our platform to assay the specificity of several predicted epitopic peptides from the SARS-CoV-2 genome against the common allotype HLA-A*02:01, which provides a convenient approach to experimentally validate peptide/HLA binding. Combined with previously described multiplexed pMHC-I multimer (Bentzen and Hadrup, 2017; Overall et al., 2020) and nanoparticle methods (Ichikawa et al., 2020), our system can be leveraged for the development of antigen libraries to monitor and expand polyclonal T cell specificities in various research and clinical settings.
Data Availability
The transgene sequences for single-chain HLA and TAPBPR expression constructs are available on GenBank, under accession numbers MN692667, MN692668, MN692669 and MN419959.
Author Contributions
S.M.O., N.G.S. and A.W.B. designed research. S.M.O. and G.I.M. performed cell culture, protein purification, peptide binding, MHC tetramerization and flow cytometry experiments. J.T.R. and A.W.B. performed glycan mapping analysis. S.M.O. and N.G.S. wrote the manuscript, with feedback from all authors.
Supplementary Material
Acknowledgments
We are grateful to Bari-Holmes-Nazario and the IBSC Flow Cytometry Facility, to Phillip Berman for access to protein production facilities in the UCSC Department of Bioengineering (supported through NIH grant R01AI113893), and to Andrew McShan and Santrupti Nerli for help with figure preparation. This research was supported by grants from NIAID [R01AI143997], NIGMS [R35GM125034] to N.G.S. and from NIGMS [R01GM115489] to A.W.B.
Author Notes: Edited by Eva Petersen
Conflict of interest
S.M.O. and N.G.S. are listed as co-inventors in a Provisional US Patent Application related to this work.
References
- Altman J.D., Moss P.A., Goulder P.J. et al. (1996) Science, 274, 94–96. [DOI] [PubMed] [Google Scholar]
- Anjanappa R., Garcia-Alai M., Kopicki J.D. et al. (2020) Nat. Commun., 11, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A.H., Hoppes R., Linnemann C. et al. (2008) Proc. Natl. Acad. Sci. U. S. A., 105, 3825–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber L.D., Patel T.P., Percival L. et al. (1996) J. Immunol., 156, 3275–3284. [PubMed] [Google Scholar]
- Baum L.G. and Cobb B.A. (2017) Glycobiology, 27, 619–624. [DOI] [PubMed] [Google Scholar]
- Bentzen A.K. and Hadrup S.R. (2017) Cancer Immunol. Immunother., 66, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethune M.T., Li X.H., Yu J. et al. (2018) Proc. Natl. Acad. Sci. U. S. A., 115, E10702–E10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum J.S., Wearsch P.A. and Cresswell P. (2013) Annu. Rev. Immunol., 31, 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle L.H., Hermann C., Boname J.M. et al. (2013) Proc. Natl. Acad. Sci. U. S. A., 110, 3465–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne G., O’Rourke S.M., Alexander D.L. et al. (2018) PLOS Biol., 16, e2005817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. and Bouvier M. (2007) EMBO J., 26, 1681–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresswell P., Bangia N., Dick T. et al. (1999) Immunol. Rev., 172, 21–28. [DOI] [PubMed] [Google Scholar]
- Fairhead M. and Howarth M. (2015) Methods Mol. Biol. Clifton NJ, 1266, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garboczi D.N., Hung D.T. and Wiley D.C. (1992) Proc. Natl. Acad. Sci., 89, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain R.N. and Margulies D.H. (1993) Annu. Rev. Immunol., 11, 403–450. [DOI] [PubMed] [Google Scholar]
- Gnjatic S., Nishikawa H., Jungbluth A.A. et al. (2006) Adv. Cancer Res., 95, 1–30. [DOI] [PubMed] [Google Scholar]
- Hadrup S.R. and Schumacher T.N. (2010) Cancer Immunol. Immunother., 59, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.H., Connolly J.M., Gould K.G. et al. (2010) Trends Immunol., 31, 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann C., Hateren A., van Trautwein N. et al. (2015a) eLife, 4, e09617.26439010 [Google Scholar]
- Hermann C., Trowsdale J. and Boyle L.H. (2015b) Tissue Antigens, 85, 155–166. [DOI] [PubMed] [Google Scholar]
- Ichikawa J., Yoshida T., Isser A. et al. (2020) Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res., 26, 3384–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilca F.T., Drexhage L.Z., Brewin G. et al. (2019a) Cell Rep., 29, 1621–1632e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilca F.T., Neerincx A., Wills M.R., et al. (2018) Proc. Natl. Acad. Sci. U. S. A., 115, E9353–E9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Natarajan K., Boyd L.F. et al. (2017) Science, 358, 1064–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Natarajan K. and Margulies D.H. (2019) Adv. Exp. Med. Biol., 1172, 21–62. [DOI] [PubMed] [Google Scholar]
- Jo S., Kim T., Iyer V.G. et al. (2008) J. Comput. Chem., 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Johnson L.A., Heemskerk B., Powell D.J. et al. (2006) J. Immunol., 177, 6548–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurewicz M.M., Willis R.A., Ramachandiran V. et al. (2019) Anal. Biochem., 584, 113328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V., Paul S., Andreatta M. et al. (2017) J. Immunol., 199, 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. and Ziff E. (1988) Nature, 336, 646–651. [DOI] [PubMed] [Google Scholar]
- Luimstra J.J., Garstka M.A., Roex M.C.J. et al. (2018) J. Exp. Med., 215, 1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mage M.G., Lee L., Ribaudo R.K. et al. (1992) Proc. Natl. Acad. Sci. U. S. A., 89, 10658–10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShan A.C., Devlin C.A., Overall S.A. et al. (2019) Proc. Natl. Acad. Sci. U. S. A., 116, 25602–25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShan A.C., Natarajan K., Kumirov V.K. et al. (2018) Nat. Chem. Biol., 14, 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz A., Anjanappa R., Wagner C. et al. (2019) Sci. Immunol., 4, eaav0860. [DOI] [PubMed] [Google Scholar]
- Morozov G.I., Zhao H., Mage M.G. et al. (2016) Proc. Natl. Acad. Sci. U. S. A., 113, E1006–E1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neerincx A., Hermann C., Antrobus R. et al. (2017) eLife, 6, e23049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerli S. and Sgourakis N.G. (2020) bioRxiv, 2020.03.23.004176. [Google Scholar]
- Overall S.A., Toor J.S., Hao S. et al. (2020) Nat. Commun., 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham P., Alpert B.N., Orr H.T. et al. (1977) J. Biol. Chem., 252, 7555–7567. [PubMed] [Google Scholar]
- Reddington S.C. and Howarth M. (2015) Curr. Opin. Chem. Biol., 29, 94–99. [DOI] [PubMed] [Google Scholar]
- Rossjohn J., Gras S., Miles J.J. et al. (2015) Annu. Rev. Immunol., 33, 169–200. [DOI] [PubMed] [Google Scholar]
- Saini S.K., Schuster H., Ramnarayan V.R. et al. (2015) Proc. Natl. Acad. Sci. U. S. A., 112, 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberger W., Garcia-Beltran W.F., Dugan H. et al. (2015) PLOS ONE, 10, e0145324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittnaegel M., Hoffmann E., Imhof-Jung S. et al. (2016) Mol. Cancer Ther., 15, 2130–2142. [DOI] [PubMed] [Google Scholar]
- Sliz P., Michielin O., Cerottini J.C. et al. (2001) J. Immunol., 167, 3276–3284. [DOI] [PubMed] [Google Scholar]
- Steger K., Brady J., Wang W. et al. (2015) J. Biomol. Screen., 20, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. and Tampé R. (2017) Science, 358, 1060–1064. [DOI] [PubMed] [Google Scholar]
- Wearsch P.A., Peaper D.R. and Cresswell P. (2011) Proc. Natl. Acad. Sci. U. S. A., 108, 4950–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooster A.L., Anderson T.S. and Lowe D.B. (2019) J. Immunol. Methods, 464, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan A. and Lennarz W.J. (2005) J. Biol. Chem., 280, 3121–3124. [DOI] [PubMed] [Google Scholar]
- Yu Y.Y.L., Netuschil N., Lybarger L. et al. (2002) J. Immunol. Baltim. Md 1950, 168, 3145–3149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The transgene sequences for single-chain HLA and TAPBPR expression constructs are available on GenBank, under accession numbers MN692667, MN692668, MN692669 and MN419959.