

Abstract
Introduction.
The emergence of SARS-CoV-2 has taken humanity off guard. Following an outbreak of SARS-CoV in 2002, and MERS-CoV about 10 years later, SARS-CoV-2 is the third coronavirus in less than 20 years to cross the species barrier and start spreading by human-to-human transmission. It is the most infectious of the three, currently causing the COVID-19 pandemic. No treatment has been approved for COVID-19. We previously proposed targets that can serve as binding sites for antiviral drugs for multiple coronaviruses, and here we set out to find current drugs that can be repurposed as COVID-19 therapeutics.
Aim.
To identify drugs against COVID-19, we performed an in silico virtual screen with the US Food and Drug Administration (FDA)-approved drugs targeting the RNA-dependent RNA polymerase (RdRP), a critical enzyme for coronavirus replication.
Methodology.
Initially, no RdRP structure of SARS-CoV-2 was available. We performed basic sequence and structural analysis to determine if RdRP from SARS-CoV was a suitable replacement. We performed molecular dynamics simulations to generate multiple starting conformations that were used for the in silico virtual screen. During this work, a structure of RdRP from SARS-CoV-2 became available and was also included in the in silico virtual screen.
Results.
The virtual screen identified several drugs predicted to bind in the conserved RNA tunnel of RdRP, where many of the proposed targets were located. Among these candidates, quinupristin is particularly interesting because it is expected to bind across the RNA tunnel, blocking access from both sides and suggesting that it has the potential to arrest viral replication by preventing viral RNA synthesis. Quinupristin is an antibiotic that has been in clinical use for two decades and is known to cause relatively minor side effects.
Conclusion.
Quinupristin represents a potential anti-SARS-CoV-2 therapeutic. At present, we have no evidence that this drug is effective against SARS-CoV-2 but expect that the biomedical community will expeditiously follow up on our in silico findings.
Keywords: sequence analysis, protein evolution, broadly neutralizing antivirals, docking
Introduction
Humanity is facing a global pandemic caused by a previously unknown coronavirus that recently crossed the species barrier [1] and is spreading by human-to-human transmission [2]. This novel coronavirus, SARS-CoV-2 (also known as 2019-nCoV), is a close relative of SARS-CoV [3], which caused an outbreak in 2002–2003 [4]. SARS-CoV had a high mortality rate of 10 %, but was limited to about 8000 cases in total [4]. MERS-CoV, another coronavirus that started an outbreak in 2012, has so far resulted in >2500 cases with an even higher mortality rate of ~35 % [5]. SARS-CoV-2, the cause of the ongoing COVID-19 pandemic, has proved to be the most infectious of these coronaviruses. As of 15 April 2020, it has caused over 2 million known infections and >130 000 deaths worldwide [6]. No vaccine or antiviral treatment for COVID-19 exists.
The positive-strand RNA genome of a coronavirus such as SARS-CoV and MERS-CoV encodes ~25 different protein products [7]. Four of these proteins have a structural function and assemble to form the capsid that encapsulates the viral genome. Upon infection, the viral RNA genome is released into a host cell, and by using the host’s translational machinery, the viral proteins are expressed. Several of these non-structural proteins (NSPs) form the replicase–transcriptase complex that works to replicate the RNA genome. Key proteins in the replicase–transcriptase complex are the RNA-dependent RNA polymerase (RdRP), the helicase and the exonuclease [8]. New copies of the viral genome and more structural proteins pack into new virions that are eventually released from the cell, on task to infect more cells. If the key proteins in the replicase–transcriptase complex can be prevented from replicating the RNA genome, new virions will not form. Recently we identified regions in the key proteins in MERS-CoV and SARS-CoV that could serve as targets for antiviral drugs [7]. We took an evolutionary approach to identify regions of at least five consecutive sites not just conserved in sequence but also in structural properties with low conformational flexibility to reduce the risk of a target being displayed for only a fraction of the time [7]. These targets represent potential drug-binding sites located in protein regions crucial for viral fitness and, therefore, conserved on long evolutionary time scales. Our study identified two to four such targets in the protease (NSP5), replicase (NSP7), helicase (NSP13) and exonuclease (NSP14), with nine targets identified in the RdRP (NSP12) [7].
In this work, we included SARS-CoV-2 in our analysis. We generated a conformational ensemble based on the 3D structure of the RdRP from SARS-CoV (PDB ID: 6NUR [9]), which is closely related and highly similar to the RdRP from SARS-CoV-2. The conformational ensemble was used for a virtual screen of the US Food and Drug Administration (FDA)-approved drugs against the conserved targets in RdRP. As the RdRP structure of SARS-CoV-2 just became available, we performed a second round of virtual screens with the FDA-approved drugs against the recently resolved RdRP conformation (PDB ID: 6M71 [10]). Altogether, these different screens revealed several drugs that are expected to inhibit RdRP catalytic activity. These drugs can, therefore, be repurposed as anti-COVID-19 therapeutics. Among the drug candidates, quinupristin, an intravenous antibiotic that has been in clinical practice against severe bacterial infections for a couple of decades and has a rather limited scope of side effects [11], appears to be particularly promising. Quinupristin is predicted to bind across the RNA tunnel of RdRP, suggesting that it may be able to inhibit viral RNA synthesis and thus block SARS-CoV-2 replication.
Methods
Phylogenetic analysis
A multiple sequence alignment of RdRP from SARS-CoV-2 (accession number: YP_009724389.1, range 4398–5324) and the RdRP sequences previously used to determine the target motifs [7] was constructed using mafft with L+INS+ i in Jalview [12]. The multiple sequence alignment was used to build a phylogenetic tree using PhyML with 100 bootstraps [13]. The best model of evolution for the dataset was determined by smart model selection [14]. The resulting phylogeny was rooted at mid-point.
SARS-CoV-2 conservation
To determine the level of conservation of the proteins with target motifs, blastp searches with the regions from the SARS-CoV orf1ab polyprotein that represent NSP5 (protease), NSP7 (replicase), NSP12 (RdRP), NSP13 (helicase) and NSP14 (exonuclease) from [7] against SARS-CoV-2 in the nr database were performed, resulting in five sequence datasets. Each dataset was aligned using muscle [15] at EBI [16], resulting in a multiple sequence alignment (MSA) and its corresponding percentage identity matrix. The percentage identity matrices showed the overall conservation across all SARS-CoV-2 sequences in each dataset. The conservation of each target motif was extracted from the MSAs through visualization in Jalview [12].
Target surface accessibility
The surface accessibility of the target motifs was determined using PyMol v. 1.8.4 [17] with the PyMol script FindSurfaceResidues.py [18] (default settings) from existing protein structures of SARS-CoV. The structures with PDB IDs 1UJ1 [19], 6NUR [9], 6JYT [20] and 5C8S [21] were used to represent NSP5 (protease), NSP7 (replicase) and NSP12 (RdRP), NSP13 (helicase) and NSP14 (exonuclease), respectively.
Protein sequence and structure analysis
A blastp search with the SEQRES part of RdRP from SARS-CoV in PDB ID 6NUR [9] was performed against SARS-CoV-2 in the Refseq_protein database to determine the sequence similarity between SARS-CoV-2 and the actual part of RdRP from SARS-CoV that was included in the PDB structure. The differing sites were visualized in their structural context based on PDB ID 6NUR.
Virtual screening of FDA approved drugs
Molecular dynamics simulations and preparation of protein conformations
The cryo-EM structure of SARS-CoV NSP12 (RdRP) bound to the NSP7 and NSP8 co-factors was taken from the Protein Data Bank (PDB ID: 6NUR) [9]. To incorporate the inherent flexibility, protein conformations were generated with all-atom molecular dynamics (MD) simulation. The system was prepared using the CHARMM-GUI web interface [22]. Standard MD simulation procedure was followed [23]. The protein was solvated with TIP3 water in a cubic box and neutralized with the NaCl ions added at a concentration of 0.15 mg l−1. The solvated system (protein, water and ions) contained ~276 000 atoms. All-atom MD simulations were performed with the CHARMM36m force field [24] using NAMD 2.13 [25]. The particle mesh Ewald (PME) [26] method was used to calculate the long-range ionic interactions. The covalent bonds involving hydrogen atoms were constrained by SHAKE [27]. A 10 000-step minimization and a 100 ps equilibration run were performed using a 1 fs time step. An NPT (constant pressure/temperature) production run was performed for 100 ns with a 2 fs time step at 300 K temperature and 1 atm pressure. A Nose–Hoover Langevin piston (with 50 fs period and 25 fs decay) was used for pressure control and the Langevin temperature coupling (with 1 ps−1 friction coefficient) was used for temperature control. Visualization and the PDB frame extraction from the trajectory were performed with VMD [28]. To prepare the receptor structures for virtual screening, 200 protein conformations were extracted from a 100 ns MD simulation. The PDB files were converted to pdbqt format using AutoDockTools 4.2 [29].
Preparation of drug files and virtual screening
The library of FDA-approved drugs was obtained from the e-Drugs3d web interface from Chemoinformatic Tools and Databases [30]. This database contains 1930 FDA-approved compounds in SDF format. Open Babel [31] was used to convert the SDF structures to pdbqt format with added polar hydrogen atoms. For virtual screening on the ensemble of SARS-CoV RdRP conformations, a docking configuration box was made by enclosing the RNA-binding tunnel region containing the evolutionarily conserved targets. Since all PDB conformations were aligned, the same configuration box could be used for docking in batch. Separately, all 1930 drugs were screened against the SARS-CoV-2 structure of RdRP (PDB ID: 6M71 [10]) using rigid docking. To further improve the binding scores of the rigid docking, flexible screenings were performed on the top ten hits by making the side chains flexible for residues Arg553, Arg555, Lys621, Asp623 and Ser814 in the RdRP tunnel region. Similarly, another round of flexible screenings was performed against the SARS-CoV-2 structure of RdRP using the top 10 hits from the ensemble docking. Vina from AutoDockTools 4.2 [29] was used to perform docking and screening. Top hits were sorted and ranked based on their binding energy scores using custom scripts.
Results and discussion
Previously we identified potential antiviral targets in the key proteins in coronavirus such as MERS-CoV and SARS-CoV [7] that are also conserved in the novel coronavirus SARS-CoV-2. RdRP was found to have the greater amount of surface-accessible targets that were strategically located in the vicinity of the active site and RNA tunnel. RdRPs are established antiviral drug targets. To find drugs that could potentially inhibit RdRP in SARS-CoV-2, and consequently be used as a therapeutic for COVID-19, four different in silico screens of FDA-approved drugs were performed targeting the RNA tunnel.
Phylogenetic analysis
The phylogeny of RdRP closely resembles the previous phylogeny [7], with SARS-CoV-2 as a sister to SARS-CoV and bat coronavirus-48, a SARS-related coronavirus isolated from a bat in Bulgaria in 2008 [32] (Fig. 1). The placement of SARS-CoV-2 is in agreement with the coronavirus phylogeny used to name the novel virus [3]. The phylogeny is overall well supported, but the clade formed by SARS-CoV and bat coronavirus-48 has low bootstrap support (54 %). Based on branch lengths, SARS-CoV-2 is more similar to SARS-CoV than to bat coronavirus-48.
Fig. 1.

Phylogenetic tree of RdRP. The tree was inferred with PhyML using LG+G+I+F as the best model of evolution and 100 bootstraps, shown at the nodes. The tree was visualized and rooted at mid-point with FigTree.
Expanded analysis of the targets in coronaviral proteins with an emphasis on RdRP
We found that all target motifs identified previously from other coronaviruses [7] were conserved in the SARS-CoV-2 reference sequence. In the comparison of all SARS-CoV-2 sequences, one amino acid substitution was found in one motif each for RdRP, helicase and exonuclease (Table 1). The overall conservation of the SARS-CoV-2 proteins for protease (NSP5), replicase (NSP7), RdRP (NSP12), helicase (NSP13) and exonuclease (NSP14) is close to 100 % (Fig. S1, available in the online version of this article).
Table 1.
Antiviral targets
|
Protein |
Target |
Sequence motif* and SARS-CoV-2 numbering† |
PDB ID‡, range |
|---|---|---|---|
|
Protease |
NSP5 : 1 |
3406-GSCGS-3387 |
1UJ1 : 143–147 |
|
Protease |
NSP5 : 2 |
346-AWLYAA-3474 |
1UJ1 : 206–211 |
|
Replicase |
NSP7 : 1 |
3866-KCTSWLL-3873 |
6NUR_C: 7–14 |
|
Replicase |
NSP7 : 2 |
3875-VLQQL-3879 |
6NUR_C: 16–20 |
|
RdRP |
NSP12 : 1 |
4599-LDNQDLNG-4606 |
6NUR_A: 207–214 |
|
RdRP |
NSP12 : 2 |
4610-DFGDF-4614 |
6NUR_A: 218–222 |
|
RdRP |
NSP12 : 3 |
4891-DKSAG-4895 |
6NUR_A: 499–503 |
|
RdRP |
NSP12 : 4 |
4958-MTNRQ-4962 |
6NUR_A: 566–570 |
|
RdRP |
NSP12 : 5 |
5047-LANEC AQVL-5055 |
6NUR_A: 655–663 |
|
RdRP |
NSP12 : 6 |
5070-GGTSSGD-5076 |
6NUR_A: 678–684 |
|
RdRP |
NSP12 : 7 |
5223-YPDPSR-5228 |
6NUR_A: 831–836 |
|
RdRP |
NSP12 : 8 |
5241-KTDGT-5245 |
6NUR_A: 849–853 |
|
RdRP |
NSP12 : 9 |
5259-YPLTK-5263 |
6NUR_A: 867–871 |
|
Helicase |
NSP13 : 1 |
5334-SQTSLR-5339 |
6JYT: 10–15 |
|
Helicase |
NSP13 : 2 |
5685-NAL P E-5689 |
6JYT: 361–365 |
|
Helicase |
NSP13 : 3 |
5725-DPAQLP-5730 |
6JYT: 401–406 |
|
Helicase |
NSP13 : 4 |
5859-SSQGS-5863 |
6JYT: 535–539 |
|
Exonuclease |
NSP14 : 1 |
6192-AHVAS-6196 |
5C8S_B: 267–271 |
|
Exonuclease |
NSP14 : 2 |
6201-MTRCLA-6206 |
5C8S_B: 276–280 |
|
Exonuclease |
NSP14 : 3 |
6349-H A FHT-6353 |
5C8S_B: 424–428 |
|
Exonuclease |
NSP14 : 4 |
6402-CNLGG-6406 |
5C8S_B: 477–481 |
*Sequence motifs in the targets provided by Rahaman and Siltberg-Liberles [7].
†Residues shown in bold are surface accessible. Residues that are not 100 % conserved across the SARS-CoV-2 strains are underlined.
For a motif to be considered a drug target it must be surface-accessible. The number of residues that are surface-accessible for protease, replicase, RdRP, helicase and exonuclease is 4, 4, 34, 20 and 17, respectively (Table 1). RdRP was expected to have the most surface-accessible residues because it has nine different motifs, compared to two each for protease and replicase, and four each for helicase and exonuclease (Table 1). The targets for RdRP are clustered in and around the RNA tunnel (Fig. 2). Replicase (NSP7) acts as a cofactor for RdRP [9] and its targets are located at the interface between the two proteins (Fig. 2). The targets in helicase (NSP13) and exonuclease (NSP14) are surface accessible, whereas those in protease (NSP5) are mostly buried and appear poorly accessible to small molecules (Table 1). In summary, RdRP appears to be the ideal target with the highest number of surface-accessible and strategically placed drug-binding sites. Furthermore, RdRPs are emerging as antiviral targets for RNA viruses [33–35].
Fig. 2.

Sequence and structure comparison of SARS-CoV RdRP based on PDB ID 6NUR to SARS-CoV-2 RdRP (grey), shown as ribbon and as surface representation from the front (a), the back (b) and the front with its cofactors, NSP7 (orange) and NSP8 (two chains: blue, green) (c). Previously proposed target regions are colour coded (targets 1 to 9 are represented by yellow, green, blue, purple, red, orange, pink, teal and brown, respectively). The active site/tunnel (from here on referred to as the RNA tunnel: yellow circle) is open and ready to perform its function. The part of the RdRP sequence from SARS-CoV included in PDB ID 6NUR is 96 % identical to the corresponding sequence from SARS-CoV-2 according to blast. The differing residues are shown as ball and stick (black). Protein structure visualized by BIOVIA Discovery Studio Visualizer v. 16.1.
Sequence comparison of RdRP in SARS-CoV-2 to SARS-CoV (based on PDB ID: 6NUR) reveals that these are 96 % identical in amino acid sequence (Fig. S2) and structural analysis shows that the nonconserved amino acid residues are located away from the RNA tunnel that represents an essential element for its catalytic activity (Fig. 2). Importantly, the target regions, in the critical area around the RNA tunnel, are far from the nonconserved amino acids between SARS-CoV and SARS-CoV-2 (Fig. 2).
Virtual screening of RdRP for drug binding
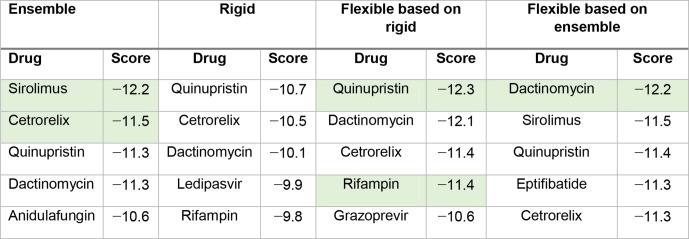
Ensembled-based molecular docking was performed for the 1930 FDA-approved drugs [30] against 200 conformations of SARS-CoV RdRP (based on PDB ID: 6NUR [9]). The top-scoring drugs generally had good binding scores on multiple protein conformations. A ranked list is prepared for distinct drugs with their best scores for the drug–protein conformations. Rigid molecular docking was performed for the 1930 FDA-approved drugs [30] against 1 conformation of SARS-CoV-2 RdRP (PDB ID: 6M71 [10]). Flexible docking, where the side chains of Lys551, Arg553, Arg555, Lys621, Asp623 and Ser814 in the RNA tunnel of RdRP in SARS-CoV-2 were kept flexible, was performed for the top 10 hits from the rigid docking and from the ensemble docking, respectively. Several drugs are identified in all the different docking rounds (Table 2).
Table 2.
Top-scoring drugs from the different docking rounds*
|
*The lowest (best) score for the five top-ranked drugs are shown in green.
The top-ranked candidate based on the virtual screens is the antibiotic quinupristin (Table 3). The in silico screens suggest that quinupristin binds across the RNA tunnel (Fig. 3), potentially blocking access for nucleoside triphosphates (NTPs) to enter the active site on one side and for RNA on the other. Without NTPs, RNA replication cannot occur. If the RdRP function can be inhibited, this may block viral replication. The second and third candidates, dactinomycin and sirolimus (rapamycin), both block the RNA tunnel, but have cytotoxic and immunosuppressive properties, respectively, preventing their use as antimicrobials [36, 37]. The fourth candidate, cetrorelix, inhibits the effects of gonadotropin-releasing hormone, is used during fertility treatments, and may have undesirable side effects for some populations. Cetrorelix also binds across the RNA tunnel. The fifth candidate, rifampin, is a broad-spectrum antibiotic that is often used to treat mycobacterial infections such as tuberculosis [37]. Our in silico screen places rifampin on the side of the RNA tunnel, in the vicinity of the RNA product/template hybrid exit (Fig. 4).
Table 3.
Top five hits from virtual screening with FDA-approved drugs against RdRP
|
Final rank and Best score* |
Drug name |
DrugBank† description |
|---|---|---|
|
1 |
Quinupristin |
An antibiotic effective against Gram-positive bacteria; commonly used in combination with dalfopristin |
|
Score: −12.3 |
||
|
2 |
Dactinomycin |
An antibiotic that inhibits bacterial transcription; has cytotoxic properties; used for chemotherapy |
|
Score: −12.2 |
||
|
3 |
Sirolimus |
A macrolide from Streptomyces hygroscopicus with immunosuppressive, antifungal and antineoplastic properties |
|
Score: −12.2 |
||
|
4 |
Cetrorelix |
A man-made hormone that blocks the effects of gonadotropin-releasing hormone |
|
Score: −11.5 |
||
|
5 |
Rifampin |
A broad-spectrum antibiotic known to inhibit DNA-dependent RNA polymerase activity by forming a stable complex with the enzyme |
|
Score: −11.4 |
Fig. 3.

Surface representations of RdRP with quinupristin. The top-ranked RdRP SARS-CoV (grey) conformation from the ensemble docking (PDB ID: 6NUR) is shown with (right) and without (left) quinupristin (red) from the front (a) and the back (b). The top-ranked RdRP SARS-CoV-2 (gold) from the flexible docking round (PDB ID: 6M71) with (right) and without (left) quinupristin (red) from the front (c). Quinupristin is buried in the RNA tunnel, blocking access for NTP and RNA. Previously proposed target regions are colour coded (targets 1 to 9 are represented by yellow, green, blue, purple, red (buried, not shown), orange, pink, teal and brown, respectively). Protein structure and drug complexes visualized by BIOVIA Discovery Studio Visualizer v. 16.1.
Fig. 4.

Surface representations of RdRP with rifampin. The top-ranked RdRP SARS-CoV-2 (grey) conformation from the flexible docking (PDB ID: 6M71) is shown with (right) and without (left) rifampin (red) from the front (a) and the back (b). Previously proposed target regions are colour coded (targets 1 to 9 are represented by yellow, green, blue, purple, red (buried, not shown), orange, pink, teal and brown, respectively). Protein structure and drug complexes visualized by BIOVIA Discovery Studio Visualizer v. 16.1.
Our five best-ranked candidates include two antibiotics that directly target RNA-binding and ribosomal regions in bacterial proteins such as the DNA-dependent RNA polymerase (rifampin) [38] or riboproteins such as the 50S unit of the ribosome (quinupristin) (Fig. 5) [39]. While the RNA polymerase and the ribosome are two diverse biomolecules, two common denominators are RNA and functionality that depend on a tunnel, two traits shared with the coronavirus RdRP. Rifampin and quinupristin prevent bacterial gene expression and protein translation, respectively. These antibiotics were not designed to act on the human host. Rifampin’s mechanism of action is to inhibit bacterial DNA-dependent RNA polymerase by binding to the side of the DNA/RNA channel that leads to the active site [38]. Rifampin binds away from the active site of the bacterial RNA polymerase in an area that is not conserved in eukaryotes [40]. Rifampin has good bioavailability and low toxicity, but is frequently used in combination with other antimicrobials to avoid antibiotic resistance [41]. Quinupristin binds to the prokaryotic 50S subunit of the ribosome, which is smaller than the eukaryotic 60S subunit and only partially homologous [42]. Quinupristin, together with dalfopristin, is part of the two-antibiotic mixture Synercid that has proven to be as safe in adults ≥65 years of age as in younger adults [43], which is important because COVID-19 has posed a particularly high risk of mortality to the elderly. Quinupristin and dalfopristin are streptogramin antibiotics known to act in concert to inhibit bacterial protein synthesis. Quinupristin and dalfopristin have been found to bind in, and block, the ribosomal exit tunnel in the 50S bacterial ribosome [39].
Fig. 5.

Quinupristin and rifampin bound to their intended drug targets based on co-crystalized X-ray structures. Quinupristin (green) has been suggested to inhibit the bacterial 50S ribosomal subunit by blocking the exit channel (a) (PDB ID: 1sm1 [39]). The bacterial 50S is a ribozyme and consists of protein (blue) and RNA (orange). Rifampin (blue) binds to one side of the channel leading up to the active site of the bacterial RNA polymerase (pink), blocking the path of elongation (b) (PDB ID: 1ynn [52]). Protein structure and drug complexes visualized by BIOVIA Discovery Studio Visualizer v. 16.1.
Currently, Synercid is used against methicillin-resistant forms of Staphylococcus aureus (MRSA) and other severe bacterial infections [44]. As an antibiotic, the synergistic effect of quinupristin and dalfopristin is needed for it to be effective. Combinations of Synercid with rifampin have been found to have an even stronger synergistic effect against MRSA and macrolide–lincosamide–streptogramin B-resistant (MLSB) isolates [45]. It is possible that a combination of quinupristin (or Synercid) and rifampin are more effective together as an antiviral against COVID-19 because, based on the in silico results, they may bind to different regions of the RNA tunnel in the coronavirus RdRP (Figs 3 and 5). It must be noted that we do not know if quinupristin or rifampin will bind to RdRP in vitro or in vivo, and the efficacy of either one or both in combination is yet to be demonstrated.
Conclusion
Molecular docking predictions have become increasingly useful, not only due to the accuracy in energy functions as well as advancement in computational speed, but also because of recent developments in cryo-EM technology. The structure of SARS-CoV RdRP that we used was the first coronavirus RdRP structure and it was released last year [9]. Currently, we have two similar RdRP structures, one for SARS-CoV and one for SARS-CoV-2 [10]. Without structural representatives, molecular docking is not possible. Molecular docking is frequently used for in silico screens of many drugs against a protein target as part of the initial steps of drug discovery [46]. Drug repurposing is a subfield of drug discovery that is focused on the identification of known drugs that can potentially bind to new targets. Drug repurposing often means lower risk of failure and a shorter preclinical phase [47]. Virtual, in silico, predictions are increasingly being used to guide the initial selections of compounds for in vitro analysis. Given the limited number of compounds in the database, it is possible that none of the drugs in the database will bind to our target in vivo. However, if we are to look for a best-binding compound in the database of FDA-approved drugs, these predictions are reasonably good. We used three types of docking (ensemble, rigid and flexible) to perform in silico screens. Ensemble docking better describes the nature of a protein’s structure by considering a conformational ensemble, but it is computationally expensive [48]. Rigid docking considers only one static conformation of the protein, but it is computationally attractive. Flexible docking allows for some residues explore their conformational space during the docking and can be used to improve binding scores. Notably, all three types returned quinupristin as a top-ranked candidate.
The in silico predicted drug candidates presented here can be considered for in vitro and in vivo tests and we hope that further exploration of such compounds and their analogues will allow researchers to quickly identify much-needed antiviral therapeutics for COVID-19. It is important to note that other coronaviruses that are yet unknown to humans could still pose future threats [46]. Under a different scenario, the potential broadly neutralizing antiviral target regions (Table 1) can be further pursued by a wider range of drug-like compounds in other libraries for future long-term drug development purposes. By utilizing data (genomic, structural, medical, etc.), viral precision medicine can be utilized to avoid playing catch-up with the next emerging coronavirus.
Given the urgency of the COVID-19 pandemic, we focused this screen on the library of already clinically used FDA-approved drugs aiming for drug repurposing. We chose to focus on RdRP because it is a known antiviral target for other RNA viruses such as hepatitis C virus [35]. Since this study began, it has been shown that MERS-CoV replication can be controlled by remdesivir in rhesus monkeys [5]. Remdesivir is an RdRP inhibitor that was developed for Ebola [49]. Remdesivir was not shown to be effective against Ebola, but it is currently included in the SOLIDARITY clinical trial for coronavirus treatments that the World Health Organization (WHO) recently announced [50]. If quinupristin, and perhaps rifampin, are indeed able to inhibit RdRP, these are RdRP inhibitors that could be evaluated rapidly as therapeutics for COVID-19. Lastly, a recent study found that 50 % of hospitalized COVID-19 patients who did not survive had secondary infections [51]. Perhaps these antibiotic candidates will have benefits that go beyond the COVID-19 infection itself.
Supplementary Data
Funding information
This work received no specific grant from any funding agency.
Acknowledgements
We would like to thank Dr Mark Gomelsky, Dr Yuk-Ching Tse-Dinh, Dr Tim Collins and Dr Laura Serbus for helpful discussions and feedback and Janelle Nunez-Castilla for proofreading. We would also like to thank the Instructional and Research Computing Center (IRCC) at Florida International University for providing the high-performance computing resources for the molecular simulations and virtual screening reported in this paper.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: FDA, US Food and Drug Administration; MD, molecular dynamics; MLSB, macrolide–lincosamide–streptogramin B-resistant; MRSA, methicillin-resistantforms of Staphylococcus aureus; MSA, multiple sequence alignment; NSP, non structural protein; NTP, nucleoside triphosphate; PDB, Protein Data Bank; PME, particle mesh Ewald; RdRP, RNA dependent RNA Polymerase; WHO, World Health Organization.
Two supplementary figures are available with the online version of this article.
References
- 1.Salata C, Calistri A, Parolin C, Palù G. Coronaviruses: a paradigm of new emerging zoonotic diseases. Pathog Dis. 2019;77 doi: 10.1093/femspd/ftaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. The Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020:1–9. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Wit E, van Doremalen N, Falzarano D, Munster VJ. Sars and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Wit E, Feldmann F, Cronin J, Jordan R, Okumura A, et al. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci U S A. 2020;117:6771–6776. doi: 10.1073/pnas.1922083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahaman J, Siltberg-Liberles J. Avoiding regions symptomatic of conformational and functional flexibility to identify antiviral targets in current and future coronaviruses. Genome Biol Evol. 2016;8:3471–3484. doi: 10.1093/gbe/evw246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fehr AR, Perlman S. Coronaviruses: Methods and Protocols. New York: Springer; 2015. Coronaviruses: An overview of their replication and pathogenesis; pp. 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirchdoerfer RN, Ward AB. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun. 2019;10 doi: 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Y, Yan L, Huang Y, Liu F, Zhao Y, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020:eabb7498. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stein GE. Safety of newer parenteral antibiotics. Clin Infect Dis. 2005;41:S293–S302. doi: 10.1086/431671. [DOI] [PubMed] [Google Scholar]
- 12.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 14.Lefort V, Longueville J-E, Gascuel O. Sms: smart model selection in PhyML. Mol Biol Evol. 2017;34:2422–2424. doi: 10.1093/molbev/msx149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edgar RC. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madeira F, Park YM, Lee J, Buso N, Gur T, et al. The EMBL-EBI search and sequence analysis tools Apis in 2019. Nucleic Acids Res. 2019;47:W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeLano WL. The PyMOL molecular graphics system, Schrödinger, LLC
- 18.Vertrees J. FindSurfaceResidues - PyMOLWiki. https://pymolwiki.org/index.php/FindSurfaceResidues (accessed 19 March 2020)
- 19.Yang H, Yang M, Ding Y, Liu Y, Lou Z, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci U S A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia Z, Yan L, Ren Z, Wu L, Wang J, et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47:6538–6550. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Y, Wu L, Shaw N, Gao Y, Wang J, et al. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc Natl Acad Sci U S A. 2015;112:9436–9441. doi: 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, et al. CHARMM-GUI input generator for NAMD, GROMACS, amber, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput. 2016;12:405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandhaus S, Chapagain PP, Tse-Dinh Y-C. Discovery of novel bacterial topoisomerase I inhibitors by use of in silico docking and in vitro assays. Sci Rep. 2018;8:1–9. doi: 10.1038/s41598-018-19944-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods. 2016 doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang W, Hardy DJ, Phillips JC, MacKerell AD, Schulten K, et al. High-Performance scalable molecular dynamics simulations of a polarizable force field based on classical drude oscillators in NAMD. J Phys Chem Lett. 2011;2:87–92. doi: 10.1021/jz101461d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darden T, Perera L, Li L, Pedersen L, Lee P. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7:R55–R60. doi: 10.1016/S0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- 27.Elber R, Ruymgaart AP, Hess B. Shake parallelization. Eur Phys J Spec Top. 2011;200:211–223. doi: 10.1140/epjst/e2011-01525-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 29.Trott O, Olson AJ. Autodock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and Multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pihan E, Colliandre L, Guichou J-F, Douguet D. e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinformatics. 2012;28:1540–1541. doi: 10.1093/bioinformatics/bts186. [DOI] [PubMed] [Google Scholar]
- 31.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, et al. Open Babel: an open chemical toolbox. J Cheminform. 2011;3 doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drexler JF, Gloza-Rausch F, Glende Jörg, Corman VM, Muth D, et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010;84:11336–11349. doi: 10.1128/JVI.00650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asthana S, Shukla S, Vargiu AV, Ceccarelli M, Ruggerone P, et al. Different molecular mechanisms of inhibition of bovine viral diarrhea virus and hepatitis C virus RNA-dependent RNA polymerases by a novel benzimidazole. Biochemistry. 2013;52:3752–3764. doi: 10.1021/bi400107h. [DOI] [PubMed] [Google Scholar]
- 34.Velkov T, Carbone V, Akter J, Sivanesan S, Li J, et al. The RNA-Dependent-RNA polymerase, an emerging antiviral drug target for the Hendra virus. Curr Drug Targets. 2014;15:103–113. doi: 10.2174/1389450114888131204163210. [DOI] [PubMed] [Google Scholar]
- 35.Bhatia HK, Singh H, Grewal N, Natt NK. Sofosbuvir: a novel treatment option for chronic hepatitis C infection. J Pharmacol Pharmacother. 2014;5:278–284. doi: 10.4103/0976-500X.142464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, et al. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104:901–912. doi: 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 39.Harms JM, Schlünzen F, Fucini P, Bartels H, Yonath A. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2004;2:4–10. doi: 10.1186/1741-7007-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma C, Yang X, Lewis PJ. Bacterial transcription as a target for antibacterial drug development. Microbiol Mol Biol Rev. 2016;80:139–160. doi: 10.1128/MMBR.00055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee C-Y, Huang C-H, Lu P-L, Ko W-C, Chen Y-H, et al. Role of rifampin for the treatment of bacterial infections other than mycobacteriosis. J Infect. 2017;75:395–408. doi: 10.1016/j.jinf.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 42.Graifer D, Karpova G. Roles of ribosomal proteins in the functioning of translational machinery of eukaryotes. Biochimie. 2015;109:1–17. doi: 10.1016/j.biochi.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 43.Pfizer SYNERCID- quinupristin and dalfopristin injection, powder, lyophilized, for solution. http://labeling.pfizer.com/ShowLabeling.aspx?id=712 (accessed 25 March 2020)
- 44.Holmes NE, Tong SYC, Davis JS, van Hal SJ, Hal S. Treatment of methicillin-resistant Staphylococcus aureus: vancomycin and beyond. Semin Respir Crit Care Med. 2015;36:017–030. doi: 10.1055/s-0034-1397040. [DOI] [PubMed] [Google Scholar]
- 45.Brown J, Freeman BB. Combining Quinupristin/Dalfopristin with other agents for resistant infections. Ann Pharmacother. 2004;38:677–685. doi: 10.1345/aph.1D323. [DOI] [PubMed] [Google Scholar]
- 46.Cheng VCC, Lau SKP, Woo PCY, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20:660–694. doi: 10.1128/CMR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pushpakom S, Iorio F, Eyers PA, Jane Escott K, Hopper S, et al. Drug repurposing: progress, challenges and recommendations. Nat Publ Gr. 2018 doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 48.Maximova T, Moffatt R, Ma B, Nussinov R, Shehu A. Principles and overview of sampling methods for modeling macromolecular structure and dynamics. PLoS Comput Biol. 2016;12:e1004619. doi: 10.1371/journal.pcbi.1004619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tchesnokov E, Feng J, Porter D, Götte M. Mechanism of inhibition of ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses. 11:326. doi: 10.3390/v11040326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kupferschmidt K. Who launches global megatrial of the four most promising coronavirus treatments. Science. 2020 doi: 10.1126/science.abb8497. [DOI] [Google Scholar]
- 51.Zhou F, Yu T, Du R, Fan G, Liu Y, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Campbell EA, Pavlova O, Zenkin N, Leon F, Irschik H, et al. Structural, functional, and genetic analysis of sorangicin inhibition of bacterial RNA polymerase. Embo J. 2005;24:674–682. doi: 10.1038/sj.emboj.7600499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.